Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2020; 16(16):3133-3148. doi:10.7150/ijbs.49520 This issue Cite
Research Paper
Regulation of energy metabolism by combination therapy attenuates cardiac metabolic remodeling in heart failure
Yuting Huang1,2*, Kai Zhang1,3*, Miaomiao Jiang2, Jingyu Ni1,2, Jingrui Chen1,2, Lan Li1,2, Jie Deng1,2, Yan Zhu2, Jingyuan Mao1,3, Xiumei Gao2 ![]() , Guanwei Fan1,2,3
, Guanwei Fan1,2,3 ![]()
1. First Teaching Hospital of Tianjin University of Traditional Chinese Medicine, Tianjin, People's Republic of China.
2. State Key Laboratory of Modern Chinese Medicine, Tianjin University of Traditional Chinese Medicine, Tianjin, People's Republic of China.
3. Tianjin Key Laboratory of Translational Research of TCM Prescription and Syndrome, Tianjin, People's Republic of China.
*These authors contributed equally to this work.
Received 2020-6-15; Accepted 2020-9-26; Published 2020-10-16
Abstract

Cardiac metabolic remodeling is recognized as an important hallmark of heart failure (HF), while strategies that target energy metabolism have therapeutic potential in treating HF. Shen-Fu formula (S-F) is a standardized herbal preparation frequently used in clinical practice and is a promising combinatorial therapy for HF-related metabolic remodeling. Herein, we performed an untargeted multi-omics analysis using transcriptomics, proteomics, and metabolomics on HF mice induced by transverse aortic constriction (TAC). Integrated and pathway-driven analyses were used to reveal the therapeutic targets associated with S-F treatment. The cardioprotective effect and potential mechanism of S-F were verified by the results from echocardiography, hemodynamics, histopathology, and biochemical assays. As a result, S-F significantly alleviated myocardial fibrosis and hypertrophy, thus reducing the loss of heart function during adverse cardiac remodeling in TAC mice. Integrated omics analysis showed that S-F synergistically mediated the metabolic flexibility of fatty acids and glucose in cardiac energy metabolism. These effects of S-F were confirmed by the activation of AMP-activated protein kinase (AMPK) and its downstream targets in the failing heart. Collectively, our results demonstrated that S-F suppressed cardiac metabolic remodeling through activating AMPK-related pathways via energy-dependent mechanisms.
Keywords: cardiovascular systems, energy metabolism, biomarker, multi-omics, combination therapy
Introduction
Heart failure (HF) is a shared chronic stage in several cardiovascular diseases. In China, because of the aging population and the increase in cardiovascular morbidity, the prevalence of HF is rapidly rising and the disease burden remains a challenge [1]. According to the 2017 ACC/AHA/HFSA Focused Update Guidelines for the Management of HF, the commonly used treatment for HF includes Class I A recommended ACEI, β-blockers, and aldosterone receptor antagonists [2]. However, these drugs do not significantly improve the five-year survival rate of HF patients and may show a ceiling effect. Therefore, there is a need to discover new therapeutic targets and strategies in HF research.
Energy deficiency and myocardial energy metabolism dysfunction are the main signs of HF [3]. As early as 2004, Van Bilsen proposed the concept of cardiac metabolic remodeling [4]. It occurs mainly due to the switch in cardiac substrates from fatty acids to glucose, which eventually leads to a deficiency in high-energy phosphate and a decrease in energy production. Subsequent studies have shown that changes in cardiac energy metabolism in HF mainly involve energy-starvation, substrate utilization, mitochondrial dysfunction, and insulin resistance [5-8]. A direct correlation has been suggested between energy deficit and overall disease severity in HF. Therefore, the modulation of cardiac energy metabolism is an effective way to treat HF. However, since HF is multifactorial, it is difficult to alter the cardiac metabolic network imbalance even with the use of combined single-target drugs. Therefore, combination therapies containing multiple drugs are a new promising approach in the treatment of HF.
Shen-Fu formula (S-F) is a patented herbal preparation consisting of extracts from Panax Ginseng and Radix Aconitum carmichaelii [9]. Previous studies have defined the composition of S-F [10, 11]. Over the past 30 years, S-F has been widely used in clinical practice in preventing and treating HF. The active ingredients of S-F are ginsenosides and aconitum alkaloids, including ginsenoside R1 (1.15 mg/g), Re (7.68 mg/g), Rg1 (3.28 mg/g), Rb1 (11.39 mg/g), Rb2 (3.64 mg/g), Rb3 (0.57 mg/g), Rd (2.11 mg/g), F2 (1.45 mg/g), Rg3 (0.24 mg/g), as well as benzoylaconine (0.17 mg/g), benzoylhypaconine (0.72 mg/g), and benzoylmesaconine (1.03 mg/g). This herbal preparation has been shown to exhibit a range of pharmacological activities, including potentiating myocardial contractility [12], improving cardiac ejection efficiency [13], promoting antioxidant activities [14], and inhibiting inflammatory response [15]. A previous Meta-analysis indicated that S-F combined with routine treatment could significantly improve heart function in HF patients. Besides, S-F showed better effect on clinical effect rate, mortality, heart rate, NT-proBNP and 6-minute walk distance [16]. Research using animal models of HF reveals that both ginsenosides and aconitum alkaloids can regulate cardiac energy metabolism dysfunction in ischemic myocardium [17, 18]. Our previous study showed that S-F treatment significantly improved cardiac function and alleviated ventricular remodeling in HF mice induced by transverse aortic constriction (TAC). However, the molecular mechanisms of S-F in HF treatment are not well understood.
In this study, multi-omics profiles were integrated with multidimensional data including physiological heart function, hemodynamic assessment, histopathological examination, and mitochondrial function to elucidate the mechanism of S-F on energy metabolism in TAC-induced HF mice. This study established that S-F counteracted cardiac metabolic remodeling by regulating fatty acid and glucose metabolism, and this presents new insights into the energy-dependent mechanism of S-F in the treatment of HF.
Materials and Methods
Animals and treatment
Adult male C57 mice (22-24 g, 8 weeks) were purchased from Beijing Vital River Laboratory Animal Technology Co., Ltd. (Certificate number: SCXK [Jing] 2016-0001). Mice were kept in a 12 h dark/light cycle at a temperature of 21 ± 2 °C and 30~70% humidity. They were fed with standard laboratory diet and water. Before surgery, mice were anesthetized using tribromoethanol (1.5%, 1 ml/100 g, i.p.). The mice were subjected to pressure overload through TAC as previously described [19]. Briefly, the aortic arch was exposed through an intercostal incision. A 5-0 nylon suture was placed around the aortic arch between the brachiocephalic trunk and the left common carotid artery. A pre-sterilized, blunted 27-gauge needle was then placed within the nylon knot, adjacent to the aorta. After the knot was tightened, the needle was removed from the knot, leaving the aorta permanently constricted. Then the sternum and the skin incision were sutured back in layers with a 5-0 suture. Sham-operated mice received similar surgical procedures but without aortic coarctation. Four weeks after TAC, S-F was administered intragastrically at doses that were equivalent to human doses of 2.5 g, 5 g, and 10 g crude herbs/day, respectively, for 28 days. For the positive control, valsartan was administered by gavage at 10.4 mg/kg. Animal trials were performed in three batches of mice. The total number of animals in each group: sham group (n = 40), TAC group (n = 52), valsartan group (n = 46), 2.5g S-F group (n = 29), 5g S-F group (n = 31), and 10g S-F group (n = 51) (See Supplementary methods for further details). The animal study protocol was approved by the Local Institutional Animal Care and Use Committee (TCM-LAEC20170049) and conformed to the guidelines of the Care and Use of Laboratory Animals published by the Ministry of Science and Technology of China. All experiments complied with the Institutional Animal Care and Use Committee guidelines and were approved by the Animal Care Committee of Tianjin University of Chinese Medicine.
Echocardiography and hemodynamic measurements
Transthoracic echocardiography was used to detect and record the cardiac function of mice before, and 4 weeks after, S-F was administered. From the long-axis parasternal view, the cardiac cycle was recorded by M-mode ultrasound. Besides, the left ventricle ejection fraction (EF), fractional shortening (FS), interventricular septum thickness at systole (IVS; s) and diastole (IVS; d), left ventricular volume at systole (LV Vol; s) and diastole (LV Vol; d), left ventricular internal systolic (LVID;s) and diastolic (LVID; d) diameters, and left ventricular (LV) mass were measured and analyzed. Hemodynamic characteristics were tested using a 1.4-Fr micro-manometer-tipped catheter (Millar Instruments, Houston, TX, USA) combined with an echocardiography Vevo 2100 Imaging System (VisualSonics, Toronto, ON, Canada).
Histological and immunofluorescent assessments
Heart samples were excised, washed, and fixed with 4% phosphate-buffered formalin for 48 h. Tissues embedded in wax were cut into 5 mm slices. The sections were stained with hematoxylin and eosin (HE), Masson, and wheat germ agglutinin (WGA) for pathological morphological analysis. The nucleus was localized using 4′,6-diamidino-2-phenylindole (DAPI), and fluorescence images were captured using an inverted fluorescence microscope (ECLIPSE, Nikon, Tokyo, Japan).
Mitochondrial oxygen consumption rate and ultrastructure
Myocardial mitochondria were extracted using a specialized mitochondrial extraction kit (Solarbio Life Sciences, Beijing, China). The isolated mitochondria were added into the seahorse 24-well plate at the concentration of 40 μg/50 μl ~ 60 μg/50 μl, and centrifuged at 2000 g for 20 min. Oligomycin, carbonyl cyanide 4-trifluoromethoxy phenylhydrazone (FCCP), rotenone/antimycin A were added to the well-incubated probe plate. The mitochondrial respiratory oxygen consumption rate (OCR) was measured at 37 °C using XFe24 seahorse cell energy metabolism analyzer (Seahorse Bioscience, Billerica, MA, USA).
The myocardial mitochondria ultrastructure was observed under a transmission electron microscope (TEM) (H-7650, HITACHI, Ibaraki, Japan). The myocardial tissue used for TEM imaging was derived from the left ventricle of the fresh heart. Tissue samples were fixed in 1% OsO4 for 30 min, incubated in 4% uranyl acetate, dehydrated in graded ethanol, and embedded in epoxy resin. The sections were double-stained with 4% uranyl acetate and lead citrate.
Transcriptomics analysis
Total RNA from the mouse heart tissue was extracted using a Trizol reagent kit (Invitrogen, Carlsbad, CA, USA). For quality control, RNA integrity was checked on an Agilent 2100 BioAnalyzer (Agilent Technologies, Santa Clara, CA, USA). Oligo (dT) beads were used to isolate the poly mRNA from the total RNA. The enriched mRNA was fragmented and reverse transcribed into cDNA using random primers. The constructed library was sequenced using an Illumina HiSeqTM 2500 (Illumina, San Diego, CA, USA) by Gene Denovo Biotechnology Co. (Guangzhou, China). Expression of transcripts was quantified using RNA-Seq by expectation-maximization (RSEM) software. Transcripts with an absolute fold-change greater than 1.5 and false detection rate (FDR) below 0.05 were classified as differentially expressed. The differentially expressed genes (DEGs) were further subjected to KEGG pathways enrichment analysis. For details, see Supplementary methods.
Proteomics analysis
Heart tissues were transferred into a lysis buffer (2% SDS, 7M urea, 1 mg/ml protease inhibitor cocktail), vortexed, and allowed to lyse for 30 min on ice. The samples were homogenized, centrifuged, reduced with TCEP, and alkylated with iodoacetamide. Protein solutions were digested with trypsin and labeled with TMT tags according to the manufacturer's instructions (TMT10plex™ Isobaric Label Reagent Set, Thermo Fisher Scientific, Waltham, MA, USA). The peptide mixture was fractionated using high pH separation on an Ultimate 3000 UPLC system (Thermo Fisher Scientific, Waltham, MA, USA), and further analyzed on an Easy-nLC 1000 system connected to a Q Exactive Hybrid Quadrupole-Orbitrap system (Thermo Fisher Scientific, Waltham, MA, USA). Protein identification was performed using Proteome Discoverer 1.2 (Thermo Fisher Scientific, Waltham, MA, USA) and the Mascot search engine (Matrix Science, London, UK). Proteins with fold changes > 1.2 or < 0.83 and significance level p < 0.05 were considered to be differentially expressed. Differentially expressed proteins (DEPs) were further annotated against KEGG databases to determine their functions. For details, see Supplementary methods.
Metabolomics analysis
Plasma samples (100 μl) were prepared by adding 3 volumes of methanol and 20 μl of internal standard (2-Chloro-L-phenylalanine) to precipitate the proteins. After centrifugation at 13000 g for 15 min, 200 μl of the supernatant was transferred to LC-MS vials. Quality control (QC) samples were prepared by mixing an equal aliquot of the supernatants from each plasma sample. The samples were analyzed using a UHPLC system (1290, Agilent Technologies, Santa Clara, CA, USA) coupled to a Q Exactive Hybrid Quadrupole-Orbitrap system (Thermo Fisher Scientific, Waltham, MA, USA). Data pre-treatment procedures were performed using XCMS software operated in the R computing environment. The processed data files were imported into the SIMCA 14.1 software package (Umetrics, Umeå, Sweden) for multivariate data analysis. Metabolites selected as biomarkers were identified based on the variable importance of project (VIP) threshold of 1 and the two-tailed p-values calculated by student's t-test. Pathway enrichment analysis was performed using the MetaboAnalyst and DAVID toolkits. For details, see Supplementary methods.
Quantitative real-time PCR (qRT-PCR)
qRT-PCR was performed using a cDNA Reverse Kit and an SYBR Green PCR Master (Roche, Basle, Switzerland) according to the manufacturer's instruction. Semi-quantitative analysis of relative gene expression was performed on a CFX96TM Real-Time PCR Detection System (Bio-Rad, Redmond, WA, USA). GAPDH was used as the housekeeping gene to standardize the amount of mRNA expressed. Details of the 27 mRNAs detected in this study as well as the primer sequences are listed in Table S1.
Western blot analysis
Heart tissues were homogenized in cold lysis buffer (PMSF: RIPA = 1:100). For western-blotting, the extracted protein was separated on 4-20% SurePAGE® gradient gels (GenScript, Nanjing, China) and then transferred to polyvinylidene fluoride (PVDF) membranes (Thermo Fisher Scientific, Waltham, MA, USA). The membranes were blocked with 5% skim milk powder and incubated with the primary antibody overnight. Immunoblot band intensities were quantified using NIH ImageJ software. Detailed descriptions of the detected proteins can be found in Table S2.
18F-FDG PET cardiac glucose metabolism
F-18-fluorodeoxyglucose (18F-FDG) (150 uCi) was injected into the tail vein of mice. The 18F-FDG uptakes of the myocardium were expressed as the standardized uptake value (SUV). The mice underwent a 20-min image acquisition in the prone position 45 min after S-F treatment. 18F-FDG positron emission tomography (PET) coupled with a cardiac PET scanner (Inveon PET/CT, Siemens, Erlangen, Germany) was used to visualize cardiac glucose metabolism [20].
Results
S-F exerts a protective effect on myocardial contraction and diastolic function in TAC-induced heart failure mice
TAC mice showed significant systolic and diastolic dysfunction and significant decrease in EF and FS, following 8 weeks of modeling (Fig. 1B, C). Besides, there was a significant increase in IVS, LVID and LV Vol (Fig. S1). There were observable changes in the ventricular filling velocity of TAC mice evidenced by decreased mitral inflow E and A wave ratio (E/A) and prolonged isovolumic relaxation time (IVRT) (Fig. 1D, E). Compared with the TAC group, the EF, FS and E/A levels in the valsartan group and S-F groups were significantly increased, while IVRT, IVS, LVID, and LV Vol were significantly decreased.
S-F attenuates transverse aortic constriction (TAC) induced cardiac dysfunction. (A) Representative M-mode echocardiographic images of mice that underwent the sham operation, TAC only, or TAC followed by treatment with S-F or valsartan. (B-E) Echocardiography-derived indices of left ventricle ejection fraction (EF) (B), fractional shortening (FS) (C), mitral inflow E and A wave ratio (E/A) (D), and isovolumic relaxation time (IVRT) (E). Heart weight/body weight ratio (HW/BW) (F), heart weight/tibia length ratio (HW/TL) (G), lung weight/body weight ratio (LW/BW) (H), and left ventricular (LV) mass (I) were measured after 4 weeks of S-F treatment. (J-L) Hemodynamic characteristics assessed by measuring stroke volume (SV) (J), maximum rise/fall rate (dp/dt max) (K), and minimum rise/fall rate (dp/dt min) (L) of left ventricular. Data are shown as mean ± SD. *p < 0.05, **p < 0.01, compared with the sham group; #p < 0.05, ##p < 0.01, compared with the TAC group, evaluated by one-way ANOVA with Dunnett's post hoc test.
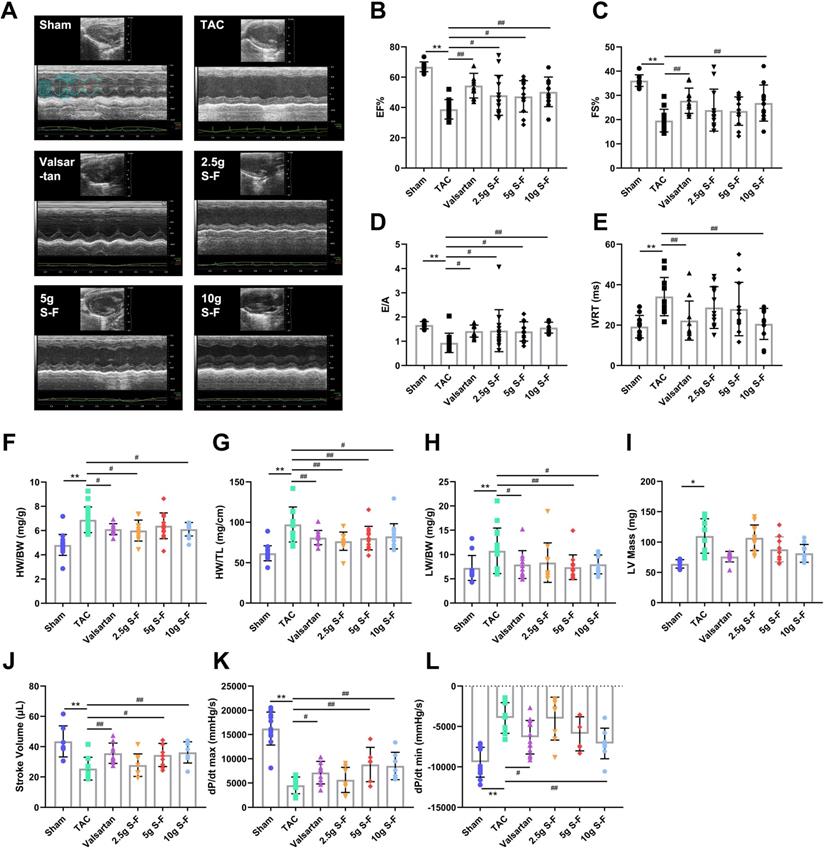
TAC mice showed a larger heart size compared to hearts of the sham mice. Besides, there was a significant increase in heart weight to body weight (HW/BW) ratio, heart weight to tibia length (HW/TL) ratio, lung weight to body weight (LW/BW) ratio, and LV mass (Fig. 1F-I). S-F and valsartan significantly reduced myocardial enlargement and decreased the values of HW/BW, HW/TL and LW/BW in TAC mice.
Besides, hemodynamic results revealed that S-F and valsartan treatment induced a significant increase in stroke volume (SV), maximum rise/fall rate (dp/dt max), and minimum rise/fall rate (dp/dt min) in TAC mice (Fig. 1J-L), with the effects of 10g S-F being the most significant. These results indicated that S-F significantly counteracted the systolic and diastolic dysfunction, cardiac hypertrophy and ventricular remodeling induced by pressure load.
S-F alleviates myocardial tissue necrosis, myocardial fibrosis, and cardiac hypertrophy
HE staining (Fig. 2A) revealed severe pathological alternations in the myocardium architecture of TAC mice, including myocardial nuclear aggregation, disordered cell arrangement, and diffuse edema compared with that of sham mice. After 4 weeks of treatment, the S-F and valsartan groups showed alleviated myocardial necrosis compared with the model group. As shown in Fig. 2B, the myocardial cells in TAC mice were disorderly arranged, the area of collagen fibers and the degree of myocardial fibrosis was significantly increased. Moreover, TAC mice showed increased staining of collagen I and III when compared to sham mice (Fig. 2C, D). S-F treatment reduced the collagen fiber area, and the myocardial cells were structurally intact and neatly arranged. WGA staining (Fig. 2E) showed that the myocardial cell gap widened, the cell membrane disappeared, and the area of myocardial cells increased significantly in TAC mice compared with sham mice. In contrast, S-F treatment narrowed the myocardial cell gap, the cell membrane was relatively intact, and the myocardial cell area was significantly reduced (Fig. 2F).
S-F alleviates myocardial tissue necrosis, myocardial fibrosis, and cardiac hypertrophy. (A, B) Representative histological images of HE staining results (A) and Masson staining (B) on heart sections from all the experimental groups. (C, D) Representative immunofluorescence images of myocardium sections stained for collagen I (C) and collagen III (D). (E) Wheat germ agglutinin (WGA) staining followed by (F) quantitative analysis of relative cell surface area. (G-I) Expression of myocardial extracellular matrix factors MMP-2 (G) MMP-9 (H) and ST2 (I) in plasma was determined by ELISA. Values shown are mean ± SD. *p < 0.05, **p < 0.01, compared with the sham group; #p < 0.05, ##p < 0.01, compared with the TAC group, analysis was performed using one-way ANOVA with Dunnett's post hoc test. MMP, matrix metalloproteinase; ST2, growth stimulation expressed gene 2 protein.
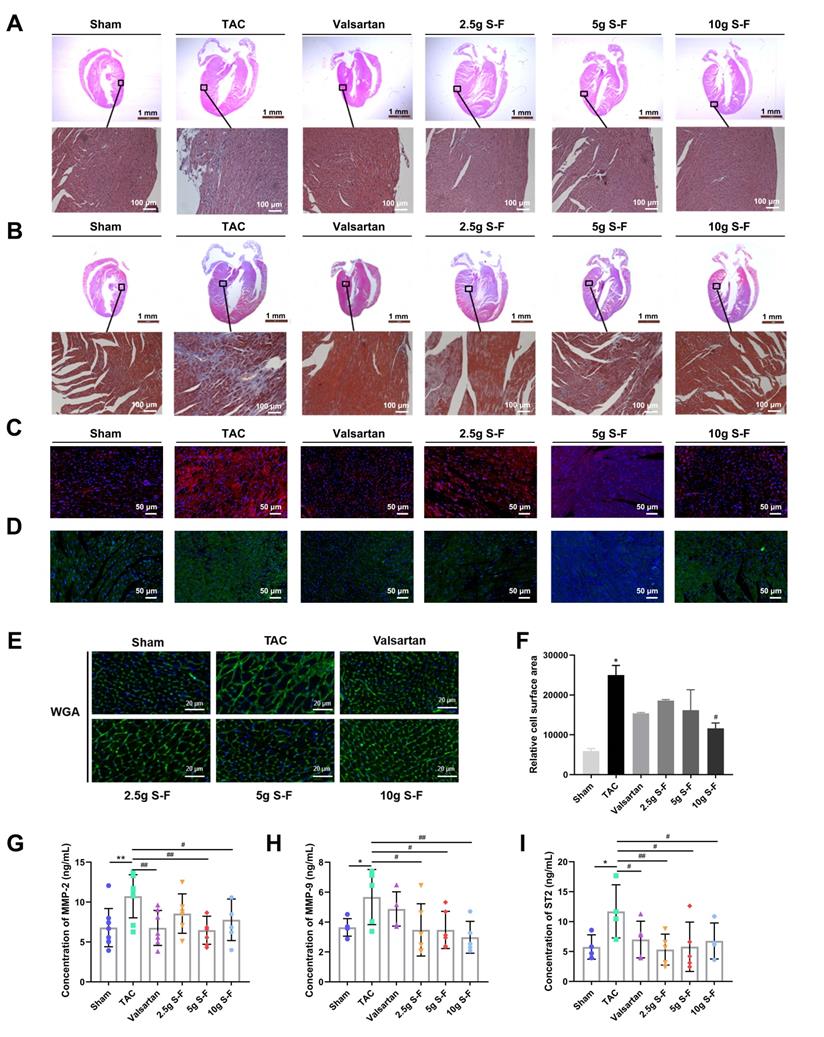
The plasma levels of myocardial extracellular matrix factors (MMP) and the expression of genes related to myocardial fibrosis and cardiac hypertrophy was determined. TAC mice showed increased levels of MMP-2, MMP-9, and ST2 in plasma (Fig. 2G-I), increased mRNA expression of atrial natriuretic peptide (ANP), brain natriuretic peptide (BNP), collagen I, fibronectin, β-myosin heavy chain (β-MHC) in myocardial tissue, and decreased expression of smooth muscle actin (SMA) and α-myosin heavy chain (α-MHC) (Fig. S2), when compared with sham mice. S-F showed a regulatory effect on the above abnormal indicators, suggesting that it decreases the degree of myocardial tissue necrosis, myocardial fibrosis, and cardiac hypertrophy, thereby improving cardiac pathology.
S-F improves the oxygen consumption rate (OCR) and enhances the structural and functional integrity of myocardial mitochondria
TAC mice showed a significant decline in basal, maximal and ATP-linked OCRs compared to sham mice (Fig. 3A). S-F treatment resulted in enhanced OCRs in basal (Fig. 3B), maximal (Fig. 3C) and ATP-linked respiration (Fig. 3D). To further investigate the effect of S-F on mitochondrial function, the protein and mRNA expression levels of five mitochondrial complexes were measured. The expression of complexes I, II, III, IV and V (Fig. 3E and Fig. S3) were significantly decreased in TAC mice, and these abnormities were significantly restored by S-F treatment. In comparison, valsartan only had a significant effect on the basal OCR and the levels of complex I and V.
S-F enhances mitochondrial oxygen consumption rate (OCR) and maintains mitochondrial structure. (A) OCR curves of myocardial mitochondria. Oligomycin, carbonyl cyanide 4-trifluoromethoxy phenylhydrazone (FCCP), rotenone and antimycin A were injected sequentially at the indicated time points into each well containing myocardial mitochondria. Mitochondrial function was assessed by the basal OCR (B), maximal OCR (C), and ATP-linked OCR (D). (E) Western blot analysis was performed to determine expression of mitochondrial complex I (NADH dehydrogenase ubiquinone flavoprotein 2, NDUFV2), complex II (succinate dehydrogenase subunit B, SDHB), complex III (ubiquinol-cytochrome C reductase binding protein, UQCRB), complex IV (cytochrome C oxidase subunit II, MT-CO2), and complex V (ATP synthase subunit delta, ATP5D). Values shown are mean ± SD. *p < 0.05, **p < 0.01, compared with the sham group; #p < 0.05, ##p < 0.01, compared with the TAC group, evaluated by one-way ANOVA with Dunnett's post hoc test. (F) Representative transmission electron microscopy images showing the mitochondrial network in myocardial tissue in all experimental groups.
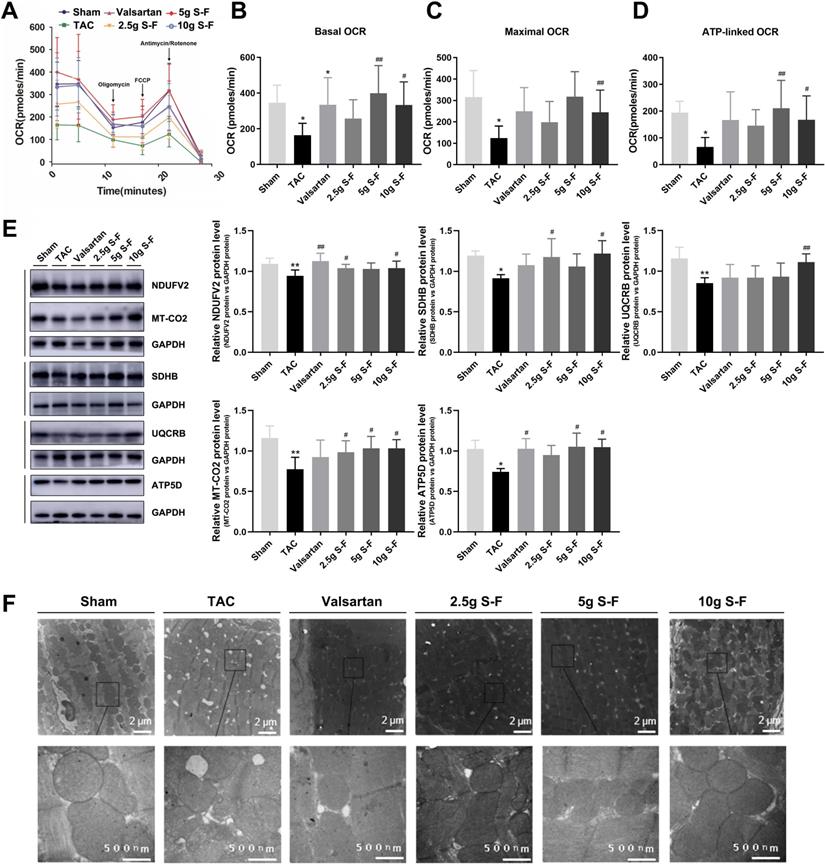
Ultrastructural aspects of the mitochondria in myocardium cells revealed structural disorders with vacuolar degeneration and edematous myocardial fibers in the TAC group (Fig. 3F). Upon treatment with 10g S-F, the mitochondria were neatly arranged, there was a reduction in the mitochondrial edema, and the mitochondria structure became relatively clear and intact. These results indicated that S-F played an anti-HF effect by improving the mitochondrial OCRs, regulating mitochondrial function and structure, and alleviating mitochondrial energy metabolism dysfunction.
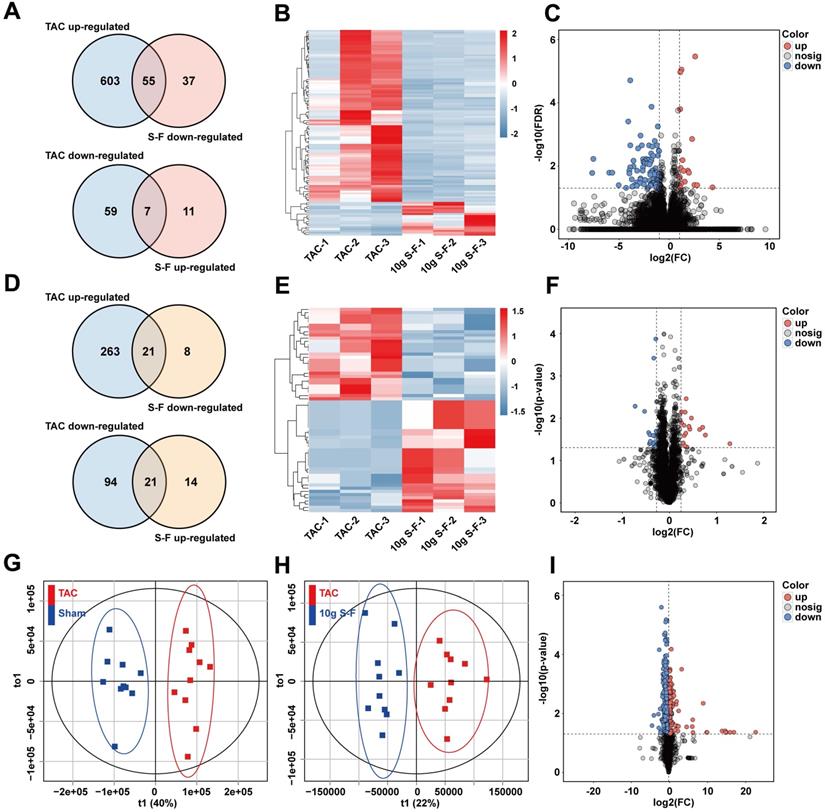
S-F mediates abnormalities in energy metabolic pathways at the transcript, protein, and metabolite levels in TAC mice
Untargeted multi-omics analysis using transcriptomics, proteomics, and metabolomics on the sham, TAC, and 10g S-F treated mice were performed to obtain a comprehensive view of the complex mechanisms of S-F. A total of 772 genes were identified as differentially expressed genes (DEGs) at the transcriptome level, out of which 724 were altered in TAC mice relative to sham mice, while 110 DEGs were restored through S-F treatment (Fig. 4A). Principal component analysis (PCA) of transcriptome data showed that samples from the TAC group were distinct from those in the sham and S-F group (Fig. S4A). Upon comparing the S-F group with the TAC model, 92 DEGs were found to be significantly down-regulated, while 18 DEGs were significantly up-regulated. These results were visualized in the hierarchically clustered heatmap (Fig. 4B) and volcano plot (Fig. 4C). At the proteome level, a total of 421 proteins were differentially expressed. The Venn diagram in Fig. 4D shows the number of differentially expressed proteins (DEPs) in the TAC group compared to the S-F group. Among the 64 proteins that were significantly affected by S-F, 29 were down-regulated and 35 were up-regulated. The corresponding heatmap and volcano plot are shown in Fig. 4E, F. Besides, the metabolic profiles of the plasma samples collected from the three groups are shown in the PCA scores plot (Fig. S4D, E). Accurate discrimination between sham and TAC groups were attained using orthogonal partial least squares discriminant analysis (OPLS-DA) (Fig. 4G, 2-component model, R2X=0.59, R2Y=0.93, Q2Y=0.80). Mice in the S-F group were distinct from those in the TAC group, evidenced by the robust OPLS-DA model (Fig. 4H, 2-component model, R2X=0.56, R2Y=0.89, Q2Y=0.50). A total of 721 metabolites were found to be differentially expressed metabolites (DEMs) in the positive and negative ion modes (Fig. S4F). A total of 336 DEMs were down-regulated and 117 DEMs were up-regulated after S-F treatment (Fig. 4I).
Signatures of S-F treatment at transcript, protein, and metabolite levels. (A) Venn-diagrams showing the number of differentially expressed genes (DEGs) that were up-regulated in the TAC group (compared with the sham group) and down-regulated by S-F (compared with the TAC group), and DEGs that were down-regulated in TAC mice and up-regulated by S-F. (B) Heatmap representation of hierarchical clustering of DEGs that were significantly reversed by S-F treatment. (C) Volcano plot showing the distribution of DEGs in the 10g S-F group compared to the TAC group. (D) Venn-diagrams showing the number of differentially expressed proteins (DEPs) in TAC mice compared to sham mice, and DEPs in S-F treated mice compared to TAC ones. (E) Heatmap of DEPs that were significantly reversed by S-F treatment. (F) Volcano plot of DEPs in the 10g S-F group compared to the TAC group. (G) Plot of OPLS-DA scores derived from plasma metabolic profiling of the TAC and sham groups (negative ion detection mode). (H) Plot of OPLS-DA scores of the 10g S-F group versus those of the TAC group (negative ion mode). (I) Volcano plot showing the distribution of differentially expressed metabolites in the 10g S-F group compared to the TAC group. The number of animals: transcriptomics analysis (n = 3 in each group), proteomics analysis (n = 3 in each group), metabolomics analysis (n = 10 in each group). OPLS-DA, orthogonal partial least squares discriminant analysis.
To evaluate the potential function of DEGs and DEPs, pathway enrichment analysis based on the KEGG database was performed. As shown in Fig. 5A, B, both DEGs and DEPs were significantly enriched in pathways related to energy metabolism, such as fatty acid metabolism and carbohydrate metabolism. Interestingly, the DEGs and DEPs involved in energy metabolic pathway were mainly distributed in the expression patterns of profiles 5 and 2 in short time-series expression miner (STEM) analysis (Fig. 5C, D). This suggested that S-F had a significant regulatory effect on energy metabolism in HF mice.
Network analysis of transcriptome and proteome. (A, B) KEGG pathway enrichment analysis of DEGs (A) and DEPs (B). Rich factor refers to the ratio of the number of DEGs (or DEPs) in the pathway entry to the total number of genes (or proteins) in the pathway entry. Pathways with p-value < 0.05 were considered significantly enriched. (C, D) Short time-series expression miner (STEM) clustering on DEGs (C) and DEPs (D). Each square represents expression trend of gene (or protein) in the sham, TAC and 10g S-F groups. The black polygonal line in the square indicates the overall expression profile of all DEG (or DEP) in the corresponding set. The profile ID and the number of DEGs and DEPs in each profile are shown on the top of the square. Colored profiles are those that are statistically significant (p < 0.05). (E) Combined analysis of DEGs and DEPs in TAC mice relative to sham mice. (F) Combined analysis of DEGs and DEPs in S-F treated mice relative to TAC ones. The omics datasets were merged based on the gene symbol. Directed p-values are defined as -log10 (p-value) times the direction of the effect. Red dots in the Q3 or Q7 quadrants indicate co-regulated entities that were consistently up-regulated or down-regulated. Blue dots in the Q2 and Q8 quadrants represent genes with significant changes in the transcriptome, while orange dots in the Q4 and Q6 quadrants represent significant genes in the proteome. (G) Interaction networks of 68 DEGs and DEPs involved in energy metabolism. Within networks, nodes are colored according to the number of connections. (H) Pathway enrichment analysis of the above DEGs and DEPs ranked by the -log10 (p-value). DEGs, differentially expressed genes; DEPs, differentially expressed proteins.
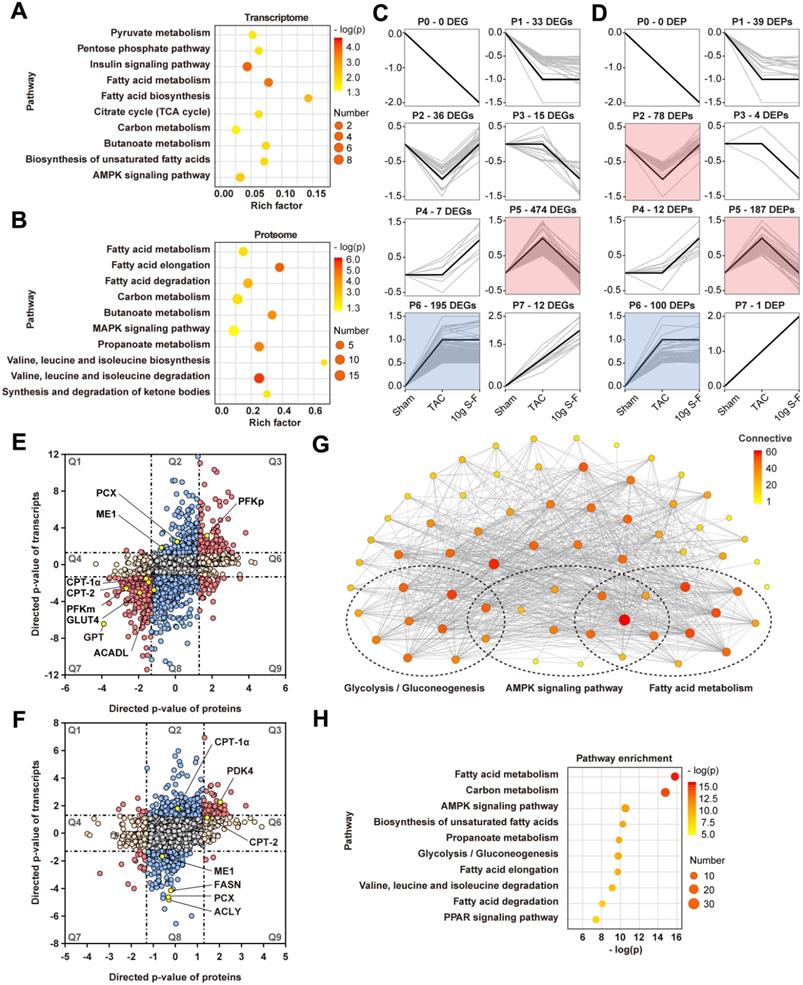
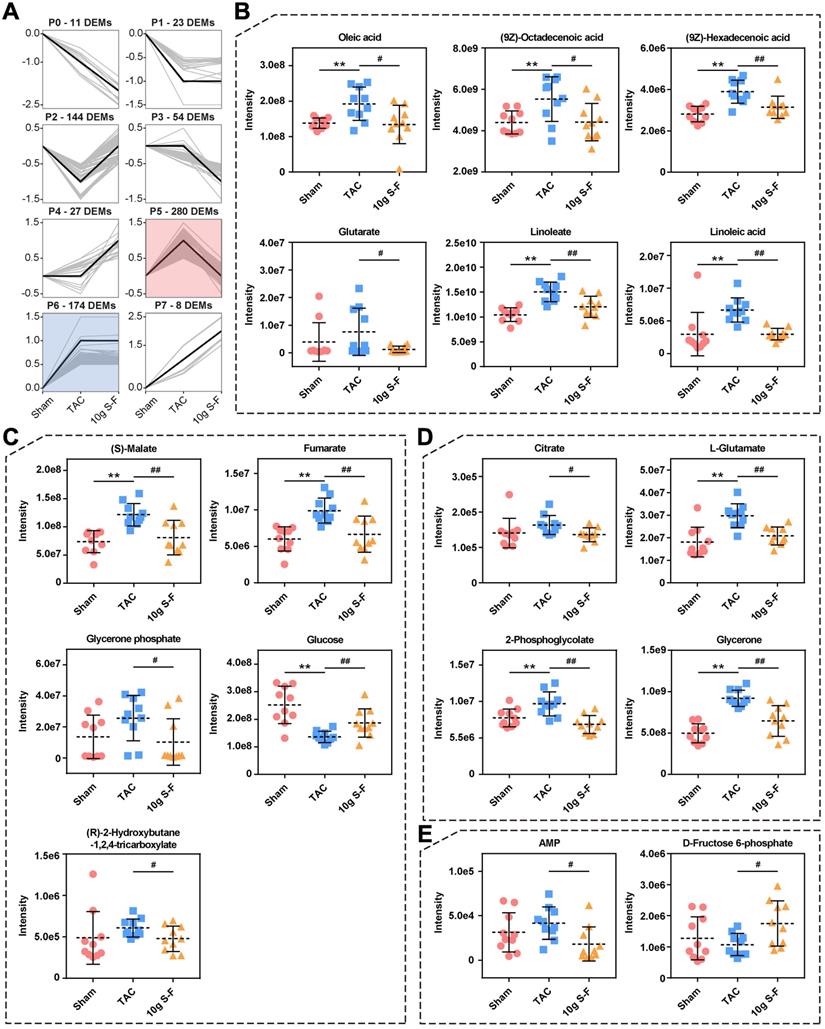
Furthermore, the transcriptome and proteome data were combined to identify genes that were altered in both omics layers. A total of 3418 genes were found to be co-regulated genes in TAC mice, out of which 96 were up-regulated (Fig. 5E, Q3) and 200 were down-regulated (Fig. 5E, Q7). After S-F treatment, a total of 63 genes differed significantly in protein abundance and transcript status (Fig. 5F). Significantly differentially expressed genes of energy metabolism in transcriptomics and proteomics included CPT-1α, CPT-2, ACADL, and FASN, which are involved in fatty acid metabolism; and ME1, PCX, PDK4, PFKm, and GLUT4, which are related to pyruvate and glucose metabolism. Moreover, an interaction network between 68 mRNAs and proteins involved in the energy metabolic pathway was established (Fig. 5G). They were found to be significantly enriched in fatty acid and glucose metabolism, as well as AMP-activated protein kinase (AMPK) and peroxisome proliferator-activated receptor (PPAR) signaling pathways (Fig. 5H). Meanwhile, metabolomics studies showed significant changes in the level of key metabolites related to energy metabolism, such as oleic acid, (9Z)-octadecenoic acid, (9Z)-hexadecenoic acid, glutarate, linoleate, and linoleic acid in the fatty acid metabolic pathways (Fig. 6B), (S)-malate, fumarate, glucose, and glycerone phosphate in the glucose metabolic pathways (Fig. 6C), as well as AMP and D-fructose 6-phosphate in the AMPK signaling pathway (Fig. 6E). Similarly, the significantly altered metabolites were mainly concentrated on profile 5 in STEM analysis (Fig. 6A), thus revealing the significant effect of S-F on energy metabolism at the downstream metabolic status. These results suggested that S-F caused synergistic effects on energy metabolism in HF mice at the transcript, protein, and metabolite levels. Fatty acid metabolism and glucose metabolism were identified as the most influenced metabolic pathways associated with S-F treatment.
Alterations in metabolome in response to S-F treatment. (A) STEM clustering on differentially expressed metabolites in the sham, TAC and 10g S-F groups. (B-E) Significantly altered metabolites in the energy metabolic pathways related to S-F treatment, including oleic acid, (9Z)-octadecenoic acid, (9Z)-hexadecenoic acid, glutarate, linoleate, and linoleic acid in the fatty acid metabolic pathways (B), (S)-malate, fumarate, glycerone phosphate, glucose, and (R)-2-hydroxybutane-1,2,4-tricarboxylate in the pyruvate and glucose metabolic pathways (C), citrate, L-glutamate, 2-phosphoglycolate, and glycerone in carbon metabolism (D), and AMP, D-fructose 6-phosphate in the AMPK signaling pathway (E). Values shown are mean ± SD. *p < 0.05, **p < 0.01, compared with the sham group; #p < 0.05, ##p < 0.01, compared with the TAC group.
Validation of the regulatory effect of S-F on glucose and fatty acid metabolism
The 18F-FDG PET method was used to compare the cardiac glucose uptake of mice in the sham group, model group and 10g S-F group (Fig. 7A). In the TAC group, the mean glucose uptake SUV significantly increased in the 8th week compared with that in the sham group (6.03 vs 0.87). However, S-F was found to significantly restore the abnormal glucose uptake in the failing heart 4 weeks after treatment (Fig. 7B).
Validation of the effect of S-F on glucose and fatty acid metabolism in TAC mice. (A) Differential F-18-fluorodeoxyglucose (18F-FDG) positron emission tomography (PET) uptake with PET/CT in cardiac muscle of mice. High signal density is colored in red, and low signal density is colored in blue and green. (B) Quantitative analysis of myocardial glucose uptake based on standardized uptake values (SUVs). Signals were measured from three biological replicates, and three technical repeats for each replicate. (C) qRT-PCR analysis of key genes that regulate glucose metabolism in heart tissues, including pyruvate dehydrogenase kinase 4 (PDK4), pyruvate/alanine aminotransferases (GPT), glucose transporter 4 (GLUT4), and phosphofructokinase muscle (PFKm). (D) qRT-PCR analysis of mRNA levels of genes that regulate fatty acid oxidation, including carnitine palmitoyl transferase II (CPT-2), carnitine palmitoyl transferase-1β (CPT-1β), long-chain acyl-CoA dehydrogenase (ACADL), and peroxisome proliferator-activated receptor γ (PPARγ). (E) mRNA levels of PPARγ coactivators (PGC-1α and PGC-1β), and estrogen-related receptors (ERRα and ERRβ). (F) Western blot analysis of the expression of AMP-activated protein kinase α (AMPKα), fatty acid translocase (CD36), CPT-1α, PGC-1α, PDK4 and GLUT4. Data are shown as mean ± SD. *p < 0.05, **p < 0.01, compared with the sham group; #p < 0.05, ##p < 0.01, compared with the TAC group, evaluated by one-way ANOVA with Dunnett's post hoc test.
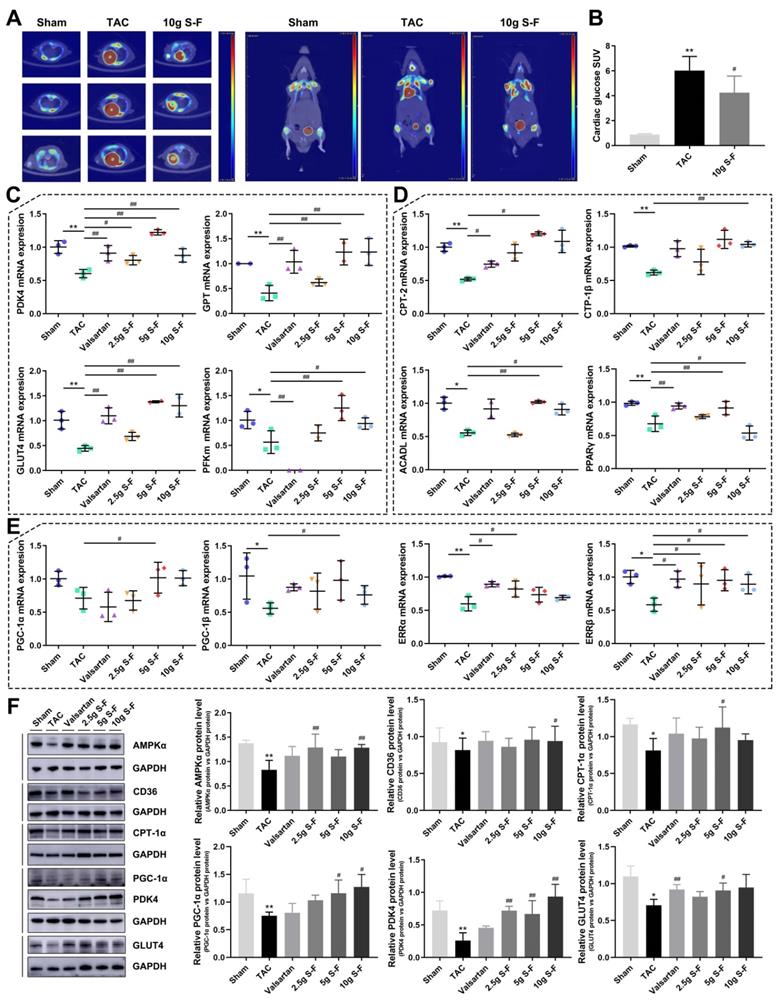
The key transcription factors related to glucose and fatty acid metabolism were measured to establish the potential effects of S-F in the energy remodeling of HF. As shown in Fig. 7C and Fig. S5, the expression of pyruvate dehydrogenase kinase 4 (PDK4), pyruvate/alanine aminotransferases (GPT), transcripts encoding glucose transporters (GLUT4), phosphofructokinase muscle (PFKm), pyruvate carboxylase (PCX), and monocarboxylate pyruvate transporter (MCT1) were found to be reduced in the failing heart. These alterations confirmed impairment in pyruvate transport, glucose uptake and metabolism. Consistent with the decreased expression of transcription factors in glucose metabolism, mRNA levels of key enzymes of fatty acid oxidation (Fig. 7D) including carnitine palmitoyl transferase II (CPT-2), carnitine palmitoyl transferase-1β (CPT-1β), long-chain acyl-CoA dehydrogenase (ACADL), and PPARγ were significantly decreased in TAC mice. Meanwhile, the transcript levels of PPARγ coactivator 1 (PGC-1α and PGC-1β), two master regulators of mitochondrial biogenesis and metabolism, and estrogen-related receptors (ERRα and ERRβ) were reduced in TAC mice (Fig. 7E).
S-F treatment significantly affected the metabolic transcripts associated with glucose and fatty acid metabolism. The expression of key genes in glucose metabolism and fatty acid oxidation including PDK4, GPT, GLUT4, PFKm, PCX, CPT-2, CPT-1β, ACADL, and PPARγ were up-regulated in varying degrees following S-F treatment (Fig. 7C, D). The expression of key mitochondrial transcription factor coactivators, such as PGC-1α, PGC-1β, ERRα and ERRβ, was also up-regulated after S-F treatment (Fig. 7E). These results indicated that S-F synergistically mediated the abnormalities in glucose and fatty acid metabolic pathways in TAC mice.
Western blot analysis was performed to detect the protein levels of AMPKα, CD36, CPT-1α, PGC-1α, PDK4, GLUT4, and PPARα (see Fig. 7F and Fig. S5). The expression of these proteins was reported to be down-regulated in TAC mice and was significantly restored by S-F treatment, but the effect of valsartan treatment was not significant. Among them, the AMPK-related signaling pathway was most significantly affected by S-F treatment.
Discussion
Pressure overload triggers complex biological responses that result in HF which is characterized by cardiac remodeling [21, 22]. In the current study, remarkable cardiac remodeling and heart dysfunction occurred in TAC mice, evidenced by significantly increased cardiac fibrosis and hypertrophy, as well as diminished echocardiographic features. S-F and valsartan treatment exhibited a protective function against myocardial fibrosis and necrosis which contributed to cardiac remodeling. Furthermore, both S-F and valsartan effectively counteracted the systolic and diastolic dysfunction, improved cardiac hemodynamics, reduced myocardial enlargement, and even down-regulated the levels of extracellular matrix markers in TAC mice. These findings indicated significant improvement in heart function and inhibition of cardiac remodeling in TAC mice after S-F treatment, and this supported the therapeutic value of S-F in HF. Comparatively, the main advantage of S-F over the positive control valsartan is at the regulation of energy metabolism as well as mitochondrial function.
Mounting evidence suggests that impaired energy metabolism and mitochondrial function contribute to cardiac remodeling leading to HF [23]. The efficiency of oxygen utilization is relatively low during HF, exhibiting an energy deficit (containing 30-40% less ATP compared with a healthy heart) due to changes in energy substrate and a gradual decline in mitochondrial oxidative phosphorylation [24]. In mouse hearts with systolic dysfunction induced by TAC, reduced mitochondrial OCRs and abnormal mitochondrial morphology were observed. Decreased mitochondrial respiration due to downregulation of mitochondrial complexes may be an important factor in linking contractile function and energy metabolism to the progression of HF [25]. S-F was found to significantly ameliorate mitochondrial dysfunction by enhancing OCRs and maintaining the mitochondrial structure. This suggests that the regulation of mitochondrial energy metabolism is one of the potential mechanisms of S-F action.
Alterations in energy substrate metabolism are recognized as important hallmarks in the cardiac remodeling process [26]. Approximately 70% to 90% of cardiac ATP originates from fatty acids metabolism. The remaining 10% to 30% comes from glucose and lactate metabolism, as well as small amounts of ketones and amino acids [3, 27]. During cardiac metabolic remodeling, energy metabolism is reprogrammed toward increased utilization of glucose and with significant downregulation of fatty acid oxidation [28]. Even though glucose metabolism consumes less oxygen, it produces fewer amounts of energy compared with fatty acid oxidation, causing the heart to remain in an energy-starved state [24]. Recent evidence suggests that maintaining fatty acid oxidation during pressure overload prevents increased dependence on glucose and further suppresses the effects of glucose on pathological growth [29, 30]. Although the mechanistic link between fatty acid metabolism and contractile function remains controversial, promoting fatty acid utilization has been shown to be a potential therapeutic approach towards decreasing the severity of HF [29, 31, 32]. In this study, abnormalities in key genes involved in fatty acid oxidation and transport were significantly restored after S-F treatment. This regulatory effect of S-F on fatty acid utilization was also confirmed through the multi-omics study. Integrated and pathway-driven analysis identified that fatty acid metabolism was the major pathway associated with S-F. Besides, the downstream metabolome showed a significant decrease in the plasma levels of the free fatty acids in TAC mice after S-F treatment.
Besides fatty acid utilization, S-F was found to have a regulatory effect on glucose metabolism. In the presence of systolic dysfunction, cardiac glucose uptake was found to be increased in TAC rats. Similar findings have been reported in previous studies [33, 34]. While glucose uptake into cardiac myocytes is increased, their subsequent entry into the mitochondrial is decreased. This results in a marked reduction in glucose oxidation and an increase in glycolysis [6]. The impaired glucose metabolism that parallels systolic dysfunction might be due in part to mitochondrial dysfunction or reduced expression of GPT, PFKm, PDK4, and GLUT4 involved in glucose oxidation. Another factor that may influence glucose oxidation is the fact that pyruvate may be channeled into anaplerotic pathways [35]. Higher levels of ME1 in mice hearts subjected to TAC and elevated abundance of (S)-malate and fumarate in mice plasma were reported. Increased anaplerotic flux via pyruvate carboxylation by ME1 may lead to a decrease in oxidation, which reduces the efficiency of ATP synthesis and exacerbates pathological remodeling [36, 37]. After 4 weeks of S-F treatment, the plasma levels of (S)-malate in TAC mice was significantly decreased, the transcript levels of ME1 were down-regulated, and the expression of PDK4, GPT, GLUT4, and PFKm was up-regulated. These alterations were accompanied by a reduction in cardiac glucose uptake in the S-F treated mice. Taken together, these results suggest that S-F may regulate the balance in glucose and fatty acids utilization, thus maintaining metabolic flexibility in energy metabolism.
AMPK pathway is one of the key signaling pathways that responded to S-F treatment. Activation of AMPK in HF may induce a wide range of effects that coordinately improve cardiac function and alleviate metabolic remodeling [38]. Specifically, increased AMPK activity could stimulate both fatty acid and glucose utilization, thereby restoring energy supply [39]. On the one hand, AMPK can stimulate fatty acids entry into the mitochondria and subsequent oxidation [40]. This increases the translocation of fatty acid translocase CD36 to the sarcolemma, promoting cardiomyocyte uptake of fatty acids [41]. Activated AMPK further inhibits the production of malonyl-CoA and activates CPT-1, while CPT-1 controls the rate-limiting step of mitochondrial fatty acid β-oxidation [42]. On the other hand, AMPK mediates the translocation of GLUT4 from the cytosol to the membrane and phosphorylates phosphofructokinase, thereby promoting glucose utilization as a cardioprotective response [43]. Furthermore, AMPK may also improve mitochondrial biogenesis in a long-term manner by activating PGC-1α [44]. PGC-1α further binds to and coactivates PPAR and ERR leading to increased fatty acid uptake and oxidation [45]. This study showed that S-F activated the AMPK signaling pathway and significantly up-regulated the levels of CD36, CPT-1α, PGC-1α, PPARα, ERRα, ERRβ, GLUT4 and PFKm in TAC mice. These results were validated in omics studies, western-blot analysis and qRT-PCR results, suggesting that S-F synergistically mediated the metabolic flexibility of fatty acids and glucose in cardiac energy metabolism, with AMPK pathway being the most impacted pathway.
This study was not without limitations. First, although S-F can modulate glucose uptake and utilization, this mechanism needs to be further explored. A significant increase in GLUT4 expression was observed in S-F treated mice, however, GLUT1 showed no significant change (data not shown). This was inconsistent with the reduction in cardiac glucose uptake and was attributed to the integrated effects of multiple components in S-F. Second, energy metabolic pathways that were the most relevant to the synergistic effects of S-F treatment were validated. Integrated omics analysis showed that S-F exerted an effect on branched chain amino acids and insulin-related pathways; however, this effect requires to be studied further. Finally, the relatively small-scale sample sets of omics studies could have possibly introduced selection bias. Therefore, large-sample sets examining the potential markers associated with S-F treatment should be conducted.
Conclusion
In conclusion, the key strength of this study is in its comprehensive design which reveals the energy-dependent mechanism of S-F through global multi-omics profiling. This study demonstrates that S-F can improve cardiac function, ameliorate mitochondrial dysfunction, and further alleviate metabolic remodeling in the failing heart. This is exhibited in the integrated omics analysis, which shows that S-F causes synergistic effects on fatty acids and glucose metabolism. The potential mechanism underlying these effects may be related to the regulation of AMPK-related signaling pathways. These findings provide new insights into the mechanism of S-F and provide evidence for the cardioprotective effect of S-F in the treatment of HF.
Abbreviations
HF: heart failure; S-F: Shen-Fu formula; TAC: transverse aortic constriction; EF: ejection fraction; FS: fractional shortening; IVS: interventricular septum thickness; LV Vol: left ventricular volume; LVID: left ventricular internal diameters; OCR: oxygen consumption rate; VIP: variable importance of project; 18F-FDG: F-18-fluorodeoxyglucose; SUV: standardized uptake value; PET: positron emission tomography; E/A: E and A wave ratio; IVRT: isovolumic relaxation time; HW/BW: heart weight to body weight ratio; HW/TL: heart weight to tibia length ratio; LW/BW: lung weight to body weight ratio; SV: stroke volume; dp/dt max: maximum rise/fall rate; dp/dt min: minimum rise/fall rate; MMP: matrix metalloproteinase; ST2: growth stimulation expressed gene 2 protein; ANP: atrial natriuretic peptide; BNP: brain natriuretic peptide; MHC: myosin heavy chain; SMA: smooth muscle actin; PCA: principal component analysis; OPLS-DA: orthogonal partial least squares discriminant analysis; DEGs: differentially expressed genes; DEPs: differentially expressed proteins; DEMs: differentially expressed metabolites; STEM: short time-series expression miner; AMPK: AMP-activated protein kinase; PPAR: peroxisome proliferator-activated receptor; PGC: PPARγ coactivator; GPT: pyruvate/alanine aminotransferases; PDK4: pyruvate dehydrogenase kinase 4; GLUT4: transcripts encoding glucose transporter 4; PFKm: phosphofructokinase muscle; PCX: pyruvate carboxylase; MCT: monocarboxylate pyruvate transporter; CPT: carnitine palmitoyl transferase; ACADL: long-chain acyl-CoA dehydrogenase; ERR: estrogen-related receptor.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
This work was supported by grants from the National Key Subject of Drug Innovation (2019ZX09201005-007), the National Key R&D Program of China (2018YFC1704500), the National Natural Science Foundation of China (81774050), Tianjin Science Foundation for Distinguished Young Scholars (17JCJQJC46200), and Tianjin Municipal Health and Health Committee (2019062).
Author Contributions
Yuting Huang, Kai Zhang, Jingyu Ni, Xiumei Gao and Guanwei Fan participated in the conceptualization and design of the study. Miaomiao Jiang and Jingrui Chen participated in the image analysis. Lan Li and Jie Deng participated in the acquisition and verification of biomarker assay data. Yan Zhu participated in the statistical analysis of the data. All authors were involved in interpretation of the study analyses. Yuting Huang and Kai Zhang participated in drafting the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hu X, Tang ZH, Ma SY, Yu YS, Chen XH, Zang GQ. Tripartite motif-containing protein 7 regulates hepatocellular carcinoma cell proliferation via the DUSP6/p38 pathway. Biochem Biophys Res Commun. 2019;511:889-895
2. Yancy CW, Jessup M, Bozkurt B. et al. 2017 ACC/AHA/HFSA focused update of the 2013 ACCF/AHA guideline for the management of heart failure: a report of the American college of cardiology/American heart association task force on clinical practice guidelines and the heart failure society of America. J Am Coll Cardiol. 2017;70:776-803
3. Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. 2013;113:709-724
4. van Bilsen M, Smeets PJ, Gilde AJ, van der Vusse GJ. Metabolic remodelling of the failing heart: the cardiac burn-out syndrome? Cardiovasc Res. 2004;61:218-226
5. Bertero E, Maack C. Metabolic remodelling in heart failure. Nat Rev Cardiol. 2018;15:457-470
6. Garcia-Ropero A, Santos-Gallego CG, Zafar MU, Badimon JJ. Metabolism of the failing heart and the impact of SGLT2 inhibitors. Expert Opin Drug Metab Toxicol. 2019;15:275-285
7. Zhou B, Tian R. Mitochondrial dysfunction in pathophysiology of heart failure. J Clin Invest. 2018;128:3716-3726
8. Riehle C, Abel ED. Insulin signaling and heart failure. Circ Res. 2016;118:1151-1169
9. Wei HM, Wu HJ, Yu W, Yan X, Zhang XY. Shenfu decoction as adjuvant therapy for improving quality of life and hepatic dysfunction in patients with symptomatic chronic heart failure. J Ethnopharmacol. 2015;169:347-355
10. Chen YF, Yu RY, Jiang L. et al. A comprehensive and rapid quality evaluation method of traditional chinese medicine decoction by integrating UPLC-QTOF-MS and UFLC-QQQ-MS and its application. Molecules. 2019;24:374
11. Song YL, Zhang N, Shi SP. et al. Large-scale qualitative and quantitative characterization of components in Shenfu injection by integrating hydrophilic interaction chromatography, reversed phase liquid chromatography, and tandem mass spectrometry. J Chromatogr A. 2015;1407:106-118
12. Luo J, Min S, Wei K, Cao J. Ion channel mechanism and ingredient bases of Shenfu Decoction's cardiac electrophysiological effects. J Ethnopharmacol. 2008;117:439-445
13. Wu H, Dai ZF, Liu X. et al. Pharmacodynamic evaluation of Shenfu injection in rats with ischemic heart failure and its effect on small molecules using matrix-assisted laser desorption/Ionization-mass spectrometry imaging. Front Pharmacol. 2019;10:1424
14. Ji JF, Jiao WZ, Cheng Y, Yan H, Su F, Chi LL. ShenFu preparation protects AML12 cells against palmitic acid-induced injury through inhibition of both JNK/Nox4 and JNK/NFkappaB pathways. Cell Physiol Biochem. 2018;45:1617-1630
15. Xia ZY, Liu XY, Zhan LY, He YH, Luo T, Xia Z. Ginsenosides compound (shen-fu) attenuates gastrointestinal injury and inhibits inflammatory response after cardiopulmonary bypass in patients with congenital heart disease. J Thorac Cardiovasc Surg. 2005;130:258-264
16. Song WT, Cheng FF, Xu L, Lin CR, Liu JX. Chinese medicine shenfu injection for heart failure: a systematic review and meta-analysis. Evid Based Complement Alternat Med. 2012;2012:713149
17. Li L, Pan CS, Yan L. et al. Ginsenoside Rg1 ameliorates rat myocardial ischemia-reperfusion injury by modulating energy metabolism pathways. Front Physiol. 2018;9:78
18. Zhou GH, Tang LY, Zhou XD, Wang T, Kou ZZ, Wang ZJ. A review on phytochemistry and pharmacological activities of the processed lateral root of Aconitum carmichaelii Debeaux. J Ethnopharmacol. 2015;160:173-193
19. Sansbury BE, DeMartino AM, Xie Z. et al. Metabolomic analysis of pressure-overloaded and infarcted mouse hearts. Circ Heart Fail. 2014;7:634-642
20. Chung YH, Lu KY, Chiu SC. et al. Early imaging biomarker of myocardial glucose adaptations in high-fat-diet-induced insulin resistance model by using (18)F-FDG PET and [U-(13)C]glucose nuclear magnetic resonance tracer. Contrast Media Mol Imaging. 2018;2018:8751267
21. Glasenapp A, Derlin K, Wang Y. et al. Multimodality imaging of inflammation and ventricular remodeling in pressure overload heart failure. J Nucl Med. 2020;61:590-596
22. Schirone L, Forte M, Palmerio S. et al. A Review of the molecular mechanisms underlying the development and progression of cardiac remodeling. Oxid Med Cell Longev. 2017;2017:3920195
23. Lai L, Leone TC, Keller MP. et al. Energy metabolic reprogramming in the hypertrophied and early stage failing heart: a multisystems approach. Circ Heart Fail. 2014;7:1022-1031
24. De Jong KA, Lopaschuk GD. Complex energy metabolic changes in heart failure with preserved ejection fraction and heart failure with reduced ejection fraction. Can J Cardiol. 2017;33:860-871
25. Tuomainen T, Tavi P. The role of cardiac energy metabolism in cardiac hypertrophy and failure. Exp Cell Res. 2017;360:12-18
26. Lopaschuk GD, Ussher JR, Folmes CD, Jaswal JS, Stanley WC. Myocardial fatty acid metabolism in health and disease. Physiol Rev. 2010;90:207-258
27. Yang KC, Kyle JW, Makielski JC, Dudley SC Jr. Mechanisms of sudden cardiac death: oxidants and metabolism. Circ Res. 2015;116:1937-1955
28. Rosca MG, Tandler B, Hoppel CL. Mitochondria in cardiac hypertrophy and heart failure. J Mol Cell Cardiol. 2013;55:31-41
29. Kolwicz SC Jr, Olson DP, Marney LC, Garcia-Menendez L, Synovec RE, Tian R. Cardiac-specific deletion of acetyl CoA carboxylase 2 prevents metabolic remodeling during pressure-overload hypertrophy. Circ Res. 2012;111:728-738
30. Choi YS, de Mattos AB, Shao D. et al. Preservation of myocardial fatty acid oxidation prevents diastolic dysfunction in mice subjected to angiotensin II infusion. J Mol Cell Cardiol. 2016;100:64-71
31. Chess DJ, Lei B, Hoit BD, Azimzadeh AM, Stanley WC. Effects of a high saturated fat diet on cardiac hypertrophy and dysfunction in response to pressure overload. J Card Fail. 2008;14:82-88
32. Lau E, Cao Q, Lam MPY. et al. Integrated omics dissection of proteome dynamics during cardiac remodeling. Nat Commun. 2018;9:120
33. Kolwicz SC Jr, Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res. 2013;113:603-616
34. Tuunanen H, Engblom E, Naum A. et al. Free fatty acid depletion acutely decreases cardiac work and efficiency in cardiomyopathic heart failure. Circulation. 2006;114:2130-2137
35. Turer A, Altamirano F, Schiattarella GG. et al. Remodeling of substrate consumption in the murine sTAC model of heart failure. J Mol Cell Cardiol. 2019;134:144-153
36. Pound KM, Sorokina N, Ballal K. et al. Substrate-enzyme competition attenuates upregulated anaplerotic flux through malic enzyme in hypertrophied rat heart and restores triacylglyceride content: attenuating upregulated anaplerosis in hypertrophy. Circ Res. 2009;104:805-812
37. Des Rosiers C, Labarthe F, Lloyd SG, Chatham JC. Cardiac anaplerosis in health and disease: food for thought. Cardiovasc Res. 2011;90:210-219
38. Beauloye C, Bertrand L, Horman S, Hue L. AMPK activation, a preventive therapeutic target in the transition from cardiac injury to heart failure. Cardiovasc Res. 2011;90:224-233
39. Kim TT, Dyck JR. Is AMPK the savior of the failing heart? Trends Endocrinol Metab. 2015;26:40-48
40. Arumugam S, Sreedhar R, Thandavarayan RA, Karuppagounder V, Watanabe K. Targeting fatty acid metabolism in heart failure: is it a suitable therapeutic approach? Drug Discov Today. 2016;21:1003-1008
41. Kim TT, Dyck JR. The role of CD36 in the regulation of myocardial lipid metabolism. Biochim Biophys Acta. 2016;1861:1450-1460
42. Hoppel CL, Tandler B, Fujioka H, Riva A. Dynamic organization of mitochondria in human heart and in myocardial disease. Int J Biochem Cell Biol. 2009;41:1949-1956
43. Li X, Liu J, Lu QG. et al. AMPK: a therapeutic target of heart failure-not only metabolism regulation. Biosci Rep. 2019;39:BSR20181767
44. Rowe GC, Jiang A, Arany Z. PGC-1 coactivators in cardiac development and disease. Circ Res. 2010;107:825-838
45. Zaha VG, Young LH. AMP-activated protein kinase regulation and biological actions in the heart. Circ Res. 2012;111:800-814
Author contact
![]() Corresponding authors: Prof. Dr. Guanwei Fan. First Teaching Hospital of Tianjin University of Traditional Chinese Medicine, 88 Changling Road, Xiqing District, Tianjin 300193, People's Republic of China. Tel.: +86-22-2798-7795. E-mail: guanwei.fanedu.cn; Prof. Dr. Xiumei Gao. Tianjin University of Traditional Chinese Medicine, 10 Poyanghu Road, Jinghai District, Tianjin 301617, People's Republic of China. Tel.: +86-22-5959-6158. E-mail: gaoxiumeiedu.cn.
Corresponding authors: Prof. Dr. Guanwei Fan. First Teaching Hospital of Tianjin University of Traditional Chinese Medicine, 88 Changling Road, Xiqing District, Tianjin 300193, People's Republic of China. Tel.: +86-22-2798-7795. E-mail: guanwei.fanedu.cn; Prof. Dr. Xiumei Gao. Tianjin University of Traditional Chinese Medicine, 10 Poyanghu Road, Jinghai District, Tianjin 301617, People's Republic of China. Tel.: +86-22-5959-6158. E-mail: gaoxiumeiedu.cn.