Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Material and methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2023; 19(10):3077-3098. doi:10.7150/ijbs.83392 This issue Cite
Research Paper
Inhibition of KMO Ameliorates Myocardial Ischemia Injury via Maintaining Mitochondrial Fusion and Fission Balance
Qiong Lai1#, Lingling Wu1#, Shuhong Dong1, Xiaozhou Zhu1, Zhaoyang Fan1, Junping Kou1, Fuming Liu2 ![]() , Boyang Yu1
, Boyang Yu1 ![]() , Fang Li1
, Fang Li1 ![]()
1. Jiangsu Key Laboratory of TCM Evaluation and Translational Research, Research Center for Traceability and Standardization of TCMs, School of Traditional Chinese Pharmacy, China Pharmaceutical University, 639 Longmian Road, Nanjing, 211198, China.
2. Jiangsu Province Hospital of Chinese Medicine, Affiliated Hospital of Nanjing University of Chinese Medicine, Nanjing, 210029, China.
#These authors contributed equally: Qiong Lai, Lingling Wu
Received 2023-2-9; Accepted 2023-5-23; Published 2023-6-12
Abstract

Looking for early diagnostic markers and therapeutic targets is the key to ensuring prompt treatment of myocardial ischemia (MI). Here, a novel biomarker xanthurenic acid (XA) was identified based on metabolomics and exhibited high sensitivity and specificity in the diagnosis of MI patients. Additionally, the elevation of XA was proved to induce myocardial injury in vivo by promoting myocardial apoptosis and ferroptosis. Combining metabolomics and transcriptional data further revealed that kynurenine 3-monooxygenase (KMO) profoundly increased in MI mice, and was closely associated with the elevation of XA. More importantly, pharmacological or heart-specific inhibition of KMO obviously suppressed the elevation of XA and profoundly ameliorated the OGD-induced cardiomyocytes injury and the ligation-induced MI injury. Mechanistically, KMO inhibition effectively restrained myocardial apoptosis and ferroptosis by modulating mitochondrial fission and fusion. In addition, virtual screening and experimental validation were adopted to identify ginsenoside Rb3 as a novel inhibitor of KMO and exhibited great cardioprotective effects by regulating mitochondrial dynamical balance. Taken together, targeting KMO may provide a new approach for the clinical treatment of MI through maintaining mitochondrial fusion and fission balance, and ginsenoside Rb3 showed great potential to be developed as a novel therapeutic drug targeting KMO.
Keywords: Myocardial ischemia, Xanthurenic acid, Kynurenine 3-monooxygenase, Mitochondrial fusion and fission, Ginsenoside Rb3
Introduction
Cardiovascular disease (CVD) is responsible for a staggering ~25 to 30% of mortality worldwide [1], and it is estimated that the number of world people dying from myocardial infarction is about 23.6 million, which may produce immense health and economic burdens in the world [2]. Myocardial ischemia (MI) is an initial and prominent factor of myocardial infarction, caused by a decrease in myocardial oxygen supply and/or an increase in myocardial oxygen demand [3-5]. Timely treatment of myocardial ischemia can reduce rates of myocardial injury and prevent it from progressing into myocardial infarction, heart failure, and some other cardiovascular diseases [6, 7]. However, roughly 75% of ischemic episodes have few symptoms or signs (i.e., subclinical ischemia), and the heart always suffers irreversibly impairment without conscious awareness [8-10]. When the myocardium suffers ischemic damage, it can only be targeted by timely reperfusion therapy in clinics. Unfortunately, the paradoxical myocardial reperfusion injury may extend the final infarct size [11], and it requires adjunctive treatment strategies beyond reperfusion to improve clinical outcomes. Therefore, looking for more accurate early diagnostic markers, effective potential therapeutic targets, and drugs are essential to ensure prompt medical attention to myocardial ischemia.
Metabolomics technology provides a way to offer insight into the disease progression through discovering potential metabolic markers and elucidating the undiscovered relevant metabolic pathways [12-14]. There are numerous studies that have proved the successful application of metabolomics in discovering biomarkers for cardiovascular diseases [15-18]. Hence, metabolomics is a good way to excavate the diagnostic biomarkers for myocardial ischemia. In addition, this might be a good attempt to combine metabolomics with transcriptomics for screening new targets for the treatment of myocardial ischemia as well.
In this study, experimental animal mice and clinical participants were performed to identify the potential metabolic biomarkers of myocardial ischemia using a combination of untargeted and targeted metabolomics analysis. The present study found that the level of metabolite xanthurenic acid (XA) was significantly elevated in the early stage of myocardial ischemia in mice and patients. More importantly, xanthurenic acid showed great potential in the diagnosis of myocardial ischemia patients. Subsequently, a functional metabolomics strategy was adopted to reveal the potential function of xanthurenic acid, as well as discover its related metabolic enzyme. Our results demonstrated that inhibition of kynurenine 3-monooxygenase (KMO) profoundly ameliorated myocardial injury by maintaining mitochondrial fusion and fission balance, which may reveal a novel potential therapeutic strategy for myocardial ischemia. Besides, ginsenoside Rb3 was screened as the novel inhibitor of KMO, and it also exhibited an excellent therapeutic effect on myocardial ischemia by maintaining mitochondrial fusion and fission balance.
Material and methods
Reagent
Ginsenoside Rb3 with high purity (> 98 %) was purchased from ChemFaces Biochemical Co., Ltd. (Wuhan, China). Ultrapure water was obtained from a Milli-Q purification system (Milford, MA, USA). Methyl thiazolyl tetrazolium (MTT) was purchased from Amersco (Washington, USA). N-acetyl-L-cysteine (NAC) was obtained from Sigma-Aldrich (St. Louis, MO, USA). Lactate dehydrogenase (LDH) assay kits (A020-2-2) were obtained from Nanjing Jiancheng Bioengineering Institute (Nanjing, China). Brain natriuretic peptide (BNP) kit (JEB-12888), high-sensitivity C-reactive protein (hs-CRP) kit (JEB-12578), tumor necrosis factor-α (TNF-α) kit (JEB-12474), malondialdehyde (MDA) kit (JEB-12601), and nitric oxide (NO) assay kit (JEB-12547) were obtained from Nanjing Jin Yibai Biological Technology Co. Ltd.
Animals and MI model
Eight- to 10-week-old male C57BL/6 (wild-type) mice were obtained from the Model Animal Research Centre of Yangzhou University and kept in SPF level animal lab. All procedures and protocols were administrated according to the National Institutes of Health Guidelines for the Care and Use of Laboratory Animals and approved by the Animal Ethics Committee of China Pharmaceutical University (Nanjing, China) (Approval No. SYXK2021-0010). All the animal studies were designed to produce equally sized groups using randomized and blinded analysis. Mice were killed using CO2 at the end of the experiments. The myocardial ischemia model was induced by left anterior descending coronary artery ligation (CAL) [19]. Briefly, mice were anesthetized with a single intraperitoneal injection of ketamine (60 mg/kg) and xylazine hydrochloride (100 mg/kg). Then the mice were connected to the artificial ventilator until the operation is completed within 3 min. The heart was exposed via a left thoracotomy, and a slipknot was tied around the left anterior descending coronary artery 3-4 mm from its origin utilizing a 6-0 silk suture. Sham-operated mice were administrated with the same surgical procedures without ligating the left anterior descending coronary artery. ST-segment elevation was used to demonstrate the success of the myocardial ischemia through an electrocardiogram monitor. In the drug administration group, the drug was dissolved in normal saline and administered by gavage or intraperitoneal injection according to the experimental measurement. Serum, plasma, and urine samples were obtained after overnight fasting and stored at -80 ºC for metabolite analyses. And all organs were washed with normal saline after removal and immediately stored at -80 ºC. The experimental groups of this study as shown in Supplementary Figure S1 (n = 7-13 / group).
Human samples
A total of 64 patients with myocardial ischemia and normal persons were enrolled from Jiangsu Province Hospital of Chinese Medicine (normal persons: 23, myocardial ischemia patients: 41). Patients with liver disease, active vasculitis, gastrointestinal pathology, cancer, or other diseases were excluded from the study. Informed consent was acquired from all participants. This study was approved by the Ethical Committee (No. 2019NL-089-02). The specific patient characteristics are represented in Supplementary Table S1. Urine and serum samples were obtained after overnight fasting and stored at -80 ºC for metabolite analyses.
Echocardiographic measurement
An echocardiographic system consisting of a Vevo 2100 Imaging System (Visual Sonics, Toronto, Canada) equipped with a 30 MHz transducer was used to assess the cardiac function of experimental mice. The mice were transferred to dorsal recumbency and placed on the imaging platform when the mice were fully anesthetized by 2.5% isoflurane in O2 gas [20]. Indicators of cardiac function measured in this study are as follows: Left ventricular (LV) ejection fraction (EF), LV fractional shortening (FS), stroke volume (SV), LV septum thickness; diastole (IVS;d), LV diameter; diastole (LVID;d), LV mass (corrected).
Histopathologic examination
The tissues were fixed in 4% paraformaldehyde solution, embedded in paraffin, sliced into pieces 5 μm thick, and stained with hematoxylin and eosin (H&E) and Masson's trichrome staining. To detect collagen fibers, the tissue sections embedded in paraffin (2-3 mm) were stained in 0.1% Sirius-red F3B in saturated picric acid. The histopathological changes were detected by an optical microscope [21]. All histopathologic evaluation was performed by two pathologists in a double-blinded fashion.
Immunohistochemistry
After fixed in the 4% paraformaldehyde and embedded in paraffin, the tissues were sectioned at 4 µm thicknesses, deparaffinized and rehydrated in PBS, and incubated with 3% hydrogen peroxide to block endogenous peroxidase activity. And then, the sections were incubated for 1 h at 37 °C with blocking liquid (Beyotime, Shanghai, China). The primary antibodies were dropwise added to the sections and kept at 4 °C for 24 h. After washing, sections were incubated with the HRP-conjugated secondary antibody (1:200, Biogot Technology, Nanjing, China) at 37 °C for 1 h. After incubated with DAB, counterstained with hematoxylin, a light microscope (DX45, Olympus Microsystems Ltd., Japanese) was applied to observe the dehydrated sections and finally imaged at 400X magnification [22].
Untargeted Metabolomics Analysis
HPLC-Q-TOF MS analysis
Serum, plasma, and urine were collected and immediately frozen at -80 ºC until analyses. Metabolite separation was conducted using Agilent Technologies 6530 Accurate-Mass Q-TOF LC/MS (USA). The mobile phases are water + 0.1% formic acid (eluent A) and acetonitrile + 0.1% formic acid (eluent B). TSK-GEL Amide-80 column (150×2.0 mm i.d., 5 μm) was adopted to analyze biological samples. The specific methods of sample pretreatment and the conditions of liquid phase analysis referred to our previous research [23].
Method assessment
The robustness and repeatability of the HPLC-Q-TOF MS analysis were examined by preparing mixed quality control samples (QC) [24]. QC test samples were pre-treated with reference to the test samples and randomly injected throughout the test sequence list. The unsupervised PCA analysis (Supplementary Figure S2A) of QC samples and the overlap of total ion chromatograms (TICs) (Supplementary Figure S2B) verified that the current analytical methods have good repeatability and robustness. Meanwhile, the RSD% values of retention time and peak area of five different ions extracted from TICs of QC sample were statistically lower than 4% and 19% respectively, which further verified the good reproducibility of the experimental method and instrument stability (Supplementary Table S2). In addition, the representative total ion chromatograms (TICs) of plasma, urine, and serum samples of each group were shown in Supplementary Figure S2C.
Data processing
The raw data obtained from the Q-TOF LC/MS ESI+ analysis was subjected to data preprocessing using R software, and the data were further screened according to the 80% principle. The processed data were imported into SIMCA-P 14.1 (Umetrics, Sweden) for orthogonal partial least-squares discriminant analysis (OPLS-DA) and PCA analysis. The metabolites with significant differences were screened by p-value < 0.05 and variables importance in the projection (VIP) value > 1.0.
Metabolite identification
The structural information of the screened differential metabolite ions was matched using the online databases HMDB, METLIN, and MassBank.
Targeted analysis by LC-MS/MS
Pretreatment of mice and clinical samples
50 μL of urine, serum, and six kinds of organ tissue grinds were taken, added 140 μL of methanol and 10 μL of internal standard solution (p-aminosalicylic acid) for protein precipitation. After high-speed centrifugation, the supernate was taken for further analysis.
LC-MS/MS analysis
The analysis was carried out on a triple quadrupole LC-MS/MS system. The mobile phases were consistent with untargeted metabolomics analysis. Chromatographic separation of all test samples was conducted on a TSK-GEL Amide-80 column (150×2.0 mm i.d., 5 μm). Therein, the gradient program of 2-arachidonoylglycerophosphocholine in serum starts with 80-75% B at 0-3 min, 75-60% B at 3-8 min, 60-50% B at 8-10 min, 50% B at 10-12 min, and then back to initial conditions, with 5 min for equilibration. Besides, the gradient program of fructoselysine, 7-methylguanine, and xanthurenic acid in urine and six kinds of organ tissues start with 70-65% B at 0-3 min, 65-60% B at 3-8 min, 60-50% B at 8-10 min, 50% B at 10-22 min, and then back to initial conditions, with 5 min for equilibration. The flow rate was 0.2 mL/min and the injection volume was 5 μL.
All metabolites were identified in the multiple-reaction-monitoring (MRM) of positive ion mode (ESI+), and metabolite identification and quantification were performed with standard substance of fructoselysine, 7-methylguanine, xanthurenic acid, and 2-arachidonoylglycerophosphocholine. Masshunter software was used for data acquisition and processing. The target LC-MS/MS method has been verified in linearity, precision, accuracy, extraction recovery, matrix effects, and mass spectrometry parameters, as shown in Supplementary Table S3-4. The precision and accuracy were below 15% and the linear curve fitted well, which reflected the high reliability and accuracy of these methods (Supplementary Figure S3).
RNA-seq and genome-wide transcriptome analysis
Total RNA was extracted from heart tissues using TRIzol (Invitrogen) and then purified. The raw data was handled through an in-house Perl scripts and the reads including the adapter are removed to obtain clean data. And then, STAR (v2.5.1b) was used to establish the index of the reference genome, align the pairs of clean reads to the reference genome, and htseqv0 6.0 was used to count the readings corresponding to each gene. EdgeR R package (3.12.1) was used to analyze and screen differentially expressed genes, and clusterProfiler R package was used in GO and KEGG pathway analysis [25].
Construction and infection of recombinant AAV-KMO
Serotype 9 adeno-associated virus (pAAV9) vectors carrying a specific sequence inhibiting KMO (PGMAAV-10261) and negative control (NC) (PGMAAV-10261) were constructed by Genomeditech Co. (Shanghai, China). Control virus particles and pAAV9-KMO (×1011 pfu/mL) were injected directly into the left ventricular free wall of mice using a 30-gauge needle. At the third week after injection, after verifying the successful genetic transfection of AAV in vivo using immunohistochemical methods, we performed sham and CAL surgery on experimental mice and sacrificed them two weeks later.
Immunofluorescence staining
The frozen sections of animal tissues were taken out and placed at room temperature, washed with phosphate buffered saline, and fixed with 4% paraformaldehyde, and then the tissue sections were permeabilized and sealed. Mfn2 (1: 200, Santa Cruz, #sc-515647), OPA1 (1:200, Proteintech, #27733-1-AP), p-Drp1 (1: 200, Cell Signaling Technology, #3455), Pink1 (1:200, Abcam, #ab216144) and Parkin (1:200, Abcam, #ab15494) antibodies were added to incubate overnight at 4 ºC and fluorescent secondary antibodies were added to incubate for 2 hours. DAPI (Beyotime Biotechnology, Shanghai, China) was used to dye the nucleus, and finally, a confocal laser scanning microscope (LSM700, Zeiss, Jena, Germany) was used for fluorescence observation and photography. In addition, the cells were washed with ice-cold PBS, and the intracellular mitochondria were localized with the MitoTracker® Deep Red. The subsequent procedure of cell immunofluorescence staining is consistent with animal tissues.
The mitochondrial structure in the image was separated and processed. Image J was used to calculate the mitochondrial length, aspect ratio (AR), and form factor (FF) values. AR describes changes in mitochondrial length, while FF describes changes in length and degree of branching. Average mitochondrial length, AR, and FF values were calculated from five random images for each experiment, with smaller values representing increased mitochondrial debris [26].
Quantification of mitochondrial DNA (mtDNA) content
The total DNA of H9c2 cells was extracted by using FastPure®Cell/Tissue DNA Isolation Mini Kit (DC102, Vazyme, Nanjing, China). mtDNA content was measured by real-time fluorescence quantitative PCR. Real-time PCR was performed on the Mastercycler EP Realplex PCR system (Bio-Rad) and the relative mtDNA expression levels were normalized to that of β-globin using the 2-ΔΔCT method. The primers used in this study were shown below: mtDNA (Sense: 5'-GAUUACAGUCAGCAAUAUA-3'; Antisense: 5'-GATGGGGCCGGTAGGTCGATAAAGGAG-3'); β-globin (Sense: 5'-CCAATCTGCTCACACAGG-3'; Antisense: 5'-CACCTTTCCCCACAGG-3') [27, 28].
Western blot analysis
As previously described [29], proteins were extracted from cells and hearts and quantified by a BCA protein assay kit. The same amounts of proteins (35 μg) were separated through 10%-15% SDS-PAGE and transferred to polyvinylidene fluoride (PVDF) membranes (Millipore Corporation, Billerica, Ma, USA) via electroblotting. After blocking, the primary antibody was added and incubated at 4 ℃ overnight: cleave caspase3 (1:2000, Cell Signaling Technology, #9664), FHC (1:1000, Affinity, #DF6278), FLC (1:1000, Abclone, #A21962), GPX4 (1: 1000, Proteintech, #67763-1-Ig), ACSL4 (1:1000, Abclone, #A6826), KMO (1:1000, Proteintech, #10698-1-AP), Mfn2 (1: 1000, Santa Cruz, #sc-515647), OPA1 (1:1000, Proteintech, #27733-1-AP), Phospho-Drp1 (1: 1000, Cell Signaling Technology, #3455), β-actin (1:1000, ZenBio, #250136), Drp1 (1:1000, Abcam, #ab184247), Pink1 (1:1000, Abcam, #ab216144), Parkin (1:1000, Abcam, #ab77924), LC3B (1:1000, Cell Signaling Technology, #43566), P62 (1:1000, Abcam, #ab109012), Beclin 1 (1:1000, Proteintech, #11306-1-AP). And then peroxidase-conjugated secondary antibody (1:10000, #BS13278, #BS12478, Bioworld, Louis Park, MN, USA) was added. Finally, bands were examined by an ECL reagent (Vazyme Biotech, Nanjing, China) and analyzed with Image Lab™ Software (version 4.1, Bio-Rad).
Transmission electron microscopy
Mouse hearts were cut into small pieces and placed in 2.5% glutaraldehyde for fixation, followed by continued fixation in 0.1 M cacodylate buffer for 1 h. The sections were dehydrated in acetone and embedded in Epon. Subsequently, ultrathin sections were stained with uranyl acetate and lead citrate and observed by LEO 906 (Zeiss) electron microscope operating at 60 kV. And digital electron micrographs were randomly taken at 5000x and 1700x magnification for each group of this study, respectively [30].
OGD injury model in vitro
The rat H9c2 cells (Cell Bank of the Chinese Academy of Sciences, Shanghai, China) were cultured in DMEM (Gibco, Grand Island, NY, USA) including 10% fetal bovine serum (FBS), 100 μg/mL streptomycin, 100 U/mL penicillin. The oxygen and glucose deprived (OGD) method was used to simulate ischemic injury in vitro. And the specific operation was to incubate the cells with glucose-free DMEM and culture in an incubator with a hypoxic environment of 37 °C, 94% N2, 1% O2, and 5% CO2. And cell viability of H9c2 cells was determined using 3-(4,5-dime thylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) method (Beyotime, Nanjing, China).
siRNA transfection
siRNA targeting specific proteins and control siRNA were transfected into H9c2 cells according to the manufacturer's instructions [31]. Briefly, cells were transfected with 20 nM KMO siRNA (Genomeditech, Shanghai, China) diluted with Hyper Transfection Super (ThikarrowTM Scientific Groups, Nanjing, China). 48 h after transfection, cells were collected and the KMO siRNA efficiency was measured by western blotting. The siRNA oligos sequence of KMO was shown below:
Rat-KMO-si-1 (Sense: 5'-GAUUACAGUCAGCAAUAUA-3'; Antisense: 5'-UAUAUUGCUGACUGUAAUC-3'); Rat-KMO-si-2 (Sense: 5'-GCUUCCAACGCAUAGUGAU-3'; Antisense: 5'-AUCACUAUGCGUUGGAAGC-3'); Rat-KMO-si-3 (Sense: 5'-GUCGCCUUCACCAGAAUAA-3'; Antisense: 5'-UUAUUCUGGUGAAGGCGAC-3').
Molecular Docking
The Autodock Vina was used to investigate the interactions between the compounds and the KMO protein. The protein structure of KMO (PDB ID: 5X68) was obtained from the PDB database (https://www.rcsb.org/) in PDB format. The crystal structure of receptor molecules Ginsenoside Rb3, Astragaloside IV, Salvianic acid C, Ginsenoside Rg3, Cryptotanshinone, and Tanshinone IIA was downloaded from TCMSP (https://tcmsp-e.com/) with mol2 format. Subsequently, the structure was pretreated with open-source Pymol™, including removing the co-crystallized ligand and selecting water molecules and cofactors. The docking process was finished via Autodock Vina. The accurate prediction of active sites was predicted by using the online-database Protein Plus (https://proteins.plus/) [32].
Surface plasmon resonance (SPR) analysis
The Surface Plasmon Resonance (SPR) method was used in conjunction with Biacore T200 (GE Healthcare, Uppsala, Sweden) to measure the binding affinities at 25 ºC. Recombinant human Kynurenine 3-hydroxylase (KMO) protein (OriGene Technologies Inc., Rockville, MD, USA) was immobilized on the CM5 sensor chip (GE Healthcare, Uppsala, Sweden) by amine-coupling method to achieve the target density of 10000 resonance units (RU). The PBS-P (10 mM sodium phosphate, 150 mM NaCl, 0.005% Tween-20, pH 7.4) was used as a running buffer. Then different concentrations of the compounds (0.1953125 µM, 0.390625 µM, 0.78125 µM, 1.5625 µM, 3.125 µM) were injected at a flow rate of 30 μL/min. Chip platforms were washed with a running buffer and 50% DMSO. The binding affinities were analyzed with the Biacore evaluation software (T200 Version 2.0) by curve fitting using a 1:1 binding model [33].
Statistical analysis
All data were represented as mean ± SD. GraphPad Prism 8 software (San Diego, CA, USA) was mainly used for data statistical analysis. All data were tested for normal distribution and equal variance. Sample sizes subjected to statistical analysis at least n = 5 per group. Comparisons between two groups were made using Student's two-tailed t-test, and one-way analysis of variance (ANOVA) was used for multiple comparisons (followed by Bonferroni's post hoc or Holm-Sidak's multiple comparisons tests). Differences were considered significant at p < 0.05. Besides, receiver operating characteristic (ROC) and area under the curve (AUC) analysis were used to evaluate the sensitivity, specificity, and Youden's index of metabolic markers.
Results
Comprehensive metabolites profiles in the serum, plasma, and urine of early stage myocardial ischemia
Coronary artery ligation (CAL) was performed to induce myocardial ischemia in mice. CAL successfully contributed to cardiac dysfunction and subsequently myocardial damage in mice. Cardiac contractile and diastolic function showed a significant decrease (Supplementary Figure S4A, Supplementary Table S5). Simultaneously, morphological damage in heart tissues became increasingly severe after 14-21 days (Supplementary Figure S4B). Thus, the time period of ischemia 14-21 days was adopted in our further research in MI mice.
Serum, plasma, and urine samples collected from the MI mice were performed with metabolomics analysis to systematically excavate metabolites with significant changes at the early stage of myocardial ischemia. A definite separation among sham, model-2w, and model-3w groups was observed in the PCA and OPLS-DA patterns in serum and urine samples (Supplementary Figure S5). A total of 52 differential metabolites (VIP > 1 and p-value < 0.05) were detected from the comparison of sham versus MI mice (model 2-3w) (Table 1). There was a distinct change of metabolites in various body fluids of MI mice. The metabolic pathways related to myocardial ischemia were further revealed based on screening results of the above differential metabolites, respectively (Figure 1A).
Statistical analysis of 52 differential metabolites from the comparison of Sham versus MI mice
| Trend | Sample | m/z | Rt(min) | VIP | p-value | FC | Identification |
|---|---|---|---|---|---|---|---|
| 2-3W↑ | Urine | 162.1120 | 14.33 | 1.38 | 1.27E-12 | 2.92 | L-isoleucyl-L-proline |
| 149.1066 | 4.46 | 1.37 | 2.70E-12 | 5.40 | L-Isoleucine | ||
| 283.1393 | 15.72 | 1.36 | 2.74E-12 | 3.49 | Prolylhydroxyproline | ||
| 283.1610 | 14.46 | 1.36 | 4.25E-10 | 2.58 | L-Valine | ||
| 344.2272 | 2.58 | 1.35 | 2.69E-09 | 4.73 | Xanthurenic acid | ||
| 136.0477 | 14.74 | 1.33 | 2.61E-07 | 7.37 | Creatine | ||
| 213.1226 | 13.24 | 1.32 | 8.87E-07 | 14.18 | L-prolyl-L-proline | ||
| 439.1370 | 10.34 | 1.30 | 1.65E-05 | 2.89 | 5-Methylcytidine | ||
| 371.2266 | 2.94 | 1.26 | 4.32E-06 | 5.82 | 3-Hydroxykynurenine | ||
| 555.2141 | 9.58 | 1.21 | 8.18E-04 | 2.66 | 7-Methylguanine | ||
| 148.0036 | 14.17 | 1.13 | 3.19E-04 | 7.65 | Taurine | ||
| 259.1028 | 11.86 | 1.09 | 4.35E-04 | 3.43 | Uridine | ||
| 332.1543 | 16.10 | 1.33 | 3.92E-05 | 1.91 | N-Acetylglutamine | ||
| 200.1281 | 2.10 | 1.20 | 1.41E-03 | 1.66 | 5-Acetamidovalerate | ||
| 268.1538 | 1.99 | 1.19 | 2.14E-03 | 1.76 | 4-O-Methylmelleolide | ||
| Serum | 808.5825 | 24.83 | 5.52 | 2.67E-02 | 26.98 | PC(16:0/22:5(7Z,10Z,13Z,16Z,19Z)) | |
| 522.3566 | 16.15 | 3.78 | 2.06E-02 | 1.64 | 2-Oleoylglycerophosphocholine | ||
| 786.6025 | 29.29 | 3.62 | 6.70E-04 | +∞ | 1,2-dioleoyl-sn-glycero-3-phosphocholine | ||
| 760.5875 | 24.76 | 3.42 | 4.70E-04 | 173.47 | PC(16:0/18:1(9Z)) | ||
| 544.3421 | 14.72 | 2.91 | 4.22E-02 | 1.72 | 2-Arachidonoylglycerophosphocholine | ||
| 782.5698 | 29.28 | 2.73 | 3.66E-02 | 8.62 | PA(18:2(9Z,12Z)/24:1(15Z)) | ||
| 808.5838 | 29.22 | 2.31 | 1.35E-03 | 50.85 | PA(24:0/20:4(5Z,8Z,11Z,14Z)) | ||
| 123.0783 | 15.44 | 2.09 | 1.60E-02 | 11.54 | Nicotinic acid | ||
| 149.0233 | 26.94 | 1.83 | 6.28E-03 | 3.99 | 1-Methyladenine | ||
| 784.5832 | 29.31 | 1.63 | 1.86E-02 | 3.82 | PA(24:1(15Z)/18:1(9Z)) | ||
| 810.6021 | 29.27 | 1.63 | 3.76E-03 | +∞ | PA(22:2(13Z,16Z)/22:1(13Z)) | ||
| 56.0530 | 27.37 | 1.57 | 3.84E-02 | 1.35 | Acrolein | ||
| 78.9985 | 31.59 | 1.11 | 7.01E-03 | 1.21 | Pyridine | ||
| 194.9650 | 29.98 | 1.02 | 5.12E-03 | 1.98 | Platinum | ||
| 2-3W↓ | Urine | 176.0210 | 14.55 | 1.37 | 8.43E-13 | 0.57 | N4-Acetylaminobutanal |
| 133.0607 | 3.09 | 1.36 | 1.32E-10 | 0.22 | N-Methyl-4-pyridone-3-carboxamide(4PY) | ||
| 286.1392 | 16.48 | 1.34 | 3.42E-10 | 0.51 | Urea | ||
| 287.1417 | 16.47 | 1.33 | 1.13E-10 | 0.54 | Glycylprolylhydroxyproline | ||
| 359.1914 | 15.84 | 1.32 | 2.17E-10 | 0.37 | Fructoselysine | ||
| 251.0367 | 14.04 | 1.29 | 1.32E-09 | 0.56 | 2,8-Dihydroxyquinoline-beta-D-glucuronide | ||
| 151.1436 | 4.37 | 1.25 | 3.02E-08 | 0.43 | Creatinine | ||
| 238.0927 | 11.98 | 1.17 | 1.03E-06 | 0.40 | Biopterin | ||
| 323.0736 | 15.05 | 1.16 | 2.10E-06 | 0.51 | Indoleacetaldehyde | ||
| 222.0965 | 13.37 | 1.12 | 3.55E-08 | 0.54 | N2-Acetyl-L-aminoadipate | ||
| 129.0656 | 14.92 | 1.23 | 4.21E-08 | 0.75 | 2'-O-Methyladenosine | ||
| 267.1190 | 14.26 | 1.21 | 3.55E-07 | 0.64 | Selenocysteine | ||
| 214.1297 | 2.03 | 1.10 | 1.01E-05 | 0.72 | Isovalerylsarcosine | ||
| Plasma | 303.2261 | 17.68 | 1.84 | 7.71E-08 | 0.84 | Linoleic acid | |
| 414.2581 | 23.44 | 1.83 | 7.71E-06 | 0.68 | 3α,12α-Dihydroxy-5β-chol-6-en-24-oic Acid | ||
| 381.2965 | 19.70 | 1.80 | 4.53E-09 | 0.57 | MG(0:0/18:0/0:0) | ||
| 379.2774 | 17.43 | 1.78 | 7.48E-05 | 0.80 | MG(18:1(9Z)/0:0/0:0) | ||
| 866.5846 | 13.84 | 1.75 | 9.21E-03 | 0.91 | LysoPE(0:0/18:0) | ||
| 351.2245 | 17.34 | 1.67 | 1.56E-06 | 0.88 | MG(0:0/18:1(11Z)/0:0) | ||
| 684.5405 | 16.76 | 1.66 | 4.51E-03 | 0.49 | MG(0:0/16:0/0:0) | ||
| 80.9482 | 0.47 | 1.74 | 1.19E-04 | 0.55 | D-Tagatose | ||
| Serum | 113.9639 | 29.33 | 1.29 | 4.67E-03 | 0.62 | Hydroxidodioxidosulfidosulfate | |
| 128.9509 | 29.33 | 1.10 | 4.41E-03 | 0.57 | Selenite |
VIP: variables importance in the projection values;
FC: fold change;
Rt: retention time;
m/z: mass-to-charge ratio;
Xanthurenic acid is identified as the metabolic marker of MI. A. Metabolite pathway of MI. Compared with the sham group, elevated metabolites in MI mice were labeled red, and the reduced metabolites were labeled green. B. Results of heat map analysis of the four potential metabolic biomarkers. Red represents high content and green represents low content (n = 6). C. Changes in the content of xanthurenic acid (XA), 2-arachidonoylglycerophosphocholine, 7-methylguanine, and fructoselysine in sham and MI model groups mice during CAL 3 weeks (n = 7-10). D. Changes in the content of xanthurenic acid, 2-arachidonoylglycerophosphocholine in normal healthy persons (n = 23) and MI patients (n = 41). All metabolites in biological samples were detected by LC-MS. E. Evaluation of sensitivity and specificity of xanthurenic acid and 2-arachidonoylglycerophosphocholine in the diagnosis of MI patients. Analysis of receiver operating characteristic (ROC) curves based on the targeted quantitative results of metabolites in clinical samples. Data are presented as mean ± SD. #p<0.05, ##p<0.01 vs. sham group or normal group.
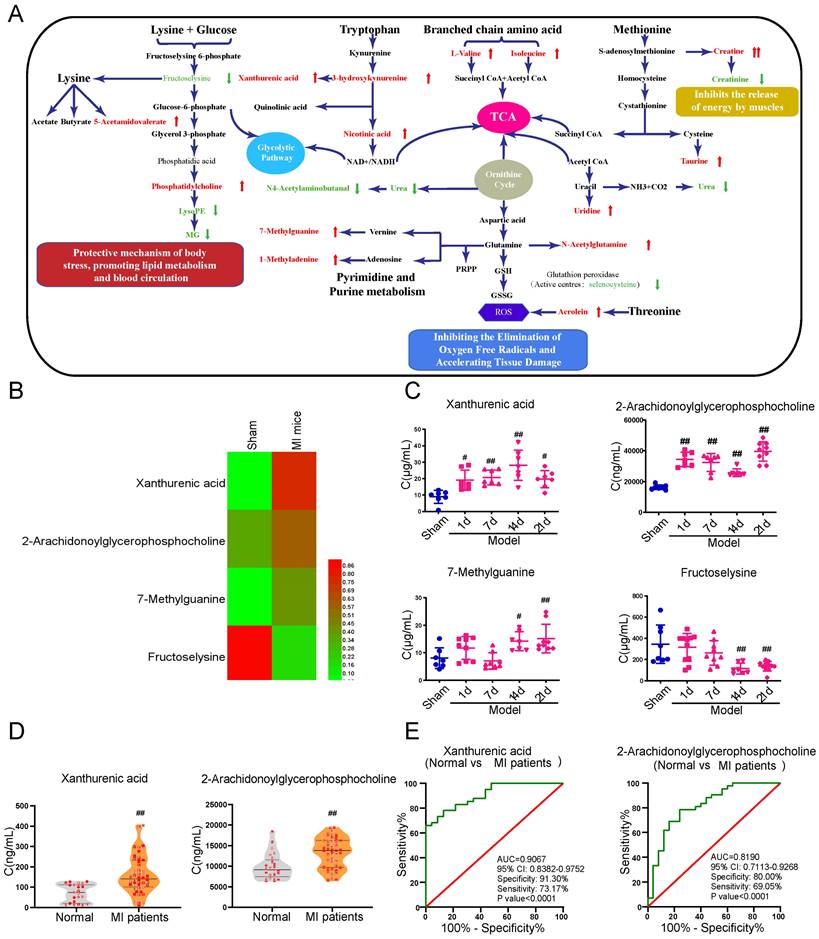
Xanthurenic acid was identified as the metabolic marker of myocardial ischemia
In addition to a majority of biomarkers known to be closely associated with MI, such as urea, taurine, and methionine, some new biomarkers were also discovered and showed different changes in the early stage of MI (Figure 1B). The metabolites xanthurenic acid, 2-arachidonoylglycerophoshphocholine, 7-methylguanine, and fructoselysine were subsequently performed for targeted analysis using a tandem LC-MS-based approach in MI mice. The levels of metabolites xanthurenic acid and 2-arachidonoylglycerophoshphocholine were significantly increased only 1 day after myocardial ischemia in mice (Figure 1C). And then, the change of these two metabolites was further verified in clinical MI patients and normal healthy persons (Figure 1D). Additionally, ROC analyses were used to assess the specificity and sensitivity of xanthurenic acid and 2-arachidonoylglycerophoshphocholine in the clinical diagnosis of MI, the results showed that xanthurenic acid had a higher AUC value and a better correlation with MI compared with 2-arachidonoylglycerophoshphocholine (Figure 1E).
Elevation of xanthurenic acid levels aggravated myocardial ischemia injury
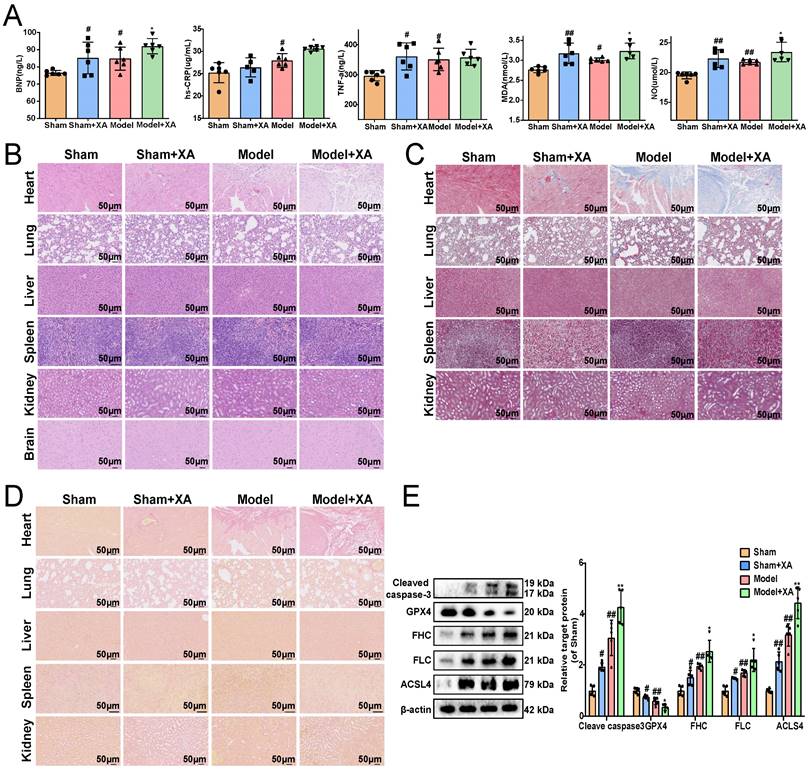
To further revealed the association between the elevated levels of xanthurenic acid and MI, xanthurenic acid was injected intraperitoneally in mice. We found that the elevation of xanthurenic acid levels further increased the content of BNP, hs-CRP, MDA, TNF-α, and NO in the serum of MI mice (Figure 2A). Histopathological examination results showed that treatment with xanthurenic acid for 8 days could lead to cardiomyocyte necrosis, myocardial fibrosis, and inflammatory cell infiltration in the heart tissue of mice with MI. We also observed that elevated levels of xanthurenic acid slightly increased the morphological damage and fibrosis of lung tissue in mice (Figure 2B-D, Supplementary Figure S6A-C). However, treatment with xanthurenic acid has no significant impact on other major organs. Besides, the elevation of xanthurenic acid was found to enhance the expression of FHC, FLC, ACSL-4, and cleaved caspase-3, and reduce GPX4 expression, which demonstrated its effect closely associated with the activation of ferroptosis and apoptosis in MI mice (Figure 2E).
Elevation of xanthurenic acid level aggravates myocardial ischemia injury. A. Serum biochemical indicators contents include BNP, hs-CRP, TNF-α, MDA, and NO in sham and MI model groups mice administered xanthurenic acid (XA), and were detected by ELISA (i.p. 100 mg/kg) (n = 6). B-D. Histological analysis of heart, lung, liver, spleen, kidney, and brain slices by hematoxylin and eosin (H&E), masson's trichrome, and sirius red staining (n = 5) (All images were 200X magnification except for spleen was 400X magnification; the lines marked in all figures represent 50 µm). E. Western blots and quantitative results of cleaved caspase-3, GPX4, FHC, FLC, and ACSL4 in each group mice heart (n = 5). Data are presented as mean ± SD. #p < 0.05, ##p < 0.01 vs. sham group, *p < 0.05, **p < 0.01 vs. model group.
KMO overexpression is a key factor leading to the elevation of xanthurenic acid in myocardial ischemia
Xanthurenic acid is mainly involved in the metabolism of tryptophan. To explore the tissue specificity of xanthurenic acid, the level of xanthurenic acid was measured in six kinds of organs. Interestingly, the results indicated that xanthurenic acid increased most significantly in heart tissue (Figure 3A). Based on the above results, we further screened specific genes of MI mice in the heart tissues by transcriptomics. A total of 1249 differential genes were screened between MI mice and the sham operation group, as shown in Figure 3B. The top 50 differential genes obtained from the comparison of MI (CAL 2-3w) and sham mice were shown in Table 2. The analysis of KEGG enrichment of different genes revealed that abnormal energy metabolism and oxidative stress mainly occurred in MI mice (Figure 3B).
Top 50 differential genes from the comparison of Sham versus MI mice
| Gene_name | log2FoldChange | pvalue | padj |
|---|---|---|---|
| Col6a5 | -5.106587534 | 3.55E-29 | 6.26E-25 |
| Cpz | -3.483426785 | 2.64E-13 | 2.33E-09 |
| Cthrc1 | -5.34211155 | 3.10E-11 | 1.82E-07 |
| Trem2 | -2.156973613 | 1.74E-10 | 7.69E-07 |
| Anxa8 | -3.887173854 | 3.59E-10 | 1.27E-06 |
| KMO | -5.094372156 | 3.71E-10 | 3.35E-06 |
| Timp1 | -3.094041602 | 6.84E-09 | 2.01E-05 |
| Fam46b | -1.784008184 | 2.35E-08 | 5.91E-05 |
| Aebp1 | -2.524560266 | 3.42E-08 | 7.53E-05 |
| Tpsb2 | -3.54076012 | 4.18E-08 | 8.18E-05 |
| Pou3f1 | -3.204860229 | 5.98E-08 | 0.000105357 |
| Slit2 | -1.071752038 | 8.87E-08 | 0.000142211 |
| Ildr2 | -2.637516087 | 1.04E-07 | 0.000152154 |
| Muc16 | -3.771936259 | 1.28E-07 | 0.000173951 |
| Adamts16 | -6.895028006 | 1.70E-07 | 0.000214091 |
| Fam180a | -3.278666239 | 1.83E-07 | 0.000214356 |
| Gpnmb | -3.340546 | 1.96E-07 | 0.000214356 |
| Ephb2 | -2.344274809 | 2.07E-07 | 0.000214356 |
| Igfbp6 | -1.795467822 | 3.09E-07 | 0.000303038 |
| Lox | -2.859616809 | 4.09E-07 | 0.000367232 |
| Cma1 | -3.020266678 | 4.52E-07 | 0.000379345 |
| Stac2 | -3.097904733 | 5.88E-07 | 0.000437574 |
| AC131720.3 | 3.274126978 | 6.14E-07 | 0.000437574 |
| Gpr176 | -3.52628993 | 6.20E-07 | 0.000437574 |
| Bnc2 | -2.431069226 | 6.21E-07 | 0.000437574 |
| Hoxc4 | -7.014916091 | 7.06E-07 | 0.000478496 |
| Marco | -6.929200672 | 8.24E-07 | 0.000538217 |
| Igkv14-126 | -7.263531139 | 8.79E-07 | 0.000553553 |
| P4ha3 | -3.846042675 | 1.06E-06 | 0.000646957 |
| Flrt2 | -1.362220675 | 1.31E-06 | 0.000725623 |
| Ighg2c | -6.167310901 | 1.32E-06 | 0.000725623 |
| Cemip | -3.915662582 | 1.32E-06 | 0.000725623 |
| Il33 | -1.94796866 | 1.64E-06 | 0.000878322 |
| Gm29216 | 1.699469385 | 2.02E-06 | 0.001047794 |
| Adra2a | -3.57009819 | 2.29E-06 | 0.001104086 |
| Steap2 | -2.009127292 | 2.32E-06 | 0.001104086 |
| Nfasc | -3.167991854 | 2.32E-06 | 0.001104086 |
| Postn | -2.92357388 | 2.53E-06 | 0.001147203 |
| Krt8 | -2.464194121 | 2.54E-06 | 0.001147203 |
| Wnt4 | -3.777285394 | 2.66E-06 | 0.001170355 |
| Grem2 | -4.589361908 | 2.90E-06 | 0.001246291 |
| Lgi2 | -2.238131931 | 3.13E-06 | 0.001302165 |
| Itgbl1 | -1.907181088 | 3.22E-06 | 0.001302165 |
| Nox4 | -2.759132495 | 3.28E-06 | 0.001302165 |
| Myof | -1.733420986 | 3.32E-06 | 0.001302165 |
| Clec4d | -3.548458422 | 3.45E-06 | 0.00132027 |
| Bnc1 | -4.101519638 | 3.71E-06 | 0.001389835 |
| Lrrn4 | -3.899149928 | 3.98E-06 | 0.001460898 |
| Saa3 | -7.135474939 | 4.67E-06 | 0.00164523 |
| Myrf | -2.301255295 | 4.81E-06 | 0.00164523 |
KMO overexpression is a key factor leading to the elevation of xanthurenic acid in MI. A. The level of xanthurenic acid in the different tissues of MI mice after 2 weeks' CAL (n = 5). Xanthurenic acid is one of the metabolites involved in the tryptophan metabolic pathway and the content of xanthurenic acid in the tissues were quantitatively analyzed by LC-MS. B. The heart tissues of sham and MI model groups mice were collected for transcriptome analysis (Sham: n = 3, Model 2-3w: n = 6). Volcano plot and KEGG pathway enrichment analysis of differential genes between sham, model 2-3w. Red dots in volcanoes represent genes whose expression is up-regulated, and green dots in volcanoes represent genes whose expression is down-regulated. C. The expressions of KAT1 and KMO in heart tissues of sham and MI model groups mice, and were determined by the immunohistochemical staining (n = 5) (400X magnification, the lines marked in all figures represent 50 µm). D. The expression level of KMO in the different tissues of sham and MI model groups mice after 2 weeks' CAL, and were measured by immunohistochemical staining (n = 5) (400X magnification, the lines marked in all figures represent 50 µm). Image J software was used for area statistics. E. Effects of KMO inhibitor UPF-648 on the contents of xanthurenic acid in urine samples of MI model groups mice after 2 weeks' CAL (n = 6-7) (1 mg/kg/day and 2 mg/kg/day). F. Effects of cardiac-specific knockdown of KMO expression through infection of recombinant AAV-KMO on the contents of xanthurenic acid in urine samples of MI model groups mice after 2 weeks' CAL (n = 6). Data are presented as mean ± SD. #p < 0.05, ##p < 0.01 vs. sham or sham + AAV-NC group, *p < 0.05, **p < 0.01 vs. model or model + AAV-NC group; ns means no significance.
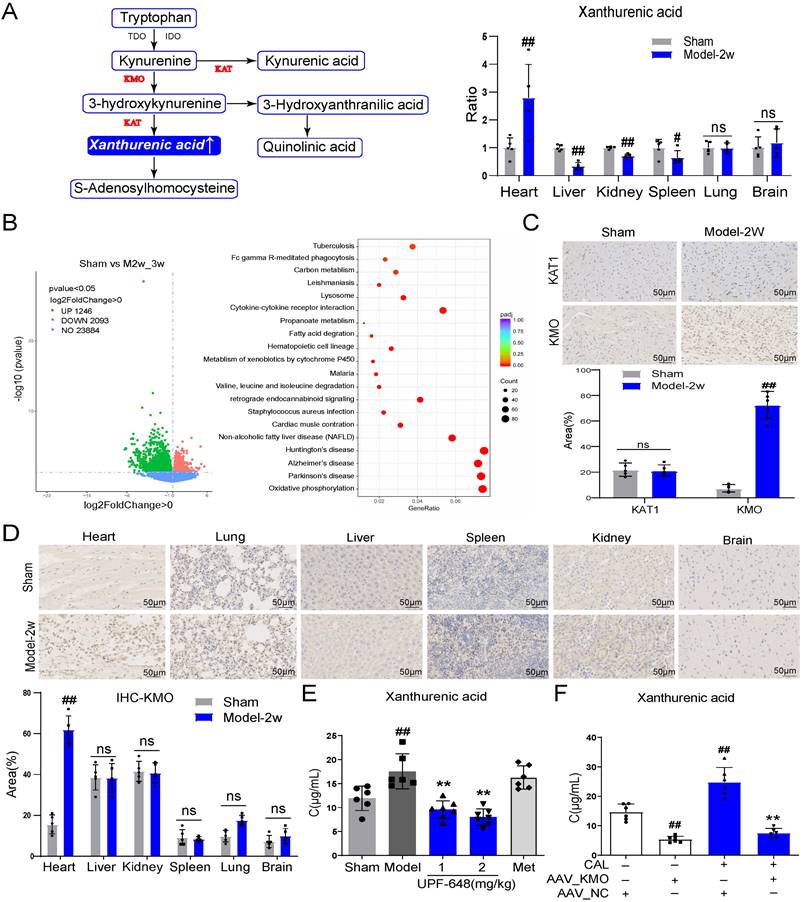
Based on the above transcriptome results, the related regulatory enzymes of xanthurenic acid were further investigated. Transcriptional data from cardiac tissue revealed that KMO and kynurenine aminotransferase 1 (KAT1) changed prominently in MI mice (CAL 2-3w). It's worth noting that, KMO appeared in the top 10 differential genes from the comparison of sham versus MI mice (Table 2). Meanwhile, immunohistochemical results of heart tissues showed that the expression of KMO dramatically increased in MI mice, while KAT1 expression did not change (Figure 3C). Combined with the results of gene and protein expression, KMO might be the key regulatory metabolic enzyme leading to the abnormal elevation of xanthurenic acid in MI. Moreover, we further found that the expression of KMO only profoundly elevated in the heart tissues of MI mice, while no obvious impacts were observed in other major organs (Figure 3D). Additionally, the inhibitor of KMO (UPF-648) and recombinant AAV-KMO were further adopted and the results showed that KMO inhibition could effectively prevent the abnormal increase of xanthurenic acid (Figure 3E-F). All these data suggested that the primary cause of increased metabolite xanthurenic acid in MI might be the overexpression of KMO.
Inhibition of KMO effectively alleviated myocardial ischemia injury
Firstly, the inhibitor of KMO (UPF-648) was used to explore the role of KMO in vitro and in vivo. We found that UPF-648 could successfully inhibit the expression of KMO (Supplementary Figure S7A-B), and it could effectively increase the cell viability as well as decrease the LDH release in OGD-induced cardiomyocytes injury (Supplementary Figure S8A-B). Meanwhile, UPF-648 could ameliorate the histological features and reduce the serum level of BNP, hs-CRP, MDA, nitric oxide (NO), and TNF-α in MI mice (Supplementary Figure S9A-B). Also, the transmission electron microscopy results showed that UPF-648 could significantly alleviate myocardial ultrastructure damage, reduce cellular edema and promote the maintenance of the mitochondrial matrices (Supplementary Figure S9C).
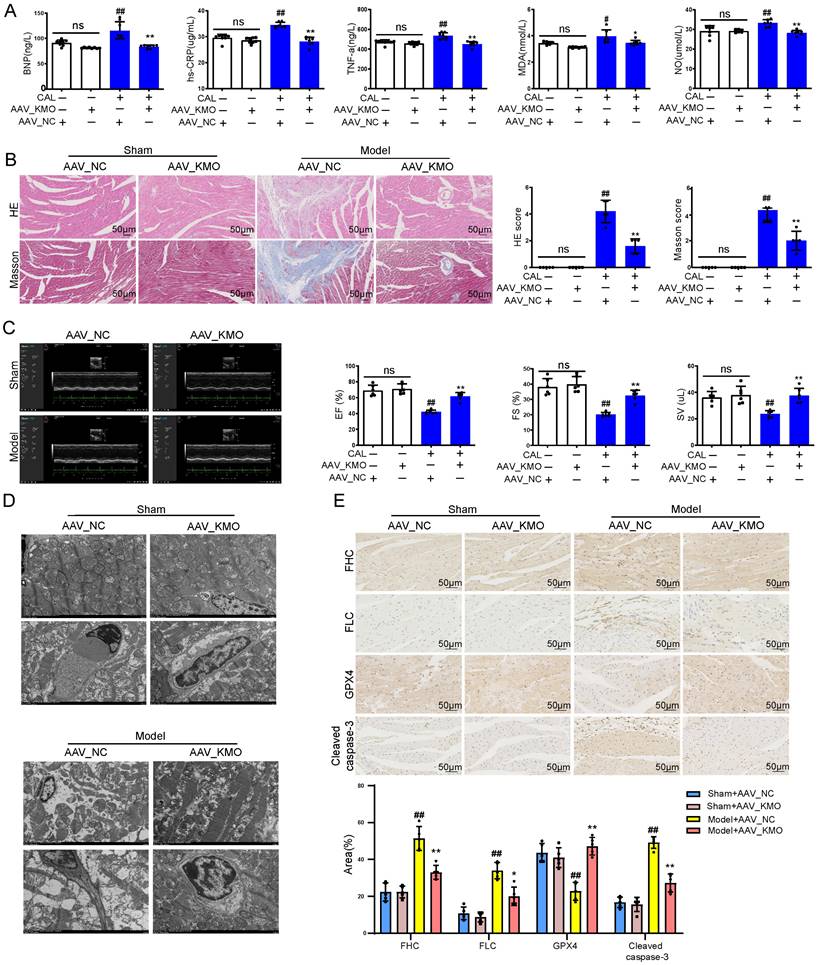
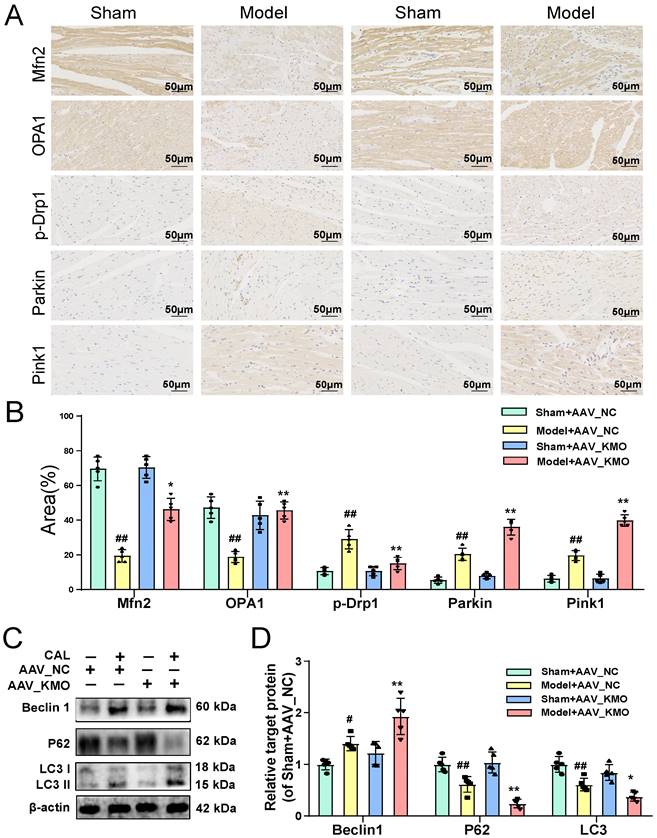
Then cardiac-specific KMO knockdown mice were constructed to further explore the function of KMO in MI injury. Inhibition of cardiac expression of KMO was achieved by intramyocardial injection of AAV9 loading the specific sequence targeting KMO (AAV-KMO) or an AAV-negative control (AAV-NC) in mice. Immunohistochemical analysis confirmed that the knockdown rate of KMO in heart tissue was over 50% (Supplementary Figure S7C). Based on the above results, KMO inhibition profoundly ameliorated the biochemical indexes (BNP, hs-CRP, MDA, NO, and TNF-α) associated with myocardial damage in serum (Figure 4A), as well as improved the pathological damage of myocardial tissue (Figure 4B). What's more, it showed an evident effect on improvement in cardiac function, such as LV ejection fraction, LV fractional shortening, and stroke volume (Figure 4C, Supplementary Table S6). We also found that inhibition of KMO in MI mice significantly ameliorated myocardial ultrastructure damage and mitochondrial morphology (Figure 4D). Moreover, we found that KMO inhibition could significantly inhibit the activation of ferroptosis and apoptosis in MI injury (Figure 4E). All these findings suggested that KMO inhibition could observably improve cardiac performance in MI injury.
Cardiac-specific knockdown of KMO effectively alleviates myocardial injury in MI. A. Serum biochemical indicators contents include BNP, hs-CRP, TNF-α, MDA, and NO in sham and MI model groups mice with cardiac-specific knockdown of KMO expression and were detected by ELISA (n = 7-8). B. Histological analysis of heart slices was shown by H&E and Masson's trichrome staining in sham and the MI model groups mice with cardiac-specific knockdown of KMO expression (n = 5) (200X magnification, scale bar, 50 µm). C. Representative echocardiographic graphs are shown. The echocardiographic parameters were measured, including ejection fraction (EF), fractional shortening (FS), and stroke volume (SV) in sham and the MI model groups mice with cardiac-specific knockdown of KMO expression (n = 6). D. Representative transmission electron microscope images of heart tissues with cardiac-specific knockdown of KMO expression (n = 3). E. Representative immunohistochemical staining of FHC, FLC, GPX4, and cleaved caspase-3 in sham and MI model groups mice heart with cardiac-specific knockdown of KMO expression (n = 5) (400X magnification, scale bar, 50 µm). Data are presented as mean ± SD. #p < 0.05, ##p < 0.01 vs. sham + AAV-NC group, *p < 0.05, **p < 0.01 vs. model + AAV-NC group; ns means no significance.
Inhibition of KMO prevents myocardial damage by maintaining mitochondrial fusion and fission balance
Next, we investigated the specific mechanisms of KMO inhibition in improving MI injury in vitro and in vivo. Previous researches reported that KMO is a protein localized to the outer mitochondrial membrane [34]. Thus, this study first investigated the effect of inhibition of KMO on mitochondrial function. The detection of mitochondrial copy number found that KMO inhibitors significantly reversed the trend of OGD-induced reduction in mtDNA number (Figure 5A). Meanwhile, the KMO inhibitor markedly up-regulated the expression of OPA1, Mfn2 and inhibited phosphorylation of Drp1 (Ser616) in OGD-induced cardiomyocytes injury and MI injury (Figure 5B-E, Supplementary Figure S10 and Figure S11A-B). And the inhibition of KMO further activated the expression of mitochondrial autophagy Pink1 and Parkin in OGD-induced cardiomyocytes injury (Figure 5F, Supplementary Figure S11C).
Inhibition of KMO reduces OGD-induced cardiomyocyte injury by regulating mitochondrial fission/fusion. A. RT-PCR analysis of mtDNA content in the OGD-induced H9c2 cells injury model treated with KMO inhibitor UPF648 (2 µmol/L) (n = 5). B. Western blotting analysis of Mfn2, OPA1, p-Drp1, and Drp1 in the OGD-induced H9c2 cells injury model treated with KMO inhibitor UPF648 (2 µmol/L) (n = 5). C - F. Representative immunofluorescence images of Mfn2, OPA1, p-Drp1, Parkin, and Pink1 in the OGD-induced H9c2 cells injury model treated with KMO inhibitor UPF648 (2 µmol/L) (n = 5). G-I. The average aspect ratio and form factor of the mitochondria labeled with MitoTracker® Deep Red were measured. Smaller mitochondrial length, aspect ratio, and form factor values represent increased mitochondrial fragmentation (n = 5). J. Western blotting analysis of Beclin1, P62, and LC3I/II in the OGD-induced H9c2 cells injury model treated with KMO inhibitor UPF648 (2 µmol/L) (n = 5). K. RT-PCR analysis of mtDNA content in the OGD-induced H9c2 cells injury model treated with siRNA-KMO or siRNA-NC (n = 5). L. Western blotting analysis of Mfn2, OPA1, p-Drp1, and Drp1 in the OGD-induced H9c2 cells injury model treated with siRNA-KMO or siRNA-NC (n = 5). M. Representative immunofluorescence images of Parkin, and Pink1 in the OGD-induced H9c2 cells injury model treated with siRNA-KMO or siRNA-NC (n = 5). N. Western blotting analysis of Beclin1, P62, and LC3I/II in the OGD-induced H9c2 cells injury model treated with siRNA-KMO or siRNA-NC (n = 5). Data are presented as mean ± SD. #p < 0.05, ##p < 0.01 vs. control group or control + siRNA-NC group, *p < 0.05, **p < 0.01 vs. OGD group or OGD + siRNA-NC group; ns means no significance.
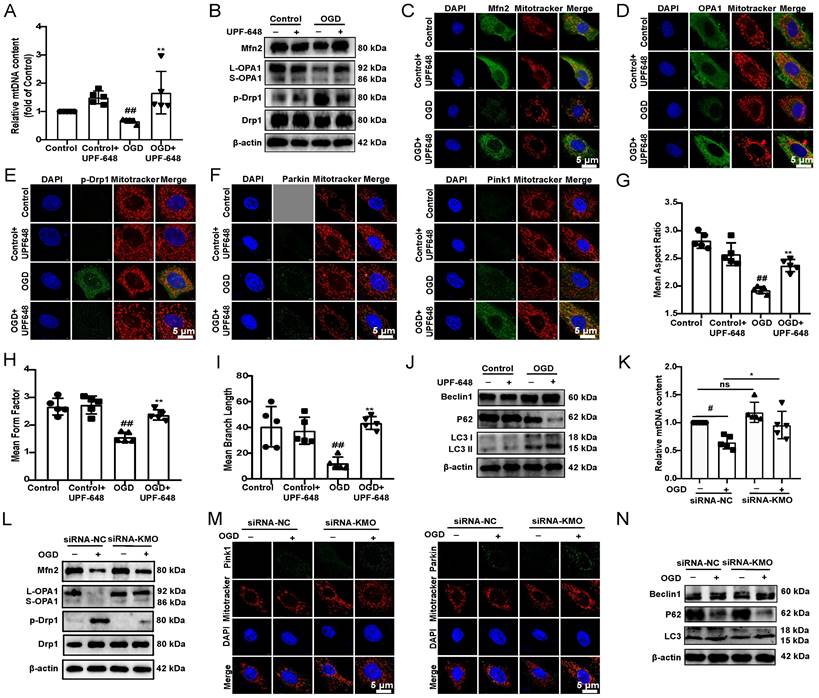
Besides, the results of immunofluorescence further indicated that the inhibitor of KMO (UPF-648) evidently ameliorated the fragmentation of mitochondria in the OGD injury model (Figure 5G-I). Interestingly, the KMO inhibition also caused an increase in its downstream autophagy proteins LC3II and Beclin1 and decreased expression of P62 (Figure 5J, Supplementary Figure S11D). Moreover, KMO-siRNA not only rescued the reduction in mtDNA content caused by OGD, also prevented OGD-induced cardiomyocytes injury by maintaining mitochondrial fusion/fission balance (Figure 5K-N, Supplementary Figure S11E-G). Meanwhile, we further validated these findings in CAL-induced MI injury using cardiac-specific knockdown of KMO. The result indicated that cardiac-specific knockdown of KMO promoted the fusion/fission balance of mitochondria and further accelerated the progress of autophagy to reduce damaged mitochondria (Figure 6). All the above data suggested that maintaining mitochondrial fusion and fission balance is indeed an important way for KMO inhibition in preventing myocardial ischemia injury.
Cardiac-specific knockdown of KMO prevents myocardial injury via maintaining mitochondrial fusion/fission balance. A-B. The expressions of Mfn2, OPA1, p-Drp1, Parkin, and Pink1 were detected by immunohistochemistry in heart tissue of sham and MI model groups mice with cardiac-specific knockdown of KMO expression (n = 5) (400X magnification, scale bar, 50 µm). C-D. Western blots and quantitative results of Beclin1, P62, and LC3I/II in heart tissue of sham and MI model groups mice with cardiac-specific knockdown of KMO expression (n = 5). Data are presented as mean ± SD. #p < 0.05, ##p < 0.01 vs. sham + AAV-NC, *p < 0.05, **p < 0.01 vs. model + AAV-NC; ns means no significance.
Ginsenoside Rb3 was screened as a novel KMO inhibitor and ameliorated myocardial ischemia injury
The above results suggested that KMO might serve as a potential target for the treatment of MI injury, thus, molecular docking technology was further conducted to screen novel KMO inhibitors based on the Traditional Chinese Medicine Systems Pharmacology Database and Analysis Platform. The virtual screening results were sorted according to the predicted binding free energy, which ranged from -5.8 to -13.0 kcal/mol. Protein-ligand interaction analysis showed that hydrogen bonds and hydrophobic interaction were the main modes of interaction between these compounds and KMO. In these compounds, Ginsenoside Rb3 showed the strongest binding affinity with human KMO, suggesting that ginsenoside Rb3 might be a new potential KMO inhibitor (Table 3). Meanwhile, the molecular docking results revealed that ginsenoside Rb3 bound to the binding pocket, which might form hydrophobic interactions with Arg40, Ser53, and Gln283 of KMO (the docking pose was shown in Figure 7A). To further confirm whether ginsenoside Rb3 directly targeted KMO, the surface plasmon resonance (SPR) technology was performed. Ginsenoside Rb3 exhibited a strong binding affinity for KMO with an estimated equilibrium dissociation constant of 1.956 μM (Figure 7B). Besides, the interaction pattern between compounds with the top 5 affinities of KMO except for ginsenoside Rb3 was shown in Supplementary Figure S13. Subsequently, the inhibitory effect of ginsenoside Rb3 on KMO was verified in vivo and in vitro. The results suggested that the expression and enzymatic activity of KMO were significantly inhibited by ginsenoside Rb3 in both myocardial tissues and cardiomyocytes (Figure 7C-E).
The docking results between compounds with KMO
| Protein-ligand | AutoDock score | Hydrogen-bond interaction | Hydrophobic interaction |
|---|---|---|---|
| Ginsenoside Rb3 | -13.0 kcal/ mol | Gly16 Gly17 Leu18 Val19 Gly20 Arg40 Ser53 Arg111 Asn115 Gly168 Tyr194 Glu196 Thr198 Thr134 Asp276 Phe278 Leu279 Leu280 Pro281 Ala282 Gln283 | Arg40 Ser53 Gln283 |
| Astragaloside IV | -11.0 kcal/ mol | Leu18 Val19 Ser53 Arg111 Gly168 Asp304 | Ala39 leu136 Asp167 Ala169 Tyr170 |
| Salvianic acid C | -10.1 kcal/ mol | Arg61 Ser287 His307 Val310 GLU322 Arg349 | Phe293 His307 Ile309 Phe313 Gln315 Ala319 |
| Ginsenoside Rg3 | -9.2 kcal/ mol | Ala39 Arg135 Asp167 Thr172 Asp304 Ile309 Gly314 Gln315 Gly316 | Ile14 Asp167 Ala169 Tyr170 |
| Cryptotanshinone | -9.0 kcal/ mol | Glu322 Arg349 | His307 Ile309 Asn318 Asp353 |
| Tanshinone IIA | -8.9 kcal/ mol | Glu322 Arg349 | His307 Ile309 Ala319 |
| Emodin | -8.9 kcal/ mol | Ser59 ARG61 Gln315 Glu322 Arg349 | Ile309 Gln315 Ala319 |
| Tanshinol A | -8.7 kcal/ mol | Arg61 Phe313 Gln315 Glu322 Arg349 | His307 Ile309 Ala319 |
| Salvianic acid D | -8.7 kcal/ mol | Ile286 Ile309 Gly316 Met317 | Ala57 Tyr194 Pro311 |
| Hydroxytanshinone IIA | -8.6 kcal/ mol | Ile309 Gly316 | Leu56 Ile224 Pro311 |
| Notoginsenoside R1 | -8.5 kcal/ mol | Leu136 Asp167 Thr172 Tyr194 Gln283 Pro284 Ile286 Ile309 | Phe295 |
| Apigenin | -8.4 kcal/ mol | Ser287 His307 Arg349 | Phe293 Ile309 |
| Calycosin | -8.3 kcal/ mol | Ser357 Asp358 | Ile224 Leu226 Asp358 Leu359 |
| Formononetin | -8.3 kcal/ mol | Thr236 Ser357 Asp358 | Leu226 Pro311 Asp358 Leu359 |
| Luteolin | -8.3 kcal/ mol | Ser53 Pro284 Ile286 | Ala169 Tyr194 Thr136 Phe238 Ile286 |
| Ginsenoside Rb1 | -8.2 kcal/ mol | Gly17 Leu18 Val19 Gly20 Arg40 Thr43 Ser53 Arg111 Glu112 Ala169 | Tyr194 Leu280 Gln283 |
| Rosmarinic acid | -8.2 kcal/ mol | Ser53 Ala57 Ile286 Asp304 Ile309 Met317 Asn318 | Met285 Pro311 |
| Neotanshinone A | -7.9 kcal/ mol | Arg61 Glu322 Arg349 | Ile309 Gln315 Asn318 Ala319 |
| Tanshinol B | -7.4 kcal/ mol | Ser59 Gln315 Ser357 | Gln104 Ile106 Gln315 Ala319 |
| Salvianic acid A | -6.5 kcal/ mol | Tyr37 Ala39 Leu136 Asp167 Tyr170 Thr172 | Leu136 Asp167 |
| Caffeic acid | -6.4 kcal/ mol | Ser53 Asp276 Leu279 Leu280 Pro281 | Tyr194 Glu196 Leu279 Leu280 |
| Protocatechuic acid | -6.4 kcal/ mol | Tyr37 Glu38 Ala39 Leu136 Tyr170 Thr172 | Ile14 Leu136 Asp167 |
| 3,4-Dihydroxyphenyl lactic acid (DLA) | -6.1 kcal/ mol | Tyr37 Ala39 Leu136 Asp167 Thr172 | Ala39 Arg135 Leu136 Asp167 |
| Protocatechualdehyde | -5.8 kcal/ mol | Glu38 Ala39 His134 Leu136 Thr172 | Ile14 Leu136 Asp167 |
Ginsenoside Rb3 is screened as a novel KMO inhibitor and ameliorated cardiac injury in MI. A. A molecular docking model carried out by Autodock vina revealed that ginsenoside Rb3 bonded to the KMO. B. SPR analysis of the binding affinity of ginsenoside Rb3 and KMO protein. Apparent equilibrium dissociation constants (Kd) were calculated as the ratio of Kd/Ka. The Kd (mol/L) value between Ginsenoside Rb3 and KMO was 1.951x10-6 mol/L. C-D. Representative western blot results in the analysis of KMO in heart tissues of MI model group mice and H9c2 cells (n = 5). E. The enzyme activity of KMO in sham and MI model group mice treated with ginsenoside Rb3 (n = 10-13, one-way ANOVA followed by Bonferroni post hoc test). F-J. HE staining and Masson staining cross-sections of heart tissues (n = 5) (200X magnification, scale bar, 50 µm) and representative echocardiographic graphs were shown (n =5-6), respectively. K-L. Serum biochemical indicators contents include BNP and LDH in sham and MI model groups mice treated with ginsenoside Rb3, and were detected by ELISA (n = 8-12). M. Representative transmission electron microscope images of heart tissues in sham and MI model groups mice treated with ginsenoside Rb3 (n = 3). Data are presented as mean ± SD. #p < 0.05, ##p < 0.01 vs. sham or control group, *p < 0.05, **p < 0.01 vs. model or OGD group; ns means no significance.
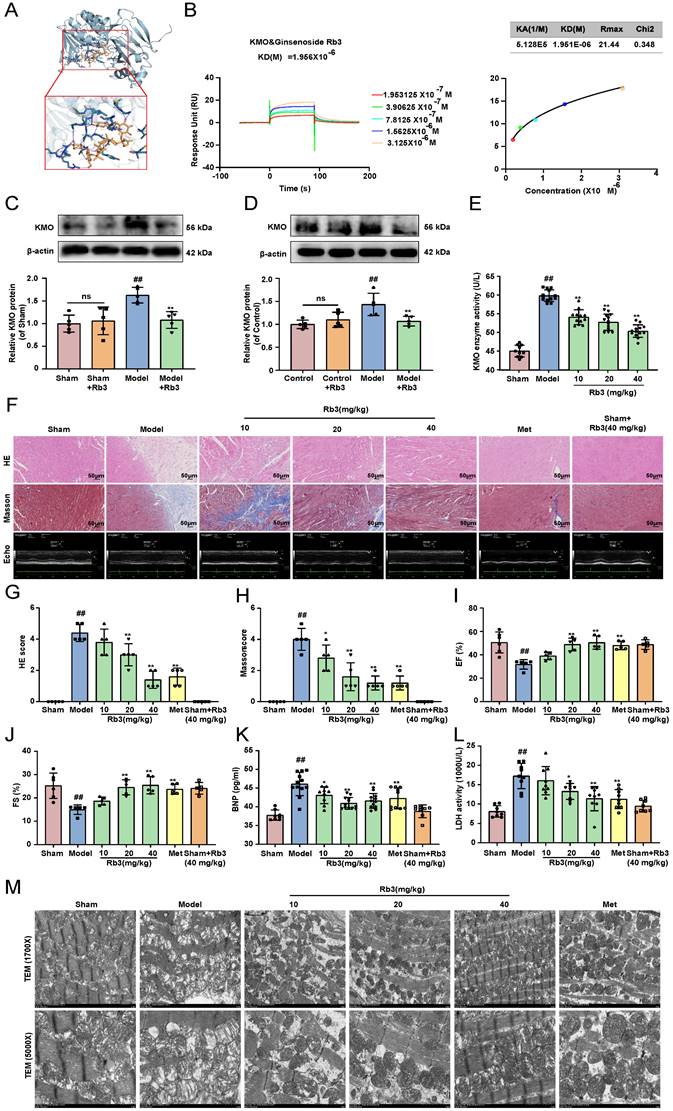
We next investigated the effect of ginsenoside Rb3 on the myocardial ischemia-induced cardiac injury. Ginsenoside Rb3 was orally administrated at doses of 10, 20, and 40 mg/kg/day for 8 days after CAL for one week. As illustrated in Figure 7F-J, ginsenoside Rb3 could effectively improve cardiac pathological injury and functions (LV ejection fraction and LV fractional shortening), as well as significantly ameliorate the biochemical indexes (BNP and LDH) related to myocardial injury in serum (Figure 7K-L). Simultaneously, it also exhibited a prominent effect on ameliorating myocardial ultrastructure damage and mitochondrial morphology (Figure 7M). In addition, ginsenoside Rb3 could also significantly increase cell viability in the OGD-induced cardiomyocytes injury (Supplementary Figure S8C). All the above results indicated that ginsenoside Rb3 may be a novel KMO inhibitor and is effective in improving the cardiac damage of MI.
Ginsenoside Rb3 attenuated myocardial ischemia injury through regulating KMO mediated mitochondrial fusion and fission
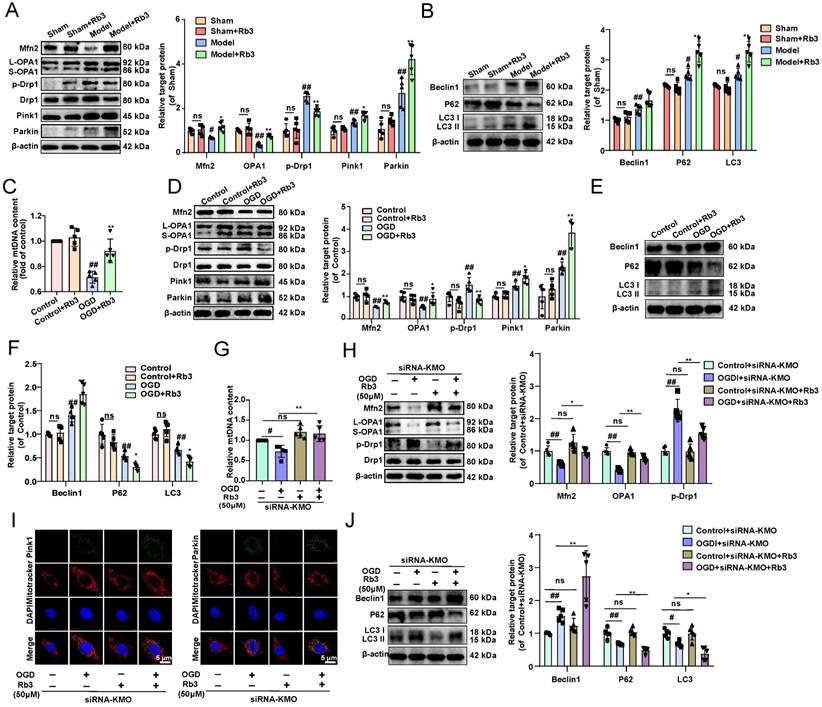
What's more, the effect of ginsenoside Rb3 on mitochondrial fusion and fission was investigated in vitro and in vivo. And we found that ginsenoside Rb3 could significantly reversed the trend of OGD-induced reduction in mtDNA number, as well as increase the expression of Mfn2, and OPA1 and decrease the phosphorylation of Drp1 in OGD-induced cardiomyocytes injury and MI injury. In addition, mitochondrial autophagy proteins of Pink1 and Parkin as well as the downstream proteins Beclin1, P62, and LC3II were further altered by ginsenoside Rb3 administration (Figure 8A-F, Supplementary Figure S14). These results consisted of the effect of the inhibitor of KMO and cardiac-specific KMO knockdown in MI mice. Besides, KMO-siRNA evidently enhanced the regulation of ginsenoside Rb3 on mtDNA number, mitochondrial fusion/fission and further promoted mitochondrial autophagy (Figure 8G-J). Taken together, all results indicated that ginsenoside Rb3 could attenuate MI injury through maintaining mitochondrial fusion and fission balance and KMO might exert a critical role in its effect.
Ginsenoside Rb3 attenuates MI through regulating KMO mediated mitochondrial fusion/fission. A. Western blots and quantitative results of Mfn2, OPA1, p-Drp1, Drp1, Parkin, and Pink1 in sham and MI model groups mice treated with ginsenoside Rb3 (n = 5). B. Western blots and quantitative results of Beclin1, P62, and LC3I/II in sham and MI model groups mice treated with ginsenoside Rb3 (n = 5). C. RT-PCR analysis of mtDNA content in the OGD-induced H9c2 cells injury model treated with ginsenoside Rb3 (n = 5). D. Western blots and quantitative results of Mfn2, OPA1, p-Drp1, Drp1, Parkin, and Pink1 in the OGD-induced H9c2 cells injury model treated with ginsenoside Rb3 (n = 5). E-F. Western blots and quantitative results of Beclin1, P62, and LC3I/II in the OGD-induced H9c2 cells injury model treated with ginsenoside Rb3 (n = 5). G. RT-PCR analysis of mtDNA content in the OGD-induced H9c2 cells injury model treated with siRNA-KMO or ginsenoside Rb3 (n = 5). H. Western blots and quantitative results of Mfn2, OPA1, p-Drp1, and Drp1 in the OGD-induced H9c2 cells injury model treated with siRNA-KMO or ginsenoside Rb3 (n = 5). I. Representative immunofluorescence images of Pink1 and Parkin in the OGD-induced H9c2 cells injury model treated with siRNA-KMO or ginsenoside Rb3 (n = 5). J. Western blots and quantitative results of Beclin1, P62, and LC3I/II in the OGD-induced H9c2 cells injury model treated with siRNA-KMO or ginsenoside Rb3 (n = 5). Data are presented as mean ± SD. #p < 0.05, ##p < 0.01 vs. sham or control or control + siRNA-KMO group, *p < 0.05, **p < 0.01 vs. model or OGD or OGD + siRNA-KMO group; ns means no significance.
Discussion
In our present study, high throughput metabolomics analysis showed that xanthinic acid may be a new metabolic marker of myocardial ischemia. Furthermore, the combined metabolic and transcriptional data demonstrated that KMO is a key enzyme regulating the abnormal elevation of xanthurenic acid in MI. Pharmacological or heart-specific inhibition of KMO significantly attenuated OGD-induced cardiomyocytes injury, as well as ligation induced myocardial ischemic injury.
Early identification and intervention of MI could delay the progression into myocardial infarction, heart failure, and some other cardiovascular diseases. It is of great significance to find relevant biomarkers for clinical diagnosis and treatment. Evaluation of myocardial pathological injury and cardiac function at different ischemic time points showed that significant myocardial injury occurred around 2-3 weeks after ischemia. And our study further revealed systemic metabolic alterations and constructed a metabolic network map of MI through untargeted metabolomics. Significant changes were found in urea, taurine, hemolysase, niacin, xanthurenic acid, 2-arachidonylglycerophospcholine, fructose-lysine, and 7-methylguanine in MI mice. Among them, urea [35, 36], LysoPE [37, 38], niacin [39], and taurine [40] have been clinically reported to be highly associated with myocardial infarction and other cardiovascular diseases. And it also included many metabolites whose specific functions in MI are still unclear, such as xanthurenic acid, 2-arachidonylglycerophospcholine, 7-methylguanine, fructoysine, etc. Additionally, targeted metabolomics was conducted to identify potential metabolic markers in MI mice and patients. And we found that both xanthurenic acid and 2-arachidonylphosphocholine significantly elevated at the early stage after MI injury. This is basically consistent with the target verification results of MI clinical specimens. Then, the sensitivity and specificity of these two potential metabolic markers were evaluated, and xanthurenic acid showed great potential as a diagnostic marker for MI.
Xanthurenic acid is mainly involved in the tryptophan/cyuridine metabolic pathway associated with neuropsychiatric and immune diseases. In recent years, aberrant activation of the Trp/Kynurenine pathway has been found to be highly associated with inflammation and cardiovascular diseases [41, 42]. Of note, 3-hydroxykynurenine/xanthurenic acid was associated with increased mortality in patients with heart failure [43]. However, it is unclear how xanthurenic acid participates in the pathologic processes of MI. To investigate the function of xanthurenic acid in vivo, its effects on six major organs were observed. And we found that elevation of xanthurenic acid significantly induced myocardial injury and fibrosis in sham mice but had no obvious effect on other organs. To further explore how xanthurenic acid promoted myocardial injury, we investigated its effects on two important cardiomyocyte death pathways, including apoptosis and ferroptosis [44]. And the results suggested that high xanthurenic acid levels could trigger myocardial injury and aggravate the pathology of myocardial infarction, which might be related to the activation of cardiomyocyte apoptosis and ferroptosis.
Based on the above results, transcriptomic analysis was further carried out to screen out the specific gene clusters of MI and to further explore the key regulatory enzymes of xanthurenic acid in heart tissue. Enrichment analysis showed that MI-specific genes were mainly involved in oxidative phosphorylation, tricarboxylic acid cycle (TCA), electron transport chain, and nucleoside triphosphate metabolism. Oxidative phosphorylation occurs mainly in the inner mitochondrial membrane of eukaryotic cells or the cytoplasm of prokaryotic cells during biochemical processes. It is the energy released by substances in the body through the oxidation process of the respiratory chain and supplies ADP and inorganic phosphate to synthesize ATP [45], which is consistent with TCA [46], and both play a key role in the body's energy metabolism. And our results indicated that abnormal energy metabolism mainly occurred in myocardial ischemia. Combined with previous transcriptome data and the expression results in different tissues of MI mice, we found that KMO expression significantly increased only in heart tissues. Meanwhile, further application of KMO inhibitors and AAV technology demonstrated that KMO inhibition significantly reduced the increase of xanthurenic acid in vivo. These results suggested that the significant increase in KMO expression might be a key factor in the abnormal increase of xanthurenic acid content in MI.
Previous studies have shown that KMO is a potential therapeutic target for several neurological diseases, such as Alzheimer's disease (AD) and Huntington's disease (HD) [47, 48]. But the relationship between KMO and MI injury is unclear. This study firstly demonstrated that KMO inhibition effectively ameliorated myocardial ischemia injury. Interestingly, heart-specific inhibition of KMO significantly prevented myocardial ferroptosis and apoptosis. These results further confirmed that KMO might serve as a new therapeutic target for MI injury. KMO is a protein specifically localized on the outer mitochondrial membrane (OMM) [49]. Over-activation of KMO has been found to induce ROS production and mitochondrial dysfunction in human primary neurons [50]. Notably, mitochondrial dysfunction is a key pathological mechanism in cardiovascular diseases, especially in the development of MI injury. The majority (about 95%) of ATP consumed by the heart comes from oxidative metabolism in mitochondria, which accounts for about 1/3 of the volume of adult cardiomyocytes [51]. Meanwhile, a previous study has found that KMO was involved in the regulation of mitochondrial dynamics and mitochondrial autophagy in Drosophila melanogaster under physiological conditions [34]. Therefore, we investigated the effect of KMO inhibition on mitochondrial fission and fusion. And we found that the expression of Drp1 (pSer616) decreased, and the expression of Mfn2 and L-OPA1/S-OPA1 markedly increased after KMO inhibition, which suggested that KMO inhibition prevented mitochondrial excessive fission and promoted mitochondrial fusion. Modest mitochondrial fission inhibition and accelerated mitochondrial fusion have been shown to protect the heart from ischemic damage [52].
What's more, the imbalance of mitochondrial fusion and fission leading to mitochondrial fragmentation is the premise of mitochondrial autophagy [53, 54]. In mammals, Pink1/Parkin pathway is a classical way to regulate mitochondrial autophagy [55]. The activation of the Pink1/Parkin pathway could promote downstream autophagosomes to bind to damaged mitochondria, thereby clearing these mitochondria [56, 57]. Subsequently, inhibition KMO has been demonstrated to significantly activate the Pink1/Parkin pathway of mitophagy, as well as accelerate the occurrence of its downstream autophagy. Above results indicated that KMO inhibition was essential in the protection of myocardial ischemia injury by maintaining mitochondrial fusion and fission balance.
Given the critical role of KMO in myocardial ischemia injury, potential KMO inhibitors were further screened by molecular docking and SPR techniques. And we found that ginsenoside Rb3 had a certain binding affinity with KMO, and its inhibitory effect on KMO was verified. Ginsenoside Rb3, the main active component in ginseng, has been widely used to treat cardiovascular diseases [58, 59]. Recent studies have reported that ginsenoside Rb3 can regulate mitochondrial function and exert anti-apoptotic and antioxidant effects [13, 59]. Regarding the role of KMO in MI, whether ginsenoside Rb3 could exert cardioprotective effect via KMO remained to be further elucidated. And we observed that ginsenoside Rb3 significantly inhibited mitochondrial fission and accelerated mitochondrial fusion and autophagy, whereas, KMO inhibition could significantly enhance the effect of ginsenoside Rb3. These findings might indicate that KMO is essential or even a key functional protein for the cardioprotection of ginsenoside Rb3.
However, there are still some limitations in our current study. Only one metabolomics technology cannot cover all endogenous metabolites in organisms, and LC-MS alone may lose some key information. Therefore, it is necessary to further combine NMR, GC-MS, and other technologies to explain the metabolic characteristics of MI from a wider range of metabolic pathways. Additionally, many specific metabolites were selected, but only four were targeted for identification. Although key regulatory enzymes regulating the significant increase of XA were found, we only used inhibitors of KMO and AAV-shRNA to initially explore its heart-related function, and further cardiac conditional knockout mice are needed to confirm.
Conclusion
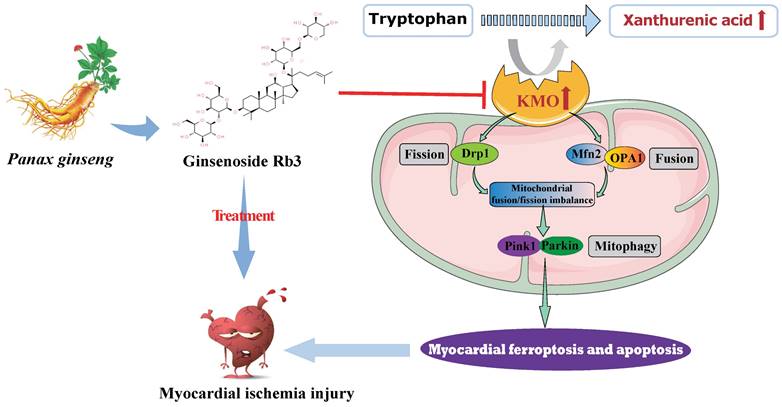
In summary, metabolomics was adopted in this study to reveal metabolite signatures and the potential mechanisms closely related to MI injury, and four potential metabolic markers were verified at animal and clinical levels. Notably, we firstly identified the key role of xanthurenic acid in MI. What's more, we interestingly found that KMO overexpression was the key factor leading to the elevation of xanthurenic acid, and inhibition of KMO could effectively alleviate myocardial injury by regulating mitochondrial fission/fusion and appropriately activating the occurrence of autophagy. These observations will not only provide a novel metabolic marker for MI injury but also indicate that targeting KMO may represent an unrecognized therapeutic intervention for MI and other related cardiovascular diseases in the future. Meanwhile, ginsenoside Rb3 was screened as a novel inhibitor of KMO, and it was also found to have an excellent therapeutic effect on MI injury through regulating KMO mediated mitochondrial fusion and fission, which showed the great clinical potential to be seen as a candidate for the treatment of clinical MI (Figure 9).
Schematic illustration of inhibition of KMO alleviated myocardial ischemic injury through regulating mitochondrial fission and fusion, and ginsenoside Rb3 was screened as a novel inhibitor of KMO.
Abbreviations
MI: myocardial ischemia; CAL: coronary artery ligation; HPLC-Q/TOF-MS: high-performance liquid chromatography and quadrupole time-of-flight mass spectrometry; KAT1: kynurenine aminotransferase 1; KMO: kynurenine 3-monooxygenase; CVD: cardiovascular disease; XA: xanthurenic acid; SV: stroke volume; IVSd: interventricular septum in diastole; LVIDd: left ventricle interior diameter in diastole; LVPWd: left ventricle posterior wall in diastole; LV Vold: left ventricular volume in diastole; LVPEP: left ventricular pre-ejection period; LVET: left ventricular ejection time; EF: ejection fraction; FS: fractional shortening; LV Mass: left ventricular mass; PEP: pre-ejection periodquality; QC: control samples; TICs: total ion chromatograms; OPLS-DA: orthogonal partial least-squares discriminant analysis; MRM: multiple-reaction-monitoring; pAAV9: Serotype 9 adeno-associated virus; NC: negative control; Mfn2: mitofusin-2; OPA1: Optic Atrophy 1; Drp1: Dynamin-relatedprotein1; DAPI: 4':6-Diamidino-2-phenylindole; FHC: Ferritin heavy chain; FLC: Ferritin light chain; GPX4: Glutathione peroxidase; Pink1: PTEN induced putative kinase 1; AR: aspect ratio; FF: form factor; DMEM: Dulbecco's modified eagle medium; FBS: fetal bovine serum; OGD: oxygen and glucose deprivation; SPR: Surface Plasmon Resonance; ROC: receiver operating characteristic; AUC: area under curve; BNP: B type natriuretic peptide; hs-CRP: hypersensitive C-reactive protein; MDA: Malondialdehyde; TNF-α: tumor necrosis factor alpha; LDH: lactate dehydrogenase; NO: nitric oxide; TCA: tricarboxylic acid cycling; AD: Alzheimer's disease; HD: Huntington's disease; OMM: outer mitochondrial membrane; ROS: reactive oxygen species.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We thank Xiaonan Ma (School of Basic Medicine and Clinical Pharmacy, China Pharmaceutical University) for the help with the optical microscope.
Funding
This research work was supported by the National Natural Science Foundation of China (No. 82274231, 82104437, 81973506), the Natural Science Foundation of Jiangsu Province (BK20210431), the China Postdoctoral Science Foundation (2021M693519 and 2022T150728), State Key Laboratory for Chemistry and Molecular Engineering of Medicinal Resources (Guangxi Normal University), (CMEMR2023-B12) and Young Talent Support Project of Jiangsu Association for Science and Technology (TJ-2022-025) and Qinglan Project of Jiangsu Province.
Ethical approval and consent to participate
The animal study was approved by the Animal Ethics Committee of China Pharmaceutical University and the Laboratory Animal Management Committee of Jiangsu Province (Approval No. SYXK2021-0010). The human study was approved by the Ethical Committee (No. 2019NL-089-02, Jiangsu Province Hospital of Chinese Medicine, Registered 27 September 2019) and it was executed in conformity with the Declaration of Helsinki (as revised in 2013). Before the trial began, the patients signed a written informed consent form.
Author contributions
FL, FML, and BYY conceived the study. QL and LLW performed the metabolomic analysis. QL, SHD and XZZ established the animal model and performed the experiments. XZZ, ZYF, FML, and JPK collected the samples. QL, LLW, SHD, and XZZ interpreted the data. QL and ZYF aided in the data analysis and wrote the paper. All authors read and approved the final paper.
Competing Interests
The authors have declared that no competing interest exists.
References
1. NCD countdown 2030. Worldwide trends in non-communicable disease mortality and progress towards sustainable development goal target 3.4. Lancet. 2018;392:1072-88
2. White HD, Chew DP. Acute myocardial infarction. Lancet. 2008;372:570-84
3. Deedwania PC, Carbajal EV. Role of myocardial oxygen demand in the pathogenesis of silent ischemia during daily life. Am J Cardiol. 1992;70:19F-24F
4. Favero G, Franceschetti L, Buffoli B, Moghadasian MH, Reiter RJ, Rodella LF. et al. Melatonin: Protection against age-related cardiac pathology. Ageing Res Rev. 2017;35:336-49
5. Gouveia M, Xia K, Colón W, Vieira SI, Ribeiro F. Protein aggregation, cardiovascular diseases, and exercise training: Where do we stand? Ageing Res Rev. 2017 40
6. Doenst T, Haverich A, Serruys P, Bonow RO, Kappetein P, Falk V. et al. PCI and CABG for treating stable coronary artery disease: JACC review topic of the week. J Am Coll Cardiol. 2019;73:964-76
7. Conti CR, Bavry AA, Petersen JW. Silent ischemia: clinical relevance. J Am Coll Cardiol. 2012;59:435-41
8. Cecchi AC, Dovellini EV, Marchi F, Pucci P, Santoro GM, Fazzini PF. Silent myocardial ischemia during ambulatory electrocardiographic monitoring in patients with effort angina. J Am Coll Cardiol. 1983;1:934-9
9. Gutterman DD. Silent myocardial ischemia. Circ J. 2009;73:785-97
10. Verbree-Willemsen L, Grobben RB, van Waes JA, Peelen LM, Nathoe HM, van Klei WA. et al. Causes and prevention of postoperative myocardial injury. Eur J Prev Cardiol. 2019;26:59-67
11. Yellon DM, Hausenloy DJ. Myocardial reperfusion injury. N Engl J Med. 2007;357:1121-35
12. Chen DQ, Cao G, Chen H, Liu D, Su W, Yu XY. et al. Gene and protein expressions and metabolomics exhibit activated redox signaling and wnt/β-catenin pathway are associated with metabolite dysfunction in patients with chronic kidney disease. Redox Biol. 2017;12:505-21
13. Chen X, Wang Q, Shao M, Ma L, Guo D, Wu Y. et al. Ginsenoside Rb3 regulates energy metabolism and apoptosis in cardiomyocytes via activating PPARα pathway. Biomed Pharmacother. 2019;120:109487
14. Johnson CH, Ivanisevic J, Siuzdak G. Metabolomics: beyond biomarkers and towards mechanisms. Nat Rev Mol Cell Biol. 2016;17:451-9
15. Sabatine MS, Liu E, Morrow DA, Heller E, McCarroll R, Wiegand R. et al. Metabolomic identification of novel biomarkers of myocardial ischemia. Circulation. 2005;112:3868-75
16. Shah SH, Kraus WE, Newgard CB. Metabolomic profiling for the identification of novel biomarkers and mechanisms related to common cardiovascular diseases: form and function. Circulation. 2012;126:1110-20
17. Tang WHW, Wang Z, Cho L, Brennan DM, Hazen SL. Diminished global arginine bioavailability and increased arginine catabolism as metabolic profile of increased cardiovascular risk. J Am Coll Cardiol. 2009;53:2061-7
18. Zhang L, Wei T-T, Li Y, Li J, Fan Y, Huang F-Q. et al. Functional metabolomics characterizes a key role for acetylneuraminic acid in coronary artery diseases. Circulation. 2018;137:1374-90
19. Gao E, Lei YH, Shang X, Huang ZM, Zuo L, Boucher M. et al. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ Res. 2010;107:1445-53
20. Guo Y, Sui JY, Kim K, Zhang Z, Qu XA, Nam YJ. et al. Cardiomyocyte homeodomain-interacting protein kinase 2 maintains basal cardiac function via extracellular signal-regulated kinase signaling. Circulation. 2019;140:1820-33
21. Guo H, Zhang Z, Su Z, Sun C, Zhang X, Zhao X. et al. Enhanced anti-tumor activity and reduced toxicity by combination andrographolide and bleomycin in ascitic tumor-bearing mice. Eur J Pharmacol. 2016;776:52-63
22. Zou XH, Li HM, Wang S, Leski M, Yao YC, Yang XD. et al. The effect of 3-hydroxybutyrate methyl ester on learning and memory in mice. Biomaterials. 2009;30:1532-41
23. Lai Q, Yuan G, Wang H, Liu Z, Kou J, Yu B. et al. Metabolomic profiling of metoprolol-induced cardioprotection in a murine model of acute myocardial ischemia. Biomed Pharmacother. 2020;124:109820
24. Peron G, Sut S, Dal Ben S, Voinovich D, Dall'Acqua S. Untargeted UPLC-MS metabolomics reveals multiple changes of urine composition in healthy adult volunteers after consumption of curcuma longa L. extract. Food Res Int. 2020;127:108730
25. Gao F, Kataoka M, Liu N, Liang T, Huang ZP, Gu F. et al. Therapeutic role of miR-19a/19b in cardiac regeneration and protection from myocardial infarction. Nat Commun. 2019;10:1802
26. Xu T, Dong Q, Luo Y, Liu Y, Gao L, Pan Y. et al. Porphyromonas gingivalis infection promotes mitochondrial dysfunction through Drp1-dependent mitochondrial fission in endothelial cells. Int J Oral Sci. 2021;13:28
27. Wan S, Cui Z, Wu L, Zhang F, Liu T, Hu J. et al. Ginsenoside Rd promotes omentin secretion in adipose through TBK1-AMPK to improve mitochondrial biogenesis via WNT5A/Ca2+ pathways in heart failure. Redox Biol. 2023;60:102610
28. Canseco DC, Kimura W, Garg S, Mukherjee S, Bhattacharya S, Abdisalaam S. et al. Human ventricular unloading induces cardiomyocyte proliferation. J Am Coll Cardiol. 2015;65:892-900
29. Pang LZ, Ju AC, Zheng XJ, Li F, Song YF, Zhao Y. et al. YiQiFuMai powder injection attenuates coronary artery ligation-induced myocardial remodeling and heart failure through modulating MAPKs signaling pathway. J Ethnopharmacol. 2017;202:67-77
30. Pereira AHM, Cardoso AC, Consonni SR, Oliveira RR, Saito A, Vaggione MLB. et al. MEF2C repressor variant deregulation leads to cell cycle re-entry and development of heart failure. EBioMedicine. 2020;51:102571
31. Gong SS, Liu JC, Wan SY, Yang WW, Zhang YY, Yu BY. et al. Schisandrol A attenuates myocardial ischemia/reperfusion-induced myocardial apoptosis through upregulation of 14-3-3. Oxid Med Cell Longev. 2021;2021:5541753
32. Yu R, Chen L, Lan R, Shen R, Li P. Computational screening of antagonists against the SARS-CoV-2 (COVID-19) coronavirus by molecular docking. Int J Antimicrob Agents. 2020;56:106012
33. Huang Y, Yu SH, Zhen WX, Cheng T, Wang D, Lin JB. et al. Tanshinone I, a new EZH2 inhibitor restricts normal and malignant hematopoiesis through upregulation of MMP9 and ABCG2. Theranostics. 2021;11:6891-904
34. Maddison DC, Alfonso-Núñez M, Swaih AM, Breda C, Campesan S, Allcock N. et al. A novel role for kynurenine 3-monooxygenase in mitochondrial dynamics. PLoS Genet. 2020;16:e1009129
35. Wang P, Yao J, Xie Y, Luo M. Gender-specific predictive markers of poor prognosis for patients with acute myocardial infarction during a 6-month follow-up. J Cardiovasc Transl Res. 2020;13:27-38
36. Di Tanna GL, Wirtz H, Burrows KL, Globe G. Evaluating risk prediction models for adults with heart failure: A systematic literature review. PLoS One. 2020;15:e0224135
37. Marcinkiewicz-Siemion M, Ciborowski M, Ptaszynska-Kopczynska K, Szpakowicz A, Lisowska A, Jasiewicz M. et al. LC-MS-based serum fingerprinting reveals significant dysregulation of phospholipids in chronic heart failure. J Pharm Biomed Anal. 2018;154:354-63
38. Tang HY, Wang CH, Ho HY, Wu PT, Hung CL, Huang CY. et al. Lipidomics reveals accumulation of the oxidized cholesterol in erythrocytes of heart failure patients. Redox Biol. 2018;14:499-508
39. Zambon A, Zhao XQ, Brown BG, Brunzell JD. Effects of niacin combination therapy with statin or bile acid resin on lipoproteins and cardiovascular disease. Am J Cardiol. 2014;113:1494-8
40. Oudit GY, Trivieri MG, Khaper N, Husain T, Wilson GJ, Liu P. et al. Taurine supplementation reduces oxidative stress and improves cardiovascular function in an iron-overload murine model. Circulation. 2004;109:1877-85
41. Niinisalo P, Raitala A, Pertovaara M, Oja SS, Lehtimäki T, Kähönen M. et al. Indoleamine 2,3-dioxygenase activity associates with cardiovascular risk factors: the Health 2000 study. Scand J Clin Lab Invest. 2008;68:767-70
42. Wirleitner B, Rudzite V, Neurauter G, Murr C, Kalnins U, Erglis A. et al. Immune activation and degradation of tryptophan in coronary heart disease. Eur J Clin Invest. 2003;33:550-4
43. Lund A, Nordrehaug JE, Slettom G, Solvang S-EH, Pedersen EKR, Midttun Ø. et al. Plasma kynurenines and prognosis in patients with heart failure. PLoS One. 2020;15:e0227365
44. Li N, Jiang W, Wang W, Xiong R, Wu X, Geng Q. Ferroptosis and its emerging roles in cardiovascular diseases. Pharmacol Res. 2021;166:105466
45. Sahoo A, Lee B, Boniface K, Seneschal J, Sahoo SK, Seki T. et al. MicroRNA-211 regulates oxidative phosphorylation and energy metabolism in human vitiligo. J Invest Dermatol. 2017;137:1965-74
46. Martínez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. 2020;11:102
47. Chang KH, Wu YR, Chen YC, Chen CM. Plasma inflammatory biomarkers for Huntington's disease patients and mouse model. Brain Behav Immun. 2015;44:121-7
48. Giorgini F, Huang S-Y, Sathyasaikumar KV, Notarangelo FM, Thomas MAR, Tararina M. et al. Targeted deletion of kynurenine 3-monooxygenase in mice: A new tool for studying kynurenine pathway metabolism in periphery and brain. The J Biol Chem. 2013;288:36554-66
49. Hirai K, Kuroyanagi H, Tatebayashi Y, Hayashi Y, Hirabayashi-Takahashi K, Saito K. et al. Dual role of the carboxyl-terminal region of pig liver L-kynurenine 3-monooxygenase: mitochondrial-targeting signal and enzymatic activity. J Biochem. 2010;148:639-50
50. Castellano-Gonzalez G, Jacobs KR, Don E, Cole NJ, Adams S, Lim CK. et al. Kynurenine 3-monooxygenase activity in human primary neurons and effect on cellular bioenergetics identifies new neurotoxic mechanisms. Neurotox Res. 2019;35:530-41
51. Hall AR, Burke N, Dongworth RK, Kalkhoran SB, Dyson A, Vicencio JM. et al. Hearts deficient in both Mfn1 and Mfn2 are protected against acute myocardial infarction. Cell Death Dis. 2016;7:e2238
52. Zheng Z, Lei C, Liu H, Jiang M, Zhou Z, Zhao Y. et al. A ROS-responsive liposomal composite hydrogel integrating improved mitochondrial function and pro-angiogenesis for efficient treatment of myocardial infarction. Adv Healthc Mater. 2022: e2200990.
53. Wai T, Langer T. Mitochondrial dynamics and metabolic regulation. Trends Endocrinol Metab. 2016;27:105-17
54. Quiles JM, Gustafsson ÅB. The role of mitochondrial fission in cardiovascular health and disease. Nat Rev Cardiol. 2022;19:723-36
55. Gladkova C, Maslen SL, Skehel JM, Komander D. Mechanism of parkin activation by PINK1. Nature. 2018;559:410-4
56. Sciarretta S, Maejima Y, Zablocki D, Sadoshima J. The role of autophagy in the heart. Annu Rev Physiol. 2018 80
57. Li G, Yang J, Yang C, Zhu M, Jin Y, McNutt MA. et al. PTENα regulates mitophagy and maintains mitochondrial quality control. Autophagy. 2018;14:1742-60
58. Chen X, Liu T, Wang Q, Wang H, Xue S, Jiang Q. et al. Synergistic effects of ginsenoside Rb3 and ferruginol in ischemia-induced myocardial infarction. Int J Mol Sci. 2022 23
59. Sun J, Yu X, Huangpu H, Yao F. Ginsenoside Rb3 protects cardiomyocytes against hypoxia/reoxygenation injury via activating the antioxidation signaling pathway of PERK/Nrf2/HMOX1. Biomed Pharmacother. 2019;109:254-61
Author contact
![]() Corresponding authors: Fuming Liu, Affiliated Hospital of Nanjing University of Chinese Medicine, Nanjing, 210029, Jiangsu, China, E-mail: doctor.liufumingcom; Boyang Yu, Jiangsu Key Laboratory of TCM Evaluation and Translation Research, Research Center for Traceability and Standardization of TCMs, School of Traditional Chinese Pharmacy, China Pharmaceutical University, Email: boyangyu59com; Fang Li, Jiangsu Key Laboratory of TCM Evaluation and Translational Research, Research Center for Traceability and Standardization of TCMs, School of Traditional Chinese Pharmacy, China Pharmaceutical University, 639 Longmian Road, Nanjing, 211198, China. Email: lifangcpucom.
Corresponding authors: Fuming Liu, Affiliated Hospital of Nanjing University of Chinese Medicine, Nanjing, 210029, Jiangsu, China, E-mail: doctor.liufumingcom; Boyang Yu, Jiangsu Key Laboratory of TCM Evaluation and Translation Research, Research Center for Traceability and Standardization of TCMs, School of Traditional Chinese Pharmacy, China Pharmaceutical University, Email: boyangyu59com; Fang Li, Jiangsu Key Laboratory of TCM Evaluation and Translational Research, Research Center for Traceability and Standardization of TCMs, School of Traditional Chinese Pharmacy, China Pharmaceutical University, 639 Longmian Road, Nanjing, 211198, China. Email: lifangcpucom.