Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Methods
Results
Discussion
Conclusion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(13):5859-5873. doi:10.7150/ijbs.119895 This issue Cite
Research Paper
TGF-βI/FERMT2/COL6A1 Reciprocal Loop Drives Tumor-Stroma Crosstalk and Promotes Peritoneal Metastasis in Gastric Cancer
Chao He1,#, Zheng Zhou1,#, Jiayue Ye2,#, Xiangliu Chen3, Yan Yang1, Xinguang Jin1, Quan Zhou1, ![]() , Lisong Teng1,
, Lisong Teng1, ![]()
1. Department of Surgical Oncology, The First Affiliated Hospital, School of Medicine, Zhejiang University, Hangzhou, China.
2. Department of Thoracic Surgery, the First Affiliated Hospital, Zhejiang University School of Medicine, Hangzhou, China.
3. Department of Gastric Surgery, Zhejiang Cancer Hospital, Hangzhou Institute of Medicine (HIM), Chinese Academy of Sciences, Hangzhou, Zhejiang, China.
# These authors contribute equally to this work.
Received 2025-6-19; Accepted 2025-9-6; Published 2025-9-12
Abstract

Background: Peritoneal metastasis (PM) is a frequent and fatal progression in advanced gastric cancer (GC), shaped by intricate interactions between tumor cells and the tumor microenvironment. Among these, gastric cancer-associated fibroblasts (GCAFs) are key mediators of tumor progression, yet the molecular regulators underlying tumor-stroma crosstalk remain poorly defined.
Methods: We combined bulk and single-cell transcriptomics, functional assays, proteomics, and in vivo models to dissect the role of FERMT2 in modulating GC-GCAF interactions and its contribution to peritoneal dissemination.
Results: FERMT2 is highly expressed in CAFs and positively correlates with both CAF infiltration and activation in GC. Functionally, FERMT2 maintains the myofibroblastic phenotype of GCAFs by acting as a competing endogenous RNA (ceRNA) for ZEB2, thereby promoting α-SMA transcription. FERMT2 also drives GCAF-derived secretion of transforming growth factor-beta 1 (TGF-β1), which in turn induces FERMT2 expression in GC cells, enhancing their migration, invasion, and resistance to anoikis. In parallel, tumor-derived FERMT2 upregulates COL6A1 and facilitates its transfer to GCAFs via exosomes, amplifying TGF-β signaling and reinforcing CAF activation. Intracellular COL6A1 sustains the pro-metastatic phenotype of GCAFs. Together, these interactions constitute a TGF-β1/FERMT2/COL6A1 positive feedback loop that fuels tumor-stroma crosstalk and promotes peritoneal dissemination in GC.
Conclusion: This study identifies a reciprocal regulatory loop involving FERMT2, TGF-β1, and COL6A1, which promotes tumor-stroma interaction and peritoneal dissemination, suggesting a potential therapeutic target for advanced gastric cancer.
Keywords: gastric cancer, peritoneal metastasis, cancer-associated fibroblasts, tumor microenvironment, exosomes
Introduction
Gastric cancer ranks as the fifth most commonly diagnosed malignancy and the third leading cause of cancer-related mortality worldwide (1). The five-year survival rate for GC remains alarmingly low, primarily due to the challenges of early detection and the limited efficacy of curative resection (2). Despite advances in surgical techniques and adjuvant therapies, the prognosis for patients with advanced GC is still poor (3). Notably, at least 33.9% of GC cases present with distant metastasis at the time of diagnosis (4), and approximately 14% of patients exhibit peritoneal metastasis at the time of initial surgery, a condition associated with a median survival of less than six months (5). These dismal outcomes highlight the urgent need to elucidate the molecular mechanisms underlying PM in GC. Understanding the key drivers and adverse prognostic factors of PM could pave the way for the development of targeted therapies and novel strategies for its prevention and clinical management.
Malignant behaviors in tumors are profoundly influenced by the complex interplay between cancer cells and the tumor microenvironment (TME) (6,7). Tumor biology is therefore dictated not only by the characteristics of neoplastic epithelial cells but also by the regulatory signals from stromal components (8). FERMT2, also known as Kindlin-2 or PLEKHC1, is a crucial member of the Kindlin family of focal adhesion proteins and has emerged as a key player in the pathogenesis and progression of various malignancies (9). As a pivotal regulator of integrin signaling, FERMT2 promotes tumor progression by modulating various cellular components within the TME. In breast cancer, FERMT2 enhances tumor-associated macrophage (TAM) infiltration by upregulating the expression and secretion of colony-stimulating factor 1 (CSF-1) from tumor cells (10). In pancreatic cancer, FERMT2 is highly expressed in pancreatic stellate cells, where it drives tumor progression through the release of pro-inflammatory cytokines (11). In oral squamous cell carcinoma (OSCC), FERMT2 regulates the expression and secretion of extracellular matrix proteins such as SPARC and COL4A1 in CAFs, promoting epithelial-mesenchymal transition (EMT) and M2 macrophage polarization (12). Collectively, these findings underscore FERMT2 as a critical regulator of TME remodeling across multiple tumor types, highlighting its potential as a therapeutic target for disrupting tumor-stroma crosstalk.
CAFs represent one of the most prominent stromal cell populations in various solid tumors, including GC (13). Within the TME, CAFs play a central role in tumor progression by regulating extracellular matrix (ECM) remodeling, promoting tumor growth and metastasis, facilitating immune evasion, and contributing to therapy resistance (14). Consequently, targeting CAF function to disrupt their bidirectional communication with cancer cells and reverse the fibrotic TME holds considerable promise for improving therapeutic outcomes in GC. Our previous bioinformatic analyses revealed that FERMT2 is predominantly expressed in stromal cells, especially fibroblasts, across multiple cancer types, with its expression strongly correlating with the infiltration and activation of CAFs within the TME (15). This suggests that FERMT2 may facilitate GC progression and peritoneal metastasis by regulating CAF-mediated ECM remodeling.
In this study, we aim to systematically elucidate the regulatory role of FERMT2 in the crosstalk between GC cells and GCAFs, and to investigate the molecular mechanisms by which FERMT2 influences GCAF-driven peritoneal metastasis in GC.
Methods
Isolation of CAFs and NFs
CAFs and normal fibroblasts (NFs) were isolated from tumor and matched adjacent non-tumor tissues of gastric adenocarcinoma patients. All specimens were pathologically confirmed. Tissues were rinsed with 1% Penicillin-Streptomycin solution and minced into 1 mm³ pieces, followed by digestion with 1 mg/mL collagenase type IV at 37°C for 1 hour. The digestion was stopped with DMEM supplemented with 20% FBS. The cell suspension was filtered, centrifuged, and cultured in DMEM with 20% FBS. Fibroblast identity was confirmed by Western blotting and immunofluorescence.
Cell lines
Two human gastric cancer cell lines (AGS and HGC-27), along with the non-cancerous gastric epithelial cell line GES-1 (to evaluate the role of exosomes or secreted proteins derived from non-cancerous cells in regulating the behavior of GCAFs), were obtained from the Cell Bank of the Chinese Academy of Sciences (Shanghai, China). Cells were maintained in RPMI-1640 medium with 10% FBS at 37°C in a 5% CO₂ incubator. NFs and GCAFs were cultured similarly in DMEM with 10% FBS.
Transwell assay for migration and invasion
AGS or HGC-27 cells (2 × 10⁴) were seeded in serum-free DMEM with or without Matrigel (Corning, USA diluted 1:8) in the upper chambers of 24-well transwell inserts. FERMT2-knockdown or wild-type GCAFs (1 × 10⁵) were seeded in the lower chambers. After 24 hours, cells on the underside of the membrane were fixed, stained with crystal violet, and counted.
EdU cell proliferation assay
GCAFs (5 × 10⁴) were seeded in 6-well plates. EdU (20 μM) was added for 2 hours, and cells were fixed, permeabilized, and stained using the Click™ EdU Kit (Beyotime, China). Nuclei were counterstained with Hoechst 33342.
Wound healing assay
GCAFs (1 × 10⁵) were seeded in 6-well plates. After reaching 90% confluence, a scratch was made, and cells were incubated in serum-free medium. Wound closure was monitored at 0 and 24 hours.
Analysis of FERMT2 in the gastric tumor microenvironment
TCGA, TARGET, and GTEx datasets were obtained from UCSC Xena (https://xenabrowser.net). EPIC scores and CAF infiltration levels were calculated from gene expression data using the IOBR R package (v0.99.0). Spearman's rank correlation between FERMT2 expression and CAF infiltration across cancer types was assessed using the 'corr.test' function in the psych R package (v2.1.6), with P < 0.05 considered statistically significant. In gastric cancer, the expression of FERMT2 in CAFs and other cellular subsets within the tumor microenvironment was analyzed using single-cell RNA-seq data obtained from the TISCH database (http://tisch.comp-genomics.org/home/).
Single-cell RNA-seq data processing
Single-cell RNA sequencing data from primary gastric cancer lesions (GSE210347) were obtained from the GEO database and analyzed using the Seurat R package (version 4.3.0.1). Cells with fewer than 200 detected genes or over 20% mitochondrial gene content were excluded, and only cells with 200 to 7,000 unique features were retained. Gene expression data were normalized using the LogNormalize method with a scale factor of 10,000. The top 2,000 highly variable genes were identified using the variance-stabilizing transformation method for downstream analysis. Data scaling was performed with regression of mitochondrial gene effects. Batch correction was carried out using the Harmony R package (version 0.1.1), followed by dimensionality reduction with principal component analysis (PCA), uniform manifold approximation and projection (UMAP), and t-distributed stochastic neighbor embedding (t-SNE). Cell clustering was performed using a resolution parameter of 0.5, and cell types were annotated based on canonical marker gene expression. For further analysis, CAFs were selected and stratified into FERMT2-positive and FERMT2-negative groups based on FERMT2 expression levels.
Cell-cell communication analysis
Intercellular communication was analyzed using the CellChat R package (version 1.6.1). Preprocessed single-cell RNA-seq data, including normalized gene expression and cell metadata, were used to define distinct cell populations, incorporating FERMT2 expression classification. Communication probabilities were computed for each cell type pair, and low-abundance interactions were excluded based on minimum cell number thresholds. The resulting interaction networks were visualized using circular plots, illustrating both the number and strength of inferred interactions.
Pseudotime analysis
Developmental trajectories of CAFs were analyzed using the Monocle3 R package (version 1.3.7). CAFs were extracted from the Seurat object, and a compatible Cell Data Set (CDS) was constructed using the raw count matrix, cell metadata, and gene annotations. Trajectories were constructed using principal component analysis, and pseudotime values were calculated for each cell to infer differentiation paths. Gene expression dynamics were assessed and plotted across pseudotime to identify stage-specific transcriptional changes.
Gene Set Enrichment Analysis (GSEA)
GSEA was performed using the singleseqgset R package (version 0.1.2.9000) to evaluate pathway-level alterations among distinct CAF populations. CAF subsets were analyzed based on FERMT2 expression, and enrichment analysis was conducted using Hallmark gene sets from MSigDB via the msigdbr package (version 7.5.1). In parallel, transcriptomic data from TCGA-STAD were analyzed by stratifying samples into high and low FERMT2 expression groups, and GSEA was conducted to identify CAF-related pathways significantly associated with FERMT2 expression.
Immunohistochemistry
Paraffin-embedded sections were incubated at 65°C for 2 hours, then deparaffinized, rehydrated, and subjected to antigen retrieval. Primary antibodies were applied at volumes proportional to tissue size and incubated at 37°C for 1 hour. After washing, HRP-conjugated goat anti-rabbit IgG polymers were added and incubated for 20 minutes at 37°C. DAB substrate was then applied for 2-5 minutes, followed by hematoxylin counterstaining.
Immunofluorescence
Cells were seeded on confocal dishes 24 hours prior to staining. The next day, cells were fixed with 4% paraformaldehyde for 15 minutes, permeabilized with 0.5% Triton X-100 for 20 minutes, and blocked with 5% BSA for 30 minutes. Primary antibody (1:200) was added and incubated overnight at 4°C. After washing with PBST, cells were incubated with secondary antibody (1:200) for 1 hour at room temperature in the dark. DAPI was added for nuclear staining, followed by a 5-minute incubation. Samples were mounted with antifade reagent.
Cell culture supernatant non-targeted proteomics sequencing
Cells were seeded in 20 cm culture dishes and cultured in serum-free medium. Upon reaching 80% confluence, the supernatant was collected, centrifuged at 3500 × g for 5 minutes at 4°C to remove debris, and frozen in liquid nitrogen for 5 minutes before storage at -80°C. Sequencing was performed by Ouyi Biological Company (Shanghai, China).
Dual-luciferase reporter assay
Plasmids for ZEB2 overexpression, α-SMA promoter-driven luciferase reporter, and their respective controls were constructed by Genomeditech (Shanghai, China). GCAFs were co-transfected with 500 ng of ZEB2 plasmid or empty vector, 500 ng of α-SMA reporter plasmid or pGL3-basic, and 25 ng of internal control plasmid (pRL-TK). After 48 hours, cells were lysed, and luciferase activity was measured using the Dual-Luciferase® Reporter Assay Kit (Yeasen Biotech, Shanghai, China) with a SpectraMax i3x microplate reader (Molecular Devices, Austria).
Western blot assay
Cells were lysed in RIPA buffer (Beyotime, China) containing phosphatase and protease inhibitors. After centrifugation, the supernatants were used for protein analysis. Protein concentrations were determined by BCA assay, and 20 μg of protein were loaded onto SDS-PAGE gels, transferred to PVDF membranes, and incubated with primary antibodies overnight at 4°C. Membranes were then incubated with HRP-conjugated secondary antibodies and developed using an enhanced chemiluminescence kit (Yeasen Biotech, China). A full list of antibodies is provided in Table S1.
Cell transfection
Small interfering RNAs (siRNAs) and negative controls (NC) targeting specific genes were synthesized by Qingke Biotech (Guangzhou, China). siRNAs were transfected into GCAFs using Polyplus transfection reagent (France). For stable gene silencing, lentiviral vectors encoding short hairpin RNAs (shRNAs) targeting FERMT2, which were packaged and purified by Genomeditech (Shanghai, China), were transfected into GCAFs and selected with puromycin for two weeks. siRNA, shRNA and miR-RNA sequences applied in this study were shown in Table S2.
ELISA assay
TGF-β1 levels in cell culture supernatants were quantified using a Human TGF-β1 ELISA Kit (Boster Biological Technology, Wuhan, China). Samples were acid-activated, neutralized, and then added to a 96-well plate pre-coated with a TGF-β1-specific antibody. Following incubation and biotin-labeled detection antibody addition, avidin-HRP conjugate was added, and color development was achieved using TMB substrate. Absorbance was measured at 450 nm, and concentrations were calculated from a standard curve.
Exosomes extraction
Exosomes were isolated from cell culture supernatants using the BeyoExo™ Enhanced Exosome Isolation Kit (Beyotime, China). The supernatant was centrifuged at 500 × g for 5 minutes at 4°C to remove cell debris, followed by a 10,000 × g centrifugation for 1 hour to eliminate large vesicles. The supernatant was filtered through a 0.22 μm membrane and concentrated 10-fold using ultrafiltration. For every 1 mL of concentrated supernatant, 190 μL of BeyoExo™ reagent was added, mixed, and incubated overnight at 4°C. The mixture was centrifuged the next day, and the exosome pellet was collected.
Animal experiment: in vivo xenograft model
An intraperitoneal dissemination model was established using 4-week-old female BALB/c nude mice, which were randomly assigned to four groups (n = 5 per group): (1) OE-NC AGS + sh-NC CAFs, (2) OE-NC AGS + sh-FERMT2 CAFs, (3) OE-FERMT2 AGS + sh-NC CAFs, and (4) OE-FERMT2 AGS + sh-FERMT2 CAFs. Each mouse received an intraperitoneal injection of a mixture containing 5 × 10⁶ AGS cells and 5 × 10⁶ GCAFs (a total of 1 × 10⁷ cells), suspended in 200 μL of serum-free DMEM and Matrigel (1:1). The suspension of tumor cells was injected intraperitoneally, approximately 0.5 cm above the midpoint of the inguinal ligament.
To assess the role of tumor-derived exosomes in peritoneal metastasis, exosomes (50 μg in 200 μL PBS) isolated from conditioned media of AGS cells overexpressing FERMT2 or carrying a control vector were injected into the peritoneal cavity of BALB/c nude mice. Additionally, the TGF-β receptor kinase inhibitor SB-431542 (10 μM, diluted in 1 μL DMSO) was administered intraperitoneally on days 1, 3, 5, and 7 after tumor cell injection, in a volume of 200 μL PBS per dose. After 40 days, the mice were euthanized. Intraperitoneal tumor nodules were collected, photographed, weighed, and subjected to statistical analysis. The nodules were then fixed in paraffin and sectioned for IHC staining. All animal procedures were approved by the Ethics Committee of the First Affiliated Hospital of Zhejiang University (Approval No.2025-116).
Statistics analysis
All experiments were performed in a minimum of three independent replicates. Data are expressed as the mean ± standard error. Statistical comparisons between two groups were carried out using the two-tailed Student's t-test. One-way ANOVA followed by Bonferroni's post-hoc test was applied to assess differences among three or more groups. A P-value less than 0.05 was considered statistically significant ((nsp>0.05; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001).
Results
FERMT2 expression is positively associated with CAF infiltration and maintains myofibroblastic phenotype in GCAFs
To investigate the stromal characteristics of GC, we analyzed TCGA-STAD transcriptomic data using the EPIC algorithm and observed significantly higher CAF infiltration in tumor tissues compared to adjacent normal tissues (Fig. 1A). Our prior single-cell transcriptomic study across multiple cancer types identified FERMT2 as predominantly expressed in stromal components, particularly in CAFs (15). Consistent with this, Spearman correlation analysis across TCGA pan-cancer cohorts revealed a strong positive association between FERMT2 expression and CAF infiltration in multiple tumor types (Fig. 1B). In GC specifically, FERMT2 expression correlated positively with CAF and endothelial cell infiltration, while associations with other immune subsets were minimal or absent (Fig. 1C).
FERMT2 expression correlates with CAF infiltration and sustains the myofibroblastic phenotype of GCAFs. (A) EPIC algorithm analysis of TCGA-STAD and GTEx datasets showing a significantly higher proportion of CAFs in gastric cancer tissues compared with normal tissues. (B) Correlation heatmap depicting positive associations between FERMT2 expression and CAF infiltration across multiple TCGA cancer types. (C) In TCGA-STAD, FERMT2 expression exhibits the second strongest correlation with CAFs infiltration among all analyzed cell types. (D) Single-cell RNA-seq datasets (GSE154352, GSE167297) reveal predominant FERMT2 expression in fibroblast clusters within gastric cancer tissues. (E) Representative IHC images from the HPA database showing FERMT2 expression in gastric cancer tissues. (F) GSEA demonstrating significant enrichment of fibroblast proliferation and migration gene signatures in tumors with high FERMT2 expression (TCGA-STAD cohort). (G) Immunoblot analysis of FERMT2 in GCAFs with stable FERMT2 knockdown. (H) EdU incorporation assay showing reduced proliferation in FERMT2-depleted GCAFs. (I) Wound-healing assay demonstrating impaired migration following FERMT2 knockdown. (J) Immunofluorescence staining showing decreased α-SMA (red) and FAP (green) levels upon FERMT2 knockdown; nuclei counterstained with DAPI. (K) Western blot analysis showing reduced α-SMA and FSP expression in FERMT2-depleted GCAFs. (L) Pearson correlation analysis in TCGA-STAD showing a strong positive association between FERMT2 and ZEB2 (r = 0.78). (M) miR-200a and miR-138 simultaneously target FERMT2 and ZEB2, suppressing α-SMA and FSP expression. (N) Silencing either FERMT2 or ZEB2 reduces expression of the other, as well as α-SMA, FSP, and vimentin in GCAFs. (O) TargetScan-predicted miR-138-5p binding sites within the 3′UTRs of both FERMT2 and ZEB2. (P) Dual-luciferase reporter assay showing that ZEB2 overexpression activates α-SMA promoter activity. (Q) Immunoblot showing upregulated α-SMA, FAP, and FSP in ZEB2-overexpressing GCAFs. (R) Violin plot of scRNA-seq data showing enrichment of α-SMA expression in CAF_myo compared with other CAF subtypes and NFs. (S) Collagen contraction assay showing reduced contractility in FERMT2-knockdown GCAFs; contraction quantified using ImageJ. (Statistical significance: ns, p > 0.05; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.)
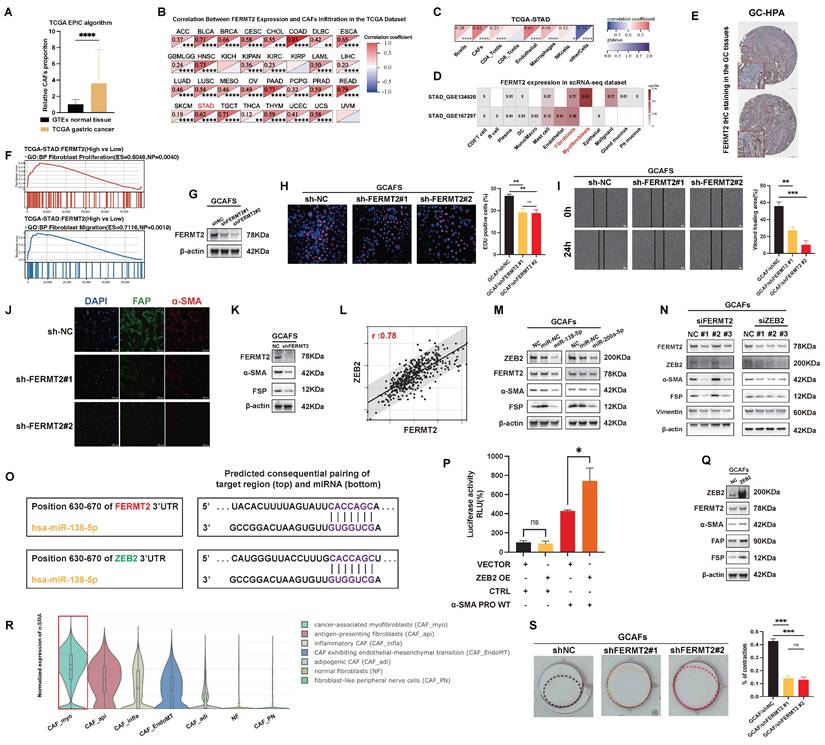
Single-cell RNA-sequencing data from the TISCH database further confirmed that FERMT2 expression was enriched in gastric CAFs (Fig. 1D). Supporting this, immunohistochemical data from the Human Protein Atlas (HPA) showed FERMT2 staining was largely restricted to stromal regions in gastric cancer tissues (Fig. 1E). Collectively, these findings establish FERMT2 as a stromal-enriched gene, closely associated with CAF infiltration, highlighting its potential as a CAF marker and functional regulator.
To explore its functional role, we stratified TCGA-STAD samples by FERMT2 expression and observed that high FERMT2 levels were associated with transcriptomic signatures related to fibroblast proliferation and migration (Fig. 1F). We next generated stable FERMT2 knockdown GCAF lines and confirmed effective silencing at the protein level (Fig. 1G). Functional assays revealed that FERMT2 silencing impaired both migratory and proliferative capacities of GCAFs (Fig. 1H-I). Immunofluorescence staining demonstrated reduced expression of CAF activation markers, including FAP and α-SMA, upon FERMT2 depletion (Fig. 1J), which was corroborated by Western blot analysis (Fig. 1K). Together, these data implicate FERMT2 as a key regulator of CAF activation and the acquisition of tumor-promoting traits.
Activation of CAFs refers to the transition from a quiescent to a functionally active state, marked by changes in cellular function and phenotype. A hallmark of CAF activation is the acquisition of a myofibroblastic phenotype, characterized by increased α-SMA expression and cytoskeletal remodeling that enhances contractility and ECM remodeling (16,17). Within the TCGA-STAD cohort, FERMT2 expression showed a strong positive correlation with ZEB2, a transcription factor linked to myofibroblastic transition (Fig. 1L).
In GCAFs, transfection with miR-138 or miR-200a led to downregulation of both FERMT2 and ZEB2, accompanied by reduced expression of α-SMA and FAP (Fig. 1M). To assess whether FERMT2 and ZEB2 function in a competing endogenous RNA network, we performed reciprocal knockdowns. Silencing FERMT2 led to reduced ZEB2 protein levels, and vice versa (Fig. 1N). TargetScan analysis identified a shared miR-138-5p binding site in the 3′UTRs of both FERMT2 and ZEB2 (Fig. 1O), supporting the hypothesis of post-transcriptional competition.
Using dual-luciferase reporter assays, we confirmed that ZEB2 binds to the ACTA2 (α-SMA) promoter and transcriptionally activates its expression (Fig. 1P). ZEB2 overexpression in GCAFs enhanced α-SMA protein levels and upregulated additional CAF activation markers, including FAP and FSP (Fig. 1Q). Single-cell RNA-sequencing (scRNA-seq) data from the Cancer-Associated Fibroblast Atlas revealed that α-SMA expression is predominantly enriched in the myofibroblastic CAF subset (CAF_myo) (Fig. 1R), consistent with the phenotype observed in our study. Notably, contractile activity was significantly impaired in FERMT2-depleted GCAFs (Fig. 1S).
Taken together, these findings indicate that FERMT2 sustains the myofibroblastic phenotype of GCAFs by functioning as a ceRNA for ZEB2. Through ZEB2-mediated transcriptional activation of α-SMA, FERMT2 facilitates the activation, migration, and contractility of GCAFs, thereby contributing to tumor-stroma crosstalk and a pro-tumorigenic microenvironment.
FERMT2⁺ CAFs display distinct transcriptional states and enhanced crosstalk with tumor cells via TGF-β signaling
We interrogated the gastric cancer scRNA-seq dataset (Luo, 2022) to characterize GCAF heterogeneity based on FERMT2 expression. GCAFs were stratified into FERMT2⁺ and FERMT2⁻ subpopulations (Fig. 2A). Pseudotime trajectory analysis revealed a progressive upregulation of both FERMT2 and FAP along the differentiation axis, suggesting a role for FERMT2 in driving GCAF activation or phenotypic transitions (Fig. 2B). Notably, the FERMT2 double-negative group (FERMT2⁻ GCAFs / FERMT2⁻ GC cells) exhibited the fewest and weakest cell-cell interactions (Fig. 2C). In addition, FERMT2⁻ GCAFs showed markedly diminished secretory capacity and reduced TGF-β pathway activity (Fig. 2D), implying impaired secretion of key factors such as TGF-β1 and attenuated support for tumor progression.
FERMT2⁺ CAFs exhibit distinct transcriptional programs and enhanced communication with cancer cells via TGF-β signaling. (A) Schematic overview of the analysis pipeline using the gastric cancer single-cell RNA-seq dataset (Luo et al., 2022). Fibroblast clusters were isolated for pseudotime, CellChat, GSEA, and NicheNet analyses based on FERMT2 expression. (B) Pseudotime analysis of CAF populations showing dynamic expression patterns of FAP and FERMT2 along the trajectory. UMAP plot illustrates clustering and annotation of CAFs in different states. (C) Cell-cell communication analysis using CellChat reveals that FERMT2⁺ CAFs exhibit more interactions and stronger signaling intensity with cancer cells than FERMT2⁻ CAFs. Right panels: Quantification of interaction numbers and signaling strength between FERMT2⁺/⁻ CAFs and cancer cells, with or without FERMT2 expression. (D) GSEA comparing FERMT2⁺ versus FERMT2⁻ CAFs reveals enrichment of pro-tumorigenic pathways, including TGF-β, WNT, and EMT signaling. (E) NicheNet analysis identifies key ligands involved in CAF-cancer cell communication. Heatmaps and dot plots display ligand activity (AUPR), expression changes in CAFs, and predicted regulatory potential of target genes. Notably, TGF-β1 is significantly upregulated in FERMT2⁺ CAFs. (F) Volcano plot of untargeted proteomic analysis of CM from GCAFs after FERMT2 knockdown versus control (sh-NC). Downregulation of secreted TGF-β1 and other associated proteins indicates FERMT2-dependent regulation of secretion.
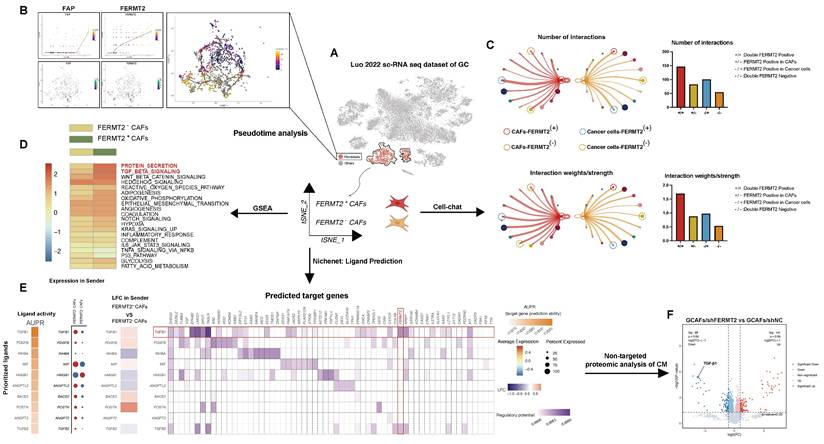
NicheNet analysis identified TGF-β1 as the most significantly altered ligand between FERMT2⁺ and FERMT2⁻ GCAFs (Fig. 2E), a finding validated by untargeted proteomics of conditioned media from GCAFs with or without FERMT2 knockdown (Fig. 2F). Collectively, these results suggest that FERMT2 promotes GCAF activation and augments communication with gastric cancer cells, likely through enhanced secretion of pro-tumorigenic mediators such as TGF-β1.
FERMT2+ GCAFs enhance gastric cancer cell migration, invasion, and anoikis resistance via TGF-β1 secretion
Our previous work demonstrated that gastric cancer cells detached from the extracellular matrix activate autocrine TGF-β1 signaling, leading to upregulation of FERMT2, increased fibronectin (FN) expression, and enhanced cell-cell adhesion to facilitate extracellular matrix deposition (18). Beyond this autocrine axis, TGF-β1 is also abundantly secreted by CAFs within the tumor microenvironment, acting in a paracrine fashion (19). To investigate this further, we established a co-culture system comprising gastric cancer cells and GCAFs. This included two experimental models: one to evaluate the impact of GCAFs on gastric cancer cell protein expression, and another to assess their effects on malignant phenotypes such as migration, invasion (Fig. 3A).
FERMT2-expressing CAFs enhance gastric cancer cell migration, invasion, and anoikis resistance via TGF-β1 secretion. (A) Schematic illustration of the transwell co-culture system used to assess the impact of GCAFs on protein expression in GC cells (3 μm pores) and on cancer cell migration and invasion (8 μm pores). (B) Western blot analysis of FN and FERMT2 expression in AGS and HGC-27 cells co-cultured with or without GCAFs for 24 and 48 hours. (C) Migration and invasion assays showing significant promotion of AGS and HGC-27 cell motility by GCAFs. (D) ELISA analysis of TGF-β1 levels in conditioned media from control and FERMT2-knockdown GCAFs. (E) Western blot showing FN, FERMT2, and N-cadherin expression in AGS and HGC-27 cells cultured with sh-NC or sh-FERMT2 GCAFs, with or without TGF-β1 neutralizing antibody (5 μg/mL). (F) Transwell migration and invasion assays demonstrating reduced GCAF-induced motility following FERMT2 knockdown or TGF-β1 blockade. (G) Anoikis resistance assay showing that AGS and HGC-27 spheroids co-cultured with sh-NC or sh-FERMT2 GCAFs, with or without TGF-β1 antibody, exhibit differential survival under super-low attachment conditions, evaluated by Calcein/PI staining. (H) In vivo tumorigenicity assay showing representative images and quantification of intraperitoneal tumors formed by AGS cells (OE-NC or OE-FERMT2) co-injected with sh-NC or sh-FERMT2 GCAFs into nude mice. (Statistical significance: ns, p > 0.05; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.)
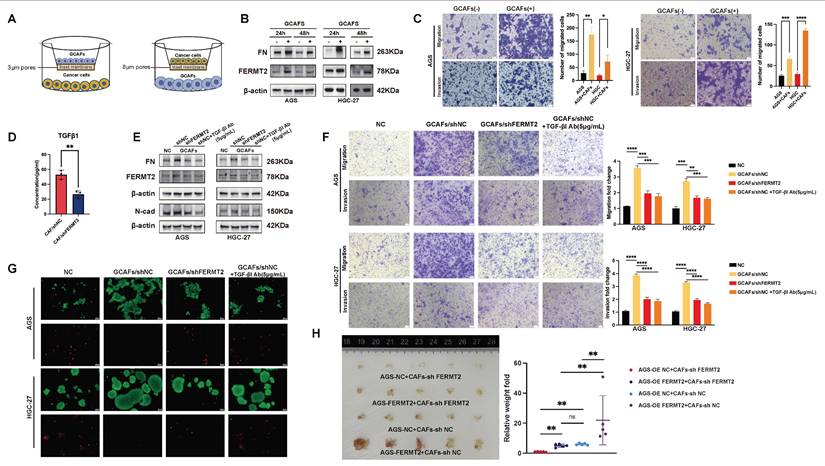
As predicted by NicheNet analysis, TGF-β1 emerged as the top-ranked differential ligand between FERMT2⁺ and FERMT2⁻ GCAFs, with FERMT2 among its downstream targets (Fig. 2E, red rectangle). In line with this prediction, co-culture of gastric cancer cells with GCAFs markedly increased FERMT2 and FN protein levels (Fig. 3B), and significantly promoted migration and invasion in both AGS and HGC-27 cell lines (Fig. 3C). ELISA confirmed that FERMT2 knockdown in GCAFs significantly reduced TGF-β1 secretion into the extracellular milieu (Fig. 3D). Notably, co-culture with shNC-GCAFs, as opposed to shFERMT2-GCAFs, robustly upregulated FERMT2 and FN in gastric cancer cells and enhanced their migratory and invasive potential—effects that were abolished by a TGF-β1 neutralizing antibody (Fig. 3E, F).
Given the pivotal roles of FERMT2 and FN in promoting anoikis resistance in gastric cancer (18), we next examined whether GCAF-derived TGF-β1 contribute to anchorage-independent survival. Gastric cancer cells co-cultured with control GCAFs (shNC-GCAFs) showed markedly enhanced survival under matrix detachment compared with those co-cultured with FERMT2-deficient GCAFs (Fig. 3G). Consistently, in vivo, these cells developed more extensive peritoneal dissemination in BALB/c nude mice (Fig. 3H). Together, these findings demonstrate that FERMT2 promotes GCAF-mediated paracrine secretion of TGF-β1, which in turn drives the invasive behavior and anoikis resistance of gastric cancer cells.
FERMT2 regulates exosomal transfer of COL6A1 to enhance TGF-β signaling and CAF activation
In preceding sections, we showed that FERMT2 promotes GCAF activation and facilitates their interaction with gastric cancer cells. However, intercellular communication within the tumor microenvironment is bidirectional. In breast cancer, for example, tumor-derived FERMT2 promotes TAM infiltration by upregulating CSF1 secretion (10). To investigate whether a similar mechanism exists in gastric cancer, we established a co-culture model to examine the influence of tumor cells on GCAFs (Fig. 4A). Both co-culture and treatment with GC cell-derived conditioned media (CM) showed that AGS cells overexpressing FERMT2 markedly upregulated FERMT2 and CAF activation markers in GCAFs and NFs, whereas FERMT2 knockdown in HGC-27 cells attenuated these effects (Fig. 4B).
FERMT2 is a critical mediator of integrin activation, known to regulate cell adhesion and focal adhesion signaling (20). Proteomic profiling of CM from gastric cancer cells with differential FERMT2 expression revealed significant enrichment of the ECM-receptor interaction pathway in the FERMT2-overexpressing group (Fig. 4C). Among the top 20 differentially secreted proteins, COL6A1 was prominently associated with this pathway (Fig. 4D, E). Moreover, gene ontology analysis indicated a striking enrichment of extracellular exosome-related components in the FERMT2-overexpressing CM, suggesting a role for exosome-mediated communication (Fig. 4F).
Transmission electron microscopy confirmed the characteristic morphology of exosomes isolated from CM (Fig. 4G), and expression of canonical markers TSG101 and CD63 validated their identity (Fig. 4H). FERMT2 overexpression significantly increased COL6A1 protein levels in AGS cells (Fig. 4I) and enhanced its loading into exosomes (Fig. 4J). Co-culture with FERMT2-overexpressing AGS cells also elevated COL6A1 expression in GCAFs compared to controls or normal gastric epithelial cells (Fig. 4K).
COL6A1 has been implicated in potentiating TGF-β signaling and tumor metastasis (21). In line with this, co-culture with FERMT2-overexpressing AGS cells markedly increased the expression of TGF-β1, ZEB2, FERMT2, and CAF activation markers in GCAFs (Fig. 4L, M). Similar effects were induced by exosomes from FERMT2-overexpressing cells, which also elevated COL6A1 and TGF-β1 levels in GCAFs (Fig. 4N).
To determine whether COL6A1 acts extracellularly or intracellularly, we supplemented GCAF cultures with recombinant COL6A1 protein. This failed to induce CAF activation or internalization (Fig. 4O). In contrast, PKH67-labeled exosomes transferred Cy3-labeled COL6A1 into GCAFs (Fig. 4P), supporting an intracellular delivery mechanism via exosomes. Thus, COL6A1 may be transferred from GC cells to CAFs via exosomes, serving as key mediators of genetic communication between cancer cells and the stroma.
FERMT2 regulates exosomal transfer of COL6A1, enhancing TGF-β signaling and CAF activation. (A) Schematic illustration of the transwell co-culture system (3 μm pores) used to assess the impact of GC cells on protein expression in GCAFs. (B) Immunoblot analysis of FERMT2, α-SMA, FAP, and FSP in NFs and GCAFs co-cultured with AGS-NC, AGS-FERMT2, HGC-shNC, HGC-shFERMT2, or with CM from AGS-NC or AGS-FERMT2 cells. (C) KEGG pathway enrichment analysis of differentially expressed proteins in AGS-FERMT2 CM versus AGS-NC CM. (D) Functional pathway distribution of differentially expressed proteins, highlighting COL6A1 involvement in ECM-receptor interactions. (E) Top 25 up- and downregulated proteins in AGS-FERMT2 CM versus AGS-NC CM. (F) GO analysis showing the most enriched pathways in AGS-FERMT2 CM versus AGS-NC CM. (G) Transmission electron microscopy (TEM) images of exosomes from AGS-NC and AGS-FERMT2 cells. (H) Immunoblot analysis of exosomal markers (TSG101, CD63) and the endoplasmic reticulum marker (Calnexin) in exosomes from AGS-NC and AGS-FERMT2 cells. (I) COL6A1 expression in lysates of AGS-NC and AGS-FERMT2 cells. (J) Immunoblot showing COL6A1 and TSG101 expression in exosomes from AGS-NC and AGS-FERMT2 cells. (K-M) Western blot analysis of COL6A1 (K), TGF-β1 (L), and ZEB2, FERMT2, α-SMA, FAP, and FSP (M) in GCAFs co-cultured with GES-1, AGS-NC, or AGS-FERMT2 cells. (N) Immunoblot analysis of TGF-β1, COL6A1, FERMT2, and ZEB2 in GCAFs treated with exosomes from AGS-NC or AGS-FERMT2 cells. (O) Western blot showing COL6A1, FERMT2, ZEB2, α-SMA, FAP, and FSP in GCAFs treated with rhCOL6A1 at different concentrations for 24 h. (P) Co-localization of exosomes and COL6A1 in GCAFs after 24 h incubation with double-labeled exosomes (PKH67, green; COL6A1, Cy3, red; nuclei, DAPI, blue). Scale bar, 100 μm. (Q) Western blot showing COL6A1, FERMT2, ZEB2, α-SMA, FAP, and FSP expression in GCAFs after COL6A1 knockdown. (R, S) Western blot analysis of Smad2/3, COL6A1, ZEB2, α-SMA, and FSP after siRNA-mediated silencing of Smad2/3 (R) and assessment of Smad2/3-mediated TGF-β1 signaling on COL6A1 and FERMT2 expression (S) in GCAFs. (T) Western blot showing TGF-β1, COL6A1, and ZEB2 in GCAFs treated with exosomes from AGS-NC or AGS-FERMT2 cells, with or without si-COL6A1. (U) Transwell migration and invasion assays of GCAFs treated with exosomes from AGS-NC or AGS-FERMT2 cells, with or without COL6A1 knockdown. (V) COL6A1 and Smad2/3 expression in AGS cells with FERMT2 overexpression or knockdown of Smad2. (W) Collagen gel contraction assay of GCAFs treated with exosomes from indicated GC cells, with or without COL6A1 knockdown; contraction quantified using ImageJ. (X, Y) In vivo tumorigenicity assays showing representative images and quantification of intraperitoneal tumors formed by AGS cells co-injected with GCAFs, with or without tumor-derived exosomes, into nude mice. (Z) Immunohistochemical staining of peritoneal metastatic nodules from BALB/c nude mice. (Statistical significance: ns, p > 0.05; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.)
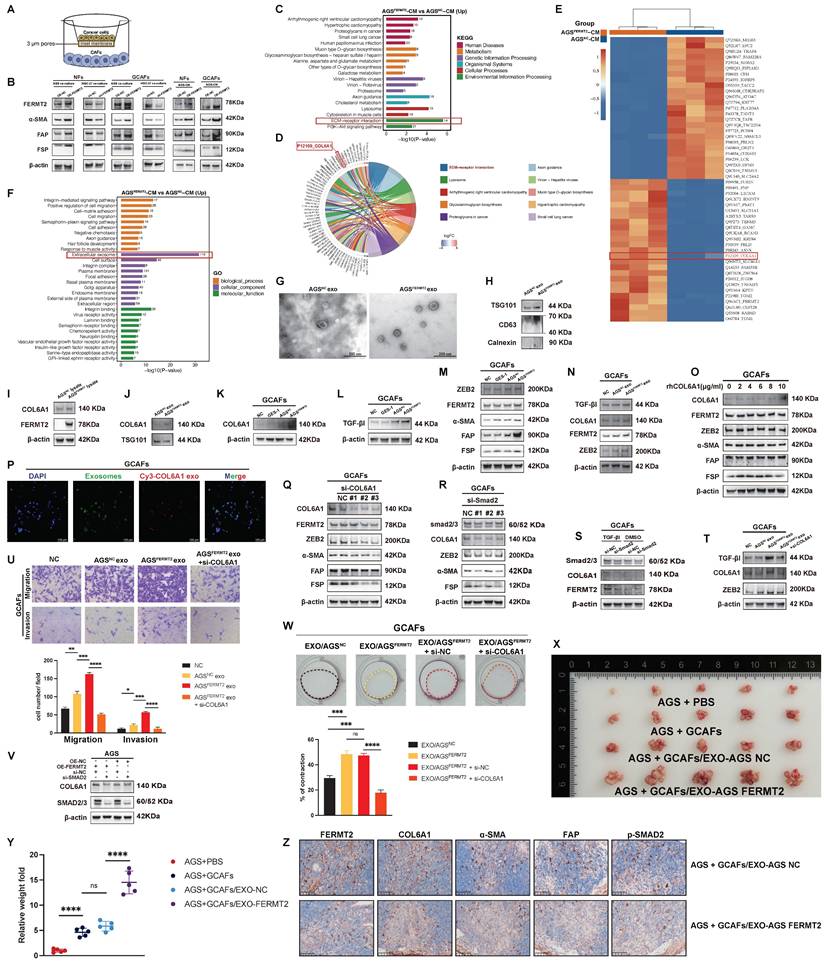
Knockdown of COL6A1 in GCAFs reduced the expression of ZEB2, FERMT2, and CAF activation markers (Fig. 4Q). Silencing SMAD2—a core TGF-β signaling effector—similarly decreased ZEB2 and CAF marker expression (Fig. 4R). Interestingly, SMAD2 knockdown also lowered COL6A1 levels, suggesting a positive feedback loop in which COL6A1 enhances TGF-β autocrine signaling to sustain its own expression (Fig. 4S). Moreover, COL6A1 depletion partially reversed the upregulation of TGF-β1 in GCAFs (Fig. 4T), of which the exosome-induced migration and invasion were attenuated in the absence of COL6A1 (Fig. 4U).
FERMT2 (also known as Kindlin-2) has been reported to regulate both actin-binding capacity and the activity of TGF-β receptor 1 (TGFβR1) (22). In our study, SMAD2 knockdown abolished the FERMT2-induced upregulation of COL6A1 (Fig. 4V), indicating that this regulation is dependent on enhanced TGF-β signaling. Exosomes derived from FERMT2-overexpressing gastric cancer cells increased the contractile activity of GCAFs, whereas COL6A1 silencing in GCAFs markedly attenuated this effect (Fig. 4W). In vivo, tumors formed following injection of AGS-FERMT2-derived exosomes displayed a stronger peritoneal metastatic capacity, along with markedly higher staining intensities of FERMT2, COL6A1, phosphorylated SMAD2/3, and CAF activation markers (α-SMA and FAP), compared with those formed with AGS-NC-derived exosomes (Fig. 4X-Z). Collectively, these findings demonstrate that gastric cancer cells deliver COL6A1 to GCAFs via exosomes, thereby amplifying TGF-β signaling and sustaining CAF activation.
Characterization of COL6A1 and ZEB2 expression in gastric cancer and their association with peritoneal metastasis and survival
We first examined the expression of COL6A1 across different stages of GC. COL6A1 expression was significantly higher in advanced stages compared to earlier stages (Fig. 5A). Moreover, COL6A1 levels were elevated in tumor tissues relative to adjacent normal tissues (Fig. 5B). Survival analysis revealed that high ZEB2 expression was associated with poorer survival outcomes. Although COL6A1 expression did not reach statistical significance in survival analysis, there was a trend towards higher expression in patients with worse outcomes (Fig. 5C). Correlation analysis further indicated a positive association between COL6A1 and TGF-β1 expression (r = 0.59), suggesting a potential link between COL6A1 and TGF-β signaling in GC (Fig. 5D).
Characterization of COL6A1 and ZEB2 expression in gastric cancer and their association with peritoneal metastasis and survival. (A) Expression of COL6A1 across different stages of gastric cancer. (B) COL6A1 expression in gastric cancer tissues compared to normal adjacent tissues. (C) Kaplan-Meier survival analysis comparing overall survival in gastric cancer patients with high versus low expression of ZEB2 and COL6A1. (D) Correlation analysis between COL6A1 and TGF-β1 expression in gastric cancer tissues. (E) Expression of ZEB2, COL6A1, and TGF-β1 in GCAFs following FERMT2 overexpression or ZEB2 knockdown. (F) UMAP plot showing cell type clustering in gastric cancer tissues, including both peritoneal metastasis and primary tumor tissues. (G) Comparison of COL6A1 and ZEB2 expression in epithelial cells from primary tumors and peritoneal metastasis tissues. (H) Comparison of COL6A1 and ZEB2 in fibroblasts from primary tumors and peritoneal metastasis tissues. (I) Violin plots illustrating the normalized expression of COL6A1 and ZEB2 across different CAF subtypes. (J) Expression of COL6A1, ZEB2, TGFβ-RI, and FSP in GCAFs treated with either FERMT2 overexpression or control, and ZEB2 knockdown or control. (K) Western blot analysis showing COL6A1 and TGFβ-RI expression in GCAFs after ZEB2 overexpression or control treatment. (L) Schematic showing the administration of TGF-β receptor kinase inhibitor SB-431542 into the peritoneal cavity of nude mice after tumor cell injection. (M, N) Representative images and quantification of intraperitoneally injected tumors formed by indicated cell mixtures and tumor-derived exosomes, with or without TGF-β receptor kinase inhibitor, in nude mice. (O) Schematic model summarizing the findings of this study. (Statistical significance: ns, p > 0.05; *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001.)
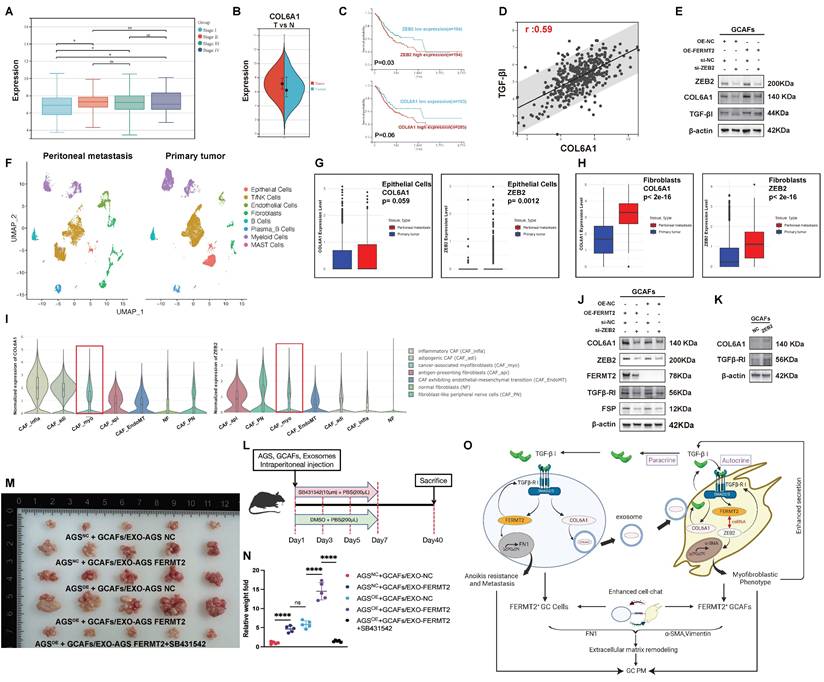
We next explored the molecular regulation of COL6A1 and ZEB2 expression. Overexpression of FERMT2 led to an increase in ZEB2, COL6A1, and TGF-β1 protein levels. Silencing ZEB2 abolished the FERMT2-induced upregulation of COL6A1 and TGF-β1, indicating that ZEB2 is essential for the FERMT2-mediated regulation of these proteins (Fig. 5E).
scRNA-seq of GC samples revealed distinct cell clusters within the TME, with clear separation between primary tumor and peritoneal metastasis tissues (Fig. 5F). Further analysis of COL6A1 and ZEB2 expression in various cell populations showed that both COL6A1 and ZEB2 were more highly expressed in epithelial cells and fibroblasts from peritoneal metastases compared to primary tumors (Fig. 5G, H).
Additionally, the Single Cell Cancer-Associated Fibroblasts Atlas highlighted that both COL6A1 and ZEB2 were enriched in the CAF_myo subset (Fig. 5I). In GCAFs, overexpression of FERMT2 significantly upregulated COL6A1 expression, which was partially reversed by ZEB2 knockdown. This suggests that FERMT2 regulates COL6A1 expression via a ZEB2-dependent mechanism. A similar trend was observed for TGFβ-RI expression (Fig. 5J). Moreover, ZEB2 overexpression in GCAFs increased the protein levels of both COL6A1 and TGFβ-RI, indicating that ZEB2-induced COL6A1 upregulation is mediated by the upregulation of TGFβ-RI (Fig. 5K).
To assess the role of TGF-β receptor kinase inhibitor SB-431542 in peritoneal metastasis, which was administered intraperitoneally on days 1, 3, 5, and 7 after tumor cell injection (Fig. 5L). And we found the treatment with SB431542 inhibited peritoneal tumor formation ability in AGS cells together with GCAFs (Fig. 5M, N). Finally, the proposed model of this study is illustrated in Fig. 5O.
Discussion
The role of FERMT2 in CAF activation has remained incompletely defined. MicroRNAs (miRNAs), a class of small non-coding RNAs, modulate gene expression by binding to the 3′ untranslated region (3′-UTR) of target mRNAs, thereby influencing multiple stages of tumorigenesis.
MiR-138-5p has been reported to suppress cancer cell functions through diverse mechanisms, ultimately impeding tumor progression (23,24). For instance, miR-138-5p delivered via mesenchymal stem cell (MSC)-derived exosomes targets SIRT1, attenuating dermal fibroblast proliferation and migration while reducing NF-κB, α-SMA, and TGF-β1 expression (25). Likewise, the miR-200 family shapes the tumor microenvironment by targeting ZEB2 to inhibit epithelial-mesenchymal transition (EMT) (26,27), and miR-200b directly suppresses FERMT2, thereby impacting cytoskeletal remodeling and adhesion (28). Notably, in oral squamous cell carcinoma, FERMT2 upregulates ZEB2 to promote tumor cell migration and invasion (29).
In parallel, studies in pulmonary fibrosis have shown that ZEB2 deletion reduces α-SMA and vimentin expression, thereby reversing the fibrotic phenotype (30). In the tumor microenvironment, CAFs—major stromal constituents—express markers including FAP, vimentin, S100A4, and α-SMA, and secrete matrix metalloproteinases such as MMP9 to remodel the extracellular matrix (31). Among subsets of CAFs, myofibroblastic CAFs (CAF_myo) display elevated α-SMA levels, heightened contractile capacity, and a stronger propensity for ECM remodeling (32).
Our findings identify a previously uncharacterized regulatory circuit in which FERMT2 functions as a ceRNA for ZEB2 in GCAFs, counteracting repression by miR-200a and miR-138. This axis enhances ZEB2 expression, leading to α-SMA upregulation and augmented contractility, thereby sustaining the myofibroblastic CAF phenotype. Such phenotypic reprogramming facilitates the transition of CAFs from a quiescent to an active state and plays a critical role in fostering peritoneal metastasis in gastric cancer.
Once functionally activated, CAFs secrete a wide range of signaling molecules, including cytokines, chemokines, proteins, and exosomes, collectively forming a distinct "secretome" that actively shapes the tumor microenvironment (33,34). Among these factors, TGF-β1 is produced not only by tumor cells but also by stromal components such as CAFs (35,36). TGF-β1 plays a pivotal role in sustaining cancer stemness (37-39), while also driving EMT processes in cancer cells (40,41), which enhances resistance to anoikis and therapeutic interventions and contributes to metastatic progression.
This secretome mediates intricate intercellular communication among CAFs, cancer cells, immune cells, and endothelial cells through both autocrine and paracrine signaling pathways (19). In this study, we found that TGF-β1 secreted by CAFs was taken up by gastric cancer cells via TGFβ-RI, leading to the activation of the downstream SMAD2/3-FERMT2 signaling cascade. Notably, the upregulation of FERMT2, in turn, enhanced the expression of TGFβ-RI, thereby further amplifying TGF-β1 signaling and establishing a positive feedback loop. This TGF-β1-mediated paracrine signaling also promoted the expression of FN in gastric cancer cells, which enhanced cell-cell adhesion and ECM deposition, ultimately contributing to increased resistance to anoikis.
However, intercellular communication within the tumor immune microenvironment is not unidirectional (42); rather, cancer cells can reciprocally modulate the functional state of CAFs through various mechanisms, including the secretion of exosomes. Enhanced TGF-β1 signaling in cancer cells promotes the expression of COL6A1 and increases exosome release, facilitating the transfer of COL6A1-enriched exosomes to GCAFs. Upon uptake, GCAFs exhibiting elevated COL6A1 levels further intensify their TGF-β1 secretion, as previously reported (21), leading to the acquisition of a myofibroblastic phenotype characterized by upregulated α-SMA and vimentin expression via TGF-β1 autocrine signaling. This phenotypic transition promotes ECM remodeling, thereby contributing to the establishment of a pro-metastatic microenvironment. Moreover, activated CAFs exhibit upregulated expression of FERMT2 and ZEB2, and secrete increased levels of TGF-β1, which in turn enhances the anoikis resistance and migratory potential of gastric cancer cells. Collectively, this reciprocal positive feedback loop amplifies TGF-β1 signaling between CAFs and cancer cells, ultimately promoting peritoneal metastasis in gastric cancer.
In this study, we demonstrate that exosomes efficiently transport COL6A1 to GCAFs. This finding supports the role of exosomes as effective carriers of intercellular communication, capable of traversing the cell membrane and delivering cargo such as COL6A1 to specific subcellular compartments (43). In contrast, recombinant COL6A1 failed to exhibit similar internalization in GCAFs, suggesting that its ineffective uptake may be due to structural differences compared to exosome-derived COL6A1. During the packaging and secretion of exosomes, proteins may undergo specific modifications, such as glycosylation or phosphorylation, which can enhance their interaction with cell surface receptors, thereby facilitating endocytosis (44). Exosomal COL6A1 is likely recognized and internalized through receptor-mediated endocytosis, directing it to particular intracellular compartments. However, recombinant COL6A1 may lack these modifications, leading to its inefficient internalization. Furthermore, exosome uptake typically occurs via clathrin-mediated or caveolae-dependent endocytosis, both of which are receptor-specific processes (45). The failure of recombinant COL6A1 to interact with these receptors likely explains its inability to effectively enter the cells.
Previous studies have shown that CAF-derived IL-33 promotes peritoneal dissemination by activating the ST2L-ERK1/2-SP1-ZEB2 axis, thereby inducing EMT in gastric cancer cells (46). Moreover, immune cells within ascitic fluid can drive TAMs to shift from a CTShigh to a C1Qhigh phenotype, leading to increased PD-L1 and NECTIN2 expression and facilitating immune evasion through the C1q-complement pathway (47). These observations underscore that disrupting intercellular communication within the gastric cancer immune microenvironment may represent a promising strategy to limit peritoneal metastasis.
In this context, our study identifies FERMT2 as a pivotal regulator, orchestrating both the maintenance of the myofibroblastic phenotype in CAFs and the enhancement of anoikis resistance in gastric cancer cells. Collectively, these findings suggest that therapeutic targeting of FERMT2 could disrupt the pro-metastatic CAF-cancer cell axis, thereby suppressing peritoneal dissemination and potentially improving clinical outcomes in gastric cancer.
Meanwhile, given the central role of the FERMT2-TGF-β1-COL6A1 axis in promoting tumor-stroma interaction and peritoneal dissemination, targeting this pathway may offer novel therapeutic opportunities. Neutralization of TGF-β1 with monoclonal antibodies or receptor kinase inhibitors has shown promise in preclinical models to suppress CAF activation and tumor progression (48,49). In addition, inhibiting tumor-derived exosome biogenesis or uptake may disrupt the intercellular communication that reinforces this feedback loop (50). Strategies aimed at reprogramming or depleting CAFs, such as FAP-targeted therapies or vitamin D analogs, also hold potential for modulating the tumor microenvironment and impairing metastatic spread (51). These approaches warrant further investigation to determine their efficacy in the context of gastric cancer.
Conclusion
In gastric cancer cells, FERMT2 enhances TGF-β1 signaling by upregulating TGFβ-RI, thereby driving the expression of FN and COL6A1. FERMT2 further promotes the secretion of COL6A1-enriched exosomes, which are taken up by GCAFs to reinforce their myofibroblastic phenotype. This, in turn, augments TGF-β1 signaling within GCAFs, while GCAF-derived TGF-β1 promotes gastric cancer cell migration, invasion, and anoikis resistance. Collectively, our findings delineate a TGF-β1/FERMT2/COL6A1 positive feedback loop that sustains reciprocal activation between tumor cells and GCAFs, thereby driving peritoneal metastasis. Targeting this pro-metastatic circuit may provide a novel therapeutic avenue to impede gastric cancer progression.
Abbreviations
PM: Peritoneal metastasis; GC: Gastric cancer; CAFs: Cancer-associated fibroblasts; GCAFs: Gastric cancer-associated fibroblasts; TGF-β1: Transforming growth factor-beta 1; TME: Tumor microenvironment; TAM: Tumor-associated macrophage; CSF-1: Colony-stimulating factor 1; OSCC: Oral squamous cell carcinoma; EMT: Epithelial-mesenchymal transition; ECM: Extracellular matrix; NFs: Normal fibroblasts; siRNAs: Small interfering RNAs; NC: Negative controls; shRNAs: Short hairpin RNAs; PCA: Principal component analysis; UMAP: Uniform manifold approximation and projection; t-SNE: t-distributed stochastic neighbor embedding; CDS: Cell data set; GSEA: Gene set enrichment analysis; TMAs: Tissue microarrays; IHC: Immunohistochemical; HPA: Human protein atlas; scRNA-seq: Single-cell RNA sequencing; CAF_myo: CAFs with a myofibroblastic phenotype; FN: Fibronectin; CM: Conditioned media; BP: Biological process; TGFβR1: TGF-β receptor 1; miRNAs: MicroRNAs; 3'-UTR: 3' untranslated region; MSC: Mesenchymal stem cell; ceRNA: Competitive endogenous RNA.
Supplementary Material
Supplementary tables.
Acknowledgements
We sincerely thank Dr. Xiangliu Chen for generously providing the fibroblast cell lines that were essential for this study. We also gratefully acknowledge BioRender for their invaluable assistance in creating the abstract diagram used in this work (created in BioRender, https://BioRender.com/qd4ejw4).
Funding
The project was supported by the Department of Science and Technology of Zhejiang Province, "Pioneer" and "Leading Goose" R&D Program of Zhejiang (NO.2024C03146), the Regional Diagnosis and Treatment Center Project by the National Health Commission (NO. JBZX-201903), and National Natural Science Foundation of China (No. 82302886).
Author contributions
Chao He: Conceptualization, Writing - original draft, Validation, Visualization, Review. Zheng Zhou: Review and editing. Jiayue Ye: Review and editing. Xiangliu Chen: Methodology. Yan Yang: Data curation. Xinguang Jin: Review. Quan Zhou: Supervision, Review and editing. Lisong Teng: Supervision, Review and editing.
Data availability
The publicly available single-cell sequencing data used in this study can be accessed through the GEO database. Primary data are available upon request.
Ethics declarations
All authors approved and directly participated in the planning, execution and/or analysis of the data presented here. And this study was reviewed and approved by the Ethics Committee of the First Affiliated Hospital of Zhejiang University.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. Ca-cancer J Clin. 2018;68:394-424
2. Smyth EC, Nilsson M, Grabsch HI, van Grieken NC, Lordick F. Gastric cancer. Lancet. 2020;396:635-48
3. Sexton RE, Al Hallak MN, Diab M, Azmi AS. Gastric cancer: a comprehensive review of current and future treatment strategies. Cancer Metastasis Rev. 2020;39:1179-203
4. Arjani S, Jeon H, Chadha B, Yousuf H, Castellucci E. Leptomeningeal carcinomatosis in gastric cancer: A Review. Gastric Cancer. 2025;28:311-25
5. Thomassen I, van Gestel YR, van Ramshorst B, Luyer MD, Bosscha K, Nienhuijs SW. et al. Peritoneal carcinomatosis of gastric origin: a population-based study on incidence, survival and risk factors. Int J Cancer. 2014;134:622-8
6. Propper DJ, Balkwill FR. Harnessing cytokines and chemokines for cancer therapy. Nat Rev Clin Oncol. 2022;19:237-53
7. Pereira BA, Vennin C, Papanicolaou M, Chambers CR, Herrmann D, Morton JP. et al. CAF Subpopulations: A New Reservoir of Stromal Targets in Pancreatic Cancer. Trends Cancer. 2019;5:724-41
8. Xiao Y, Yu D. Tumor microenvironment as a therapeutic target in cancer. Pharmacol Therapeut. 2021;221:107753
9. Qi L, Yu Y, Chi X, Xu W, Lu D, Song Y. et al. Kindlin-2 interacts with α-actinin-2 and β1 integrin to maintain the integrity of the Z-disc in cardiac muscles. FEBS Lett. 2015;589:2155-62
10. Sossey-Alaoui K, Pluskota E, Bialkowska K, Szpak D, Parker Y, Morrison CD. et al. Kindlin-2 Regulates the Growth of Breast Cancer Tumors by Activating CSF-1-Mediated Macrophage Infiltration. Cancer Res. 2017;77:5129-41
11. Yoshida N, Masamune A, Hamada S, Kikuta K, Takikawa T, Motoi F. et al. Kindlin-2 in pancreatic stellate cells promotes the progression of pancreatic cancer. Cancer Lett. 2017;390:103-14
12. Ma X, Zhao D, Liu S, Zuo J, Wang W, Wang F. et al. FERMT2 upregulation in CAFS enhances EMT of OSCC and M2 macrophage polarization. Oral Dis. 2024;30:991-1003
13. Sahai E, Astsaturov I, Cukierman E, DeNardo DG, Egeblad M, Evans RM. et al. A framework for advancing our understanding of cancer-associated fibroblasts. Nat Rev Cancer. 2020;20:174-86
14. Hosein AN, Brekken RA, Maitra A. Pancreatic cancer stroma: an update on therapeutic targeting strategies. Nat Rev Gastroenterol Hepatol. 2020;17:487-505
15. Wu GH, He C, Che G, Zhou Z, Chen BY, Wu HM. et al. The role of FERMT2 in the tumor microenvironment and immunotherapy in pan-cancer using comprehensive single-cell and bulk sequencing. Heliyon. 2024;10:e30505
16. Kalluri R. The biology and function of fibroblasts in cancer. Nat Rev Cancer. 2016;16:582-98
17. Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. 2014;15:786-801
18. He C, Zhou Z, Yang Y, Zhu S, Wang H, Teng L. FERMT2 drives anoikis resistance and peritoneal metastasis by enhancing extracellular matrix deposition in gastric cancer. Gastric Cancer. 2025;28:409-421
19. Song M, He J, Pan QZ, Yang J, Zhao J, Zhang YJ. et al. Cancer-Associated Fibroblast-Mediated Cellular Crosstalk Supports Hepatocellular Carcinoma Progression. Hepatology. 2021;73:1717-35
20. Larjava H, Plow EF, Wu C. Kindlins: essential regulators of integrin signalling and cell-matrix adhesion. EMBO Rep. 2008;9:1203-8
21. Zhang Y, Liu Z, Yang X, Lu W, Chen Y, Lin Y. et al. H3K27 acetylation activated-COL6A1 promotes osteosarcoma lung metastasis by repressing STAT1 and activating pulmonary cancer-associated fibroblasts. Theranostics. 2021;11:1473-92
22. Zhan J, Song J, Wang P, Chi X, Wang Y, Guo Y. et al. Kindlin-2 induced by TGF-β signaling promotes pancreatic ductal adenocarcinoma progression through downregulation of transcriptional factor HOXB9. Cancer Lett. 2015;361:75-85
23. Bai X, Shao J, Zhou S, Zhao Z, Li F, Xiang R. et al. Inhibition of lung cancer growth and metastasis by DHA and its metabolite, RvD1, through miR-138-5p/FOXC1 pathway. J Exp Clin Cancer Res. 2019;38:479
24. Li D, She J, Hu X, Zhang M, Sun R, Qin S. The ELF3-regulated lncRNA UBE2CP3 is over-stabilized by RNA-RNA interactions and drives gastric cancer metastasis via miR-138-5p/ITGA2 axis. Oncogene. 2021;40:5403-15
25. Zhao W, Zhang R, Zang C, Zhang L, Zhao R, Li Q. et al. Exosome Derived from Mesenchymal Stem Cells Alleviates Pathological Scars by Inhibiting the Proliferation, Migration and Protein Expression of Fibroblasts via Delivering miR-138-5p to Target SIRT1. Int J Nanomedicine. 2022;17:4023-38
26. Korpal M, Lee ES, Hu G, Kang Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J Biol Chem. 2008;283:14910-4
27. Park SM, Gaur AB, Lengyel E, Peter ME. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008;22:894-907
28. Zhang HF, Zhang K, Liao LD, Li LY, Du ZP, Wu BL. et al. miR-200b suppresses invasiveness and modulates the cytoskeletal and adhesive machinery in esophageal squamous cell carcinoma cells via targeting Kindlin-2. Carcinogenesis. 2014;35:292-301
29. Ren W, Gao L, Qiang C, Li S, Zheng J, Wang Q. et al. Kindlin-2-mediated upregulation of ZEB2 facilitates migration and invasion of oral squamous cell carcinoma in a miR-200b-dependent manner. Am J Transl Res. 2018;10:2529-41
30. Wu Q, Gui W, Jiao B, Han L, Wang F. miR-138 inhibits epithelial-mesenchymal transition in silica-induced pulmonary fibrosis by regulating ZEB2. Toxicology. 2021;461:152925
31. Bueno-Urquiza LJ, Godínez-Rubí M, Villegas-Pineda JC, Vega-Magaña AN, Jave-Suárez LF, Puebla-Mora AG. et al. Phenotypic Heterogeneity of Cancer Associated Fibroblasts in Cervical Cancer Progression: FAP as a Central Activation Marker. Cells. 2024;13:560
32. Gao Y, Li J, Cheng W, Diao T, Liu H, Bo Y. et al. Cross-tissue human fibroblast atlas reveals myofibroblast subtypes with distinct roles in immune modulation. Cancer Cell. 2024;42:1764-1783.e10
33. Mao X, Xu J, Wang W, Liang C, Hua J, Liu J. et al. Crosstalk between cancer-associated fibroblasts and immune cells in the tumor microenvironment: new findings and future perspectives. Mol Cancer. 2021;20:131
34. Huang TX, Tan XY, Huang HS, Li YT, Liu BL, Liu KS. et al. Targeting cancer-associated fibroblast-secreted WNT2 restores dendritic cell-mediated antitumour immunity. Gut. 2022;71:333-44
35. Rodeck U, Bossler A, Graeven U, Fox FE, Nowell PC, Knabbe C. et al. Transforming growth factor beta production and responsiveness in normal human melanocytes and melanoma cells. Cancer Res. 1994;54:575-81
36. Calon A, Tauriello DVF, Batlle E. TGF-beta in CAF-mediated tumor growth and metastasis. Semin Cancer Biol. 2014;25:15-22
37. Khan SU, Fatima K, Malik F. Understanding the cell survival mechanism of anoikis-resistant cancer cells during different steps of metastasis. Clin Exp Metastasis. 2022;39:715-26
38. Yang K, Xie Y, Xue L, Li F, Luo C, Liang W. et al. M2 tumor-associated macrophage mediates the maintenance of stemness to promote cisplatin resistance by secreting TGF-β1 in esophageal squamous cell carcinoma. J Transl Med. 2023;21:26
39. Kaowinn S, Seo EJ, Heo W, Bae JH, Park EJ, Lee S. et al. Cancer upregulated gene 2 (CUG2), a novel oncogene, promotes stemness-like properties via the NPM1-TGF-β signaling axis. Biochem Biophys Res Commun. 2019;514:1278-84
40. Wang LN, Zhang ZT, Wang L, Wei HX, Zhang T, Zhang LM. et al. TGF-β1/SH2B3 axis regulates anoikis resistance and EMT of lung cancer cells by modulating JAK2/STAT3 and SHP2/Grb2 signaling pathways. Cell Death Dis. 2022;13:1-12
41. Ko H. Geraniin inhibits TGF-β1-induced epithelial-mesenchymal transition and suppresses A549 lung cancer migration, invasion and anoikis resistance. Bioorg Med Chem Lett. 2015;25:3529-34
42. Chen X, Song E. Turning foes to friends: targeting cancer-associated fibroblasts. Nat Rev Drug Discov. 2019;18:99-115
43. Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367:eaau6977
44. Lötvall J, Hill AF, Hochberg F, Buzás EI, Di Vizio D, Gardiner C. et al. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J Extracell Vesicles. 2014;3:26913
45. Huang Y, Kanada M, Ye J, Deng Y, He Q, Lei Z. et al. Exosome-mediated remodeling of the tumor microenvironment: From local to distant intercellular communication. Cancer Lett. 2022;543:215796
46. Zhou Q, Wu X, Wang X, Yu Z, Pan T, Li Z. et al. The reciprocal interaction between tumor cells and activated fibroblasts mediated by TNF-α/IL-33/ST2L signaling promotes gastric cancer metastasis. Oncogene. 2020;39:1414-28
47. Li Y, Jiang L, Chen Y, Li Y, Yuan J, Lu J. et al. Specific lineage transition of tumor-associated macrophages elicits immune evasion of ascitic tumor cells in gastric cancer with peritoneal metastasis. Gastric Cancer. 2024;27:519-38
48. Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y. et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature. 2018;554:544-8
49. Peng D, Fu M, Wang M, Wei Y, Wei X. Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol Cancer. 2022;21:104
50. Wortzel I, Dror S, Kenific CM, Lyden D. Exosome-Mediated Metastasis: Communication from a Distance. Dev Cell. 2019;49:347-60
51. Sherman MH, Yu RT, Engle DD, Ding N, Atkins AR, Tiriac H. et al. Vitamin D receptor-mediated stromal reprogramming suppresses pancreatitis and enhances pancreatic cancer therapy. Cell. 2014;159:80-93
Author contact
![]() Corresponding authors: Lisong Teng: lstengedu.cn. Quan Zhou: zhouquanzqedu.cn.
Corresponding authors: Lisong Teng: lstengedu.cn. Quan Zhou: zhouquanzqedu.cn.