Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
The PI3K/Akt/mTOR Pathway: A...
IR-Remodeled Liver...
Pathway-Driven Metabolic...
Direct Regulation of Immune...
PI3K/Akt/mTOR Pathway-Mediated...
Molecular Remodeling of HCC...
Summary and Outlook
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2025; 21(14):6025-6041. doi:10.7150/ijbs.120657 This issue Cite
Review
The PI3K/Akt/mTOR Pathway: Immuno-Metabolic Orchestration in IR/MASH-Associated Hepatocellular Carcinoma
Jian Zhao1, Yuehua Zhang2, Zhigong Wei3, Kai Li1, Lei Sun2 ![]() , Dan Li4
, Dan Li4 ![]() , Yongsheng Wang1
, Yongsheng Wang1 ![]()
1. Thoracic Oncology Ward, Cancer Center, and State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, Chengdu, Sichuan, 610041, P.R. China.
2. West China School of Public Health and West China Fourth Hospital, Sichuan University, Chengdu, Sichuan 610041, P.R. China.
3. Department of Biotherapy, Cancer Center and State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, P.R. China.
4. Institute of Respiratory Health, Frontiers Science Center for Disease-related Molecular Network, and Precision Medicine Research Center, Precision Medicine Key Laboratory of Sichuan Province, West China Hospital, Sichuan University, Chengdu, Sichuan 610041, P.R. China.
Received 2025-6-30; Accepted 2025-8-15; Published 2025-9-27
Abstract

Insulin resistance (IR) and Metabolic Dysfunction-Associated Steatohepatitis (MASH) are key drivers of hepatocellular carcinoma (HCC), yet the mechanisms underlying their induction of an immunosuppressive tumor microenvironment (TME) require elucidation. This review posits that the PI3K/Akt/mTOR signaling pathway acts as the central integrator of this process, becoming fundamentally rewired—or “imprinted”—by the unique pathological context of IR/MASH-HCC. We highlight how this “imprinted” pathway integrates disparate pathological signals to precisely direct tumor metabolic reprogramming, TME immune landscape remodeling, and the metabolic-dependent regulation of immune cells. We particularly dissect the synergistic amplification of pathway-mediated immune evasion (including PD-L1 upregulation and EMT) by the IR/MASH microenvironment. This integrated framework, which conceptualizes the pathway as the central processing unit of a uniquely aggressive immuno-metabolic phenotype, not only illuminates the unique biology of IR/MASH-HCC but also provides new insights and a theoretical basis for the clinical translation of targeting the PI3K/Akt/mTOR pathway—encompassing novel combination strategies and biomarker development—to foster more effective clinical interventions.
Keywords: Hepatocellular Carcinoma, PI3K/Akt/mTOR Signaling Pathway, Insulin Resistance, MASH, Tumor Immune Microenvironment, Metabolic Reprogramming
Introduction
Hepatocellular Carcinoma (HCC), comprising 80-90% of primary liver cancers, is a major cause of cancer-related mortality worldwide, with a particularly poor prognosis in its advanced stages [1]. Although viral hepatitis remains the leading global cause, the incidence of HCC associated with metabolic disorders is rising dramatically in industrialized and westernized regions, posing a formidable challenge [2,3]. Metabolic dysfunction-associated fatty liver disease (MAFLD), a term that has replaced the former nomenclature of non-alcoholic fatty liver disease (NAFLD), has a global prevalence of approximately 25% and has become the primary cause of chronic liver disease and HCC in many countries [4-6]. MAFLD can progress to Metabolic Dysfunction-Associated Steatohepatitis (MASH), a condition accompanied by inflammation and fibrosis that is a high-risk state for cirrhosis and HCC [7]. Insulin resistance (IR), a core pathophysiological feature of MAFLD and type 2 diabetes (T2DM), is also a confirmed independent risk factor for HCC [8-10], contributing to a pro-tumorigenic “oncogenic microenvironment” through multiple pathways, including hepatic lipotoxicity, chronic low-grade inflammation, oxidative stress, accelerated fibrosis, and direct stimulation of proliferative pathways [11,12]. This confluence of metabolic insults establishes a uniquely pro-tumorigenic landscape, yet a central question remains largely unanswered: How does a systemic metabolic disease like IR translate into a specific, aggressive, and immune-evasive cancer phenotype at the molecular level? This review posits that the answer lies with a single, indispensable protagonist: the Phosphoinositide 3-kinase (PI3K)/Protein Kinase B (Akt)/mammalian Target of Rapamycin (mTOR) signaling pathway, which serves as the critical nexus between systemic metabolic dysfunction and the local tumor immuno-metabolic landscape.
The pathogenesis of HCC is a complex process involving the dysregulation of several key signaling networks, and pathways such as Wnt/β-catenin, MAPK, and Notch are undeniably critical drivers in a broader context [13,14]. However, this review asserts that the PI3K/Akt/mTOR pathway occupies a unique and central position specifically within the etiology of MASH-driven HCC. Its centrality is not arbitrary; it is based on a unique convergence of qualifications that no other oncogenic pathway possesses. First, unlike its counterparts, the PI3K/Akt/mTOR cascade is a primary downstream effector of insulin and insulin-like growth factor (IGF) signaling [15], positioning it as a direct molecular “sensor” for the hallmark metabolic disturbances of MASH—namely, IR and the resultant chronic hyperinsulinemia [10]. While other pathways contribute to cell proliferation, the PI3K/Akt/mTOR pathway uniquely translates these specific systemic metabolic insults into direct pro-tumorigenic action within the hepatocyte. Second, its downstream effects are profoundly dual-faceted, directly shaping the two core pillars of MASH-HCC progression. On one hand, it orchestrates the profound metabolic reprogramming essential for cancer cell survival, including the enhancement of the Warburg effect and potent stimulation of de novo lipogenesis (DNL) [16,17]. On the other hand, it is intricately linked to the regulation of the immune landscape, modulating the expression of immune checkpoints like PD-L1 [18,19] and actively shaping an immunosuppressive tumor microenvironment through its metabolic outputs [20]. Therefore, in the specific subtype of HCC that arises from a backdrop of metabolic disease, the PI3K/Akt/mTOR pathway functions as a central coordinator at the center of a “perfect storm.” It is simultaneously: 1) The Sensor, directly activated by the disease's defining feature; 2) The Engine, driving the metabolic reprogramming required for tumor growth; and 3) The Conductor, orchestrating the immunosuppressive TME through metabolic warfare and checkpoint regulation. It is this unparalleled role as the principal integrator of metabolism and immunity that justifies its selection as the central focus of this review, offering the most relevant lens through which to understand and potentially target MASH-driven HCC.
Despite its importance, few existing reviews have systematically elucidated from an integrated perspective how the PI3K/Akt/mTOR pathway, within the specific pathological context of IR/MASH, acts as a central coordinator to precisely orchestrate the complex “immuno-metabolic-signal” interaction network. Clarifying this unique perspective is crucial for understanding the pathological mechanisms of IR/MASH-related HCC, identifying therapeutic targets, and guiding clinical strategies.
To address this critical gap, this review will comprehensively depict the central coordinating role of this pathway. We will delve into its “molecular imprinting” in the IR/MASH context, its role in driving metabolic reprogramming and shaping the TME, the “metabolic-dependent” regulation of immune cells, key feedback loops, and the synergistic amplification of immune evasion mechanisms. Furthermore, based on these mechanisms, we will analyze the molecular remodeling of the HCC therapeutic response, assess the challenges, and propose novel precision combination therapy strategies. This work therefore aims not only to synthesize existing data but to provide a novel, multidimensional molecular framework for understanding the unique biology of IR/MASH-driven HCC and to offer a theoretical basis for developing more precise and effective therapeutic strategies.
Ultimately, by reframing the PI3K/Akt/mTOR pathway not as a simple linear cascade but as a centrally 'imprinted' immuno-metabolic coordinator, this review provides a unifying framework to reconceptualize the pathogenesis of MASH-driven HCC. It moves beyond a mere catalog of individual molecular events to construct an integrated logic that explains the disease's unique biological aggressiveness and immune resistance. This perspective is therefore not merely academic; it offers a fundamental basis for overcoming current therapeutic challenges and for the rational design of next-generation combination strategies tailored to this growing and distinct patient population.
The PI3K/Akt/mTOR Pathway: A Double-Edged Sword of Physiological Regulation and Pathological Dysregulation
The PI3K/Akt/mTOR signaling cascade is a highly conserved and vital intracellular network in eukaryotic cells. It integrates extracellular signals (e.g., growth factors like insulin, IGF-1) and intracellular cues (e.g., nutrients, energy status) to precisely regulate a wide range of activities including metabolism, growth, proliferation, survival, migration, and angiogenesis [16,21]. Its core components include the lipid kinase PI3K, the serine/threonine protein kinase Akt (also known as PKB), and mTOR, which forms two functionally distinct complexes: mTORC1 and mTORC2 [22,23]. Physiologically, this pathway is a key mediator of insulin signaling, essential for maintaining glucose homeostasis [9,24]. The tumor suppressor Phosphatase and tensin homolog (PTEN) is the most critical negative regulator of the pathway, terminating the signal by dephosphorylating PIP3 [25,26]. Notably, PTEN's activity is itself precisely regulated by the cellular redox state, with its active site cysteine residue (Cys124) being highly sensitive to inactivation by reactive oxygen species (ROS), a feature of profound importance in pathological states [27].
However, in the pathological environment of IR/MASH, pathway dysregulation presents a central paradox: despite upstream signaling being impeded by inflammatory and lipotoxic insults that impair insulin receptor substrate (IRS) proteins [28-30], downstream branches of the pathway can bypass this blockade to achieve sustained, aberrant activation. This profound and durable rewiring of the pathway's signaling logic, we term 'molecular imprinting'. In this context, molecular imprinting refers to the decoupling of the PI3K/Akt/mTOR pathway from its physiological upstream inputs and negative feedback controls, rendering it constitutively active and hyper-responsive to the specific pathological cues of the MASH microenvironment. A key mechanism of this imprinting is the sustained functional inhibition of PTEN by ROS or lipid peroxidation products [31,32]. Concurrently, the pathway's internal negative feedback loops, such as the classic S6K→IRS1 loop, are “hijacked” by pro-inflammatory signals. This decoupling of upstream signal blockade and downstream sustained activation, driven by the failure of negative feedback mechanisms, is the core feature that defines its molecular 'imprinting' [33].
In HCC tumor cells, this pathway's aberrant activation is even more prevalent and complex. The driving factors now include not only the aforementioned environmental factors from MASH but also a host of cell-intrinsic genetic and epigenetic alterations, such as loss-of-function mutations in the PTEN gene itself [34], mutations in other pathway components, and the overexpression of upstream receptor tyrosine kinases (RTKs). Furthermore, specific inflammatory-oncogenic circuits reinforce this sustained activation. For instance, in the context of obesity-driven HCC, the pro-inflammatory cytokine IL-6, in concert with androgen receptor signaling, can induce the expression of the cell cycle-related kinase (CCRK), which in turn establishes a positive feedback loop to drive mTORC1 activation [35]. These cell-intrinsic changes, superimposed upon the sustained stimuli from the MASH microenvironment (high insulin/IGF, inflammation, oxidative stress), collectively result in a strong and persistent aberrant activation of the PI3K/Akt/mTOR pathway.
Crucially, this 'imprinted' failure of PI3K pathway feedback is not a static feature but the central dynamic engine that propels the malignant progression from MASH to HCC. This progression can be conceptualized as a disastrous cascade initiated by the imprinted pathway: 1. Initiation (Decoupling and Priming): In the MASH stage, sustained PTEN inhibition locks the PI3K/Akt pathway into a state of chronic, low-intensity activation, relentlessly driving pro-tumorigenic shifts [36]. 2. Promotion (Survival, Senescence, and Microenvironment Corruption): Sustained Akt activation confers a survival advantage while paradoxically inducing a pro-tumorigenic Senescence-Associated Secretory Phenotype (SASP) that corrupts the local microenvironment [37-39]. 3. Progression (Genomic Instability): The combination of continuous proliferative pressure and a ROS-laden microenvironment creates a perfect storm for the accumulation of DNA damage and mutations [40]. 4. Transformation (The Final Hit): This process facilitates the acquisition of critical genetic lesions (e.g., in TP53, CTNNB1) that liberate a hepatocyte clone from all remaining restraints [41]. Thus, the 'imprinted' failure of PI3K pathway feedback acts as the master initiator of this disastrous cascade, sequentially navigating a damaged hepatocyte through survival, microenvironment corruption, and genomic instability, ultimately culminating in malignant transformation. At this point, the PI3K pathway, a 'double-edged sword' for maintaining homeostasis, has been fully tipped to its pathological side, becoming a fatal engine of hepatocarcinogenesis. This transformation, however, does not occur in a vacuum; it is forged by the unique pathological pressures of the IR-remodeled liver microenvironment, which lays the foundation for this profound pathway dysregulation.
IR-Remodeled Liver Microenvironment: Laying the Foundation for Pathway Dysregulation
By triggering a cascade of systemic and local pathophysiological changes, IR profoundly reshapes the liver's tissue architecture, metabolic homeostasis, and immune balance, thereby constructing a unique, highly pro-tumorigenic microenvironment for PI3K/Akt/mTOR pathway dysregulation. Understanding the key components of this microenvironment is crucial for comprehending how the pathway is “imprinted”.
At the metabolic level, a toxic milieu of lipids and their byproducts directly interferes with PI3K/Akt signaling, laying the primary foundation for its 'imprinting'. The core of IR is the reduced responsiveness of target tissues to insulin, leading to an influx of FFAs into the liver and enhanced hepatic DNL [10,42]. The key culprits are excessively accumulated lipotoxic molecules, such as saturated fatty acids (SFAs) [43], ceramides [44], and diacylglycerols (DAGs) [15], which disrupt the PI3K/Akt/mTOR pathway by interfering with IRS function or inhibiting Akt phosphorylation.
At the immuno-inflammatory level, the chronic inflammation of MASH provides a continuous barrage of signals that hijack and sustain PI3K/Akt pathway activity. Lipotoxicity, ROS, and gut-derived factors activate innate immune cells like Kupffer cells, which release pro-inflammatory cytokines such as TNF-α and IL-6 [31,45]. These factors not only amplify IR but also remodel the immune cell landscape towards an immunosuppressive phenotype, characterized by M2-like TAMs, exhausted Teffs, and an increase in Tregs and MDSCs [45-47].
At the oxidative stress level, a state of severe oxidative stress provides the most direct mechanism for 'imprinting' the pathway by functionally inactivating its key negative regulator, PTEN. ROS in MASH are derived from various sources, including mitochondrial dysfunction and NADPH oxidase (NOX) activation [10,36,48]. Critical ROS mediators and lipid peroxidation products can directly oxidize or modify PTEN, while chronic oxidative stress can deplete antioxidant systems, ultimately leading to sustained PTEN inactivation [49].
At the matrix remodeling level, the fibrotic landscape provides a source of potent growth factors that offer another layer of sustained, external activation for the PI3K/Akt pathway. Chronic inflammation and hepatocyte injury activate hepatic stellate cells (HSCs), leading to liver fibrosis [50]. This altered physical environment exacerbates immunosuppression through growth factors (e.g., TGF-β, HGF) secreted by activated HSCs [51].
In summary, the MASH microenvironment is not a passive backdrop but an active forger. The convergence of lipotoxic interference, inflammatory hijacking, direct PTEN inactivation by ROS, and sustained growth factor signaling collectively and profoundly 'imprint' the PI3K/Akt/mTOR pathway, transforming it from a physiological regulator into a core pathological engine. Once imprinted, this dysregulated pathway becomes a master conductor, orchestrating a profound reprogramming of tumor cell metabolism, as we will discuss next.
Pathway-Driven Metabolic Reprogramming and Its Immune Shaping of the TME
In the specific pathological context of IR-driven HCC, the PI3K/Akt/mTOR pathway functions as the central driver of a profound metabolic reprogramming. Its impact extends beyond merely satisfying the tumor's own anabolic demands; it actively remodels the entire functional landscape of the TME by orchestrating the release and consumption of key metabolites. This process can be conceptualized as the tumor leveraging metabolic alterations as a weapon to establish an immunosuppressive niche [52]. Pathway activation is particularly prominent in the IR environment, driven by complex factors including the common loss of function of the tumor suppressor PTEN, strong stimulation from persistent hyperinsulinemia/IGF signaling, and even direct pathway activation by high-fat diet-induced Akt palmitoylation, providing a direct metabolic link [53].
Firstly, the activated PI3K/Akt/mTOR pathway promotes the Warburg effect. By stabilizing the key transcription factor HIF-1α, possibly in synergy with MYC, it upregulates GLUT1 and key glycolytic enzymes (e.g., HK2, LDHA) [15]. This process is greatly amplified in the MASH liver, where pre-existing hyperglycemia provides abundant substrate, while the hypoxic and chronic inflammatory signals within the MASH liver may synergize with pathway activation to further stabilize HIF-1α, pushing glycolysis to its limit and causing a massive accumulation of lactate in the TME [31].
Secondly, the pathway orchestrates a profound shift in anabolic metabolism, with a particular focus on lipids. A primary effect is the potent drive for de novo lipogenesis (DNL) and cholesterol synthesis. By phosphorylating and activating the master transcriptional regulator SREBP1, the pathway upregulates key lipogenic genes like FASN, ACC, and SCD1 [54]. This pathway-driven DNL is superimposed upon a liver already pathologically burdened by steatosis, creating a far more severe lipotoxic environment. Tumor cells actively secrete these newly synthesized lipids, transforming the microenvironment's metabolic landscape. Specifically, this leads to the release of saturated fatty acids (SFAs), such as palmitate, and monounsaturated fatty acids (MUFAs), like oleate [55]. These secreted lipids are potent, differential regulators of immune function. The accumulation of SFAs is known to be directly toxic to effector T cells, inducing a form of cell death known as lipoapoptosis [46]. In contrast, these lipids can promote the polarization of macrophages towards an immunosuppressive M2 phenotype [56] and provide fuel for the pro-tumorigenic functions of MDSCs and Tregs, which rely on fatty acid oxidation (FAO) [57,58].
Thirdly, the pathway is deeply involved in remodeling amino acid metabolism. It upregulates key amino acid transporters (e.g., SLC7A5/LAT1, SLC1A5/ASCT2) to enhance the uptake of essential amino acids like glutamine, which supports both rapid proliferation and sustained mTORC1 activation [59,60]. However, this increased consumption by tumor cells, combined with the inflammatory environment and the enrichment of M2-like TAMs (with high expression of IDO and ARG1), can lead to the severe local depletion of other critical amino acids, such as tryptophan and arginine [61].
These metabolic alterations converge to create a hostile TME through three primary mechanisms. First, resource competition, wherein tumor cells monopolize glucose via the Warburg effect, leading to mTORC1 inhibition and functional exhaustion in effector T cells [46]. Second, direct metabolic suppression, where secreted metabolites act as signaling molecules. Lactate, for instance, inhibits T cell function through various means, including cytoplasmic acidification and histone lactylation [62,63]. Simultaneously, specific SFAs are directly lipotoxic to these T cells. In stark contrast, these same metabolites—lactate and fatty acids—can fuel the function and polarization of immunosuppressive cells like M2-TAMs and Tregs [47]. Third, nutrient deprivation, primarily of key amino acids such as tryptophan and arginine by enzymes like IDO and ARG1, further cripples the anti-tumor T cell response [64] (Figure 1).
PI3K/Akt/mTOR pathway dysregulation by the IR/MASH oncogenic microenvironment fuels HCC metabolic reprogramming. In hepatocellular carcinoma (HCC) cells, the insulin resistance (IR) and Metabolic Dysfunction-Associated Steatohepatitis (MASH) microenvironment—characterized by hyperinsulinemia, inflammatory cytokines (TNF-α, IL-6), saturated fatty acids (SFAs), and reactive oxygen species (ROS)—dysregulates PI3K/Akt/mTOR signaling. This occurs via mechanisms including impaired IRS signaling and ROS-mediated PTEN inactivation. These events promote hyperactivation of Akt (by PDK1 and mTORC2) and subsequently mTORC1. Activated mTORC1 drives metabolic reprogramming towards enhanced glycolysis (via HIF-1α) and de novo lipogenesis (via SREBP1c), fueling HCC progression and shaping the tumor microenvironment. Phosphorylation is denoted by (P).
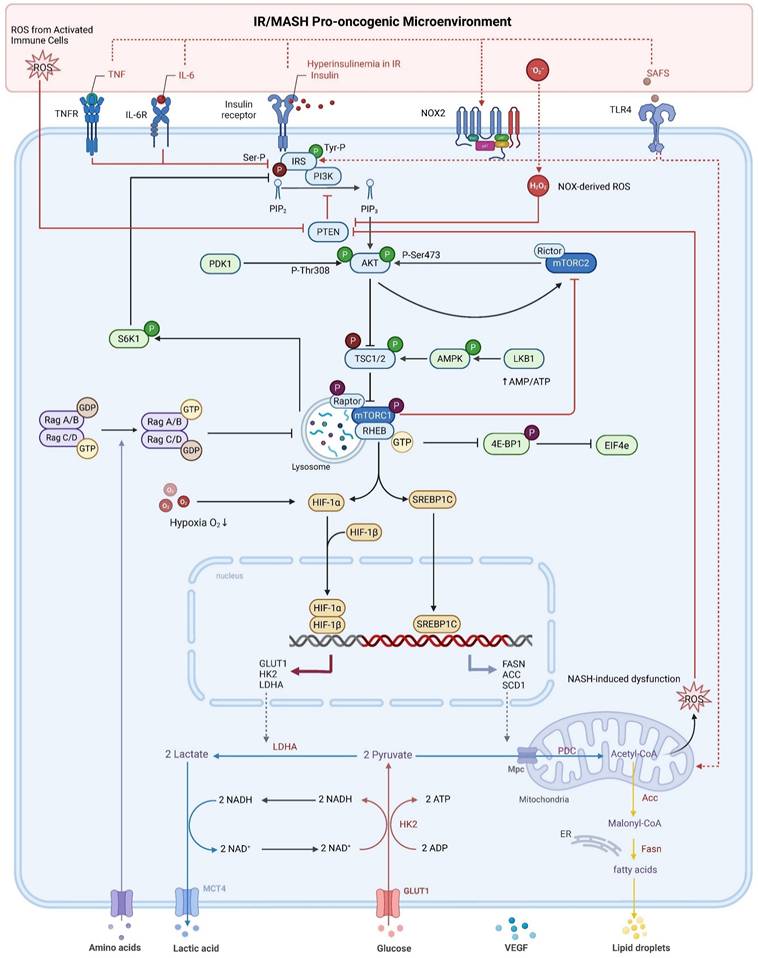
Perhaps no single mechanism better exemplifies this coordinated immuno-metabolic crosstalk than the recently identified inflammatory-oncogenic circuit involving the cell cycle-related kinase (CCRK). This circuit acts as a potent upstream activator of mTORC1 signaling in obesity-associated HCC, and is cooperatively induced by obesity-driven pro-inflammatory signals (the IL-6/STAT3 axis) and androgen receptor (AR) signaling—providing a molecular explanation for the male predominance in MASH-HCC. Mechanistically, activated CCRK drives the mTORC1 pathway, which in turn orchestrates a perfect two-pronged assault. On the metabolic front, it amplifies DNL by activating SREBP1. On the immune front, it stimulates the secretion of G-CSF, leading to the robust recruitment of highly immunosuppressive polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs) into the liver [35]. This “inflammatory-CCRK-mTORC1” axis thus represents a powerful molecular bridge that translates the inflammatory and hormonal milieu of MASH directly into coordinated metabolic reprogramming and profound immunosuppression.
Direct Regulation of Immune Cells by the PI3K/Akt/mTOR Pathway: Metabolic Dependence and Bidirectional Feedback
The influence of the PI3K/Akt/mTOR pathway is not confined to tumor cells; its signaling status within various immune cells in the tumor microenvironment (TME) directly dictates their functional phenotypes, and its activity is itself highly dependent on the local metabolic milieu [60]. This “metabolic dependence” of intracellular pathway regulation, coupled with a complex bidirectional feedback network involving other TME components, collectively shapes the immune landscape of IR-driven HCC.
Metabolic Environment-Shaped Pathway Activity and Functional Choices in Immune Cells
The unique metabolic features of the TME (e.g., low glucose, high lactate, altered lipid profiles) profoundly influence the activity of the PI3K/Akt/mTOR and related pathways (e.g., AMPK) within infiltrating immune cells. For instance, in tumor-associated macrophages (TAMs), lipids enriched in the TME can drive their polarization towards an M2-like, pro-tumorigenic phenotype by specifically activating the internal PI3Kγ signaling pathway [57,65]. High lactate may also promote M2 polarization [56]. In contrast, the function of effector T cells (Teffs) is highly dependent on mTORC1-driven glycolysis [59,66]. The low glucose in the TME directly inhibits mTORC1 activity [46,67], while high lactate can further suppress the PI3K/Akt/mTORC1 pathway [62,68], collectively leading to impaired Teff function and exhaustion [69]. This is a key mechanism driving T cell exhaustion, severely limiting their persistence and efficacy (including that of CAR-T cells) in solid tumors [70]. In contrast, regulatory T cells (Tregs) exhibit greater metabolic adaptability, such as the ability to utilize lactate [71], allowing them to thrive. This differential response and dependence of various immune cell pathways on the TME's metabolism is a major reason for the formation of the immunosuppressive landscape.
Feedback Regulation of Tumor Cell PI3K Pathway by TME Cells and Amplification in the IR Context
Immune and stromal cells in the TME are not passive bystanders; they actively secrete various factors that “feedback” to tumor cells, sustaining the activation of pro-survival/proliferative pathways like PI3K/Akt/mTOR. This feedback is particularly critical in IR/MASH-driven HCC, as the chronic inflammatory and fibrotic environment provides abundant cellular (e.g., activated HSCs, TAMs) and molecular sources for these signals. Key feedback mechanisms include growth factors secreted by activated HSCs and TAMs, such as HGF (activating PI3K/Akt via c-Met) [72] and EGF (activating PI3K/Akt via EGFR and mediating drug resistance) [73]. These signals constitute a major external push for maintaining sustained pathway activation in the HSC/TAM-rich IR/MASH environment. Additionally, the inherent chronic inflammation of IR/MASH continuously releases pro-inflammatory cytokines like TNF-α and IL-6 [74,75]. These cytokines act on tumor cells, activating NF-κB and STAT3 pathways, which can then engage in crosstalk with the PI3K/Akt pathway to synergistically enhance cell survival and proliferation [76,77]. (Figure 2). This vicious cycle of metabolic control and pro-tumorigenic feedback creates a pre-conditioned battleground where the tumor's core immune evasion strategies are not just active but are poised for synergistic amplification.
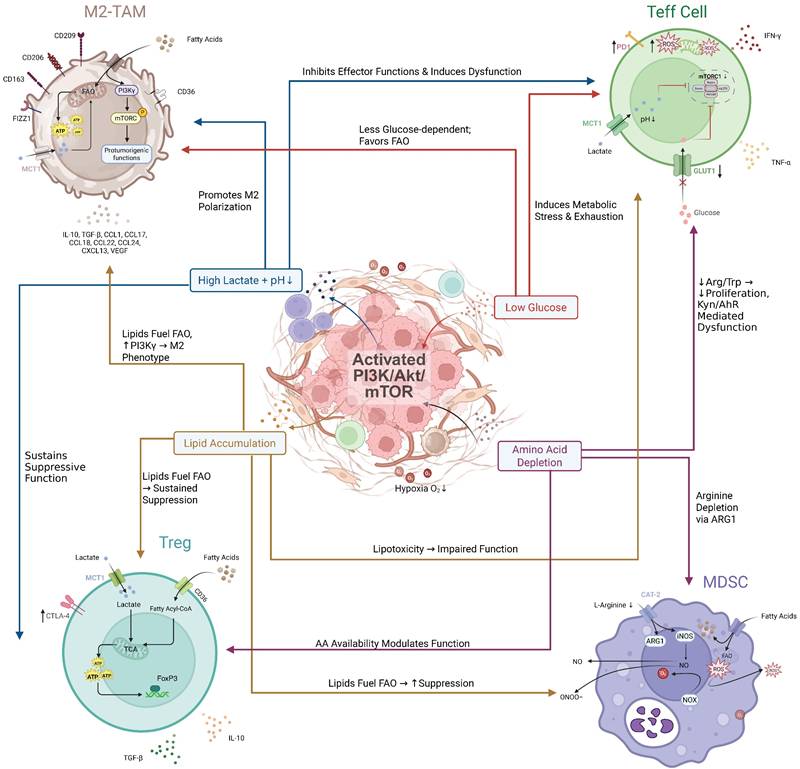
HCC-shaped metabolic landscape of the TME dictates immune cell fate and function. HCC cells with activated PI3K/Akt/mTOR signaling actively remodel the tumor microenvironment (TME) by consuming nutrients and releasing metabolites. This creates a metabolically hostile milieu characterized by high lactate, low glucose, lipid accumulation, and amino acid depletion. These distinct metabolic features differentially affect infiltrating immune cells: Effector T cells (Teffs) are metabolically crippled by glucose deprivation and lactate/lipid toxicity, leading to exhaustion. Regulatory T cells (Tregs) exhibit metabolic flexibility, utilizing lactate and fatty acids to fuel their suppressive functions. M2-like Tumor-Associated Macrophages (M2-TAMs) are polarized by lipids and lactate towards a pro-tumorigenic phenotype. Myeloid-Derived Suppressor Cells (MDSCs) thrive in the lipid-rich environment, supporting their potent immunosuppressive activities. Collectively, the HCC-orchestrated metabolic reprogramming of the TME creates a deeply immunosuppressive landscape.
PI3K/Akt/mTOR Pathway-Mediated Immune Evasion: Synergistic Amplification in the IR/MASH Microenvironment
Aberrant activation of the PI3K/Akt/mTOR pathway within HCC cells orchestrates a complex immune evasion program. In IR/MASH-driven HCC, this evasion appears significantly enhanced, not as a simple consequence of pathway activation, but as a result of a profound “synergistic amplification” between pathway-mediated mechanisms and the unique pathological microenvironment (chronic inflammation, hypoxia, fibrosis, etc.) [52,78,79]. The IR/MASH microenvironment acts as an active participant, and its unique combination of metabolic and inflammatory pressures interacts with core intracellular signaling pathways to amplify key immune evasion strategies, endowing IR/MASH-HCC with unique “stealth” and “counter-attack” capabilities [80]. This section will focus on how two key immune evasion mechanisms—PD-L1 upregulation and epithelial-mesenchymal transition (EMT)—are markedly enhanced by this synergy.
Synergistic Upregulation of PD-L1: A Convergence of Intrinsic and Extrinsic Signals
The synergistic upregulation of PD-L1 is a prime example of this amplification, driven primarily by the convergence of intrinsic and extrinsic signals. Programmed death-ligand 1 (PD-L1) is a key immune checkpoint that enables immune escape by inhibiting T cell function [81]. The PI3K/Akt/mTOR pathway is a core driver of PD-L1 expression in HCC, primarily by activating downstream transcription factors like hypoxia-inducible factor-1α (HIF-1α), nuclear factor-κB (NF-κB), and signal transducer and activator of transcription 3 (STAT3) to promote the transcription of CD274 (the gene encoding PD-L1) [82]. It can also regulate its expression through non-coding RNAs or post-translational modifications.
In IR/MASH-driven HCC, PD-L1 expression is further amplified because the MASH microenvironment's key features—hypoxia and chronic inflammation—are themselves potent external inducers of these same transcription factors. Tissue remodeling and metabolic dysfunction-induced hypoxia directly stabilize and activate HIF-1α [83-85]. Simultaneously, chronic inflammation, driven by lipotoxicity, DAMPs, gut-derived LPS, and a plethora of pro-inflammatory cytokines (e.g., TNF-α, IL-6), continuously activates NF-κB and STAT3 pathways in various liver cells [86-88]. Consequently, signals from the tumor's intrinsic PI3K/Akt/mTOR pathway converge with signals from the external MASH microenvironment, all activating the same set of critical transcription factors (HIF-1α, NF-κB, STAT3) [89]. This multi-source signal convergence, coupled with potential pathway crosstalk (e.g., IL-6/STAT3 and PI3K/Akt interaction) [77] and altered chromatin accessibility [90], leads to a supra-additive amplification of PD-L1 transcription. This synergistically driven high level of PD-L1 dramatically enhances the capacity of IR/MASH-HCC to directly inhibit T cells and evade immune surveillance.
Synergistic Promotion of EMT: From a Duet to a Self-Reinforcing Loop
The promotion of EMT represents a more complex form of synergy, evolving from a duet of signaling pathways into a self-reinforcing feedback loop. Epithelial-mesenchymal transition (EMT) endows tumor cells with invasive, metastatic, and therapy-resistant properties and is regulated by a network of EMT-inducing transcription factors (EMT-TFs) like Snail, Slug, Twist, and ZEB [91]. In HCC, the transforming growth factor-β (TGF-β) pathway and the PI3K/Akt/mTOR pathway are two key upstream drivers of EMT [20,72]. The PI3K/Akt pathway promotes EMT by inhibiting GSK3β-mediated degradation of Snail, thereby stabilizing it [92].
In IR/MASH-driven HCC, the EMT process is also significantly enhanced by synergy with the microenvironment, where the key factor is high TGF-β signaling from liver fibrosis. Late-stage MASH is often accompanied by significant fibrosis, where activated hepatic stellate cells (HSCs) are the main source of TGF-β [93]. This potent, sustained external TGF-β signal forms a strong synergy with the tumor's intrinsically activated PI3K/Akt pathway, manifested through both crosstalk, as TGF-β can activate PI3K/Akt via its non-SMAD pathways [94], and convergence on the same downstream EMT-TFs [95]. This dual-pronged assault results in a more robust and sustained induction of the EMT phenotype.
Furthermore, this synergistic network is amplified by a third crucial element: the intrinsic, pro-tumorigenic signaling of PD-L1 itself. Beyond its well-established role as an immune checkpoint, emerging evidence reveals that tumor cell-intrinsic PD-L1 can function as a signal transducer to directly promote cancer progression [96]. In various cancer models, high PD-L1 expression has been shown to directly activate the PI3K/Akt pathway [97]. Mechanistically, PD-L1-activated Akt then promotes EMT through the canonical pathway: it phosphorylates and inhibits glycogen synthase kinase 3β (GSK3β), thereby preventing the degradation of the master EMT transcription factor, Snail [98]. Crucially, this pathway does not operate in a single direction. A bidirectional regulatory relationship exists where key EMT-TFs, such as ZEB1 and TWIST1, can bind to the PD-L1 promoter region, upregulating its transcription [99,100]. This establishes a potent positive feedback loop (“PD-L1 → PI3K/Akt → EMT → PD-L1”), which locks the tumor into a highly aggressive, metastatic, and simultaneously immune-evasive state, further amplifying the pro-invasive signals from both the fibrotic microenvironment and the tumor's intrinsic PI3K/Akt dysregulation.
Other Synergistic Mechanisms and an Integrated Perspective
Beyond the synergistic enhancement of PD-L1 and EMT, other immune evasion mechanisms mediated by the PI3K/Akt/mTOR pathway may also be amplified in the unique pathological environment of IR/MASH. A compelling example is the direct recruitment of potent immunosuppressive cells. As previously detailed in Section 4, the inflammatory-driven CCRK-mTORC1 axis provides a compelling example of direct immunosuppressive cell recruitment, wherein mTORC1-stimulated G-CSF secretion leads to the robust influx of polymorphonuclear myeloid-derived suppressor cells (PMN-MDSCs) into the liver, actively constructing an immune-evasive niche [35]. Other potential synergistic interactions may include pathway-driven VEGF secretion, which, superimposed on the high TGF-β background and hypoxia [101] of the MASH environment, collectively constructs a more complex immunosuppressive cytokine milieu. The chronic metabolic stress of the IR/MASH environment may also select for tumor clones that are more dependent on PI3K/Akt survival signals, reinforcing their resistance to apoptosis [11]. As for the net effect of autophagy regulation on immune evasion, it is more complex and requires further investigation.
In summary, the unique microenvironment of IR/MASH is not merely a passive background but actively engages in profound synergistic interactions with the aberrantly activated PI3K/Akt/mTOR signaling pathway within tumor cells. This synergy, mainly manifested through signal pathway convergence and crosstalk, directly amplifies the intensity and durability of key immune evasion mechanisms, particularly PD-L1-mediated T cell inhibition and EMT-mediated cell phenotype remodeling. This 'double-hit' on the immune system, driven by the unique synergy within the MASH microenvironment, likely underpins the notoriously aggressive phenotype and therapeutic resistance of this HCC subtype [102], posing significant challenges for current immunotherapies (Figure 3).
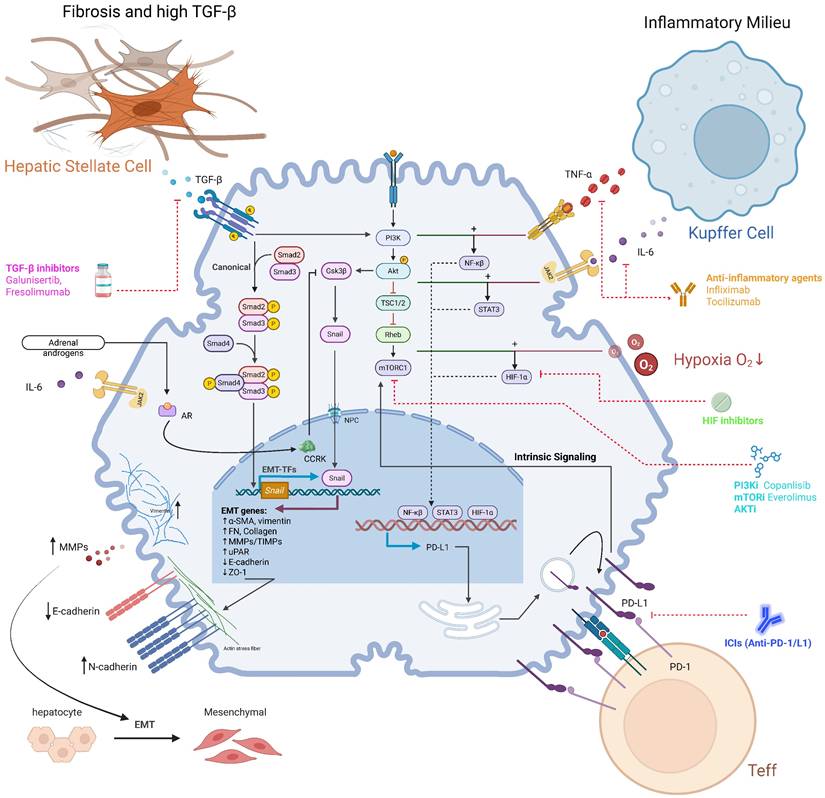
Synergy between intrinsic PI3K/Akt/mTOR signaling and the IR/MASH microenvironment amplifies immune evasion and reveals therapeutic targets. In HCC cells, intrinsic PI3K/Akt/mTOR signaling converges with extrinsic cues from the IR/MASH microenvironment to drive robust immune evasion. (A) Synergy in PD-L1 Upregulation: The MASH microenvironment, characterized by an inflammatory milieu (e.g., TNF-α, IL-6) and hypoxia, synergizes with the PI3K/Akt/mTOR pathway to enhance the activity of transcription factors (NF-κB, STAT3, HIF-1α), leading to supra-additive upregulation of PD-L1 and subsequent Teff inhibition. (B) Synergy in EMT Promotion: The fibrotic MASH microenvironment, rich in TGF-β, activates the canonical SMAD pathway. This synergizes with the PI3K/Akt pathway's stabilization of the EMT-TF Snail, driving a robust EMT program. This network is further amplified by PD-L1 intrinsic signaling and the inflammatory-oncogenic CCRK axis, which is co-activated by IL-6 and androgens via the androgen receptor (AR). (C) Therapeutic Opportunities: This complex network reveals multiple points for therapeutic intervention, including PI3K/mTOR inhibitors, ICIs, anti-inflammatory agents, HIF inhibitors, and TGF-β inhibitors, highlighting the potential for mechanism-based combination strategies.
Molecular Remodeling of HCC Therapeutic Response by the IR/MASH Microenvironment and Novel Clinical Translation Strategies
The unique molecular mechanisms detailed previously—the “imprinting” of the PI3K/Akt/mTOR pathway and the “synergistic amplification” of immune evasion—are not just of academic interest; they profoundly impact the response to current HCC therapies and provide a clear rationale for the clinical challenges observed in the MASH-HCC subtype.
The clinical significance of the PI3K/Akt/mTOR pathway in hepatocellular carcinoma (HCC) is underscored by its high frequency of dysregulation and its profound impact on patient prognosis. Aberrant activation of this pathway is one of the most common molecular events in HCC, with upregulation of mTOR signaling reported in approximately 40-50% of cases [54,103]. While precise prevalence data for the MASH-HCC subtype is still emerging, the pathway's role is strongly implicated, as the core metabolic drivers of MASH, such as insulin resistance and hyperinsulinemia, are potent upstream activators of PI3K/Akt signaling [15]. Furthermore, genomic analyses of MASH-related HCC cohorts have identified somatic mutations in key pathway components, including activating mutations in PIK3CA and loss-of-function mutations in the tumor suppressor PTEN [34]. Clinically, activation of the PI3K/Akt pathway is not a benign event; it serves as a notable risk factor for earlier tumor recurrence and is consistently associated with a more aggressive tumor phenotype and poor patient prognosis [17].
Given its central role, the PI3K/Akt/mTOR pathway has been an intensely investigated therapeutic target for HCC [104]. However, the clinical journey of its inhibitors has been challenging. Initial enthusiasm was tempered by the disappointing results of large, randomized phase III trials, most notably the EVOLVE-1 study, where the mTOR inhibitor everolimus failed to improve overall survival in patients with advanced HCC who had progressed on sorafenib [105]. In stark contrast, a significant survival benefit has been consistently observed in a specific clinical setting: for HCC patients undergoing liver transplantation, an immunosuppressive regimen based on mTOR inhibitors (sirolimus or everolimus) has been shown to improve both overall and recurrence-free survival [106]. The current therapeutic rationale has therefore shifted. The PI3K/Akt/mTOR pathway is now recognized as a critical escape route and a key mechanism of acquired resistance to multi-kinase inhibitors like sorafenib [107]. Consequently, the focus of ongoing clinical development has moved towards novel, more specific inhibitors and, crucially, towards combination strategies aiming to overcome these resistance mechanisms [104].
Challenges to the Efficacy and Safety of PI3K/Akt/mTOR Inhibitors
The clinical application of PI3Ki in HCC is inherently challenging, and these challenges are significantly exacerbated in the IR/MASH context where single-agent efficacy is generally limited and often accompanied by significant toxic side effects [104]. The reasons for this failure are a direct and predictable consequence of the “molecular imprinting” of the pathway.
Firstly, metabolic disorders mediate drug resistance. Persistent hyperinsulinemia in the IR state is a potent physiological activator of the PI3K/Akt pathway and can directly antagonize the efficacy of upstream PI3K or Akt inhibitors through an “insulin feedback” effect [108]. More critically, the pathway has bypass routes that render it insensitive to standard inhibitors. MASH-associated lipotoxicity, such as the saturated fatty acid palmitate (PA), has been shown to activate Akt through mechanisms independent of the classic PI3K-PIP3 axis (e.g., via ZDHHC17/24-mediated palmitoylation), providing a novel explanation for the poor efficacy of PI3Ki in a lipid-rich MASH microenvironment [109].
Secondly, there is an overlap of metabolic toxicity. Common side effects of PI3Ki (especially PI3Kα and pan-PI3K/mTOR inhibitors) include hyperglycemia and hyperlipidemia [110]. For example, hyperglycemia was a common adverse event in the VICTORIA study (NCT02730923) evaluating the mTOR inhibitor vistusertib [111]. In IR/MASH-HCC patients with pre-existing severe metabolic disorders, the risk and severity of these side effects are likely increased, complicating clinical management and potentially compromising treatment continuity.
Challenges to the Efficacy of Immune Checkpoint Inhibitors (ICIs)
Although ICI-based combination therapies have significantly improved the prognosis for some advanced HCC patients [112], a growing body of clinical evidence suggests that the IR/MASH-HCC subtype may have a distinct and often poorer response. Subgroup analyses of multiple clinical trials and real-world studies indicate that non-viral/MASH-HCC patients may have worse overall survival (OS), progression-free survival (PFS), and/or objective response rates (ORR) when treated with ICIs compared to viral HCC patients [113,114]. The mechanisms of “synergistically amplified immune evasion” discussed in this review provide a solid molecular basis for understanding this clinical challenge, which manifests on multiple levels:
- Broad Upregulation of PD-L1 and the “PD-L1 Paradox”: The MASH microenvironment drives widespread PD-L1 expression on tumor and immune cells through lipotoxicity [79], chronic inflammation [115], and hypoxia [116], creating a formidable immunosuppressive barrier. Even IgA+ cells accumulating in the MASH liver highly express PD-L1 [117]. However, this high expression does not always translate to a good response, as a “PD-L1 paradox” may exist where ICIs could be ineffective or even harmful in this context [118].
- EMT and Fibrosis-Mediated Immune Exclusion: The fibrotic, high-TGF-β MASH microenvironment potently drives EMT in HCC [119]. EMT is strongly associated with an “immune-excluded” TME phenotype characterized by reduced CD8+ T cell infiltration [120] and the dense fibrotic matrix also acts as a physical barrier, preventing T cell access to tumor cells [121], a major cause of primary resistance to ICIs.
- Hostile Metabolic Microenvironment: The MASH-HCC TME, with its high lactate [122] and abnormal lipid accumulation, directly inhibits effector T cell functions while supporting immunosuppressive Tregs and M2-TAMs [123]. This renders T cells ineffective even after the PD-1/PD-L1 brake is released.
- Deeply Entrenched Immunosuppressive Cell Network: The MASH TME favors the enrichment of multiple immunosuppressive cell populations (M2-TAMs, Tregs, MDSCs) [3,124,125]. The recruitment of these cells is actively driven by tumor-intrinsic signaling; for example, as previously noted, the CCRK-mTORC1 axis induces G-CSF secretion, leading to the accumulation of potent T cell-suppressive PMN-MDSCs [35].
Precision Combination Therapy Strategies for IR/MASH-HCC
Faced with the complex dysregulation of the PI3K/Akt/mTOR pathway and the unique challenges it poses to existing therapies in IR/MASH-driven HCC, developing mechanism-based precision combination strategies is urgently needed. Table 1 provides a structured overview of these strategies, linking them directly to the core pathological mechanisms discussed in this review.
Mechanism-Based Rationale for Combination Therapies in IR/MASH-HCC.
| Therapeutic Strategy | Targeted Pathological Mechanism | Core Mechanistic Rationale | Specific Molecular Targets / Processes | Example Combinations | Potential Challenges |
|---|---|---|---|---|---|
| PI3Ki + ICI | Dismantle “Synergistic Amplification” of Immune Evasion | Reprogram the immunosuppressive TME; reduce PD-L1 expression; target inhibitory immune cells. | PI3Kδ/γ in Tregs/TAMs; Tumor PD-L1 (synergistically upregulated); T-cell metabolic fitness. | Anti-PD-(L)1 Ab + PI3Kδ/γ inhibitors | Context-dependent effects on PD-L1; managing overlapping toxicities. |
| PI3Ki + Metabolic Modulator | Reverse “Molecular Imprinting” of the Pathway | Correct the systemic/local metabolic milieu that sustains pathway activation; prevent insulin feedback resistance. | Systemic IR (HOMA-IR); AMPK activation; mTOR signaling; SGLT2. | PI3Ki + GLP-1RAs, SGLT2i, or Metformin | Lack of preclinical data in relevant models; potential for drug-drug interactions. |
| ICI + TME Modulator | Overcome the Hostile Tumor Microenvironment | Break down physical and metabolic barriers to T-cell infiltration and function. | TGF-β-driven fibrosis/EMT; Hypoxia-driven lipogenesis; MDSC recruitment. | Anti-PD-(L)1 Ab + Anti-TGF-β Ab, HIF-2α inhibitors, or CCR2/5 inhibitors | Identifying dominant suppressive axis in individual patients. |
| Triple Therapy (PI3Ki + ICI + TME Modulator) | Mount a Multi-pronged Attack on Deep-seated Resistance | Simultaneously attack the core pillars of MASH-HCC: imprinted signaling, immune checkpoints, and key TME drivers. | PI3K pathway, PD-1/PD-L1 axis, AND a key TME driver (e.g., TGF-β, hypoxia). | PI3Kδ/γ inhibitor + Anti-PD-L1 Ab + Anti-TGF-β Ab | Significant risk of cumulative toxicity; complex biomarker development. |
Combination with Metabolic Modulators: “Un-imprinting the Pathway”. A promising direction is to combine PI3Ki with drugs that improve systemic IR and the core pathology of MASH, aiming to “un-imprint” the pathway and restore sensitivity. These include novel antidiabetic agents like GLP-1 Receptor Agonists (GLP-1RAs) and SGLT2 Inhibitors (SGLT2i), which show potential in improving MASLD-related liver outcomes [126-129] and may prevent the insulin feedback effect common in PI3Ki therapy [130,131]. Another key agent is Metformin, a classic AMPK activator whose potential to inhibit mTOR signaling suggests its value as a combination partner [132,133]. However, its effect when combined with ICIs requires cautious evaluation, as some studies have reported associations with poorer outcomes [134]. The critical challenge remains the lack of preclinical studies in well-defined IR/MASH-HCC models to validate these synergistic effects.
PI3Ki + ICIs: Dismantling Coordinated Immune Evasion. Positioning PI3Ki as a “sensitizer” or “synergistic partner” for ICIs is a core strategy to overcome the “synergistically amplified” immune evasion in IR/MASH-HCC. The rationale is multidimensional. PI3Ki can broadly remodel the immune microenvironment, particularly by targeting PI3Kδ and PI3Kγ isoforms, which are central to regulating immunosuppressive populations like MDSCs and TAMs [104]. Given that the IR/MASH-HCC TME is typically enriched with these cells, selective PI3Ki (especially PI3Kδ/γ inhibitors) hold immense promise for dismantling the immunosuppressive landscape and significantly enhancing ICI efficacy [135-138]. Despite promising preclinical data, a stark lack of published data directly evaluating these combinations in relevant IR/MASH-HCC models remains a critical evidence gap [139].
Triple-Drug Strategies: A Forward-Looking Approach. Given the complexity of IR/MASH-HCC and the potential for deep-seated resistance to dual therapies [104], exploring “triple therapy” by adding a third agent to the PI3Ki + ICI backbone is a logical future direction. The third agent should be selected to target key drivers of the microenvironment, such as anti-TGF-β drugs to overcome fibrosis or HIF inhibitors to counteract hypoxia [121,140]. While theoretically attractive for IR/MASH-HCC, these strategies are currently in the conceptual and very early exploratory stages and require rigorous validation in sophisticated preclinical models and the development of biomarkers to guide individualized combination strategies.
Summary and Outlook
Despite recent progress in HCC treatment, effectively addressing its molecular heterogeneity, particularly in subtypes associated with metabolic disorders, remains a major hurdle for precision oncology [13]. IR and its related conditions, MAFLD/MASH, are key drivers of the growing global HCC burden, urgently requiring a deeper understanding of their unique pathogenic mechanisms. This review has focused on the signaling hub PI3K/Akt/mTOR, systematically proposing an integrated view: in IR-driven HCC, this pathway is not merely an independent signaling module but a central coordinator, “imprinted” by the unique IR/MASH pathological microenvironment. It then precisely orchestrates tumor-intrinsic metabolic reprogramming, immune cell functional remodeling, and complex tumor-microenvironment interactions, ultimately driving tumor progression and immune evasion.
This integrated understanding not only reveals the unique biological complexity of IR-driven HCC but also provides new dimensions for future therapeutic strategies. However, the clinical efficacy of existing PI3Ki in HCC, especially in patients with an IR/MASH background, has often been disappointing [105]. This is likely because preclinical research and drug development have failed to fully consider the special role of the pathway as a “coordinator” within the complex, “imprinted” IR/MASH microenvironment. These challenges stem from the pathway's significant resilience and compensatory mechanisms [141], the significant metabolic side effects of its inhibition [142], and the specific interferences presented by the IR/MASH environment itself.
Therefore, future research and therapeutic development must move beyond single-target thinking and adopt a new paradigm. We must recognize that MASH-HCC is not just another etiology of liver cancer; it is a distinct biological entity defined by the central, coordinating role of the “imprinted” PI3K/Akt/mTOR pathway. This paradigm shift requires a fundamental re-evaluation of our entire research and clinical development pipeline:
- Rebuilding Preclinical Models: To effectively evaluate new strategies, the research framework itself needs innovation. Standard cell lines or simple xenografts are inadequate. The field urgently needs preclinical models that faithfully recapitulate the “perfect storm” of human IR/MASH-driven HCC: systemic metabolic dysregulation (IR), chronic inflammation, fibrosis, and a functional immune system [118].
- Defining Composite Biomarkers: Moving beyond single-gene mutation analysis is critical. A true predictive biomarker for MASH-HCC must be a “composite” panel that captures: 1) host IR/MASH status; 2) tumor PI3K/Akt/mTOR pathway “imprinting” features; and 3) TME “synergistic evasion” features [143].
- Designing Smarter Clinical Trials: Future clinical trials must prospectively use IR/MASH etiology as a key stratification factor and incorporate co-primary endpoints that measure not only tumor response but also dynamic changes in the metabolic and immune microenvironment.
- Developing Rational Combination Therapies: Based on the pathway's coordinating role and synergy with the microenvironment, exploring rational combinations is crucial, including combinations of PI3Ki with ICIs, metabolic interventions, or other microenvironment modulators.
In conclusion, examining the PI3K/Akt/mTOR pathway within an integrated immuno-metabolic-signal framework for IR/MASH-driven HCC not only deepens our understanding of the pathophysiology of this important HCC subtype but also provides a critical theoretical basis and a fresh perspective for overcoming current therapeutic dilemmas. Although the challenges are immense, a deeper investigation into this complex regulatory network and the development of more intelligent intervention strategies hold the promise of finally translating a deep mechanistic understanding into tangible clinical benefits for this large and growing patient population.
Abbreviations
ACC: Acetyl-CoA Carboxylase; Akt: Protein Kinase B; AMPK: AMP-activated Protein Kinase; AR: Androgen Receptor; ARG1: Arginase 1; CCRK: Cell Cycle-Related Kinase; DNL: De Novo Lipogenesis; DAGs: Diacylglycerols; EGF: Epidermal Growth Factor; EMT: Epithelial-Mesenchymal Transition; EMT-TFs: EMT-inducing Transcription Factors; FAO: Fatty Acid Oxidation; FASN: Fatty Acid Synthase; G-CSF: Granulocyte-Colony Stimulating Factor; GLP-1RAs: GLP-1 Receptor Agonists; GLUT1: Glucose Transporter 1; HCC: Hepatocellular Carcinoma; HGF: Hepatocyte Growth Factor; HIF-1α: Hypoxia-Inducible Factor-1α; HK2: Hexokinase 2; HOMA-IR: Homeostatic Model Assessment for Insulin Resistance; HSCs: Hepatic Stellate Cells; ICIs: Immune Checkpoint Inhibitors; IDO: Indoleamine 2,3-Dioxygenase; IR: Insulin Resistance; IRS: Insulin Receptor Substrate; LDHA: Lactate Dehydrogenase A; MAFLD: Metabolic Dysfunction-Associated Fatty Liver Disease; MASH: Metabolic Dysfunction-Associated Steatohepatitis; MCTs: Monocarboxylate Transporters; MDSCs: Myeloid-Derived Suppressor Cells; mTOR: mammalian Target of Rapamycin; mTORC1: mTOR Complex 1; mTORC2: mTOR Complex 2; NF-κB: Nuclear Factor-κB; ORR: Objective Response Rate; OS: Overall Survival; PD-L1: Programmed Death-Ligand 1; PFS: Progression-Free Survival; PI3K: Phosphoinositide 3-kinase; PI3Ki: PI3K/Akt/mTOR inhibitors; PMN-MDSCs: Polymorphonuclear Myeloid-Derived Suppressor Cells; PPARs: Peroxisome Proliferator-Activated Receptors; PTEN: Phosphatase and Tensin Homolog; ROS: Reactive Oxygen Species; RTKs: Receptor Tyrosine Kinases; SASP: Senescence-Associated Secretory Phenotype; SGLT2i: Sodium-Glucose Cotransporter 2 Inhibitors; STAT3: Signal Transducer and Activator of Transcription 3; TAMs: Tumor-Associated Macrophages; Teff: Effector T cells; TGF-β: Transforming Growth Factor-β; TME: Tumor Microenvironment; Tregs: Regulatory T cells; VEGF: Vascular Endothelial Growth Factor.
Acknowledgements
This work was supported by the Chinese NSFC under Grant 81872489.
Author contributions
Jian Zhao: Conceptualization, Investigation, Visualization, Writing - Original Draft. Yuehua Zhang: Conceptualization, Investigation, Visualization, Writing - Original Draft. Zhigong Wei: Investigation, Writing - Review & Editing. Kai Li: Writing - Review & Editing. Yongsheng Wang: Conceptualization, Supervision, Writing - Review & Editing, Funding acquisition. Dan Li: Conceptualization, Supervision, Writing - Review & Editing. Lei Sun: Conceptualization, Supervision, Writing - Review & Editing. All authors have given approval to the final version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA A Cancer J Clinicians. 2021May;71(3):209-49
2. Llovet JM, Pinyol R, Kelley RK, El-Khoueiry A, Reeves HL, Wang XW. et al. Molecular pathogenesis and systemic therapies for hepatocellular carcinoma. Nat Cancer. 2022Apr;3(4):386-401
3. Pfister D, Núñez NG, Pinyol R, Govaere O, Pinter M, Szydlowska M. et al. NASH limits anti-tumour surveillance in immunotherapy-treated HCC. Nature. 2021Apr;592(7854):450-6
4. Riazi K, Azhari H, Charette JH, Underwood FE, King JA, Afshar EE. et al. The prevalence and incidence of NAFLD worldwide: a systematic review and meta-analysis. The Lancet Gastroenterology & Hepatology. 2022 Sept;7(9):851-61
5. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease—Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016 July;64(1):73-84
6. Estes C, Razavi H, Loomba R, Younossi Z, Sanyal AJ. Modeling the epidemic of nonalcoholic fatty liver disease demonstrates an exponential increase in burden of disease. Hepatology. 2018Jan;67(1):123-33
7. Stefan N, Häring H-U, Cusi K. Non-alcoholic fatty liver disease: causes, diagnosis, cardiometabolic consequences, and treatment strategies. The Lancet Diabetes & Endocrinology. 2019Apr;7(4):313-24
8. Anstee QM, Mantovani A, Tilg H, Targher G. Risk of cardiomyopathy and cardiac arrhythmias in patients with nonalcoholic fatty liver disease. Nat Rev Gastroenterol Hepatol. 2018 July;15(7):425-39
9. Tilg H, Adolph TE, Moschen AR. Multiple Parallel Hits Hypothesis in Nonalcoholic Fatty Liver Disease: Revisited After a Decade. Hepatology. 2021Feb;73(2):833-42
10. Foerster F, Gairing SJ, Müller L, Galle PR. NAFLD-driven HCC: Safety and efficacy of current and emerging treatment options. Journal of Hepatology. 2022Feb;76(2):446-57
11. Apostolo D, Ferreira LL, Vincenzi F, Vercellino N, Minisini R, Latini F. et al. From MASH to HCC: the role of Gas6/TAM receptors. Front Immunol. 2024;15:1332818
12. Choi J, Corder NLB, Koduru B, Wang Y. Oxidative stress and hepatic Nox proteins in chronic hepatitis C and hepatocellular carcinoma. Free Radical Biology and Medicine. 2014 July;72:267-84
13. Hepatocellular carcinoma. Nat Rev Dis Primers. 2021 Jan;7(1):7.
14. Zheng J, Wang S, Xia L, Sun Z, Chan KM, Bernards R. et al. Hepatocellular carcinoma: signaling pathways and therapeutic advances. Sig Transduct Target Ther. 2025Feb;10(1):35
15. Chiefari E, Mirabelli M, La Vignera S, Tanyolaç S, Foti DP, Aversa A. et al. Insulin Resistance and Cancer: In Search for a Causal Link. IJMS. 2021Oct;22(20):11137
16. Alzahrani AS. PI3K/Akt/mTOR inhibitors in cancer: At the bench and bedside. Seminars in Cancer Biology. 2019Dec;59:125-32
17. Tian L-Y, Smit DJ, Jücker M. The Role of PI3K/AKT/mTOR Signaling in Hepatocellular Carcinoma Metabolism. IJMS. 2023Jan;24(3):2652
18. Scheau C, Badarau IA, Costache R, Caruntu C, Mihai GL, Didilescu AC. et al. The Role of Matrix Metalloproteinases in the Epithelial-Mesenchymal Transition of Hepatocellular Carcinoma. Analytical Cellular Pathology. 2019Nov;2019:1-10
19. Giannelli G, Koudelkova P, Dituri F, Mikulits W. Role of epithelial to mesenchymal transition in hepatocellular carcinoma. Journal of Hepatology. 2016Oct;65(4):798-808
20. Shen M, Xu Z, Xu W, Jiang K, Zhang F, Ding Q. et al. Inhibition of ATM reverses EMT and decreases metastatic potential of cisplatin-resistant lung cancer cells through JAK/STAT3/PD-L1 pathway. J Exp Clin Cancer Res. 2019Dec;38(1):149
21. Bang J, Jun M, Lee S, Moon H, Ro SW. Targeting EGFR/PI3K/AKT/mTOR Signaling in Hepatocellular Carcinoma. Pharmaceutics. 2023Aug;15(8):2130
22. Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017Apr;169(2):361-71
23. Liu GY, Sabatini DM. mTOR at the nexus of nutrition, growth, ageing and disease. Nat Rev Mol Cell Biol. 2020Apr;21(4):183-203
24. Bo T, Gao L, Yao Z, Shao S, Wang X, Proud CG. et al. Hepatic selective insulin resistance at the intersection of insulin signaling and metabolic dysfunction-associated steatotic liver disease. Cell Metabolism. 2024May;36(5):947-68
25. Li YZ, Di Cristofano A, Woo M. Metabolic Role of PTEN in Insulin Signaling and Resistance. Cold Spring Harb Perspect Med. 2020Aug;10(8):a036137
26. Chalhoub N, Baker SJ. PTEN and the PI3-Kinase Pathway in Cancer. Annu Rev Pathol Mech Dis. 2009Feb;4(1):127-50
27. Trinh VH, Nguyen Huu T, Sah DK, Choi JM, Yoon HJ, Park SC. et al. Redox Regulation of PTEN by Reactive Oxygen Species: Its Role in Physiological Processes. Antioxidants. 2024Feb;13(2):199
28. Copps KD, White MF. Regulation of insulin sensitivity by serine/threonine phosphorylation of insulin receptor substrate proteins IRS1 and IRS2. Diabetologia. 2012Oct;55(10):2565-82
29. Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-Mediated Inhibition of Insulin Receptor Tyrosine Kinase Activity in TNF-α- and Obesity-Induced Insulin Resistance. Science. 1996Feb;271(5249):665-70
30. Li T, Wang G, Asrih M, Jornayvaz FR. Inflammation as a potential link between nonalcoholic fatty liver disease and insulin resistance. J Endocrinol. 2013 Sept;218(3):R25-36
31. Bessone F, Razori MV, Roma MG. Molecular pathways of nonalcoholic fatty liver disease development and progression. Cell Mol Life Sci. 2019Jan;76(1):99-128
32. Shearn CT, Smathers RL, Backos DS, Reigan P, Orlicky DJ, Petersen DR. Increased carbonylation of the lipid phosphatase PTEN contributes to Akt2 activation in a murine model of early alcohol-induced steatosis. Free Radical Biology and Medicine. 2013Dec;65:680-92
33. Carracedo A, Pandolfi PP. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene. 2008 Sept;27(41):5527-41
34. Rebouissou S, Nault J-C. Advances in molecular classification and precision oncology in hepatocellular carcinoma. Journal of Hepatology. 2020Feb;72(2):215-29
35. Sun H, Yang W, Tian Y, Zeng X, Zhou J, Mok MTS. et al. An inflammatory-CCRK circuitry drives mTORC1-dependent metabolic and immunosuppressive reprogramming in obesity-associated hepatocellular carcinoma. Nat Commun. 2018Dec;9(1):5214
36. Che L, Stevenson CK, Plas DR, Wang J, Du C. BRUCE liver-deficiency potentiates MASLD/MASH in PTEN liver-deficient background by impairment of mitochondrial metabolism in hepatocytes and activation of STAT3 signaling in hepatic stellate cells. bioRxiv. 2024 Sept. 2024 09.13.611500
37. Xu X, Ye L, Araki K, Ahmed R. mTOR, linking metabolism and immunity. Seminars in Immunology. 2012Dec;24(6):429-35
38. Villanueva A, Chiang DY, Newell P, Peix J, Thung S, Alsinet C. et al. Pivotal Role of mTOR Signaling in Hepatocellular Carcinoma. Gastroenterology. 2008Dec;135(6):1972-1983.e11
39. Manning BD, Toker A. AKT/PKB Signaling: Navigating the Network. Cell. 2017Apr;169(3):381-405
40. Liu Q, Turner KM, Alfred Yung WK, Chen K, Zhang W. Role of AKT signaling in DNA repair and clinical response to cancer therapy. Neuro-Oncology. 2014Oct;16(10):1313-23
41. Nishida N, Kudo M. Oxidative Stress and Epigenetic Instability in Human Hepatocarcinogenesis. Dig Dis. 2013;31(5-6):447-53
42. Khaznadar F, Khaznadar O, Petrovic A, Hefer M, Gjoni F, Gjoni S. et al. MAFLD Pandemic: Updates in Pharmacotherapeutic Approach Development. CIMB. 2024 June;46(7):6300-14
43. Ilan Y. Compounds of the sphingomyelin-ceramide-glycosphingolipid pathways as secondary messenger molecules: new targets for novel therapies for fatty liver disease and insulin resistance. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2016 June;310(11):G1102-17
44. Bikman BT, Summers SA. Ceramides as modulators of cellular and whole-body metabolism. J Clin Invest. 2011Nov;121(11):4222-30
45. Arrese M, Cabrera D, Kalergis AM, Feldstein AE. Innate Immunity and Inflammation in NAFLD/NASH. Dig Dis Sci. 2016May;61(5):1294-303
46. Sugiura A, Rathmell JC. Metabolic Barriers to T Cell Function in Tumors. J Immunol. 2018Jan;200(2):400-7
47. Zeng W, Li F, Jin S, Ho P-C, Liu P-S, Xie X. Functional polarization of tumor-associated macrophages dictated by metabolic reprogramming. J Exp Clin Cancer Res. 2023 Sept;42(1):245
48. Liang S, Kisseleva T, Brenner DA. The Role of NADPH Oxidases (NOXs) in Liver Fibrosis and the Activation of Myofibroblasts. Front Physiol. 2016;7:17
49. Nguyen Huu T, Park J, Zhang Y, Duong Thanh H, Park I, Choi JM. et al. The Role of Oxidative Inactivation of Phosphatase PTEN and TCPTP in Fatty Liver Disease. JCM. 2013Dec;12(1):120
50. Tsuchida T, Lee YA, Fujiwara N, Ybanez M, Allen B, Martins S. et al. A simple diet- and chemical-induced murine NASH model with rapid progression of steatohepatitis, fibrosis and liver cancer. Journal of Hepatology. 2018Aug;69(2):385-95
51. Chen Q, Zhang Z, Huang Q, Chen H, Hong W, Lin T. et al. HK1 from hepatic stellate cell-derived extracellular vesicles promotes progression of hepatocellular carcinoma. Nat Metab. 2022Oct;4(10):1306-21
52. Pinter M, Pinato DJ, Ramadori P, Heikenwalder M. NASH and Hepatocellular Carcinoma: Immunology and Immunotherapy. Clin Cancer Res. 2023Feb;29(3):513-20
53. Du D, Liu C, Qin M, Zhang X, Xi T, Yuan S. et al. Metabolic dysregulation and emerging therapeutical targets for hepatocellular carcinoma. Acta Pharmaceutica Sinica B. 2022Feb;12(2):558-80
54. Matter MS, Decaens T, Andersen JB, Thorgeirsson SS. Targeting the mTOR pathway in hepatocellular carcinoma: Current state and future trends. Journal of Hepatology. 2014Apr;60(4):855-65
55. Iturbe-Rey S, Maccali C, Arrese M, Aspichueta P, Oliveira CP, Castro RE. et al. Lipotoxicity-driven metabolic dysfunction-associated steatotic liver disease (MASLD). Atherosclerosis. 2025Jan;400:119053
56. Puthenveetil A, Dubey S. Metabolic reprograming of tumor-associated macrophages. Ann Transl Med. 2020Aug;8(16):1030-1030
57. Lanahan SM, Wymann MP, Lucas CL. The role of PI3Kγ in the immune system: new insights and translational implications. Nat Rev Immunol. 2022Nov;22(11):687-700
58. Huang B, Song B, Xu C. Cholesterol metabolism in cancer: mechanisms and therapeutic opportunities. Nat Metab. 2020Feb;2(2):132-41
59. Liu C, Chapman NM, Karmaus PWF, Zeng H, Chi H. mTOR and metabolic regulation of conventional and regulatory T cells. J Leukoc Biol. 2015May;97(5):837-47
60. Powell JD, Pollizzi KN, Heikamp EB, Horton MR. Regulation of Immune Responses by mTOR. Annu Rev Immunol. 2012Apr;30(1):39-68
61. Li M, Yang Y, Xiong L, Jiang P, Wang J, Li C. Metabolism, metabolites, and macrophages in cancer. J Hematol Oncol. 2023;16:80
62. Hao Z-N, Tan X-P, Zhang Q, Li J, Xia R, Ma Z. Lactate and Lactylation: Dual Regulators of T-Cell-Mediated Tumor Immunity and Immunotherapy. Biomolecules. 2024Dec;14(12):1646
63. Zhu W, Fan C, Hou Y, Zhang Y. Lactylation in tumor microenvironment and immunotherapy resistance: New mechanisms and challenges. Cancer Letters. 2025 Sept;627:217835
64. Zappasodi R, Serganova I, Cohen IJ, Maeda M, Shindo M, Senbabaoglu Y. et al. CTLA-4 blockade drives loss of Treg stability in glycolysis-low tumours. Nature. 2021Mar;591(7851):652-8
65. Luo Q, Zheng N, Jiang L, Wang T, Zhang P, Liu Y. et al. Lipid accumulation in macrophages confers protumorigenic polarization and immunity in gastric cancer. Cancer Science. 2020Nov;111(11):4000-11
66. Raynor JL, Chi H. Nutrients: Signal 4 in T cell immunity. J Exp Med. 2024Mar;221(3):e20221839
67. Hu W, Li F, Liang Y, Liu S, Wang S, Shen C. et al. Glut3 overexpression improves environmental glucose uptake and antitumor efficacy of CAR-T cells in solid tumors. J Immunother Cancer. 2025Jan;13(1):e010540
68. Luby A, Alves-Guerra M-C. Targeting Metabolism to Control Immune Responses in Cancer and Improve Checkpoint Blockade Immunotherapy. Cancers. 2021Nov;13(23):5912
69. Li W, Pan X, Chen L, Cui H, Mo S, Pan Y. et al. Cell metabolism-based optimization strategy of CAR-T cell function in cancer therapy. Front Immunol. 2023;14:1186383
70. Gumber D, Wang LD. Improving CAR-T immunotherapy: Overcoming the challenges of T cell exhaustion. eBioMedicine. 2022Mar;77:103941
71. O'Sullivan D, Pearce EL. Targeting T cell metabolism for therapy. Trends in Immunology. 2015Feb;36(2):71-80
72. Ding W, You H, Dang H, LeBlanc F, Galicia V, Lu SC. et al. Epithelial-to-Mesenchymal Transition of Murine Liver Tumor Cells Promotes Invasion†,‡. Hepatology. 2010 Sept;52(3):945-53
73. Steinway SN, Dang H, You H, Rountree CB, Ding W. The EGFR/ErbB3 Pathway Acts as a Compensatory Survival Mechanism upon c-Met Inhibition in Human c-Met+ Hepatocellular Carcinoma. PLoS ONE. 2015;10(5):e0128159
74. Duan Y, Pan X, Luo J, Xiao X, Li J, Bestman PL. et al. Association of Inflammatory Cytokines With Non-Alcoholic Fatty Liver Disease. Front Immunol. 2022;13:880298
75. Markman B, Dienstmann R, Tabernero J. Targeting the PI3K/Akt/mTOR Pathway - Beyond Rapalogs. Oncotarget. 2010Nov;1(7):530-43
76. Park S, Zhao D, Hatanpaa KJ, Mickey BE, Saha D, Boothman DA. et al. RIP1 Activates PI3K-Akt via a Dual Mechanism Involving NF-κB-Mediated Inhibition of the mTOR-S6K-IRS1 Negative Feedback Loop and Down-regulation of PTEN. Cancer Res. 2009May;69(10):4107-11
77. Xu J, Lin H, Wu G, Zhu M, Li M. IL-6/STAT3 Is a Promising Therapeutic Target for Hepatocellular Carcinoma. Front Oncol. 2021;11:760971
78. Monti E, Vianello C, Leoni I, Galvani G, Lippolis A, D'Amico F. et al. Gut Microbiome Modulation in Hepatocellular Carcinoma: Preventive Role in NAFLD/NASH Progression and Potential Applications in Immunotherapy-Based Strategies. Cells. 2025Jan;14(2):84
79. Pipitone RM, Lupo G, Zito R, Javed A, Petta S, Pennisi G. et al. The PD-1/PD-L1 Axis in the Biology of MASLD. IJMS. 2024Mar;25(7):3671
80. Llovet JM, Willoughby CE, Singal AG, Greten TF, Heikenwälder M, El-Serag HB. et al. Nonalcoholic steatohepatitis-related hepatocellular carcinoma: pathogenesis and treatment. Nat Rev Gastroenterol Hepatol. 2023Aug;20(8):487-503
81. Yi M, Zheng X, Niu M, Zhu S, Ge H, Wu K. Combination strategies with PD-1/PD-L1 blockade: current advances and future directions. Mol Cancer. 2022Jan;21(1):28
82. Yu J, Ling S, Hong J, Zhang L, Zhou W, Yin L. et al. TP53/mTORC1-mediated bidirectional regulation of PD-L1 modulates immune evasion in hepatocellular carcinoma. J Immunother Cancer. 2023Nov;11(11):e007479
83. Chen T-H, Lin S-H, Lee M-Y, Wang H-C, Tsai K-F, Chou C-K. Mitochondrial alterations and signatures in hepatocellular carcinoma. Cancer Metastasis Rev. 2025Mar;44(1):34
84. Wang X, de Carvalho Ribeiro M, Iracheta-Vellve A, Lowe P, Ambade A, Satishchandran A. et al. Macrophage-Specific Hypoxia-Inducible Factor-1α Contributes to Impaired Autophagic Flux in Nonalcoholic Steatohepatitis. Hepatology. 2019Feb;69(2):545-63
85. Foglia B, Sutti S, Cannito S, Rosso C, Maggiora M, Autelli R. et al. Hepatocyte-Specific Deletion of HIF2α Prevents NASH-Related Liver Carcinogenesis by Decreasing Cancer Cell Proliferation. Cellular and Molecular Gastroenterology and Hepatology. 2022;13(2):459-82
86. Kouroumalis E, Tsomidis I, Voumvouraki A. Pathogenesis of Hepatocellular Carcinoma: The Interplay of Apoptosis and Autophagy. Biomedicines. 2023Apr;11(4):1166
87. Jin K, Liu Y, Shi Y, Zhang H, Sun Y, Zhangyuan G. et al. PTPROt aggravates inflammation by enhancing NF-κB activation in liver macrophages during nonalcoholic steatohepatitis. Theranostics. 2020;10(12):5290-304
88. Willy JA, Young SK, Stevens JL, Masuoka HC, Wek RC. CHOP links endoplasmic reticulum stress to NF-κB activation in the pathogenesis of nonalcoholic steatohepatitis. MBoC. 2015 June;26(12):2190-204
89. Shi Y. Regulatory mechanisms of PD-L1 expression in cancer cells. Cancer Immunol Immunother. 2018Oct;67(10):1481-9
90. Han Y, Liu D, Li L. PD-1/PD-L1 pathway: current researches in cancer. Am J Cancer Res. 2020;10(3):727-42
91. Lamouille S, Xu J, Derynck R. Molecular mechanisms of epithelial-mesenchymal transition. Nat Rev Mol Cell Biol. 2014Mar;15(3):178-96
92. Hao L, Li S, Deng J, Li N, Yu F, Jiang Z. et al. The current status and future of PD-L1 in liver cancer. Front Immunol. 2023;14:1323581
93. Xu F, Liu C, Zhou D, Zhang L. TGF-β/SMAD Pathway and Its Regulation in Hepatic Fibrosis. J Histochem Cytochem. 2016Mar;64(3):157-67
94. Wendt MK, Allington TM, Schiemann WP. Mechanisms of the Epithelial-Mesenchymal Transition by TGF-β. Future Oncol. 2009Oct;5(8):1145-68
95. Gonzalez DM, Medici D. Signaling mechanisms of the epithelial-mesenchymal transition. Sci Signal. 2014 Sept;7(344):re8
96. Yadollahi P, Jeon Y-K, Ng WL, Choi I. Current understanding of cancer-intrinsic PD-L1: regulation of expression and its protumoral activity. BMB Rep. 2021Jan;54(1):12-20
97. Almozyan S, Colak D, Mansour F, Alaiya A, Al-Harazi O, Qattan A. et al. PD-L1 promotes OCT4 and Nanog expression in breast cancer stem cells by sustaining PI3K/AKT pathway activation. Intl Journal of Cancer. 2017Oct;141(7):1402-12
98. Wang H, Fang R, Wang X-F, Zhang F, Chen D-Y, Zhou B. et al. Stabilization of Snail through AKT/GSK-3β signaling pathway is required for TNF-α-induced epithelial-mesenchymal transition in prostate cancer PC3 cells. European Journal of Pharmacology. 2013Aug;714(1-3):48-55
99. Zhang Y-C, Zhang Y-T, Wang Y, Zhao Y, He L-J. What role does PDL1 play in EMT changes in tumors and fibrosis? Front Immunol. 2023;14:1226038
100. Jiang Y, Zhan H. Communication between EMT and PD-L1 signaling: New insights into tumor immune evasion. Cancer Letters. 2020Jan;468:72-81
101. Qiang M, Chen Z, Liu H, Dong J, Gong K, Zhang X. et al. Targeting the PI3K/AKT/mTOR pathway in lung cancer: mechanisms and therapeutic targeting. Front Pharmacol. 2025;16:1516583
102. Baj J, Kołodziej M, Kobak J, Januszewski J, Syty K, Portincasa P. et al. Significance of Immune and Non-Immune Cell Stroma as a Microenvironment of Hepatocellular Carcinoma—From Inflammation to Hepatocellular Carcinoma Progression. IJMS. 2024 Sept;25(19):10233
103. Ally A, Balasundaram M, Carlsen R. et al. Comprehensive and integrative genomic characterization of hepatocellular carcinoma. Cell. 2017;169:1327-41
104. Sun EJ, Wankell M, Palamuthusingam P, McFarlane C, Hebbard L. Targeting the PI3K/Akt/mTOR Pathway in Hepatocellular Carcinoma. Biomedicines. 2021Nov;9(11):1639
105. Zhu AX, Kudo M, Assenat E, Cattan S, Kang Y-K, Lim HY. et al. Effect of Everolimus on Survival in Advanced Hepatocellular Carcinoma After Failure of Sorafenib. JAMA. 2014 July;312(1):57
106. Toso C, Merani S, Bigam DL, Shapiro AMJ, Kneteman NM. Sirolimus-based immunosuppression is associated with increased survival after liver transplantation for hepatocellular carcinoma†. Hepatology. 2010Apr;51(4):1237-43
107. Narci K, Kahraman DC, Koyas A, Ersahin T, Tuncbag N, Atalay RC. Context dependent isoform specific PI3K inhibition confers drug resistance in hepatocellular carcinoma cells. BMC Cancer. 2022Dec;22(1):320
108. Hopkins BD, Pauli C, Du X, Wang DG, Li X, Wu D. et al. Suppression of insulin feedback enhances the efficacy of PI3K inhibitors. Nature. 2018Aug;560(7719):499-503
109. Chen J, Chen J, Huang J, Li Z, Gong Y, Zou B. et al. HIF-2α upregulation mediated by hypoxia promotes NAFLD-HCC progression by activating lipid synthesis via the PI3K-AKT-mTOR pathway. Aging. 2019Dec;11(23):10839-60
110. Kurma K, Zeybek Kuyucu A, Roth GS, Sturm N, Mercey-Ressejac M, Abbadessa G. et al. Effect of Novel AKT Inhibitor Vevorisertib as Single Agent and in Combination with Sorafenib on Hepatocellular Carcinoma in a Cirrhotic Rat Model. IJMS. 2022Dec;23(24):16206
111. Heudel P, Frenel J-S, Dalban C, Bazan F, Joly F, Arnaud A. et al. Safety and Efficacy of the mTOR Inhibitor, Vistusertib, Combined With Anastrozole in Patients With Hormone Receptor-Positive Recurrent or Metastatic Endometrial Cancer. JAMA Oncol. 2022 July;8(7):1001
112. Giraud J, Chalopin D, Blanc J-F, Saleh M. Hepatocellular Carcinoma Immune Landscape and the Potential of Immunotherapies. Front Immunol. 2021;12:655697
113. Wu YL, Cappuyns S, Loh A, Sun S, Lewis S, Sung MW. et al. Impact of underlying liver disease on unresectable hepatocellular carcinoma treated with immune checkpoint inhibitors. BJC Rep. 2024Jan;2(1):8
114. Costante F, Airola C, Santopaolo F, Gasbarrini A, Pompili M, Ponziani FR. Immunotherapy for nonalcoholic fatty liver disease-related hepatocellular carcinoma: Lights and shadows. WJGO. 2022 Sept;14(9):1622-36
115. Brown ZJ, Ruff SM, Pawlik TM. The effect of liver disease on hepatic microenvironment and implications for immune therapy. Front Pharmacol. 2023;14:1225821
116. Fu Y, Mackowiak B, Feng D, Lu H, Guan Y, Lehner T. et al. MicroRNA-223 attenuates hepatocarcinogenesis by blocking hypoxia-driven angiogenesis and immunosuppression. Gut. 2023Oct;72(10):1942-58
117. Shalapour S, Lin X-J, Bastian IN, Brain J, Burt AD, Aksenov AA. et al. Inflammation-induced IgA+ cells dismantle anti-liver cancer immunity. Nature. 2017Nov;551(7680):340-5
118. Febbraio MA, Reibe S, Shalapour S, Ooi GJ, Watt MJ, Karin M. Preclinical Models for Studying NASH-Driven HCC: How Useful Are They? Cell Metabolism. 2019Jan;29(1):18-26
119. Xin X, Cheng X, Zeng F, Xu Q, Hou L. The Role of TGF-β/SMAD Signaling in Hepatocellular Carcinoma: from Mechanism to Therapy and Prognosis. Int J Biol Sci. 2024;20(4):1436-51
120. Sukowati CHC, El-Khobar KE, Tiribelli C. Immunotherapy against programmed death-1/programmed death ligand 1 in hepatocellular carcinoma: Importance of molecular variations, cellular heterogeneity, and cancer stem cells. WJSC. 2021 July;13(7):795-824
121. Roy AM, Iyer R, Chakraborty S. The extracellular matrix in hepatocellular carcinoma: Mechanisms and therapeutic vulnerability. Cell Reports Medicine. 2023 Sept;4(9):101170
122. Li X, Yang Y, Zhang B, Lin X, Fu X, An Y. et al. Lactate metabolism in human health and disease. Sig Transduct Target Ther. 2022 Sept;7(1):305
123. Yang J, He J, Feng Y, Xiang M. Obesity contributes to hepatocellular carcinoma development via immunosuppressive microenvironment remodeling. Front Immunol. 2023;14:1166440
124. Li Z, Duan D, Li L, Peng D, Ming Y, Ni R. et al. Tumor-associated macrophages in anti-PD-1/PD-L1 immunotherapy for hepatocellular carcinoma: recent research progress. Front Pharmacol. 2024;15:1382256
125. Li C, Xiong L, Yang Y, Jiang P, Wang J, Li M. et al. Sorafenib enhanced the function of myeloid-derived suppressor cells in hepatocellular carcinoma by facilitating PPARα-mediated fatty acid oxidation. Mol Cancer. 2025Jan;24(1):34
126. Kanwal F, Kramer JR, Li L, Yang Y-X, Cao Y, Yu X. et al. GLP-1 Receptor Agonists and Risk for Cirrhosis and Related Complications in Patients With Metabolic Dysfunction-Associated Steatotic Liver Disease. JAMA Intern Med. 2024Nov;184(11):1314
127. Wang L, Berger NA, Kaelber DC, Xu R. Association of GLP-1 Receptor Agonists and Hepatocellular Carcinoma Incidence and Hepatic Decompensation in Patients With Type 2 Diabetes. Gastroenterology. 2024 Sept;167(4):689-703
128. Engström A, Wintzell V, Melbye M, Svanström H, Eliasson B, Gudbjörnsdottir S. et al. Association of glucagon-like peptide-1 receptor agonists with serious liver events among patients with type 2 diabetes: A Scandinavian cohort study. Hepatology. 2024 June;79(6):1401-11
129. Mao X, Zhang X, Lai R, Cheung K-S, Yuen M-F, Cheung R. et al. GLP-1 RA and Reduced Liver and Non-Liver Complications in Adults with T2D and MASLD: A Target Trial Emulation Study. Clin Mol Hepatol. 2025 Apr DOI: 10.3350/cmh. 2024 1096
130. Jojima T, Wakamatsu S, Kase M, Iijima T, Maejima Y, Shimomura K. et al. The SGLT2 Inhibitor Canagliflozin Prevents Carcinogenesis in a Mouse Model of Diabetes and Non-Alcoholic Steatohepatitis-Related Hepatocarcinogenesis: Association with SGLT2 Expression in Hepatocellular Carcinoma. IJMS. 2019Oct;20(20):5237
131. Rao H, An X, Qu X, Yu J, Xie J, Ke J. et al. SGLT2i delays c-Myc-induced HCC progression via targeting mTOR. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease. 2025 June;1871(5):167805
132. Tsai P-C, Huang C-F, Yeh M-L, Hsieh M-H, Kuo H-T, Hung C-H. et al. Metformin and statins reduce hepatocellular carcinoma risk in chronic hepatitis C patients with failed antiviral therapy. Clin Mol Hepatol. 2024 July;30(3):468-86
133. Zhang R, Wang N, Fan B, Zhang J. Potentiation of Sorafenib's Action by Berberine via Suppression of the mTOR Signaling Pathway in Human Hepatoma Cells. Nutrition and Cancer. 2025May;77(4-5):553-65
134. Rimini M, Montes M, Amadeo E, Vitiello F, Kudo M, Tada T. et al. Impact of metformin, statin, aspirin and insulin on the prognosis of uHCC patients receiving first line Lenvatinib or Atezolizumab plus Bevacizumab. Sci Rep. 2024Aug;14(1):20200
135. Johnson Z, Tarantelli C, Civanelli E, Cascione L, Spriano F, Fraser A. et al. IOA-244 is a Non-ATP-competitive, Highly Selective, Tolerable PI3K Delta Inhibitor That Targets Solid Tumors and Breaks Immune Tolerance. Cancer Res Commun. 2023Apr;3(4):576-91
136. Cai H, Zhang Y, Wang J, Gu J. Defects in Macrophage Reprogramming in Cancer Therapy: The Negative Impact of PD-L1/PD-1. Front Immunol. 2021;12:690869
137. Li J, Xue J, Liu T, Feng Y, Xu N, Huang J. et al. Phase Ib study of the oral PI3Kδ inhibitor linperlisib in patients with advanced solid tumors. Int J Clin Oncol. 2025Feb;30(2):241-51
138. Hu N, Li H, Tao C, Xiao T, Rong W. The Role of Metabolic Reprogramming in the Tumor Immune Microenvironment: Mechanisms and Opportunities for Immunotherapy in Hepatocellular Carcinoma. IJMS. 2024May;25(11):5584
139. Carneiro BA, Jotte RM, Gabrail NY, Wentzel K, Huang F, Chaturvedi S. et al. Safety and Efficacy of Copanlisib in Combination with Nivolumab: A Phase Ib Study in Patients with Advanced Solid Tumors. Cancer Res Commun. 2025Mar;5(3):444-57
140. Ding M, Zhang S, Guo Y, Yao J, Shen Q, Huang M. et al. Tumor Microenvironment Acidity Triggers Lipid Accumulation in Liver Cancer via SCD1 Activation. Mol Cancer Res. 2022May;20(5):810-22
141. Rozengurt E, Soares HP, Sinnet-Smith J. Suppression of Feedback Loops Mediated by PI3K/mTOR Induces Multiple Overactivation of Compensatory Pathways: An Unintended Consequence Leading to Drug Resistance. Mol Cancer Ther. 2014Nov;13(11):2477-88
142. Sivendran S, Agarwal N, Gartrell B, Ying J, Boucher KM, Choueiri TK. et al. Metabolic complications with the use of mTOR inhibitors for cancer therapy. Cancer Treatment Reviews. 2014Feb;40(1):190-6
143. Shen Q, He Y, Qian J, Wang X. Identifying tumor immunity-associated molecular features in liver hepatocellular carcinoma by multi-omics analysis. Front Mol Biosci. 2022;9:960457
Author contact
![]() Corresponding authors: Lei Sun, MD, West China School of Public Health and West China Fourth Hospital, Sichuan University, Chengdu, Sichuan 610041, P.R. China. Tel: +86 13668105208; Email: 395364912com. Dan Li, MD, Institute of Respiratory Health, Frontiers Science Center for Disease-related Molecular Network, and Precision Medicine Research Center, Precision Medicine Key Laboratory of Sichuan Province, West China Hospital, Sichuan University, Chengdu, China. Email: lidancn. Yongsheng Wang, MD, Department of Thoracic Oncology, Cancer Center and State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, Chengdu, China. Email: wangysedu.cn; Tel: +86 18980602258.
Corresponding authors: Lei Sun, MD, West China School of Public Health and West China Fourth Hospital, Sichuan University, Chengdu, Sichuan 610041, P.R. China. Tel: +86 13668105208; Email: 395364912com. Dan Li, MD, Institute of Respiratory Health, Frontiers Science Center for Disease-related Molecular Network, and Precision Medicine Research Center, Precision Medicine Key Laboratory of Sichuan Province, West China Hospital, Sichuan University, Chengdu, China. Email: lidancn. Yongsheng Wang, MD, Department of Thoracic Oncology, Cancer Center and State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, Chengdu, China. Email: wangysedu.cn; Tel: +86 18980602258.