Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Key Features of the...
3. Mechanisms by Which PFME...
4. Impact of LC Progression on...
5. Clinical Significance and...
6. Discussion and Future...
7. Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2026; 22(4):1920-1949. doi:10.7150/ijbs.127307 This issue Cite
Review
Reprogrammed Fibrotic Niche Fuels Lung Cancer Initiation and Reciprocal Remodeling
Zhufeng Hu1#, Wu Dan1#, Mengran Xi2#, Zhengyuan Fang3, Kunlun Feng4, Jie Mei5, Zhang Ting1 ![]() , Baojun Liu1
, Baojun Liu1 ![]() , Zhiwen Luo1,6,7,8,9
, Zhiwen Luo1,6,7,8,9 ![]()
1. Department of Integrative Medicine, Huashan Hospital Affiliated to Fudan University, Shanghai 200040, China.
2. City University of Hong Kong, China.
3. Department of Gastrointestinal Surgery, Renji Hospital, Shanghai 200127, China.
4. First Clinical College, Shandong University of Traditional Chinese Medicine, China.
5. The First Clinical Medicine College, Nanjing Medical University, Nanjing, 211166, China.
6. Department of Sports Medicine, Huashan Hospital Affiliated to Fudan University, Shanghai 200040, China.
7. Fudan Zhangjiang Institute, Fudan University, Shanghai, China.
8. Fudan University - Dr. Kong Joint Research Center for Sports Medicine and Health Footwear, Fudan University Institute of Sports Medicine (Jinqiao Laboratory), Shanghai, China.
9. Department of Orthopaedics, Jiaxing Key Laboratory of Basic Research and Clinical Translation on Orthopedic Biomaterials, The Second Affiliated Hospital of Jiaxing University (Sports Hospital of Jiaxing), 1518 North Huancheng Road, Jiaxing 314000, China.
#Equal contribution.
Received 2025-10-25; Accepted 2026-1-8; Published 2026-1-22
Abstract

Pulmonary Fibrosis (PF), an end-stage manifestation of interstitial lung diseases, is associated with largely unfavorable prognoses. Lung cancer (LC), a leading cause of nationally cancer-related mortality with progressively increasing incidence, exhibits pathological interconnections with PF. The chronic remodeling of the pulmonary microenvironment—including cellular components, extracellular matrix (ECM), inflammatory cytokine networks, and metabolic reprogramming—represents the core pathogenic mechanism underlying PF-LC comorbidity. This review systematically elaborates how the fibrotic microenvironment promotes malignant transformation of lung cancer via chronic inflammation, increased matrix stiffness, immunosuppressive regulation, and epigenetic modulation. Furthermore, we investigate the bidirectional crosstalk by which LC progression reciprocally modulates fibrotic processes. Finally, we integrate current clinical challenges and propose novel therapeutic strategies targeting the fibrotic microenvironment to address this lethal pathophysiological synergy.
Keywords: pulmonary fibrotic microenvironment, lung cancer, cancer-associated fibroblasts, mechanotransduction, metabolic reprogramming, microbiome dysbiosis
1. Introduction
PF, the end-stage manifestation of interstitial lung diseases, is characterized by aberrant activation and proliferation of fibroblasts accompanied by massive ECM deposition, leading to destructive remodeling of lung architecture. Notably, the PF microenvironment shares striking similarities with the tumor microenvironment (TME), particularly in ECM remodeling, chronic inflammation, and immunosuppression(1). For instance, activated myofibroblasts in PF and cancer-associated fibroblasts (CAFs) in TME both drive pathological progression via secretion of transforming growth factor (TGF)-β, interleukin-6 (IL-6), and other profibrotic cytokines(2-5). However, the PF microenvironment exhibits unique protumorigenic features: compared to the dynamic ECM remodeling in TME, PF displays significantly higher and irreversible ECM crosslinking, resulting in sustained elevation of matrix stiffness. This biomechanical stress activates the yes-associated protein (YAP)/transcriptional coactivator with PDZ-binding motif (TAZ) mechanical signaling pathway, driving alveolar epithelial cells (AECs) toward epithelial-mesenchymal transition (EMT)—a process associated with elevated genomic instability compared to normal lung tissue(6-10).
Idiopathic pulmonary fibrosis (IPF), the most prevalent and progressive PF subtype with unknown etiology, high symptom burden, and poor prognosis, is marked by persistent TGF-β signaling abnormalities in its microenvironment. Beyond promoting fibrosis, TGF-β signaling via Smad3-dependent pathways induces infiltration of immunosuppressive cells (e.g., regulatory T cells and M2 macrophages)(11-13). While this immunosuppressive landscape shares functional parallels with Programmed cell death-1 (PD-1)/Programmed death-ligand 1 (PD-L1)-mediated T cell exhaustion in TME, PF-associated myeloid cells (e.g., macrophages) predominantly drive fibrotic rather than oncogenic processes(14,15). Non-IPF PF variants, often arising from autoimmune diseases or environmental exposures, frequently progress to progressive pulmonary fibrosis (PPF) in 30-50% of cases, exhibiting comparable disease burdens and outcomes to IPF(16). A critical distinction lies in the hypoxic microenvironment of PF lungs, where hypoxia-inducible factor (HIF)-1α upregulation elevates PD-L1 expression in epithelial cells, causing CD8+ T cell exhaustion—a mechanism analogous to hypoxia-driven immune evasion in TME. However, PF-associated hypoxic zones are more extensive and persistent, potentially accelerating multifocal lung carcinogenesis(17-20). These pathobiological overlaps and divergences underscore the need to dissect PF-TME crosstalk for developing comorbidity-specific therapeutics.
Lung cancer remains the leading cause of cancer-related mortality globally, with incidence rates steadily rising(21). Epidemiological evidence strongly implicates PF, particularly IPF, as a significant risk factor for lung carcinogenesis, showing a 3.5- to 7.3-fold increased lung cancer risk even after adjusting for smoking exposure, a disparity most pronounced in Asian populations(22-27). This robust association may stem from synergistic microenvironmental evolution: while the PF microenvironment provides a pro-cancerous "soil" (e.g., elevated matrix stiffness, inflammatory cytokines), lung cancer cells reciprocally recruit fibroblasts via connective tissue growth factor (CTGF) and lysyl oxidase like 2 (LOXL2) secretion, forming a feedforward "fibrosis-tumor" loop distinct from the unidirectional stromal regulation in TMEs(1,28-31). Mechanistic studies reveal that chronic inflammation in PF microenvironments generates high reactive oxygen species (ROS) levels, inducing TP53 mutations in AECs and directly promoting carcinogenesis(32,33). Notably, patients with concurrent lung cancer and PF exhibit 1.51-fold higher all-cause mortality compared to IPF patients without malignancy(27), potentially attributed to fibroblast activation via miR-21-mediated crosstalk between CAFs and myofibroblasts, which synergistically drives ECM stiffening and chemoresistance—a phenomenon rarely observed in TMEs of isolated lung cancer(34,35). Therapeutic targeting of this axis shows promise: antifibrotic treatment (e.g., nintedanib) reduces all-cause mortality by 39% in IPF patients with comorbid lung cancer(27), while its fibroblast growth factor receptor (FGFR) signaling inhibition confers dual anti-fibrotic and anticancer effects(36). These findings underscore the potential of targeting PF-TME convergent pathways for synergistic therapeutic benefits over conventional oncology approaches.
Despite these advances, critical knowledge gaps persist: 1. How do fibrotic microenvironments uniquely drive lung cancer initiation and early evolution through chronic inflammation, genomic instability, epigenetic reprogramming, biomechanical signaling, immunosuppression, and stem cell niches? 2. How do tumor-derived factors (e.g., pro-fibrotic cytokines, metabolic reprogramming) remodel fibrotic microenvironments during cancer progression, exacerbating therapeutic resistance and poor outcomes through bidirectional feedback loops? This review systematically dissects PF-microenvironment (PFME)-TME interactions, emphasizing PFME-driven carcinogenic mechanisms and TME-mediated fibrotic reinforcement. We further identify actionable targets at the intersection of these pathologies for novel therapeutic strategies.
2. Key Features of the PFME
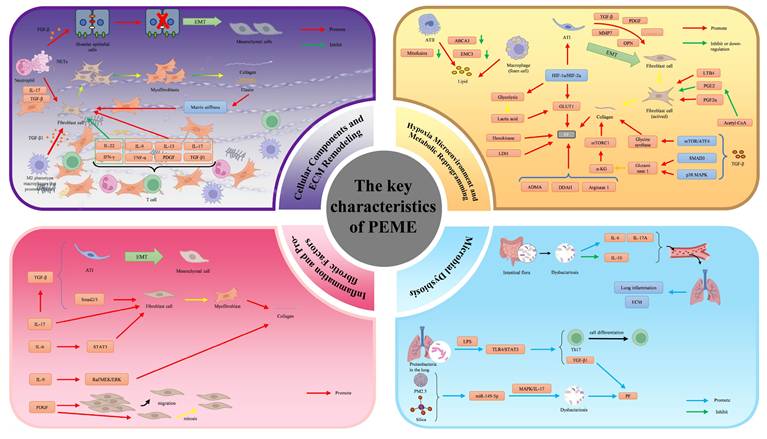
The key characteristics of the PFME encompass cellular components, ECM remodeling, inflammatory networks, hypoxia, metabolic reprogramming, and microbiome dysbiosis. These features interact synergistically to drive fibrotic progression while creating a "soil" for lung cancer initiation and early evolution through mechanisms such as chronic inflammation, genomic instability, mechanical signaling, and immunosuppression. Below is a brief overview of each characteristic, with emphasis on their direct associations with lung carcinogenesis. (Fig. 1).
Schematic diagram of the key characteristic of the PFME. The key characteristics of the pulmonary fibrotic microenvironment encompass cellular components, ECM remodeling, inflammation and pro-fibrotic factor networks, hypoxic microenvironment, metabolic reprogramming, and microbiome dysbiosis. Cellular components of the pulmonary fibrotic microenvironment primarily consist of fibroblasts, inflammatory cells, and epithelial cells. Fibroblasts differentiate into myofibroblasts. Excessive myofibroblasts remain persistently activated in fibrotic lungs and secrete ECM components such as collagen type I and α-SMA. Inflammatory cells generate various inflammatory cytokines and pro-fibrotic factors, thereby driving fibrosis. Alveolar epithelial cells undergo EMT, which promotes the development of PF. The ECM is remodeled by various cells. Increased matrix stiffness further promotes the progression of PF. Hypoxia induces alveolar epithelial cells to secrete pro-fibrotic factors and can affect fibrosis through metabolic alterations. Metabolism in PF is reprogrammed, which further promotes PF. Inflammatory cytokines and pro-fibrotic factors synergistically drive disease progression through complex interactive networks. In PF, structural and functional dysbiosis of the gut and lung microbiota exacerbates the inflammatory and fibrotic processes via multidimensional mechanisms. Copyrighted from the BioRender platform.
2.1 Cellular Components
The cellular components within the PFME, including fibroblasts, inflammatory cells, and epithelial cells, drive fibrosis through abnormal activation and intercellular crosstalk, while directly contributing to lung cancer initiation.
2.1.1 Activated Fibroblasts/Myofibroblasts
Activated myofibroblasts are the core effector cells of PF, promoting fibrosis by secreting ECM components (such as collagen I) and expressing the α-SMA contractile apparatus(37). In the PFME, fibroblasts secrete TGF-β to activate CAFs, thereby establishing a pro-tumor microenvironment(5). CAF subtypes exhibit heterogeneity. Different CAF subtypes promote malignant cell proliferation, metabolism, stemness, chemotherapy resistance, EMT, invasion, and migration through paracrine signaling, ECM remodeling, and/or metabolite exchange(38). Myofibroblast-like CAFs (myCAFs) differentiate upon TGF-β induction, highly express α-SMA and fibroblast activating protein (FAP), and dominate excessive ECM production, forming a physical barrier that promotes treatment resistance, but may suppress early tumors via collagen cross-linking; Inflammatory CAFs (iCAFs) rely on IL-1 signaling to secrete inflammatory factors such as IL-6 and CXCL-1, recruit myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs) to suppress immunity, and exhibit dynamic transformation with myCAFs; Antigen-presenting CAFs (apCAFs) present antigens via MHC class II molecules, which can either activate Tregs to enhance immunosuppression or trigger CD4+ T cell anti-tumor responses in specific cancer types; Tumor-suppressive CAFs (rCAFs) secrete Meflin and Asporin to inhibit ECM cross-linking and the TGF-β pathway, suppressing tumor growth in the early stage but decreasing with disease progression(39). For example, CAFs induce resistance to almonertinib in patients with non-small cell lung cancer (NSCLC) by activating the YAP/TAZ signaling pathway(40).
2.1.2 Inflammatory Cells
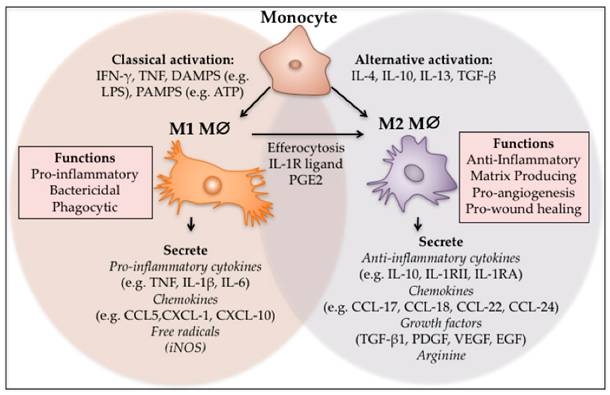
Inflammatory cells promote malignant transformation in PF through chronic inflammatory signals. Macrophages polarize into M2 phenotypes (such as M2a, M2c, M2d), secrete high levels of TGF-β1, induce fibroblast activation and suppress immune responses, creating a microenvironment permissive for lung cancer growth(41). Additionally, macrophages secrete cytokines such as tumor necrosis factor-α (TNF-α), IL-1β, cyclooxygenase-2 (COX-2), vascular endothelial growth factor (VEGF), etc., mediating immunosuppression, immune evasion, and metastasis(42) (Fig. 2).
M1 and M2 polarisation of macrophages. Monocytes can be classically or alternatively activated to form M1 and M2 macrophages respectively. M1 macrophages can also differentiate into M2 macrophages through local cues and after efferocytosis. The M1 phenotype is pro-inflammatory, phagocytic and bactericidal, while the M2 macrophages act to switch off inflammation and regulate re-vascularisation and wound closure. (Note. From Hesketh et al.(41) (2017) under the terms of the Creative Commons Attribution International License (CC BY 4.0).).
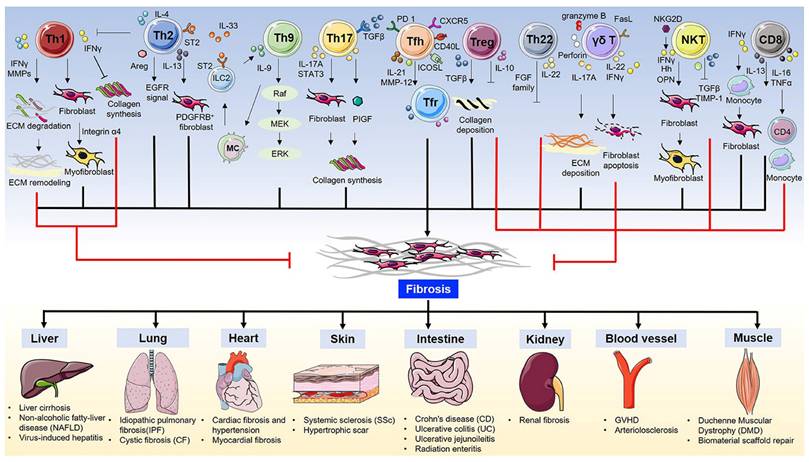
T cells play a key role in regulating fibrosis through diverse subtypes and functional polarization. T cell subsets (such as Th2, Th17) drive fibrosis by secreting factors such as IL-13 and IL-17, while suppressing anti-tumor immunity(43) (Fig. 3). For example, CD4+ T cells can upregulate PD-1 and secrete IL-17A and TGF-β1 via the signal transducer and activator of transcription 3 (STAT3) pathway, promoting PF(44).
T cells in fibrosis and fibrotic diseases. (Note. From Zhang et al.(43) (2020) under the terms of the Creative Commons Attribution International License (CC BY 4.0).).
Neutrophils bridge the progression of fibrosis and lung cancer by releasing toxic mediators and forming neutrophil extracellular traps (NETs). By forming neutrophil extracellular traps, neutrophils directly activate fibroblasts and induce epithelial damage and EMT, and generate sustained chronic inflammation mediated by TGF-β and IL-17 signaling, whose components (such as MPO) exacerbate oxidative stress, increase the risk of DNA mutations in lung epithelial cells, thereby directly driving lung cancer initiation(45).
2.1.3 Epithelial Cells
Repeated injury and alterations in alveolar epithelial cells (AECs), including proliferation and hyperplasia, have been shown to weaken epithelial-mesenchymal crosstalk, leading to aberrant activation of AECs and epithelial cell senescence—all closely associated with pulmonary fibrosis (PF) progression(46-48). Existing evidence indicates that PF is caused by repeated injury to AECs, triggering aberrant interactions between epithelial cells and fibroblasts (49,50). Apoptosis of Type II alveolar epithelial cells (ATII) and inactivation of the Hippo pathway enhance YAP/TAZ signaling, promoting aberrant regeneration (10). This is not only the core mechanism of PF but also provides a basis for the formation of the lung cancer stem cell niche, directly linked to early tumorigenesis. This will be discussed in detail in subsequent sections.
2.2 ECM Remodeling
Pathological epithelial remodeling and ECM accumulation are the core pathological features of PF. The ECM is primarily composed of collagen, elastin, glycoproteins, and proteoglycans, and its remodeling process is closely related to fibroblasts(51). TGF-β signaling drives fibroblast proliferation and differentiation into myofibroblasts, which secrete collagen excessively(41). As mentioned earlier, macrophages, T cells, and epithelial cells jointly promote ECM crosslinking and deposition via paracrine signals (such as IL-6 and TGF-β). The mechanotransduction pathways that play a key role in PF progression include Rho-associated protein kinase (ROCK), myocardin-related transcription factor (MRTF), YAP, and TAZ(7,52,53). Increased matrix stiffness activates the YAP/TAZ pathway via integrins, inducing fibroblast activation and fibrosis(7). This process forms a positive feedback loop: stiffness enhancement → YAP/TAZ activation → collagen secretion → further hardening. Increased ECM stiffness can hinder the penetration of drugs and immune cells and provide a niche for tumor progression(54). This suggests the dual role of ECM stiffness in tumor-fibrosis crosstalk.
2.3 Inflammation and Pro-fibrotic Factor Network
In the pathological progression of PF, inflammatory cytokines and pro-fibrotic factors synergistically drive disease advancement through intricate interaction networks. TGF-β exposure during lung tissue injury induces EMT(55). Additionally, TGF-β promotes fibroblast-to-myofibroblast differentiation via the Smad2/3 pathway, enhancing collagen and fibronectin deposition(56). IL-6 accelerates fibroblast proliferation by activating the signal transducer and activator of transcription 3 (STAT3) signaling cascade(57). IL-9 stimulates collagen deposition through the Raf/MEK/ERK pathway(58). IL-17 directly activates fibroblasts and myofibroblasts, inhibits autophagy, and induces EMT in epithelial cells by promoting TGF-β production(59). The PDGF family induces fibroblast migration while exerting anti-apoptotic and mitogenic effects on ECM-producing fibroblasts and myofibroblasts, thereby accelerating fibrotic disease progression(60). These pro-fibrotic factors activate fibroblasts or enhance collagen deposition via distinct signaling pathways to advance PF. Conversely, fibroblasts or ECM remodeling exert feedback regulation on this pro-fibrotic factor network (Fig. 1).
2.4 Hypoxic Microenvironment
Hypoxia-induced lung tissue injury activates hypoxia-responsive pathways, with core regulators HIF-1α/HIF-2α playing pivotal roles in both IPF and lung cancer. Hypoxia induces alveolar epithelial cells to secrete pro-fibrotic factors such as TGF-β and PDGF, activating fibroblasts through enhanced glycolysis, upregulation of glucose transporter 1 (GLUT1), ECM deposition, mitochondrial ROS/tricarboxylic acid (TCA) metabolite-mediated macrophage activation, and induction of alveolar epithelial apoptosis or EMT, thereby driving IPF fibrosis(61,62). Simultaneously, hypoxia upregulates matrix metalloproteinases 7 (MMP7)/MMP14 to promote lung cancer invasion/metastasis, activates Warburg metabolism for tumor survival, and suppresses immunity via lactate and other metabolites(62). IPF and lung cancer share HIFs-MMPs pathways and metabolic mechanisms, suggesting targeted therapies could simultaneously intervene in fibrosis and tumor progression.
2.5 Metabolic Reprogramming
In PF, metabolic reprogramming drives fibrotic progression through coordinated dysregulation of lipid, amino acid, carbohydrate metabolism, and the TCA cycle.
Lipid metabolism in PF is characterized by impaired surfactant production and ectopic lipid accumulation. Alveolar type II (ATII) cells downregulate lipid synthesis genes (ABCA3) and cholesterol metabolism genes, reducing surfactant and promoting cholesterol deposition(63-66). Loss of mitochondrial or endoplasmic reticulum proteins (mitofusins, EMC3) further disrupts lipid homeostasis(67,68). Foam macrophages exhibit a pro-fibrotic phenotype via CD36-mediated fatty acid uptake, while inhibiting CD36 alleviates fibrosis(69-71). Key lipid mediators regulate fibrosis. LTB4 enhances collagen deposition (blockade reduces fibrosis), PGE2 inhibits fibroblast proliferation (reduced in advanced PF), and PGF2α drives fibrosis independently of TGF-β (72-74).
Amino acid metabolism supports collagen biosynthesis in PF. TGF-β upregulates glycine synthesis, glutaminase 1, and α-KG to stabilize collagen(75-77). Upregulation of arginase 1 in IPF drives proline synthesis, promotes collagen accumulation, and targeting this reduces fibrosis(78,79). Asymmetric dimethylarginine (ADMA) accumulates due to dimethylarginine dimethylaminohydrolase (DDAH) inhibition, impairs NO production, and promotes fibrosis(80).
Hypoxia stabilizes HIF-1α, enhances glycolysis and GLUT1 expression, and promotes fibrosis (70). TGF-β upregulates GLUT1 and LDH, increases lactate production; acidic pH activates TGF-β-mediated myofibroblast differentiation(81-83). Overexpressed hexokinase/LDH in IPF fibroblasts support fibrosis, and hexokinase inhibition (e.g., lonidamine) has anti-fibrotic effects(81,84).
The TCA cycle is disrupted, altering bioenergetics and epigenetics. Mitochondrial acetyl-CoA reduces histone acetylation, inhibits COX-2/PGE2, and promotes fibrosis(85). Glutamine-derived α-KG activates mTORC1 to stabilize collagen and modulates anti-apoptotic XIAP via Jumonji domain-containing protein D3 (JMJD3)/ubiquitously transcribed tetratricopeptide repeat, chromosome X (UTX) to maintain fibroblasts (77,86).
2.6 Microbiome Dysbiosis
Microbiota dysbiosis plays a key role in the progression of PF through the gut-lung axis and local lung ecology. Its pro-fibrotic mechanisms involve multidimensional pathways such as immune imbalance, metabolite signaling, and epithelial barrier damage.
At the gut-lung axis level, structural dysbiosis of the gut microbial community is a key driver of PF. Specifically, the reduction of Akkermansia muciniphila, which has immunomodulatory functions, disrupts immune homeostasis, leading to increased levels of systemic pro-inflammatory cytokines (such as IL-6, IL-17A) and suppression of anti-inflammatory cytokines (such as IL-10), thereby exacerbating lung inflammation and fibrosis via the circulatory system(87). Additionally, the gut microbiota promotes M2 polarization of macrophages and Th cell imbalance by downregulating short-chain fatty acids and upregulating lipopolysaccharide (LPS), and accelerates PF progression by targeting core PF pathways (TGF-β, YAP/TAZ, metabolic reprogramming)(88).
In the local lung, microbiome dysbiosis is also crucial, typically characterized by the enrichment of Gram-negative bacteria (e.g., Proteobacteria). LPS released by these bacteria activates Toll-like receptor 4 (TLR4) signaling, driving the TLR4/STAT3 phosphorylation cascade to promote Th17 cell differentiation and expression of the pro-fibrotic factor TGF-β1(99,100). Environmental factors (such as PM2.5, silica) can disrupt lung microbial homeostasis and induce abnormal miRNA expression, exacerbating dysbiosis via the MAPK/IL-17 pathway and forming a feedforward loop that accelerates fibrosis(89,90).
Insufficient expression of antimicrobial peptides due to primary immunodeficiencies (such as TLR5 deficiency), as well as sex differences, also modulate microbiome balance and affect PF susceptibility(91,92).
3. Mechanisms by Which PFME Promotes Lung Carcinogenesis
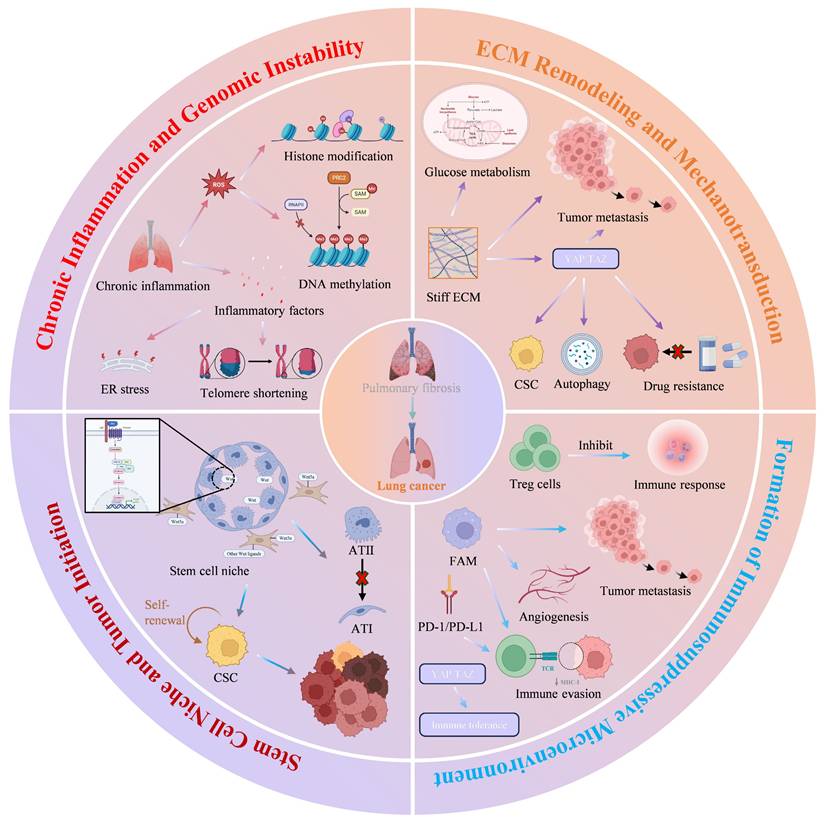
The process by which the PFME drives lung cancer development is a complex, multidimensional, multi-stage synergistic process, and its core mechanism is illustrated in Figure 4 (Fig. 4). As the key pathological basis for the progression from PF to lung cancer, PFME reshapes the pulmonary microenvironment and drives malignant transformation through four core links. First, chronic inflammation, as an initiating factor, induces genomic instability via ROS-mediated oxidative stress, immunosuppression, and endoplasmic reticulum stress, laying the "initiation" foundation for the malignant transformation of alveolar epithelial cells. Second, ECM remodeling and mechanotransduction, by sensing changes in matrix stiffness through the YAP/TAZ signaling pathway, drive tumor cell proliferation, drug resistance, metabolic reprogramming, and immune escape, dominating the "progression-metastasis" stage. Third, construction of an immunosuppressive microenvironment, relying on the synergistic effects of Tregs, M2-polarized tumor-associated macrophages (TAMs), and the PD-1/PD-L1 axis, weakens anti-tumor immune responses and assists tumor "colonization-immune escape". Fourth, abnormal regulation of the ATII stem cell niche, which maintains stem cell stemness and drives malignant transformation via Wnt signaling, providing "seed" cells for lung cancer occurrence. These mechanisms do not exist in isolation but rather through signal crosstalk and stage overlap, collectively constituting a complete pathological network of PFME-promoted lung cancer occurrence. The following will focus on the above core links and analyze their molecular mechanisms and functional effects one by one.
Schematic diagram of the mechanism by which PFME promotes lung carcinogenesis. Chronic inflammation, as a core pathological feature of PF, induces genomic instability through multidimensional mechanisms, providing a critical driving force for the malignant transformation of lung epithelial cells. ECM remodeling increases matrix stiffness, activates mechanical signaling, and reprograms glucose metabolic pathways in tumor cells, thereby promoting malignant progression, drug resistance, and metastasis of cancer. The formation of an immunosuppressive microenvironment exerts a significant impact on the malignant progression of cancer. The aberrant expression of Tregs, M2-type macrophages, and the immune checkpoint PD-1/PD-L1 in PFME contributes to the formation of an immunosuppressive microenvironment, influencing cancer progression. ATII lung stem cells exhibit active Wnt signaling and form a Wnt signaling niche. The Wnt niche drives the proliferative potential and disease progression of lung adenocarcinoma. Copyrighted from the BioRender platform.
3.1 Chronic Inflammation and Genomic Instability
Chronic inflammation, as a core pathological feature of PF, induces genomic instability through multidimensional mechanisms, providing a key driving force for the malignant transformation of alveolar epithelial cells during the initiation and promotion stages of lung cancer development. Chronic inflammation mediates oxidative stress and immunosuppression via ROS, laying the histological foundation for the malignant transformation of alveolar epithelial cells.
In the PFME, chronic inflammation continuously promotes the generation of ROS and inflammatory mediators(93). ROS generated by cells drives multidimensional pathological processes through oxidative stress, including DNA damage, abnormal activation of signaling pathways, epigenetic alterations, cell phenotype transformation, and metabolic reprogramming, ultimately leading to the occurrence and progression of pulmonary fibrosis and lung cancer(32,94). ROS generated by cells triggers oxidative stress: on one hand, it directly induces epithelial cell apoptosis, activates pro-fibrotic signaling pathways (such as TGF-β/MAPK), upregulates the synthesis of pre-fibrotic cytokines, generates myCAFs, and causes lung tissue damage and lays the "foundation" for fibrosis; on the other hand, excessive ROS alters DNA methylation patterns and regulates specific histone modifications, leading to abnormal gene expression profiles and driving multi-stage carcinogenesis progression(32). For example, ROS can induce p53 mutations, opening the "gate" for cellular malignant transformation(33). Additionally, ROS damages mitochondrial DNA and the respiratory chain, resulting in reduced ATP synthesis and increased ROS, forming a vicious cycle(95). In terms of metabolism, ROS induces the Warburg effect via HIF-1α stabilization to support the energy demands of tumor cells(94). Excess ROS leads to immune cell dysfunction by oxidizing key immune molecules and inhibits signal presentation between dendritic cells (DCs) and T cells(95). ROS also drives M1 macrophage polarization, secreting TNF-α and IL-1β to exacerbate inflammation, while inhibiting M2 macrophage function and hindering ECM degradation(96). Meanwhile, inflammation impairs DNA repair mechanisms, further exacerbating genomic instability and providing a molecular basis for subsequent clonal selection(97).
Notably, PF-specific endoplasmic reticulum (ER) stress forms a positive feedback loop with inflammatory signals, amplifying the risk of genetic damage during the initiation stage. In PF, oxidative stress in the ER leads to protein misfolding and promotes ER stress, further leading to excessive ROS(98). ROS induced by ER stress causes DNA double-strand breaks; under its sustained action, the function of the DNA-dependent protein kinase catalytic subunit (DNA-PKcs)-non-homologous end joining (NHEJ) pathway is inhibited, reducing the efficiency of DNA double-strand break repair, and leading to the accumulation of genomic instability and mutation burden(99). Furthermore, when ROS accumulation exceeds the repair capacity, markers of genomic instability such as telomere shortening accelerate clonal selection during the promotion stage. Telomere dysfunction synergizes with chronic inflammation to accelerate genomic collapse, promoting clonal expansion during the promotion stage. Telomere shortening is driven by mutations such as telomerase reverse transcriptase (TERT)/ telomerase RNA component (TERC)(100-102). Telomere shortening promotes IFN-γ secretion by activating the cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS)-stimulator of interferon genes (STING) pathway, which not only enhances the inflammatory microenvironment but also remodels cell cycle regulation via interferon signaling, forcing cells to accelerate clonal selection and expansion under the "stress" of genomic instability(103).
3.2 ECM Remodeling and Mechanotransduction
In the PFME, ECM remodeling and mechanotransduction drive the escalation of malignant cellular phenotypes through the YAP/TAZ signaling pathway during the progression and metastasis stages of lung cancer development.
As transcriptional co-activators, YAP/TAZ regulate key pathways in cytoplasm-nucleus shuttling: they form complexes with TEA domain family members (TEAD), bind to distal enhancer regions, promote histone acetylation and recruitment of transcriptional machinery, and target the regulation of cell cycle and anti-apoptotic pathways, providing molecular support for tumor cell proliferation(104,105). In fibrotic lung tissue, spindle-shaped myofibroblasts highly express TAZ, and their pro-fibrotic effects are partially mediated by the transcriptional target plasminogen activator inhibitor-1 (PAI-1), which is not regulated by matrix stiffness and independent of TGF-β signaling, creating a "mechanical microenvironment" for tumor cell invasion(7).
After cells sense increased ECM stiffness via integrins, the Rho-ROCK pathway mediates increased F-actin tension, promoting YAP/TAZ nuclear translocation and activation; this process is independent of the Hippo pathway, and YAP/TAZ still respond to ECM stiffness even in the absence of LATS1/2(6,106,107). More critically, YAP/TAZ drive malignant progression by sensing ECM mechanical signals. They maintain cancer stem cell (CSC) traits and increase basal autophagy levels, thereby enhancing chemoresistance of cancer cells(108,109). YAP/TAZ also promote the progression and metastasis of lung cancer in vivo(110). YAP/TAZ bind to TEAD to directly activate PD-L1 gene transcription and secrete chemokines such as C-C motif chemokine ligand 2 (CCL2) and CCL5, attracting MDSCs and Tregs, suppressing CD8+ T cell function, and promoting the expression of T cell exhaustion markers (PD-1, TIM-3)(111). Studies have shown that increased ECM stiffness reprograms the glucose metabolism of tumor cells and coordinates intercellular metabolic communication, thereby enhancing tumor activity(112). Additionally, intrafibrillar crosslinks in the ECM may contribute to lung cancer metastasis(113). In summary, the ECM-mechanical signaling axis comprehensively drives the evolution of lung cancer from local progression to distant metastasis, ranging from "mechanical microenvironment construction" to "metabolic-immune reprogramming."
3.3 Formation of the Immunosuppressive Microenvironment
The synergistic effects of Tregs, M2-polarized TAMs, and the PD-1/PD-L1 axis construct an immunosuppressive microenvironment during the colonization and immune escape stages of lung cancer development.
Tregs exhibit a "stage-specific functional switch" in the progression of IPF: in the early stage, they drive EMT and promote fibrosis by secreting factors such as PDGF and TGF-β, providing a "fibrotic soil" for tumor cell colonization; in the late stage, they indirectly maintain lung homeostasis by enhancing epithelial repair and inhibiting excessive fibroblast accumulation(114,115). Flow cytometry and single-cell sequencing confirm that in advanced PF, the proportion and hyperfunction of Tregs increase. Their accumulation, by secreting cytokines such as IL-10 and TGF-β, promotes the polarization of alveolar macrophages into the M2 type and inhibits effector T cell activation, while also inducing CD8+ T cell exhaustion, thereby forming an immunosuppressive microenvironment conducive to lung cancer growth(116,117).
YAP/TAZ further enhance immune tolerance by recruiting MDSCs, while promoting TAMs to secrete IL-6 to enhance immunosuppression, forming a "fibrosis-immunosuppression" vicious cycle(111). As key effector cells in tumor progression, M2-polarized TAMs in PF promote tumor angiogenesis by secreting VEGF, inhibit the activity of T cells and natural killer (NK) cells, express PD-L1/PD-L2 and recruit Tregs, assisting tumor cell colonization and growth from both vascular support and immune escape aspects(42).
Dysregulation of the PD-1/PD-L1 axis plays a central role in immune escape. The proportion of PD-L1-positive tumor cells is positively correlated with tumor severity and prognosis(118). In IPF patients, the expression of PD-1/PD-L1 is abnormally elevated in lung tissues and peripheral blood. The mechanism is that the PD-1/PD-L1 signal inhibits excessive T cell activation under physiological conditions to maintain immune tolerance(119-121)(Fig. 5). However, in the TME, cancer cells upregulate PD-L1 to inhibit excessive T cell activation and cytokine secretion, and evade cytotoxic attacks by CD8+ T cells(122,123). The activation of the PD-1/PD-L1 checkpoint in the PFME synergizes with PF-driven expansion of Tregs and M2 TAMs, weakening anti-tumor immune responses during the immune escape stage and clearing obstacles for the survival and proliferation of lung cancer cells.
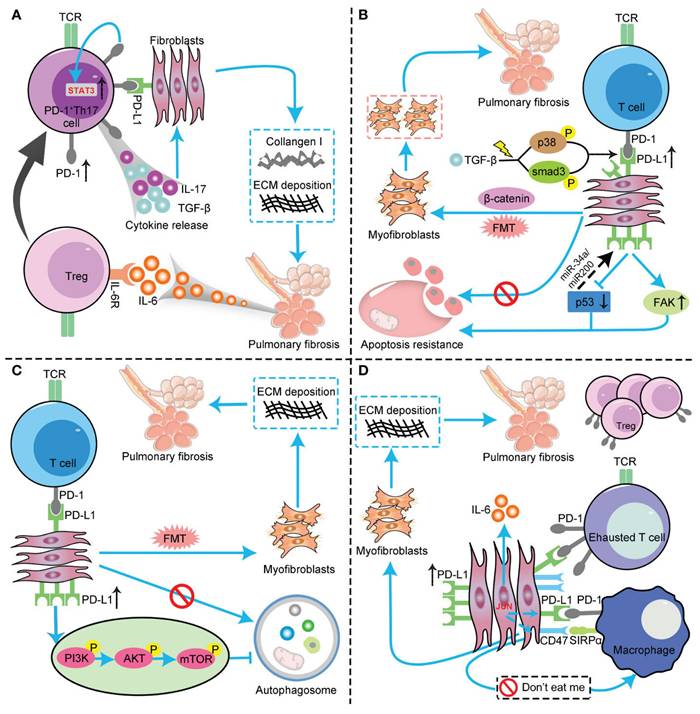
The profibrotic role of the PD-1/PD-L1 axis in IPF through interaction with multiple cell types and pathways. (A) PD-L1 up-regulation on Th17 T cells promotes pulmonary fibrosis through STAT3-mediated IL-17 and TGF-β production; (B) PD-L1 up-regulation on lung fibroblasts promotes pulmonary fibrosis via p53, FAK, Smad3, and β-catenin signaling pathways. On the one hand, PD-L1 up-regulation on lung fibroblasts may cause myofibroblasts to apoptosis-resistance and evasion phagocytosis via macrophages by inhibiting the p53 pathway and activating the FAK pathway, ultimately leading to excessive proliferation of myofibroblasts to trigger IPF. On the other hand, PD-L1 mediates lung fibroblast to myofibroblast transition (FMT) through Smad3 and β-catenin signaling pathways, thus promoting pulmonary fibrosis; (C) PD-L1 up-regulation on lung fibroblasts could induce myofibroblasts proliferation and ECM deposition through inhibiting autophagy, and eventually promotes pulmonary fibrosis; (D) PD-L1 up-regulation on lung fibroblasts promotes pulmonary fibrosis by inhibiting adaptive immunity. JUN upregulates the expression levels of PD-L1 and CD47 in fibroblasts and dormant macrophages. As a result, the above cells are converted into exhausted T cells and quiescent macrophages. In this context, myofibroblasts can evade immune clearance and resist macrophage-induced phagocytosis. In addition, JUN can also directly regulate IL-6 at the chromatin level, leading to inhibitory adaptive immune responses— primarily T cell exhaustion and upregulation of activated Tregs. FAK, focal adhesion kinase; TCR, T cell receptor; AKT, protein kinase B; PI3K, phosphoinositide 3-kinase; mTOR, mammalian target of rapamycin. (Note. From Jiang et al.(120) (2022) under the terms of the Creative Commons Attribution International License (CC BY 4.0).).
3.4 Stem Cell Niche and Tumor Initiation
Abnormal regulation of the ATII stem cell niche provides the "seed" for the formation of CSCs during the initiation stage of lung cancer development.
All stem cells, including ATII stem cells, are regulated by their local microenvironment or niche(124). In normal alveolar repair, transient activation of Wnt signaling promotes ATII differentiation; while in the PFME, continuously activated Wnt signaling locks stem cell stemness and drives malignant transformation. Normal alveolar epithelial repair relies on the differentiation of ATII into ATI, but Hippo pathway inactivation in the PFME can elevate nuclear YAP/TAZ levels, enhance the alveolar regenerative capacity of the ATI cell lineage, and instead reduce bleomycin-induced fibrosis(10). Conversely, excessive Hippo pathway activation drives aberrant differentiation of ATII stem cells, disrupts the homeostasis of alveolar epithelial repair, and increases the risk of PF and lung cancer(10,125).
Adjacent mesenchymal fibroblasts of ATII stem cells secrete ligands such as Wnt5a to form a single-cell-scale Wnt signaling niche, preventing ATII stem cells from differentiating into ATI and maintaining their stemness(125). This stemness maintenance mechanism acts as a "pusher" for tumor initiation when aberrant (e.g., Wnt/β-catenin signaling dysregulation). The Wnt pathway is a core pathway for stem cell self-renewal and carcinogenesis. Dysregulated Wnt/β-catenin signaling promotes the renewal, proliferation, and differentiation of cancer stem cells, playing a key role in tumorigenesis and therapeutic responses(126). Studies have shown that aberrant activation of the Wnt pathway can promote the renewal or proliferation of colorectal cancer stem cells(127-129). In lung cancer, however, it manifests as "malignant transformation of the alveolar stem cell niche": its Wnt-producing microenvironment drives enhanced stemness and increased proliferative potential of lung adenocarcinoma cells, while small-molecule inhibitors targeting key Wnt post-translational modifications can reduce tumor growth and significantly decrease lung cancer cell proliferation(128). The Wnt/β-catenin pathway serves as one of the core mechanisms of lung cancer immune escape by inhibiting T cell infiltration, recruiting immune cells, and metabolic regulation(130). This suggests that aberrant stem cell niche is a key link in lung cancer initiation, and disrupting this pathway may simultaneously intervene in fibrosis and tumorigenesis.
4. Impact of LC Progression on the PFME
The progression of LC not only drives malignant transformation but also reversely remodels the PFME, creating a vicious cycle that exacerbates both diseases. This section details how LC progression impacts PFME across three key stages: First, during the tumor growth phase, cancer cells secrete pro-fibrotic factors and remodel the stroma; second, during the invasion-metastasis phase, which is characterized by ECM stiffening, mechanotransduction, and metabolic reprogramming; finally, during the therapeutic intervention phase, radiotherapy, chemotherapy, and targeted therapy may induce or exacerbate PFME. These effects collectively amplify PFME abnormalities, which in turn fuel further LC progression (Fig. 6).
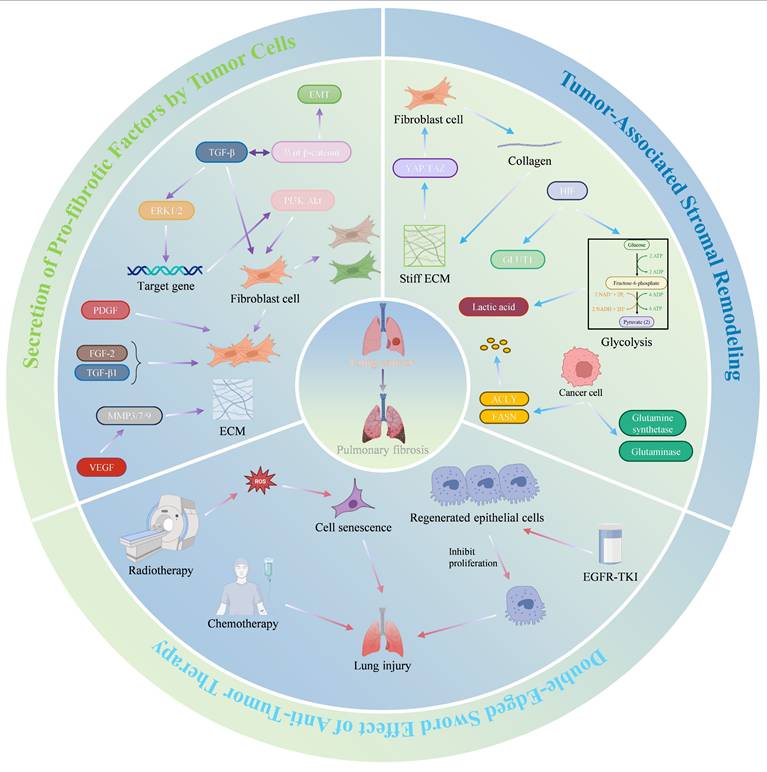
Schematic diagram of the impact of LC progression on the PFME. Crosstalk between the Wnt/β-catenin pathway and TGF-β can stimulate the proliferation and differentiation of myofibroblasts. Meanwhile, tumor cells secrete pro-fibrotic factors such as PDGF, VEGF, and FGF. Tumor-associated stromal remodeling can promote the progression of PF. Increased ECM stiffness activates the YAP/TAZ pathway, driving fibroblast activation and fibrosis, thus forming a feedback loop. Metabolic changes in tumor cells also promote PF progression. Additionally, tumor-targeted therapies, such as radiotherapy, chemotherapy, and targeted therapy, may also increase the potential risk of exacerbating PF. Copyrighted from the BioRender platform.
4.1 Secretion of Pro-fibrotic Factors by Tumor Cells
Lung PF is closely linked to the Wnt/β-catenin signaling pathway. This pathway regulates molecules associated with tissue invasion, such as matrix metalloproteinases (MMPs), laminins, and cyclin D1, which drive EMT. However, the most critical function of the Wnt/β-catenin pathway may lie in its crosstalk with TGF-β(131). Aberrant activation of this pathway has been observed in cancers such as lung cancer and mesothelioma(132). Specifically, TGF-β may activate extracellular signal-regulated kinases 1 and 2 (ERK1/2), while its target genes activate other signaling pathways, including the phosphatidylinositol 3-kinase (PI3K)/protein kinase B (Akt) pathway, which regulates proliferation and apoptosis. The role of PI3K in TGF-β-induced proliferation and differentiation of myofibroblasts has been demonstrated(133).
Furthermore, tyrosine kinase receptor ligands, including PDGF, VEGF, and fibroblast growth factor (FGF), are abnormally expressed in lung cancer and PF(134). PDGF signaling plays a critical role in tumor growth, angiogenesis, and lymphangiogenesis in cancer(135). For instance, studies using A549 cells as a NSCLC model revealed that crenolanib (a PDGF receptor inhibitor) dose-dependently suppresses cell proliferation and induces apoptosis(136). PDGF also regulates VEGF expression via autocrine mechanisms in NSCLC and facilitates CAF recruitment to tumor masses(135,137). Beyond its angiogenic role, VEGF directly acts on cancer cells to modulate the growth, migration, and production of pro-angiogenic factors(138). FGF influences NSCLC cell proliferation, therapeutic sensitivity, and apoptosis(139).
Lung cancer cells promote PF progression by secreting tyrosine kinase receptor ligands such as PDGF, VEGF, and FGF—pro-fibrotic factors implicated in PF. Evidence indicates that dysregulated PDGF expression plays a pivotal role in PF development. In bleomycin-induced PF models, PDGF-A/B/C mRNA expression is significantly upregulated(140,141). PDGF stimulates lung fibroblast proliferation, thereby driving PF progression(142). TGF-β1 enhances PF by inducing FGF-2 release and upregulating FGFR1/2, while FGF-2 synergizes with TGF-β1 to promote fibroblast proliferation in IPF patients(142-144). VEGF orchestrates extracellular matrix remodeling by inducing matrix metalloproteinase (MMP3/7/9) expression, and anti-VEGF gene therapy attenuates inflammation and fibrosis in bleomycin models(145,146). Collectively, these tumor-derived pro-fibrotic factors synergistically advance PF progression, suggesting that targeting these factors may represent a promising therapeutic strategy.
Collectively, these tumor-derived pro-fibrotic factors act as key drivers of PFME fibrosis during the tumor growth phase, primarily via Wnt/β-catenin-TGF-β crosstalk and PI3K/Akt signaling to activate myofibroblasts.
4.2 Tumor-Associated Stromal Remodeling
The stromal components within the TME undergo remodeling to promote tumor progression. Interactions between lung cancer cells and fibroblasts have been extensively reported in previous studies. Two primary subtypes of CAFs—myofibroblast CAFs (myoCAFs) and inflammatory CAFs (iCAFs)—have been described in multiple cancer types(147,148). Analysis of human tissues revealed that α-SMA-positive myoCAFs correlate with poor prognosis across various cancers and resistance to immunotherapy in NSCLC(148,149). ECM production and remodeling represent critical functions of fibroblasts in all tissues(150). Tumor-driven activation of the Wnt/β-catenin pathway mediates TGF-β crosstalk, stimulating myofibroblast proliferation and differentiation, promoting collagen deposition, and increasing ECM stiffness(131). Additionally, pro-fibrotic factors secreted by lung cancer cells, such as PDGF, VEGF, and FGF, further enhance myofibroblast proliferation and differentiation(134). Myofibroblasts reciprocally drive malignant progression by secreting collagen, increasing ECM stiffness, and activating the YAP/TAZ pathway. Studies have demonstrated that YAP/TAZ-mediated mechanotransduction activates fibroblast activation and fibrosis(7). Furthermore, Smad3 facilitates CAF generation via macrophage-myofibroblast transformation(151). The interplay between lung cancer cells and myofibroblasts in TME remodeling not only advances lung cancer progression but also promotes PF, highlighting potential therapeutic targets.
A hallmark of cancer is metabolic reprogramming. Tumor cells alter carbohydrate, lipid, and protein metabolism to support proliferation. Hypoxia, a defining feature of the TME, upregulates HIFs during hypoxic responses, enhancing glycolysis and upregulating GLUT1 to promote PF(61,152). The hypoxic microenvironment also suppresses anti-tumor immunity via metabolites such as lactate(61). Lactate's pro-fibrotic role—mediated by reduced extracellular pH to amplify TGF-β-driven myofibroblast differentiation—has been previously described(81,82). Cancer cells increase de novo lipogenesis by upregulating enzymes such as ATP-citrate lyase (ACLY) and fatty acid synthase (FASN)(153). Amino acid metabolism reshapes the TME and modulates immune cell function, either enhancing or inhibiting immune surveillance(154). In tumor cells, enzymes associated with glutamine metabolism, such as glutamine synthetase and glutaminase, exhibit elevated expression levels correlated with tumor severity and poor prognosis(155). Glutamine metabolism also impacts immune cell function within the TME, influencing immune surveillance dynamics(156). While the effects of lipid and amino acid metabolic reprogramming on PF have been discussed earlier, these processes collectively accelerate PF development. Their interplay within TME components drives coordinated progression of both tumors and PF.
Thus, during the invasion-metastasis phase, tumor-stromal interactions and metabolic reprogramming collaboratively reshape PFME into a pro-fibrotic, pro-tumor niche.
4.3 Double-Edged Sword Effect of Anti-Tumor Therapy
There are multiple treatment modalities for tumors, but some approaches, such as radiotherapy and chemotherapy, carry an increased risk of exacerbating PF. Radiotherapy remains a cornerstone of definitive (curative) and palliative treatment for many malignancies. Patients receiving definitive radiotherapy typically have locally advanced, surgically unresectable, or medically inoperable malignant tumors. For patients whose final treatment is surgery or chemotherapy, radiotherapy is often used as adjuvant/consolidation therapy. However, chemotherapy as an anti-tumor treatment frequently causes lung injury(157). Approximately 9-30% of cancer patients receiving thoracic tumor radiotherapy develop radiation-induced PF, which may be associated with radiation-induced cellular senescence(158). Radiation-induced cellular senescence is linked to free radical reactions. The presence of oxygen enhances free radical-mediated cellular damage due to the formation of various ROS. Free radicals/ROS disrupt the structure and function of DNA, lipids, and proteins through ionization and/or reactions, leading to metabolic and functional alterations that ultimately result in senescence or cell death(158). Chemotherapy also induces lung injury, with its toxic effects generally dose-dependent(159). The mechanisms of PF caused by chemotherapy are often drug-specific. For example, vincristine promotes the transdifferentiation of fibroblasts into myofibroblasts via the P38 and ERK signaling pathways, thereby inducing PF(160). In cancer patients treated with cisplatin, apoptotic-resistant lung fibroblasts exist, and their anti-apoptotic mechanism is associated with casein kinase 2 (CK2)/X-ray repair complementing defective repair in Chinese hamster cells 1 (XRCC1)-dependent DNA repair activity, contributing to the development and progression of PF(161).
Targeted therapy is also a viable option for tumors but may be linked to PF progression. EGFR mutation-positive lung cancer patients benefit from survival advantages with EGFR-tyrosine kinase inhibitors (EGFR-TKIs), yet EGFR-TKIs can cause lung injury(162)(Table 1). EGFR plays a critical role in epithelial cell maintenance; thus, EGFR-TKIs may impair epithelial cell growth and migration, alter cytokine expression, and induce the recruitment of inflammatory cells, leading to subsequent tissue damage. Suzuki et al. reported that gefitinib treatment enhances fibrosis in bleomycin-induced mice, with the mechanism related to reduced proliferation of regenerative epithelial cells(163). This suggests that EGFR-TKIs should be used cautiously in cancer patients with PF.
Incidence of class-specific adverse events associated with EGFR-TKIs for treatment of NSCLC.
| EGFR-TKIs | n | Incidence of rash (%) | Incidence of Diarrhea (%) | AST/ALT Elevation (%) | Incidence of ILD (%) |
|---|---|---|---|---|---|
| Gefitinib | 580 | 70.0 | 45.5 | 38.3 | 3.4 |
| Erlotinib | 276 | 92.4 | 51.1 | 36.6 | 4.0 |
| Afatinib | 160 | 88.0 | 91.0 | 10.0 | 1.3 |
| Osimertinib | 279 | 58.0 | 58.0 | 7.5 | 4.0 |
ILD: interstitial lung diseases. (Note. Modified from Ohmori et al.(162) (2021) under the terms of the Creative Commons Attribution International License (CC BY 4.0).)
These findings highlight that in the therapeutic intervention phase, radiotherapy, chemotherap or targeted therapy (e.g., EGFR-TKIs) inadvertently exacerbate PFME damage through ROS, cellular senescence, or epithelial injury.
In summary, LC progression impacts the PFME through core mechanisms across three distinct stages. During the tumor growth phase, LC secretes pro-fibrotic factors via Wnt/β-catenin-TGF-β-PI3K/Akt crosstalk to stimulate myofibroblast proliferation. During the invasion-metastasis phase, tumor stromal remodeling and metabolic reprogramming stiffen the matrix and amplify fibrosis. During the therapeutic phase, radiotherapy, chemotherapy, or targeted therapy induce ROS, senescence, or epithelial injury, leading to PFME deterioration. These effects form a feedback loop (Fig. 6). LC-driven PFME remodeling further promotes tumor growth and fibrosis, highlighting the bidirectional crosstalk between LC and PFME. This aligns with the preceding description, where PFME initiates LC, and LC progression conversely exacerbates PFME, jointly driving disease severity.
5. Clinical Significance and Translational Medicine Perspective
5.1 Diagnostic and Prognostic Biomarkers
The diagnostic and prognostic biomarkers for lung cancer and PF are well-defined; given that the interconnections and interactions between PF and lung cancer have been elucidated in the preceding content, their diagnostic and prognostic biomarkers are interrelated (Table 2).
Summary of Key Biomarkers
| Biomarker | Association Type | Associated Diseases | Main Functions/Mechanisms | Clinical Research | Reference |
|---|---|---|---|---|---|
| MMP-7 | Fibrosis-related | lung adenocarcinoma and NSCLC | Secreted proteolytic enzymes degrade ECM components; promote tumor cell invasion, metastasis, and angiogenesis | NSCLC tissues showed a positive expression rate of 68.89% (vs. 14% in normal lung tissues); the iMVD in the positive group was 46.2±6.77 (vs. 30.8±7.54 in the negative group, P<0.001) | (164-166) |
| KL-6 | Fibrosis-related | lung adenocarcinoma and TR-ILD | MUC1, expressed in ATII cells, regulates the stemness and paclitaxel resistance of lung adenocarcinoma through the MUC1-EGFR-NF-κB/MAPK signaling pathway | IPF combined with lung cancer patients exhibit poor prognosis with elevated KL-6; Nintedanib treatment stabilizes KL-6 levels at 24 months | (167-172) |
| CEA | Tumor-related | LC and ILD | CEA family members, with elevated serum levels, are associated with both malignant and non-malignant diseases (e.g., PF) | Serum CEA levels show a downward trend in PF, lung cancer, and other patients; elevated levels are associated with both malignant and non-malignant diseases | (173-175) |
| CYFRA21-1 | Tumor-related | LC and IPF | Alveolar epithelial injury releases products that reflect epithelial damage and repair | Serum CYFRA21-1 levels in IPF patients are associated with disease progression/mortality and localized to proliferative epithelium | (176) |
| PD-L1 | Tumor-related | LC and IPF | Immune suppressor molecules, inhibit T cell activation; upregulated in IPF via EMT-induced PF; PD-1/PD-L1 inhibitor targets | IPF patients exhibit PD-L1 upregulation in fibrotic lung tissue and serum; the PD-L1-high (≥80%) subgroup in the M7824 trial achieved an ORR of 85.7% | (177-179) |
5.1.1 Association of Fibrosis-related Biomarkers with Lung Cancer Prognosis
MMP-7 belongs to fibrosis-related biomarkers and is a secreted proteolytic enzyme that can degrade various ECM components and other substrates, thereby promoting tumor cell invasion and metastasis. Studies have shown that high expression of MMP-7 in lung adenocarcinoma is an independent predictor of the high incidence of air space diffusion in lung adenocarcinoma, and multivariate analysis has revealed that MMP-7 expression is associated with tumor behavior and poor prognosis(164). Compared with non-neoplastic lung tissues, the mRNA expression level of MMP-7 is higher in NSCLC tissues, further suggesting the association between MMP-7 and lung cancer prognosis(165). Statistically, the positive expression rate of MMP-7 in NSCLC tissues is 68.89%, which is significantly higher than 14% in normal lung tissues. Additionally, the mean microvessel density (iMVD) of MMP-7-positive lung cancer tissues is 46.2 ± 6.77, significantly higher than 30.8 ± 7.54 in the negative group (t = 9.641, P < 0.001), indicating that MMP-7 promotes tumor angiogenesis(166).
Krebs von den Lungen-6 (KL-6) plays an important role in various types of ILDs and can be used to assess the fibrotic process and the severity of disease progression(167). KL-6 is a mucin 1 (MUC1) typically expressed on the surface of human ATⅡ cells(168). MUC1 is highly expressed in patients with lung adenocarcinoma and is associated with higher tumor infiltration. Furthermore, MUC1-EGFR collaborates with IL-6 by activating the nuclear factor kappa-B (NF-Κb) and MAPK pathways to regulate the stemness and paclitaxel resistance of lung adenocarcinoma(169). Studies have indicated that serum KL-6 can serve as a biomarker for lung cancer treatment-related interstitial lung disease (TR-ILD), with high KL-6 levels (cut-off value: >436 U/mL) being an independent risk factor for severe TR-ILD(170). Excessively high serum KL-6 concentrations in patients with IPF combined with lung cancer are associated with poor prognosis; after 24 months of Nintedanib treatment, most patients' pulmonary function parameters and serum KL-6 levels tend to stabilize(171). Serum KL-6 levels are significantly elevated in lung cancer patients both with and without ILD. ILD has a significant impact on the prognosis of lung cancer patients. However, in patients without ILD, elevated KL-6 levels may still be associated with poor prognosis(172). This suggests that KL-6 may act as a trans-disease state pan-fibrosis-tumor microenvironment activation biomarker, reflecting prognostic risk independent of the pathological background of ILD.
5.1.2 Association of Tumor-related Biomarkers with PF
Carcinoembryonic antigen (CEA) is an important tumor marker for colorectal cancer and several other malignancies. The human CEA family comprises 29 genes, of which 18 are expressed; 7 belong to the CEA subfamily, and 11 belong to the pregnancy-specific glycoprotein subfamily(173). A study systematically comparing serum CEA levels across patients with different diseases found that serum CEA levels showed a decreasing trend in patients with PF, pancreatic cancer, uremia, chronic obstructive pulmonary disease, colon cancer, Alzheimer's disease, rectal cancer, and lung cancer, indicating that elevated serum CEA levels are associated with both cancer and non-cancerous diseases(174). Serum CEA levels are frequently elevated in patients with interstitial lung disease (ILD), and the risk of cancer development in ILD patients increases with rising serum CEA levels, suggesting an interconnection between CEA and PF(175).
Cytokeratin 19 fragment (CYFRA21-1) is a keratin degradation product released from damaged alveolar epithelium and is primarily used as a tumor marker with significant diagnostic value for lung cancer. A prospective observational study revealed that serum CYFRA21-1 concentrations in IPF patients correlate with disease progression and mortality, and CYFRA21-1 is localized to the proliferative epithelium in IPF lung tissues, indicating that CYFRA21-1 serves as a marker of epithelial damage and renewal, potentially becoming an important biomarker for the prognosis and treatment of IPF patients(176).
Additionally, PD-L1, an immune tumor marker, is an immune inhibitory molecule that suppresses T cell activation, leading to tumor progression(177). One study found that PD-L1 expression is upregulated in the fibrotic lung tissues and serum of IPF patients; this upregulated PD-L1 increases vimentin levels by inhibiting vimentin ubiquitination, stimulates EMT of alveolar epithelial cells, and thereby induces PF(178). In summary, these tumor-related markers may serve as prognostic indicators for PF in the future, potentially improving clinical diagnosis and treatment for patients.
5.2 Treatment Challenges and Strategy Optimization
5.2.1 Efficacy and Safety of Immune Checkpoint Inhibitors in Fibrosis-Lung Cancer Patients
Immune checkpoint inhibitors (ICIs) revitalize antitumor immune responses by disrupting inhibitory immune checkpoint molecules such as PD-1 and cytotoxic T lymphocyte antigen 4 (CTLA-4). Currently, there are two main types of approved immune checkpoint inhibitors: those targeting CTLA-4 and those targeting the PD-1 and its ligand PD-L1 pathway(180). PD-1 can promote PF through the STAT3 pathway, and silencing PD-1 reduces PF(44). PD-L1 increases vimentin levels by inhibiting vimentin ubiquitination, stimulates EMT of alveolar epithelial cells, and thus induces PF; silencing PD-L1 reduces PF(178). Additionally, studies have confirmed the potential benefits of targeting the PD-1/PD-L1 pathway in treating PF and indicated that this pathway is associated with immune regulation(121). This undoubtedly demonstrates the feasibility of using ICIs to treat PF.
Currently, clinical studies on ICIs in patients with fibrosis-lung cancer are relatively limited. In an Asian study, 6 patients with mild idiopathic interstitial pneumonia (IIP)-NSCLC received nivolumab treatment(181). After 12 weeks of treatment, none of the 6 patients experienced drug-related non-hematological grade 3/4 or hematological grade 4 adverse events. Furthermore, no patients developed pneumonia of any grade, and 3 cases achieved partial response. This supports the use of ICIs in patients with mild fibrosis-lung cancer and warrants further exploration and development.
However, while enhancing normal immune responses, ICIs may also amplify the antitumor effects of cellular immunity, leading to immune tolerance imbalance and immune-related adverse events (irAEs). IrAEs can involve all organs throughout the body and are common; data from one study show that more than 80% of patients using ICIs developed irAEs in some systems(182,183). In targeted therapy with ICIs for lung cancer, immune-related pneumonia must be considered. Studies have found that the risk of developing immune checkpoint inhibitor-related pneumonia (CIP) is higher in ILD patients using ICIs compared to those without ILD, although these studies indicate that CIP is not associated with increased mortality(184-186). In summary, in addition to investigating whether ICIs can improve disease progression in IPF patients, future studies should also clarify their role in prognosis.
5.2.2 Potential Anticancer Effects of Antifibrotic Drugs
Currently approved drugs for PF treatment, such as nintedanib and pirfenidone, also exhibit effects in lung cancer. Nintedanib is a multi-target tyrosine kinase inhibitor (TKI) that primarily alleviates inflammation and fibrosis in lung connective tissues by inhibiting multiple signaling pathways, including vascular endothelial growth factor receptor (VEGFR), FGFR, and platelet-derived growth factor receptor (PDGFR). Notably, TKIs are targeted drugs for cancer treatment; thus, nintedanib is also approved for use in combination with docetaxel as an effective second-line option for advanced NSCLC patients(187). In a randomized phase III trial involving 243 chemotherapy-refractory advanced NSCLC patients with IPF, the results showed that the median progression-free survival was 14.6 months in the nintedanib plus chemotherapy group versus 11.8 months in the chemotherapy group, with median progression-free survival times of 6.2 months and 5.5 months, respectively, indicating that combination chemotherapy improves overall survival in NSCLC patients(188).
Pirfenidone is a pleiotropic molecule that inhibits TGF-β, collagen synthesis, and fibroblast proliferation, and mediates tissue repair(189,190). Miuri et al. observed that the incidence of lung cancer in IPF patients treated with pirfenidone was significantly lower than in the non-pirfenidone IPF patient group(191). Additionally, experimental data indicate that the combination of pirfenidone and cisplatin may increase the mortality of CAFs and tumor cells in preclinical models of non-small cell lung cancer(192).
5.2.3 Optimization of Combination Therapy Strategies
Combination therapy of antifibrotic drugs with ICIs is beneficial. Studies have shown that in patients with IPF combined with squamous cell carcinoma, the addition of nintedanib can prevent atezolizumab-induced pneumonia or acute exacerbation of IPF(193). Another study demonstrated that pirfenidone combined with PD-L1 inhibitors in treating lung cancer-fibrosis mouse models significantly delayed tumor growth, reduced PF, and improved mouse survival rates compared to PD-L1 inhibitors alone(194). When using antifibrotic drugs combined with ICIs to treat lung cancer patients with PF, considering antifibrotic therapy followed by immunotherapy may be a favorable option. This is because ECM stiffness can reduce the efficacy of ICIs, whereas antifibrotic drugs can lower ECM stiffness, thereby enhancing the efficiency of T cell infiltration by subsequent ICIs(195).
5.3 Key Therapeutic Targets and Intervention Strategies
5.3.1 Strategies Targeting the Dual-Edged Sword Effect of TGF-β
TGF-β plays a dual role in the fibrotic and lung cancer microenvironments—suppressing inflammation in the early stage and driving fibrosis and immune escape in the late stage(3). Given the pivotal role of TGF-β in fibrosis and tumor progression, along with its complex dual effects, developing precise intervention strategies that can circumvent its detrimental effects while harnessing its beneficial roles represents a critical focus and challenge in current research. TGF-β activates fibroblasts through the canonical Smad pathway, promoting collagen deposition, which is its primary role in PF. In the development and progression of lung cancer, however, TGF-β exhibits a dual-edged sword effect. In the early stages of tumorigenesis, TGF-β acts as a tumor suppressor by inhibiting proliferation and inducing apoptosis(196). However, the immunosuppressive microenvironment regulated by TGF-β can indirectly promote tumor escape(197). For example, TGF-β controls the development and function of the innate immune system by inhibiting NK cells and regulating the proliferation of macrophages, antigen-presenting DCs, and granulocytes. It also suppresses the differentiation of CTLs, Th1, and Th2 cells, and even modulates B cell proliferation and differentiation(198).
Therefore, effective intervention in the TGF-β signaling pathway is crucial for the simultaneous management of lung cancer and PF. Galunisertib, an oral small-molecule inhibitor of TGF-β receptor I (TβRI) kinase, specifically downregulates SMAD2 phosphorylation to inhibit activation of the canonical pathway. However, prolonged continuous exposure to galunisertib leads to cardiotoxicity(199). The Phase Ib/II trial of Galunisertib combined with Nivolumab showed that there were no dose-limiting toxicities in the Phase Ib, and common treatment-related adverse events (TRAEs) in the Phase II NSCLC cohort (n=25) were itching (36%), fatigue (32%), and decreased appetite (28%). There was only one case of Grade 3 immune-related encephalitis, no Grade 4/5 TRAEs, and safety was comparable to monotherapy(200). Furthermore, the median overall survival of this trial was 11.99 months and the median progression-free survival was 5.26 months; compared with which, the efficacy was superior in the TGF-β high-expression group(200). Additionally, some TβRI kinase inhibitors exhibit cardiotoxicity and cutaneous toxicity at high doses(201). Thus, there is a need to identify precise interventions targeting TGF-β and its downstream pathways or multi-targeted drugs.
Second-generation PD-1/PD-L1 inhibitors, such as M7824 and SHR-1701, have been developed to simultaneously block PD-1/PD-L1 and TGF-β/TGF-βR. M7824 is a bifunctional fusion protein composed of a monoclonal antibody against PD-L1 fused to the extracellular domain of human TβRII. It specifically binds to both PD-L1 and TGF-β. Studies show that M7824 inhibits tumor growth and metastasis by activating both the innate and adaptive immune systems, with effects superior to those of anti-PD-L1 antibodies or TGF-β traps alone(202). Furthermore, in vitro and in vivo treatment of NSCLC cells with M7824 reduces TGF-β1-mediated mesenchymal features, including expression of mesenchymal markers, proliferation inhibition, and chemoresistance(203). Another study indicates that SHR-1701 overcomes resistance to PD-1/PD-L1 inhibitors in lung cancer patients caused by lymphocyte recovery disorders(204). A phase I expansion trial has verified the safety and efficacy of M7824. Among 80 patients with advanced NSCLC who had failed platinum-based chemotherapy (and had not previously received immunotherapy), the total objective response rate (ORR) was 21.3%, with an ORR of 25% at the recommended phase II dose (1200 mg). Notably, in the PD-L1-high expression subgroup (≥ 80%), a significant response was achieved (ORR = 85.7%), with a total TRAE rate of 69% and no treatment-related deaths(179). These results demonstrate that M7824 is tolerable and exhibits therapeutic potential in platinum-resistant NSCLC, particularly in patients with high PD-L1 expression.
5.3.2 Matrix Stiffness Regulation: Lysyl Oxidase (LOX) Inhibitors and YAP/TAZ Inhibitors
Matrix stiffness is a core mechanical feature of the fibrotic-lung cancer microenvironment. Targeting ECM cross-linking enzymes (e.g., the LOX family) can reverse pro-tumorigenic mechanical signals. The LOX family comprises five members: LOX, LOXL1, LOXL2, LOXL3, and LOXL4. These are copper-dependent enzymes in the extracellular space that play a key role in ECM cross-linking but also have other intracellular functions associated with fibrosis and carcinogenesis(205). LOX catalyzes collagen/elastin cross-linking, increasing matrix stiffness, which activates YAP/TAZ signaling to promote PF and lung cancer development(7,110,206). LOXL2 promotes tumor cell survival and chemoresistance, regulates cell adhesion, motility, and invasion, and remodels the TME. Growing evidence indicates that high LOXL2 expression correlates with tumor grade, poor prognosis, and reduced survival(207). Thus, the LOX family represents a promising target.
LOX inhibitors have been developed and are undergoing clinical trials. Simtuzumab, a monoclonal antibody against LOXL2, failed to improve lung function in IPF patients when used alone. In a phase II trial comparing simtuzumab with placebo in IPF patients, no difference in progression-free survival was observed between the two groups(208). This may be related to simtuzumab's anti-angiogenic effects but lack of inhibition of LOXL2 catalytic activity, collagen cross-linking, or tissue stiffening(209). Although simtuzumab cannot be used alone for PF treatment, its anti-angiogenic properties suggest potential synergistic effects when combined with nintedanib, though further experiments are required. In contrast, PXS-5505 is a potent inhibitor. PXS-5505 is a novel small-molecule pan-LOX inhibitor. In a bleomycin-induced mouse lung model, PXS-5505 normalized collagen/elastin cross-linking, reducing PF to normal levels(210). Additionally, PXS-5505 decreases chemotherapy-induced connective tissue formation and stiffness in pancreatic cancer, with its mechanism of enhancing chemotherapeutic efficacy linked to reduced matrix stiffness(211). These findings suggest that PXS-5505 can disrupt the ECM barrier to enhance drug efficacy, and it may be combined with PD-1 antibodies or other drugs in the future to synergistically enhance anti-fibrotic and anti-tumor effects.
The role of the YAP/TAZ pathway has been elucidated earlier and it is a potential therapeutic target. Verteporfin is a small-molecule inhibitor of YAP-TEAD that specifically interferes with the formation of the YAP-TEAD protein complex and can significantly suppress YAP-dependent malignant tumor phenotypes(111). Verteporfin inhibits the proliferation of non-small cell lung cancer (NSCLC) cells and induces apoptosis by inhibiting the expression of Anoctamin 1 protein and blocking the EGFR-STAT3 signaling pathway, and it is effective against gefitinib-resistant cells with good safety(212). IAG933 is also a YAP-TEAD inhibitor that demonstrates significant antitumor activity in various cancer models (including lung cancer), especially with better efficacy when combined with EGFR, Kirsten rat sarcoma 2 viral oncogene homolog (KRAS), or MEK inhibitors(213). Although direct clinical trial data on YAP/TAZ inhibitors are relatively limited, the existing preclinical data has demonstrated that this pathway can effectively inhibit cancer.
5.3.3 Synergistic Strategies of Exercise Intervention in Anti-Cancer and Anti-Fibrosis
Recent studies on exercise have consistently demonstrated its role in promoting health and combating cancer. As a non-pharmacological intervention, exercise training can reshape the TME and fibrotic microenvironment through multi-system regulation, exhibiting unique advantages in inhibiting tumor progression and fibrosis. First, in terms of prevention, exercise reduces the risk of lung cancer development; second, it improves patients' quality of life; third, it synergistically enhances the efficacy of other therapies. The biological basis of exercise in lung cancer anti-cancer intervention primarily includes regulating the tumor immune microenvironment, inhibiting tumor angiogenesis, promoting tumor cell apoptosis, and modulating the ECM(214). Its anti-fibrotic effects mainly involve directly inhibiting fibroblast activation, reducing the expression of pro-fibrotic factors such as TGF-β, and slowing collagen deposition(215).
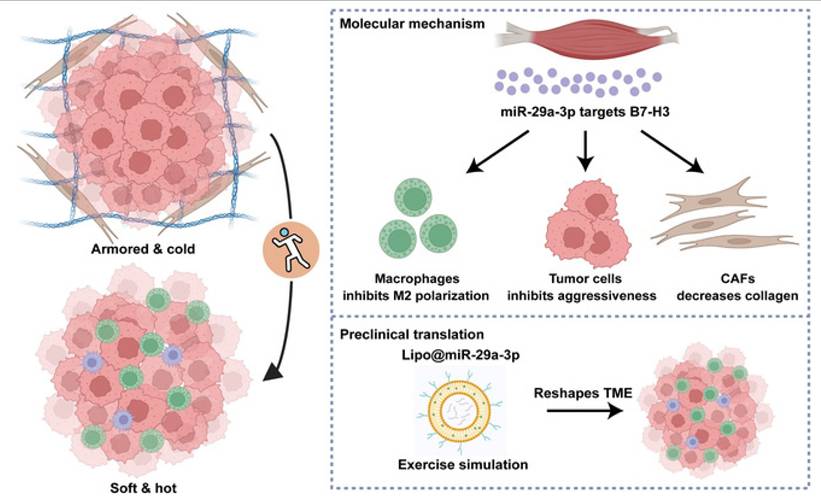
Exercise-induced miR-29a-3p can reduce ECM stiffness, increase T cell infiltration, and enhance immune response to therapy(216). Studies have reported that miR-29a-3p is an exercise-responsive miRNA that inhibits B7 homolog 3 (B7-H3) expression to suppress macrophage M2 polarization, tumor cell invasiveness, and collagen synthesis by fibroblasts. Additionally, this study developed a novel biomaterial, lipo@miR-29a-3p, to mimic the benefits of exercise(217)(Fig. 7). Furthermore, exercise can regulate THSD7B expression, which exhibits positive prognostic implications and tumor-suppressive functions across cancers(218). Objectives, safety, feasibility, and efficacy of exercise training in the context of early-stage and advanced lung cancer have been demonstrated. Exercise training in early-stage lung cancer patients (preoperative and postoperative) and advanced lung cancer patients can improve exercise tolerance, prevent complications, and enhance quality of life(219,220). Studies have also shown that exercise serves as a rehabilitation strategy for PF patients, significantly improving their exercise capacity, lung function, and cardiorespiratory endurance(221). Thus, integrating reasonable exercise into anti-cancer and anti-fibrotic strategies contributes to improved patient prognosis and quality of life.
Schematic overview of the current study. Exercise-responsive miR-29a-3p can make armored and cold tumors turn into soft and hot tumors, which greatly increases its therapeutic significance in clinical practice. Mechanistically, miR-29a-3p targets B7-H3 expression to inhibit tumor cell aggressiveness, macrophage M2 polarization, and collagen synthesis of fibroblasts. Translationally, a novel biomaterial lipo@miR-29a-3p was developed to simulate the benefits of exercise. Copyrighted from the BioRender platform. (Note. From Mei et al.(217) (2025) under the terms of the Creative Commons Attribution International License (CC BY 4.0).).
In summary, the multi-dimensional regulatory roles of targeting TGF-β dual effects, matrix stiffness modulation, and exercise intervention collectively represent a new paradigm for fibrosis-lung cancer treatment. Exercise reshapes the tumor immune microenvironment, inhibits abnormal ECM remodeling, and modulates the tumor metabolic microenvironment, synergizing with targeted strategies. Combined with spatiotemporal selective inhibition, mechanoresponsive drugs, and intelligent combination strategies, these approaches are expected to break through current treatment bottlenecks and achieve a transition from "anti-fibrosis + anti-cancer" linear intervention to "microenvironment reversal + tumor eradication" systemic breakthroughs.
6. Discussion and Future Directions
6.1 Controversies and Unresolved Questions
6.1.1 Does the Fibrotic Microenvironment Directly Induce Lung Cancer, or Act Only as a "Cancer-Promoting Soil"?
There is ongoing debate as to whether the PFME directly induces LC or merely acts as a "cancer-promoting soil." The "direct carcinogenesis" hypothesis is supported by genomic damage and epigenetic drivers. Chronic inflammatory factors in the PFME (e.g., IL-6, TNF-α) and ROS can directly induce TP53 gene mutations and DNA damage in alveolar epithelial cells (32,33,110). In metaplastic and bronchiolar epithelial samples from LC-IPF patients, allelic loss of the candidate tumor suppressor gene fragile histidine triad (FHIT) is more common than in IPF patients without LC(222). Additionally, specific point mutations at codon 12 of the KRAS gene have been detected in LC-IPF patients(223). Interestingly, this mutation has not been identified among the numerous KRAS mutations observed in lung cancer tissues(224). Similar to IPF, miR-21 is overexpressed in LC and serves as an independent negative prognostic factor for overall survival in NSCLC patients(225). These findings support the "direct carcinogenesis" hypothesis.
The "cancer-promoting soil" hypothesis is supported by arguments prioritizing immune escape and contradictory observations. Infiltration of Tregs and M2-type macrophages in the PFME weakens immune surveillance, allowing pre-existing premalignant cell clones to expand and promoting angiogenesis and metastasis in cancer cells(41,42,117). Aberrant PD-1/PD-L1 expression in IPF patients suppresses excessive T cell activation and cytokine secretion to maintain immune tolerance to self-antigens, thereby assisting cancer cells in evading immune surveillance(120,122,123). Furthermore, studies have found that due to promoter hypermethylation, the relative expression of SMAD4 (a known tumor suppressor gene) in LC tissues is significantly lower in LC-IPF patients compared to LC patients without IPF(226,227). These studies suggest that the microenvironment may primarily promote the survival of mutated cells rather than directly inducing mutations.
Of course, it is also possible that both hypotheses coexist, with one predominating under certain conditions. Future research is needed to resolve this controversy to better understand the link between PF and LC and improve patient care. Future studies could construct humanized PF organoid models to clarify the oncogenic contributions of microenvironmental components via genetic editing (e.g., clustered regularly interspaced short palindromic repeats (CRISPR) knockout of ROS-generating enzymes). Clinically, longitudinal tracking of free DNA (cfDNA) mutation dynamics in the bronchoalveolar lavage fluid of PF patients could correlate with the timing of lung cancer onset. If direct carcinogenesis dominates, early genetic intervention strategies (e.g., antioxidants) should be developed; if the "cancer-promoting soil" hypothesis prevails, enhanced immune monitoring (e.g., regular assessment of CD8+ T cell function) should be prioritized.
6.1.2 Heterogeneity of Lung Cancer Impact Across Different Types of Fibrosis
Different types of fibrosis exert distinct effects on lung cancer, primarily including IPF, autoimmune-related PF, and occupational pneumoconiosis. IPF is a chronic, progressive, fibrotic interstitial lung disease with lesions confined to the lungs, predominantly affecting middle-aged and elderly individuals. The prevalence of LC in IPF patients ranges from 2.7% to 48%, significantly higher than in the general population(134) (Table 3). As previously discussed, IPF pathogenesis is primarily driven by TGF-β/Smad signaling, high ECM stiffness, and hypoxia. Autoimmune-related PF, exemplified by rheumatoid arthritis-associated interstitial lung disease (RA-ILD), is a PF linked to autoimmune processes. Studies indicate that RA is associated with a >50% increased risk of lung cancer, while RA-ILD patients represent a particularly high-risk group with a ~3-fold increased risk(228). RA-ILD may arise from immune recognition of post-translationally modified proteins (induced by smoking or other lung injuries), leading to secondary joint disease characteristic of RA(229). Additionally, RA-ILD results from environmental-genetic-immune interactions that expose modified antigens, generate anti-citrullinated protein antibodies (ACPA), and drive chronic inflammation, ultimately progressing to usual interstitial pneumonia (UIP)-like pathological changes(230). Pneumoconiosis refers to a group of lung diseases caused by inhalation of respirable particles (typically < 5 μm in diameter) reaching the terminal airways and alveoli, with silicosis as a representative example. Silicosis is an occupational lung fibrosis characterized by diffuse nodular PF caused by long-term inhalation of free silica dust in occupational settings. Studies have reported a correlation between silicosis and increased lung cancer risk, with an odds ratio (OR) of 1.47(24). The pathophysiology of silicosis involves silica particle deposition in alveoli. Macrophage phagocytosis of these particles triggers inflammation, stimulates fibroblast proliferation, and collagen production; additionally, silicosis is associated with autoimmunity(231).
Prevalence of LC in IPF patients.
| Study | Number of IPF patients | Number of Patients with LC-IPF | Prevalance of LC (%) |
|---|---|---|---|
| Nagai | 99 | 31 | 31.3 |
| Matsusitha | 20 | ND | 48.2 |
| Park | 281 | 63 | 22.4 |
| Le Jeune | 1064 | 29 | 2.7 |
| Ozawa | 103 | 21 | 20.4 |
| Lee | 1685 | 70 | 6.8 |
| Kreuter | 265 | 42 | 16 |
| Tomasetti | 181 | 23 | 13 |
| Yoon | 1108 | 27 | 2.8 |
| Kato | 632 | 70 | 11.1 |
ND: not determined. (Note. From Ballester et al.(134) (2019) under the terms of the Creative Commons Attribution International License (CC BY 4.0).)
Given the distinct mechanisms of different fibrosis types, do differences in immune cell infiltration patterns lead to variations in pro-tumorigenic factors? Do the physical properties of particles in occupational PF influence lung cancer subtypes? Furthermore, do these differences impact clinical management? For example, anti-fibrotic drugs (e.g., nintedanib) are effective for IPF-associated LC but may exacerbate immune imbalance in autoimmune PF. Addressing these questions may require multi-omics comparisons and organoid model construction. Mechanistically, spatial transcriptomic sequencing of LC tissues from IPF, RA-ILD, and pneumoconiosis patients could identify subtype-specific pathways. Clinically, disease-specific PF-LC co-culture systems could screen for type-dependent therapeutic targets, facilitating the development of subtype-specific monitoring guidelines.
6.1.3 Impact of Spatiotemporal Heterogeneity of the Fibrotic Microenvironment on Lung Cancer
The PFME is spatiotemporally heterogeneous, with molecular and cellular compositions varying across regions (spatial) and disease stages (temporal), forming a dynamically evolving pathological niche. Spatially, the fibrotic region can be divided into an active fibrotic frontier (activated fibroblasts with high collagen triple helix repeat containing 1 (CTHRC1)/FAP expression) and an end-stage fibrotic zone (extensive collagen deposition with low cell activity). Temporally, the progression spans from alveolar epithelial injury (loss of ATI cells, proliferation of ATII cells) to fibroblast activation, macrophage phenotype switching (fatty acid binding protein 4 (FABP4)+ to secreted phosphoprotein 1 (SPP1)+), and finally end-stage fibrosis(232).
Mechanisms by which PFME spatiotemporal heterogeneity impacts LC development include EMT, immune microenvironment remodeling, and molecular gradient formation. Keratin 5 (KRT5) -/KRT17+ abnormal basal-like cells spatially co-localize with activated fibroblasts, promoting MMP7 and collagen, type I, alpha 1 (COL1A1) expression(233-235). This enhances ECM remodeling, providing a physical scaffold for LC invasion and promoting EMT. SPP1+ macrophages are enriched during fibrotic progression, potentially activating pro-fibrotic and immunosuppressive pathways via TGF-β(236,237), thereby inhibiting anti-tumor immunity and promoting LC immune escape. Pseudo-time trajectory analysis of alveolar regions from homeostasis to end-stage fibrosis reveals dynamic changes in transcription factors such as SRY-related high-mobility-group box 4 (SOX4)/SOX9(235,238), which drive the maintenance of LC stem cell properties and enhance chemoresistance.
In summary, spatially, the active frontier zone primarily promotes invasion (via increased EMT and MMP production), while the end-stage zone primarily promotes hypoxia adaptation (via increased HIF-α factor production and angiogenesis). Temporally, early stages are driven by epithelial injury and clonal selection (KRAS/TP53 mutations), whereas late stages are dominated by immune suppression promoting metastasis (enrichment of SPP1+ macrophages)(232). Could this spatiotemporal heterogeneity lead to differences in treatment response? From a spatiotemporal perspective, early-stage therapy should prioritize anti-fibrosis (e.g., nintedanib), mid-stage therapy should target fibroblasts (e.g., FAP inhibitors), and late-stage therapy should focus on immune combination approaches (e.g., anti-PD-L1 + anti-SPP1 drugs). Additionally, matrix degradation strategies (e.g., LOX inhibitors) should be considered in advanced stages. Future studies could utilize spatial multi-omics and dynamic imaging to analyze PFME spatiotemporal heterogeneity and develop location-targeted drug delivery systems.
In conclusion, resolving these controversies will advance PF-LC research from "phenomenological association" to "mechanistic intervention," providing a theoretical framework for personalized prevention and treatment.
6.2 Technological Drivers of Research Opportunities
Emerging technologies are redefining the research paradigm of pulmonary fibrosis-lung cancer microenvironment (PF-LCME) studies, transitioning from single-cell resolution to spatiotemporal dynamic dimensions and providing revolutionary tools for dissecting complex interaction networks.
6.2.1 Single-Cell Sequencing and Spatial Multi-Omics
Single-cell sequencing and spatial multi-omics are currently playing pivotal roles. Single-cell multi-omics aids in resolving cellular heterogeneity. Single-cell RNA sequencing (scRNA-seq) has identified three distinct epithelial cell subtype populations in IPF, characterized by conducting airway basal cells and goblet cells, as well as additional atypical transitional cells that contribute to pathological processes in IPF(63). Additionally, scRNA-seq has identified a highly enriched KRT5⁻/KRT17⁺ pathologically ECM-producing epithelial cell population in PF lungs(233). Furthermore, single-cell analysis has revealed the role of FAP+ CAF (also called CAF-S1) in immunotherapy resistance(148). Single-cell atlas analysis has identified distinct cell types in advanced NSCLC, with significant heterogeneity across patients in terms of cell composition, chromosomal structure, developmental trajectories, intercellular signaling networks, and phenotypic dominance(239). Single-cell sequencing of LC and PF patients can determine potential intercellular interactions between the two conditions.
Spatial multi-omics facilitates the localization of interaction hotspots. Spatial transcriptomics has uncovered dysregulated molecular niches associated with distal lung remodeling in PF, informing other spatial transcriptomic studies(232). One study combined spatial transcriptomics and scRNA-seq to reveal that spatially and transcriptionally distinct fibroblast lineages converge into transcriptionally consistent myofibroblasts, identifying secreted frizzled-related protein 1 (SFRP1) as a regulator of TGF-β1-driven fibroblast phenotypes during fibrogenesis(240). This advances the development of therapeutic interventions to limit or reverse fibroblast focus formation. Future applications of spatial transcriptomics, spatial proteomics, and spatial metabolomics could reveal a ternary coupling of collagen deposition-lactate metabolism-immune suppression in fibrotic regions.
6.2.2 Organoids and Co-Culture Models
Organoids are miniature organ models formed by self-organization of stem cells (e.g., pluripotent stem cells, adult stem cells) or tissue-derived cells under three-dimensional culture conditions, exhibiting structural and functional features similar to genuine organs. They are primarily applied in disease modeling, drug screening, and regenerative medicine. Co-culture involves culturing two or more cell types in the same system to simulate intercellular interactions (e.g., paracrine signaling, physical contact), with key applications in TME simulation, neural circuit construction, and host-pathogen interactions. The combined use of these two approaches is currently driving the development of high-fidelity disease models and precision medicine.
In PF-related research, Yao et al. analyzed interactions between primary fibroblasts and ATⅡ cells in organoid models(241). They found that mucin 5B (MUC5B) was associated with ATⅡ cells in IPF patient samples. Their model demonstrated that fibrotic primary fibroblasts induce impaired differentiation of AT2 cells via the STAT3 signaling pathway, a phenomenon also observed in IPF patients. This model can be used for mechanistic studies and drug development. In cancer-related research, Esther et al. used co-cultures of colon cancer cells and CAFs to recapitulate the mesenchymal-like invasive characteristics of colorectal cancer(242), aiding in understanding the relationship between CAFs and cancer cell invasiveness.
To better understand PF-LC interactions, future studies could use organoids and co-culture models to simulate the fibrosis-tumor interface. For example, constructing three-dimensional fibrosis-tumor organoids by co-culturing IPF patient-derived lung fibroblasts with lung cancer cells to recapitulate ECM stiffness and hypoxia gradients. Alternatively, developing fibrosis-tumor co-culture systems on chips to parallel-test 120 drug combinations for high-throughput drug screening.
6.2.3 Artificial Intelligence (AI) Predictive Models
The advent of AI has brought numerous conveniences to scientific research. AI applications in medicine primarily branch into two categories: virtual and physical. The virtual branch includes informatics methods ranging from deep learning for information management to health management system control, as well as tools that actively guide clinicians in treatment decisions. The physical branch is best represented by robots that assist elderly patients or attending physicians, including nanoscale targeted robots—an innovative drug delivery system(243).
High-resolution computed tomography (HRCT), a more sensitive imaging technique, is considered a core diagnostic tool for interstitial lung diseases(244). Park et al. used AI to analyze HRCT lung segmentation in 647 ILD patients, achieving an accuracy rate of 98%(245). Two studies demonstrated excellent performance in IPF diagnosis, with diagnostic accuracy approaching expert levels (78.9%)(246,247). Additionally, Ren et al. used predictive AI methods to identify TRAF2 and NCK interacting kinase (TNIK) as an anti-fibrotic target and generated a small-molecule TNIK inhibitor, INS018_055(248). AI also excels in lung cancer diagnosis and prognosis, with various models achieving diagnostic accuracy rates exceeding 70%, underscoring its value in these areas(249).
Future applications of AI could involve analyzing HRCT images to extract fibrotic region texture features and constructing lung cancer risk models, using deep learning algorithms to automatically segment honeycombing regions and predict canceration probability. Furthermore, AI might integrate multi-omics data—for example, combining single-cell data, spatial transcriptomics, and clinical information to build heterogeneous networks, identify key driver nodes, and construct graph neural networks. AI could even dynamically predict changes in serum or other biomarkers to adjust treatment regimens.
In summary, the synergistic application of single-cell and spatial omics, organoid models, multi-omics integration, and AI is progressively unraveling the "black box" of the PF-LC microenvironment. Future efforts should focus on technological standardization, clinical translation validation, and ethical norms to advance these tools from the laboratory to personalized diagnostics and therapeutics, ultimately improving patient outcomes.
6.3 Translational Medicine Challenges
6.3.1 Balancing the Conflicting Demands of Anti-Fibrotic and Anti-Tumor Therapies
The cross-application of anti-fibrotic therapy and anti-tumor therapy has emerged as a core direction in the comprehensive management of tumors in recent years. However, their complex interactions—the coexistence of synergy and antagonism—pose a key challenge in clinical practice. This dual characteristic necessitates breakthroughs in three dimensions: mechanistic elucidation, toxicity control, and strategy optimization—to drive the transformation of combined therapy from "empirical attempts" to "precision-designed" approaches.
The interaction between anti-fibrotic therapy and anti-tumor therapy is essentially a dynamic game involving multiple signaling pathways and cell types within the TME, with its efficacy highly dependent on the functional positioning and temporal characteristics of therapeutic targets in the TME. TGF-β acts as a "double-edged sword" in the TME: in the early stages of tumorigenesis, it exerts anti-tumor effects by inhibiting proliferation, inducing apoptosis, and suppressing cell immortalization; however, as the tumor progresses, its anti-tumor effects gradually diminish, and it instead promotes tumor deterioration through mechanisms such as EMT, migration, and immune suppression(250). Consequently, while TGF-β inhibitors reduce fibrosis, they may also weaken the "natural anti-tumor" function of TGF-β in early-stage tumors. For example, in hepatocellular carcinoma models, TGF-β inhibitors, while alleviating fibrosis, may abrogate the early anti-tumor inhibitory effects of TGF-β, leading to rapid tumor progression(196). This contradiction highlights that the clinical application of TGF-β inhibitors must be strictly restricted to "progressive tumors dominated by fibrosis" rather than early-stage or low-fibrosis tumors. Anti-angiogenic drugs reduce abnormal tumor angiogenesis by inhibiting VEGF signaling. Theoretically, they can improve drug delivery efficiency and enhance immune cell infiltration through "vascular normalization." However, excessive inhibition of VEGF damages normal vascular endothelial cells, inducing pulmonary hypoperfusion and chronic ischemia, which exacerbates tumor hypoxia, promotes EMT, and drives metastasis—as observed with bevacizumab(251).
The combined application of anti-fibrotic and anti-tumor therapies requires attention to additive toxicity, which necessitates risk reduction through multi-dimensional strategies. ICIs exert anti-tumor effects by relieving T cell suppression, but their activation of the immune system may also attack normal tissues, inducing irAEs. Although studies have suggested that pirfenidone combined with ICIs is safe in patients with IPF and NSCLC, only four reported cases cannot fully confirm its safety(252). The optimal approach is to evaluate the patient's pulmonary immune microenvironment status using multi-parameter biomarkers (e.g., serum KL-6, IL-6 levels in bronchoalveolar lavage fluid) before combination therapy to avoid a "1+1>2" toxic effect.
Optimizing the sequence of anti-fibrotic and anti-tumor therapies requires a foundation in the dynamic evolutionary characteristics of the TME, with the core focus being precise control over "when to intervene," "how to switch," and "how long to maintain." For patients with high fibrosis severity and tumors in a relatively quiescent stage, anti-fibrotic therapy should be prioritized. Anti-fibrotic drugs reduce ECM stiffness, minimize abnormal vascular leakage, and thereby improve subsequent anti-tumor efficacy. For "dual-driver" patients where fibrosis and tumor progression coexist, alternating use of anti-fibrotic and anti-tumor drugs can avoid long-term cumulative toxicity from single-agent therapy. The timing window for treatment sequencing must be adjusted based on the real-time status of the TME. Dynamic monitoring via imaging techniques (e.g., multi-organ ultrasound elastography to assess ECM stiffness) or molecular biomarkers (e.g., serum hyaluronic acid, fibroblast growth factor) is necessary to prevent loss of efficacy due to premature treatment switching.
The interaction between anti-fibrotic and anti-tumor therapies is essentially a microcosm of TME complexity. Their clinical application demands breaking through the linear "either-or" mindset and shifting to a "dynamic balance" systems perspective. In the future, with advancements in technologies such as single-cell sequencing and spatial transcriptomics, we aim to more precisely parse the interaction networks of various cell types in the TME. Combined with AI-driven treatment response prediction models, this will ultimately enable "personalized" precision combination therapy, representing not only a breakthrough direction for anti-tumor treatment but also the best practice of the translational medicine concept "from bench to bedside."
6.3.2 Organ-Specific Drug Delivery Systems
Targeted delivery to the lungs is a key strategy to mitigate systemic toxicity and improve therapeutic efficacy. Cutting-edge technologies include nanoparticle-based targeted delivery, responsive smart delivery, and inhaled local delivery.
Progress has been made in nanoparticle technology for PF. Gold nanoparticles loaded with imatinib significantly enhance the anti-fibrotic efficacy of imatinib, inhibiting fibroblast and macrophage proliferation(253). Studies have shown that intratracheal administration of pirfenidone nanoparticles results in significantly higher lung pirfenidone levels compared to pirfenidone solution. Nanoparticles maintain sustained delivery of pirfenidone in the lungs and enhance its anti-fibrotic effects(254). Advances in nanoparticle targeting for LC have also been reported. A nanostructured lipid nanocarrier-based system (NLCS) has been proposed for efficient tumor-targeted local delivery of anti-tumor drugs and specific siRNA mixtures, specifically targeting lung cancer cells to effectively inhibit tumor growth and prevent adverse effects on healthy organs (255).
Additionally, progress has been made in responsive smart delivery systems. Wang et al. developed a gold nanoparticle-based smart responsive delivery system that precisely releases paclitaxel in the weakly acidic tumor microenvironment, reducing damage to normal tissues and side effects(256). Li et al. developed a folic acid-modified ultrasound-responsive drug delivery system for delivering siRNA encoding signal regulatory protein alpha (SIRPα) mRNA and immune adjuvant Fe₃O₄ nanoparticles(257). Under ultrasound conditions, nanobubbles effectively transfect macrophages, inhibiting SIRPα mRNA and protein expression, promoting macrophage phagocytosis, and synergistically reversing M2 polarization to enhance immunotherapy in NSCLC.
Currently, inhaled local delivery methods such as dry powder inhalation and aerosol gels are less commonly used in PF and LC. However, given that inhaled administration delivers drugs directly to the lungs with rapid absorption, quick onset, no first-pass hepatic metabolism, and minimal side effects, future research could explore the role of inhaled local delivery systems in PF and LC.
Balancing anti-fibrotic and anti-tumor therapies requires overcoming the dual challenges of mechanism interaction and toxicity management, and organ-specific delivery systems provide revolutionary tools for this purpose. Future efforts should focus on precise sequential therapy, smart responsive carriers, and clinical translation standards to drive a paradigm shift from "conflicting therapies" to "synergistic eradication."
7. Conclusion
The interaction between the PFME and LC is a multidimensional, dynamically evolving vicious cycle. This review systematically elucidates its core mechanisms: the fibrotic microenvironment drives lung cancer initiation through chronic inflammation, genomic mutations, epigenetic reprogramming, mechanical signaling, immune suppression, and stem cell niches; conversely, tumor cells promote fibrosis progression by secreting pro-fibrotic factors, remodeling the ECM, and reprogramming metabolism. This bidirectional interaction not only explains the high prevalence and poor prognosis of their co-occurrence but also provides novel perspectives for targeted therapy.
Interdisciplinary research is critical to breaking through current therapeutic bottlenecks. Single-cell and spatial multi-omics technologies have revealed the cellular interaction networks within the microenvironment; organoid and organ-on-a-chip models have enabled dynamic simulation of pathological interfaces; and AI-driven multi-modal data integration is driving innovation in personalized treatment strategies. However, translational medicine still faces significant challenges: How to balance the conflicting effects of anti-fibrotic and anti-tumor therapies? How to achieve organ-selective drug delivery? Resolving these issues requires deep integration of bioengineering, computational science, and clinical medicine.
Looking to the future, precision medicine strategies should focus on the following directions, with deeper integration of mechanistic insights and technological innovations to address the complexity of fibrosis-lung cancer crosstalk:
First, real-time microenvironment monitoring and spatiotemporal dynamic intervention: Beyond developing multi-target mechanically responsive nanodrugs, the next step involves applying in vivo imaging techniques to dynamically track the fibrosis-tumor interface and determine optimal drug administration timing. For example, the best time to administer inhibitors is when both ECM stiffness and myofibroblast activation reach thresholds that promote tumor growth; continuous biopsies combined with scRNA-seq to observe temporal changes in cell states can dynamically adjust treatment regimens. If scRNA-seq reveals a surge in pro-inflammatory macrophages during anti-fibrotic therapy, combining it with colony-stimulating factor-1 receptor (CSF-1R) inhibitors to suppress this immunosuppressive subset can enhance efficacy.
Second, multi-omics-guided predictive stratified therapy: After molecular classification of "fibrosis-lung cancer" based on spatial transcriptomic and radiomic features, proteomics and metabolomics data can further refine treatment strategies for different subtypes. Specifically, tumors with a "sclerotic" microenvironment (characterized by LOXL2 expression and low hyaluronan levels) may be more sensitive to combined LOXL2 inhibition and hyaluronidase therapy, while "inflammatory" subtypes (with elevated IL-6 and TNF-α levels) may respond better to anti-fibrotic therapies.
Third, intelligent treatment systems with closed-loop optimization: A reinforcement learning model should be developed and trained to integrate multi-dimensional patient data—including clinical parameters, molecular features, and real-time physiological signals—to dynamically adjust combinations of anti-fibrotic and immunotherapy. For instance, if LOXL2 inhibition induces pneumonia, the model can intelligently recommend reducing anti-PD-1 dosage to balance efficacy and toxicity. These systems will also account for inter-patient variability; for example, a smoker with EGFR mutations may require a different immunotherapy backbone compared to a non-smoker, a decision guided by predictive models trained on large-scale multi-omics datasets.
Fourth, targeting the stem cell-niche axis to disrupt fibrosis-tumor crosstalk: ATII stem cells and their derived microenvironment are critical drivers of both fibrosis and cancer. Future interventions may include: (1) Niche disruption: Using antibody-drug conjugates targeting ATII cell surface markers (e.g., surfactant protein C) to selectively deplete cancer-prone stem cells while sparing normal progenitors; (2) Epigenetic reprogramming: Applying small-molecule inhibitors of histone methyltransferases or DNA methyltransferases to reverse abnormal gene expression caused by excessive ROS; (3) Metabolic reprogramming: Blocking glutamine metabolism in ATII-derived fibroblasts to starve the TME of energy required for both fibrosis progression and tumor growth.
Fifth, overcoming treatment resistance through combinatorial innovation: Beyond sequential anti-fibrotic and anti-tumor therapies, exploring synergistic combinations is key. Examples include: (1) Immunotherapy + anti-fibrosis: PD-1 inhibitors may fail in fibrotic tumors due to physical exclusion by dense ECM; co-administration with collagen-degrading enzymes or MMP activators can degrade ECM components (e.g., collagen), enhancing immune cell infiltration; (2) Targeted therapy + metabolic reprogramming: EGFR inhibitors (e.g., gefitinib) can be paired with PFKFB3 inhibitors to block aerobic glycolysis in both tumor cells and CAFs, depriving tumors of metabolic support; (3) Radiotherapy + fibrosis reversal: Hypo-fractionated radiotherapy can induce immunogenic cell death in tumor cells, while low-dose anti-TGF-β antibodies mitigate radiation-induced lung fibrosis.
Sixth, improving prognosis and quality of life through exercise: In addition to conventional treatments, reasonable exercise helps improve lung function. Exercise needs to be designed with individualized regimens. For example, for patients with poor physical performance status, exercise can start with low-intensity resistance training; patients with severe pulmonary fibrosis need to avoid high-intensity interval training to prevent hypoxemia exacerbation caused by hyperventilation; meanwhile, real-time monitoring technologies can be combined to dynamically adjust exercise regimens, ensuring that patients are not harmed. Additionally, the combined application of exercise with existing therapies (e.g., anti-fibrotic drugs,ICIs) can be further explored in the future. For instance, whether pre-exercise conditioning can enhance the tumor infiltration rate of PD-1 inhibitors, or whether the combination of exercise and MMP activators can accelerate ECM remodeling to improve drug delivery efficiency, providing a scientific basis for integrating "exercise prescriptions" into precision oncology management.
Finally, promoting clinical research progress using human-relevant models: To accelerate clinical translation, organoids and microphysiological systems should recapitulate the spatiotemporal dynamic evolution of fibrosis-lung cancer crosstalk. For example, "fibroblast-tumor organoids" co-cultured with patient-derived ECM and immune cells can simulate the transition from fibrosis to malignancy and test combinatorial therapies in a context-dependent manner.
In summary, unraveling the fibrosis-lung cancer vicious cycle demands a holistic approach that integrates real-time microenvironmental insights, multi-omics-driven personalization, intelligent therapeutic decision-making, and innovative niche-targeted strategies. Only through such deep mechanistic understanding and technological synergy can we transform the paradigm from managing co-morbid disease to achieving genuine "radical reversal"—restoring normal lung architecture, eliminating malignant clones, and preventing recurrence.
Acknowledgements
Funding
This work was financially supported by National Natural Science Foundation of China (82102634), National Natural Science Foundation of China (82474261), National Natural Science Foundation of China (82505803), Health Shanghai Initiative Special Fund (Medical-Sports Integration, JKSHZX-2022-02), Shenzhen “San-Ming” Project of Medicine (SZSM202211019), Fund of Fudan University - Dr. Kong Joint Research Center for Sports Medicine and Health Footwear (250025HZ010).
Author contributions
Zhufeng Hu#, Wu Dan#, and Mengran Xi# contributed equally to this work.
- Conceptualization: Zhiwen Luo, Baojun Liu, Zhang Ting
- Methodology: Zhufeng Hu, Wu Dan, Mengran Xi
- Investigation: Zhengyuan Fang, Kunlun Feng, Jie Mei
- Data Curation: Zhufeng Hu, Wu Dan, Mengran Xi
- Formal Analysis: Zhengyuan Fang, Jie Mei
- Visualization: Zhufeng Hu, Mengran Xi
- Resources: Kunlun Feng, Baojun Liu, Zhang Ting
- Writing - Original Draft: Zhufeng Hu, Wu Dan, Mengran Xi
- Writing - Review & Editing: Zhiwen Luo, Baojun Liu, Zhang Ting
- Supervision: Zhiwen Luo, Baojun Liu, Zhang Ting
- Project Administration: Zhiwen Luo, Baojun Liu
- Funding Acquisition: Zhiwen Luo, Baojun Liu, Zhang Ting
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kinoshita T, Goto T. Molecular Mechanisms of Pulmonary Fibrogenesis and Its Progression to Lung Cancer: A Review. Int J Mol Sci. 2019Mar22;20(6):1461
2. Fletcher CD. Myofibroblastic tumours: an update. Verh Dtsch Ges Pathol. 1998;82:75-82
3. Peng D, Fu M, Wang M, Wei Y, Wei X. Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol Cancer. 2022Apr23;21(1):104
4. Wang L, Cao L, Wang H, Liu B, Zhang Q, Meng Z. et al. Cancer-associated fibroblasts enhance metastatic potential of lung cancer cells through IL-6/STAT3 signaling pathway. Oncotarget. 2017Sept29;8(44):76116-28
5. Roberts AB, Wakefield LM. The two faces of transforming growth factor beta in carcinogenesis. Proc Natl Acad Sci U S A. 2003July22;100(15):8621-3
6. Dupont S, Morsut L, Aragona M, Enzo E, Giulitti S, Cordenonsi M. et al. Role of YAP/TAZ in mechanotransduction. Nature. 2011June8;474(7350):179-83
7. Liu F, Lagares D, Choi KM, Stopfer L, Marinković A, Vrbanac V. et al. Mechanosignaling through YAP and TAZ drives fibroblast activation and fibrosis. Am J Physiol Lung Cell Mol Physiol. 2015Feb15;308(4):L344-357
8. Noguchi S, Saito A, Mikami Y, Urushiyama H, Horie M, Matsuzaki H. et al. TAZ contributes to pulmonary fibrosis by activating profibrotic functions of lung fibroblasts. Sci Rep. 2017Feb14;7:42595
9. Thrash HL, Pendergast AM. Multi-Functional Regulation by YAP/TAZ Signaling Networks in Tumor Progression and Metastasis. Cancers (Basel). 2023Sept24;15(19):4701
10. Warren R, Lyu H, Klinkhammer K, De Langhe SP. Hippo signaling impairs alveolar epithelial regeneration in pulmonary fibrosis. Elife. 2023May11;12:e85092
11. Hu HH, Chen DQ, Wang YN, Feng YL, Cao G, Vaziri ND. et al. New insights into TGF-β/Smad signaling in tissue fibrosis. Chem Biol Interact. 2018Aug25;292:76-83
12. Bommireddy R, Doetschman T. TGFbeta1 and Treg cells: alliance for tolerance. Trends Mol Med. 2007Nov;13(11):492-501
13. Tang PMK, Nikolic-Paterson DJ, Lan HY. Macrophages: versatile players in renal inflammation and fibrosis. Nat Rev Nephrol. 2019Mar;15(3):144-58
14. Bhattacharya M, Ramachandran P. Immunology of human fibrosis. Nat Immunol. 2023Sept;24(9):1423-33
15. Zhou BW, Liu HM, Xu F, Jia XH. The role of macrophage polarization and cellular crosstalk in the pulmonary fibrotic microenvironment: a review. Cell Commun Signal. 2024Mar9;22(1):172
16. Koudstaal T, Funke-Chambour M, Kreuter M, Molyneaux PL, Wijsenbeek MS. Pulmonary fibrosis: from pathogenesis to clinical decision-making. Trends Mol Med. 2023Dec;29(12):1076-87
17. Li X, Cai H, Cai Y, Zhang Q, Ding Y, Zhuang Q. Investigation of a Hypoxia-Immune-Related Microenvironment Gene Signature and Prediction Model for Idiopathic Pulmonary Fibrosis. Front Immunol. 2021;12:629854
18. Yang L, Gilbertsen A, Xia H, Benyumov A, Smith K, Herrera J. et al. Hypoxia enhances IPF mesenchymal progenitor cell fibrogenicity via the lactate/GPR81/HIF1α pathway. JCI Insight. 2023Feb22;8(4):e163820
19. Rashid M, Zadeh LR, Baradaran B, Molavi O, Ghesmati Z, Sabzichi M. et al. Up-down regulation of HIF-1α in cancer progression. Gene. 2021Sept25;798:145796
20. McKinney EF, Lee JC, Jayne DRW, Lyons PA, Smith KGC. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature. 2015July30;523(7562):612-6
21. Siegel RL, Giaquinto AN, Jemal A. Cancer statistics, 2024. CA Cancer J Clin. 2024;74(1):12-49
22. Karampitsakos T, Sampsonas F, Herazo-Maya JD, Tzouvelekis A. Management of patients with idiopathic pulmonary fibrosis and lung cancer: challenges in clinical practice. Curr Opin Pulm Med. 2023Sept1;29(5):416-26
23. Sun X, Lei S, Zhao H, Guo L, Wang Y. Mortality-related risk factors of idiopathic pulmonary fibrosis: a systematic review and meta-analysis. J Thorac Dis. 2024Dec31;16(12):8338-49
24. Krabbe J, Steffens KM, Drießen S, Kraus T. Lung cancer risk and occupational pulmonary fibrosis: systematic review and meta-analysis. Eur Respir Rev. 2024Jan31;33(171):230224
25. Ozawa Y, Suda T, Naito T, Enomoto N, Hashimoto D, Fujisawa T. et al. Cumulative incidence of and predictive factors for lung cancer in IPF. Respirology. 2009July;14(5):723-8
26. Brown SAW, Dobelle M, Padilla M, Agovino M, Wisnivesky JP, Hashim D. et al. Idiopathic Pulmonary Fibrosis and Lung Cancer. A Systematic Review and Meta-analysis. Ann Am Thorac Soc. 2019Aug;16(8):1041-51
27. Karampitsakos T, Spagnolo P, Mogulkoc N, Wuyts WA, Tomassetti S, Bendstrup E. et al. Lung cancer in patients with idiopathic pulmonary fibrosis: A retrospective multicentre study in Europe. Respirology. 2023Jan;28(1):56-65
28. Chen L, Li S, Li W. LOX/LOXL in pulmonary fibrosis: potential therapeutic targets. J Drug Target. 2019Aug;27(7):790-6
29. Cano A, Eraso P, Mazón MJ, Portillo F. LOXL2 in Cancer: A Two-Decade Perspective. Int J Mol Sci. 2023Sept21;24(18):14405
30. Ihn H. Pathogenesis of fibrosis: role of TGF-beta and CTGF. Curr Opin Rheumatol. 2002Nov;14(6):681-5
31. Chu CY, Chang CC, Prakash E, Kuo ML. Connective tissue growth factor (CTGF) and cancer progression. J Biomed Sci. 2008Nov;15(6):675-85
32. Vancheri C, Failla M, Crimi N, Raghu G. Idiopathic pulmonary fibrosis: a disease with similarities and links to cancer biology. Eur Respir J. 2010Mar;35(3):496-504
33. Xing Y, Wei X, Liu Y, Wang MM, Sui Z, Wang X. et al. Autophagy inhibition mediated by MCOLN1/TRPML1 suppresses cancer metastasis via regulating a ROS-driven TP53/p53 pathway. Autophagy. 2022Aug;18(8):1932-54
34. Kunita A, Morita S, Irisa TU, Goto A, Niki T, Takai D. et al. MicroRNA-21 in cancer-associated fibroblasts supports lung adenocarcinoma progression. Sci Rep. 2018June11;8(1):8838
35. Huang Y, He Y, Li J. MicroRNA-21: a central regulator of fibrotic diseases via various targets. Curr Pharm Des. 2015;21(17):2236-42
36. Thakkar D, Singh S, Wairkar S. Advanced Delivery Strategies of Nintedanib for Lung Disorders and Beyond: A Comprehensive Review. AAPS PharmSciTech. 2024July1;25(6):150
37. Singh SR, Hall IP. Airway myofibroblasts and their relationship with airway myocytes and fibroblasts. Proc Am Thorac Soc. 2008Jan1;5(1):127-32
38. Cheng PSW, Zaccaria M, Biffi G. Functional heterogeneity of fibroblasts in primary tumors and metastases. Trends Cancer. 2025Feb;11(2):135-53
39. Yamamoto Y, Kasashima H, Fukui Y, Tsujio G, Yashiro M, Maeda K. The heterogeneity of cancer-associated fibroblast subpopulations: Their origins, biomarkers, and roles in the tumor microenvironment. Cancer Sci. 2023Jan;114(1):16-24
40. Wei G, Nie Y, Sun M, Zhou W, Zhao H, Chen F. et al. Cancer-associated fibroblasts induce almonertinib resistance in non-small cell lung cancer. J Transl Med. 2025Jan10;23(1):42
41. Hesketh M, Sahin KB, West ZE, Murray RZ. Macrophage Phenotypes Regulate Scar Formation and Chronic Wound Healing. Int J Mol Sci. 2017July17;18(7):1545
42. Rodríguez-Morales P, Franklin RA. Macrophage phenotypes and functions: resolving inflammation and restoring homeostasis. Trends Immunol. 2023Dec;44(12):986-98
43. Zhang M, Zhang S. T Cells in Fibrosis and Fibrotic Diseases. Front Immunol. 2020;11:1142
44. Celada LJ, Kropski JA, Herazo-Maya JD, Luo W, Creecy A, Abad AT. et al. PD-1 up-regulation on CD4+ T cells promotes pulmonary fibrosis through STAT3-mediated IL-17A and TGF-β1 production. Sci Transl Med. 2018Sept26;10(460):eaar8356
45. Yan S, Li M, Liu B, Ma Z, Yang Q. Neutrophil extracellular traps and pulmonary fibrosis: an update. J Inflamm (Lond). 2023Jan19;20(1):2
46. Königshoff M, Bonniaud P. Live and let die: targeting alveolar epithelial cell proliferation in pulmonary fibrosis. Am J Respir Crit Care Med. 2014Dec15;190(12):1339-41
47. Zhang C, Zhu X, Hua Y, Zhao Q, Wang K, Zhen L. et al. YY1 mediates TGF-β1-induced EMT and pro-fibrogenesis in alveolar epithelial cells. Respir Res. 2019Nov8;20(1):249
48. Lehmann M, Hu Q, Hu Y, Hafner K, Costa R, van den Berg A. et al. Chronic WNT/β-catenin signaling induces cellular senescence in lung epithelial cells. Cell Signal. 2020June;70:109588
49. Selman M, Pardo A. Idiopathic pulmonary fibrosis: an epithelial/fibroblastic cross-talk disorder. Respir Res. 2002;3(1):3
50. Wuyts WA, Agostini C, Antoniou KM, Bouros D, Chambers RC, Cottin V. et al. The pathogenesis of pulmonary fibrosis: a moving target. Eur Respir J. 2013May;41(5):1207-18
51. Schiller HB, Fernandez IE, Burgstaller G, Schaab C, Scheltema RA, Schwarzmayr T. et al. Time- and compartment-resolved proteome profiling of the extracellular niche in lung injury and repair. Mol Syst Biol. 2015July14;11(7):819
52. Zhou Y, Huang X, Hecker L, Kurundkar D, Kurundkar A, Liu H. et al. Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J Clin Invest. 2013Mar;123(3):1096-108
53. Huang X, Yang N, Fiore VF, Barker TH, Sun Y, Morris SW. et al. Matrix stiffness-induced myofibroblast differentiation is mediated by intrinsic mechanotransduction. Am J Respir Cell Mol Biol. 2012Sept;47(3):340-8
54. Huang J, Zhang L, Wan D, Zhou L, Zheng S, Lin S. et al. Extracellular matrix and its therapeutic potential for cancer treatment. Signal Transduct Target Ther. 2021Apr23;6(1):153
55. Kasai H, Allen JT, Mason RM, Kamimura T, Zhang Z. TGF-beta1 induces human alveolar epithelial to mesenchymal cell transition (EMT). Respir Res. 2005June9;6(1):56
56. Lv Q, Wang J, Xu C, Huang X, Ruan Z, Dai Y. Pirfenidone alleviates pulmonary fibrosis in vitro and in vivo through regulating Wnt/GSK-3β/β-catenin and TGF-β1/Smad2/3 signaling pathways. Mol Med. 2020May24;26(1):49
57. Han D, Gong H, Wei Y, Xu Y, Zhou X, Wang Z. et al. Hesperidin inhibits lung fibroblast senescence via IL-6/STAT3 signaling pathway to suppress pulmonary fibrosis. Phytomedicine. 2023Apr;112:154680
58. Guo X, Cen Y, Wang J, Jiang H. CXCL10-induced IL-9 promotes liver fibrosis via Raf/MEK/ERK signaling pathway. Biomed Pharmacother. 2018Sept;105:282-9
59. Senoo S, Higo H, Taniguchi A, Kiura K, Maeda Y, Miyahara N. Pulmonary fibrosis and type-17 immunity. Respir Investig. 2023Sept;61(5):553-62
60. Klinkhammer BM, Floege J, Boor P. PDGF in organ fibrosis. Mol Aspects Med. 2018Aug;62:44-62
61. Romero Y, Aquino-Gálvez A. Hypoxia in Cancer and Fibrosis: Part of the Problem and Part of the Solution. Int J Mol Sci. 2021Aug3;22(15):8335
62. Selman M, Pardo A. Role of epithelial cells in idiopathic pulmonary fibrosis: from innocent targets to serial killers. Proc Am Thorac Soc. 2006June;3(4):364-72
63. Xu Y, Mizuno T, Sridharan A, Du Y, Guo M, Tang J. et al. Single-cell RNA sequencing identifies diverse roles of epithelial cells in idiopathic pulmonary fibrosis. JCI Insight. 2016Dec8;1(20):e90558
64. Rindlisbacher B, Schmid C, Geiser T, Bovet C, Funke-Chambour M. Serum metabolic profiling identified a distinct metabolic signature in patients with idiopathic pulmonary fibrosis - a potential biomarker role for LysoPC. Respir Res. 2018Jan10;19(1):7
65. Ruwisch J, Sehlmeyer K, Roldan N, Garcia-Alvarez B, Perez-Gil J, Weaver TE. et al. Air Space Distension Precedes Spontaneous Fibrotic Remodeling and Impaired Cholesterol Metabolism in the Absence of Surfactant Protein C. Am J Respir Cell Mol Biol. 2020Apr;62(4):466-78
66. Gunasekara L, Schürch S, Schoel WM, Nag K, Leonenko Z, Haufs M. et al. Pulmonary surfactant function is abolished by an elevated proportion of cholesterol. Biochim Biophys Acta. 2005Oct15;1737(1):27-35
67. Chung KP, Hsu CL, Fan LC, Huang Z, Bhatia D, Chen YJ. et al. Mitofusins regulate lipid metabolism to mediate the development of lung fibrosis. Nat Commun. 2019July29;10(1):3390
68. Tang X, Snowball JM, Xu Y, Na CL, Weaver TE, Clair G. et al. EMC3 coordinates surfactant protein and lipid homeostasis required for respiration. J Clin Invest. 2017Dec1;127(12):4314-25
69. Parks BW, Black LL, Zimmerman KA, Metz AE, Steele C, Murphy-Ullrich JE. et al. CD36, but not G2A, modulates efferocytosis, inflammation, and fibrosis following bleomycin-induced lung injury. J Lipid Res. 2013Apr;54(4):1114-23
70. Nie Y, Hu Y, Yu K, Zhang D, Shi Y, Li Y. et al. Akt1 regulates pulmonary fibrosis via modulating IL-13 expression in macrophages. Innate Immun. 2019Oct;25(7):451-61
71. Reyfman PA, Walter JM, Joshi N, Anekalla KR, McQuattie-Pimentel AC, Chiu S. et al. Single-Cell Transcriptomic Analysis of Human Lung Provides Insights into the Pathobiology of Pulmonary Fibrosis. Am J Respir Crit Care Med. 2019June15;199(12):1517-36
72. Li K, Zhao J, Wang M, Niu L, Wang Y, Li Y. et al. The Roles of Various Prostaglandins in Fibrosis: A Review. Biomolecules. 2021May24;11(6):789
73. Izumo T, Kondo M, Nagai A. Effects of a leukotriene B4 receptor antagonist on bleomycin-induced pulmonary fibrosis. Eur Respir J. 2009Dec;34(6):1444-51
74. Bärnthaler T, Theiler A, Zabini D, Trautmann S, Stacher-Priehse E, Lanz I. et al. Inhibiting eicosanoid degradation exerts antifibrotic effects in a pulmonary fibrosis mouse model and human tissue. J Allergy Clin Immunol. 2020Mar;145(3):818-833.e11
75. Selvarajah B, Azuelos I, Platé M, Guillotin D, Forty EJ, Contento G. et al. mTORC1 amplifies the ATF4-dependent de novo serine-glycine pathway to supply glycine during TGF-β1-induced collagen biosynthesis. Sci Signal. 2019May21;12(582):eaav3048
76. Bernard K, Logsdon NJ, Benavides GA, Sanders Y, Zhang J, Darley-Usmar VM. et al. Glutaminolysis is required for transforming growth factor-β1-induced myofibroblast differentiation and activation. J Biol Chem. 2018Jan26;293(4):1218-28
77. Ge J, Cui H, Xie N, Banerjee S, Guo S, Dubey S. et al. Glutaminolysis Promotes Collagen Translation and Stability via α-Ketoglutarate-mediated mTOR Activation and Proline Hydroxylation. Am J Respir Cell Mol Biol. 2018Mar;58(3):378-90
78. Endo M, Oyadomari S, Terasaki Y, Takeya M, Suga M, Mori M. et al. Induction of arginase I and II in bleomycin-induced fibrosis of mouse lung. Am J Physiol Lung Cell Mol Physiol. 2003Aug;285(2):L313-321
79. Kitowska K, Zakrzewicz D, Königshoff M, Chrobak I, Grimminger F, Seeger W. et al. Functional role and species-specific contribution of arginases in pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008Jan;294(1):L34-45
80. Pullamsetti SS, Savai R, Dumitrascu R, Dahal BK, Wilhelm J, Konigshoff M. et al. The role of dimethylarginine dimethylaminohydrolase in idiopathic pulmonary fibrosis. Sci Transl Med. 2011June15;3(87):87ra53
81. Judge JL, Lacy SH, Ku WY, Owens KM, Hernady E, Thatcher TH. et al. The Lactate Dehydrogenase Inhibitor Gossypol Inhibits Radiation-Induced Pulmonary Fibrosis. Radiat Res. 2017July;188(1):35-43
82. Kottmann RM, Kulkarni AA, Smolnycki KA, Lyda E, Dahanayake T, Salibi R. et al. Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-β. Am J Respir Crit Care Med. 2012Oct15;186(8):740-51
83. Andrianifahanana M, Hernandez DM, Yin X, Kang JH, Jung MY, Wang Y. et al. Profibrotic up-regulation of glucose transporter 1 by TGF-β involves activation of MEK and mammalian target of rapamycin complex 2 pathways. FASEB J. 2016Nov;30(11):3733-44
84. Yin X, Choudhury M, Kang JH, Schaefbauer KJ, Jung MY, Andrianifahanana M. et al. Hexokinase 2 couples glycolysis with the profibrotic actions of TGF-β. Sci Signal. 2019Dec17;12(612):eaax4067
85. Coward WR, Watts K, Feghali-Bostwick CA, Knox A, Pang L. Defective histone acetylation is responsible for the diminished expression of cyclooxygenase 2 in idiopathic pulmonary fibrosis. Mol Cell Biol. 2009Aug;29(15):4325-39
86. Bai L, Bernard K, Tang X, Hu M, Horowitz JC, Thannickal VJ. et al. Glutaminolysis Epigenetically Regulates Antiapoptotic Gene Expression in Idiopathic Pulmonary Fibrosis Fibroblasts. Am J Respir Cell Mol Biol. 2019Jan;60(1):49-57
87. Keshavarz Aziziraftar S, Bahrami R, Hashemi D, Shahryari A, Ramezani A, Ashrafian F. et al. The beneficial effects of Akkermansia muciniphila and its derivatives on pulmonary fibrosis. Biomed Pharmacother. 2024Nov;180:117571
88. Zhang Q, Luo T, Yuan D, Liu J, Fu Y, Yuan J. Qi-Long-Tian capsule alleviates pulmonary fibrosis development by modulating inflammatory response and gut microbiota. Funct Integr Genomics. 2023Feb22;23(1):64
89. Jia Q, Li Q, Wang Y, Zhao J, Jiang Q, Wang H. et al. Lung microbiome and transcriptome reveal mechanisms underlying PM2.5 induced pulmonary fibrosis. Sci Total Environ. 2022July20;831:154974
90. Jia Q, Wang H, Wang Y, Xue W, Jiang Q, Wang J. et al. Investigation of the mechanism of silica-induced pulmonary fibrosis: The role of lung microbiota dysbiosis and the LPS/TLR4 signaling pathway. Sci Total Environ. 2024Feb20;912:168948
91. Chioma OS, Mallott E, Shah-Gandhi B, Wiggins Z, Langford M, Lancaster AW. et al. Low Gut Microbial Diversity Augments Estrogen-Driven Pulmonary Fibrosis in Female-Predominant Interstitial Lung Disease. Cells. 2023Feb28;12(5):766
92. Sakamachi Y, Wiley E, Solis A, Johnson CG, Meng X, Hussain S. et al. Toll-Like-Receptor 5 protects against pulmonary fibrosis by reducing lung dysbiosis. bioRxiv. 2024 Apr 30. 2024 04.30.591719
93. Azad N, Rojanasakul Y, Vallyathan V. Inflammation and lung cancer: roles of reactive oxygen/nitrogen species. J Toxicol Environ Health B Crit Rev. 2008Jan;11(1):1-15
94. Moloney JN, Cotter TG. ROS signalling in the biology of cancer. Semin Cell Dev Biol. 2018Aug;80:50-64
95. Glorieux C, Liu S, Trachootham D, Huang P. Targeting ROS in cancer: rationale and strategies. Nat Rev Drug Discov. 2024Aug;23(8):583-606
96. Liu S, Huang B, Cao J, Wang Y, Xiao H, Zhu Y. et al. ROS fine-tunes the function and fate of immune cells. Int Immunopharmacol. 2023June;119:110069
97. Klapp V, Álvarez-Abril B, Leuzzi G, Kroemer G, Ciccia A, Galluzzi L. The DNA Damage Response and Inflammation in Cancer. Cancer Discov. 2023July7;13(7):1521-45
98. Saha P, Talwar P. Idiopathic pulmonary fibrosis (IPF): disease pathophysiology, targets, and potential therapeutic interventions. Mol Cell Biochem. 2024Sept;479(9):2181-94
99. Xiang Z, Hou G, Zheng S, Lu M, Li T, Lin Q. et al. ER-associated degradation ligase HRD1 links ER stress to DNA damage repair by modulating the activity of DNA-PKcs. Proc Natl Acad Sci U S A. 2024Sept10;121(37):e2403038121
100. Armanios MY, Chen JJL, Cogan JD, Alder JK, Ingersoll RG, Markin C. et al. Telomerase mutations in families with idiopathic pulmonary fibrosis. N Engl J Med. 2007Mar29;356(13):1317-26
101. Tsakiri KD, Cronkhite JT, Kuan PJ, Xing C, Raghu G, Weissler JC. et al. Adult-onset pulmonary fibrosis caused by mutations in telomerase. Proc Natl Acad Sci U S A. 2007May1;104(18):7552-7
102. Kropski JA, Blackwell TS, Loyd JE. The genetic basis of idiopathic pulmonary fibrosis. Eur Respir J. 2015June;45(6):1717-27
103. Ebata H, Loo TM, Takahashi A. Telomere Maintenance and the cGAS-STING Pathway in Cancer. Cells. 2022June17;11(12):1958
104. Piccolo S, Dupont S, Cordenonsi M. The biology of YAP/TAZ: hippo signaling and beyond. Physiol Rev. 2014Oct;94(4):1287-312
105. Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B. et al. Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat Cell Biol. 2015Sept;17(9):1218-27
106. Aragona M, Panciera T, Manfrin A, Giulitti S, Michielin F, Elvassore N. et al. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell. 2013Aug29;154(5):1047-59
107. Sansores-Garcia L, Bossuyt W, Wada KI, Yonemura S, Tao C, Sasaki H. et al. Modulating F-actin organization induces organ growth by affecting the Hippo pathway. EMBO J. 2011May10;30(12):2325-35
108. Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C. et al. The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell. 2011Nov11;147(4):759-72
109. Song S, Honjo S, Jin J, Chang SS, Scott AW, Chen Q. et al. The Hippo Coactivator YAP1 Mediates EGFR Overexpression and Confers Chemoresistance in Esophageal Cancer. Clin Cancer Res. 2015June1;21(11):2580-90
110. Lau AN, Curtis SJ, Fillmore CM, Rowbotham SP, Mohseni M, Wagner DE. et al. Tumor-propagating cells and Yap/Taz activity contribute to lung tumor progression and metastasis. EMBO J. 2014Mar3;33(5):468-81
111. Zhang P, Zhan Y. [Research Advances in Targeting the YAP/TAZ Signaling Pathway to Improve Cancer Immunotherapy]. Zhongguo Fei Ai Za Zhi. 2025Mar20;28(3):221-9
112. Bertero T, Oldham WM, Grasset EM, Bourget I, Boulter E, Pisano S. et al. Tumor-Stroma Mechanics Coordinate Amino Acid Availability to Sustain Tumor Growth and Malignancy. Cell Metab. 2019Jan8;29(1):124-140.e10
113. Narciso M, Martínez Á, Júnior C, Díaz-Valdivia N, Ulldemolins A, Berardi M. et al. Lung Micrometastases Display ECM Depletion and Softening While Macrometastases Are 30-Fold Stiffer and Enriched in Fibronectin. Cancers (Basel). 2023Apr21;15(8):2404
114. Wang F, Xia H, Yao S. Regulatory T cells are a double-edged sword in pulmonary fibrosis. Int Immunopharmacol. 2020July;84:106443
115. Boveda-Ruiz D, D'Alessandro-Gabazza CN, Toda M, Takagi T, Naito M, Matsushima Y. et al. Differential role of regulatory T cells in early and late stages of pulmonary fibrosis. Immunobiology. 2013Feb;218(2):245-54
116. Unterman A, Zhao AY, Neumark N, Schupp JC, Ahangari F, Cosme C. et al. Single-Cell Profiling Reveals Immune Aberrations in Progressive Idiopathic Pulmonary Fibrosis. Am J Respir Crit Care Med. 2024Aug15;210(4):484-96
117. Astarita JL, Dominguez CX, Tan C, Guillen J, Pauli ML, Labastida R. et al. Treg specialization and functions beyond immune suppression. Clin Exp Immunol. 2023Mar16;211(2):176-83
118. Onodera K, Notsuda H, Kumata S, Watanabe T, Watanabe Y, Suzuki T. et al. PD-L1 expression correlates with the oncological severity and prognosis of early-stage lung cancer. Surg Today. 2025Nov;55(11):1635-43
119. Wang B, Bai W, Ma H, Li F. Regulatory Effect of PD1/PD-Ligand 1 (PD-L1) on Treg Cells in Patients with Idiopathic Pulmonary Fibrosis. Med Sci Monit. 2021Jan2;27:e927577
120. Jiang A, Liu N, Wang J, Zheng X, Ren M, Zhang W. et al. The role of PD-1/PD-L1 axis in idiopathic pulmonary fibrosis: Friend or foe? Front Immunol. 2022;13:1022228
121. Ni K, Liu M, Zheng J, Wen L, Chen Q, Xiang Z. et al. PD-1/PD-L1 Pathway Mediates the Alleviation of Pulmonary Fibrosis by Human Mesenchymal Stem Cells in Humanized Mice. Am J Respir Cell Mol Biol. 2018June;58(6):684-95
122. Yi M, Niu M, Xu L, Luo S, Wu K. Regulation of PD-L1 expression in the tumor microenvironment. J Hematol Oncol. 2021Jan7;14(1):10
123. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012Mar22;12(4):252-64
124. Morrison SJ, Spradling AC. Stem cells and niches: mechanisms that promote stem cell maintenance throughout life. Cell. 2008Feb22;132(4):598-611
125. Nabhan AN, Brownfield DG, Harbury PB, Krasnow MA, Desai TJ. Single-cell Wnt signaling niches maintain stemness of alveolar type 2 cells. Science. 2018Mar9;359(6380):1118-23
126. Zhang Y, Wang X. Targeting the Wnt/β-catenin signaling pathway in cancer. J Hematol Oncol. 2020Dec4;13(1):165
127. Merlos-Suárez A, Barriga FM, Jung P, Iglesias M, Céspedes MV, Rossell D. et al. The intestinal stem cell signature identifies colorectal cancer stem cells and predicts disease relapse. Cell Stem Cell. 2011May6;8(5):511-24
128. Tammela T, Sanchez-Rivera FJ, Cetinbas NM, Wu K, Joshi NS, Helenius K. et al. A Wnt-producing niche drives proliferative potential and progression in lung adenocarcinoma. Nature. 2017May18;545(7654):355-9
129. Vermeulen L, De Sousa E Melo F, van der Heijden M, Cameron K, de Jong JH, Borovski T. et al. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nat Cell Biol. 2010May;12(5):468-76
130. Muto S, Enta A, Maruya Y, Inomata S, Yamaguchi H, Mine H. et al. Wnt/β-Catenin Signaling and Resistance to Immune Checkpoint Inhibitors: From Non-Small-Cell Lung Cancer to Other Cancers. Biomedicines. 2023Jan12;11(1):190
131. Chilosi M, Poletti V, Zamò A, Lestani M, Montagna L, Piccoli P. et al. Aberrant Wnt/beta-catenin pathway activation in idiopathic pulmonary fibrosis. Am J Pathol. 2003May;162(5):1495-502
132. Mazieres J, He B, You L, Xu Z, Jablons DM. Wnt signaling in lung cancer. Cancer Lett. 2005May10;222(1):1-10
133. Conte E, Fruciano M, Fagone E, Gili E, Caraci F, Iemmolo M. et al. Inhibition of PI3K prevents the proliferation and differentiation of human lung fibroblasts into myofibroblasts: the role of class I P110 isoforms. PLoS One. 2011;6(10):e24663
134. Ballester B, Milara J, Cortijo J. Idiopathic Pulmonary Fibrosis and Lung Cancer: Mechanisms and Molecular Targets. Int J Mol Sci. 2019Jan30;20(3):593
135. Noskovičová N, Petřek M, Eickelberg O, Heinzelmann K. Platelet-derived growth factor signaling in the lung. From lung development and disease to clinical studies. Am J Respir Cell Mol Biol. 2015Mar;52(3):263-84
136. Wang P, Song L, Ge H, Jin P, Jiang Y, Hu W. et al. Crenolanib, a PDGFR inhibitor, suppresses lung cancer cell proliferation and inhibits tumor growth in vivo. Onco Targets Ther. 2014;7:1761-8
137. Shikada Y, Yonemitsu Y, Koga T, Onimaru M, Nakano T, Okano S. et al. Platelet-derived growth factor-AA is an essential and autocrine regulator of vascular endothelial growth factor expression in non-small cell lung carcinomas. Cancer Res. 2005Aug15;65(16):7241-8
138. Frezzetti D, Gallo M, Maiello MR, D'Alessio A, Esposito C, Chicchinelli N. et al. VEGF as a potential target in lung cancer. Expert Opin Ther Targets. 2017Oct;21(10):959-66
139. Suzuki T, Yasuda H, Funaishi K, Arai D, Ishioka K, Ohgino K. et al. Multiple roles of extracellular fibroblast growth factors in lung cancer cells. Int J Oncol. 2015Jan;46(1):423-9
140. Zhuo Y, Zhang J, Laboy M, Lasky JA. Modulation of PDGF-C and PDGF-D expression during bleomycin-induced lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2004Jan;286(1):L182-188
141. Gurujeyalakshmi G, Hollinger MA, Giri SN. Pirfenidone inhibits PDGF isoforms in bleomycin hamster model of lung fibrosis at the translational level. Am J Physiol. 1999Feb;276(2):L311-318
142. Hetzel M, Bachem M, Anders D, Trischler G, Faehling M. Different effects of growth factors on proliferation and matrix production of normal and fibrotic human lung fibroblasts. Lung. 2005;183(4):225-37
143. Xiao L, Du Y, Shen Y, He Y, Zhao H, Li Z. TGF-beta 1 induced fibroblast proliferation is mediated by the FGF-2/ERK pathway. Front Biosci (Landmark Ed). 2012June1;17(7):2667-74
144. Yu Z hong, Wang D ding, Zhou Z you, He S lian, Chen A an, Wang J. Mutant soluble ectodomain of fibroblast growth factor receptor-2 IIIc attenuates bleomycin-induced pulmonary fibrosis in mice. Biol Pharm Bull. 2012;35(5):731-6
145. Hamada N, Kuwano K, Yamada M, Hagimoto N, Hiasa K, Egashira K. et al. Anti-vascular endothelial growth factor gene therapy attenuates lung injury and fibrosis in mice. J Immunol. 2005July15;175(2):1224-31
146. McKeown S, Richter AG, O'Kane C, McAuley DF, Thickett DR. MMP expression and abnormal lung permeability are important determinants of outcome in IPF. Eur Respir J. 2009Jan;33(1):77-84
147. Elyada E, Bolisetty M, Laise P, Flynn WF, Courtois ET, Burkhart RA. et al. Cross-Species Single-Cell Analysis of Pancreatic Ductal Adenocarcinoma Reveals Antigen-Presenting Cancer-Associated Fibroblasts. Cancer Discov. 2019Aug;9(8):1102-23
148. Kieffer Y, Hocine HR, Gentric G, Pelon F, Bernard C, Bourachot B. et al. Single-Cell Analysis Reveals Fibroblast Clusters Linked to Immunotherapy Resistance in Cancer. Cancer Discov. 2020Sept;10(9):1330-51
149. Hanley CJ, Mellone M, Ford K, Thirdborough SM, Mellows T, Frampton SJ. et al. Targeting the Myofibroblastic Cancer-Associated Fibroblast Phenotype Through Inhibition of NOX4. J Natl Cancer Inst. 2018Jan1;110(1):109-20
150. Bonnans C, Chou J, Werb Z. Remodelling the extracellular matrix in development and disease. Nat Rev Mol Cell Biol. 2014Dec;15(12):786-801
151. Tang PCT, Chung JYF, Xue VWW, Xiao J, Meng XM, Huang XR. et al. Smad3 Promotes Cancer-Associated Fibroblasts Generation via Macrophage-Myofibroblast Transition. Adv Sci (Weinh). 2022Jan;9(1):e2101235
152. Heng WS, Gosens R, Kruyt FAE. Lung cancer stem cells: origin, features, maintenance mechanisms and therapeutic targeting. Biochemical Pharmacology. 2019Feb1;160:121-33
153. Merino Salvador M, Gómez de Cedrón M, Moreno Rubio J, Falagán Martínez S, Sánchez Martínez R, Casado E. et al. Lipid metabolism and lung cancer. Crit Rev Oncol Hematol. 2017Apr;112:31-40
154. Yoo HC, Yu YC, Sung Y, Han JM. Glutamine reliance in cell metabolism. Exp Mol Med. 2020Sept;52(9):1496-516
155. Jin J, Byun JK, Choi YK, Park KG. Targeting glutamine metabolism as a therapeutic strategy for cancer. Exp Mol Med. 2023Apr;55(4):706-15
156. Wang B, Pei J, Xu S, Liu J, Yu J. A glutamine tug-of-war between cancer and immune cells: recent advances in unraveling the ongoing battle. J Exp Clin Cancer Res. 2024Mar8;43(1):74
157. Hanania AN, Mainwaring W, Ghebre YT, Hanania NA, Ludwig M. Radiation-Induced Lung Injury: Assessment and Management. Chest. 2019July;156(1):150-62
158. He Y, Thummuri D, Zheng G, Okunieff P, Citrin DE, Vujaskovic Z. et al. Cellular senescence and radiation-induced pulmonary fibrosis. Transl Res. 2019July;209:14-21
159. Abid SH, Malhotra V, Perry MC. Radiation-induced and chemotherapy-induced pulmonary injury. Curr Opin Oncol. 2001July;13(4):242-8
160. Xu H, Yang J, Tu M, Weng J, Xie M, Zhou Z. et al. Vincristine Promotes Transdifferentiation of Fibroblasts Into Myofibroblasts via P38 and ERK Signal Pathways. Front Pharmacol. 2022;13:901000
161. Im J, Nho RS. Fibroblasts from patients with idiopathic pulmonary fibrosis are resistant to cisplatin-induced cell death via enhanced CK2-dependent XRCC1 activity. Apoptosis. 2019June;24(5-6):499-510
162. Ohmori T, Yamaoka T, Ando K, Kusumoto S, Kishino Y, Manabe R. et al. Molecular and Clinical Features of EGFR-TKI-Associated Lung Injury. Int J Mol Sci. 2021Jan14;22(2):792
163. Suzuki H, Aoshiba K, Yokohori N, Nagai A. Epidermal growth factor receptor tyrosine kinase inhibition augments a murine model of pulmonary fibrosis. Cancer Res. 2003Aug15;63(16):5054-9
164. Yamada K, Kadota K, Fujimoto S, Yoshida C, Ibuki E, Ishikawa R. et al. MMP-7 expression is associated with a higher rate of tumor spread through air spaces in resected lung adenocarcinomas. Lung Cancer. 2023Jan;175:125-30
165. Safranek J, Holubec L, Topolcan O, Pesta M, Klecka J, Vodicka J. et al. Expression of mRNA MMP-7 and mRNA TIMP-1 in non-small cell lung cancer. Anticancer Res. 2007;27(4C):2953-6
166. Xiao XY, Lang XP. Correlation Between MMP-7 and bFGF Expressions in Non-small Cell Lung Cancer Tissue and Clinicopathologic Features. Cell Biochem Biophys. 2015Nov;73(2):427-32
167. Soccio P, Moriondo G, d'Alessandro M, Scioscia G, Bergantini L, Gangi S. et al. Role of BAL and Serum Krebs von den Lungen-6 (KL-6) in Patients with Pulmonary Fibrosis. Biomedicines. 2024Jan24;12(2):269
168. Gao Y, Du T, Yang L, Wu L. Research progress of KL-6 in respiratory system diseases. Crit Rev Clin Lab Sci. 2024Nov;61(7):599-615
169. Xu H, Du Z, Li Z, Liu X, Li X, Zhang X. et al. MUC1-EGFR crosstalk with IL-6 by activating NF-κB and MAPK pathways to regulate the stemness and paclitaxel-resistance of lung adenocarcinoma. Ann Med. 2024Dec;56(1):2313671
170. Park HK, Yoon CS, Na YO, Lee JK, Oh HJ, Park HY. et al. Serum KL-6 levels predict the occurrence and severity of treatment-related interstitial lung disease in lung cancer. Sci Rep. 2023Oct23;13(1):18126
171. d'Alessandro M, Bergantini L, Cameli P, Pieroni M, Refini RM, Sestini P. et al. Serum Concentrations of KL-6 in Patients with IPF and Lung Cancer and Serial Measurements of KL-6 in IPF Patients Treated with Antifibrotic Therapy. Cancers (Basel). 2021Feb9;13(4):689
172. Miyazaki K, Kurishima K, Kagohashi K, Kawaguchi M, Ishikawa H, Satoh H. et al. Serum KL-6 levels in lung cancer patients with or without interstitial lung disease. J Clin Lab Anal. 2010;24(5):295-9
173. Hammarström S. The carcinoembryonic antigen (CEA) family: structures, suggested functions and expression in normal and malignant tissues. Semin Cancer Biol. 1999Apr;9(2):67-81
174. Hao C, Zhang G, Zhang L. Serum CEA levels in 49 different types of cancer and noncancer diseases. Prog Mol Biol Transl Sci. 2019;162:213-27
175. Dai H, Liu J, Liang L, Ban C, Jiang J, Liu Y. et al. Increased lung cancer risk in patients with interstitial lung disease and elevated CEA and CA125 serum tumour markers. Respirology. 2014July;19(5):707-13
176. Molyneaux PL, Fahy WA, Byrne AJ, Braybrooke R, Saunders P, Toshner R. et al. CYFRA 21-1 Predicts Progression in Idiopathic Pulmonary Fibrosis: A Prospective Longitudinal Analysis of the PROFILE Cohort. Am J Respir Crit Care Med. 2022June15;205(12):1440-8
177. Wang X, Teng F, Kong L, Yu J. PD-L1 expression in human cancers and its association with clinical outcomes. Onco Targets Ther. 2016;9:5023-39
178. Li Q, Deng MS, Wang RT, Luo H, Luo YY, Zhang DD. et al. PD-L1 upregulation promotes drug-induced pulmonary fibrosis by inhibiting vimentin degradation. Pharmacol Res. 2023Jan;187:106636
179. Paz-Ares L, Kim TM, Vicente D, Felip E, Lee DH, Lee KH. et al. Bintrafusp Alfa, a Bifunctional Fusion Protein Targeting TGF-β and PD-L1, in Second-Line Treatment of Patients With NSCLC: Results From an Expansion Cohort of a Phase 1 Trial. J Thorac Oncol. 2020July;15(7):1210-22
180. Dougan M, Pietropaolo M. Time to dissect the autoimmune etiology of cancer antibody immunotherapy. J Clin Invest. 2020Jan2;130(1):51-61
181. Fujimoto D, Morimoto T, Ito J, Sato Y, Ito M, Teraoka S. et al. A pilot trial of nivolumab treatment for advanced non-small cell lung cancer patients with mild idiopathic interstitial pneumonia. Lung Cancer. 2017Sept;111:1-5
182. Cramer P, Bresalier RS. Gastrointestinal and Hepatic Complications of Immune Checkpoint Inhibitors. Curr Gastroenterol Rep. 2017Jan;19(1):3
183. Hofmann L, Forschner A, Loquai C, Goldinger SM, Zimmer L, Ugurel S. et al. Cutaneous, gastrointestinal, hepatic, endocrine, and renal side-effects of anti-PD-1 therapy. Eur J Cancer. 2016June;60:190-209
184. Tasaka Y, Honda T, Nishiyama N, Tsutsui T, Saito H, Watabe H. et al. Non-inferior clinical outcomes of immune checkpoint inhibitors in non-small cell lung cancer patients with interstitial lung disease. Lung Cancer. 2021May;155:120-6
185. Dobre IA, Frank AJ, D'Silva KM, Christiani DC, Okin D, Sharma A. et al. Outcomes of Patients With Interstitial Lung Disease Receiving Programmed Cell Death 1 Inhibitors: A Retrospective Case Series. Clin Lung Cancer. 2021Sept;22(5):e738-44
186. Kanai O, Kim YH, Demura Y, Kanai M, Ito T, Fujita K. et al. Efficacy and safety of nivolumab in non-small cell lung cancer with preexisting interstitial lung disease. Thorac Cancer. 2018July;9(7):847-55
187. Reck M, Kaiser R, Mellemgaard A, Douillard JY, Orlov S, Krzakowski M. et al. Docetaxel plus nintedanib versus docetaxel plus placebo in patients with previously treated non-small-cell lung cancer (LUME-Lung 1): a phase 3, double-blind, randomised controlled trial. Lancet Oncol. 2014Feb;15(2):143-55
188. Otsubo K, Kishimoto J, Ando M, Kenmotsu H, Minegishi Y, Horinouchi H. et al. Nintedanib plus chemotherapy for nonsmall cell lung cancer with idiopathic pulmonary fibrosis: a randomised phase 3 trial. Eur Respir J. 2022Dec;60(6):2200380
189. Hisatomi K, Mukae H, Sakamoto N, Ishimatsu Y, Kakugawa T, Hara S. et al. Pirfenidone inhibits TGF-β1-induced over-expression of collagen type I and heat shock protein 47 in A549 cells. BMC Pulm Med. 2012June13;12:24
190. Lin X, Yu M, Wu K, Yuan H, Zhong H. Effects of pirfenidone on proliferation, migration, and collagen contraction of human Tenon's fibroblasts in vitro. Invest Ophthalmol Vis Sci. 2009Aug;50(8):3763-70
191. Miura Y, Saito T, Tanaka T, Takoi H, Yatagai Y, Inomata M. et al. Reduced incidence of lung cancer in patients with idiopathic pulmonary fibrosis treated with pirfenidone. Respir Investig. 2018Jan;56(1):72-9
192. Mediavilla-Varela M, Boateng K, Noyes D, Antonia SJ. The anti-fibrotic agent pirfenidone synergizes with cisplatin in killing tumor cells and cancer-associated fibroblasts. BMC Cancer. 2016Mar2;16:176
193. Yamakawa H, Oba T, Ohta H, Tsukahara Y, Kida G, Tsumiyama E. et al. Nintedanib allows retreatment with atezolizumab of combined non-small cell lung cancer/idiopathic pulmonary fibrosis after atezolizumab-induced pneumonitis: a case report. BMC Pulm Med. 2019Aug22;19(1):156
194. Qin W, Zou J, Huang Y, Liu C, Kang Y, Han H. et al. Pirfenidone facilitates immune infiltration and enhances the antitumor efficacy of PD-L1 blockade in mice. Oncoimmunology. 2020Sept22;9(1):1824631
195. Shen XT, Xie SZ, Zheng X, Zou TT, Hu BY, Xu J. et al. Cirrhotic-extracellular matrix attenuates aPD-1 treatment response by initiating immunosuppressive neutrophil extracellular traps formation in hepatocellular carcinoma. Exp Hematol Oncol. 2024Feb22;13(1):20
196. Moon H, Han KH, Ro SW. Pro-tumorigenic roles of TGF-β signaling during the early stages of liver tumorigenesis through upregulation of Snail. BMB Rep. 2017Dec;50(12):599-600
197. Travis MA, Sheppard D. TGF-β activation and function in immunity. Annu Rev Immunol. 2014;32:51-82
198. Sanjabi S, Oh SA, Li MO. Regulation of the Immune Response by TGF-β: From Conception to Autoimmunity and Infection. Cold Spring Harb Perspect Biol. 2017June1;9(6):a022236
199. Herbertz S, Sawyer JS, Stauber AJ, Gueorguieva I, Driscoll KE, Estrem ST. et al. Clinical development of galunisertib (LY2157299 monohydrate), a small molecule inhibitor of transforming growth factor-beta signaling pathway. Drug Des Devel Ther. 2015;9:4479-99
200. Nadal E, Saleh M, Aix SP, Ochoa-de-Olza M, Patel SP, Antonia S. et al. A phase Ib/II study of galunisertib in combination with nivolumab in solid tumors and non-small cell lung cancer. BMC Cancer. 2023July28;23(1):708
201. Colak S, Ten Dijke P. Targeting TGF-β Signaling in Cancer. Trends Cancer. 2017Jan;3(1):56-71
202. Lan Y, Zhang D, Xu C, Hance KW, Marelli B, Qi J. et al. Enhanced preclinical antitumor activity of M7824, a bifunctional fusion protein simultaneously targeting PD-L1 and TGF-β. Sci Transl Med. 2018Jan17;10(424):eaan5488
203. David JM, Dominguez C, McCampbell KK, Gulley JL, Schlom J, Palena C. A novel bifunctional anti-PD-L1/TGF-β Trap fusion protein (M7824) efficiently reverts mesenchymalization of human lung cancer cells. Oncoimmunology. 2017;6(10):e1349589
204. Cheng B, Ding K, Chen P, Ji J, Luo T, Guo X. et al. Anti-PD-L1/TGF-βR fusion protein (SHR-1701) overcomes disrupted lymphocyte recovery-induced resistance to PD-1/PD-L1 inhibitors in lung cancer. Cancer Commun (Lond). 2022Jan;42(1):17-36
205. Chen W, Yang A, Jia J, Popov YV, Schuppan D, You H. Lysyl Oxidase (LOX) Family Members: Rationale and Their Potential as Therapeutic Targets for Liver Fibrosis. Hepatology. 2020Aug;72(2):729-41
206. Fogelgren B, Polgár N, Szauter KM, Ujfaludi Z, Laczkó R, Fong KSK. et al. Cellular fibronectin binds to lysyl oxidase with high affinity and is critical for its proteolytic activation. J Biol Chem. 2005July1;280(26):24690-7
207. Barker HE, Cox TR, Erler JT. The rationale for targeting the LOX family in cancer. Nat Rev Cancer. 2012July19;12(8):540-52
208. Raghu G, Brown KK, Collard HR, Cottin V, Gibson KF, Kaner RJ. et al. Efficacy of simtuzumab versus placebo in patients with idiopathic pulmonary fibrosis: a randomised, double-blind, controlled, phase 2 trial. Lancet Respir Med. 2017Jan;5(1):22-32
209. Bell JA, Davies ER, Brereton CJ, Vukmirovic M, Roberts JJW, Lunn K. et al. Spatial transcriptomic validation of a biomimetic model of fibrosis enables re-evaluation of a therapeutic antibody targeting LOXL2. Cell Rep Med. 2024Sept17;5(9):101695
210. Yao Y, Findlay A, Stolp J, Rayner B, Ask K, Jarolimek W. Pan-Lysyl Oxidase Inhibitor PXS-5505 Ameliorates Multiple-Organ Fibrosis by Inhibiting Collagen Crosslinks in Rodent Models of Systemic Sclerosis. Int J Mol Sci. 2022May16;23(10):5533
211. Chitty JL, Yam M, Perryman L, Parker AL, Skhinas JN, Setargew YFI. et al. A first-in-class pan-lysyl oxidase inhibitor impairs stromal remodeling and enhances gemcitabine response and survival in pancreatic cancer. Nat Cancer. 2023Sept;4(9):1326-44
212. Jeong SB, Das R, Kim DH, Lee S, Oh HI, Jo S. et al. Anticancer effect of verteporfin on non-small cell lung cancer via downregulation of ANO1. Biomed Pharmacother. 2022Sept;153:113373
213. Chapeau EA, Sansregret L, Galli GG, Chène P, Wartmann M, Mourikis TP. et al. Direct and selective pharmacological disruption of the YAP-TEAD interface by IAG933 inhibits Hippo-dependent and RAS-MAPK-altered cancers. Nat Cancer. 2024July;5(7):1102-20
214. Luo Z, Wan R, Liu S, Feng X, Peng Z, Wang Q. et al. Mechanisms of exercise in the treatment of lung cancer - a mini-review. Front Immunol. 2023;14:1244764
215. Zhao Z, Zhu Y, Wan D. Exercise and tissue fibrosis: recent advances in therapeutic potential and molecular mechanisms. Front Endocrinol (Lausanne). 2025;16:1557797
216. Luo Z, Mei J, Wang X, Wang R, He Z, Geffen Y. et al. Voluntary exercise sensitizes cancer immunotherapy via the collagen inhibition-orchestrated inflammatory tumor immune microenvironment. Cell Rep. 2024Sept24;43(9):114697
217. Mei J, Luo Z, Cai Y, Wan R, Qian Z, Chu J. et al. Altered Atlas of Exercise-Responsive MicroRNAs Revealing miR-29a-3p Attacks Armored and Cold Tumors and Boosts Anti-B7-H3 Therapy. Research (Wash D C). 2025;8:0590
218. Liu W, Li L, Bai X, Zhang M, Lv W, Ma Y. et al. Osteosarcoma Cell-Derived Migrasomes Promote Macrophage M2 Polarization to Aggravate Osteosarcoma Proliferation and Metastasis. Advanced Science [Internet]. 2025 May [cited. 2025 July 14];12(17). Available from: https://advanced.onlinelibrary.wiley.com/doi/10.1002/advs.202409870
219. Cavalheri V, Granger CL. Exercise training as part of lung cancer therapy. Respirology. 2020Nov;25(Suppl 2):80-7
220. Himbert C, Klossner N, Coletta AM, Barnes CA, Wiskemann J, LaStayo PC. et al. Exercise and lung cancer surgery: A systematic review of randomized-controlled trials. Crit Rev Oncol Hematol. 2020Dec;156:103086
221. Gao Y, Zhao L, Yang Z, He K, Zhang T, Yi J. Efficacy of exercise in patients with pulmonary fibrosis: A systematic review and meta-analysis. Medicine (Baltimore). 2022Dec2;101(48):e31789
222. Uematsu K, Yoshimura A, Gemma A, Mochimaru H, Hosoya Y, Kunugi S. et al. Aberrations in the fragile histidine triad (FHIT) gene in idiopathic pulmonary fibrosis. Cancer Res. 2001Dec1;61(23):8527-33
223. Takahashi T, Munakata M, Ohtsuka Y, Nisihara H, Nasuhara Y, Kamachi-Satoh A. et al. Expression and alteration of ras and p53 proteins in patients with lung carcinoma accompanied by idiopathic pulmonary fibrosis. Cancer. 2002Aug1;95(3):624-33
224. Karachaliou N, Mayo C, Costa C, Magrí I, Gimenez-Capitan A, Molina-Vila MA. et al. KRAS Mutations in Lung Cancer. Clinical Lung Cancer. 2013May1;14(3):205-14
225. Vancheri C. Common pathways in idiopathic pulmonary fibrosis and cancer. Eur Respir Rev. 2013Sept1;22(129):265-72
226. Takenaka K, Gemma A, Yoshimura A, Hosoya Y, Nara M, Hosomi Y. et al. Reduced transcription of the Smad4 gene during pulmonary carcinogenesis in idiopathic pulmonary fibrosis. Mol Med Rep. 2009;2(1):73-80
227. Schutte M, Hruban RH, Hedrick L, Cho KR, Nadasdy GM, Weinstein CL. et al. DPC4 gene in various tumor types. Cancer Res. 1996June1;56(11):2527-30
228. Brooks RT, Luedders B, Wheeler A, Johnson TM, Yang Y, Roul P. et al. The Risk of Lung Cancer in Rheumatoid Arthritis and Rheumatoid Arthritis-Associated Interstitial Lung Disease. Arthritis Rheumatol. 2024Dec;76(12):1730-8
229. Paulin F, Doyle TJ, Fletcher EA, Ascherman DP, Rosas IO. Rheumatoid Arthritis-Associated Interstitial Lung Disease and Idiopathic Pulmonary Fibrosis: Shared Mechanistic and Phenotypic Traits Suggest Overlapping Disease Mechanisms. Rev Invest Clin. 2015;67(5):280-6
230. Sullivan DI, Ascherman DP. Rheumatoid Arthritis-Associated Interstitial Lung Disease (RA-ILD): Update on Prevalence, Risk Factors, Pathogenesis, and Therapy. Curr Rheumatol Rep. 2024Dec;26(12):431-49
231. Pollard KM. Silica, Silicosis, and Autoimmunity. Front Immunol. 2016;7:97
232. Vannan A, Lyu R, Williams AL, Negretti NM, Mee ED, Hirsh J. et al. Spatial transcriptomics identifies molecular niche dysregulation associated with distal lung remodeling in pulmonary fibrosis. Nat Genet. 2025Mar;57(3):647-58
233. Habermann AC, Gutierrez AJ, Bui LT, Yahn SL, Winters NI, Calvi CL. et al. Single-cell RNA sequencing reveals profibrotic roles of distinct epithelial and mesenchymal lineages in pulmonary fibrosis. Sci Adv. 2020July;6(28):eaba1972
234. Adams TS, Schupp JC, Poli S, Ayaub EA, Neumark N, Ahangari F. et al. Single-cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv. 2020July;6(28):eaba1983
235. Mayr CH, Santacruz D, Jarosch S, Bleck M, Dalton J, McNabola A. et al. Spatial transcriptomic characterization of pathologic niches in IPF. Sci Adv. 2024Aug9;10(32):eadl5473
236. Morse C, Tabib T, Sembrat J, Buschur KL, Bittar HT, Valenzi E. et al. Proliferating SPP1/MERTK-expressing macrophages in idiopathic pulmonary fibrosis. Eur Respir J. 2019Aug;54(2):1802441
237. Kramerova I, Kumagai-Cresse C, Ermolova N, Mokhonova E, Marinov M, Capote J. et al. Spp1 (osteopontin) promotes TGFβ processing in fibroblasts of dystrophin-deficient muscles through matrix metalloproteinases. Hum Mol Genet. 2019Oct15;28(20):3431-42
238. Street K, Risso D, Fletcher RB, Das D, Ngai J, Yosef N. et al. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics. 2018June19;19(1):477
239. Wu F, Fan J, He Y, Xiong A, Yu J, Li Y. et al. Single-cell profiling of tumor heterogeneity and the microenvironment in advanced non-small cell lung cancer. Nat Commun. 2021May5;12(1):2540
240. Mayr CH, Sengupta A, Asgharpour S, Ansari M, Pestoni JC, Ogar P. et al. Sfrp1 inhibits lung fibroblast invasion during transition to injury-induced myofibroblasts. Eur Respir J. 2024Feb;63(2):2301326
241. Yao Y, Ritzmann F, Miethe S, Kattler-Lackes K, Colakoglu B, Herr C. et al. Co-culture of human AT2 cells with fibroblasts reveals a MUC5B phenotype: insights from an organoid model. Mol Med. 2024Nov23;30(1):227
242. Strating E, Verhagen MP, Wensink E, Dünnebach E, Wijler L, Aranguren I. et al. Co-cultures of colon cancer cells and cancer-associated fibroblasts recapitulate the aggressive features of mesenchymal-like colon cancer. Front Immunol. 2023;14:1053920
243. Hamet P, Tremblay J. Artificial intelligence in medicine. Metabolism. 2017 Apr;69S:S36-40
244. Soffer S, Morgenthau AS, Shimon O, Barash Y, Konen E, Glicksberg BS. et al. Artificial Intelligence for Interstitial Lung Disease Analysis on Chest Computed Tomography: A Systematic Review. Acad Radiol. 2022Feb;29(Suppl 2):S226-35
245. Park B, Park H, Lee SM, Seo JB, Kim N. Lung Segmentation on HRCT and Volumetric CT for Diffuse Interstitial Lung Disease Using Deep Convolutional Neural Networks. J Digit Imaging. 2019Dec;32(6):1019-26
246. Walsh SLF, Calandriello L, Silva M, Sverzellati N. Deep learning for classifying fibrotic lung disease on high-resolution computed tomography: a case-cohort study. Lancet Respir Med. 2018Nov;6(11):837-45
247. Christe A, Peters AA, Drakopoulos D, Heverhagen JT, Geiser T, Stathopoulou T. et al. Computer-Aided Diagnosis of Pulmonary Fibrosis Using Deep Learning and CT Images. Invest Radiol. 2019Oct;54(10):627-32
248. Ren F, Aliper A, Chen J, Zhao H, Rao S, Kuppe C. et al. A small-molecule TNIK inhibitor targets fibrosis in preclinical and clinical models. Nat Biotechnol. 2025Jan;43(1):63-75
249. Huang S, Yang J, Shen N, Xu Q, Zhao Q. Artificial intelligence in lung cancer diagnosis and prognosis: Current application and future perspective. Semin Cancer Biol. 2023Feb;89:30-7
250. Jj L. The Dual Role of TGFβ in Human Cancer: From Tumor Suppression to Cancer Metastasis. ISRN molecular biology [Internet]. 2012 Dec 24 [cited 2025 July 23]. 2012 Available from: https://pubmed.ncbi.nlm.nih.gov/27340590/
251. Fan Y, Cheng H, Liu Y, Liu S, Lowe S, Li Y. et al. Metformin anticancer: Reverses tumor hypoxia induced by bevacizumab and reduces the expression of cancer stem cell markers CD44/CD117 in human ovarian cancer SKOV3 cells. Front Pharmacol. 2022;13:955984
252. Yamamoto Y, Yano Y, Kuge T, Okabe F, Ishijima M, Uenami T. et al. Safety and effectiveness of pirfenidone combined with carboplatin-based chemotherapy in patients with idiopathic pulmonary fibrosis and non-small cell lung cancer: A retrospective cohort study. Thorac Cancer. 2020Nov;11(11):3317-25
253. Codullo V, Cova E, Pandolfi L, Breda S, Morosini M, Frangipane V. et al. Imatinib-loaded gold nanoparticles inhibit proliferation of fibroblasts and macrophages from systemic sclerosis patients and ameliorate experimental bleomycin-induced lung fibrosis. J Control Release. 2019Sept28;310:198-208
254. Trivedi R, Redente EF, Thakur A, Riches DWH, Kompella UB. Local delivery of biodegradable pirfenidone nanoparticles ameliorates bleomycin-induced pulmonary fibrosis in mice. Nanotechnology. 2012Dec21;23(50):505101
255. Taratula O, Kuzmov A, Shah M, Garbuzenko OB, Minko T. Nanostructured lipid carriers as multifunctional nanomedicine platform for pulmonary co-delivery of anticancer drugs and siRNA. J Control Release. 2013Nov10;171(3):349-57
256. Wang Z, Zhao Y, Yang Z, Li X, Xing H, Qu W. et al. Construction of Intelligent Responsive Drug Delivery System and Multi-Mode Imaging Based on Gold Nanodots. Macromol Rapid Commun. 2022May;43(10):e2200034
257. Li M, Li Y, Zheng J, Ma Z, Zhang J, Wu H. et al. Ultrasound-responsive nanocarriers with siRNA and Fe3O4 regulate macrophage polarization and phagocytosis for augmented non-small cell lung cancer immunotherapy. J Nanobiotechnology. 2024Oct7;22(1):605
Author contact
![]() Corresponding authors: Zhang Ting, E-mail: cczthd18com; Baojun Liu, E-mail: liubaojunorg.cn; Zhiwen Luo, E-mail: zhiwen.luo_fudancom.
Corresponding authors: Zhang Ting, E-mail: cczthd18com; Baojun Liu, E-mail: liubaojunorg.cn; Zhiwen Luo, E-mail: zhiwen.luo_fudancom.