Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Molecular and Metabolic Drivers...
Role of Copper Homeostasis and...
Clinical Application of Copper...
Limits and Challenges
Future Directions
Conclusions
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2026; 22(6):2842-2868. doi:10.7150/ijbs.126580 This issue Cite
Review
Role of Copper Homeostasis and Cuproptosis in Cardiovascular Disease: Molecular Insights and Metabolic Perspectives
Xianzhe Yu1#, Dou Yuan1,2#, Yabo Wang1, Leibo Wang3, Lingling Zhu4,5 ![]() , Qi An1
, Qi An1 ![]() , Yunfei Ling1
, Yunfei Ling1 ![]()
1. Department of Cardiovascular Surgery, West China Hospital of Sichuan University, No. 37 GuoXue Xiang, Chengdu, Sichuan 610041, China.
2. Department of Cardiovascular Surgery, Chengdu Shang Jin Nan Fu Hospital, West China Hospital of Sichuan University, Chengdu, Sichuan 610041, China.
3. Institute of Human Genetics, Jena University Hospital, Jena 07747, Germany.
4. Department of Medical Oncology, West China Hospital of Sichuan University, No. 37 GuoXue Xiang, Chengdu, Sichuan 610041, China.
5. Lung Cancer Center/Lung Cancer Institute, West China Hospital of Sichuan University, No. 37 GuoXue Xiang, Chengdu, Sichuan 610041, China.
#These authors contributed equally: Xianzhe Yu, Dou Yuan.
Received 2025-10-11; Accepted 2026-2-5; Published 2026-2-18
Abstract

Copper is an essential trace element; however, its homeostasis is frequently disrupted in cardiovascular diseases, which are a leading cause of mortality worldwide. The recent discovery of cuproptosis—a copper-dependent form of regulated cell death (RCD)—has provided a crucial mechanistic link between this imbalance and cardiomyocyte loss. In this review, we synthesize the current understanding of how dysregulated copper metabolism and cuproptosis drive the pathogenesis of major cardiovascular conditions, including myocardial ischemia/reperfusion (I/R) injury, anthracycline-induced cardiotoxicity, atherosclerosis, diabetic cardiomyopathy (DCM), and sepsis-induced cardiac dysfunction, through pathways such as mitochondrial dysfunction, oxidative stress, and inflammation. We further evaluated emerging therapeutic strategies that target copper homeostasis—including chelators, chaperone inhibitors, and ionophores—and critically analyzed the translational challenges they face, such as off-target effects and preclinical model limitations. Advancing our knowledge of cardiac copper biology holds significant promise for the development of novel and precise therapeutic approaches for cardiovascular diseases.
Keywords: copper, cuproptosis, copper homeostasis, cardiovascular disease, mitochondrial dysfunction
Introduction
Cell death is a critical driver of cardiovascular disease pathogenesis, contributing to tissue damage, dysfunction, and inflammation [1]. Among the various regulated cell death (RCD) pathways, a novel form—cuproptosis—has recently emerged, uniquely triggered by disruptions in copper metabolism [2]. Mechanistically distinct from other forms of cell death, cuproptosis is characterized by the direct binding of copper to lipoylated enzymes in the mitochondrial tricarboxylic acid (TCA) cycle, leading to proteotoxic stress and subsequent cell death, independent of classical apoptotic signaling [3].
Copper, an essential trace element, acts as a cofactor for numerous metabolic enzymes and is vital for physiological processes, including antioxidant defense, mitochondrial respiration, and energy metabolism [4]. Cellular function relies on a tightly regulated copper metabolic network that maintains copper concentrations within a narrow range. Copper deficiency impairs copper-dependent enzymes, whereas copper overload promotes excessive reactive oxygen species (ROS) generation, leading to oxidative damage to lipids, proteins, and DNA [5, 6]. This oxidative stress can activate classical apoptotic pathways, a process termed copper-induced apoptosis, which is mediated by DNA/membrane damage and the caspase/Bcl-2/p53 pathway and can be inhibited by caspase inhibitors [7]. In contrast, cuproptosis represents a separate copper-dependent pathway in which excess copper directly targets mitochondrial metabolism, inducing cell death through aggregated lipoylated proteins and iron-sulfur (Fe-S) cluster loss, independent of apoptotic signaling [3, 8]. Copper-induced oxidative stress, although a key modulator of cellular damage, is not a direct cell death pathway. It is a cellular stress state resulting from copper-driven ROS accumulation, which can facilitate apoptosis or other death modalities, such as ferroptosis, and can be alleviated by antioxidants [9, 10].
Given that cellular function depends critically on precise copper homeostasis, any disturbance in this balance can have severe pathological consequences [11]. As a novel RCD modality involving multiple signaling and metabolic pathways, cuproptosis has been implicated in various cardiovascular conditions, including cardiomyopathy, heart failure (HF), and myocardial injury [12]. Therefore, this review aims to summarize the regulation of copper homeostasis in cardiomyocytes, explore cuproptosis-related targets in cardiovascular diseases, and discuss the translational potential of therapies targeting this pathway.
Molecular and Metabolic Drivers of Cuproptosis
Cells undergoing cuproptosis exhibit unique biochemical, genetic, metabolic, and morphological features that distinguish them from other recognized forms of cell death, such as apoptosis, pyroptosis, necroptosis, and ferroptosis [13, 14] (Table 1). Apoptosis is a caspase-dependent process activated by intrinsic (e.g., mitochondrial outer membrane permeabilization) or extrinsic (e.g., death receptor activation) stimuli. Key cardiac triggers include hypoxia, oxidative stress, and neurohormonal overload [15]. Its morphological hallmarks include cell shrinkage, chromatin condensation, and formation of apoptotic bodies [15]. Ferroptosis is driven by iron-dependent lipid peroxidation, primarily initiated by glutathione (GSH) depletion or inactivation of GSH peroxidase 4 (GPX4). It is a key contributor to myocardial ischemia/reperfusion (I/R) injury [16]. Morphologically, it is associated with distinct mitochondrial changes, such as mitochondrial reticulum shrinkage and fragmentation [3]. Although cuproptosis can intersect with pathways such as apoptosis and ferroptosis, its initiation and execution are uniquely co-regulated by interconnected metabolic pathways involving copper, GSH, and lipids, which are particularly critical in cardiomyocytes [17]. Given these distinct mechanisms, cuproptosis has been implicated as a contributing factor in the pathogenesis of diverse cardiovascular diseases, including dilated cardiomyopathy, HF, and atherosclerosis [12].
Molecular intersections and distinctions between cuproptosis and other regulated cell-death pathways
| Cell death pathway | Primary trigger | Morphological features | Biochemical characteristics | Core execution mechanism | Key molecules/ hallmarks | Key genes | Ref. |
|---|---|---|---|---|---|---|---|
| Cuproptosis | Excess copper ions | Mitochondrial shrinkage and plasma membrane rupture | Cu2+ binds to lipoacylated DLAT and induces disulphide bond-dependent aggregation of lipoylated DLAT | Copper-binding to lipoylated TCA cycle proteins, leading to protein aggregation and proteotoxic stress. | Copper, FDX1, and lipoylated DLAT | FDX1 | [265] |
| Apoptosis | Death receptor signaling, DNA damage, and cellular stress | Apoptotic bodies' chromatin condensation, DNA fragmentation, and cell shrinkage | DNA fragmentation, and the dismantling of cellular components | Caspase activation, DNA fragmentation, and the dismantling of cellular components. | Caspases, BCL-2 family, and cytochrome c | BCL-2, Bak, and caspases | [266] |
| Ferroptosis | Iron-dependent generation of lipid peroxidation | Small mitochondria and elevated mitochondrial membrane density. | Decreased expression of GSH and GPX4, increased divalent iron and lipid peroxidation | Inactivation of GPX4, leading to lethal accumulation of lipid peroxides. | Iron, lipid peroxides, and GPX4 | GPX4 | [267] |
| Pyroptosis | Pathogen Shigella infection | Cell swelling, plasma membrane leakage, and chromatin condensation | Caspase-1 activation dependent, GSDMD cleavage and inflammatory factor release | Pore formation in the plasma membrane, causing inflammatory cell lysis. | Gasdermin D, inflammasome, and IL-1β | GSDMD, NLRP 3, and caspases-1/4/5 | [268] |
| Necroptosis | Tumor necrosis factor receptor signaling and caspase-8 inhibition | Disintegration of plasma membrane, swelling of organelles, and spillage of cellular contents | Activation of RIPK1, RIPK4, and MLKL and a decrease in ATP levels | RIPK1/RIPK3/MLKL-mediated plasma membrane rupture. | RIPK3 and phosphorylated MLKL | RIPK3 and MLKL | [269] |
Heart Copper Metabolism and Cuproptosis
Regulation of Copper Homeostasis in the Cardiovascular System
Copper homeostasis is a tightly regulated process that is essential for cardiovascular health and balances systemic absorption, distribution, utilization, and excretion. This equilibrium is particularly critical in high-energy-demand tissues, such as the heart, where copper serves as an indispensable cofactor for key metabolic enzymes and mitochondrial function [18, 19]. Intracellularly, copper buffering by molecules such as GSH and metallothionein (MT) prevents ROS generation via Fenton reactions [20]. Specific chaperones then deliver copper to target proteins; for instance, the copper chaperone for superoxide dismutase (CCS) activates Cu/Zn superoxide dismutase 1 (SOD1), which is a key antioxidant enzyme in vascular cells [21]. Notably, CCS expression is regulated by copper levels, increasing during deficiency and degrading under excess conditions [22].
The cardiovascular roles of copper are multifaceted. As the catalytic core of cytochrome c oxidase (complex IV, CCO), it is fundamental to mitochondrial adenosine triphosphate (ATP) production [23]. It also activates other crucial enzymes, including extracellular SOD3, ferroxidase ceruloplasmin (CP) (vital for iron metabolism), and lysyl oxidase (LOX) [24]. LOX crosslinks collagen and elastin, providing structural integrity and elasticity to the heart and vasculature [25]. The critical dependence on copper is underscored by pathologies arising from its dysregulation. For example, copper restriction impairs CCO assembly and function, leading to reduced mitochondrial ATP production and oxygen consumption [26]. Beyond metabolism and structure, copper promotes angiogenesis by stabilizing hypoxia-inducible factor-1α (HIF-1α) via transporters such as Copper Transporter 1 (CTR1; Solute Carrier Family 31 Member A1, SLC31A1) and ATPase Copper Transporting Alpha (ATP7A), thereby enhancing pro-angiogenic gene expression [27, 28].
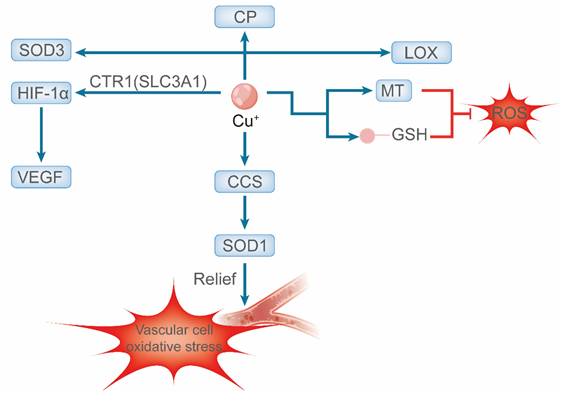
Given these essential functions, serum copper levels are closely associated with cardiometabolic risk factors, including dyslipidemia, type 2 diabetes, and obesity, and are regarded as predictive indicators of cardiovascular disease risk [29]. In summary, disruptions in copper homeostasis—whether arising from deficiency, excess, or dysfunction of molecular chaperones and transporters—are fundamentally implicated in the pathogenesis of various cardiovascular diseases [30] (Figure 1).
Schematic representation of copper-mediated regulation of vascular redox homeostasis. Intracellularly, Cu⁺ acts as an essential cofactor that activates secretory enzymes (e.g., CP and LOX) is delivered to SOD1 via CCS to combat superoxide and stabilizes HIF-1α to promote VEGF expression. Cellular redox balance is maintained by copper buffers (MT) and antioxidants (GSH).
Copper Homeostasis Imbalance and Cardiovascular Disease
Copper overload induces cardiovascular injury through multiple interconnected mechanisms. Systemically, excess copper catalyzes Fenton-like reactions, generating excessive ROS that cause oxidative damage to lipids, proteins, and DNA [31]. It concurrently impairs antioxidant defenses (e.g., by inhibiting catalase) [32] and promotes inflammation by elevating pro-inflammatory cytokine levels and reducing nitric oxide (NO) bioavailability [33]. At the cellular level, these insults trigger distinct pathogenic pathways. In endothelial cells (ECs), copper overload disrupts mitochondrial dynamics, increases fission and ROS production, and ultimately leads to cell death [34] In cardiomyocytes, copper promotes hypertrophy and inflammation [35], disrupts fatty acid and lipid metabolism, upregulates autophagy-associated proteins and genes, and impedes calcium ion uptake by the sarcoplasmic reticulum, thereby inhibiting myocardial contractility [36]. Chronic copper overload is a potent driver of pathological cardiac remodeling, primarily by inducing sustained oxidative stress. The accumulation of copper catalyzes excessive ROS production, which directly damages mitochondrial integrity and function and activates pro-fibrotic signaling pathways—most notably transforming growth factor-beta (TGF-β) [37]. These structural and cellular alterations culminate in adverse outcomes. The resulting myocardial fibrosis disrupts the normal electrical conduction system of the heart, creating a substrate for electrophysiological instability. This, combined with the accompanying hypertrophic and neurohormonal changes, significantly increases the susceptibility to severe arrhythmias, including ventricular tachycardia and atrial fibrillation [38, 39]. Elevated serum copper levels, often observed in aging and diabetes, are both a driver and a biomarker of vascular damage, forming a vicious cycle that accelerates vascular aging and increases the risk of cardiovascular disease [40, 41]. Recent bioinformatic studies have further underscored its clinical relevance by highlighting a pronounced cuproptosis signature in myocardial infarction, involving key regulators such as SLC31A1 and ferredoxin-1 (FDX1) [42].
In contrast, copper deficiency promotes cardiovascular pathology through distinct mechanisms, primarily mitochondrial dysfunction and impaired structural integrity [43]. Deficiency disrupts the assembly and function of CCO by impairing copper-delivery proteins (e.g., COX17, SCO1/SCO2), severely reducing ATP production [44]. The resultant energy crisis and diminished SOD activity increase oxidative stress, promote low-density lipoprotein (LDL) oxidation [45], and disrupt calcium homeostasis, leading to diastolic dysfunction and HF [46].
In the myocardium, a reduction in LOX activity disrupts the connective tissue architecture and impairs contractile function [47]. Conversely, elevated LOX activity promotes pathological cardiac remodeling by driving excessive collagen crosslinking and deposition. This leads to myocardial fibrosis, increased tissue stiffness, impaired angiogenesis, and ultimately contributes to HF [48, 49]. These defects ultimately lead to extensive structural and functional pathological alterations. The heart undergoes concentric hypertrophy, characterized by thickened ventricular walls without cavity enlargement, resembling pressure overload hypertrophy [50].
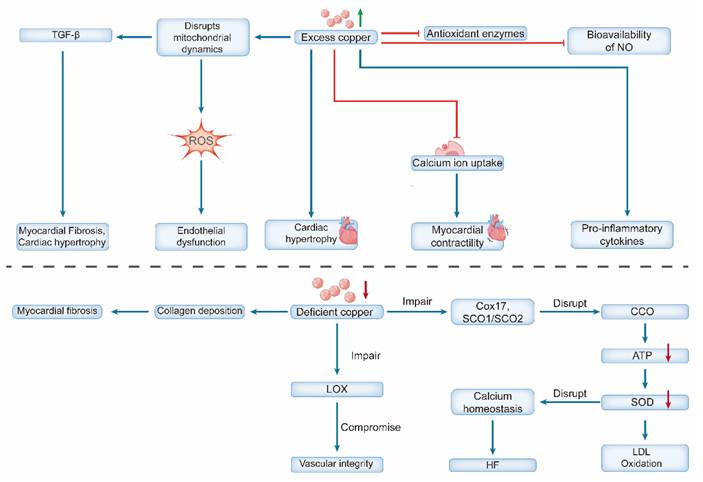
A critical aspect of copper deficiency is that its effects are often reversible. Copper supplementation in both animal models and humans (e.g., patients with SCO2 mutations) can restore CCO activity, reverse hypertrophy and fibrosis, and significantly improve cardiac function [51, 52]. This reversibility underscores the crucial and dynamic role of copper in maintaining cardiovascular health (Figure 2).
Dual effects of copper dyshomeostasis on cardiovascular pathophysiology. Copper excess triggers oxidative stress (by impairing antioxidant enzymes and NO bioavailability) and disrupts Ca²⁺ homeostasis, leading to endothelial dysfunction, cardiac hypertrophy, and contractile impairment. Copper deficiency compromises cuproenzyme function, causing mitochondrial dysfunction (via impaired CCO), reduced antioxidant defense (via SOD downregulation), and structural defects (via reduced LOX activity leading to fibrosis). Collectively, these changes promote heart failure.
Copper Metabolism in Cuproptosis
Cuproptosis is a unique form of RCD driven by the cytotoxic accumulation of copper, with its upstream regulation and execution being critically dependent on the mitochondrial reductase FDX1 [53]. It reduces Cu²⁺ to the more reactive Cu⁺ and concurrently supplies electrons for the biosynthesis of the lipoyl moiety on key mitochondrial enzymes, such as Dihydrolipoamide Acetyltransferase (DLAT) [54]. This lipoylation creates high-affinity Cu + binding sites. Subsequent copper binding induces aberrant oligomerization of these proteins, disrupting the TCA cycle and initiating cell death [55].
The FDX1-mediated process culminates in profound proteotoxic stress driven by the aggregation of lipoylated mitochondrial proteins and the concomitant destabilization of Fe-S cluster proteins, which together lead to acute mitochondrial failure [3]. The copper ionophore elesclomol exhibits a paradoxical dual role that is critically dependent on the cellular context. In the presence of FDX1 and adequate copper, cuproptosis is induced by delivering extracellular Cu²⁺ into cells [56]. Conversely, under conditions of copper deficiency or FDX1 loss, elesclomol shifts to a protective function, restoring mitochondrial respiration by delivering copper to CCO [57]. This duality highlights the precise metabolic context required for cuproptosis. The physiological importance of copper import is further underscored by the essential role of the standard importer SLC31A1, as evidenced by reduced cardiac copper levels in SLC31A1-deficient mice [50, 58].
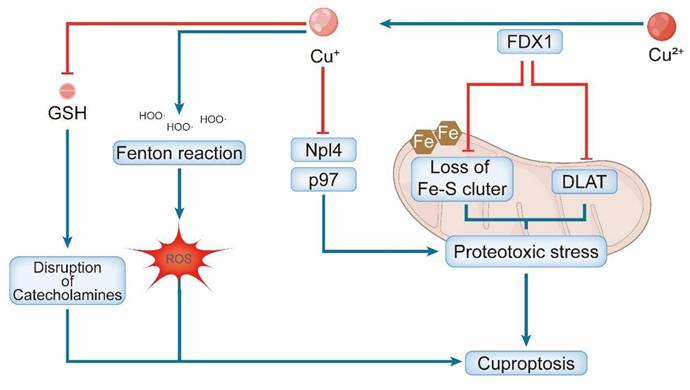
Furthermore, cuproptosis is amplified by secondary mechanisms. Copper ions can participate in Fenton-like reactions, generating ROS that cause oxidative damage to DNA and lipids [59]. Additionally, copper may inhibit the p97-Npl4 complex, which is essential for protein degradation, thereby exacerbating underlying proteotoxic stress [60]. GSH depletion and copper-catalyzed oxidation of catecholamines generate toxic quinones and ROS, which synergistically contribute to cuproptosis [3]. These parallel pathways collectively intensify the proteotoxic and oxidative burdens, ultimately leading to irreversible cell death (Figure 3).
Schematic representation of copper-induced cuproptosis. Excess copper is reduced by FDX1 to generate Cu⁺, which drives cell death via two primary mechanisms: (1) Cu⁺-mediated aggregation of lipoylated TCA cycle proteins (e.g., DLAT) and loss of Fe-S clusters, and (2) Cu⁺-induced disruption of the Npl4/p97 protein quality-control system. Both pathways converge to induce fatal proteotoxic stress in the mitochondria. Elevated Cu⁺ levels also deplete glutathione (GSH) and promote ROS generation, further contributing to cellular toxicity.
Crosstalk between Ferroptosis and Cuproptosis
Ferroptosis, an iron-dependent form of RCD driven by GSH depletion and GPX4 inactivation, is potently accelerated by copper through multi-pronged mechanisms that converge on iron-dependent lipid peroxidation [61, 62]. Copper promotes ferroptosis primarily by dismantling the core GPX4-centered antioxidant defense system. It depletes the intracellular pool of GSH, which is GPX4's essential cofactor, through the formation of Cu-GSH complexes and catalyzing its oxidation to glutathione disulfide (GSSG) [63, 64]. More critically, copper can directly bind to the cysteine residues of GPX4, triggering its autophagic degradation and consequent loss of activity, which impairs the cell's ability to repair lipid peroxidation [65].
Fe²⁺ acts as a key catalyst driving lipid peroxidation. The Fenton reaction generates highly reactive hydroxyl radicals. These radicals directly target polyunsaturated fatty acids within cell membranes, thereby initiating and propagating the chain reaction of lipid peroxidation [66]. This pro-ferroptotic effect is significantly amplified by the intricate crosstalk between copper and iron metabolism. Copper modulates post-translational regulation of iron homeostasis, potentially by inhibiting Fe-S cluster biosynthesis, and transcriptionally upregulates key iron uptake genes, such as transferrin receptor 1. These actions collectively lead to an expansion of the intracellular labile iron pool, thereby supplying more catalysts for Fenton reactions and intensifying lipid peroxidation [67, 68]. In cardiovascular diseases, the ferroptosis and cuproptosis pathways exhibit significant crosstalk. Copper stress promotes iron-dependent lipid peroxidation, whereas ferroptosis-derived mtROS enhance cuproptosis by accelerating DLAT oligomerization [69]. This interplay creates a vicious cycle of cardiomyocyte and vascular cell death, exacerbating cellular injury and pathological remodeling.
The clinical significance of the copper-ferroptosis axis is underscored by bioinformatic analyses revealing the co-dysregulation of ferroptosis and cuproptosis signatures in human diseases [70]. The shared dysregulation of key genes (e.g., POR, SLC7A5, and STAT3) in conditions such as sepsis-induced cardiomyopathy highlights the interplay between these metal-dependent death pathways [71]. Furthermore, the copper transporter ATP7A has been identified as a novel ferroptosis regulator, as its deficiency downregulates the cystine transporter SLC7A11, impairing GSH synthesis and sensitizing cells to ferroptosis [72]. Collectively, these findings illustrate an intricate network in which copper ions modulate cell fate by targeting multiple nodes of the ferroptotic pathway, presenting a promising avenue for therapeutic interventions.
Chemotherapy-induced Cardiac Insult: Copper Homeostasis Disruption
Chemotherapy-induced cardiotoxicity encompasses a range of cardiovascular complications, including cardiomyopathy, HF, and arrhythmias. Among the key causative agents, anthracyclines, such as doxorubicin (DOX), are particularly notable, as the severe cardiac damage they induce—characterized by ventricular dilatation, interstitial fibrosis, and progression to HF—significantly limits their clinical utility [73]. DOX-induced cardiotoxicity is driven by established mechanisms, such as oxidative stress, and centrally by dysregulated copper homeostasis and cuproptosis. Disruption of this critical ion balance impairs essential cardiac functions, including the maintenance of myocardial structure, electrical conduction, and contractile performance [74, 75].
This detrimental cascade begins with pathological intracellular copper accumulation. Mechanistically, DOX stabilizes the copper importer SLC31A1 (CTR1) by inhibiting its proteasomal degradation and downregulating the copper efflux transporter ATPase Copper Transporting Beta (ATP7B), leading to a net increase in cellular copper [17, 76]. The cuproptosis pathway is triggered in this copper-rich environment. DOX upregulates the expression and activity of FDX1, a reductase that converts accumulated Cu²⁺ to the more reactive Cu⁺ [77]. Furthermore, DOX upregulates the expression of key mitochondrial enzymes, including DLAT [78]. The increased abundance of lipoylated proteins, combined with the elevated labile copper pool induced by DOX, creates a permissive environment that drives cuproptosis. Cu⁺ ions directly bind to the lipoyl groups, inducing aberrant oligomerization of DLAT and other TCA cycle enzymes, promoting the loss of Fe-S cluster proteins. This cascade results in irreversible proteotoxic stress, ultimately leading to cardiomyocyte death [74]. The pivotal role of FDX1 in this pathway is underscored by experimental evidence that genetic ablation of FDX1 confers significant protection against DOX-induced cardiotoxicity in mice, preserving the left ventricular ejection fraction [76]. The intrinsic oxidative damage mediated by DOX is further amplified by Cu⁺-catalyzed Fenton-like reactions. This compounded oxidative insult exacerbates mitochondrial membrane damage, respiratory chain dysfunction and DNA damage [79, 80].
Physiological Copper Metabolism
Copper homeostasis is maintained through a tightly coordinated process involving systemic absorption, distribution, and excretion. Dietary copper absorption occurs primarily in the stomach, duodenum, and small intestine [81]. In this process, Cu²⁺ is first reduced to Cu⁺ by metalloreductases, such as duodenal cytochrome B (DCYTB) and six-transmembrane epithelial antigen of the prostate (STEAP). The resulting Cu⁺ is then transported to the apical membrane of intestinal cells via copper importers, including CTR1/SLC31A1, CTR2, and divalent metal transporter 1 (DMT1) [82]. Subsequently, the copper-transporting ATPase ATP7A facilitates copper efflux from these cells into the portal circulation, a process that is upregulated during copper deficiency to enhance absorption [83]. ATP7A, which mediates dietary copper absorption in the intestine, and ATP7B, which promotes hepatic copper excretion, exert complementary functions that are critical for maintaining systemic copper homeostasis [84].
The liver is the central hub for systemic copper regulation. Dietary copper, which is bound to albumin, enters hepatocytes via the portal vein [85]. Within the liver, the closely related ATPase ATP7B performs two critical location-dependent functions: under normal conditions, it loads copper onto CP in the Golgi apparatus for secretion into the bloodstream, and during copper excess, it traffics to the biliary canaliculus to expel excess copper into the bile [86, 87]. This biliary pathway is the primary and irreversible route of copper elimination [88]. The entire system is dynamic; high copper intake downregulates intestinal absorption and upregulates biliary excretion, whereas deficiency states promote intestinal uptake and reduce biliary loss to conserve copper [89].
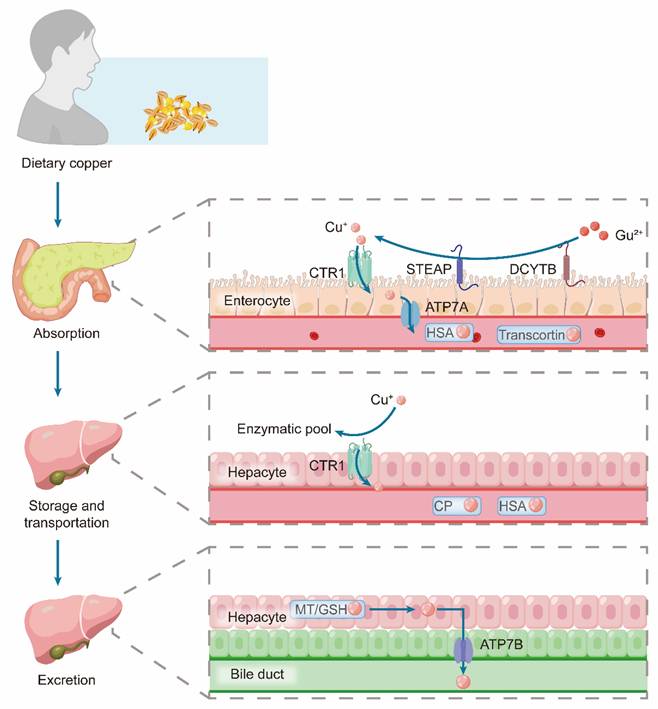
At the cellular level, a sophisticated chaperone network minimizes cytotoxic free copper levels. Upon entry via CTR1, copper is buffered by GSH or MTs [90]. The cytoplasmic chaperone Antioxidant 1 (ATOX1) then distributes copper to the ATP7A and ATP7B transporters in the trans-Golgi network for incorporation into cuproenzymes [91]. Under copper overload, these transporters relocate to vesicles or the plasma membrane to mediate the efflux [92]. Beyond its cytoplasmic role, ATOX1 can translocate to the nucleus and function as a copper-dependent transcription factor, potentially regulating pathways such as HIF-1α signaling [93] (Figure 4).
Schematic representation of copper metabolism in the body, including absorption, storage, transport, and excretion. Dietary copper is absorbed by enterocytes, where Cu²⁺ is reduced and transported via proteins such as CTR1, STEAP, DCYTB, and ATP7A, and then carried in circulation by HSA and Transcortin. In hepatocytes, copper is taken up via CTR1 for incorporation into enzymatic pools or is transported by CP and HSA. Excretion occurs through the bile duct, with ATP7B and MT/GSH mediating copper transport into bile.
Additional chaperones target copper to specific organelles. CCS delivers copper to SOD1 in the cytoplasm and intermembrane space, whereas COX17 relays it to the mitochondria [83]. Within the mitochondria, copper is passed through secondary chaperones (SCO1, SCO2, and COX11) for incorporation into CCO, which is essential for respiratory function [94]. Proteins such as MEMO1 further fine-tune this network by suppressing ATOX1-mediated ROS under excess copper [95]. This precise regulation is critical for cardiovascular function. In the vasculature, it stabilizes endothelial NO synthase to preserve NO bioavailability and vasodilatory function [96].
Copper Homeostasis Dysregulation and Myocardial Pathological Reprogramming
The progression of cardiovascular diseases, such as myocardial infarction and HF, is driven by core pathological processes, including structural remodeling (e.g., hypertrophy and fibrosis) and metabolic reprogramming of cardiomyocytes toward a fetal-like state [97]. A growing body of evidence has identified disrupted copper homeostasis and subsequent activation of cuproptosis as key upstream drivers of this maladaptive process [98]. Pathological stressors, such as pressure overload or myocardial infarction, create an inflammatory microenvironment that elevates local copper concentrations, partly due to its release from necrotic cells and upregulated import in immune cells [99]. Excess copper exerts a dual pathological effect. First, it directly induces cardiomyocyte loss through cuproptosis, characterized by mitochondrial lipoylated protein aggregation and respiratory chain collapse [100]. Second, profound metabolic and oxidative stress from cuproptosis acts as a potent trigger for pathological myocardial reprogramming. It forces a metabolic shift from oxidative phosphorylation to glycolysis and activates established pro-reprogramming signaling pathways, such as YAP/TAZ, which exacerbate inflammatory responses and fibrosis, thereby accelerating adverse ventricular remodeling [49, 101]. In vascular smooth muscle cells (VSMCs), it disrupts the TCA cycle and provokes ROS production, leading to cellular hypertrophy and phenotypic switching, which culminates in pathological thickening of the vascular wall [102].
Conversely, copper deficiency contributes to pathology via distinct mechanisms. It impairs the activity of SOD1, exacerbating intracellular oxidative stress and sensitizing cardiomyocytes to damage [12, 103]. Furthermore, by reducing CCO activity and compromising oxidative phosphorylation, copper deficiency compels cardiomyocytes to rely more heavily on anaerobic glycolysis, thereby reinforcing the metabolic shift that underpins pathological reprogramming [104]. In summary, both excess and deficiency of copper converge to promote myocardial reprogramming and remodeling, highlighting copper homeostasis as a critical nodal point in the pathogenesis of cardiovascular diseases.
GSH Metabolism and Cuproptosis in the Heart
GSH, a tripeptide composed of glycine, cysteine, and glutamic acid, is a pivotal hub in cellular defense, functioning as both a primary antioxidant and a crucial regulator of copper ion bioavailability [105]. Its cardioprotective role is mediated primarily through two synergistic mechanisms: direct copper chelation and maintenance of redox homeostasis [106].
Beyond simple chelation, GSH also acts as a molecular chaperone, facilitating the delivery of Cu⁺ to specific copper chaperones (e.g., ATOX1) and supporting their function, particularly in copper export [107]. Crucially, the GSH/Glutaredoxin 1 system maintains the reduced state of cysteine residues within the copper-binding motifs of the transporters ATP7A and ATP7B, which is essential for their copper-transport activity [108].
These dual roles converge to inhibit cuproptosis. Mitochondrial GSH sequesters copper, preventing its binding to lipoylated proteins, such as DLAT, thereby blocking the aberrant protein oligomerization that drives this cell death pathway [109]. Consequently, intracellular GSH depletion increases the free copper pool, promotes protein oligomerization, and heightens sensitivity to copper-mediated cell death [110].
Under cardiac stress conditions, such as I/R injury, HF, or drug toxicity, GSH depletion initiates a vicious cycle of damage. The resulting increase in free copper exacerbates oxidative stress and proteotoxicity, further depleting GSH pools and impairing its metal-binding capacity [111, 112]. This cycle is amplified by copper's ability to catalyze the oxidation of catecholamines, generating additional toxic quinones and ROS that contribute to cardiotoxicity [113].
Notably, GSH's cardioprotective role extends beyond cuproptosis. As an essential cofactor for GPX4, GSH reduces lipid peroxides and inhibits ferroptosis [114]. However, this function can be compromised during copper overload, as Cu²⁺ binding to the cysteine residues of GPX4 promotes its ubiquitination and autophagic degradation, thereby disrupting lipid peroxide metabolism and sensitizing cells to ferroptosis [65]. This finding reveals a complex interactive network between copper dyshomeostasis, oxidative stress, and distinct programmed cell death pathways involved in cardiac pathology.
Lipid Metabolism and Cuproptosis in the Heart
Copper and lipid metabolism exhibit complex bidirectional interactions that critically influence the development and progression of cardiovascular diseases [115]. Dysregulation of one pathway often propagates dysfunction in another, establishing a vicious cycle that amplifies cellular injury and ultimately drives disease progression [116].
Copper exerts a profound influence on lipid metabolism, with both deficiency and excess producing distinct but detrimental effects [117]. Copper deficiency primarily disrupts lipid homeostasis by impairing the activation of key transcription factors, sterol regulatory element-binding proteins (SREBP-1 and SREBP-2), which regulate fatty acid and cholesterol synthesis [88]. This disruption leads to suppressed lipogenesis and failure to maintain normal lipid homeostasis. Furthermore, copper deficiency concurrently promotes triglyceride hydrolysis and enhances fatty acid β-oxidation, creating a state of metabolic imbalance that contributes to pathology [118]. These disturbances, compounded by impaired mitochondrial fatty acid oxidation, ultimately contribute to dyslipidemia—characterized by elevated serum triglycerides, LDL cholesterol (LDL-C), and total cholesterol—which accelerates pathological processes such as atherosclerosis and fatty liver disease [119, 120].
Copper Overload promotes cardiomyocyte injury by inducing oxidative stress, which in turn disrupts lipid metabolism [36]. The hydroxyl radicals produced via copper-catalyzed Fenton reactions mediate oxidative damage, primarily by oxidizing LDL into oxidized LDL (ox-LDL), which serves as a key driver of atherosclerotic plaque formation [121]. Moreover, copper influences lipid signaling by modulating the activity of phosphodiesterase PDE3B, an enzyme that hydrolyzes cyclic adenosine monophosphate (cAMP) to regulate lipolysis [122]. The cuproptosis-related gene PDHA1 may serve as a node linking these pathways, regulating lipid metabolism through its interaction with the PI3K-Akt-mTOR signaling pathway [123] (Figure 5).
Dual effects of copper dyshomeostasis on lipid metabolism and atherosclerotic progression. Excess copper triggers the Fenton reaction, which induces mitochondrial lipid metabolism disorders. It also modulates PDHA1 and PI3K-Akt-mTOR signaling to promote lipid metabolism disorders and PDE3B (promoting cAMP-mediated lipolysis). Excessive lipids further disrupt copper transporters/chaperones, forming a vicious cycle. Accumulated lipids drive OX-LDL production, ultimately leading to atherosclerosis. Conversely, deficient copper impairs SREBP 1/2 activity, reducing triglyceride hydrolysis and fatty acid β-oxidation (also contributing to lipid dysregulation).
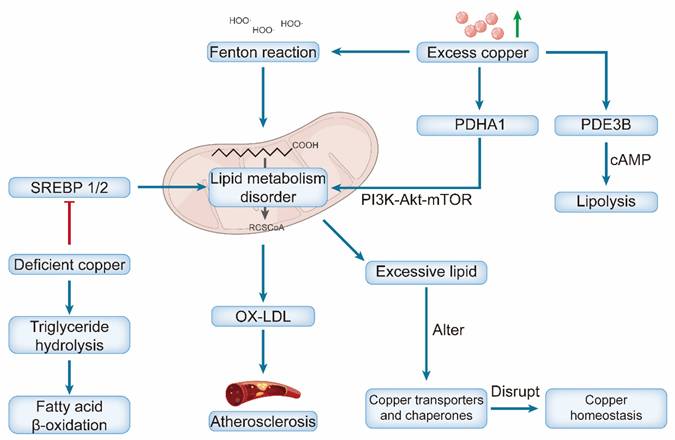
The lipid composition and fluidity of cell membranes affect the efficiency of the primary copper importer SLC31A1 (CTR1), thereby influencing the intracellular copper balance [124]. Excessive lipid accumulation disrupts copper homeostasis by altering the expression and localization of key copper transporters and chaperones [125]. The lipid microenvironment can also modulate susceptibility to cuproptosis. Under certain conditions, it may synergize with copper chaperones to facilitate efficient copper transport, thereby preventing cuproptosis [126]. More broadly, alterations in lipid metabolism, such as those mediated by short-chain fatty acids (e.g., butyric acid), can influence cell death pathways by modulating the expression of copper-related genes [127]. This establishes a feedback loop, as copper is an essential cofactor for numerous lipid metabolic enzymes; thus, initial copper deficiency can directly disrupt lipid homeostasis, which, in turn, further perturbs copper handling [116].
Central Role of Mitochondrial Metabolism in Cardiovascular Disease and Cardiac Cuproptosis
Mitochondrial metabolism serves a dual role in the cardiovascular system: it is the primary energy source for cardiomyocytes and a central integrator of overall cardiac health, and its dysfunction is a key driver of disease pathogenesis [128]. This centrality is underscored by the heart's exceptional dependence on mitochondrial oxidative phosphorylation, which supplies approximately 95% of the ATP required for contraction [23]. Consequently, mitochondrial dysfunction has been identified as a central contributor to the onset and progression of various cardiovascular diseases, including HF, myocardial ischemia/reperfusion (I/R) injury, and atherosclerosis. The progressive decline in mitochondrial function, characterized by impaired ATP synthesis, excessive ROS production, and structural abnormalities, triggers a vicious cycle involving energy crises, oxidative stress, and inflammatory responses, ultimately accelerating cardiomyocyte death [129, 130].
At the molecular level, this dysfunction mediates pathology via several interconnected pathways. The mitochondrial electron transport chain (particularly Complexes I and III) is a major site of ROS generation. Beyond directly damaging mitochondrial components, ROS oxidize LDL to form ox-LDL, which promotes atherosclerosis by driving macrophage-to-foam cell formation [131, 132]. ROS can activate pro-inflammatory pathways, such as NF-κB, thereby inducing cardiomyocyte hypertrophy, apoptosis, and myocardial fibrosis, which further exacerbate cardiac structural and functional impairments [133].
Mitochondrial calcium (Ca²⁺) homeostasis disorder represents another crucial mechanism that exhibits close crosstalk with the ROS pathway. As a key regulator of ATP synthesis, Ca²⁺ modulates enzymes such as pyruvate dehydrogenase. Abnormal Ca²⁺ influx impairs energy production and activates caspase proteins, initiating the intrinsic apoptotic pathway and accelerating cardiomyocyte loss [134, 135]. Notably, excessive ROS can further exacerbate calcium homeostasis disorders by damaging the sarcoplasmic reticulum Ca²⁺-ATPase and mitochondrial calcium uniporter, forming a malignant cascade of “excessive ROS-calcium imbalance-energy crisis” [136].
Of particular relevance, the heart is exquisitely vulnerable to copper imbalance owing to its profound reliance on mitochondrial energy production—organelles central to both copper homeostasis and the execution of cuproptosis [75, 137]. The initiation of cuproptosis critically depends on mitochondrial oxidative stress, as the oxidation of lipoyl groups on target proteins is a prerequisite for their high-affinity binding to Cu⁺, which drives toxic protein aggregation [3]. The accumulated Cu⁺ catalyzes intramitochondrial Fenton-like reactions, generating a burst of mtROS and amplifying oxidative stress. This creates feed-forward cycles that exacerbate cuproptosis: mtROS oxidize cytosolic GSH, releasing bound Cu⁺, and may upregulate the copper importer CTR1 via pathways such as NF-κB, increasing copper influx [65]. The resulting oxidative stress promotes Drp1-mediated mitochondrial fission, mPTP opening, and membrane depolarization, thereby amplifying apoptotic signaling [52, 138]. The copper-induced ROS burst, coupled with disrupted TCA cycle-derived GSH precursors, leads to irreversible GSH depletion [108, 139]. This inactivates GPX4, halting lipid peroxide clearance and driving cell death [140].
Conversely, copper deficiency impairs cardiac mitochondria via distinct mechanisms. It disrupts the copper chaperone system (e.g., COX17, SCO1/SCO2), drastically reducing CCO biosynthesis and activity. This compromises ATP production and myocardial oxygen consumption rate, which is vital for contractile function [46, 141]. Deficiency also impairs PGC-1α, the master regulator of mitochondrial biogenesis. Notably, both its loss and uncontrolled overexpression are destructive, highlighting the need for precise regulation to maintain cardiac metabolism [142, 143]. Biochemically, these failures manifest as enlarged and degenerated mitochondria with loss of cristae, driving pathological cardiac remodeling [144]. A self-amplifying vicious cycle lies at the heart of this pathology. Pre-existing mitochondrial dysfunction due to aging, diabetes, or ischemia creates a permissive environment for cuproptosis by depleting GSH and reducing SOD2 activity, thereby compromising the organelle's ability to chelate copper and neutralize mtROS [145, 146]. This expands the labile Cu⁺ pool and lowers the threshold for cuproptosis initiation. In turn, cuproptosis inflicts further mitochondrial damage, disrupting membrane integrity and metabolic pathways [147]. This reciprocal relationship establishes a bidirectional causal link that accelerates the pathogenesis of cardiovascular diseases (Figure 6).
Copper dyshomeostasis in heart disease via mitochondrial pathways. Excess copper enters via CTR1, and excess ROS reduces GSH and drives OX-LDL production, contributing to atherosclerosis, Drp1-mediated fission, mPTP opening, and ultimately cuproptosis. Copper deficiency impairs mitochondrial biogenesis (PGC-1α) and disrupts COX assembly (via COX17, SCO1, and SCO2), resulting in bioenergetic failure due to ATP deficiency.
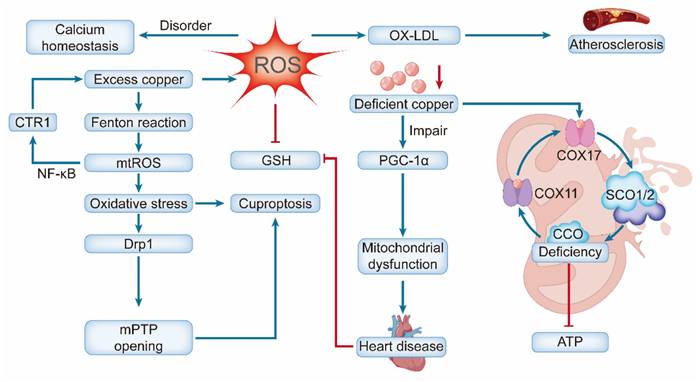
To counteract this vicious cycle, the heart is dependent on a robust mitochondrial quality-control system. This system replenishes healthy mitochondria through biogenesis, maintains network integrity via dynamics (fusion/fission), and clears damaged units through mitophagy [148]. Given their pivotal roles, targeting mitochondrial metabolism and copper homeostasis presents a promising strategy for novel therapeutic interventions in several cardiovascular diseases.
Cell-type-specific Cuproptosis in Cardiovascular System
The cardiovascular system comprises diverse cell types—including cardiomyocytes, ECs, VSMCs, and fibroblasts—each exhibiting distinct susceptibilities and responses to cuproptosis [149]. This heterogeneity is primarily determined by three interlinked factors: mitochondrial abundance, metabolic activity, and intrinsic copper handling capabilities. Consequently, cell types with high mitochondrial respiration, the primary target of copper toxicity, are disproportionately vulnerable [80].
Cardiomyocytes are the primary site of copper-induced injury because of their immense reliance on mitochondrial metabolism. The high density of mitochondria provides numerous targets for copper binding to lipoylated TCA cycle enzymes, directly disrupting the energy supply vital for contraction [75]. This intrinsic vulnerability is compounded by a weak copper export system, characterized by low expression of ATP7A/B transporters, which facilitates toxic accumulation [100]. In pathologies such as myocardial I/R injury, this confluence of factors leads to extensive cuproptosis, directly worsening systolic function [150].
ECs exhibit significant susceptibility, which is dictated more by their physiological context than their metabolic rate. Their direct contact with circulating copper and pro-oxidants (e.g., ox-LDL), combined with limited copper export capacity, creates a precarious copper balance [151, 152]. Given their role as central regulators of vascular homeostasis, EC cuproptosis can initiate widespread dysfunction, impairing barrier integrity and vasoreactivity [153].
The susceptibility of VSMCs is not fixed but is a direct function of their phenotypic state. Differentiated contractile VSMCs are relatively resistant. In contrast, synthetic, proliferative VSMCs—which dominate pathologies such as atherosclerosis—undergo a metabolic shift toward glycolysis and possess reduced antioxidant defenses, rendering them highly vulnerable to cuproptosis [154, 155]. This phenotypic targeting makes cuproptosis a potential modulator of vascular remodeling.
Fibroblasts are the most resistant cell type, protected by their low mitochondrial content and high expression of copper-buffering proteins, such as MTs [12]. However, this resilience does not render them inert. Instead, they are activated by damage-associated molecular patterns released from neighboring cardiomyocytes or ECs undergoing cuproptosis. Thus, paradoxically, fibroblast resistance fuels chronic disease progression by promoting pathological fibrosis [156].
The cell-type-specific impact of cuproptosis creates a dualistic injury pattern: it directly causes acute loss of contractile and endothelial function by eliminating cardiomyocytes and ECs, while simultaneously driving chronic maladaptive remodeling through the activation of resistant VSMCs and fibroblasts [154, 157]. This refined understanding underscores the necessity of cell-type-targeted therapeutic strategies that either protect vulnerable cells or modulate the response of resistant cells in cardiovascular diseases.
Role of Copper Homeostasis and Cuproptosis in Cardiovascular Disease
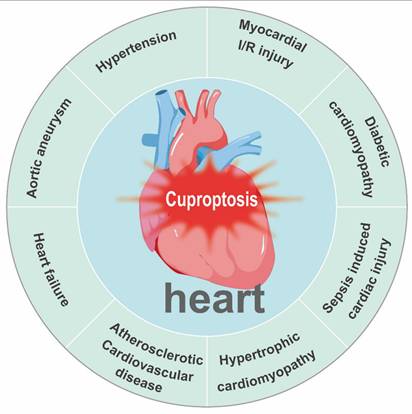
Copper acts as a quintessential “double-edged sword” in cardiovascular biology. Although it serves as an essential cofactor for critical enzymes governing antioxidant defense, energy production, and structural integrity, its dysregulation—manifesting as either deficiency or excess—is a common pathogenic thread across diverse cardiovascular conditions. These include myocardial I/R injury, diabetic cardiomyopathy (DCM), sepsis-induced cardiac injury, hypertrophic cardiomyopathy, atherosclerotic cardiovascular disease, HF, aortic aneurysm (AA), and hypertension (Figure 7). In each of these pathologies, disrupted copper homeostasis contributes to disease progression through distinct but convergent molecular mechanisms (Table 2).
Cuproptosis is associated with major cardiac and vascular diseases, including myocardial ischemia/reperfusion injury, diabetic cardiomyopathy, sepsis-induced cardiac injury, hypertrophic cardiomyopathy, atherosclerotic cardiovascular disease, heart failure, aortic aneurysm, and hypertension.
Key cuproptosis molecular changes in cardiovascular diseases
| Disease/condition | FDX1 | DLAT | SLC31A1 (CTR1) | ATP7A/B expression | Key pathological outcome | Key evidence and proposed mechanisms | Ref. |
|---|---|---|---|---|---|---|---|
| Myocardial I/R injury | ↑ | ↑ | ↑ | - | Increased cardiomyocyte cuproptosis; larger infarct | Reperfusion triggers intense oxidative stress and mitochondrial dysfunction, leading to coordinated upregulation of copper uptake and cuproptosis execution, resulting in secondary cardiomyocyte death. | [17] |
| Doxorubicin induced cardiotoxicity | ↓ | ↑ | ↑ | ATP7B ↓ | Mitochondrial injury and HF progression | Doxorubicin directly targets mitochondria, inducing severe oxidative stress and clearly activating the FDX1/DLAT-mediated cuproptosis pathway. | [74] |
| Diabetic cardiomyopathy | ↑ | ↑ | ↑ | ↓ | Impaired contractility and myocardial fibrosis | Hyperglycemia and metabolic disorders cause persistent mitochondrial stress, creating a pro-cuproptotic environment. The coordinated upregulation accelerates cardiomyocyte loss and fibrosis. | [169] |
| Sepsis-induced cardiomyopathy | ↑ | - | ↑ | Systemic inflammation and oxidative stress can activate FDX1 in the heart, leading to myocardial injury and functional impairment. | [270] | ||
| Atherosclerotic cardiovascular disease | ↑ | - | ↑ | ATP7A↑ | Endothelial dysfunction | In vascular endothelial and smooth muscle cells, inflammation and oxidative stress can activate cuproptosis, promoting cell death. | [187] |
| Heart failure | ↓ | ↑ | ↓ | ATP7A↑ | Progressive failure of the pump function | Heart failure and fibrosis of the heart were more obvious, suggesting the existence of a large number of cardiomyocyte loss. | [100] |
| Aortic aneurysm | ↓ | ↑ | - | - | Elastic fiber rupture, loss of smooth muscle cells | Cuproptosis is activated in vascular smooth muscle cells (VSMCs), leading to VSMC loss, which weakens the aortic wall. | [271] |
| Hypertension | ↓ | ↑ | ↑ | ATP7A↓ | Cardiac hypertrophy and myocardial fibrosis | Pressure overload leads to hypertrophy and mitochondrial dysfunction | [12] |
Copper Homeostasis and Cuproptosis in Myocardial ischemia/reperfusion Injury
Copper homeostasis is critically involved in the pathophysiology of myocardial I/R injury, with both deficiency and excess exacerbating damage through distinct mechanisms [158]. The acute inflammatory and oxidative stress that characterizes I/R injury is profoundly influenced by cellular copper status [159].
Copper deficiency impairs the heart's intrinsic defense mechanisms, thereby exacerbating I/R injury. It disrupts antioxidant defenses by reducing the activity of copper-dependent enzymes, such as CCS, leading to uncontrolled oxidative stress and intensified inflammatory responses [160, 161]. Chronic deficiency may elevate the baseline risk of ischemia by impairing vascular elasticity (via compromised LOX function) and promoting platelet aggregation [162]. Mechanistically, aberrant copper homeostasis during I/R impairs Fe-S cluster integrity, increases ROS production, and depletes GSH, culminating in synergistic proteotoxic and oxidative damage [160]. Supporting this, therapeutic strategies aimed at correcting deficiencies or restoring copper-dependent signaling have shown promise. In preclinical models of I/R injury, copper supplementation improves functional recovery by mitigating oxidative damage and enhancing mitochondrial bioenergetics [96]. In animal models, low-dose copper sulfate intake exerts potent antioxidant, anti-inflammatory, and anti-proliferative effects, attenuating I/R-induced tissue damage [163]. Similarly, controlled administration of copper ions can protect against tissue death by mitigating oxidative stress and inflammation, as evidenced by reduced lipid peroxidation and enhanced antioxidant reserve levels [164].
In contrast, copper overload causes damage through direct cytotoxic mechanisms. Excess copper can promote ferritin depletion and disrupt iron metabolism, thereby sensitizing cardiomyocytes to ferroptosis, a process that synergistically aggravates I/R injury [67]. This is supported by clinical observations; serum copper and CP levels rise post-myocardial infarction, indicating a stress response that, when excessive, may transition from a compensatory mechanism to a contributor of damage [165]. In summary, maintaining copper levels within a strict physiological range is paramount for mitigating I/R injury. Deficiency compromises intrinsic defensive capacity, whereas overload directly instigates oxidative and proteotoxic stress, including crosstalk with ferroptosis [166]. Therapeutic interventions must be precisely calibrated to restore homeostasis without inducing toxicity.
Copper Homeostasis and Cuproptosis in Diabetic cardiomyopathy
DCM is characterized by a pathological triad of cardiomyocyte death, interstitial fibrosis, and structural remodeling, culminating in HF [167]. Beyond the established mechanisms, a pivotal contributor to DCM pathogenesis is the disruption of systemic and cellular copper homeostasis, which primarily inflicts damage through the aberrant compartmentalization of copper within cardiac tissues [80].
The imbalance in copper metabolism in DCM presents a paradox. Systemic diabetes is associated with elevated plasma and urinary copper concentrations; however, the myocardium exhibits a marked reduction in copper content [168]. This discrepancy arises from defective myocardial copper handling, as evidenced by impaired expression and trafficking of key copper chaperones, which disrupts mitochondrial copper delivery in diabetic hearts [169]. The consequent sequestration of redox-active Cu²⁺ in the extracellular compartment and certain intracellular pools is a key mediator of cytotoxicity and cardiotoxicity in diabetes [51, 170]. Evidence from human studies and experimental models has linked cuproptosis to DCM. Mechanistically, high-glucose-induced AGEs promote cuproptosis in cardiomyocytes by upregulating SLC31A1 expression, which drives pathogenic Cu⁺ accumulation [80]. This extracellular and mislocalized intracellular copper exacerbates DCM through multiple pathways, activating the TGF-β/Smad signaling pathway and amplifying oxidative stress, collectively promoting extracellular matrix deposition and fibrotic remodeling [171].
Given that direct coronary perfusion of low-concentration copper impairs cardiac function, dysregulation of copper homeostasis presents a clear and actionable therapeutic target. Accordingly, the copper chelator triethylenetetramine (TETA) has shown efficacy in markedly improving cardiac performance in preclinical models of DCM [80]. These findings underscore the fact that the core pathology stems from dysregulated copper transport and tissue distribution, not dietary copper intake [169]. Consequently, the therapeutic promise for DCM lies not in generic copper restriction but in strategies capable of rectifying this specific copper mislocalization. Approaches such as targeted chelation or restoration of chaperone function present a rational and promising avenue for halting DCM progression.
Copper Homeostasis and Cuproptosis in Sepsis-induced Cardiac Injury
Sepsis-induced cardiac dysfunction is a primary cause of death and a major determinant of poor long-term outcomes [172]. Its pathogenesis involves excessive oxidative stress, dysregulated inflammatory cascades and profound mitochondrial damage [173]. Beyond these established factors, emerging evidence suggests that the disruption of copper homeostasis and the induction of cuproptosis are critical integrative mechanisms that potentially link systemic inflammatory-metabolic stress to direct cardiomyocyte death [174].
The relevance of this pathway is underscored by the significant dysregulation of cuproptosis-related genes in experimental models of septic cardiomyopathy [126]. The septic milieu itself—characterized by a cytokine storm and metabolic acidosis—acts as a potent upstream disruptor of copper homeostasis, priming the heart for copper-dependent injury [175]. This dysregulation is systemically reflected by a marked elevation in CP, a key copper-transporting acute-phase protein and a recognized clinical biomarker of sepsis [176]. However, the pathophysiological role of elevated CP levels may be dualistic. Although it has protective antioxidant functions, its surge may also alter copper kinetics, potentially increasing the delivery of redox-active copper to tissues or disrupting its cellular utilization, thereby paradoxically exacerbating copper-mediated toxicity in cardiomyocytes [177]. The elucidation of cuproptosis as a mechanism connecting inflammatory, metabolic, and mitochondrial damage to cardiomyocyte death opens new therapeutic avenues. Consequently, targeting this pathway—for instance, by modulating systemic copper distribution or using specific cuproptosis inhibitors—represents a promising strategy for cardioprotection in sepsis.
Copper Homeostasis and Cuproptosis in Hypertrophic Cardiomyopathy
Copper deficiency is a well-established causative factor of cardiac hypertrophy, which manifests as concentric thickening of the ventricular walls and interventricular septum, resembling pressure-overload hypertrophy [178]. This pathology is primarily driven by severe mitochondrial dysfunction, which impairs energy production and triggers compensatory hypertrophic responses [179]. This process is exacerbated by dysregulated vascular endothelial growth factor (VEGF) and ROS generated via the Fenton reaction, collectively promoting maladaptive remodeling [180].
In a pressure overload-induced cardiomyocyte hypertrophy model, COX17 deletion inactivated CCO. This mitochondrial respiratory defect likely secondarily impairs MFN1-mediated mitochondrial fusion, potentially through mechanisms involving energy depletion or elevated oxidative stress [52]. Similarly, mutations in the SCO2 gene impair mitochondrial function and can cause cardiomyopathy, underscoring the critical role of copper delivery systems [181]. Beyond bioenergetics, copper ions are essential cofactors for specific signaling pathways. For instance, copper is required for the activation of MEK1 kinase, and disruption of this copper-dependent signaling contributes directly to myofibrillar disorganization and pathological hypertrophy [182, 183].
In the context of dietary copper deficiency, supplementation can restore cardiac copper levels. This restoration leads to improved mitochondrial health, enhanced CCO activity, angiogenesis (via VEGF signaling), and attenuation of cardiac hypertrophy [35, 178]. In cases of specific chaperone deficiencies (e.g., SCO2 mutations), copper-histidine therapy can improve cardiac function by restoring copper availability [184]. Targeting the downstream effectors of copper-dependent pathways is another strategy. For instance, MEK1 inhibitors have been shown to improve cardiac function and reverse myocardial fibrosis and hypertrophy [185]. In pathological states where localized or systemic copper excess contributes to hypertrophy, copper chelators can alleviate oxidative stress and subsequent remodeling [12].
Copper Homeostasis and Cuproptosis in Atherosclerotic Cardiovascular Disease
Atherosclerosis, a chronic inflammatory disease of the arterial wall, is characterized by the accumulation of lipids, inflammatory cells, and extracellular matrix, leading to plaque formation [186]. Copper homeostasis plays a complex, context-dependent role in this pathology, with both excess and deficiency contributing to disease progression via distinct mechanisms [187, 188].
Intracellular copper accumulation is a key feature of atherosclerosis. In atherosclerotic cells, such as ox-LDL-treated macrophages, copper levels are significantly elevated (by approximately 62%), providing a catalyst for redox reactions [154]. Through the Fenton reaction, redox-cycling copper ions potently catalyze hydroxyl radical formation, causing DNA damage and propagating lipid peroxidation [3]. Serum Cu²⁺ also drives the oxidation of LDL to form copper-ox-LDL, a key ligand that binds to the LOX-1 receptor on ECs to initiate plaque formation [189]. Furthermore, copper stimulates acute-phase proteins and activates the pro-inflammatory transcription factor NF-κB via ROS generation. This cascade promotes vascular inflammation, a process further amplified by upregulated CP expression [190].
Paradoxically, copper-dependent processes can also impair VSMC accumulation within the neointima, thereby affecting plaque stability [191, 192]. Moreover, copper homeostasis critically regulates the migration of VSMCs. The release of free copper ions, facilitated by transporters such as ATP7A and ATOX1, induces neointimal thickening following vascular injury, contributing directly to lesion development [193, 194]. In human studies, the expression level of the copper transporter SLC31A1 was directly correlated with atherosclerotic plaque vulnerability [42].
The pathogenic role of copper is context-dependent and exhibits a U-shaped risk curve. As described above, excess copper drives oxidative and inflammatory injuries. Conversely, copper deficiency elevates the risk of atherosclerosis by increasing cholesterol levels and reducing the activity of the copper-dependent antioxidant enzyme SOD1. This diminishes the overall antioxidant defenses and reduces NO bioavailability, leading to endothelial dysfunction [187]. The intertwined dysregulation of copper and iron metabolism further exacerbates the disease by promoting apoptosis in lipid-rich foam cells and VSMCs, which are particularly vulnerable to metal-dependent cell death [195, 196].
Epidemiological evidence shows that a high dietary copper intake is associated with a reduced incidence of coronary heart disease [39]. In animal models of copper deficiency, copper supplementation mitigated atherosclerotic lesion progression, as evidenced by reductions in lesion area, endothelial cell death, and plasma cholesterol levels [188]. Conversely, given the pro-oxidant and pro-inflammatory effects of excess copper within plaques, preclinical studies have reported that strategies utilizing copper chelators may hold potential for attenuating plaque progression, although this requires further investigation. Additionally, copper-related DNA methylation alterations are linked to an elevated risk of acute coronary syndrome, suggesting an epigenetic role in the disease [197, 198].
Copper Homeostasis and Cuproptosis in Heart Failure
HF, a complex clinical syndrome characterized by significant functional impairment and high morbidity, stems from impaired cardiac contractility and/or ventricular filling [199]. A growing body of evidence underscores the critical role of disrupted copper homeostasis in its pathogenesis, wherein both excess and deficiency of copper significantly contribute to disease progression [30].
A meta-analysis conducted by Liu et al. [200] confirmed that elevated serum copper levels have been repeatedly are associated with an increased risk of HF. A meta-analysis of 1,504 individuals confirmed a positive association between high serum copper levels and HF [201]. Clinically, elevated copper levels and their carrier protein CP are significant predictors of adverse outcomes, including mortality, in patients with HF [202]. The underlying pathophysiology involves intracellular copper accumulation in cardiomyocytes, which directly induces cuproptosis [100]. Copper overload impairs mitochondrial electron transport, leading to excessive ROS production and a diminished antioxidant capacity. This is notably exacerbated by increased hydrogen peroxide production at the flavin site of mitochondrial complex IV [203]. The resultant oxidative damage triggers an inflammatory response characterized by the upregulation of acute-phase proteins, such as CP, further exacerbating cardiac dysfunction [204].
Paradoxically, copper deficiency worsens HF. Experimental studies have demonstrated that a copper-deficient diet exacerbates myocardial dysfunction in HF models [205]. In mouse models, copper deficiency impairs cardiac function, manifesting as reduced diastolic and systolic performance alongside a diminished inotropic response to beta-adrenergic stimulation, which compromises the heart's ability to adapt to stress [206].
This dual pathophysiology suggests the need for targeted therapeutic strategies. In states of copper overload, interventions that remove excess copper or restore normal cellular copper transport have shown promise in improving cardiac and mitochondrial function in preclinical studies [51]. In contrast, copper supplementation can fully restore cardiac function and normalize the β-adrenergic response in deficiency states [206]. In summary, maintaining precise copper homeostasis is essential for cardiac function. Therapeutic strategies that correct specific imbalances—whether through chelation in overload or supplementation in deficiency—hold significant promise for mitigating HF progression.
Copper Homeostasis and Cuproptosis in Aortic Aneurysm
AA, a life-threatening pathological dilation of the aortic wall and the ninth leading cause of death globally, arises from a complex interplay of factors, including inflammation, apoptosis, and oxidative stress [207]. Emerging evidence firmly establishes that the disruption of copper homeostasis is a significant contributor to this process, driving aortic wall degeneration through interconnected pathways involving oxidative damage, inflammation, vascular dysfunction, and metabolic defects [208].
A pivotal initiating event is the downregulation or dysfunction of the key copper exporter, ATP7A. This defect leads to intracellular copper accumulation, exacerbating local oxidative stress and promoting aneurysm progression. Part of this effect is mediated by the upregulation of miR-125b, which amplifies copper-dependent pro-inflammatory signaling [209]. The accumulated copper ions function as potent catalysts for destructive processes within the aortic wall. They directly drive lipid peroxidation and inhibit essential enzymes, compromising vascular integrity [210]. Furthermore, copper imbalance disrupts the critical equilibrium that governs NO synthesis and degradation. As NO is a central regulator of vascular tone and remodeling, this disruption promotes the pathological dilation characteristic of aneurysms [211]. Underlying mitochondrial defects, often linked to disturbances in the TCA cycle, can be worsened by copper imbalance, further contributing to the energetic deficit and cellular stress that propel AA progression [212]. The delineation of these copper-mediated pathways has revealed promising therapeutic targets. Strategies aimed at correcting copper transport (e.g., by restoring ATP7A function) or mitigating the downstream consequences of excess copper (e.g., using targeted antioxidants) could potentially slow or halt aortic wall degeneration.
Copper Homeostasis and Cuproptosis in Hypertension
Clinical and experimental evidence confirms a U-shaped relationship between copper levels and blood pressure, indicating that both copper deficiency and excess are associated with a higher prevalence of hypertension [213]. This duality reflects the distinct pathogenic mechanisms at each extreme of copper homeostasis.
On the one hand, copper deficiency promotes hypertension primarily through impaired antioxidant defense and vascular dysfunction. Deficiency inactivates the copper-dependent antioxidant enzyme SOD1, leading to increased vascular oxidative stress. This oxidative environment uncouples endothelial NO synthase, reducing NO bioavailability and impairing vasodilation—a direct pathway to increased vascular resistance [214, 215]. Furthermore, as copper is a physiological inhibitor of angiotensin-converting enzyme, its deficiency disinhibits the renin-angiotensin-aldosterone system, further exacerbating hypertension [216, 217]. Consistent with this mechanistic link, lower blood copper levels have been observed in hypertensive animal models [215].
Copper dysregulation and excess copper also contribute to disease progression. In the vasculature, cuproptosis directly induces EC death and drives VSMCs toward a pro-inflammatory synthetic phenotype. This dual insult exacerbates endothelial dysfunction and promotes pathological vascular remodeling and sclerosis, creating a vicious cycle that elevates blood pressure [198, 218]. In the heart under pressure overload, intracellular copper accumulation can trigger cardiomyocyte cuproptosis via FDX1 activation, leading to lipoylated protein aggregation and mitochondrial damage, thereby exacerbating cardiac injury [12].
Population-based studies have corroborated this complex relationship. A cross-sectional study reported demographic variations and a potential independent association between serum copper and hypertension [219]. Notably, the protective negative association between dietary copper intake and myocardial infarction appears particularly strong among patients with hypertension, underscoring the clinical importance of maintaining optimal copper status in this high-risk group [39]. In summary, precise copper homeostasis is crucial for maintaining normal vascular tone and cardiac health, and deviations in either direction contribute to the pathogenesis and complications of hypertension.
Clinical Application of Copper Targeting Strategy for Cardiovascular Disease
Targeting dysregulated copper homeostasis represents a promising frontier in cardiovascular therapy, offering a mechanistic approach that is distinct from conventional treatments. Current strategies aim to restore balance through two principal means: chelation to mitigate copper overload or supplementation to correct deficiency, both with the goal of re-establishing metabolic equilibrium. In contrast, conventional therapies, such as β-blockers, statins, and ACEIs/ARBs, primarily target neurohormonal pathways, lipid metabolism, and hemodynamics [220] (Table 3). Therefore, modulating copper homeostasis and cuproptosis addresses a novel pathogenic axis that is not directly targeted by existing first-line drugs, suggesting the potential for synergistic or personalized therapeutic strategies.
Characteristics of conventional cardiovascular drugs and copper metabolism-related drugs
| Class of agent | Representative drugs | Core mechanism | Indications | Key adverse effects | Ref. |
|---|---|---|---|---|---|
| Conventional drugs | Metoprolol (β-blocker) | Blocks β-adrenergic receptors to inhibit sympathetic nervous system excitation | Hypertension, heart failure, and arrhythmia | Bradycardia, fatigue, and bronchospasm | [272] |
| Conventional drugs | Enalapril (ACE inhibitor) | Inhibits angiotensin-converting enzyme, reducing the production of the potent vasoconstrictor angiotensin II | Hypertension, heart failure, cardiac protection after myocardial infarction, and diabetic nephropathy | Dry cough, angioedema, hyperkaliemia, and kidney damage | [273] |
| Conventional drugs | Losartan/valsartan (ARB) | Blocks the binding of angiotensin II to its receptor | Hypertension, heart failure, and diabetic nephropathy | Hyperkaliemia and kidney damage | [274] |
| Conventional drugs | Atorvastatin (statin) | Inhibits HMG-CoA reductase and lowers LDL cholesterol | Atherosclerosis, atherosclerotic cardiovascular disease (ASCVD) | Muscle soreness or myopathy, elevated liver enzymes, and risk of new-onset diabetes | [275] |
| Copper metabolism-Targeted agents | TTM (trientine tetrahydrochloride) and TETA (trientine) | Chelates labile Cu⁺ and reduces bioavailable copper | Copper metabolic disorders | Copper deficiency, bone marrow suppression, and kidney damage | [276] |
| Copper metabolism-Targeted agents | Elesclomol (copper ionophore, repurposed) | Binds to extracellular copper ions (Cu²⁺) to form complexes that enter cells | Cancer treatment | Oxidative stress-related toxicity and mitochondrial dysfunction | [58] |
Copper Chelators
Copper chelators are therapeutic agents that bind copper ions to form stable complexes, alleviating cardiovascular pathologies, primarily by reducing the labile intracellular copper pool. This inhibits cuproptosis and associated oxidative damage by preventing ROS generation [80] (Table 4). Furthermore, the reduction in bioavailable copper impairs the function of copper-dependent proteins and signaling pathways, including SOD1, copper chaperones (ATOX1, ATP7A), and transcription factors, such as HIF-1α and NF-κB [221]. The overarching cardioprotective effect is achieved through the restoration of mitochondrial integrity and function, as evidenced by the recovery of key components (e.g., SCO1, CCO, and SOD1) and upregulating the biogenesis regulator PGC-1α [51, 204]. The benefits of this approach have been validated in several disease models. Copper chelation inhibits pivotal VSMC migration in intimal hyperplasia [194] improves recovery after myocardial infarction [222] and reduces neointimal formation and atherosclerosis in ApoE-deficient mice [223]. Overall, by chelating excess copper, these agents help to restore metabolic homeostasis and protect cardiovascular cells.
Copper chelators for use in clinical trials
| Conditions | Phases | Primary Objective | Results | Enrolled | ClinicalTrials.gov identifier | Status | Organizing Location |
|---|---|---|---|---|---|---|---|
| Wilson Disease | The period of the day best correlated with 24 h urinary copper excretion | 30 | NCT06430359 | Recruiting | France | ||
| Head and Neck Cancer | II | The safety and efficacy of penicillamine (a common copper chelator) | 10 | NCT06103617 | Recruiting | China | |
| Wilson Disease | Assess copper parameters in participants with Wilson disease | 64 | NCT02763215 | Completed | United States/ Austria/Germany/ Poland/United Kingdom | ||
| Wilson Disease | The clinical efficacy and safety of trientine | 48 | NCT03299829 | Completed | Taiwan | ||
| Idiopathic Pulmonary Fibrosis | I/II | The safety of the administration of a copper chelating agent, tetrathiomolybdate | The primary endpoint is safety with secondary endpoints including change in pulmonary function, exercise capacity, and quality of life | 23 | NCT00189176 | Completed | United States |
| Psoriasis Vulgaris | II | The safety and efficacy of Tetrathiomolybdate in psoriasis therapy | 10 | NCT00113542 | Completed | United States | |
| Wilson Disease | I/II | the safety of single IV doses of UX701 | 82 | NCT04884815 | Active, not recruiting | United States/Canada/ Portugal/Spain/ United Kingdom | |
| Wilson Disease | Not Applicable | A single daily treatment with trientine is as effective or better than a patient's current maintenance therapy | The primary endpoint is the demonstration of equivalence to a patient's prior therapy. Secondary endpoints include: 1) demonstration of stability or improvement in parameters of copper metabolism; 2) improvement in adherence to therapy; and 3) no progression of liver disease | 8 | NCT01472874 | Completed | United States |
| Epithelial Ovarian Cancer | I/II | copper chelator in conjunction with cytotoxic agents to conquer platinum-resistance | 18 | NCT03480750 | Completed | Taiwan |
Tetrathiomolybdate (TTM) is a highly selective copper chelator that prevents copper accumulation and toxicity in vivo by sequestering excess copper ions [224]. TTM inhibits their uptake and delivery by chaperone proteins to downstream cuproenzymes, such as LOX, inducing functional defects in these enzymes [225]. The efficacy of TTM stems from its ability to modulate copper bioavailability across diverse cardiovascular disease models. It attenuates atherosclerosis in ApoE⁻/⁻ mice by reducing bioavailable copper and suppressing vascular inflammation [198] and protects against AAs in ATP7A-deficient mice by inhibiting endothelial ROS [209]. In the heart, TTM improves myocardial injury in diabetes and after ischemia by enhancing mitochondrial protein activity and reducing infarct size [80]. It also alleviates chronic stress-induced myocardial fibrosis by regulating the cardiac copper levels [49]. Additionally, TTM inhibits TNF-α-induced activation of NF-κB and AP-1, leading to the suppressed expression of adhesion molecules (VCAM-1 and ICAM-1) and the chemokine MCP-1. This attenuates endothelial activation and contributes to the inhibition of atherosclerotic progression [226, 227].
TETA, typically administered as a dihydrochloride salt, restores cardiac function by rectifying copper homeostasis and repairing mitochondrial integrity. Its primary mechanism involves restoring the activity of key mitochondrial enzymes, such as SOD1, CCS, and CCO, thereby re-establishing metabolic function [51]. Animal studies have validated this efficacy, showing that TETA therapy enhances cardiac pumping efficiency and normalizes the levels of copper, copper-binding proteins, and CCO in the myocardial tissue [51]. Furthermore, it ameliorates hypertrophic cardiomyopathy and reverses diabetes-induced mitochondrial damage by restoring the expression and function of crucial cardiac energy metabolic proteins [37, 228].
Small-molecule Inhibitors of Copper Chaperone Proteins
The clinical utility of conventional copper chelators is limited by dose-dependent toxicities, primarily stemming from systemic copper deficiency and off-target chelation of essential metals, such as zinc and iron. This underscores the pressing need for novel agents capable of selectively modulating intracellular copper homeostasis without systemic depletion [46]. Emerging strategies focus on precise molecular targets within copper transport machinery.
A key approach involves the development of small molecules that directly inhibit intracellular copper trafficking. The drug DCAC50 exemplifies this strategy by binding to the copper chaperones ATOX1 and CCS, thereby disrupting copper delivery and its dependent signaling without harming normal cells [229, 230]. The copper chaperone ATOX1 is a particularly compelling target in this context. Beyond its cytoplasmic transport role, ATOX1 is critically involved in vascular pathology; it promotes VSMC migration—a key process in neointimal and atherosclerotic lesion formation—and recruits inflammatory cells [93, 194]. Mechanistic insights have revealed that ATOX1 translocates to the nucleus in response to inflammatory cytokines or copper ions [93]. Moreover, in TNF-α-stimulated ECs, the copper-dependent ATOX1-TRAF4 interaction promotes nuclear translocation and initiates ROS-dependent inflammatory responses, solidifying this axis as a promising therapeutic target [45].
In contrast, a distinct pharmacological strategy employs copper ionophores, such as elesclomol. Instead of inhibiting copper flux, these agents form complexes with copper to facilitate cellular uptake. Their pro-oxidative action exploits copper to induce selective toxicity in target cells, representing a different therapeutic logic that is useful in specific contexts [231, 232]. Together, these approaches—precise inhibition of copper chaperones and conditional use of copper ionophores—represent innovative strategies for targeting copper dyshomeostasis in cardiovascular and other diseases.
Copper Ionophore
Copper ionophores are small molecules that enhance intracellular copper bioavailability by shuttling copper across the cell membrane. Primarily used to treat research overload or exploit copper toxicity in therapy, this class includes disulfiram and pyrithione [233].
Their intracellular effects are complex and context dependent. Some ionophores can increase NO levels and reduce pro-inflammatory cytokine levels, indicating their anti-inflammatory potential [234]. Others, such as members of the 8-hydroxyquinoline family, can synergize with SOD transporters to mitigate intracellular ROS, thereby protecting against oxidative stress and apoptosis [235]. A central therapeutic challenge is their lack of cell-type specificity, which leads to off-target effects [236]. To overcome this limitation, targeted delivery systems are being developed. For example, Su et al. [237] conjugated N-acetylgalactosamine to a copper ionophore to create Gal-Cu, a liver-targeting system. This design enhances copper delivery to hepatocytes while reducing exposure to nontarget organs, thereby minimizing systemic toxicity.
However, the use of ionophores remains a pharmacological double-edged sword. Cells possess adaptive homeostatic mechanisms, such as upregulation of the copper efflux transporter ATP7B, to counteract increased influx [238]. Excessive ionophore administration can overwhelm these defenses, leading to systemic copper poisoning, characterized by hepatotoxicity, nephrotoxicity, and hematopoietic suppression [239]. Therefore, the therapeutic window for ionophores is narrow, and precision in their delivery and dosing is paramount.
Limits and Challenges
Conventional copper-modulating therapies, including chelators (e.g., TTM and TETA) and the ionophore elesclomol, are fundamentally limited by their lack of specificity, which constrains their clinical utility and leads to significant adverse effects. The most critical drawback is the inability to precisely target diseased tissues or cells. Chelators non-selectively deplete systemic copper, harming healthy organs that require copper for normal function and leading to systemic deficiency [240]. Conversely, ionophores such as elesclomol lack cellular specificity, delivering copper indiscriminately and risking overload in healthy tissues [241]. Furthermore, many chelators lack selectivity for copper over other essential metals, causing off-target depletion of ions such as zinc and iron, which compounds their toxicity [242].
The non-selective action of these agents impairs the functions of vital cuproenzymes. In cardiomyocytes, a reduction in CCO activity compromises mitochondrial energy production [243]. Systemically, copper deficiency disrupts iron metabolism by impairing CP ferroxidase activity [244]. The most common clinical manifestations of this broad physiological disruption are bone marrow suppression and anemia. Anemia is multifactorial, arising from defective CCO activity in erythrocyte precursors (normocytic anemia) and, in the case of TTM, from CP dysfunction, leading to iron retention and sideroblastic features [224, 244]. Some chelators, such as D-penicillamine, also cause direct neurotoxicity, likely by crossing the blood-brain barrier and disrupting copper-dependent central nervous system enzymes, impairing energy metabolism, and increasing oxidative stress [245, 246].
These therapies have a narrow therapeutic window, indicating that their efficacy and toxicity are critically dose dependent. For example, excessive doses of the copper ionophore elesclomol can flood cells with copper, overwhelming antioxidant defenses and triggering lethal oxidative stress [247] whereas excessive copper supplementation can cause overload and ROS-mediated damage [248]. Moreover, patient-specific factors significantly alter the risk. In DCM, upregulation of the copper importer SLC31A1 makes patients exceptionally sensitive to chelators, whereby a standard dose can precipitate severe deficiency [240]. Concurrent liver dysfunction, by impairing copper excretion, further increases the risk of accumulation and adverse cardiovascular events [242, 249].
Conventional in vitro systems, such as primary cardiomyocytes and H9c2 cell lines, have significant limitations in studying copper metabolism. They lack the systemic context necessary to recapitulate inter-organ crosstalk (e.g., hepatic regulation of cardiac copper) and are inherently static, making them unsuitable for modelling the gradual pathogenesis of chronic copper imbalances. Consequently, their utility is largely confined to studies on acute toxicity or transient metabolic alterations [250, 251]. The simplified 2D microenvironment—devoid of a 3D matrix, vasculature, and key factors such as CP—further leads to dysregulated copper transporter expression and non-physiological toxicity responses [252]. Although animal models provide a more integrated physiological context, their translational value is limited by interspecies differences in copper metabolism and the difficulty in modelling human-specific gene-environment interactions [253]. A major shortcoming is their frequent failure to replicate human comorbidities (e.g., hypertension and diabetes), which profoundly alters copper homeostasis and cell death pathways. This discrepancy likely underlies the repeated failure of copper-targeted interventions in clinical translation [254]. Thus, a critical appreciation of these inherent limitations at each stage—from cell culture to animal models—is paramount for rigorously interpreting data and designing studies with genuine clinical relevance.
Future Directions
Building upon a critical understanding of the limitations inherent in current therapies and research models, the future of this field is unequivocally directed toward the development of precision strategies. The cornerstone of this approach is achieving precise spatiotemporal control over drug delivery and action, which is fundamentally dependent on guidance from a robust biomarker framework for accurate patient stratification. Future breakthroughs will depend on the deep integration of targeted delivery technologies with this biomarker system.
To overcome the drawback of the nonspecific systemic distribution exhibited by current agents, the development of delivery systems capable of precisely targeting diseased cardiac cells is paramount. Nanoparticles decorated with ligands for cardiomyocyte-specific receptors enable passive targeting (e.g., the EPR effect), active targeting, and intelligent drug release in response to the pathological microenvironment (e.g., pH and enzymes), ensuring drug enrichment and localized release at the cardiac site [255, 256]. Designing prodrugs that are inactive in the systemic circulation but are selectively activated by enzymes overexpressed in diseased tissues (e.g., cathepsins and matrix metalloproteinases) allows for the localized release of active copper-modulating agents, thereby minimizing off-target effects [257]. Utilizing tissue-specific promoters to drive the localized expression of copper transporters or chelating proteins within specific organs (e.g., the heart) allows for fine-tuned modulation of local copper homeostasis without disrupting systemic balance [258].
The clinical translation of the aforementioned precision strategies urgently requires a biomarker system capable of dynamically and specifically reflecting disruptions in copper homeostasis and their pathological consequences. Serum CP, as the primary copper transport protein, reflects systemic copper status, inflammation, and oxidative stress. Its elevation in conditions such as HF signals an underlying copper and redox imbalance [259, 260]. Non-ceruloplasmin-bound copper (NCC) represents the redox-active “free” copper fraction, which is a key driver of oxidative injury and cuproptosis. An increase in NCC may indicate an acute pathological risk, even when total copper levels are within the normal range [261]. Soluble SLC31A1 reflects the activity of cellular copper uptake via CTR1. Elevated levels may indicate increased tissue susceptibility to copper-mediated damage, particularly when cellular compensatory mechanisms, such as copper buffering or efflux, are impaired [262]. The high expression of copper-related genes, such as FDX1 and SLC31A1, in metabolic organs, such as the liver and intestine, poses a challenge in interpreting their specific roles in cardiovascular pathology, underscoring the need for tissue-specific models [263]. Integrating these copper-specific markers (e.g., CP and NCC) with established cardiac biomarkers, such as BNP and troponin, can help identify a “high copper risk” patient subgroup, thereby informing personalized treatment decisions [264]. Furthermore, dynamic monitoring of these biomarkers holds promise for elucidating the role of cuproptosis in disease progression, particularly in patients refractory to standard therapies.
The key to advancing the field of copper-targeted therapy lies in the systematic coupling of innovative spatially precise delivery technologies with a dynamic multi-level biomarker framework. Only through such an integrated strategy can the modulation of copper homeostasis be transformed from a blunt systemic intervention into a truly precise and personalized therapeutic approach for treating cardiovascular diseases. Future research must focus on facilitating the clinical validation and translation of these technologies, ultimately enabling the safe and effective use of copper for the prevention and treatment of cardiovascular diseases.
Conclusions
Copper homeostasis is a fundamental pillar in sustaining cardiovascular health, and its dysregulation is a pivotal driver of diverse cardiovascular pathologies. This review systematically unravels the intricate interplay between copper metabolism and cuproptosis, a unique copper-dependent RCD pathway, in the onset and progression of cardiovascular diseases. From myocardial I/R injury to DCM, atherosclerosis, and HF, cuproptosis mediates pathogenic processes through mechanisms including mitochondrial dysfunction, proteotoxic stress, GSH depletion, and lipid metabolism disruption.
Notably, the dual nature of copper imbalance—both deficiency and excess—contributes to cardiovascular damage via distinct yet interconnected pathways. Copper deficiency impairs mitochondrial respiration, antioxidant defense, and vascular structural integrity, whereas copper overload triggers oxidative stress, inflammatory responses, and aberrant activation of cell death signaling. Modulating copper homeostasis, either through chelation to mitigate overload or supplementation to correct deficiency, has demonstrated therapeutic potential for alleviating tissue damage and improving cardiac function in preclinical models.
The translational value of targeting cuproptosis and copper metabolism lies in the identification of key regulatory molecules (e.g., FDX1, DLAT, ATP7A/B) and potential biomarkers (e.g., serum CP and NCC) that could guide clinical decision-making. However, current challenges, such as the lack of tissue-specific targeting of copper-modulating agents and interspecies differences in preclinical models, necessitate further refinement of therapeutic strategies.
Future research on cuproptosis in cardiovascular diseases should advance along these critical paths. First, elucidating the cell-type-specific sensitivities and regulatory mechanisms of cuproptosis in major cardiac cells, including cardiomyocytes, ECs, and fibroblasts, is essential. Furthermore, understanding how copper homeostasis is dysregulated in comorbid conditions (e.g., diabetes and HF) will provide a more comprehensive pathophysiological perspective. Translational efforts should focus on developing cardiac-specific modulators of copper homeostasis, such as small-molecule inhibitors targeting the copper transporter CTR1 in cardiomyocytes. Concurrently, screening for clinically applicable biomarkers—such as serum NCC levels or cardiomyocyte-enriched microRNAs—is crucial for patient stratification and monitoring.
Acknowledgements
Funding
This work was supported by the National Natural Science Foundation of China (grant numbers 82202989, 82573498); the Noncommunicable Chronic Diseases-National Science and Technology Major Project (grant number 2023ZD050610X/2023ZD0506100 to W Li); 135 Project of State Key Laboratory of Respiratory Health and Multimorbidity, West China Hospital, Sichuan University (grant number RHM24211); and the Noncommunicable Chronic Diseases-National Science and Technology Major Project (grant numbers 2024ZD0520000, 2024ZD0520006); 135project for disciplines of excellence-Clinical Research Fund, West China Hospital, Sichuan University(grant number 2025HXFH030).
Author Contribution Statement
Xianzhe Yu, Yabo Wang, and Leibo Wang contributed to the conception of the study.
Xianzhe Yu and Lingling Zhu drafted the manuscript and prepared the figures.
Dou Yuan, Qi An and Yunfei Ling revised the manuscript.
All the authors approved the final version of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Qu Z, Pang X, Mei Z, Li Y, Zhang Y, Huang C. et al. The positive feedback loop of the NAT10/Mybbp1a/p53 axis promotes cardiomyocyte ferroptosis to exacerbate cardiac I/R injury. Redox biology. 2024;72:103145
2. Chen K, Zhou A, Zhou X, He J, Xu Y, Ning X. Cellular Trojan Horse initiates bimetallic Fe-Cu MOF-mediated synergistic cuproptosis and ferroptosis against malignancies. Science advances. 2024;10:eadk3201
3. Tsvetkov P, Coy S, Petrova B, Dreishpoon M, Verma A, Abdusamad M. et al. Copper induces cell death by targeting lipoylated TCA cycle proteins. Science (New York, NY). 2022;375:1254-61
4. Boaru DL, Leon-Oliva D, Castro-Martinez P, Garcia-Montero C, Fraile-Martinez O, García-González B. et al. Cuproptosis: Current insights into its multifaceted role in disease, cancer, and translational/therapeutic opportunities. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2025;190:118422
5. Kahlson MA, Dixon SJ. Copper-induced cell death. Science (New York, NY). 2022;375:1231-2
6. Lu Y, Li C, Liu Y, Wu T, Lu P. Association between late age-related macular degeneration and dietary intake of copper, iron, zinc and selenium: a 2005-2008 NHANES cross-sectional observational study. BMC ophthalmology. 2025;25:327
7. Guo H, Ouyang Y, Wang J, Cui H, Deng H, Zhong X. et al. Cu-induced spermatogenesis disease is related to oxidative stress-mediated germ cell apoptosis and DNA damage. Journal of hazardous materials. 2021;416:125903
8. Li K, Wu L, Wang H, Fu Z, Gao J, Liu X. et al. Apoptosis and cuproptosis Co-activated Copper-based metal-organic frameworks for cancer therapy. Journal of nanobiotechnology. 2024;22:546
9. Ni C, Li D. Ferroptosis and oxidative stress in endometriosis: A systematic review of the literature. Medicine. 2024;103:e37421
10. Deng W, Zhong L, Ye S, Luo J, Ren G, Huang J. et al. Mir22hg facilitates ferritinophagy-mediated ferroptosis in sepsis by recruiting the m6A reader YTHDC1 and enhancing Angptl4 mRNA stability. Journal of bioenergetics and biomembranes. 2024;56:405-18
11. Kunutsor SK, Dey RS, Laukkanen JA. Circulating Serum Copper Is Associated with Atherosclerotic Cardiovascular Disease, but Not Venous Thromboembolism: A Prospective Cohort Study. Pulse (Basel, Switzerland). 2021;9:109-15
12. Chen YF, Qi RQ, Song JW, Wang SY, Dong ZJ, Chen YH. et al. Sirtuin 7 ameliorates cuproptosis, myocardial remodeling and heart dysfunction in hypertension through the modulation of YAP/ATP7A signaling. Apoptosis: an international journal on programmed cell death. 2024;29:2161-82
13. Zhao G, Sun H, Zhang T, Liu JX. Copper induce zebrafish retinal developmental defects via triggering stresses and apoptosis. Cell communication and signaling: CCS. 2020;18:45
14. Tao X, Wan X, Wu D, Song E, Song Y. A tandem activation of NLRP3 inflammasome induced by copper oxide nanoparticles and dissolved copper ion in J774A.1 macrophage. Journal of hazardous materials. 2021;411:125134
15. Song X, Zhou Z, Liu J, Li J, Yu C, Zeh HJ. et al. Cytosolic cytochrome c represses ferroptosis. Cell metabolism. 2025;37:1326-43.e10
16. Zhang GY, Gao Y, Guo XY, Wang GH, Guo CX. MiR-199a-5p promotes ferroptosis-induced cardiomyocyte death responding to oxygen-glucose deprivation/reperfusion injury via inhibiting Akt/eNOS signaling pathway. The Kaohsiung journal of medical sciences. 2022;38:1093-102
17. Chen G, Wei M, Zhao CY, Zhang AY, Su JB, Ni ZR. et al. Phillygenin ameliorates myocardial ischemia-reperfusion injury by inhibiting cuproptosis via the autophagy-lysosome degradation of CTR1. Free radical biology & medicine. 2025;237:542-57
18. Blockhuys S, Celauro E, Hildesjö C, Feizi A, Stål O, Fierro-González JC. et al. Defining the human copper proteome and analysis of its expression variation in cancers. Metallomics: integrated biometal science. 2017;9:112-23
19. Li X, Ling J, Hu Q, Fang C, Mei K, Wu Y. et al. Association of serum copper (Cu) with cardiovascular mortality and all-cause mortality in a general population: a prospective cohort study. BMC public health. 2023;23:2138
20. Lei G, Sun M, Cheng J, Ye R, Lu Z, Horbath A. et al. Radiotherapy promotes cuproptosis and synergizes with cuproptosis inducers to overcome tumor radioresistance. Cancer cell. 2025;43:1076-92.e5
21. Sudhahar V, Okur MN, O'Bryan JP, Minshall RD, Fulton D, Ushio-Fukai M. et al. Caveolin-1 stabilizes ATP7A, a copper transporter for extracellular SOD, in vascular tissue to maintain endothelial function. American journal of physiology Cell physiology. 2020;319:C933-c44
22. Bertinato J, L'Abbé MR. Copper modulates the degradation of copper chaperone for Cu,Zn superoxide dismutase by the 26 S proteosome. The Journal of biological chemistry. 2003;278:35071-8
23. Ramzan R, Dolga AM, Michels S, Weber P, Culmsee C, Rastan AJ. et al. Cytochrome c Oxidase Inhibition by ATP Decreases Mitochondrial ROS Production. Cells. 2022;11:992
24. Roy S, Lutsenko S. Mechanism of Cu entry into the brain: many unanswered questions. Neural regeneration research. 2024;19:2421-9
25. Boaru DL, De Leon-Oliva D, De Castro-Martinez P, Garcia-Montero C, Fraile-Martinez O, García-González B. et al. Extracellular matrix dysregulation in aging, calcification, and cancer diseases: insights into cellular senescence, inflammation, and novel therapeutic strategies. International journal of biological sciences. 2025;21:6808-81
26. Shurygina IA, Prozorova GF, Trukhan IS, Korzhova SA, Dremina NN, Emel'yanov AI. et al. Evaluation of the Safety and Toxicity of the Original Copper Nanocomposite Based on Poly-N-vinylimidazole. Nanomaterials (Basel, Switzerland). 2021;12:16
27. Yang Z, Su W, Wei X, Pan Y, Xing M, Niu L. et al. Hypoxia inducible factor-1α drives cancer resistance to cuproptosis. Cancer cell. 2025;43:937-54.e9
28. Tan Z, Zhou B, Zheng J, Huang Y, Zeng H, Xue L. et al. Lithium and Copper Induce the Osteogenesis-Angiogenesis Coupling of Bone Marrow Mesenchymal Stem Cells via Crosstalk between Canonical Wnt and HIF-1α Signaling Pathways. Stem cells international. 2021;2021:6662164
29. Vazquez-Moreno M, Sandoval-Castillo M, Rios-Lugo MJ, Klünder-Klünder M, Cruz M, Martínez-Navarro I. et al. Overweight and Obesity Are Positively Associated with Serum Copper Levels in Mexican Schoolchildren. Biological trace element research. 2023;201:2744-9
30. Zou R, Zhang M, Zou Z, Shi W, Tan S, Wang C. et al. Single-cell transcriptomics reveals zinc and copper ions homeostasis in epicardial adipose tissue of heart failure. International journal of biological sciences. 2023;19:4036-51
31. van Rensburg MJ, van Rooy M, Bester MJ, Serem JC, Venter C, Oberholzer HM. Oxidative and haemostatic effects of copper, manganese and mercury, alone and in combination at physiologically relevant levels: An ex vivo study. Human & experimental toxicology. 2019;38:419-33
32. Khan Z, Jan R, Asif S, Farooq M, Kim KM. Exogenous GABA Enhances Copper Stress Resilience in Rice Plants via Antioxidant Defense Mechanisms, Gene Regulation, Mineral Uptake, and Copper Homeostasis. Antioxidants (Basel, Switzerland). 2024;13:700
33. Liu H, Guo H, Deng H, Cui H, Fang J, Zuo Z. et al. Copper induces hepatic inflammatory responses by activation of MAPKs and NF-κB signalling pathways in the mouse. Ecotoxicology and environmental safety. 2020;201:110806
34. Fan Y, Cheng Z, Mao L, Xu G, Li N, Zhang M. et al. PINK1/TAX1BP1-directed mitophagy attenuates vascular endothelial injury induced by copper oxide nanoparticles. Journal of nanobiotechnology. 2022;20:149
35. Bajpai AK, Gu Q, Orgil BO, Xu F, Torres-Rojas C, Zhao W. et al. Cardiac copper content and its relationship with heart physiology: Insights based on quantitative genetic and functional analyses using BXD family mice. Frontiers in cardiovascular medicine. 2023;10:1089963
36. Li Q, Liao J, Lei C, Shi J, Zhang H, Han Q. et al. Metabolomics analysis reveals the effect of copper on autophagy in myocardia of pigs. Ecotoxicology and environmental safety. 2021;213:112040
37. Farrant J, Dodd S, Vaughan C, Reid A, Schmitt M, Garratt C. et al. Rationale and design of a randomised trial of trientine in patients with hypertrophic cardiomyopathy. Heart (British Cardiac Society). 2023;109:1175-82
38. Pan M, Cheng ZW, Huang CG, Ye ZQ, Sun LJ, Chen H. et al. Long-term exposure to copper induces mitochondria-mediated apoptosis in mouse hearts. Ecotoxicology and environmental safety. 2022;234:113329
39. Wen H, Niu X, Hu L, Sun N, Zhao R, Wang Q. et al. Dietary copper intake and risk of myocardial infarction in US adults: A propensity score-matched analysis. Frontiers in cardiovascular medicine. 2022;9:942000
40. Hsiao CD, Wu HH, Malhotra N, Liu YC, Wu YH, Lin YN. et al. Expression and Purification of Recombinant GHK Tripeptides Are Able to Protect against Acute Cardiotoxicity from Exposure to Waterborne-Copper in Zebrafish. Biomolecules. 2020;10:1202
41. Chen Z, Li YY, Liu X. Copper homeostasis and copper-induced cell death: Novel targeting for intervention in the pathogenesis of vascular aging. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2023;169:115839
42. Zhou S, Wang L, Huang X, Wang T, Tang Y, Liu Y. et al. Comprehensive bioinformatics analytics and in vivo validation reveal SLC31A1 as an emerging diagnostic biomarker for acute myocardial infarction. Aging. 2024;16:8361-77
43. Hu R, Huang Y, Jiang X, Xu Y, Zheng Z, Shi Y. et al. Maternal dietary copper deficiency induces cardiomyopathy and liver injury in mice by activating autophagy. Nutrition research (New York, NY). 2024;126:1-10
44. Ramchandani D, Berisa M, Tavarez DA, Li Z, Miele M, Bai Y. et al. Copper depletion modulates mitochondrial oxidative phosphorylation to impair triple negative breast cancer metastasis. Nature communications. 2021;12:7311
45. Das A, Sudhahar V, Ushio-Fukai M, Fukai T. Novel interaction of antioxidant-1 with TRAF4: role in inflammatory responses in endothelial cells. American journal of physiology Cell physiology. 2019;317:C1161-c71
46. Guo Q, Wang J, Sun R, He Z, Chen Q, Liu W. et al. Comprehensive Construction of a Circular RNA-Associated Competing Endogenous RNA Network Identified Novel Circular RNAs in Hypertrophic Cardiomyopathy by Integrated Analysis. Frontiers in genetics. 2020;11:764
47. Tsang KM, Knutsen RH, Billington CJ Jr, Lindberg E, Steenbock H, Fu YP. et al. Copper-Binding Domain Variation in a Novel Murine Lysyl Oxidase Model Produces Structurally Inferior Aortic Elastic Fibers Whose Failure Is Modified by Age, Sex, and Blood Pressure. International journal of molecular sciences. 2022;23:6749
48. Marhuenda E, Xanthis I, Smith PO, Prakash A, Kallem T, Pandey P. et al. Mechanosensitive biochemical imprinting of the talin interaction with DLC1 regulates RhoA activity and cardiomyocyte remodeling. Science advances. 2025;11:eadt6083
49. Tu B, Song K, Zhou ZY, Lin LC, Liu ZY, Sun H. et al. SLC31A1 loss depletes mitochondrial copper and promotes cardiac fibrosis. European heart journal. 2025;46:2458-74
50. Baker ZN, Jett K, Boulet A, Hossain A, Cobine PA, Kim BE. et al. The mitochondrial metallochaperone SCO1 maintains CTR1 at the plasma membrane to preserve copper homeostasis in the murine heart. Human molecular genetics. 2017;26:4617-28
51. Zhang S, Liu H, Amarsingh GV, Cheung CCH, Wu D, Narayanan U. et al. Restoration of myocellular copper-trafficking proteins and mitochondrial copper enzymes repairs cardiac function in rats with diabetes-evoked heart failure. Metallomics: integrated biometal science. 2020;12:259-72
52. Yin W, Li R, Feng X, James Kang Y. The Involvement of Cytochrome c Oxidase in Mitochondrial Fusion in Primary Cultures of Neonatal Rat Cardiomyocytes. Cardiovascular toxicology. 2018;18:365-73
53. Schulz V, Basu S, Freibert SA, Webert H, Boss L, Mühlenhoff U. et al. Functional spectrum and specificity of mitochondrial ferredoxins FDX1 and FDX2. Nature chemical biology. 2023;19:206-17
54. Shen J, Wang L, Bi J. Bioinformatics analysis and experimental validation of cuproptosis-related lncRNA LINC02154 in clear cell renal cell carcinoma. BMC cancer. 2023;23:160
55. Lu X, Chen X, Lin C, Yi Y, Zhao S, Zhu B. et al. Elesclomol Loaded Copper Oxide Nanoplatform Triggers Cuproptosis to Enhance Antitumor Immunotherapy. Advanced science (Weinheim, Baden-Wurttemberg, Germany). 2024;11:e2309984
56. Tsvetkov P, Detappe A, Cai K, Keys HR, Brune Z, Ying W. et al. Mitochondrial metabolism promotes adaptation to proteotoxic stress. Nature chemical biology. 2019;15:681-9
57. Huang X, Wang T, Ye J, Feng H, Zhang X, Ma X. et al. FDX1 expression predicts favourable prognosis in clear cell renal cell carcinoma identified by bioinformatics and tissue microarray analysis. Frontiers in genetics. 2022;13:994741
58. Guo B, Yang F, Zhang L, Zhao Q, Wang W, Yin L. et al. Cuproptosis Induced by ROS Responsive Nanoparticles with Elesclomol and Copper Combined with αPD-L1 for Enhanced Cancer Immunotherapy. Advanced materials (Deerfield Beach, Fla). 2023;35:e2212267
59. Wang H, Zhang S, Zhang J. The copper resistance mechanism in a newly isolated Pseudoxanthomonas spadix ZSY-33. Environmental microbiology reports. 2023;15:484-96
60. Loffelmann M, Škrott Z, Majera D, Štarha P, Kryštof V, Mistrík M. Identification of novel dithiocarbamate-copper complexes targeting p97/NPL4 pathway in cancer cells. European journal of medicinal chemistry. 2023;261:115790
61. Wang X, Shen T, Lian J, Deng K, Qu C, Li E. et al. Resveratrol reduces ROS-induced ferroptosis by activating SIRT3 and compensating the GSH/GPX4 pathway. Molecular medicine (Cambridge, Mass). 2023;29:137
62. Li F, Wu X, Liu H, Liu M, Yue Z, Wu Z. et al. Copper Depletion Strongly Enhances Ferroptosis via Mitochondrial Perturbation and Reduction in Antioxidative Mechanisms. Antioxidants (Basel, Switzerland). 2022;11:2084
63. Bonet-Aleta J, Encinas-Gimenez M, Urriolabeitia E, Martin-Duque P, Hueso JL, Santamaria J. Unveiling the interplay between homogeneous and heterogeneous catalytic mechanisms in copper-iron nanoparticles working under chemically relevant tumour conditions. Chemical science. 2022;13:8307-20
64. Zhang T, Dai X, Wang H, Hu Y, Gao X, Shu X. et al. Copper-Induced Duodenal Injury: Unveiling the Dual Role of Cuproptosis and Ferroptosis via the FDX1/GPX4 Axis. The Journal of nutritional biochemistry. 2025: 110181.
65. Xue Q, Yan D, Chen X, Li X, Kang R, Klionsky DJ. et al. Copper-dependent autophagic degradation of GPX4 drives ferroptosis. Autophagy. 2023;19:1982-96
66. Timoshnikov VA, Kobzeva T, Selyutina OY, Polyakov NE, Kontoghiorghes GJ. Effective inhibition of copper-catalyzed production of hydroxyl radicals by deferiprone. Journal of biological inorganic chemistry: JBIC: a publication of the Society of Biological Inorganic Chemistry. 2019;24:331-41
67. Chen S, Chen T, Xu C, Yu X, Shi J, Yang C. et al. Iron overload exaggerates renal ischemia-reperfusion injury by promoting tubular cuproptosis via interrupting function of LIAS. Redox biology. 2025;86:103795
68. Liu G, Tang R, Wang C, Yu D, Wang Z, Yang H. et al. Bimetallic nanoconjugate hijack Fe-S clusters to drive a closed-loop cuproptosis-ferroptosis strategy for osteosarcoma inhibition. Journal of colloid and interface science. 2026;703:139052
69. Han Z, Lu X, He Y, Zhang T, Zhou Z, Zhang J. et al. Integration of bulk/scRNA-seq and multiple machine learning algorithms identifies PIM1 as a biomarker associated with cuproptosis and ferroptosis in abdominal aortic aneurysm. Frontiers in immunology. 2024;15:1486209
70. Li Y, Wang RY, Deng YJ, Wu SH, Sun X, Mu H. Molecular characteristics, clinical significance, and cancer immune interactions of cuproptosis and ferroptosis-associated genes in colorectal cancer. Frontiers in oncology. 2022;12:975859
71. Song J, Ren K, Zhang D, Lv X, Sun L, Deng Y. et al. A novel signature combing cuproptosis- and ferroptosis-related genes in sepsis-induced cardiomyopathy. Frontiers in genetics. 2023;14:1170737
72. Gao W, Huang Z, Duan J, Nice EC, Lin J, Huang C. Elesclomol induces copper-dependent ferroptosis in colorectal cancer cells via degradation of ATP7A. Molecular oncology. 2021;15:3527-44
73. Zhang J, Wang M, Ding W, Zhao M, Ye J, Xu Y. et al. Resolvin E1 protects against doxorubicin-induced cardiotoxicity by inhibiting oxidative stress, autophagy and apoptosis by targeting AKT/mTOR signaling. Biochemical pharmacology. 2020;180:114188
74. Liu Y, Shen M, Zhu S, Du Z, Lin L, Ma J. et al. Metallothionein rescues doxorubicin cardiomyopathy via mitigation of cuproptosis. Life sciences. 2025;363:123379
75. Chen YF, Qi RQ, Zhao L, Liang LR, Song JW, Zhou XY. et al. Aprocitentan mitigates doxorubicin-induced cardiotoxicity by inhibiting cuproptosis, oxidative stress, and mitochondrial impairments via the activation of sirtuin 7. International immunopharmacology. 2025;148:114141
76. El-Nablaway M, Sonpol HMA, Elewa YHA, Ali MAM, Adel M, Zayed ES. et al. Copper Chelation by Penicillamine Protects Against Doxorubicin-Induced Cardiomyopathy by Suppressing FDX1-Mediated Cuproptosis. Biomolecules. 2025;15:1320
77. Chen D, Yu W, Zhong C, Hong Q, Huang G, Que D. et al. Elabela ameliorates doxorubicin-induced cardiotoxicity by promoting autophagic flux through TFEB pathway. Pharmacological research. 2022;178:106186
78. Wei J, Lan G, Zhang W, Ran W, Wei Y, Liu X. et al. Targeting FDX1 by trilobatin to inhibit cuproptosis in doxorubicin-induced cardiotoxicity. British journal of pharmacology. 2025;182:2409-25
79. Liu J, Yuan Y, Cheng Y, Fu D, Chen Z, Wang Y. et al. Copper-Based Metal-Organic Framework Overcomes Cancer Chemoresistance through Systemically Disrupting Dynamically Balanced Cellular Redox Homeostasis. Journal of the American Chemical Society. 2022;144:4799-809
80. Huo S, Wang Q, Shi W, Peng L, Jiang Y, Zhu M. et al. ATF3/SPI1/SLC31A1 Signaling Promotes Cuproptosis Induced by Advanced Glycosylation End Products in Diabetic Myocardial Injury. International journal of molecular sciences. 2023;24:1667
81. Amin A, Khoury NC, Lacayo M, Kostanyan S. Copper Deficiency-Induced Neuropathy After Bariatric Surgery Disguised as Demyelinating Disease: A Case Report. Cureus. 2022;14:e22705
82. Zhang C, Huang T, Li L. Targeting cuproptosis for cancer therapy: mechanistic insights and clinical perspectives. Journal of hematology & oncology. 2024;17:68
83. Chen H, Li D, Zhang H, Zhang M, Lin Y, He H. et al. Mechanisms of copper metabolism and cuproptosis: implications for liver diseases. Frontiers in immunology. 2025;16:1633711
84. Singla A, Chen Q, Suzuki K, Song J, Fedoseienko A, Wijers M. et al. Regulation of murine copper homeostasis by members of the COMMD protein family. Disease models & mechanisms. 2021;14:dmm045963
85. Yamkate P, Gold RM, Twedt DC, Suchodolski JS, Steiner JM, Lidbury JA. Assessment of the intracellular distribution of copper in liver specimens from cats. PloS one. 2022;17:e0264003
86. Das S, Mohammed A, Mandal T, Maji S, Verma J, Ruturaj. et al. Polarized trafficking and copper transport activity of ATP7B: A mutational approach to establish genotype-phenotype correlation in Wilson disease. Human mutation. 2022;43:1408-29
87. Zhou D, Zi H, Yang X, Li X, Li Y, Xu A. et al. Dysfunction of ATP7B Splicing Variant Caused by Enhanced Interaction with COMMD1 in Wilson Disease. Cellular and molecular gastroenterology and hepatology. 2025;19:101418
88. Morrell A, Tallino S, Yu L, Burkhead JL. The role of insufficient copper in lipid synthesis and fatty-liver disease. IUBMB life. 2017;69:263-70
89. Yuan XZ, Yang RM, Wang XP. Management Perspective of Wilson's Disease: Early Diagnosis and Individualized Therapy. Current neuropharmacology. 2021;19:465-85
90. Santoro A, Calvo JS, Peris-Díaz MD, Krężel A, Meloni G, Faller P. The Glutathione/Metallothionein System Challenges the Design of Efficient O(2) -Activating Copper Complexes. Angewandte Chemie (International ed in English). 2020;59:7830-5
91. Maji S, Pirozzi M, Ruturaj, Pandey R, Ghosh T, Das S. et al. Copper-independent lysosomal localisation of the Wilson disease protein ATP7B. Traffic (Copenhagen, Denmark). 2023;24:587-609
92. Lasorsa A, Nardella MI, Rosato A, Mirabelli V, Caliandro R, Caliandro R. et al. Mechanistic and Structural Basis for Inhibition of Copper Trafficking by Platinum Anticancer Drugs. Journal of the American Chemical Society. 2019;141:12109-20
93. Sudhahar V, Shi Y, Kaplan JH, Ushio-Fukai M, Fukai T. Whole-Transcriptome Sequencing Analyses of Nuclear Antixoxidant-1 in Endothelial Cells: Role in Inflammation and Atherosclerosis. Cells. 2022;11:2919
94. Zhao X, Chen J, Yin S, Shi J, Zheng M, He C. et al. The expression of cuproptosis-related genes in hepatocellular carcinoma and their relationships with prognosis. Frontiers in oncology. 2022;12:992468
95. Zhang X, Walke GR, Horvath I, Kumar R, Blockhuys S, Holgersson S. et al. Memo1 binds reduced copper ions, interacts with copper chaperone Atox1, and protects against copper-mediated redox activity in vitro. Proceedings of the National Academy of Sciences of the United States of America. 2022;119:e2206905119
96. Liu X, Liu H, Wang N, Lai S, Qiu C, Gao S. et al. The interactive toxic effect of homocysteine and copper on cardiac microvascular endothelial cells during ischemia-reperfusion injury. Chemico-biological interactions. 2025;408:111387
97. Liu N, Zhen Z, Xiong X, Xue Y. Aerobic exercise protects MI heart through miR-133a-3p downregulation of connective tissue growth factor. PloS one. 2024;19:e0296430
98. Lin ZJ, Dong X, He H, Jiang JL, Guan ZJ, Li X. et al. A simplified herbal decoction attenuates myocardial infarction by regulating macrophage metabolic reprogramming and phenotypic differentiation via modulation of the HIF-1α/PDK1 axis. Chinese medicine. 2024;19:75
99. Yang S, Li X, Yan J, Jiang F, Fan X, Jin J. et al. Disulfiram downregulates ferredoxin 1 to maintain copper homeostasis and inhibit inflammation in cerebral ischemia/reperfusion injury. Scientific reports. 2024;14:15175
100. Kong B, Zheng X, Hu Y, Zhao Y, Hai J, Ti Y. et al. Sirtuin3 attenuates pressure overload-induced pathological myocardial remodeling by inhibiting cardiomyocyte cuproptosis. Pharmacological research. 2025;216:107739
101. Fang X, Ji Y, Li S, Wang L, He B, Li B. et al. Paeoniflorin attenuates cuproptosis and ameliorates left ventricular remodeling after AMI in hypobaric hypoxia environments. Journal of natural medicines. 2024;78:664-76
102. Wei W, Heng YY, Wu FF, Dong HY, Zhang PF, Li JX. et al. Sodium Tanshinone IIA Sulfonate alleviates vascular senescence in diabetic mice by modulating the A20-NFκB-NLRP3 inflammasome-catalase pathway. Scientific reports. 2024;14:17665
103. Tetteh PA, Kalvani Z, Stevens D, Sappal R, Kamunde C. Copper, cadmium, and zinc trigger multifaceted effects and interactions on cardiac mitochondrial bioenergetics and reactive oxygen species production in rainbow trout. Comparative biochemistry and physiology Toxicology & pharmacology: CBP. 2025;297:110286
104. Oliva CR, Ali MY, Flor S, Griguer CE. Copper-Induced Enhancement of Glioblastoma Tumorigenicity via Cytochrome C Oxidase. Antioxidants (Basel, Switzerland). 2025;14:142
105. Zhou J, Hao J, Wang D, Wu J, Wang Z, Lin P. et al. Ligand reaction-based fluorescent peptide probes for the detection of Cu(2+) and glutathione. Luminescence: the journal of biological and chemical luminescence. 2024;39:e4671
106. Yin J, Liu C, Guo J, Li M, Chen B, Zhang X. et al. A copper-loaded self-assembled nanoparticle for disturbing the tumor redox balance and triple anti-tumor therapy. Journal of materials chemistry B. 2024;12:3509-20
107. Zhao Y, Wang R, Hu C, Wang Y, Li Z, Yin D. et al. Complanatoside A disrupts copper homeostasis and induces cuproptosis via directly targeting ATOX1 in prostate cancer. Toxicology and applied pharmacology. 2025;496:117257
108. Xue Q, Kang R, Klionsky DJ, Tang D, Liu J, Chen X. Copper metabolism in cell death and autophagy. Autophagy. 2023;19:2175-95
109. Li SR, Bu LL, Cai L. Cuproptosis: lipoylated TCA cycle proteins-mediated novel cell death pathway. Signal transduction and targeted therapy. 2022;7:158
110. Xu W, Qian J, Hou G, Wang T, Wang J, Wang Y. et al. A hollow amorphous bimetal organic framework for synergistic cuproptosis/ferroptosis/apoptosis anticancer therapy via disrupting intracellular redox homeostasis and copper/iron metabolisms. Advanced Functional Materials. 2022;32:2205013
111. Lou Q, Yu L, Liu S, Shao C, Wan H, He Y. Calycosin-7-O-β-D-glucoside modulates copper homeostasis through SLC31A1 to mitigate cuproptosis in cerebral ischemia/reperfusion injury. Chemico-biological interactions. 2025;420:111702
112. Guo Q, Ma M, Yu H, Han Y, Zhang D. Dexmedetomidine enables copper homeostasis in cerebral ischemia/reperfusion via ferredoxin 1. Annals of medicine. 2023;55:2209735
113. Alqarni MH, Muharram MM, Alshahrani SM, Labrou NE. Copper-induced oxidative cleavage of glutathione transferase F1-1 from Zea mays. International journal of biological macromolecules. 2019;128:493-8
114. Ye Y, Chen A, Li L, Liang Q, Wang S, Dong Q. et al. Repression of the antiporter SLC7A11/glutathione/glutathione peroxidase 4 axis drives ferroptosis of vascular smooth muscle cells to facilitate vascular calcification. Kidney international. 2022;102:1259-75
115. Wang J, Zhao J, Li L, Lin X, Guo X, Peng F. et al. Association Between Peritoneal Glucose Absorption, Lipid Metabolism, and Cardiovascular Disease Risk in Nondiabetic Patients on Peritoneal Dialysis. Journal of renal nutrition: the official journal of the Council on Renal Nutrition of the National Kidney Foundation. 2025;35:196-206
116. Zhong CC, Zhao T, Hogstrand C, Chen F, Song CC, Luo Z. Copper (Cu) induced changes of lipid metabolism through oxidative stress-mediated autophagy and Nrf2/PPARγ pathways. The Journal of nutritional biochemistry. 2022;100:108883
117. Gao Y, Yu T, Ai F, Ji C, Wu Y, Huang X. et al. Bacillus coagulans XY2 ameliorates copper-induced toxicity by bioadsorption, gut microbiota and lipid metabolism regulation. Journal of hazardous materials. 2023;445:130585
118. Yang Z, Lian W, Waiho K, Zhu L, Chen A, Cheng Y. et al. Effects of copper exposure on lipid metabolism and SREBP pathway in the Chinese mitten crab Eriocheir sinensis. Chemosphere. 2022;308:136556
119. Chen L, Min J, Wang F. Copper homeostasis and cuproptosis in health and disease. Signal transduction and targeted therapy. 2022;7:378
120. Xie L, Yuan Y, Xu S, Lu S, Gu J, Wang Y. et al. Downregulation of hepatic ceruloplasmin ameliorates NAFLD via SCO1-AMPK-LKB1 complex. Cell reports. 2022;41:111498
121. Terasaki M, Yashima H, Mori Y, Saito T, Shiraga Y, Kawakami R. et al. Glucose-Dependent Insulinotropic Polypeptide Suppresses Foam Cell Formation of Macrophages through Inhibition of the Cyclin-Dependent Kinase 5-CD36 Pathway. Biomedicines. 2021;9:832
122. Whitlow TJ, Zhang Y, Ferguson N, Perez AM, Patel H, Link-Kemp JA. et al. Regulation of Atp7a RNA contributes to differentiation-dependent Cu redistribution in skeletal muscle cells. Metallomics: integrated biometal science. 2023;15:mfad042
123. Chen H, Liang K, Hou C, Piao HL. Dichloroacetic acid and rapamycin synergistically inhibit tumor progression. Journal of Zhejiang University Science B. 2023;24:397-405
124. Curnock R, Cullen PJ. Mammalian copper homeostasis requires retromer-dependent recycling of the high-affinity copper transporter 1. Journal of cell science. 2020;133:jcs249201
125. Li C, Wu Y, Li H, Wang H, Liu JX. Lipid-related metabolism during zebrafish embryogenesis under unbalanced copper homeostasis. Fish physiology and biochemistry. 2022;48:1571-86
126. Yan J, Li Z, Li Y, Zhang Y. Sepsis induced cardiotoxicity by promoting cardiomyocyte cuproptosis. Biochemical and biophysical research communications. 2024;690:149245
127. Zhang L, Huang S, Yuan Y. Butyrate inhibits the malignant biological behaviors of breast cancer cells by facilitating cuproptosis-associated gene expression. Journal of cancer research and clinical oncology. 2024;150:287
128. Dai S, Cao T, Shen H, Zong X, Gu W, Li H. et al. Landscape of molecular crosstalk between SARS-CoV-2 infection and cardiovascular diseases: emphasis on mitochondrial dysfunction and immune-inflammation. Journal of translational medicine. 2023;21:915
129. Liu Y, Huang Y, Xu C, An P, Luo Y, Jiao L. et al. Mitochondrial Dysfunction and Therapeutic Perspectives in Cardiovascular Diseases. International journal of molecular sciences. 2022;23:16053
130. Cai W, Chong K, Huang Y, Huang C, Yin L. Empagliflozin improves mitochondrial dysfunction in diabetic cardiomyopathy by modulating ketone body metabolism and oxidative stress. Redox biology. 2024;69:103010
131. Vîlcu A, Comarița IK, Constantin A, Alexandru N, Nemecz M, Safciuc F. et al. Innovative Therapy with Stem Cell-Derived Extracellular Vesicles on Cardiac Hypertrophy in an Animal Model of Atherosclerosis; Elucidation of the Molecular Mechanisms Involved in the Repair Process. Biomolecules. 2025;15:1424
132. Mollenhauer M, Bokredenghel S, Geißen S, Klinke A, Morstadt T, Torun M. et al. Stamp2 Protects From Maladaptive Structural Remodeling and Systolic Dysfunction in Post-Ischemic Hearts by Attenuating Neutrophil Activation. Frontiers in immunology. 2021;12:701721
133. Hao J, Wang H, Li B, Chen J, Wang W, Wang Y. et al. Tyrosine phosphatase Shp2 accelerated the progression of dilated cardiomyopathy via upregulating NF-κB/Src/FAK signaling induced ROS and mitophagy. Scientific reports. 2025;15:29522
134. Roginski AC, Wajner A, Cecatto C, Wajner SM, Castilho RF, Wajner M. et al. Disturbance of bioenergetics and calcium homeostasis provoked by metabolites accumulating in propionic acidemia in heart mitochondria of developing rats. Biochimica et biophysica acta Molecular basis of disease. 2020;1866:165682
135. Deshpande A, Weirauch L, Baral TK, Steier M, Borlepawar A, Kumari M. et al. Elevated levels of Letm1 drives mitochondrial dysfunction and cardiomyocyte stress-mediated apoptosis in cultured cardiomyocytes. Cell communication and signaling: CCS. 2025;23:378
136. Peng C, Zhang Y, Lang X, Zhang Y. Role of mitochondrial metabolic disorder and immune infiltration in diabetic cardiomyopathy: new insights from bioinformatics analysis. Journal of translational medicine. 2023;21:66
137. Wei T, Wang Q, Chen T, Zhou Z, Li S, Li Z. et al. The possible association of mitochondrial fusion and fission in copper deficiency-induced oxidative damage and mitochondrial dysfunction of the heart. Journal of trace elements in medicine and biology: organ of the Society for Minerals and Trace Elements (GMS). 2024;85:127483
138. Zheng YY, Tong XY, Zhang DY, Ouyang JM. Enhancement of Antioxidative and Anti-Inflammatory Activities of Corn Silk Polysaccharides After Selenium Modification. Journal of inflammation research. 2024;17:7965-91
139. Li J, Li Y, Zhao Y, Liu S, Li W, Tan H. et al. Mitigation of depleted uranium-induced mitochondrial damage by ethylmalonic encephalopathy 1 protein via modulation of hydrogen sulfide and glutathione pathways. Archives of toxicology. 2025;99:1133-41
140. Li H, Li Y, Yu Y, Ren X, Yang C, Jin W. et al. GSH exhaustion via inhibition of xCT-GSH-GPX4 pathway synergistically enhanced DSF/Cu-induced cuproptosis in myelodysplastic syndromes. Free radical biology & medicine. 2024;222:130-48
141. Cobine PA, Moore SA, Leary SC. Getting out what you put in: Copper in mitochondria and its impacts on human disease. Biochimica et biophysica acta Molecular cell research. 2021;1868:118867
142. Wu W, Huynh K, Du JC, She G, Duong T, Ziemann M. et al. Hippo pathway activation causes multiple lipid derangements in a murine model of cardiomyopathy. Biochimica et biophysica acta Molecular and cell biology of lipids. 2025;1870:159590
143. Liu ZY, Liu ZY, Lin LC, Song K, Tu B, Zhang Y. et al. Redox homeostasis in cardiac fibrosis: Focus on metal ion metabolism. Redox biology. 2024;71:103109
144. Fan X, Wei X, Wang W, Chai W, Xiao J, Han J. et al. Cardioprotective effects of the jiming formula on myocardial metabolism in Mice with myocardial infarction via the AMPK/SIRT1/PGC-1α pathway. Phytomedicine: international journal of phytotherapy and phytopharmacology. 2025;141:156727
145. Chen L, Wei J, Deng G, Xu G. Disulfidptosis-related gene in acute myocardial infarction and its diagnostic value and functions based on bioinformatics analysis and machine learning. Frontiers in cardiovascular medicine. 2025;12:1513342
146. Kowluru RA, Mohammad G. Epigenetic modifications in diabetes. Metabolism: clinical and experimental. 2022;126:154920
147. Liao Q, Deng J, Tong J, Gan Y, Hong W, Dong H. et al. p53 induces circFRMD4A to suppress cancer development through glycolytic reprogramming and cuproptosis. Molecular cell. 2025;85:132-49.e7
148. Saunders TL, Windley SP, Gervinskas G, Balka KR, Rowe C, Lane R. et al. Exposure of the inner mitochondrial membrane triggers apoptotic mitophagy. Cell death and differentiation. 2024;31:335-47
149. Katoh M, Nomura S, Yamada S, Ito M, Hayashi H, Katagiri M. et al. Vaccine Therapy for Heart Failure Targeting the Inflammatory Cytokine Igfbp7. Circulation. 2024;150:374-89
150. Liu J, Qu P, Shi J, Liang T, Gu Y, Li X. et al. Engineered Multifunctional Hydrogel Delivering Novel CBX7 Inhibitor Modulates Cuproptosis Via Liquid-Liquid Phase Separation to Restore Cardiac Function in Aged Myocardial Infarction. Advanced science (Weinheim, Baden-Wurttemberg, Germany). 2025: e11630.
151. Cho SK, Gwon S, Kim HA, Kim J, Cho SY, Kim DE. et al. Abnormal Development of Neural Stem Cell Niche in the Dentate Gyrus of Menkes Disease. International journal of stem cells. 2022;15:270-82
152. Das A, Ash D, Fouda AY, Sudhahar V, Kim YM, Hou Y. et al. Cysteine oxidation of copper transporter CTR1 drives VEGFR2 signalling and angiogenesis. Nature cell biology. 2022;24:35-50
153. Mo N, Tai C, Yang Y, Ling C, Zhang B, Wei L. et al. MT2A promotes angiogenesis in chronically ischemic brains through a copper-mitochondria regulatory mechanism. Journal of translational medicine. 2025;23:162
154. Cui Y, Chen Y, Gan N, Li M, Liao W, Zhou Y. et al. A novel cuproptosis-related diagnostic gene signature and differential expression validation in atherosclerosis. Molecular biomedicine. 2023;4:21
155. Liu Y, Zeng L, Cai Q, Zeng Y, Zheng S, Gong X. et al. Cytoskeletal-related genes function as checkpoints for the maintenance of VSMC contractile phenotype and prevent pathological remodeling in arterial diseases. Journal of advanced research. 2025
156. Zhou Y, Wang Q, Wang Q, Zhou Z, Peng X, Qu B. et al. Self-assembled copper chlorogenic acid nanoparticles: Inducing pyroptosis and cuproptosis to activate antitumor immunity. Journal of controlled release: official journal of the Controlled Release Society. 2025;384:113941
157. Lin Y, Chen K, Guo J, Chen P, Qian ZR, Zhang T. Identification of cuproptosis-related genes and immune infiltration in dilated cardiomyopathy. International journal of cardiology. 2024;399:131702
158. Xiang K, Wu H, Liu Y, Wang S, Li X, Yang B. et al. MOF-derived bimetallic nanozyme to catalyze ROS scavenging for protection of myocardial injury. Theranostics. 2023;13:2721-33
159. Han X, Jiang Z, Hou Y, Zhou X, Hu B. Myocardial ischemia-reperfusion injury upregulates nucleostemin expression via HIF-1α and c-Jun pathways and alleviates apoptosis by promoting autophagy. Cell death discovery. 2024;10:461
160. Wang Y, Wang D, Wu C, Wang B, He S, Wang H. et al. MMP 9-instructed assembly of bFGF nanofibers in ischemic myocardium to promote heart repair. Theranostics. 2022;12:7237-49
161. Sahu M, Sharma AK, Sharma G, Kumar A, Nandave M, Babu V. Facile synthesis of bromelain copper nanoparticles to improve the primordial therapeutic potential of copper against acute myocardial infarction in diabetic rats. Canadian journal of physiology and pharmacology. 2022;100:210-9
162. Gu J, Huang W, Duanmu Z, Zhuang R, Yang X. Cuproptosis and copper deficiency in ischemic vascular injury and repair. Apoptosis: an international journal on programmed cell death. 2024;29:1007-18
163. Tural K, Ozden O, Bilgi Z, Kubat E, Ermutlu CS, Merhan O. et al. The protective effect of betanin and copper on heart and lung in end-organ ischemia reperfusion injury. Bratislavske lekarske listy. 2020;121:211-7
164. Tural K, Ozden O, Bilgi Z, Kubat E, Ermutlu CS, Merhan O. et al. The protective effect of betanin and copper on spinal cord ischemia-reperfusion injury. The journal of spinal cord medicine. 2021;44:704-10
165. Yang D, Wang T, Liu J, Wang H, Kang YJ. Reverse regulation of hepatic ceruloplasmin production in rat model of myocardial ischemia. Journal of trace elements in medicine and biology: organ of the Society for Minerals and Trace Elements (GMS). 2021;64:126686
166. Chen X, Cai Q, Liang R, Zhang D, Liu X, Zhang M. et al. Copper homeostasis and copper-induced cell death in the pathogenesis of cardiovascular disease and therapeutic strategies. Cell death & disease. 2023;14:105
167. Rostami A, Palomer X, Pizarro-Delgado J, Barroso E, Valenzuela-Alcaraz B, Crispi F. et al. PPARβ/δ prevents inflammation and fibrosis during diabetic cardiomyopathy. Pharmacological research. 2024;210:107515
168. Cooper GJ, Chan YK, Dissanayake AM, Leahy FE, Keogh GF, Frampton CM. et al. Demonstration of a hyperglycemia-driven pathogenic abnormality of copper homeostasis in diabetes and its reversibility by selective chelation: quantitative comparisons between the biology of copper and eight other nutritionally essential elements in normal and diabetic individuals. Diabetes. 2005;54:1468-76
169. Attia H, Alshanwani A, Alatrouzi N, Ibrahim S, Alanteet A, Algarzae NK. et al. Carvacrol alleviates diabetic cardiomyopathy in rats: Targeting Cuproptosis pathway. Toxicology and applied pharmacology. 2025;506:117636
170. Sharma AK, Kumar A, Taneja G, Nagaich U, Deep A, Datusalia AK. et al. Combined and individual strategy of exercise generated preconditioning and low dose copper nanoparticles serve as superlative approach to ameliorate ISO-induced myocardial infarction in rats. Pharmacological reports: PR. 2018;70:789-95
171. Gong D, Lu J, Chen X, Choong SY, Zhang S, Chan YK. et al. Molecular changes evoked by triethylenetetramine treatment in the extracellular matrix of the heart and aorta in diabetic rats. Molecular pharmacology. 2006;70:2045-51
172. Kleyman A, Pisciotta W, Gaupp C, Khaliq W, Hofmaenner D, Brealey D. et al. Sepsis-induced hypocholesterolemia is linked to low cardiomyocyte membrane cholesterol and impaired catecholamine responsiveness. Critical care (London, England). 2025;29:399
173. Qu H, Wu J, Pan Y, Abdulla A, Duan Z, Cheng W. et al. Biomimetic Nanomodulator Regulates Oxidative and Inflammatory Stresses to Treat Sepsis-Associated Encephalopathy. ACS nano. 2024;18:28228-45
174. Zhao D, Zhuang J, Wang L, Wu L, Xu W, Zhao L. et al. Unveiling Key Biomarkers and Mechanisms in Septic Cardiomyopathy: A Comprehensive Transcriptome Analysis. Journal of inflammation research. 2024;17:11451-67
175. Li Q, Zhang F, Wang H, Tong Y, Fu Y, Wu K. et al. NEDD4 lactylation promotes APAP induced liver injury through Caspase11 dependent non-canonical pyroptosis. International journal of biological sciences. 2024;20:1413-35
176. Hackler J, Wisniewska M, Greifenstein-Wiehe L, Minich WB, Cremer M, Bührer C. et al. Copper and selenium status as biomarkers of neonatal infections. Journal of trace elements in medicine and biology: organ of the Society for Minerals and Trace Elements (GMS). 2020;58:126437
177. Martín Giménez VM, Bergam I, Reiter RJ, Manucha W. Metal ion homeostasis with emphasis on zinc and copper: Potential crucial link to explain the non-classical antioxidative properties of vitamin D and melatonin. Life sciences. 2021;281:119770
178. Xu Y, Xu Q, Zheng Z, Jiang X, Shi Y, Huang Y. et al. Fructose aggravates copper-deficiency-induced cardiac remodeling by inhibiting SERCA2a. The Journal of pharmacy and pharmacology. 2024;76:567-78
179. Shi L, Tan Y, Zheng W, Cao G, Zhou H, Li P. et al. CTRP3 alleviates mitochondrial dysfunction and oxidative stress injury in pathological cardiac hypertrophy by activating UPRmt via the SIRT1/ATF5 axis. Cell death discovery. 2024;10:53
180. Li H, Wang Y, Liu J, Chen X, Duan Y, Wang X. et al. Endothelial Klf2-Foxp1-TGFβ signal mediates the inhibitory effects of simvastatin on maladaptive cardiac remodeling. Theranostics. 2021;11:1609-25
181. Hill S, Deepa SS, Sataranatarajan K, Premkumar P, Pulliam D, Liu Y. et al. Sco2 deficient mice develop increased adiposity and insulin resistance. Molecular and cellular endocrinology. 2017;455:103-14
182. Grasso M, Bond GJ, Kim YJ, Boyd S, Matson Dzebo M, Valenzuela S. et al. The copper chaperone CCS facilitates copper binding to MEK1/2 to promote kinase activation. The Journal of biological chemistry. 2021;297:101314
183. Collins HE, Anderson JC, Wende AR, Chatham JC. Cardiomyocyte stromal interaction molecule 1 is a key regulator of Ca(2+) -dependent kinase and phosphatase activity in the mouse heart. Physiological reports. 2022;10:e15177
184. Pacheu-Grau D, Bareth B, Dudek J, Juris L, Vögtle FN, Wissel M. et al. Cooperation between COA6 and SCO2 in COX2 maturation during cytochrome c oxidase assembly links two mitochondrial cardiomyopathies. Cell metabolism. 2015;21:823-33
185. Fu D, Zhou J, Xu S, Tu J, Cai Y, Liu J. et al. Smilax glabra Roxb. flavonoids protect against pathological cardiac hypertrophy by inhibiting the Raf/MEK/ERK pathway: In vivo and in vitro studies. Journal of ethnopharmacology. 2022;292:115213
186. Roeters van Lennep JE, Tokgözoğlu LS, Badimon L, Dumanski SM, Gulati M, Hess CN. et al. Women, lipids, and atherosclerotic cardiovascular disease: a call to action from the European Atherosclerosis Society. European heart journal. 2023;44:4157-73
187. Chen YT, Xu XH, Lin L, Tian S, Wu GF. Identification of Three Cuproptosis-specific Expressed Genes as Diagnostic Biomarkers and Therapeutic Targets for Atherosclerosis. International journal of medical sciences. 2023;20:836-48
188. Wang N, Xu X, Li H, Feng Q, Wang H, Kang YJ. Atherosclerotic lesion-specific copper delivery suppresses atherosclerosis in high-cholesterol-fed rabbits. Experimental biology and medicine (Maywood, NJ). 2021;246:2671-8
189. El-Hajjar L, Hindieh J, Andraos R, El-Sabban M, Daher J. Myeloperoxidase-Oxidized LDL Activates Human Aortic Endothelial Cells through the LOX-1 Scavenger Receptor. International journal of molecular sciences. 2022;23:2837
190. Zhou Q, Zhang Y, Lu L, Zhang H, Zhao C, Pu Y. et al. Copper induces microglia-mediated neuroinflammation through ROS/NF-κB pathway and mitophagy disorder. Food and chemical toxicology: an international journal published for the British Industrial Biological Research Association. 2022;168:113369
191. Ballester-Servera C, Alonso J, Cañes L, Vázquez-Sufuentes P, Puertas-Umbert L, Fernández-Celis A. et al. Lysyl oxidase-dependent extracellular matrix crosslinking modulates calcification in atherosclerosis and aortic valve disease. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2023;167:115469
192. Ballester-Servera C, Alonso J, Cañes L, Vázquez-Sufuentes P, García-Redondo AB, Rodríguez C. et al. Lysyl Oxidase in Ectopic Cardiovascular Calcification: Role of Oxidative Stress. Antioxidants (Basel, Switzerland). 2024;13:523
193. Filip A, Taleb S, Bascetin R, Jahangiri M, Bardin M, Lerognon C. et al. Increased atherosclerotic plaque in AOC3 knock-out in ApoE(-/-) mice and characterization of AOC3 in atherosclerotic human coronary arteries. Frontiers in cardiovascular medicine. 2022;9:848680
194. Kohno T, Urao N, Ashino T, Sudhahar V, McKinney RD, Hamakubo T. et al. Novel role of copper transport protein antioxidant-1 in neointimal formation after vascular injury. Arteriosclerosis, thrombosis, and vascular biology. 2013;33:805-13
195. Huang L, Liu Y, Yu L, Cheng A, Cao J, Wang R. et al. Association of metal elements deposition with symptomatic carotid artery stenosis and their spatial distribution in atherosclerosis plaques. Metallomics: integrated biometal science. 2025;17:mfaf019
196. Kuang L, Chen Q, Liu Z, Wu L, Wu S. Types, Molecular Mechanisms and Potential Therapeutic Targets of Programmed Endothelial Cell Death in Atherosclerosis. Journal of cardiovascular pharmacology. 2025;86:214-226
197. Long P, Wang Q, Zhang Y, Zhu X, Yu K, Jiang H. et al. Profile of copper-associated DNA methylation and its association with incident acute coronary syndrome. Clinical epigenetics. 2021;13:19
198. Bai T, Wang L, Qiao Z, Wang Z. Cuproptosis, a potential target for the therapy of diabetic critical limb ischemia. Free radical biology & medicine. 2025;234:131-40
199. Ye F, Nelson MB, Bertoni AG, Ditzenberger GL, Duncan P, Mentz RJ. et al. Severity of functional impairments by race and sex in older patients hospitalized with acute decompensated heart failure. Journal of the American Geriatrics Society. 2022;70:3447-57
200. Liu R, Yao J, Chen K, Peng W. Association between biomarkers of zinc and copper status and heart failure: a meta-analysis. ESC heart failure. 2024;11:2546-56
201. Huang L, Shen R, Huang L, Yu J, Rong H. Association between serum copper and heart failure: a meta-analysis. Asia Pacific journal of clinical nutrition. 2019;28:761-9
202. Chrysochou E, Kanellopoulos PG, Koukoulakis KG, Sakellari A, Karavoltsos S, Minaidis M. et al. Heart Failure and PAHs, OHPAHs, and Trace Elements Levels in Human Serum: Results from a Preliminary Pilot Study in Greek Population and the Possible Impact of Air Pollution. Molecules (Basel, Switzerland). 2021;26:3207
203. Zhu SY, Zhou WQ, Niu YY, Zheng C, Liu X, Zhang YY. et al. COX17 restricts renal fibrosis development by maintaining mitochondrial copper homeostasis and restoring complex IV activity. Acta pharmacologica Sinica. 2023;44:2091-102
204. Wang D, Tian Z, Zhang P, Zhen L, Meng Q, Sun B. et al. The molecular mechanisms of cuproptosis and its relevance to cardiovascular disease. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2023;163:114830
205. Nagai S, Kondo T, Morimoto R, Hiraiwa H, Mizuno C, Nozaki A. et al. Association of trace element abnormalities and adverse outcomes in patients with acute heart failure. Journal of cardiology. 2025;86:545-551
206. Elsherif L, Wang L, Saari JT, Kang YJ. Regression of dietary copper restriction-induced cardiomyopathy by copper repletion in mice. The Journal of nutrition. 2004;134:855-60
207. Lu H, Du W, Ren L, Hamblin MH, Becker RC, Chen YE. et al. Vascular Smooth Muscle Cells in Aortic Aneurysm: From Genetics to Mechanisms. Journal of the American Heart Association. 2021;10:e023601
208. Van Den Heuvel LJF, Peeters S, Meester JAN, Coucke PJ, Loeys BL. An exploration of alternative therapeutic targets for aortic disease in Marfan syndrome. Drug discovery today. 2024;29:104023
209. Sudhahar V, Das A, Horimatsu T, Ash D, Leanhart S, Antipova O. et al. Copper Transporter ATP7A (Copper-Transporting P-Type ATPase/Menkes ATPase) Limits Vascular Inflammation and Aortic Aneurysm Development: Role of MicroRNA-125b. Arteriosclerosis, thrombosis, and vascular biology. 2019;39:2320-37
210. Pincemail J, Tchana-Sato V, Courtois A, Musumeci L, Cheramy-Bien JP, Munten J. et al. Alteration of Blood Oxidative Stress Status in Patients with Thoracic Aortic Dissection: A Pilot Study. Antioxidants (Basel, Switzerland). 2023;12:1106
211. Tsui KH, Hsiao JH, Lin LT, Tsang YL, Shao AN, Kuo CH. et al. The Cross-Communication of Cuproptosis and Regulated Cell Death in Human Pathophysiology. International journal of biological sciences. 2024;20:218-30
212. Oller J, Gabandé-Rodríguez E, Ruiz-Rodríguez MJ, Desdín-Micó G, Aranda JF, Rodrigues-Diez R. et al. Extracellular Tuning of Mitochondrial Respiration Leads to Aortic Aneurysm. Circulation. 2021;143:2091-109
213. He P, Li H, Liu C, Liu M, Zhang Z, Zhang Y. et al. U-shaped association between dietary copper intake and new-onset hypertension. Clinical nutrition (Edinburgh, Scotland). 2022;41:536-42
214. Qi J, Fu LY, Liu KL, Li RJ, Qiao JA, Yu XJ. et al. Resveratrol in the Hypothalamic Paraventricular Nucleus Attenuates Hypertension by Regulation of ROS and Neurotransmitters. Nutrients. 2022;14:4177
215. Xu H, Liu Z, Yao B, Xu Z. The impact of dietary copper intake on cardiovascular morbidity and mortality among hypertensive patients: a longitudinal analysis from NHANES (2001-2018). BMC public health. 2025;25:936
216. Wang K, Ning X, Qin C, Wang J, Yan W, Zhou X. et al. Respiratory Exposure to Copper Oxide Particles Causes Multiple Organ Injuries via Oxidative Stress in a Rat Model. International journal of nanomedicine. 2022;17:4481-96
217. Mawandji NBS, Lisboa N, Gomes KN, Vieira JM, Simão JJ, Alonso-Vale MI. et al. Chronic Copper Overload Triggers Inflammation in Mesenteric PVAT Alongside Changes in Renin-Angiotensin System-Related Pathways. Nutrients. 2025;17:2082
218. Cholewińska E, Juśkiewicz J, Majewski M, Smagieł R, Listos P, Fotschki B. et al. Effect of Copper Nanoparticles in the Diet of WKY and SHR Rats on the Redox Profile and Histology of the Heart, Liver, Kidney, and Small Intestine. Antioxidants (Basel, Switzerland). 2022;11:910
219. Zhang Z, Zhao S, Wu H, Qin W, Zhang T, Wang Y. et al. Cross-sectional study: Relationship between serum trace elements and hypertension. Journal of trace elements in medicine and biology: organ of the Society for Minerals and Trace Elements (GMS). 2022;69:126893
220. Insani WN, Whittlesea C, Wei L. Prevalence of cardiovascular drug-related adverse drug reactions consultations in UK primary care: A cross-sectional study. PloS one. 2024;19:e0307237
221. Xie W, Guo Z, Zhao L, Wei Y. The copper age in cancer treatment: from copper metabolism to cuproptosis. Progress in Materials Science. 2023;138:101145
222. Chen N, Guo L, Wang L, Dai S, Zhu X, Wang E. Sleep fragmentation exacerbates myocardial ischemia-reperfusion injury by promoting copper overload in cardiomyocytes. Nature communications. 2024;15:3834
223. Nankivell VA, Sandeman L, Stretton L, Vidanapathirana AK, Rajora MA, Chen J. et al. Theranostic porphyrin nanoparticles identify atherosclerosis via multimodal imaging and elicit atheroprotective effects. Materials today Bio. 2025;34:102202
224. Kirk FT, Munk DE, Swenson ES, Quicquaro AM, Vendelbo MH, Larsen A. et al. Effects of tetrathiomolybdate on copper metabolism in healthy volunteers and in patients with Wilson disease. Journal of hepatology. 2024;80:586-95
225. Saifi MA, Godugu C. Copper chelation therapy inhibits renal fibrosis by modulating copper transport proteins. BioFactors (Oxford, England). 2022;48:934-45
226. Wei H, Zhang WJ, Leboeuf R, Frei B. Copper induces-and copper chelation by tetrathiomolybdate inhibits-endothelial activation in vitro. Redox report: communications in free radical research. 2014;19:40-8
227. Guo H, Jing L, Xia C, Zhu Y, Xie Y, Ma X. et al. Copper Promotes LPS-Induced Inflammation via the NF-кB Pathway in Bovine Macrophages. Biological trace element research. 2024;202:5479-88
228. Reid A, Miller C, Farrant JP, Polturi R, Clark D, Ray S. et al. Copper chelation in patients with hypertrophic cardiomyopathy. Open heart. 2022;9:e001803
229. Karginova O, Weekley CM, Raoul A, Alsayed A, Wu T, Lee SS. et al. Inhibition of Copper Transport Induces Apoptosis in Triple-Negative Breast Cancer Cells and Suppresses Tumor Angiogenesis. Molecular cancer therapeutics. 2019;18:873-85
230. Kim YJ, Bond GJ, Tsang T, Posimo JM, Busino L, Brady DC. Copper chaperone ATOX1 is required for MAPK signaling and growth in BRAF mutation-positive melanoma. Metallomics: integrated biometal science. 2019;11:1430-40
231. Gale JR, Hartnett-Scott K, Ross MM, Rosenberg PA, Aizenman E. Copper induces neuron-sparing, ferredoxin 1-independent astrocyte toxicity mediated by oxidative stress. Journal of neurochemistry. 2023;167:277-95
232. Zhao J, Chen Y, Lu D, Zeng Y, Li J, Zhu J. et al. Elesclomol-Induced Copper Influx Attenuates Lung Adenocarcinoma Progression with Involvement of the ER Stress/PCK2 Axis. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2025;39:e71250
233. Almyre C, Bounaix N, Godard F, Baris OR, Cayer AL, Sardin E. et al. The copper ionophore disulfiram improves mitochondrial function in various yeast and human cellular models of mitochondrial diseases. Human molecular genetics. 2025;34:1072-85
234. Qi RQ, Chen YF, Cheng J, Song JW, Chen YH, Wang SY. et al. Elabela alleviates cuproptosis and vascular calcification in vitaminD3- overloaded mice via regulation of the PPAR-γ /FDX1 signaling. Molecular medicine (Cambridge, Mass). 2024;30:223
235. Chen L, Hu Y, Cheng Y, Wang H. A Hydroxyquinoline Polymer with Excellent Amyloidosis Inhibition and Protein Delivery Ability to Combat Amyloid-β-Mediated Neurotoxicity. Nano letters. 2024;24:12882-12890
236. Zheng P, Zhou C, Lu L, Liu B, Ding Y. Elesclomol: a copper ionophore targeting mitochondrial metabolism for cancer therapy. Journal of experimental & clinical cancer research: CR. 2022;41:271
237. Su TA, Shihadih DS, Cao W, Detomasi TC, Heffern MC, Jia S. et al. A Modular Ionophore Platform for Liver-Directed Copper Supplementation in Cells and Animals. Journal of the American Chemical Society. 2018;140:13764-74
238. Cai H, Cheng X, Wang XP. ATP7B gene therapy of autologous reprogrammed hepatocytes alleviates copper accumulation in a mouse model of Wilson's disease. Hepatology (Baltimore, Md). 2022;76:1046-57
239. Shi H, Xie Z, Zhang J, Lu F, Luo H, Guo P. et al. Cu-doped dendritic biodegradable nanoplatforms for augmenting cuproptosis and tumor-starvation therapy through mitochondrial metabolic cascade modulation. Materials today Bio. 2025;35:102477
240. Saedi S, Tan Y, Watson SE, Wintergerst KA, Cai L. Potential pathogenic roles of ferroptosis and cuproptosis in cadmium-induced or exacerbated cardiovascular complications in individuals with diabetes. Frontiers in endocrinology. 2024;15:1461171
241. Mori K, Kuramochi T, Matsusaki M, Hashiguchi Y, Okumura M, Saio T. et al. Metal-Responsive Up-Regulation of Bifunctional Disulfides for Suppressing Protein Misfolding and Promoting Oxidative Folding. Angewandte Chemie (International ed in English). 2025;64:e202502187
242. Dubey PR, Kaur G, Shukla R. Nano-mediated Management of Metal Toxicity-induced Neurodegeneration: A Critical Review. Molecular neurobiology. 2025;62:8400-19
243. Du F, Yu Q, Yan S, Zhang Z, Vangavaragu JR, Chen D. et al. Gain of PITRM1 peptidase in cortical neurons affords protection of mitochondrial and synaptic function in an advanced age mouse model of Alzheimer's disease. Aging cell. 2021;20:e13368
244. Cannas D, Loi E, Serra M, Firinu D, Valera P, Zavattari P. Relevance of Essential Trace Elements in Nutrition and Drinking Water for Human Health and Autoimmune Disease Risk. Nutrients. 2020;12:2074
245. Patwa J, Thakur A, Sharma A, Flora SJS. Monoisoamyl DMSA reduced copper-induced neurotoxicity by lowering 8-OHdG level, amyloid beta and Tau protein expressions in Sprague-Dawley rats. Metallomics: integrated biometal science. 2020;12:1428-48
246. López-Guerrero VE, Posadas Y, Sánchez-López C, Smart A, Miranda J, Singewald K. et al. A Copper-Binding Peptide with Therapeutic Potential against Alzheimer's Disease: From the Blood-Brain Barrier to Metal Competition. ACS chemical neuroscience. 2025;16:241-61
247. Liu X, Cheng L, Huang L, Li M, Shen Q, Li D. et al. EZH2 promotes endometriosis progression through estrogen receptor and TNFα expression. Frontiers in endocrinology. 2025;16:1574938
248. Yang M, Yu H, Xu K, Xie J, Zheng H, Feng R. et al. No evidence of a genetic causal relationship between ankylosing spondylitis and iron homeostasis: A two-sample Mendelian randomization study. Frontiers in nutrition. 2023;10:1047640
249. Wan R, Li W, Yang K, Li L, Wang S, Lei L. et al. Immunomodulatory and bone regenerative properties of copper/procyanidins-modified titanium surfaces. Biomaterials advances. 2025;169:214199
250. Mathur A, Ma Z, Loskill P, Jeeawoody S, Healy KE. In vitro cardiac tissue models: Current status and future prospects. Advanced drug delivery reviews. 2016;96:203-13
251. Rezvani M, Vallier L, Guillot A. Modeling Nonalcoholic Fatty Liver Disease in the Dish Using Human-Specific Platforms: Strategies and Limitations. Cellular and molecular gastroenterology and hepatology. 2023;15:1135-45
252. Morss Clyne A, Swaminathan S, Díaz Lantada A. Biofabrication strategies for creating microvascular complexity. Biofabrication. 2019;11:032001
253. Tadenev ALD, Burgess RW. Model validity for preclinical studies in precision medicine: precisely how precise do we need to be? Mammalian genome: official journal of the International Mammalian Genome Society. 2019;30:111-22
254. Curran J, Burkhoff D, Kloner RA. Beyond Reperfusion: Acute Ventricular Unloading and Cardioprotection During Myocardial Infarction. Journal of cardiovascular translational research. 2019;12:95-106
255. Liang J, Wang R, Wu H, Huang Z, Zhang R, Jiang F. Copper metabolism and cuproptosis: broad perspectives in the treatment of hepatocellular carcinoma. Frontiers in oncology. 2025;15:1555858
256. Nagaraj K. Surfactant-based drug delivery systems for cancer therapy: Advances, challenges, and future perspectives. International journal of pharmaceutics. 2025;679:125655
257. Ding Y, Huang Z, Luo Y, Lin H, Wang J, Zeng Z. et al. A fibroblast activation protein α-activatable nanoagent co-delivering diethyldithiocarbamate and copper for tumor therapy and imaging. Acta biomaterialia. 2024;187:316-27
258. Liu YT, Chen L, Li SJ, Wang WY, Wang YY, Yang QC. et al. Dysregulated Wnt/β-catenin signaling confers resistance to cuproptosis in cancer cells. Cell death and differentiation. 2024;31:1452-66
259. Stakhneva EM, Kashtanova EV, Polonskaya YV, Striukova EV, Shramko VS, Sadovski EV. et al. The Search for Associations of Serum Proteins with the Presence of Unstable Atherosclerotic Plaque in Coronary Atherosclerosis. International journal of molecular sciences. 2022;23:12795
260. Narayanan IG, Natarajan SK. Peptides derived from histidine and methionine-rich regions of copper transporter 1 exhibit anti-angiogenic property by chelating extracellular Cu. Chemical biology & drug design. 2018;91:797-804
261. Sasanian N, Bernson D, Horvath I, Wittung-Stafshede P, Esbjörner EK. Redox-Dependent Copper Ion Modulation of Amyloid-β (1-42) Aggregation In Vitro. Biomolecules. 2020;10:924
262. Liu J, Huang H, Zhang X, Shen Y, Jiang D, Hu S. et al. Unveiling the Cuproptosis in Colitis and Colitis-Related Carcinogenesis: A Multifaceted Player and Immune Moderator. Research (Washington, DC). 2025;8:0698
263. Tang J, Xu Z, Jiang M, Luo C, Li X, Jiang Y. et al. Cuproptosis is involved in small intestinal toxicity in the paraffins during pregnancy. Toxicology letters. 2025;412:85-94
264. De Feyter S, Beyens A, Callewaert B. ATP7A-related copper transport disorders: A systematic review and definition of the clinical subtypes. Journal of inherited metabolic disease. 2023;46:163-73
265. Yang W, Xiao C, Zheng J, Song J, Li XG. Copper homeostasis and cuproptosis represent emerging targets for therapeutic intervention in inflammatory diseases. Pharmacological research. 2025;221:107988
266. Mustafa M, Ahmad R, Tantry IQ, Ahmad W, Siddiqui S, Alam M. et al. Apoptosis: A Comprehensive Overview of Signaling Pathways, Morphological Changes, and Physiological Significance and Therapeutic Implications. Cells. 2024;13:1838
267. Yan HF, Zou T, Tuo QZ, Xu S, Li H, Belaidi AA. et al. Ferroptosis: mechanisms and links with diseases. Signal transduction and targeted therapy. 2021;6:49
268. Yu P, Zhang X, Liu N, Tang L, Peng C, Chen X. Pyroptosis: mechanisms and diseases. Signal transduction and targeted therapy. 2021;6:128
269. Liu X, Xie X, Ren Y, Shao Z, Zhang N, Li L. et al. The role of necroptosis in disease and treatment. MedComm. 2021;2:730-55
270. Siang W, Guoyun W, Yan F, Wenji L. Mettl1 mitigates sepsis-induced cardiomyopathy via inhibition of FDX1-dependent cuproptosis. Gene. 2025;978:149892
271. Hu X, Hu L, Si X, Feng Q, Ma Y, Liu Z. et al. Comprehensive Bioinformatics Analysis Reveals the Role of Shared Cuproptosis- and Ferroptosis-Related DEG DLD in Abdominal Aortic Aneurysm. Journal of cellular and molecular medicine. 2025;29:e70399
272. Wang Q, Wang Y, West TM, Liu Y, Reddy GR, Barbagallo F. et al. Carvedilol induces biased β1 adrenergic receptor-nitric oxide synthase 3-cyclic guanylyl monophosphate signalling to promote cardiac contractility. Cardiovascular research. 2021;117:2237-51
273. Kainat A, Phang CR, Ain NU, Agarwal B. The First Occurrence of Angioedema After Discontinuation of Angiotensin-Converting Enzyme Inhibitor. Cureus. 2021;13:e19553
274. Claesen K, Roth L, Mertens JC, Hermans K, Sim Y, Hendriks D. Pleiotropic Effects of Atorvastatin Result in a Downregulation of the Carboxypeptidase U System (CPU, TAFIa, CPB2) in a Mouse Model of Advanced Atherosclerosis. Pharmaceutics. 2021;13:1731
275. Li W, Wang D, Lin C, Cai T, Zhao M, Liang L. et al. A Meta-Analysis of the Incidence of Adverse Reactions of Statins in Various Diseases. Cardiovascular therapeutics. 2025;2025:6684099
276. Kamlin COF, T MJ, J LH, N SA. Trientine Tetrahydrochloride, From Bench to Bedside: A Narrative Review. Drugs. 2024;84:1509-18
Author contact
![]() Corresponding authors: Yunfei Ling, Associate Professor of Cardiovascular Surgery, Department of Cardiovascular Surgery, West China Hospital of Sichuan University, No. 37 GuoXue Xiang, Chengdu, Sichuan 610041, China, Tel.: +86- 18980609254, Email: lingyunfeiedu.cn. Qi An, Professor of Cardiovascular Surgery, Department of Cardiovascular Surgery, West China Hospital of Sichuan University, No. 37 GuoXue Xiang, Chengdu, Sichuan 610041, China, Tel.: +86-18980601524, Email: anqiedu.cn. Lingling Zhu, Ph.D., Associate Professor, Lung Cancer Center/Lung Cancer Institute & Department of Medical Oncology, Cancer Center, West China Hospital, Sichuan University, No. 37, Guoxue Road, Chengdu 610041, People's Republic of China, Tel.: +86-18980609626, Email: zhulingling8262023com.
Corresponding authors: Yunfei Ling, Associate Professor of Cardiovascular Surgery, Department of Cardiovascular Surgery, West China Hospital of Sichuan University, No. 37 GuoXue Xiang, Chengdu, Sichuan 610041, China, Tel.: +86- 18980609254, Email: lingyunfeiedu.cn. Qi An, Professor of Cardiovascular Surgery, Department of Cardiovascular Surgery, West China Hospital of Sichuan University, No. 37 GuoXue Xiang, Chengdu, Sichuan 610041, China, Tel.: +86-18980601524, Email: anqiedu.cn. Lingling Zhu, Ph.D., Associate Professor, Lung Cancer Center/Lung Cancer Institute & Department of Medical Oncology, Cancer Center, West China Hospital, Sichuan University, No. 37, Guoxue Road, Chengdu 610041, People's Republic of China, Tel.: +86-18980609626, Email: zhulingling8262023com.