Impact Factor ISSN: 1449-2288
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. TAM origins, states, and...
3. Three high-value metabolic...
4....
5. Disease-specific syntheses
6. Translational Toolkit:...
7. Therapeutic strategies and...
8. Clinical development...
9. Conclusion
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2026; 22(6):3087-3107. doi:10.7150/ijbs.127634 This issue Cite
Review
Metabolic-Immune Coupling in Urologic Cancers: Macrophage Reprogramming as a Therapeutic Nexus
Wenxue Huang1, ![]() , Weijia Li1,
, Weijia Li1, ![]() , Wentai Shangguan1,3, Lin Yang1, Zhuohang Li1, Boyuan Sun1, Cunzhen Ma1, Xunguo Yang1, Peidan Peng1,
, Wentai Shangguan1,3, Lin Yang1, Zhuohang Li1, Boyuan Sun1, Cunzhen Ma1, Xunguo Yang1, Peidan Peng1, ![]() , Jie Zhao4,
, Jie Zhao4, ![]() , Bisheng Cheng1,2,
, Bisheng Cheng1,2, ![]() , Peng Wu1,
, Peng Wu1, ![]()
1. Nanfang Hospital, Southern Medical University, Guangzhou, 510000, China.
2. Department of Surgery, Division of Urology, Beth Israel Deaconess Medical Center, Harvard Medical School,Boston, MA, USA.
3. Department of Urology, Second Affiliated Hospital of Naval Medical University, Shanghai, 200003, China.
4. NMPA Key Laboratory for Research and Evaluation of Drug Metabolism, Guangdong Provincial Key Laboratory of New Drug Screening, School of Pharmaceutical Sciences, Southern Medical University, Guangzhou, 510515, China.
# These authors contributed equally to this work.
Received 2025-10-31; Accepted 2026-2-13; Published 2026-2-26
Abstract

Therapeutic responsiveness in urologic cancers is gated by metabolic-immune coupling that conditions tumor-associated macrophages (TAMs). Myeloid-dominated “cold” ecosystems blunt antigen handling, phagocytosis, and trafficking, limiting the benefit of immune-checkpoint inhibitors (ICIs). This review focuses on three high-value axes that shape TAM state and niche: the lactate-pH / hypoxia-HIF-VEGF axis that enforces acidic, adenosinergic suppression and angiogenic programs; lipid rafts axis that stabilizes inhibitory hubs (e.g., PI3K-AKT/TREM2) and skews phagocytosis/antigen presentation; and ferroptosis-redox axis control that sets inflammatory versus tolerogenic set-points. The review further outlines pharmacodynamic anchors—hyperpolarized 13C-pyruvate MRI (kPL), soluble ANGPT2, and spatial NT5E/ADORA2A modules—to operationalize a bench-to-biomarker-to-bedside loop using organoid-immune co-cultures, humanized/xenograft systems, and ex vivo tumor slices. This framework prioritizes adaptive enrichment for glycolysis- or adenosine-high tumors, rational timing/sequencing with ICIs, and avoidance of global myelosuppression. Collectively, metabolism-informed TAM re-education offers a route to convert myeloid-dominated “cold” ecosystems into treatment-responsive states across urologic cancers.
Keywords: Tumor-associated macrophage, metabolic reprogramming, prostate cancer, bladder cancer, renal cell carcinoma
1. Introduction
Urologic malignancies, including prostate cancer (PCa), bladder cancer (BCa), and renal cell carcinoma (RCC), represent major contributors to global cancer incidence and mortality[1, 2]. Despite the remarkable success of ICIs across multiple cancer types, response rates remain highly variable between patients in urologic cancers[3]. A substantial fraction of tumors display an immune-excluded or non-inflamed microenvironment characterized by enrichment of immunosuppressive myeloid populations; these tumors less often benefit from immunotherapy[3]. Within this landscape, tumor-associated macrophages (TAMs) constitute a dominant and highly plastic myeloid compartment with a central role in shaping immune tone and treatment responsiveness.
Macrophage states are not fixed but are continuously instructed by tumor- and microenvironment-derived cues[4]. Among these, tumor metabolic rewiring is a major force that reshapes the tumor microenvironment (TME) and, in turn, immune-cell function[5]. The Warburg effect describes the preference of cancer cells for high-rate glycolysis even in the presence of oxygen, accompanied by lactate accumulation[6]. This metabolic configuration supports proliferative demands while driving hallmark TME features, including nutrient competition, regional hypoxia, extracellular acidosis, and enrichment of metabolites such as lactate and adenosine[7-9]. Beyond glycolysis, dysregulated lipid and iron metabolism in tumor cells intersects with the metabolic needs of immune cells, generating a multi-layered metabolic-immune coupling that can bias TAM programs toward immunosuppression[10, 11].
In this review, we posit that metabolic-immune coupling constrains therapeutic responses in urologic cancers largely by shaping TAM plasticity and niche adaptation. Urologic tumors exhibit disease-specific metabolic features and establish reciprocal metabolic interactions with immune and stromal compartments, together reinforcing an immunosuppressive milieu[12, 13]. Recent advances in single-cell transcriptomics, spatial profiling, and multi-omics integration have begun to resolve how TAM heterogeneity aligns with local metabolic constraints and how these constraints may be pharmacologically perturbed[14].
Distinct from prior reviews that discuss macrophages or cancer metabolism in isolation, this review offers a TAM-centered, clinically oriented framework for urologic malignancies by organizing current evidence into three high-value metabolic axes that repeatedly converge on macrophage function and immune contexture: the lactate-pH / hypoxia-HIF-VEGF axis, the lipid rafts axis, and the ferroptosis-redox axis. We connect these axes to spatially patterned TAM states and to practical translational questions, including which niches are plausibly rate-limiting in PCa, BCa, and RCC, what macrophage phenotypes are expected to shift upon intervention, and which pharmacodynamic readouts may confirm pathway engagement in patients. By integrating mechanistic biology with models, biomarkers, and trial considerations, we aim to provide an executable roadmap for metabolism-informed TAM reprogramming and rational immunotherapy combinations in urologic cancers.
2. TAM origins, states, and niches in urologic cancers
2.1 Origins and heterogeneity of TAMs
TAMs in urologic cancers display pronounced heterogeneity, which mainly arises from dual ontogeny—embryonically derived tissue-resident macrophages maintained by local proliferation and monocyte-derived macrophages recruited from the circulation[4, 15, 16]. Traditionally, based on in-vitro activation paradigms, macrophages were simply divided into the classically activated, pro-inflammatory M1 type and the anti-inflammatory, pro-tumor M2 type[17, 18]. In subsequent studies, M2 macrophages were further subdivided into M2a, M2b, M2c, and M2d according to the stimuli[19-22]. However, this binary taxonomy is overly simplistic within the authentic TME and fails to capture the functional diversity of TAMs. Modern single-cell transcriptomics reveals a continuum of states in urologic tumors, including phagocytic/antigen-presenting, angiogenic, immunoregulatory, and immune-stimulatory subsets.
Studies in murine PCa models indicate that, compared with resident TAMs, monocyte-derived TAMs are more prone to immunosuppressive phenotypes and promote tumor progression[23]. However, in RCC and BCa, systematic lineage-tracing data in human tissues remain limited, and the relative ontogeny contributions are yet to be clarified.
2.2 Organ-specific TAM niches
Distinct metabolic and immune contexts across urologic organs sculpt unique TAM niches and create disease-specific therapeutic challenges:
Prostate cancer. TAMs often cluster near androgen-synthesizing glands and exhibit conspicuous lipid-loaded phenotypes. Cholesterol burden drives liver X receptor (LXR) and peroxisome proliferator-activated receptor γ (PPARγ) pathways, inducing immunoregulatory TAM states. One study suggested that lipid-rich TAMs enhance tumor progression and suppress clearance by altering membrane architecture and reducing antigen-presentation capacity[24]. Androgen signaling may affect lipid metabolism, leading to lipid loading in TAMs and thereby suppressing their phagocytic activity and antigen presentation.
Bladder cancer. TAMs encounter distinctive metabolic challenges, notably acidic and adenosinergic niches generated by high lactate and purine metabolism. BCa typically shows intense glycolysis with abundant lactate production, which promotes regulatory TAM states and shapes a strongly immunosuppressive TME[25]. In such environments, TAMs tend to polarize toward an M2-like phenotype; M2 cells further suppress antitumor immunity by producing IL-10 and TGF-β. Studies show that downregulation of SPOP, an E3 ubiquitin ligase, in BCa promotes cancer stemness and macrophage recruitment/polarization via the STAT3/CCL2/IL-6 axis, creating a symbiotic relationship wherein tumor cells and TAMs mutually reinforce each other[26].
Renal cell carcinoma. Originating from metabolically active renal tubular epithelial cells, RCC harbors TAMs with unique lipid-droplet accumulation and hypoxia-inducible factor (HIF) dependence. Due to von Hippel-Lindau tumor-suppressor gene (VHL) mutations, lipid droplets accumulate and HIF pathways are constitutively activated, creating hypoxia that restricts antigen presentation and fosters pro-angiogenic TAM phenotypes. Against a background of constitutive HIF-α activation caused by VHL mutations, metabolic reprogramming in renal cancer cells profoundly influences TAM states[27]. In fumarate hydratase-deficient renal cell carcinoma (FH-deficient RCC), glycolytic activity is markedly increased due to the Warburg effect, and closely associated with resistance to ICIs. An integrated multi-omics analysis identified a glycolysis signature (Glyc.Sig) that inversely correlates with ICI efficacy, and pinpointed lactate dehydrogenase A (LDHA) as a potential combinatorial target[28].
2.3 Spatial ecology of TAMs
Spatial ecology is increasingly recognized as a major organizer of TAM heterogeneity in urologic cancers. Spatial multi-omics and high-plex imaging have shown that TAM programs are not randomly distributed but instead align with anatomically and metabolically distinct tumor compartments. Regional sampling bias can shift inferred TAM states[29]. TAMs in tumor cores often display stronger immunosuppressive features, being exposed to extreme metabolic stress—hypoxia, low pH, and nutrient deprivation—that drives HIF-1α stabilization and glycolysis; by contrast, margin-associated TAMs are shaped less by extreme hypoxia/acidosis and more by tissue-injury-associated cues, cytokine networks, and direct interaction with tumor cells undergoing invasion and remodeling. TAMs at invasive margins may retain greater antigen-presenting capacity and interact more with lymphocytes[30, 31]. TAMs adjacent to tertiary lymphoid structures (TLSs) can acquire more activated phenotypes that help initiate and sustain antitumor immunity[32]. Recognizing such spatial ecological differences is vital for therapeutic design, since TAMs at different locations can respond very differently to the same strategy.
3. Three high-value metabolic axes that shape TAM function
3.1 Lactate-pH / hypoxia-HIF-VEGF axis
As a major end-product of glycolysis, lactate accumulates in metabolically active tumors such as BCa and RCC and serves as a key metabolite shaping an immunosuppressive TME[33-35]. Lactate affects TAM function and polarization via multiple mechanisms: under hypoxia, intensified glycolysis produces abundant lactate that is exported through MCT1 and MCT4, monocarboxylate transporter proteins. Lactate accumulation and extracellular acidosis stabilize HIF-1α in TAMs, induce ARG1, VEGFA, and PD-L1, and promote polarization toward an M2-like immunosuppressive phenotype[5]. In M1 macrophages, glycolysis is an important metabolic program[36]; downregulation of macrophage glycolysis is consistently accompanied by reduced secretion of inflammatory cytokines[37]. HIF-1α is a well-studied factor that induces glycolysis and M1 polarization, and its overexpression upregulates glycolysis and the pentose phosphate pathway (PPP)[38].
Lactate is a key immunosuppressive metabolite produced by tumor cells[8, 39, 40]. Researchers argue that lactate itself, rather than downstream metabolites, drives M2 polarization[41]. In pituitary adenoma, lactate induces M2 polarization via mTORC2 and ERK pathways, after which M2 macrophages secrete CCL17 to promote invasion[42]. Similar mechanisms may exist in urologic cancers, particularly in highly glycolytic BCa subtypes. Upon polarization, macrophage function is affected by lactate: lactate downregulates the macrophage-specific vacuolar ATPase subunit ATP6V0d2, enhances HIF-2α-mediated vascular endothelial growth factor (VEGF) production, and promotes cancer growth[43]. In BCa, TAMs around necrotic cores upregulate MCT4 and CD39, suggesting a lactate-driven adenosine-mediated immunosuppression. Spatial transcriptomics corroborates the co-localization of lactate-enriched regions with immunosuppressive TAM subsets[44].
Acidic microenvironments driven by lactate can suppress antigen processing and presentation through multiple mechanisms. As reviewed by Wang et al., acidosis undermines DC/macrophage antigen presentation[45]. Furthermore, both in vitro and in vivo, macrophages are more prone to IL-4-induced M2 polarization at pH 6.8; conversely, neutralizing low pH can shift TAM phenotypes toward inflammatory states and suppress tumor progression[46]. In addition, recent reviews summarize how lactate in the TME promotes protumoral macrophage polarization through metabolic control and signaling pathways such as GPR81[39, 47].
3.2 Lipid rafts axis
Lipid metabolic rewiring is a hallmark of urologic cancers—especially PCa and RCC[48-50]. Through lipid and cholesterol metabolism, tumor cells and TAMs develop complex metabolic coupling: TAMs dynamically shape immune signal transduction by modulating lipid and cholesterol metabolism[51], and lipid rafts play a central role in determining immunosuppressive versus activating phenotypes[52, 53].
Lipid rafts, cholesterol- and sphingolipid-rich microdomains, act as signaling platforms that regulate receptor clustering, downstream phosphorylation, and vesicular transport. In TAMs, altered raft composition remodels receptor topology and biases signaling toward immunoregulatory states.
At the transcriptional level, sterol regulatory element-binding protein 1 (SREBP1) acts as a central node linking lipid availability to the immunosuppressive fitness of TAMs[54, 55]. In tumors, regulatory T cells can indirectly sustain an immunosuppressive TAM program by maintaining SREBP1-dependent lipid metabolic pathways in macrophages, thereby supporting mitochondrial integrity and survival of protumor TAM states, which provides a concrete rationale for discussing SREBP1 not only as a tumor-cell lipogenesis driver but also as a macrophage-intrinsic regulator of TAM persistence and function in lipid-rich urologic TMEs[55].
Recent studies indicate that, in hepatocellular carcinoma, cytokine-induced CD36 promotes recruitment of circulating monocytes via CCL2/CCR2/p110γ signaling, thereby expanding TAM populations; at the same time, raft stabilization is reinforced, promoting lipid accrual[56]. These raft-enriched complexes strengthen PI3K-AKT inhibitory signaling and push macrophages toward M2-like polarization[57]. Conversely, LXR-ABCA1/ABCG1-controlled cholesterol efflux governs raft cholesterol content and membrane order[58]. Importantly, tumor settings can exploit this axis in different directions. A study indicates that cancer cells drive cholesterol efflux from TAMs, leading to membrane lipid-raft depletion and a reprogrammed TAM state that favored protumor behavior[59]. This highlights that raft remodeling is pathway-selective—altering cytokine signaling and immune polarization by changing membrane organization rather than simply increasing or decreasing the density of rafts. In parallel, ABCA1-dependent membrane remodeling has been shown to regulate Toll-like receptor signaling by influencing receptor trafficking into rafts, supporting the concept that cholesterol handling can reshape innate immune responsiveness[60]. Such raft-centric reprogramming is particularly pronounced in PCa, where androgen-driven lipogenesis is closely intertwined with macrophage cholesterol metabolism[61]; thus, raft-macrophage metabolic coupling is also likely to manifest across urologic tumors. Spatial metabolomics reveals higher abundances of metabolites related to lipid synthesis and remodeling in PCa compared with normal prostate[62]. Lipid rafts are not static scaffolds but metabolically responsive decision nodes that govern TAM polarization. Targeting raft dynamics therefore emerges as a potential strategy to reprogram macrophage fate in lipid-enriched urologic tumors.
3.3 Ferroptosis-redox axis
Ferroptosis, a newly recognized form of cell death, usually features massive iron accumulation and lipid peroxidation; its occurrence is iron-dependent[63]. Ferroptosis inducers reduce intracellular antioxidant capacity and lead to accumulation of lipid reactive oxygen species (ROS), ultimately causing oxidative cell death[64, 65]. Recent research links ferroptosis to multiple diseases, including cancer, neurological disorders, ischemia-reperfusion injury, kidney injury, and hematologic conditions[63].TAMs import iron through TfR1 and DMT1 and export iron via FPN; the balance determines the level of the labile iron pool. Excess Fe2+ triggers in a Fenton-type radical chemistry using iron as the catalyst to generate ROS and initiate membrane lipid peroxidation. M2-like TAMs, which have lower GPX4 and GSH, are more sensitive to ferroptotic stress[11].
GPX4, as a central enzyme that reduces phospholipid hydroperoxides, can buffer ferroptotic stress and mitigate the toxic effects of ferroptosis. Importantly, macrophage susceptibility to GPX4 loss is activation-state dependent: alternatively activated (IL-4-driven) macrophages become highly vulnerable to ferroptosis when GPX4 is impaired, whereas inflammatory macrophages can be comparatively protected, highlighting ontogeny/activation cues as determinants of ferroptosis sensitivity[66]. Consistent with this, macrophage subsets can display heterogeneous ferroptosis susceptibility linked to differential GPX4 stability and redox wiring, suggesting that ferroptosis-based strategies may selectively pressure M2-like TAM programs but require careful control to avoid collateral impairment of myeloid antigen-handling functions[67].
From the perspective of ferroptosis substrates, ACSL4 shapes ferroptosis sensitivity by channeling polyunsaturated fatty acids into peroxidation-prone phospholipids. In tumors, cytotoxic T cell-derived IFN-γ can synergize with selective fatty acids to promote ACSL4-dependent tumor ferroptosis, providing a mechanistic basis for how immune activation and ferroptotic vulnerability can reinforce each other during immunotherapy[68]. Emerging data further suggest that ACSL4 can couple ferroptosis propensity to macrophage functional states, including contexts where ACSL4-associated lipid remodeling aligns with M1 polarization[69-71].
When immunogenic ferroptosis occurs, oxidized phospholipids and damage-associated molecular patterns (DAMPs) such as HMGB1 are released, activating macrophages and dendritic cells and promoting cross-presentation[72], potentially driving TAMs toward M1 phenotypes[73]. In PCa, specific circular RNAs can induce ferroptosis in TAMs by suppressing HSP90, thereby promoting M1-like polarization and hindering tumor progression[74, 75].
Iron metabolism is tightly linked to TAM clearance and cross-presentation capacity[76]. Iron-rich M1-like macrophages often show stronger phagocytosis, but excessive iron may drive ROS overproduction and functional impairment[77]. Under iron restriction, M2-like macrophages tend to express high ferritin and iron-export proteins, maintain low intracellular iron, and support tissue repair and remodeling[76]. Thus, tuning iron metabolism may be an effective strategy to reprogram TAM function, particularly in “cold” tumors enriched with M2-like TAMs.
This metabolic axis is especially critical in urologic cancers, which frequently involve lipid and iron dysregulation (Figure 1). In the future, combining ferroptosis inducers with ICIs may achieve synergistic clinical translation by acting on both tumor cells and TAMs (Table 1).
Three high-value metabolic axes that shape TAM function. pH reduction and HIF-VEGF axis activation driven by hypoxia and lactate, along with lipid rafts-centered metabolism promotes TAM polarization; ferroptosis triggered by ROS and specific circRNAs drives macrophage polarization and alters the antigen-presenting capacity. MCT, monocarboxylate transporter; HIF, hypoxia-inducible factor; mTORC, mechanistic target of rapamycin complex; ERK, extracellular signal-regulated kinase; ARG-1, arginase-1; VEGF-A, vascular endothelial growth factor -A; SREBP, sterol regulatory element-binding protein; FASN, fatty acid synthase; PI3K, phosphatidylinositol 3-kinase; AKT, ak strain transforming; TFR1, transferrin receptor 1; DMT1, divalent metal transporter 1; FPN, ferroportin; ROS, reactive oxygen species; HSP, heat shock protein; OxPLs, oxidized phospholipids; DAMPs, damage-associated molecular patterns.
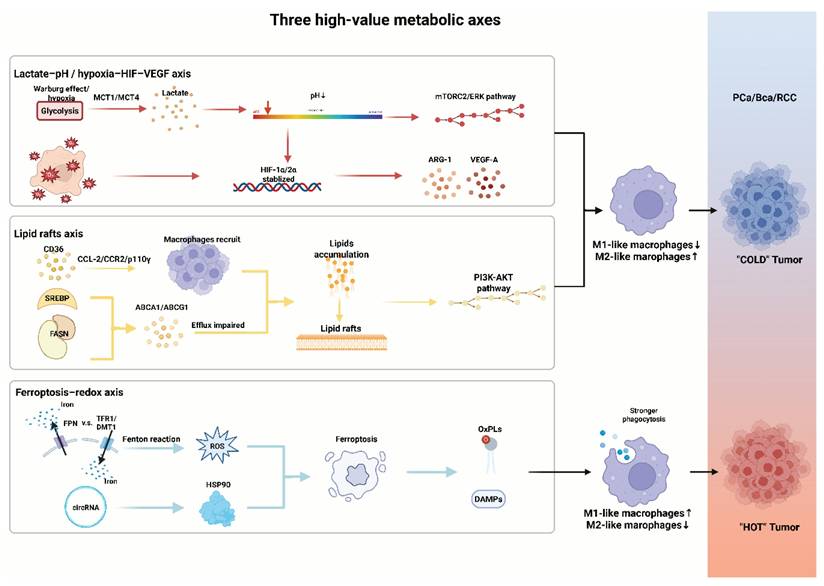
Metabolic axes that reprogram tumor-associated macrophages in urologic cancers.
| Axis | Key nodes / pathways | Targets (examples) | Expected TAM shift | Niche change | Biomarkers (PD/response) | Example agents / modalities | Future directions | Relative clinical trials | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| Lactate-pH / hypoxia-HIF-VEGF | Warburg glycolysis → lactate export (MCT1/4); extracellular acidosis; hypoxia→HIF-1α/2α→VEGF;adenosine crosstalk (CD39/CD73→A2A/A2B) | LDHA; MCT1/MCT4; HIF-2α; CD39/CD73; A2A/A2B | M2-like→M1-like re-education; ↑phagocytosis, ↑antigen presentation; ↓ARG1/IL-10 | ↓acidity (↑pH); ↓adenosine tone; transient vessel normalization; ↓hypoxia | HP-¹³C-pyruvate MRI kPL; plasma/urine lactate & adenosine panels; soluble ANGPT2 (RCC); spatial NT5E/ADORA2A modules | MCT1 inhibitor AZD3965; systemic buffering (bicarbonate/THAM); anti-CD73 (oleclumab); A2A antagonist (ciforadenant); HIF-2α inhibitor (belzutifan) | Define priming window before/with ICI; adaptive enrichment for glycolysis/adenosine-high tumors; integrate spatial PD with HP-¹³C imaging | NCT01791595NCT02531919NCT03773666NCT02655822NCT05239728 | 38414647146121 |
| Lipid rafts | Cholesterol/sphingolipid-rich microdomains;ABCA1/ABCG1 efflux;SREBP-driven lipogenesis; TREM2/PI3K-AKT inhibitory hubs | FASN; SREBP; TREM2; ABCA1/ABCG1(efflux/raft fluidity) | M2-immunoregulatory→pro-inflammatory tilt; ↓raft clustering; ↑APC function | ↓raft stability on tumor/myeloid membranes; ↓tolerogenic signaling platforms | Lipid-raft transcriptomic modules; lipidomic signatures; spatial PLIN/HILPDA context (RCC) | FASN inhibitor (denifanstat/TVB-2640); cholesterol targeting/efflux enhancers; statin/oxysterol-axis exploration | Drugging raft biology in TAMs; combine with PD-1/PD-L1 after raft destabilization; define fluidity PD | NCT05743621NCT02922764NCT04691375 | 585975165 |
| Ferroptosis-redox | Iron import (TfR1/DMT1) & export (FPN); GPX4/GSH antioxidant axis; lipid peroxidation; immunogenic ferroptosis (HMGB1/OxPLs) | System xc⁻ (SLC7A11); GPX4; iron chelation; | Context-dependent: induce immunogenic death → ↑cross-presentation/M1 shift;restrain iron overload to avoid dysfunction | ↑oxidized lipids in TME (if induced); potential ↑antigenicity; redox normalization with chelation | Oxidized phospholipids; circulating iron indices; redox gene programs; phagocytosis/ADCP function readouts | Erastin/RSL3 derivatives; GPX4 inhibitors; iron chelators (deferoxamine/deferasirox); nano-ferroptosis platforms | Dose-finding to avoid global myelosuppression; pair with ICI in biomarker-defined settings; map myeloid-specific ferroptosis sensitivity | Early-stage R&D | 131686669 |
MCT, monocarboxylate transporter; HIF, hypoxia-inducible factor; VEGF, vascular endothelial growth factor; LDHA, lactate dehydrogenase A; ANGPT, angiopoietin; RCC, renal cell carcinoma; SREBP, sterol regulatory element-binding protein; TREM2, triggering receptor expressed on myeloid cells - 2; FASN, fatty acid synthase; TFR1, transferrin receptor 1; DMT1, divalent metal transporter 1; FPN, ferroportin; OxPLS, oxidized phospholipids; TME, tumor microenvironment.
Collectively, the lactate-pH/hypoxia-HIF-VEGF axis, lipid rafts signaling, and ferroptosis-redox axis pressure form a convergent mechanistic framework through which metabolic stress is translated into durable TAM programs in urologic tumors. Across these axes, metabolites and membrane organization jointly reshape macrophage polarization, antigen handling, angiogenic output, and immune-checkpoint tone. Key gaps remain in defining how these axes intersect in space and time. Translationally, these insights argue for mechanism-anchored metabolic priming to re-educate TAMs and sensitize tumors to classic treatments, ideally guided by pharmacodynamic and spatial readouts that report on axis engagement and TAM-state transitions.
4. Metabolism-epigenetics-immunity triad in TAM plasticity
4.1 Metabolites serve as epigenetic regulators
Metabolites serve as substrates or cofactors for epigenetic modifications and directly shape TAM transcriptional programs and functional states.
In breast-cancer models, tumor-secreted arginine is taken up by TAMs and aberrantly activates polyamine biosynthesis. Excess spermidine modulates DNA demethylase activity, remodeling the epigenetic states of key immune-regulatory genes and ultimately driving macrophages toward immunosuppressive phenotypes[78].
As noted, hypoxia stabilizes HIF-1α, which induces glycolysis and M1 polarization[38]. Beyond the canonical HIF pathway, hypoxia elicits profound epigenetic reprogramming. By altering H3K4me3 distribution, hypoxia drives widespread transcription start site (TSS) switching, thereby reshaping the 5'-UTR of mRNAs. This remodeling changes the translational efficiency of key metabolic genes such as PDK1, facilitating glycolytic reprogramming and enabling cells, including TAMs, to adapt to hypoxia[79].
Microbiome-derived metabolites also participate in these feedback circuits. For example, indole-3-acetic acid produced by the commensal Parabacteroides distasonis suppresses fatty acid synthase (FASN) expression via the AhR axis, thereby promoting phospholipid remodeling and increasing ferroptosis sensitivity in BCa cells. Such metabolism-epigenetic reprogramming directly reshapes the TME and provides a new perspective for targeting microbe-host interactions[80].
4.2 Metabolically controlled transcription factors
Multiple transcription factors act as metabolic sensors connecting cellular metabolic states to TAM gene programs. HIFs are stabilized under hypoxia but are also regulated by metabolites such as succinate and fumarate, driving genes for angiogenesis and glycolysis. In FH-deficient RCC, fumarate accumulation inhibits HIF prolyl-hydroxylases, stabilizes HIF, and promotes angiogenesis and tumor progression[28].
PPARs, regulated by lipid metabolites, are highly expressed in M2-like TAMs and promote fatty-acid oxidation and oxidative phosphorylation (OXPHOS)[81]. SREBPs, as master regulators of cholesterol and fatty-acid synthesis, are activated in lipid-loaded TAMs, supporting membrane biogenesis and raft formation[82, 83]. NRF2 responds to oxidative stress and regulates antioxidant networks, protecting TAMs against ROS damage[84]. Members of the STAT family are activated by cytokines and growth factors: STAT1 drives M1-associated genes, whereas STAT3/STAT6 foster M2 polarization[81].
4.3 Metabolically guided immune outputs
Metabolic reprogramming and the resultant epigenetic changes ultimately modulate cytokine/chemokine outputs, fixing TAMs into distinct functional states that directly influence antitumor immunity.
Work by Kuang et al. in HCC demonstrates that the tumor-exported polyamine metabolite N1-acetylspermidine induces macrophage polarization toward immunosuppressive states and recruits CCR8+ regulatory T cells, thereby diminishing the efficacy of checkpoint blockade[85]. This is a prototypical case in which metabolites directly govern immune-cell function and output into specific immune patterns.
A review by Malczewski, A. B et al. details how gut microbial metabolites optimize immune responses. Short-chain fatty acids enhance CD8+ cytotoxicity and memory differentiation by inhibiting histone deacetylases and activating GPCRs, thus augmenting ICI efficacy. This indicates that metabolic signals from distant organs (the gut) can, via epigenetic mechanisms, systemically influence local tumor immunity[86].
4.4 Metabolism-epigenetics-immunity feedback loops
Within the TME, TAM metabolism-epigenetics-immunity states are not isolated; they are continuously shaped by signals from other cells and systems. Through cytokine and chemokine production, the metabolic state of TAMs forms complex feedback loops with other TME components.
As noted, inflammatory macrophages stimulate hepatocellular carcinoma cells to upregulate spermidine/spermine N1-acetyltransferase 1, inducing production and export of N1-acetylspermidine. This metabolite in turn acts on macrophages, polarizing them toward more immunosuppressive phenotypes—constituting a malignant positive feedback loop mediated by metabolites between tumor cells and TAMs that progressively exacerbates immunosuppression[85].
For example, IL-10 and TGF-β produced by M2 TAMs not only directly suppress T-cell function but also influence metabolic properties of tumor and other immune cells to promote tumor progression[5]. Likewise, tumor-derived metabolites such as lactate and adenosine further reinforce immunosuppressive TAM phenotypes, forming self-sustaining feedback circuits[25, 43, 87].
5. Disease-specific syntheses
5.1 Prostate cancer / castration-resistant prostate cancer (CRPC)
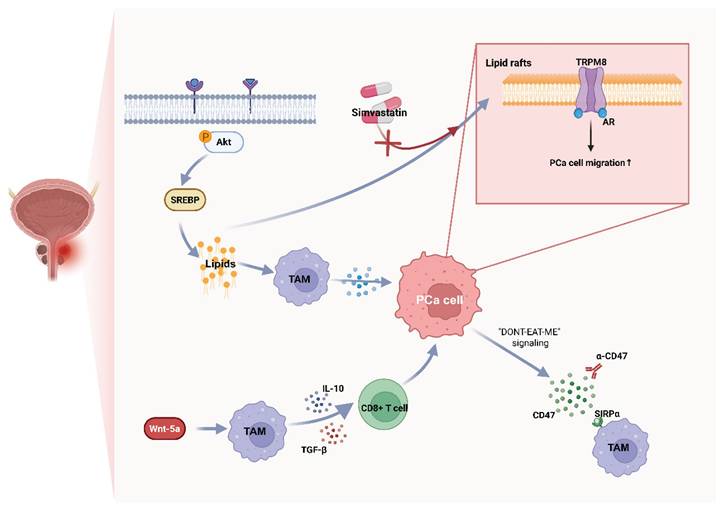
Prostate tumors—especially CRPC—exhibit high dependence on lipid synthesis and uptake. This metabolic remodeling affects tumor cells and TAMs, while cholesterol-rich lipid rafts organize immunoregulatory and motility signals on membranes[10, 88, 89].
Lipid addiction in CRPC commonly features SREBP-driven lipogenesis and upregulation of fatty-acid uptake/desaturation programs such as FASN, SCD, which support proliferation, stress tolerance, and therapeutic resistance[90]. Accumulation of cholesterol and complex lipids not only satisfies biosynthetic needs but also impacts antigen presentation and cytokine secretion in myeloid cells within the microenvironment[88, 91]. Cholesterol-enriched rafts act as “signal islands” that co-localize receptors such as AR complex, TRPM8 and kinases to promote tumor-cell survival and motility; raft disruption alters phosphorylation cascades and weakens these signals[10, 89, 92]. Such raft-organized platforms are also associated with immune evasion because raft composition modulates myeloid-receptor clustering and downstream PI3K-AKT/TREM2-linked inhibitory programs[93].
In the TME, accumulations of Myeloid-derived suppressor cells (MDSCs) and TAMs become key drivers of androgen receptor (AR)-independent resistance. For example, in microenvironments with high Wnt5a, infiltration of CD68+ macrophages increases significantly; these induced macrophages secrete IL-10 and TGF-β, directly suppressing CD8+ T-cell function and driving immunosuppression[94]—revealing a resistance pathway mediated by myeloid cells that bypasses AR signaling itself. Macrophages may also promote anti-androgen resistance via paracrine loops and AR/TREM2 signaling within myeloid cells, collectively weakening antitumor immunity and accelerating progression[95, 96].
In addition, neuroendocrine transdifferentiation (NEPC) is a common event in PCa progression and is associated with distinctive metabolic and immune features. Therapeutic pressure can drive lineage plasticity toward neuroendocrine phenotypes through factors such as activation of FOXA2, ONECUT2, and KIT, resulting in AR low/negative states and altered immune interactions[97, 98]. NEPC and advanced CRPC often upregulate “don't-eat-me” checkpoints—most notably CD47—which engages macrophage SIRPα to suppress phagocytosis; blocking CD47/SIRPα restores clearance and enhances antitumor immunity in preclinical systems[99-101]. Moreover, reverse signaling through CD47 may support PCa-cell survival under phagocytic stress, suggesting a dual role for this axis in lineage plasticity and refractory disease[102] (Figure 2).
Representative metabolic-immune coupling pathways in prostate cancer. Lipid Rafts-stabilized inhibitory signaling and AR-independent myeloid circuits Wnt-5a blunt macrophage functions; in NEPC, upregulation of CD47-SIRPα axis suppresses clearance. TAM, tumor-associated macrophage; TRPM8, transient receptor potential melastatin 8; SIRPα, signal-regulatory protein α.
5.2 Bladder cancer: glycolytic and adenosinergic immunosuppression
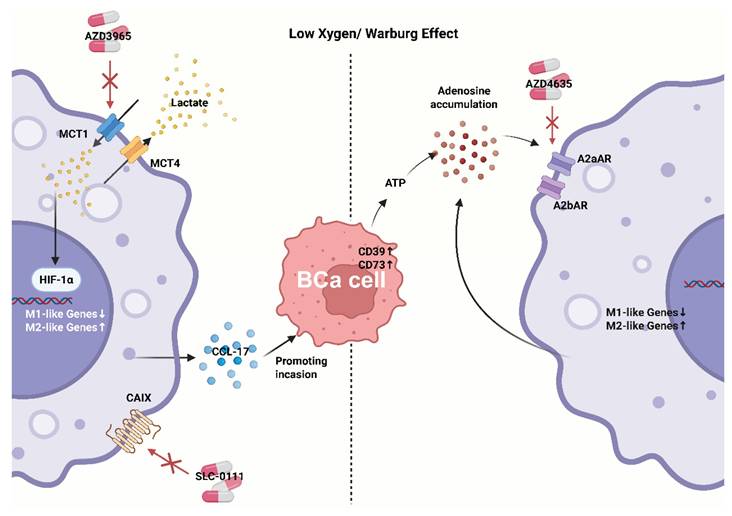
As noted, BCa is characterized by high glycolytic activity and lactate accumulation, creating an acidic, adenosinergic milieu that polarizes TAMs, weakens T/NK-cell effector programs, and maintains an immune-excluded (“cold”) phenotype[103, 104].
In BCa and other solid tumors, lactate accumulation biases myeloid cells toward suppressive states (e.g., Arg1high/IL-10high) and impairs cytotoxic lymphocytes; new studies suggest lactate consolidates immune paralysis via epigenetic reprogramming such as histone acetylation/lactylation[105, 106]. In BCa cohorts, glycolysis signatures correlate with poorer prognosis and immune imbalance, supporting glycolysis as a driver rather than a bystander[106].
Adenosinergic immunosuppression is another hallmark of the BCa TME. Under hypoxia and cellular stress, CD39 and CD73 are upregulated on tumor/stromal compartments (including BCa-derived exosomes), converting ATP to adenosine; adenosine signals through A2A/A2B receptors to raise cAMP and blunt antitumor responses[107, 108]. In the syngeneic MB49 model, pharmacologic A2B blockade slows tumor growth; recent orthotopic bladder cancer studies show that co-targeting CD73 augments anti-PD-L1 efficacy, nominating the adenosine axis as an actionable immunosuppressive hub in BCa[109, 110]. Notably, TAMs themselves are an important source of adenosine in urologic tumors, creating self-reinforcing immunosuppression[111].
Recent data indicate the potential to “warm” cold tumors by targeting these metabolic axes.
(1) Lactate control (production/transport). Inhibiting LDHA or blocking MCT1/4-mediated lactate shuttling increases metabolic stress in tumor cells while reducing immunosuppressive export[112]. The MCT1 inhibitor AZD3965 has first-in-human data, and preclinical evidence for LDH blockade is accumulating[113]. In BCa models, LDHA activity associates with invasiveness; mechanistically, combining lactate-pathway inhibitors with ICIs may restore antigen presentation and CTL function within acidic niches[104, 114].
(2) Purinergic blockade (CD39/CD73/A2A/A2B). Strategies include anti-CD39 or anti-CD73 antibodies and small-molecule A2A/A2B antagonists; across multiple tumors, such agents release the adenosine “brake” and synergize with PD-1/PD-L1 therapy, with orthotopic BCa evidence as disease-specific support[109, 115, 116].
(3) Integration with intravesical immunotherapy. BCG relies on strong innate recognition and trained-immunity-like programs; in principle, tuning lactate and adenosine could augment BCG-induced myeloid activation and cross-priming, potentially converting partial responders into inflamed responders[117-119] (Figure 3).
Representative metabolic-immune coupling pathways in bladder cancer. Acidic/adenosinergic niches suppress antigen handling and promote TAMs polarization, maintaining a "cold" phenotype despite checkpoint blockade. CAIX, carbonic anhydrase IX.
5.3 Renal cell carcinoma: HIF-centric metabolism and angiogenesis coupling
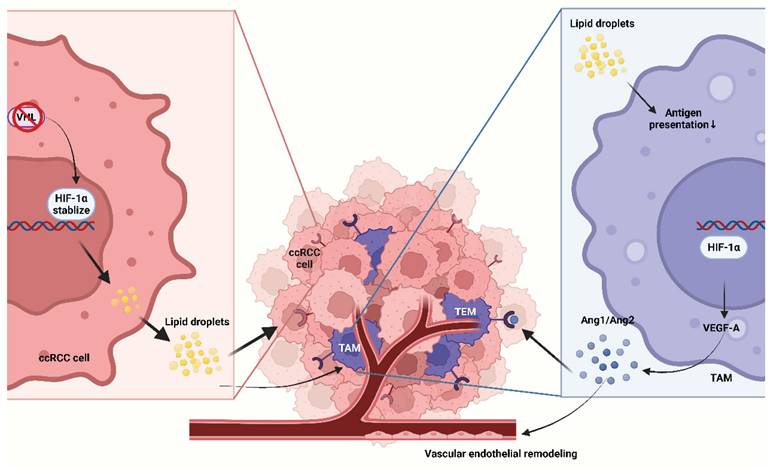
RCC—particularly the clear-cell subtype (ccRCC)—is characterized by early loss of VHL, with consequent stabilization of HIF-α, which drives profound metabolic reprogramming. This includes heightened glycolysis, diminished OXPHOS, and lipid-droplet accumulation, profoundly influencing the TME and producing a lipid-laden phenotype[120-123].
Lipid droplets serve as metabolic hallmarks. ccRCC accumulates neutral lipids and cholesteryl esters in droplets driven by HIF-regulated programs such as HILPDA/PLIN family and glutamine-dependent lipogenesis; droplets buffer redox stress and supply fatty acids for signaling and membrane remodeling[124, 125]. Lipid-rich tumor cells also alter myeloid metabolism via lipid mediators/oxylipins and impair antigen presentation; TAMs exposed to HIF-driven lipid signals readily acquire pro-angiogenic, immunoregulatory states[126-128].
TAM-angiogenesis coupling plays a central role in RCC progression. TAMs produce pro-angiogenic factors, including VEGF-A and ANGPT2; via the VEGF/ANG-TIE2 axis, they collaborate with tumor-derived angiogenic signals to remodel endothelium and recruit Tie2+ macrophages (TEMs)[129, 130]. Although neovascularization supplies oxygen and nutrients, it is typically abnormal and leaky, exacerbating hypoxia and acidosis and creating self-reinforcing loops[131]. Anti-VEGF therapy is standard-of-care in RCC, and part of its efficacy depends on effects on TAMs. Anti-VEGF/VEGFR Tyrosine kinase inhibitors (TKIs) can transiently normalize vasculature, facilitate Cytotoxic T lymphocyte (CTL) infiltration, and modulate myeloid composition—considered a mechanistic basis for ICI-TKI combinations (first-line regimens such as pembrolizumab-axitinib, pembrolizumab-lenvatinib, nivolumab-cabozantinib) that improve OS/PFS[132-135].
Targeting upstream HIF, hypoxia also enhances immune suppression and sustains TAM programs that promote aberrant vasculature, providing a mechanistic rationale to pair HIF-2α blockade with PD-1/PD-L1 inhibitors[136-138]. Cutting-edge research is now further exploring the feasibility of combining HIF-2α inhibitors with ICIs. Clinically, the phase 3 LITESPARK-005 trial established belzutifan (HIF-2α inhibitor) activity after prior PD-(L)1 and VEGF-TKI: PFS HR 0.75 (95% CI 0.63-0.90) versus everolimus, a mTOR inhibitor; due to non-proportional hazards, medians were similar (both 5.6 months), yet curves separated late and responses were higher for belzutifan[139]. On-target toxicities of HIF-2α inhibition such as anemia, hypoxia are predictable and manageable with dose modification and monitoring, an important consideration when layering with ICI. Belzutifan and Lenvatinib combination has shown promising activity post-PD-1/PD-L1 in multicenter studies and press-release analyses, meeting PFS and ORR endpoints versus cabozantinib in previously treated disease, showing combination strategies are maturing[140] (Figure 4).
Representative metabolic-immune coupling pathways in clear cell renal cell carcinomas. VHL loss leads to HIF signaling activation, manifested as lipid droplet accumulation that shapes a lipid droplet-rich clear-cell phenotype; cytokines produced by TAMs act through the VEGF/ANG-TIE2 axis and synergize with tumor cell-derived angiogenic signals to remodel the endothelium and recruit TEMs. Ang, angiopoietin; TEM, Tie2-expressing macrophage.
Disease-specific metabolism selects for distinct macrophage programs and resistance logics that can be mapped onto the three metabolic axes rather than simply reprogram TAMs in urologic cancers. Harmonizing cross-cancer TAM taxonomies remains gaps needed to be further studied. While another gaps is the limited ability to prospectively identify which metabolic niche is rate-limiting in an individual patient, especially given strong spatial heterogeneity. Translationally, these points argue for organ- and niche-tailored strategies rather than uniform TAM targeting, with patient selection and response evaluation anchored to spatially informed biomarkers.
6. Translational Toolkit: Models, Readouts, and Biomarkers
6.1 Preclinical model systems
Studying metabolism-immunity crosstalk in urologic tumors requires models that capture complex metabolic coupling between tumor and immune cells.
Organoids with immune co-cultures—co-culturing tumor organoids with autologous or syngeneic immune cells—preserve tumor genomic and spatial heterogeneity along with patient-specific metabolic and immune features. They are suitable for high-throughput screening and mechanistic work and allow controlled manipulation of lactate, cholesterol, and adenosine pathways. Recent reviews and urologic exemplars describe immune-organoid systems that incorporate myeloid components, recapitulate in-vivo settings, permit precise control of microenvironmental variables (pH, O2, metabolites), and integrate smoothly with spatial/multiplex readouts to direct TAM perturbation. Organoids have been established across PCa, BCa, and RCC[141-143]. Nonetheless, these systems simplify the immune milieu, lack full stromal/vascular signals, and cannot fully encompass the entire metabolism-immunity nexus; long-term culture may induce metabolic drift, limiting predictive value[143].
Humanized xenograft models implant human tumors into immunodeficient mice reconstituted with human immune systems, enabling in-vivo assessment of human-specific targets. They are crucial for testing strategies that target human metabolic enzymes or checkpoints, and they provide intact vasculature for combinations such as ICI, anti-CD73/A2A, and HIF-2α inhibitors[144, 145]. However, interspecies metabolic gaps, risk of GVHD, and incomplete myeloid reconstitution may impact TAM-focused studies[144].
Ex vivo precision-cut tumor slices preserve native TME architecture and composition and suit short-term readouts for metabolic interventions. Fresh slices from RCC/PCa/BCa retain multicellular structure, gradients, and local metabolite pools, enabling rapid Pharmacokinetics/ Pharmacodynamics (PK/PD) assessment of therapy-induced metabolic and immune functional changes close to the patient context; nonetheless, tissue availability and throughput constrain their use[146].
Each model has unique strengths and limitations. Selection should be tailored to the scientific question and hypothesis under test.
6.2 Readouts: multi-omics and imaging
Advanced analytics provide unprecedented insight into TAM metabolic states and functions.
Metabolic flux analysis uses stable-isotope tracers to track fluxes and reveal authentic in-situ TAM metabolism. When integrated with scRNA-seq and spatial transcriptomics, these data resolve metabolic heterogeneity among TAM subsets and across space[147-149].
Single-cell spatial multi-omics, including spatial transcriptomics and mass spectrometry imaging, offers spatial context for metabolite distributions, cell composition, and gene expression within native tissues. In urologic tumors, integrating these technologies with metabolism-immune phenotypes reveals how metabolic gradients shape TAM distribution/states (phagocytic, angiogenic, regulatory) and their proximity to metabolic niches (acidic, lipid-droplet-rich, hypoxic)[150-152].
Metabolic imaging, such as hyperpolarized MRI and specialized PET tracers, supports noninvasive, longitudinal monitoring of metabolic interventions. For example, HP 13C-pyruvate MRI quantifies real-time pyruvate to lactate conversion rate (kPL) and is advancing clinically in PCa; molecular PET beyond FDG such as hypoxia and glutamine tracers is applicable to urologic tumors[153-156].
Functional assays such as phagocytosis/ADCP rely on flow cytometry or imaging like pH-sensitive cargo and live uptake to quantify macrophage effector functions linked to antitumor immunity[157].
6.3 Biomarker development
Successful clinical integration of metabolism-immune targeting requires biomarkers that identify likely responders. Representative candidate biomarkers include ANGPT2, HP 13C-pyruvate MRI kPL, myeloid-derived IL-23, and PLIN2.
6.3.1 Plasma ANGPT2 (RCC) is a myeloid-angiogenesis coupling biomarker associated with ICI-TKI efficacy. ANGPT2 rises during anti-VEGF escape and recruits/perivascularly localizes Tie2+ macrophages, strengthening sprouting angiogenesis and immunosuppression[158, 159]. High ANGPT2 often corresponds to abnormal, leaky vasculature; on-treatment ANGPT2 decline parallels vascular normalization, facilitating CTL entry (frequently observed under VEGFR-TKI + ICI)[160]. The ANGPT2/TIE2 module resonates with TEM (Tie2+ macrophage) transcriptional features and the CSF1/IL-34-CSF1R axis, highlighting an immune-vascular network[160]. Thus, plasma ANGPT2 should be correlated with TEM density and endothelial tip-cell features in tumor slices/spatial omics and can serve as a PD readout to monitor early responses to ICI—TKI combinations. Because nonspecific inflammation can also elevate ANGPT2, it must be interpreted alongside perfusion/hypoxia imaging indices and myeloid gene modules.
6.3.2 HP 13C-pyruvate MRI kPL (PCa & BCa): Hyperpolarized (HP) 13C-pyruvate MRI is a stable-isotope molecular imaging modality that provides real-time assessment of the rate of metabolism through glycolytic pathways in human prostate cancer. HP 13C-pyruvate MRI kPL, as a live glycolytic-flux biomarker associated with “cold” to “hot” transitions, reflecting glycolytic pressure that shapes acidity/adenosine niches and TAM polarization[161]. In metastatic PCa, kPL can decline early on therapy and correlate with metabolic response; in metabolism-immunity studies, a drop in kPL can serve as an indirect readout of lactate-pathway inhibition (LDH/MCT) or ICI engagement during transition from “cold” to “hot” phenotype[161, 162]. It is suitable as a PD biomarker and early surrogate endpoint, for example, kPL decline linked to changes in urinary/blood lactate and adenosine metabolites. Although evidence for individual stratification is still accruing. Meanwhile, sensitivity is limited for lesions not dominated by glycolytic metabolism[161, 163].
6.3.3 Myeloid-derived IL-23 (CRPC): a driver of AR-independent resistance and a serum/tissue biomarker. IL-23 secreted by MDSCs sustains CRPC growth and AR program output under androgen-deprived conditions; blockade reverses this effect in animal models and human explants. Elevated IL-23 often coexists with myeloid infiltration and exhausted T cells; IHC/multiplex imaging shows pSTAT3 activation co-localizing with IL-23 in CD68+ / CD163+ myeloid cells. The IL-23 axis can link NF-κB/STAT3 inflammatory programs with residual AR-target gene expression, indicating a “myeloid—AR-independent resistance” route[164]. Therefore, IL-23 was considered to have potential for predicting anti-androgen resistance and monitoring responses to myeloid-targeted interventions, although prospective thresholds, standardized assays, and interventional trials are needed. Because IL-23 also rises in systemic inflammation, single-analyte specificity is limited; it is best interpreted alongside MDSC/TAM transcriptional signatures and circulating cytokine panels[164].
6.3.4 PLIN2 (RCC): a marker linking HIF-driven lipid-droplet biology to noninvasive urinary detection. PLIN2 coats lipid droplets and is upregulated in VHL/HIF-driven ccRCC; urinary PLIN2 shows good diagnostic performance. Histologically, PLIN2 immunocytochemistry (IHC) aligns with the “clear-cell/lipid-droplet-rich” phenotype; metabolic imaging often shows expanded lipid pools and rich perfusion (relevant for anti-angiogenic treatment selection and monitoring)[165]. PLIN2 co-upregulates with HIF targets and lipid-droplet programs, delineating a HIF-centric lipid-droplet axis[166]. It is suitable for noninvasive screening/differential diagnosis and follow-up monitoring; however, dedicated studies on predictive value for immune combinations are lacking, and combined interpretation with imaging/gene programs is most prudent[165].
6.3.5 Summary
In general, circulating biomarkers offer accessible alternatives but may not fully mirror site-specific metabolism-immunity interactions; imaging biomarkers provide whole-tumor assessment and noninvasive monitoring but may lack molecular specificity. An ideal strategy likely integrates multi-layered information to maximize predictive value while minimizing invasiveness and cost (Table 2).
Experimental platforms and pharmacodynamic readouts to operationalize metabolism-informed TAM re-education.
| Toolkit | Platform / biomarker | What it measures | Strengths | Limits | Best use cases | Ref. |
|---|---|---|---|---|---|---|
| Preclinical model systems | Organoids with immune co-cultures | Tumor-intrinsic metabolism and immune interactions under controlled pH/O2/metabolites; TAM/T-cell crosstalk | Patient-specific; perturbation-ready; scalable; integrates with imaging/spatial/multiplex | Simplified stroma/vasculature; partial myeloid fidelity; metabolic drift over time | Titrate lactate/adenosine/lipid interventions; shortlist combinations; mechanism discovery | 136 |
| Humanized xenograft models | In-vivo human tumor + human immune system; vascular and pharmacologic context | Human-specific targets; drug PK/PD; vessel effects (VEGF/HIF) | Incomplete myeloid reconstitution; interspecies metabolism; GVHD risk | Test ICI + metabolic agents (CD73/A2A, MCT1, HIF-2α) | 137 | |
| Ex vivo tumor slices | Native architecture & gradients; short-term functional PD | Patient-proximal; preserves spatial niches; rapid turnaround | Limited tissue; short culture window; throughput constraints | Bridge PK/PD to biology; visualize TAM phagocytosis/raft disruption | 139 | |
| Readouts | Metabolic flux analysis | Real-time carbon/nitrogen flux through glycolysis, TCA, FAO | Mechanistic resolution; quantifies pathway engagement | Specialized analytics; tissue requirement | Confirm on-target effect of LDH/MCT/FASN modulation | 140 142 |
| Single-cell spatial multi-omics | Cell states & niches (myeloid/TAM programs; NT5E/ADORA2A; PLIN/HILPDA) | Topographic context; heterogeneity; discovery-ready | Cost/complexity; standardization needs | Patient selection & pharmacodynamic mapping; mechanism of resistance | 143 | |
| Metabolic imaging (HP-13C MRI; PET tracers) | Live flux (kPL), hypoxia, glutamine uptake | Noninvasive; whole-tumor; longitudinal | Access & sensitivity vary; lesion heterogeneity | Early PD anchor; adaptive enrichment | 146 147 148 | |
| Functional assays (phagocytosis/ADCP) | Macrophage effector function (uptake/clearance) | Actionable, mechanistic | Assay variability; ex-vivo dependence | Go/No-Go gating for macrophage-centric combos | 150 | |
| Biomarker | Plasma ANGPT2 (RCC) | Myeloid-angiogenesis coupling; vascular tone | Accessible; links to ICI-TKI biology | Inflammation confounders; needs context imaging | Monitor RCC combos; enrich ANGPT2-high cases | 153 |
| HP-¹³C-pyruvate MRI kPL (PCa & BCa) | Pyruvate→lactate flux (glycolytic pressure) | Early, noninvasive PD; spatially comprehensive | Availability; not all lesions glycolysis-dominant | Track lactate-pathway inhibition; prime-then-ICI designs | 154 | |
| Myeloid-derived IL-23 (CRPC) | AR-independent resistance circuit from myeloid cells | Biology-anchored; tissue/serum measurable | Specificity limits in systemic inflammation | CRPC enrichment; monitor anti-myeloid strategies | 157 | |
| PLIN2 (RCC) | HIF-centric lipid-droplet program; urinary detection | Noninvasive screen/follow-up; histologic concordance | Predictive value for combos under study | RCC detection/monitoring; pair with imaging | 158 159 |
PK/PD, pharmacokinetics/ pharmacodynamics; VEGF, vascular endothelial growth factor; HIF, hypoxia-inducible factor; GVHD, graft-versus-host disease; ICI, immune checkpoint inhibitor; MCT, monocarboxylate transporter; LDH, lactate dehydrogenase; FASN, fatty acid synthase; TCA, tricarboxylic acid cycle; FAO, fatty acid oxidation; MRI, magnetic resonance imaging; ANGPT2, angiopoietin 2; RCC, renal cell carcinoma; PCA, prostate cancer; BCA, bladder cancer; CRPC, castration-resistant prostate cancer; PLIN, Perilipin.
7. Therapeutic strategies and combination
In urologic cancers, metabolic reprogramming shapes immunosuppressive ecologies—acidic, adenosinergic, and raft-stabilized—that push macrophages toward tumor-promoting states; accordingly, clinically relevant treatments increasingly turn to direct metabolic interventions and immunotherapy combinations to re-educate macrophages and restore cytotoxic programs.
7.1 Direct metabolic interventions
Because lactate and low pH blunt antigen presentation and suppress macrophage phagocytosis, lactate-pathway interference is attractive. The clinical MCT1 inhibitor AZD3965 demonstrated acceptable safety and on-target pharmacodynamics in first-in-human studies, raising intracellular lactate and remodeling tumor bioenergetics as expected; these data provide a feasible foundation for lactate relief + ICI treatment[113]. In parallel, tumor acidity buffering with systemic alkalinizing agents such as bicarbonate/THAM) has long been shown in preclinical models to increase intratumoral pH and improve drug penetration, yet widespread clinical adoption requires rigorous standardization of dose, patient selection, and safety frameworks[167-169]. Carbonic anhydrase IX (CAIX) is a transmembrane protein closely associated with the hypoxic conditions of the tumor microenvironment. Given CAIX's strong hypoxia-link and overexpression in RCC and a subset of bladder cancers, CAIX blockade remains a rational direct approach to neutralize extracellular acidity that impairs drug penetration and myeloid function[170]. The CAIX inhibitor SLC-0111 completed a phase-I trial with acceptable tolerability and pharmacokinetics across solid tumors, operationalizing pH-axis targeting in humans[171]. As previously mentioned, hypoxia and lactic acid accumulation contribute to the reinforcement of the M2-like TAM phenotype.
These treatments have the potential to ameliorate hypoxia, increase intratumoral pH and has been shown in preclinical models to improve responses to checkpoint blockade; mechanistically, such buffering is expected to relieve pH-driven suppression of macrophage phagocytosis and antigen presentation, thereby facilitating T-cell reinvigoration and potentially supporting “cold” to “hot” transitions.
Lipid and cholesterol modulation offers another promising direction because these programs stabilize suppressive signaling platforms in myeloid and tumor cells. The selective FASN inhibitor denifanstat TVB-2640 has advanced through phase I/II settings with manageable toxicity and combinability, indicating that lipid-anabolism modulation is clinically tractable and may, in principle, reduce de novo lipogenesis that supports suppressive TAM programs in lipid-rich niches; mechanistically, constraining lipid flux and perturbing cholesterol-rich rafts may dismantle myeloid suppressive circuits and enhance ICI sensitivity.[172] Classic mechanistic work in PCa shows that cholesterol targeting disrupts raft-dependent survival and motility signals—a principle applicable to macrophage-tumor crosstalk where raft clustering coordinates PI3K/AKT and immunoregulatory receptors[10].
Iron metabolism and ferroptosis modulation can be pursued with iron chelators (e.g., deferoxamine) and ferroptosis inducers (e.g., erastin derivatives). Although iron chelators are pharmacologically mature and may suppress unchecked peroxidation loops in select settings, oncology-specific schedules and on-target immune consequences require prospective validation[173]. Conversely, inducing ferroptosis, by inhibiting system xc- or GPX4, or via nano-delivery, can increase immunogenic stress and antigenicity, but effects on myeloid states are context-dependent and call for careful dose-finding and cautious integration with ICIs[63, 174-176]. This therapeutic strategy attenuates iron-catalyzed Fenton-type radical reactions, thereby reducing ROS generation and mitigating membrane lipid peroxidation. As previously discussed, while iron-rich M1-like TAMs typically exhibit enhanced phagocytic capacity, excessive iron levels can trigger ROS overproduction, leading to functional impairment. Conversely, under iron-restricted conditions, M2-like TAMs prioritize the expression of ferritin and iron exporters to maintain low intracellular iron levels, which supports their role in tissue repair and remodeling. Consequently, modulating iron metabolism and ferroptosis represents a promising strategy for reprogramming TAM functions, particularly within 'cold' tumors characterized by an abundance of M2-like TAMs.
7.2 Combination strategies
Combining metabolic interventions with ICIs shows great promise in preclinical models. The rationale is that metabolic reprogramming can convert “cold” tumors into “hot” tumors and thus enhance ICI efficacy.
HIF-2α inhibitor-Belzutifan-based combinations are leading the metabolic-ICI interface in RCC, with multiple readouts now available across lines of therapy[177]. The phase II LITESPARK-003 (NCT03634540) doublet belzutifan + cabozantinib demonstrated durable antitumor activity in both treatment-naïve and post-immunotherapy cohorts, offering a modular backbone for future ICI-layering or sequencing[177]. In another umbrella KEYMAKER-U03 Substudy 03A (NCT04626479), first-line belzutifan + pembrolizumab + lenvatinib achieved high activity; notably, Arm 4 showed ORR 77.5% (95% CI 66.8-86.1) and median PFS 31.8 months (95% CI 26.3-NR) vs the pembrolizumab + lenvatinib reference (HR 0.45, 95% CI 0.25-0.83), with anemia emerging as the expected on-target toxicity of HIF-2α blockade[178].
Adenosine pathway and ICIs combinations seek to release hypoxia-linked immunosuppression upstream of PD-1/PD-L1[179, 180]. In mCRPC, the phase II modular study AZD4635 (A2A antagonist) + durvalumab and/or oleclumab (D8731C00001/NCT04089553) established feasibility and safety with mixed efficacy, informing patient-selection strategies and biomarker development for adenosine-rich niches,[181]. In MIBC, the neoadjuvant durvalumab ± oleclumab (anti-CD73) feasibility study BLASST-2 (NCT03773666) demonstrated practicality of perioperative adenosine-axis modulation; institutional and registry pages detail design and endpoints while mature efficacy readouts are awaited. In RCC, ciforadenant (CPI-444, A2A antagonist) + atezolizumab (NCT02655822) produced signals of activity in early-phase studies, with exploratory adenosine-gene signatures associating with response—supporting a test-and-treat paradigm for adenosinergic niches[182]. The combination serves as a strategic intervention to abrogate adenosine-mediated myeloid immunosuppression, thereby shifting TAM polarization from IL-10-producing, repair-associated programs toward MHC-IIhigh inflammatory antigen-presenting phenotypes. This phenotypic reprogramming may act synergistically with PD-1/PD-L1 inhibition to facilitate the 'cold-to-hot' transformation of the tumor microenvironment in adenosine-rich tumors. Nevertheless, the clinical translation of this synergy necessitates further validation via prospective TAM characterization within specific urologic malignancy cohorts.
7.3 Considerations for clinical development
Safe translation requires giving equal weight to timing, sequencing, and dose-finding. A pragmatic approach is a stage-gated design: begin with a short metabolic run-in like MCT1 or CD73/A2A modulation to document PD effects and tolerability; proceed to add ICIs once predefined PD thresholds are met; and adaptively enrich patients by biomarkers-glycolysis high (for MCT1 ± ICI), adenosine high (for CD73/A2A ± ICI), or ANGPT2 high (for ICI with anti-angiogenic support). Dose escalation should prioritize on-target immune modulation rather than global myelosuppression, with predefined stop rules based on PD readouts (HP 13C-pyruvate MRI kPL, adenosine/lactate panels, ANGPT2) and safety signals (acid-base balance for buffering, lipid/cardiac panels for FASN inhibition, cytopenias for iron-modifying agents).
At the exploratory-decision stage, correlating soluble metabolites with spatial macrophage phenotypes in organoids/ex-vivo slices suffices; at early go/no-go nodes, test-retest HP 13C-pyruvate MRI and predefined adenosine panels can anchor decisions; and for registrational use, analytically validated thresholds and multi-analyte composites are likely required.
Broadly, successful clinical translation of metabolism-immunity combinations requires careful attention to timing and sequencing. Preclinical data suggest that in some contexts, metabolic priming should precede immunotherapy to appropriately reprogram the TME, whereas in others, concurrent dosing may yield optimal synergy. Dose-finding is also crucial, since metabolic drugs may have therapeutic windows distinct from those of ICIs; global myelosuppression may constrain feasible regimens.
Despite the rationale for combining metabolic interventions with immunotherapy, resistance is likely to emerge through several non-mutually exclusive routes. First, metabolic redundancy and plasticity can bypass single-node blockade—for instance, high or induced MCT4 expression has been associated with reduced sensitivity to the MCT1 inhibitor AZD3965, consistent with compensatory lactate export capacity[183]. Second, tumors may reactivate parallel immunosuppressive metabolite circuits, such as reinforcing the CD39-CD73-adenosine axis or shifting signaling toward alternative adenosine receptors, thereby sustaining myeloid/T-cell suppression even when one component is inhibited[184-186]. Third, interventions intended to leverage ferroptosis/redox stress can select for strengthened antioxidant defense programs, including FSP1-CoQ and GCH1-BH4-linked protection, or mitochondrial redox buffering via DHODH, potentially limiting durable immune reprogramming. Collectively, these considerations argue for incorporating on-treatment pharmacodynamic readouts, including TAM states, to distinguish target bypass from insufficient immune engagement and to guide rational next-line combinations rather than assuming uniform synergy[187-190].
As for the considerations of unique toxicities and safety considerations of metabolic interventions. A key translational caveat of metabolic-immune interventions is that many targets governing TAM states are also indispensable for normal tissue homeostasis. For example, pharmacologic HIF-2α inhibition such as belzutifan frequently induces anemia and can cause clinically significant hypoxia[191]. In parallel, blockade of lactate transport is mechanistically attractive for disrupting lactate-driven immunosuppression, yet first-in-human evaluation of the MCT1 inhibitor AZD3965 revealed dose-limiting toxicities that were largely on-target, including electroretinogram changes/retinopathy and a cardiac troponin rise, supporting the need for baseline eye/cardiac screening and longitudinal monitoring when targeting lactate shuttling in patients[113]. FASN inhibitor TVB-2640 showed a manageable safety profile dominated by reversible skin and ocular adverse events[192], while adenosine-axis blockade is generally considered tolerable, early clinical signals commonly include fatigue, pruritus, and appetite decrease[180].
This chapter translates the mechanistic framework into therapeutic options, but it also highlights why clinical benefit cannot be assumed from pathway plausibility alone. Across direct interventions and rational combinations, the shared goal is to relax metabolite-imposed constraints and shift TAMs away from suppressive or angiogenic programs toward states more compatible with antigen presentation and effector T-cell activity; nevertheless, the actual TAM subset engaged by each intervention is likely niche-restricted and remains under-validated in patient samples. Two practical limitations stand out: resistance driven by metabolic redundancy, and toxicities because many metabolic nodes are essential for normal tissues. These points motivate a trial-development mindset in which TAM-resolved pharmacodynamic endpoints, ideally with spatial context, careful sequencing with ICIs, and safety-aware dosing are treated as core design elements, enabling credible testing of whether metabolic interventions can reproducibly promote “cold” to “hot” transitions in urologic cancers.
8. Clinical development landscape and future directions
8.1 Current trial landscape
Clinically, three partly overlapping corridors have emerged for bringing metabolic control into myeloid-dominated urologic cancers, and each corridor answers a different question rather than repeating the same signal seen in combination studies.
The HIF-2α experience in ccRCC shows a single metabolic node can be drugged and sequenced around ICI/TKI without losing activity. The phase III LITESPARK-005 trial—belzutifan versus everolimus in patients who had already received PD-1/PD-L1 and VEGF-TKI—showed a PFS benefit (HR ≈ 0.75) and higher ORR for belzutifan, with anemia/hypoxia as an expected on-target pattern. The study not only tried to prove synergy, it also proved that metabolic control after immunotherapy is clinically workable in RCC[193-195]. By attenuating HIF-2α-dependent hypoxia programs, belzutifan may also dampen hypoxia-conditioned pro-angiogenic TAM/TEM-like states enriched in perivascular/hypoxic niches, which could reduce immune exclusion and facilitate CD8⁺ T-cell infiltration—i.e., a plausible “cold” to “hot” shift, particularly in combination settings[139, 196].
Studies that focus on the adenosine pathway—AZD4635 + durvalumab/oleclumab in mCRPC (NCT04089553) and ciforadenant (CPI-444) + atezolizumab in RCC/mCRPC (NCT02655822)—were designed to see whether lifting an immunometabolic brake could make an ICI-experienced microenvironment signal again. The results showed that in unselected populations, the activity was modest, but exploratory adenosine gene signatures and mandatory on-study biopsies from the AZD4635 trial pinpointed who might benefit. This corridor therefore legitimizes a filter strategy for adenosine- or lactate-heavy niches, instead of universal combination[181]. A2A receptor antagonist is expected to relieve adenosine-driven immunosuppressive signaling in macrophages, thereby potentially shifting adenosine-conditioned suppressive TAM programs toward more antigen-presenting/inflammatory states[9, 180].
First-in-human inhibition of CAIX with SLC-0111 proved that correcting extracellular acidity is pharmacologically achievable and tolerable in solid tumors, which matters for hypoxic RCC/BCa where acidity directly impairs phagocytosis and drug penetration.
In parallel, glutaminase programs—telaglenastat (CB-839) + nivolumab for RCC/melanoma/NSCLC (NCT02771626) and RCC-focused follow-ups—showed disease control and an acceptable safety profile, but only moderate antitumor activity, implying that future urologic cancers trials should either co-select for glutamine-addicted tumors or use glutaminase agents explicitly as microenvironment primers before ICI[197-199]. Cabozantinib has been reported to reduce TAM abundance/M2 polarization and promote a more inflamed microenvironment—together hypothesized to lessen myeloid suppression and facilitate CD8⁺ recruitment that still requires TAM-resolved clinical pharmacodynamic validation[200].
8.2 Failure analyses and paths to success
In recent years, some clinical trials targeting metabolism-immunity axes in urologic tumors have not met their primary endpoints. Potential causes include:
(1) Selection lagging behind mechanism. Glycolysis high or adenosine high tumors are rarely pre-specified, diluting benefit signals; pragmatic biomarker gates should be prospectively embedded to enrich patients most likely to undergo TAM reprogramming. (2) Endpoints not aligned with immunometabolic pharmacology. In ICI combinations, RECIST remains necessary but insufficient; pairing iRECIST or immune-adapted endpoints with PD matrices (adenosine/lactate panels and soluble ANGPT2) is more likely to capture macrophage-mediated “cold” to “hot” transitions before size changes occur. (3) PK/PD mismatches. Short-half-life purinergic agents or subtherapeutic alkalinization can cause under-exposure precisely during windows when antigen release and myeloid re-education should be maximal; stage-gated run-ins requiring PD proof such as kPL decline before randomization mitigate this failure mode. (4) Limited tissue access. On-treatment biopsies are sporadic, extending the learning cycle; embedding ex vivo slices and spatial profiling into early cohorts can directly visualize TAM phagocytic tone, raft disruption, and hypoxia-adenosine relief, thereby bridging PK/PD with outcomes.
A consensus, executable roadmap should establish a short-cycle, iterative loop: start with perturbation-ready models and end with fit-for-purpose decision tools. Practically, patient-derived organoids and ex vivo precision-cut slices provide controlled environments for titrating lactate, adenosine, or lipid interventions while directly imaging phagocytosis, antigen presentation, and raft organization. Readouts should include stable-isotope flux, single-cell/spatial multi-omics of myeloid states, and HP 13C-pyruvate MRI kPL as a noninvasive PD anchor that links glycolytic pressure to macrophage re-education.
Maturation of fit-for-purpose biomarkers should follow staged development: exploratory associations (organoid/slice—soluble lactate/adenosine and spatial TAM phenotypes) —analytical validation and test—retest (HP 13C-pyruvate MRI kPL, adenosine/lactate panels)—prospective clinical utility, with macrophage-centric PD thresholds gating escalation of combination therapy.
9. Conclusion
Metabolic-immune coupling is emerging as a central determinant of therapeutic outcome in urologic cancers. In this review, we synthesize a TAM-centered framework across PCa, BCa, and ccRCC by highlighting three convergent metabolic circuits—the lactate-pH/hypoxia-HIF-VEGF axis, the lipid rafts axis, and the ferroptosis-redox axis—that can impose durable immunosuppressive or pro-angiogenic macrophage programs and thereby limit responsiveness to immune checkpoint blockade and anti-angiogenic therapy. This perspective emphasizes TAMs as a tractable therapeutic node at which metabolite-imposed constraints may be relaxed to restore phagocytosis, antigen handling, and immune-permissive vascular states. Moving forward, clinical progress will possibly depend less on developing a single dominant metabolic agent and more on institutionalizing metabolism-informed, TAM-anchored trial designs: metabolic or spatial enrichment at entry such as glycolysis/high adenosine/hypoxia modules, early pharmacodynamic confirmation in organoid-immune co-cultures, humanized or ex vivo platforms, and stepwise escalation with PD-1/PD-L1 or VEGF-TKI combinations. Fit-for-purpose biomarkers, including hyperpolarized 13C-pyruvate MRI (kPL), plasma ANGPT2, spatial NT5E/ADORA2A, lipid-droplet markers such as PLIN2, should be integrated prospectively to shorten decision cycles and render negative arms informative. Establishing multicenter standards for tissue handling and spatial profiling, together with transparent sharing of metabolic-myeloid datasets across PCa, BCa and RCC, will be essential to translate macrophage reprogramming from a mechanistic concept into a predictable and durable clinical strategy.
Acknowledgements
Funding
This work was supported by National Natural Science Foundation of China (82570912,81870522 and 82173304), Guangzhou Key Research and Development Program (2023B03J1245) to Peng Wu.The National Natural Science Foundation of China (82503289); President Foundation of Nanfang Hospital, Southern Medical University (2024A028), China Postdoctoral Science Foundation (2025M782052) to Bisheng Cheng.
Author contributions
Wenxue Huang and Weijia Li: Conceptualization, Methodology, Investigation, Writing—Original Draft (equal contribution). Wentai Shangguan: Formal Analysis, Data Curation, Visualization. Lin Yang, Zhuohang Li, and Boyuan Sun: Investigation, Resources, Validation. Cunzhen Ma and Xunguo Yang: Methodology, Supervision, Writing—Review & Editing. Peidan Peng, Jie Zhao, Bisheng Cheng, and Peng Wu: Conceptualization, Supervision, Project Administration, Funding Acquisition, Writing—Review & Editing (co-corresponding authors).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A. et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin. 2021;71(3):209-49
2. Zhang N, Bian Q, Gao Y, Wang Q, Shi Y, Li X. et al. The Role of Fascin-1 in Human Urologic Cancers: A Promising Biomarker or Therapeutic Target? Technol Cancer Res Treat. 2023;22:15330338231175733
3. Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science. 2018;359(6382):1350-5
4. Cassetta L, Pollard JW. A timeline of tumour-associated macrophage biology. Nat Rev Cancer. 2023;23(4):238-57
5. Hu T, Liu CH, Lei M, Zeng Q, Li L, Tang H. et al. Metabolic regulation of the immune system in health and diseases: mechanisms and interventions. Signal Transduct Target Ther. 2024;9(1):268
6. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324(5930):1029-33
7. Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y. et al. Metabolic regulation of gene expression by histone lactylation. Nature. 2019;574(7779):575-80
8. Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V. et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature. 2014;513(7519):559-63
9. Allard B, Allard D, Buisseret L, Stagg J. The adenosine pathway in immuno-oncology. Nat Rev Clin Oncol. 2020;17(10):611-29
10. Zhuang L, Kim J, Adam RM, Solomon KR, Freeman MR. Cholesterol targeting alters lipid raft composition and cell survival in prostate cancer cells and xenografts. J Clin Invest. 2005;115(4):959-68
11. Jiang X, Stockwell BR, Conrad M. Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol. 2021;22(4):266-82
12. Kaelin WG Jr. The von Hippel-Lindau tumour suppressor protein: O2 sensing and cancer. Nat Rev Cancer. 2008;8(11):865-73
13. Linehan WM, Schmidt LS, Crooks DR, Wei D, Srinivasan R, Lang M. et al. The Metabolic Basis of Kidney Cancer. Cancer Discov. 2019;9(8):1006-21
14. Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd. et al. Comprehensive Integration of Single-Cell Data. Cell. 2019;177(7):1888-902.e21
15. Hiam-Galvez KJ, Allen BM, Spitzer MH. Systemic immunity in cancer. Nat Rev Cancer. 2021;21(6):345-59
16. Vogel A, Weichhart T. Tissue-resident macrophages - early passengers or drivers in the tumor niche? Curr Opin Biotechnol. 2023;83:102984
17. Sunakawa Y, Stintzing S, Cao S, Heinemann V, Cremolini C, Falcone A. et al. Variations in genes regulating tumor-associated macrophages (TAMs) to predict outcomes of bevacizumab-based treatment in patients with metastatic colorectal cancer: results from TRIBE and FIRE3 trials. Ann Oncol. 2015;26(12):2450-6
18. Xiang X, Wang J, Lu D, Xu X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct Target Ther. 2021;6(1):75
19. Mantovani A, Sica A, Sozzani S, Allavena P, Vecchi A, Locati M. The chemokine system in diverse forms of macrophage activation and polarization. Trends Immunol. 2004;25(12):677-86
20. Zhang Q, Sioud M. Tumor-Associated Macrophage Subsets: Shaping Polarization and Targeting. Int J Mol Sci. 2023 24(8)
21. Nakai K. Multiple roles of macrophage in skin. J Dermatol Sci. 2021;104(1):2-10
22. Arora S, Dev K, Agarwal B, Das P, Syed MA. Macrophages: Their role, activation and polarization in pulmonary diseases. Immunobiology. 2018;223(4-5):383-96
23. Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K. et al. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344(6186):921-5
24. Masetti M, Carriero R, Portale F, Marelli G, Morina N, Pandini M. et al. Lipid-loaded tumor-associated macrophages sustain tumor growth and invasiveness in prostate cancer. J Exp Med. 2022 219(2)
25. Chaudagar K, Hieromnimon HM, Khurana R, Labadie B, Hirz T, Mei S. et al. Reversal of Lactate and PD-1-mediated Macrophage Immunosuppression Controls Growth of PTEN/p53-deficient Prostate Cancer. Clin Cancer Res. 2023;29(10):1952-68
26. Li M, Cui Y, Qi Q, Liu J, Li J, Huang G. et al. SPOP downregulation promotes bladder cancer progression based on cancer cell-macrophage crosstalk via STAT3/CCL2/IL-6 axis and is regulated by VEZF1. Theranostics. 2024;14(17):6543-59
27. Kim H, Shim BY, Lee SJ, Lee JY, Lee HJ, Kim IH. Loss of Von Hippel-Lindau (VHL) Tumor Suppressor Gene Function: VHL-HIF Pathway and Advances in Treatments for Metastatic Renal Cell Carcinoma (RCC). Int J Mol Sci. 2021 22(18)
28. Liu S, Yuan Y, He J, Zhou Y, Wang Y, Ye X. et al. Integrated multi-omics analysis reveals a glycolytic signature that predicts pan-cancer immune checkpoint inhibitor response and LDHA as a combinatorial target in fumarate hydratase-deficient renal cell carcinoma. Front Immunol. 2025;16:1666121
29. Gong D, Arbesfeld-Qiu JM, Perrault E, Bae JW, Hwang WL. Spatial oncology: Translating contextual biology to the clinic. Cancer Cell. 2024;42(10):1653-75
30. Li W, Chen S, Lu J, Mao W, Zheng S, Zhang M. et al. YY1 enhances HIF-1α stability in tumor-associated macrophages to suppress anti-tumor immunity of prostate cancer in mice. Nat Commun. 2025;16(1):6261
31. Jin R, Neufeld L, McGaha TL. Linking macrophage metabolism to function in the tumor microenvironment. Nat Cancer. 2025;6(2):239-52
32. Yang L, Shangguan W, Li W, Huang W, Li Z, Sun B. et al. Role of tertiary lymphoid structures in the tumour microenvironment and immunotherapy response of renal cell carcinoma. Clin Transl Med. 2025;15(10):e70455
33. Muraoka S, Baba T, Akazawa T, Katayama KI, Kusumoto H, Yamashita S. et al. Tumor-derived lactic acid promotes acetylation of histone H3K27 and differentiation of IL-10-producing regulatory B cells through direct and indirect signaling pathways. Int J Cancer. 2025;156(4):840-52
34. Jin J, Yan P, Wang D, Bai L, Liang H, Zhu X. et al. Targeting lactylation reinforces NK cell cytotoxicity within the tumor microenvironment. Nat Immunol. 2025;26(7):1099-112
35. Xie B, Lin J, Chen X, Zhou X, Zhang Y, Fan M. et al. CircXRN2 suppresses tumor progression driven by histone lactylation through activating the Hippo pathway in human bladder cancer. Mol Cancer. 2023;22(1):151
36. Colin S, Chinetti-Gbaguidi G, Staels B. Macrophage phenotypes in atherosclerosis. Immunol Rev. 2014;262(1):153-66
37. Yuan Y, Fan G, Liu Y, Liu L, Zhang T, Liu P. et al. The transcription factor KLF14 regulates macrophage glycolysis and immune function by inhibiting HK2 in sepsis. Cell Mol Immunol. 2022;19(4):504-15
38. Wang T, Liu H, Lian G, Zhang SY, Wang X, Jiang C. HIF1α-Induced Glycolysis Metabolism Is Essential to the Activation of Inflammatory Macrophages. Mediators Inflamm. 2017;2017:9029327
39. Wang ZH, Peng WB, Zhang P, Yang XP, Zhou Q. Lactate in the tumour microenvironment: From immune modulation to therapy. EBioMedicine. 2021;73:103627
40. Jin M, Cao W, Chen B, Xiong M, Cao G. Tumor-Derived Lactate Creates a Favorable Niche for Tumor via Supplying Energy Source for Tumor and Modulating the Tumor Microenvironment. Front Cell Dev Biol. 2022;10:808859
41. Fang X, Zhao P, Gao S, Liu D, Zhang S, Shan M. et al. Lactate induces tumor-associated macrophage polarization independent of mitochondrial pyruvate carrier-mediated metabolism. Int J Biol Macromol. 2023;237:123810
42. Zhang A, Xu Y, Xu H, Ren J, Meng T, Ni Y. et al. Lactate-induced M2 polarization of tumor-associated macrophages promotes the invasion of pituitary adenoma by secreting CCL17. Theranostics. 2021;11(8):3839-52
43. Liu N, Luo J, Kuang D, Xu S, Duan Y, Xia Y. et al. Lactate inhibits ATP6V0d2 expression in tumor-associated macrophages to promote HIF-2α-mediated tumor progression. J Clin Invest. 2019;129(2):631-46
44. Lu Y, Ye F, Han X, Wang Z, Li X, Zheng L. Integrated spatial transcriptome and metabolism study reveals metabolic heterogeneity in human bladder cancer. Cancer Gene Ther. 2025
45. Wang JX, Choi SYC, Niu X, Kang N, Xue H, Killam J. et al. Lactic Acid and an Acidic Tumor Microenvironment suppress Anticancer Immunity. Int J Mol Sci. 2020 21(21)
46. El-Kenawi A, Gatenbee C, Robertson-Tessi M, Bravo R, Dhillon J, Balagurunathan Y. et al. Acidity promotes tumour progression by altering macrophage phenotype in prostate cancer. Br J Cancer. 2019;121(7):556-66
47. Llibre A, Kucuk S, Gope A, Certo M, Mauro C. Lactate: A key regulator of the immune response. Immunity. 2025;58(3):535-54
48. Jin HR, Wang J, Wang ZJ, Xi MJ, Xia BH, Deng K. et al. Lipid metabolic reprogramming in tumor microenvironment: from mechanisms to therapeutics. J Hematol Oncol. 2023;16(1):103
49. Parupathi P, Devarakonda LS, Francois E, Amjed M, Kumar A. Reprogrammed Lipid Metabolism-Associated Therapeutic Vulnerabilities in Prostate Cancer. Int J Mol Sci. 2025 26(18)
50. Heravi G, Yazdanpanah O, Podgorski I, Matherly LH, Liu W. Lipid metabolism reprogramming in renal cell carcinoma. Cancer Metastasis Rev. 2022;41(1):17-31
51. Qiao X, Hu Z, Xiong F, Yang Y, Peng C, Wang D. et al. Lipid metabolism reprogramming in tumor-associated macrophages and implications for therapy. Lipids Health Dis. 2023;22(1):45
52. Zhang S, Zhu N, Li HF, Gu J, Zhang CJ, Liao DF. et al. The lipid rafts in cancer stem cell: a target to eradicate cancer. Stem Cell Res Ther. 2022;13(1):432
53. Greenlee JD, Subramanian T, Liu K, King MR. Rafting Down the Metastatic Cascade: The Role of Lipid Rafts in Cancer Metastasis, Cell Death, and Clinical Outcomes. Cancer Res. 2021;81(1):5-17
54. Shimano H, Sato R. SREBP-regulated lipid metabolism: convergent physiology - divergent pathophysiology. Nat Rev Endocrinol. 2017;13(12):710-30
55. Liu C, Chikina M, Deshpande R, Menk AV, Wang T, Tabib T. et al. Treg Cells Promote the SREBP1-Dependent Metabolic Fitness of Tumor-Promoting Macrophages via Repression of CD8(+) T Cell-Derived Interferon-γ. Immunity. 2019;51(2):381-97.e6
56. Qin H, Xiao A, Lu Q, Li Y, Luo X, Zheng E. et al. The fatty acid receptor CD36 promotes macrophage infiltration via p110γ signaling to stimulate metastasis. J Adv Res. 2025;74:237-53
57. Cheng Y, Bai F, Ren X, Sun R, Guo X, Liu W. et al. Phosphoinositide-Binding Protein TIPE1 Promotes Alternative Activation of Macrophages and Tumor Progression via PIP3/Akt/TGFβ Axis. Cancer Res. 2022;82(8):1603-16
58. Ito A, Hong C, Rong X, Zhu X, Tarling EJ, Hedde PN. et al. LXRs link metabolism to inflammation through Abca1-dependent regulation of membrane composition and TLR signaling. Elife. 2015;4:e08009
59. Goossens P, Rodriguez-Vita J, Etzerodt A, Masse M, Rastoin O, Gouirand V. et al. Membrane Cholesterol Efflux Drives Tumor-Associated Macrophage Reprogramming and Tumor Progression. Cell Metab. 2019;29(6):1376-89.e4
60. Zhu X, Owen JS, Wilson MD, Li H, Griffiths GL, Thomas MJ. et al. Macrophage ABCA1 reduces MyD88-dependent Toll-like receptor trafficking to lipid rafts by reduction of lipid raft cholesterol. J Lipid Res. 2010;51(11):3196-206
61. Sun WJ, Liang F, Zhao H, Wang YM, Zhou ZY, Xu H. et al. Androgen induces 3'UTR shortening of de novo lipogenesis genes by alternative polyadenylation in prostate cancer cells. Sci China Life Sci. 2025
62. Andersen MK, Høiem TS, Claes BSR, Balluff B, Martin-Lorenzo M, Richardsen E. et al. Spatial differentiation of metabolism in prostate cancer tissue by MALDI-TOF MSI. Cancer Metab. 2021;9(1):9
63. Li J, Cao F, Yin HL, Huang ZJ, Lin ZT, Mao N. et al. Ferroptosis: past, present and future. Cell Death Dis. 2020;11(2):88
64. Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol. 2008;15(3):234-45
65. Friedmann Angeli JP, Schneider M, Proneth B, Tyurina YY, Tyurin VA, Hammond VJ. et al. Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol. 2014;16(12):1180-91
66. Liu Y, Luo X, Chen Y, Dang J, Zeng D, Guo X. et al. Heterogeneous ferroptosis susceptibility of macrophages caused by focal iron overload exacerbates rheumatoid arthritis. Redox Biol. 2024;69:103008
67. Piattini F, Matsushita M, Muri J, Bretscher P, Feng X, Freigang S. et al. Differential sensitivity of inflammatory macrophages and alternatively activated macrophages to ferroptosis. Eur J Immunol. 2021;51(10):2417-29
68. Liao P, Wang W, Wang W, Kryczek I, Li X, Bian Y. et al. CD8(+) T cells and fatty acids orchestrate tumor ferroptosis and immunity via ACSL4. Cancer Cell. 2022;40(4):365-78.e6
69. Cao D, Zheng J, Li Z, Yu Y, Chen Z, Wang Q. ACSL4 inhibition prevents macrophage ferroptosis and alleviates fibrosis in bleomycin-induced systemic sclerosis model. Arthritis Res Ther. 2023;25(1):212
70. Chen P, Wang D, Xiao T, Gu W, Yang H, Yang M. et al. ACSL4 promotes ferroptosis and M1 macrophage polarization to regulate the tumorigenesis of nasopharyngeal carcinoma. Int Immunopharmacol. 2023;122:110629
71. Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I. et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13(1):91-8
72. Yang Y, Wang Y, Guo L, Gao W, Tang TL, Yan M. Interaction between macrophages and ferroptosis. Cell Death Dis. 2022;13(4):355
73. Qi D, Peng M. Ferroptosis-mediated immune responses in cancer. Front Immunol. 2023;14:1188365
74. Cheng B, Wang Q, Li Z, Luo T, Xie J, Singh S. et al. Induction of ferroptosis in prostate cancer by CCDC7(19-13) via TRIM21-mediated ubiquitination of SLC7A11. Cell Death Differ. 2025
75. Cheng B, Yang J, Huang W, Luo T, Li L, Wu J. et al. Regulation of macrophage plasticity by circCCDC719-13 through HSP90 inhibition suppresses prostate cancer progression and metastasis: a translational study. Int J Surg. 2025
76. Xue Y, Wang S, Yin Y, Chai X, Zhou Z, Li H. et al. A Therapeutic System Regulating Iron Metabolism in CD63(+) Macrophage Subsets Activates Anti-tumor Immunity for Bone Metastasis Therapy. ACS Nano. 2025;19(41):36544-65
77. Gao J, Zhang X, Liu Y, Gu X. Ferroptosis in immune cells: Implications for tumor immunity and cancer therapy. Cytokine Growth Factor Rev. 2025;84:59-73
78. Zhu Y, Zhou Z, Du X, Lin X, Liang ZM, Chen S. et al. Cancer cell-derived arginine fuels polyamine biosynthesis in tumor-associated macrophages to promote immune evasion. Cancer Cell. 2025;43(6):1045-60.e7
79. Watt K, Dauber B, Szkop KJ, Lee L, Jovanovic P, Chen S. et al. Epigenetic alterations facilitate transcriptional and translational programs in hypoxia. Nat Cell Biol. 2025
80. Li W, Shangguan W, Huang W, Zhao J, Zhu Y, Xie M. et al. Gut Parabacteroides distasonis-derived Indole-3-Acetic Acid Promotes Phospholipid Remodeling and Enhances Ferroptosis Sensitivity via the AhR-FASN Axis in Bladder Cancer. Adv Sci (Weinh). 2025;12(34):e04688
81. Liu J, Paolino M. Ubiquitination in macrophage plasticity: from inflammatory to immunometabolic pathways. Front Immunol. 2025;16:1621328
82. Xu M, Jiang SY, Tang S, Zhu M, Hu Y, Li J. et al. Nuclear SREBP2 condensates regulate the transcriptional activation of lipogenic genes and cholesterol homeostasis. Nat Metab. 2025;7(5):1034-51
83. Wang Y, Xiao H, Lai L, Zheng Z. Therapeutic strategies targeting SREBP transcription factors: an update to 2024. Acta Materia Medica. 2025;4(3):437-65
84. Hegde S, Giotti B, Soong BY, Halasz L, Le Berichel J, Schaefer MM. et al. Myeloid progenitor dysregulation fuels immunosuppressive macrophages in tumours. Nature. 2025
85. Liu ZY, Wu CY, Wu RQ, Wang JC, Huang CX, Wang XY. et al. Efflux of N1-acetylspermidine from hepatoma fosters macrophage-mediated immune suppression to dampen immunotherapeutic efficacy. Immunity. 2025;58(6):1572-85.e10
86. Malczewski AB, Coward JI, Ketheesan N, Navarro S. Immunometabolism: The role of gut-derived microbial metabolites in optimising immune response during checkpoint inhibitor therapy. Clin Transl Med. 2025;15(9):e70472
87. Ding W, Mo J, Su Y, Zhang Q, Sun D, Yao X. et al. Metabolic reprogramming of tumor-associated macrophages via adenosine-A(2A)R signaling drives cross-resistance in non-small cell lung cancer. Drug Resist Updat. 2025;82:101272
88. Terry AR, Hay N. Emerging targets in lipid metabolism for cancer therapy. Trends Pharmacol Sci. 2024;45(6):537-51
89. Grolez GP, Gordiendko DV, Clarisse M, Hammadi M, Desruelles E, Fromont G. et al. TRPM8-androgen receptor association within lipid rafts promotes prostate cancer cell migration. Cell Death Dis. 2019;10(9):652
90. Wei G, Huang Y, Li W, Xie Y, Zhang D, Niu Y. et al. SREBF1-based metabolic reprogramming in prostate cancer promotes tumor ferroptosis resistance. Cell Death Discov. 2025;11(1):75
91. Sun Y, Guo W, Guo Y, Lin Z, Wang D, Guo Q. et al. Apoptosis induction in human prostate cancer cells related to the fatty acid metabolism by wogonin-mediated regulation of the AKT-SREBP1-FASN signaling network. Food Chem Toxicol. 2022;169:113450
92. Vona R, Iessi E, Matarrese P. Role of Cholesterol and Lipid Rafts in Cancer Signaling: A Promising Therapeutic Opportunity? Front Cell Dev Biol. 2021;9:622908
93. Wang Q, Wu Y, Long Y, Li R, Shi Y, Zheng Y. et al. AR(+)TREM2(+) macrophage induced pathogenic immunosuppression promotes prostate cancer progression. Nat Commun. 2025;16(1):6964
94. Guo Y, Zhang Z, Dong L, Shi X, Li X, Luo M. et al. Wnt5a augments intracellular free cholesterol levels and promotes castration resistance in prostate cancer. J Transl Med. 2025;23(1):347
95. Consiglio CR, Udartseva O, Ramsey KD, Bush C, Gollnick SO. Enzalutamide, an Androgen Receptor Antagonist, Enhances Myeloid Cell-Mediated Immune Suppression and Tumor Progression. Cancer Immunol Res. 2020;8(9):1215-27
96. Li XF, Selli C, Zhou HL, Cao J, Wu S, Ma RY. et al. Macrophages promote anti-androgen resistance in prostate cancer bone disease. J Exp Med. 2023 220(4)
97. Han M, Li F, Zhang Y, Dai P, He J, Li Y. et al. FOXA2 drives lineage plasticity and KIT pathway activation in neuroendocrine prostate cancer. Cancer Cell. 2022;40(11):1306-23.e8
98. Qian C, Yang Q, Rotinen M, Huang R, Kim H, Gallent B. et al. ONECUT2 acts as a lineage plasticity driver in adenocarcinoma as well as neuroendocrine variants of prostate cancer. Nucleic Acids Res. 2024;52(13):7740-60
99. Huang J, Liu F, Li C, Liang X, Li C, Liu Y. et al. Role of CD47 in tumor immunity: a potential target for combination therapy. Sci Rep. 2022;12(1):9803
100. Martori C, Sanchez-Moral L, Paul T, Pardo JC, Font A, Ruiz de Porras V. et al. Macrophages as a Therapeutic Target in Metastatic Prostate Cancer: A Way to Overcome Immunotherapy Resistance? Cancers (Basel). 2022 14(2)
101. Liu Y, Tan H, Dai J, Lin J, Zhao K, Hu H. et al. Targeting macrophages in cancer immunotherapy: Frontiers and challenges. J Adv Res. 2025;76:695-713
102. Chou CW, Hung CN, Chiu CH, Tan X, Chen M, Chen CC. et al. Phagocytosis-initiated tumor hybrid cells acquire a c-Myc-mediated quasi-polarization state for immunoevasion and distant dissemination. Nat Commun. 2023;14(1):6569
103. Ma Y, Sun Y, Guo H, Yang R. Tumor-associated macrophages in bladder cancer: roles and targeted therapeutic strategies. Front Immunol. 2024;15:1418131
104. Caslin HL, Abebayehu D, Pinette JA, Ryan JJ. Lactate Is a Metabolic Mediator That Shapes Immune Cell Fate and Function. Front Physiol. 2021;12:688485
105. Xu B, Liu Y, Li N, Geng Q. Lactate and lactylation in macrophage metabolic reprogramming: current progress and outstanding issues. Front Immunol. 2024;15:1395786
106. Zhang Y, Zhang X, Meng Y, Xu X, Zuo D. The role of glycolysis and lactate in the induction of tumor-associated macrophages immunosuppressive phenotype. Int Immunopharmacol. 2022;110:108994
107. Zhu Z, Shen Z, Xu C. Inflammatory pathways as promising targets to increase chemotherapy response in bladder cancer. Mediators Inflamm. 2012;2012:528690
108. Rodríguez-Pampín I, González-Pico L, Selas A, Andújar A, Prieto-Díaz R, Sotelo E. Targeting the adenosinergic axis in cancer immunotherapy: Insights into A(2A) and A(2B) receptors and novel clinical combination strategies. Pharmacol Rev. 2025;77(6):100092
109. Furriel F, Ferreira H, Sampaio-Ribeiro G, Pereira M, Eloy C, Neves B. et al. Co-targeting the CD73-adenosinergic axis enhances the anti-tumor efficacy of anti-PD-L1 immunotherapy in bladder cancer. Biomed Pharmacother. 2025;188:118188
110. Cekic C, Sag D, Li Y, Theodorescu D, Strieter RM, Linden J. Adenosine A2B receptor blockade slows growth of bladder and breast tumors. J Immunol. 2012;188(1):198-205
111. Lyu A, Fan Z, Clark M, Lea A, Luong D, Setayesh A. et al. Evolution of myeloid-mediated immunotherapy resistance in prostate cancer. Nature. 2025;637(8048):1207-17
112. Li Z, Cui J. Targeting the lactic acid metabolic pathway for antitumor therapy. Mol Ther Oncolytics. 2023;31:100740
113. Halford S, Veal GJ, Wedge SR, Payne GS, Bacon CM, Sloan P. et al. A Phase I Dose-escalation Study of AZD3965, an Oral Monocarboxylate Transporter 1 Inhibitor, in Patients with Advanced Cancer. Clin Cancer Res. 2023;29(8):1429-39
114. Massari F, Ciccarese C, Santoni M, Iacovelli R, Mazzucchelli R, Piva F. et al. Metabolic phenotype of bladder cancer. Cancer Treat Rev. 2016;45:46-57
115. Sun C, Wang B, Hao S. Adenosine-A2A Receptor Pathway in Cancer Immunotherapy. Front Immunol. 2022;13:837230
116. Augustin RC, Leone RD, Naing A, Fong L, Bao R, Luke JJ. Next steps for clinical translation of adenosine pathway inhibition in cancer immunotherapy. J Immunother Cancer. 2022 10(2)
117. Abou Chakra M, Luo Y, Duquesne I, M AOD. Update on the Mechanism of Action of Intravesical BCG Therapy to Treat Non-Muscle-Invasive Bladder Cancer. Front Biosci (Landmark Ed). 2024;29(8):295
118. Arts RJW, Carvalho A, La Rocca C, Palma C, Rodrigues F, Silvestre R. et al. Immunometabolic Pathways in BCG-Induced Trained Immunity. Cell Rep. 2016;17(10):2562-71
119. de Queiroz N, Marinho FV, de Araujo A, Fahel JS, Oliveira SC. MyD88-dependent BCG immunotherapy reduces tumor and regulates tumor microenvironment in bladder cancer murine model. Sci Rep. 2021;11(1):15648
120. Akuma M, Kim M, Zhu C, Wiljer E, Gaudreau-Lapierre A, Patterson LD. et al. Loss of VHL-mediated pRb regulation promotes clear cell renal cell carcinoma. Cell Death Dis. 2025;16(1):307
121. Golijanin B, Malshy K, Khaleel S, Lagos G, Amin A, Cheng L. et al. Evolution of the HIF targeted therapy in clear cell renal cell carcinoma. Cancer Treat Rev. 2023;121:102645
122. Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME. et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature. 1999;399(6733):271-5
123. Liao D, Johnson RS. Hypoxia: a key regulator of angiogenesis in cancer. Cancer Metastasis Rev. 2007;26(2):281-90
124. Sainero-Alcolado L, Garde-Lapido E, Snaebjörnsson MT, Schoch S, Stevens I, Ruiz-Pérez MV. et al. Targeting MYC induces lipid droplet accumulation by upregulation of HILPDA in clear cell renal cell carcinoma. Proc Natl Acad Sci U S A. 2024;121(7):e2310479121
125. Fresnedo O, Lopez-Gomez JA, Ceniceros C, Larrinaga G, Saiz A, Mosteiro L. et al. Adaptations of lipid metabolism in low-grade clear cell renal cell carcinoma are linked to cholesteryl ester accumulation. Sci Rep. 2025;15(1):24762
126. di Meo NA, Lasorsa F, Rutigliano M, Loizzo D, Ferro M, Stella A. et al. Renal Cell Carcinoma as a Metabolic Disease: An Update on Main Pathways, Potential Biomarkers, and Therapeutic Targets. Int J Mol Sci. 2022 23(22)
127. Duan Z, Luo Y. Targeting macrophages in cancer immunotherapy. Signal Transduct Target Ther.
128. Mantovani A, Allavena P, Marchesi F, Garlanda C. Macrophages as tools and targets in cancer therapy. Nat Rev Drug Discov. 2022;21(11):799-820
129. Duran CL, Borriello L, Karagiannis GS, Entenberg D, Oktay MH, Condeelis JS. Targeting Tie2 in the Tumor Microenvironment: From Angiogenesis to Dissemination. Cancers (Basel). 2021 13(22)
130. Chen L, Li J, Wang F, Dai C, Wu F, Liu X. et al. Tie2 Expression on Macrophages Is Required for Blood Vessel Reconstruction and Tumor Relapse after Chemotherapy. Cancer Res. 2016;76(23):6828-38
131. Lewis CE, Harney AS, Pollard JW. The Multifaceted Role of Perivascular Macrophages in Tumors. Cancer Cell. 2016;30(2):365
132. Dogra R, Vaishampayan U. Hypoxia Inducible Factor-2α (HIF-2α) Pathway Inhibitors. J Kidney Cancer VHL. 2025;12(3):1-15
133. Chung HJ, Kondoh C, Bae WK, Tamada S, Matsubara N, Lee HJ. et al. First-line pembrolizumab-axitinib versus sunitinib in metastatic RCC: subgroup analysis of patients enrolled in the phase 3 KEYNOTE-426 in Eastern Asia. Jpn J Clin Oncol. 2025;55(4):406-13
134. Chen X, Xu Z, Wu C, Xie L, Wang P, Liu X. Efficacy and toxicity of immune checkpoint inhibitors combination therapy for advanced renal cell carcinoma: a systematic review and network meta-analysis. Front Immunol. 2024;15:1255577
135. Narayan V, Jonasch E, Iliopoulos O, Maughan BL, Oudard S, Else T. et al. Hypoxia-inducible factor-2α (HIF-2α) inhibitor belzutifan in von Hippel-Lindau (VHL) disease-associated neoplasms: 5-year follow-up of the phase 2 LITESPARK-004 study. J Clin Oncol. 2025;43(16_suppl):4507 -
136. Chen Z, Han F, Du Y, Shi H, Zhou W. Hypoxic microenvironment in cancer: molecular mechanisms and therapeutic interventions. Signal Transduct Target Ther. 2023;8(1):70
137. Kasherman L, Siu DHW, Woodford R, Harris CA. Angiogenesis Inhibitors and Immunomodulation in Renal Cell Cancers: The Past, Present, and Future. Cancers (Basel). 2022 14(6)
138. Lee WS, Yang H, Chon HJ, Kim C. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp Mol Med. 2020;52(9):1475-85
139. Choueiri TK, Powles T, Peltola K, de Velasco G, Burotto M, Suarez C. et al. Belzutifan versus Everolimus for Advanced Renal-Cell Carcinoma. N Engl J Med. 2024;391(8):710-21
140. Albiges L, Beckermann K, Miller WH, Goh JC, Gajate P, Harris CA. et al. Belzutifan plus lenvatinib for patients (pts) with advanced clear cell renal cell carcinoma (ccRCC) after progression on a PD-1/L1 and VEGF inhibitor: Preliminary results of arm B5 of the phase 1/2 KEYMAKER-U03B study. J Clin Oncol. 2023;41(16_suppl):4553 -
141. Wang J, Tao X, Zhu J, Dai Z, Du Y, Xie Y. et al. Tumor organoid-immune co-culture models: exploring a new perspective of tumor immunity. Cell Death Discov. 2025;11(1):195
142. Wang X, Lin D, Feng N. Harnessing organoid technology in urological cancer: advances and applications in urinary system tumors. World J Surg Oncol. 2025;23(1):295
143. Wang Q, Yuan F, Zuo X, Li M. Breakthroughs and challenges of organoid models for assessing cancer immunotherapy: a cutting-edge tool for advancing personalised treatments. Cell Death Discov. 2025;11(1):222
144. Chuprin J, Buettner H, Seedhom MO, Greiner DL, Keck JG, Ishikawa F. et al. Humanized mouse models for immuno-oncology research. Nat Rev Clin Oncol. 2023;20(3):192-206
145. Kumari R, Feuer G, Bourré L. Humanized Mouse Models for Immuno-oncology Drug Discovery. Curr Protoc. 2023;3(8):e852
146. Dimou P, Trivedi S, Liousia M, D'Souza RR, Klampatsa A. Precision-Cut Tumor Slices (PCTS) as an Ex Vivo Model in Immunotherapy Research. Antibodies (Basel). 2022 11(2)
147. Antoniewicz MR. A guide to (13)C metabolic flux analysis for the cancer biologist. Exp Mol Med. 2018;50(4):1-13
148. Dong W, Rawat ES, Stephanopoulos G, Abu-Remaileh M. Isotope tracing in health and disease. Curr Opin Biotechnol. 2022;76:102739
149. Bartman CR, Faubert B, Rabinowitz JD, DeBerardinis RJ. Metabolic pathway analysis using stable isotopes in patients with cancer. Nat Rev Cancer. 2023;23(12):863-78
150. Semba T, Ishimoto T. Spatial analysis by current multiplexed imaging technologies for the molecular characterisation of cancer tissues. Br J Cancer. 2024;131(11):1737-47
151. Park YM, Lin DC. Moving closer towards a comprehensive view of tumor biology and microarchitecture using spatial transcriptomics. Nat Commun. 2023;14(1):7017
152. Nunes JB, Ijsselsteijn ME, Abdelaal T, Ursem R, van der Ploeg M, Giera M. et al. Integration of mass cytometry and mass spectrometry imaging for spatially resolved single-cell metabolic profiling. Nat Methods. 2024;21(10):1796-800
153. Deen SS, Rooney C, Shinozaki A, McGing J, Grist JT, Tyler DJ. et al. Hyperpolarized Carbon 13 MRI: Clinical Applications and Future Directions in Oncology. Radiol Imaging Cancer. 2023;5(5):e230005
154. Wodtke P, Grashei M, Schilling F. Quo Vadis Hyperpolarized (13)C MRI? Z Med Phys. 2025;35(1):8-32
155. Chen HY, de Kouchkovsky I, Bok RA, Ohliger MA, Wang ZJ, Gebrezgiabhier D. et al. Multivariate Framework of Metabolism in Advanced Prostate Cancer Using Whole Abdominal and Pelvic Hyperpolarized (13)C MRI-A Correlative Study with Clinical Outcomes. Cancers (Basel). 2025 17(13)
156. Garg P, Singhal G, Horne D, Kulkarni P, Salgia R, Singhal SS. Molecular PET imaging: Unlocking the secrets of cancer metabolism. Biochem Pharmacol. 2025:117202.
157. Cao X, Chen J, Li B, Dang J, Zhang W, Zhong X. et al. Promoting antibody-dependent cellular phagocytosis for effective macrophage-based cancer immunotherapy. Sci Adv. 2022;8(11):eabl9171
158. Wu X, Giobbie-Hurder A, Liao X, Connelly C, Connolly EM, Li J. et al. Angiopoietin-2 as a Biomarker and Target for Immune Checkpoint Therapy. Cancer Immunol Res. 2017;5(1):17-28
159. Rigamonti N, Kadioglu E, Keklikoglou I, Wyser Rmili C, Leow CC, De Palma M. Role of angiopoietin-2 in adaptive tumor resistance to VEGF signaling blockade. Cell Rep. 2014;8(3):696-706
160. Guillaume Z, Auvray M, Vano Y, Oudard S, Helley D, Mauge L. Renal Carcinoma and Angiogenesis: Therapeutic Target and Biomarkers of Response in Current Therapies. Cancers (Basel). 2022 14(24)
161. Chen HY, Aggarwal R, Bok RA, Ohliger MA, Zhu Z, Lee P. et al. Hyperpolarized (13)C-pyruvate MRI detects real-time metabolic flux in prostate cancer metastases to bone and liver: a clinical feasibility study. Prostate Cancer Prostatic Dis. 2020;23(2):269-76
162. Dutta P, Salzillo TC, Pudakalakatti S, Gammon ST, Kaipparettu BA, McAllister F. et al. Assessing Therapeutic Efficacy in Real-time by Hyperpolarized Magnetic Resonance Metabolic Imaging. Cells. 2019 8(4)
163. Nelson SJ, Kurhanewicz J, Vigneron DB, Larson PE, Harzstark AL, Ferrone M. et al. Metabolic imaging of patients with prostate cancer using hyperpolarized [1-¹³C]pyruvate. Sci Transl Med. 2013;5(198):198ra08
164. Calcinotto A, Spataro C, Zagato E, Di Mitri D, Gil V, Crespo M. et al. IL-23 secreted by myeloid cells drives castration-resistant prostate cancer. Nature. 2018;559(7714):363-9
165. Morrissey JJ, Mellnick VM, Luo J, Siegel MJ, Figenshau RS, Bhayani S. et al. Evaluation of Urine Aquaporin-1 and Perilipin-2 Concentrations as Biomarkers to Screen for Renal Cell Carcinoma: A Prospective Cohort Study. JAMA Oncol. 2015;1(2):204-12
166. Cao Q, Ruan H, Wang K, Song Z, Bao L, Xu T. et al. Overexpression of PLIN2 is a prognostic marker and attenuates tumor progression in clear cell renal cell carcinoma. Int J Oncol. 2018;53(1):137-47
167. Bogdanov A, Bogdanov A, Chubenko V, Volkov N, Moiseenko F, Moiseyenko V. Tumor acidity: From hallmark of cancer to target of treatment. Front Oncol. 2022;12:979154
168. Gillies RJ, Ibrahim-Hashim A, Ordway B, Gatenby RA. Back to basic: Trials and tribulations of alkalizing agents in cancer. Front Oncol. 2022;12:981718
169. Estrella V, Chen T, Lloyd M, Wojtkowiak J, Cornnell HH, Ibrahim-Hashim A. et al. Acidity generated by the tumor microenvironment drives local invasion. Cancer Res. 2013;73(5):1524-35
170. de Martino M, Lucca I, Mbeutcha A, Wiener HG, Haitel A, Susani M. et al. Carbonic anhydrase IX as a diagnostic urinary marker for urothelial bladder cancer. Eur Urol. 2015;68(4):552-4
171. McDonald PC, Chia S, Bedard PL, Chu Q, Lyle M, Tang L. et al. A Phase 1 Study of SLC-0111, a Novel Inhibitor of Carbonic Anhydrase IX, in Patients With Advanced Solid Tumors. Am J Clin Oncol. 2020;43(7):484-90
172. Kelly W, Diaz Duque AE, Michalek J, Konkel B, Caflisch L, Chen Y. et al. Phase II Investigation of TVB-2640 (Denifanstat) with Bevacizumab in Patients with First Relapse High-Grade Astrocytoma. Clin Cancer Res. 2023;29(13):2419-25
173. Kontoghiorghe CN, Kontoghiorghes GJ. Efficacy and safety of iron-chelation therapy with deferoxamine, deferiprone, and deferasirox for the treatment of iron-loaded patients with non-transfusion-dependent thalassemia syndromes. Drug Des Devel Ther. 2016;10:465-81
174. Bannai S, Kitamura E. Transport interaction of L-cystine and L-glutamate in human diploid fibroblasts in culture. J Biol Chem. 1980;255(6):2372-6
175. Zhou Q, Meng Y, Li D, Yao L, Le J, Liu Y. et al. Ferroptosis in cancer: From molecular mechanisms to therapeutic strategies. Signal Transduct Target Ther. 2024;9(1):55
176. Du Y, Guo Z. Recent progress in ferroptosis: inducers and inhibitors. Cell Death Discov. 2022;8(1):501
177. Choueiri TK, Merchan JR, Figlin R, McDermott DF, Arrowsmith E, Michaelson MD. et al. Belzutifan plus cabozantinib as first-line treatment for patients with advanced clear-cell renal cell carcinoma (LITESPARK-003): an open-label, single-arm, phase 2 study. Lancet Oncol. 2025;26(1):64-73
178. Choueiri TK, Powles T, Voss MH, Plimack ER, Gurney H, Song Y. et al. LITESPARK-012: pembrolizumab plus lenvatinib with or without belzutifan or quavonlimab for advanced renal cell carcinoma. Future Oncol. 2023;19(40):2631-40
179. Thompson EA, Powell JD. Inhibition of the Adenosine Pathway to Potentiate Cancer Immunotherapy: Potential for Combinatorial Approaches. Annu Rev Med. 2021;72:331-48
180. Fong L, Hotson A, Powderly JD, Sznol M, Heist RS, Choueiri TK. et al. Adenosine 2A Receptor Blockade as an Immunotherapy for Treatment-Refractory Renal Cell Cancer. Cancer Discov. 2020;10(1):40-53
181. Falchook GS, Reeves J, Gandhi S, Spigel DR, Arrowsmith E, George DJ. et al. A phase 2 study of AZD4635 in combination with durvalumab or oleclumab in patients with metastatic castration-resistant prostate cancer. Cancer Immunol Immunother. 2024;73(4):72
182. Fong L, Forde PM, Powderly JD, Goldman JW, Nemunaitis JJ, Luke JJ. et al. Safety and clinical activity of adenosine A2a receptor (A2aR) antagonist, CPI-444, in anti-PD1/PDL1 treatment-refractory renal cell (RCC) and non-small cell lung cancer (NSCLC) patients. J Clin Oncol.35(15_suppl):3004-.
183. Beloueche-Babari M, Casals Galobart T, Delgado-Goni T, Wantuch S, Parkes HG, Tandy D. et al. Monocarboxylate transporter 1 blockade with AZD3965 inhibits lipid biosynthesis and increases tumour immune cell infiltration. Br J Cancer. 2020;122(6):895-903
184. Xia C, Yin S, To KKW, Fu L. CD39/CD73/A2AR pathway and cancer immunotherapy. Mol Cancer. 2023;22(1):44
185. Evans JV, Suman S, Goruganthu MUL, Tchekneva EE, Guan S, Arasada RR. et al. Improving combination therapies: targeting A2B-adenosine receptor to modulate metabolic tumor microenvironment and immunosuppression. J Natl Cancer Inst. 2023;115(11):1404-19
186. Chen S, Akdemir I, Fan J, Linden J, Zhang B, Cekic C. The Expression of Adenosine A2B Receptor on Antigen-Presenting Cells Suppresses CD8(+) T-cell Responses and Promotes Tumor Growth. Cancer Immunol Res. 2020;8(8):1064-74
187. Zhu Y, Jiang C, He J, He C, Zhou X, Huang X. et al. Cirbp suppression compromises DHODH-mediated ferroptosis defense and attenuates hypothermic cardioprotection in an aged donor transplantation model. J Clin Invest. 2024 134(9)
188. Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I. et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575(7784):693-8
189. Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH. et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature. 2019;575(7784):688-92
190. Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Müller C, Zandkarimi F. et al. GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent Sci. 2020;6(1):41-53
191. Curry L, Soleimani M. Belzutifan: a novel therapeutic for the management of von Hippel-Lindau disease and beyond. Future Oncol. 2024;20(18):1251-66
192. Falchook G, Infante J, Arkenau HT, Patel MR, Dean E, Borazanci E. et al. First-in-human study of the safety, pharmacokinetics, and pharmacodynamics of first-in-class fatty acid synthase inhibitor TVB-2640 alone and with a taxane in advanced tumors. EClinicalMedicine. 2021;34:100797
193. Rini BI, Suarez Rodriguez C, Albiges L, Jalkanen K, De Velasco Oria GA, Burotto M. et al. LBA74 Final analysis of the phase III LITESPARK-005 study of belzutifan versus everolimus in participants (pts) with previously treated advanced clear cell renal cell carcinoma (ccRCC). Ann Oncol. 2024;35:S1262-S3
194. Gulati S, Lara PN. Belzutifan versus Everolimus in Advanced Kidney Cancer: A Commentary on LITESPARK-005 Trial from ESMO 2023. Kidney Cancer. 2024;8(1):23-4
195. Lee JL, Park SH, Kato T, Saika T, Masumori N, Oya M. et al. LBA2 Belzutifan versus everolimus for previously treated advanced clear cell renal cell carcinoma (ccRCC): Updated follow-up of the East Asian subgroup of the randomized phase III LITESPARK-005 trial. Ann Oncol. 2024;35:S1505
196. Lewis CE, De Palma M, Naldini L. Tie2-expressing monocytes and tumor angiogenesis: regulation by hypoxia and angiopoietin-2. Cancer Res. 2007;67(18):8429-32
197. Gouda MA, Voss MH, Tawbi H, Gordon M, Tykodi SS, Lam ET. et al. A phase I/II study of the safety and efficacy of telaglenastat (CB-839) in combination with nivolumab in patients with metastatic melanoma, renal cell carcinoma, and non-small-cell lung cancer. ESMO Open. 2025;10(5):104536
198. Brown JE. Glutaminase Inhibitors—Do They Have a Role in the Treatment of Metastatic Clear-Cell Renal Cell Carcinoma? JAMA Oncol. 2022;8(10):1418-9
199. Aden D, Sureka N, Zaheer S, Chaurasia JK, Zaheer S. Metabolic Reprogramming in Cancer: Implications for Immunosuppressive Microenvironment. Immunology. 2025;174(1):30-72
200. Scirocchi F, Napoletano C, Pace A, Rahimi Koshkaki H, Di Filippo A, Zizzari IG. et al. Immunogenic Cell Death and Immunomodulatory Effects of Cabozantinib. Front Oncol. 2021;11:755433
Author contact
![]() Corresponding authors: Peidan Peng: 451532841com, Jie Zhao: zhaojie_0412com, Bisheng Cheng: chengbshsysu.edu.cn, Peng Wu: doctorwupengcom.
Corresponding authors: Peidan Peng: 451532841com, Jie Zhao: zhaojie_0412com, Bisheng Cheng: chengbshsysu.edu.cn, Peng Wu: doctorwupengcom.