Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2026; 22(7):3367-3388. doi:10.7150/ijbs.128204 This issue Cite
Research Paper
LINC-EPS Protects Against Neurodegeneration by Driving a PGC-1α-Mediated Anti-Ferroptosis Program in Parkinson's Disease
Ziqi Liu1#, Ruoxun Wang1#, Xinrui Lan1, Min Shen1, Mingfeng Jiang1, Rongqing Li1, Jie Zhao1, Jing Li1, Sainan Wang1, Qicheng Wang1, Xinyi Xu1, Wei Li2 ![]() , Weijuan Gong1
, Weijuan Gong1 ![]() , Li Qian1
, Li Qian1 ![]()
1. Key Laboratory of the Jiangsu Higher Education Institutions for Nucleic Acid & Cell Fate Regulation (Yangzhou University), Faculty of Medicine, Yangzhou University, Yangzhou, Jiangsu, 225001, PR China.
2. Department of Center Laboratory, Kunshan Hospital of Chinese Medicine, Affiliated Hospital of Yangzhou University, Kunshan, Jiangsu, 215300, PR China.
# These authors contributed equally.
Received 2025-11-11; Accepted 2026-3-1; Published 2026-3-17
Abstract

Parkinson's disease (PD) is a progressive neurodegenerative disorder characterized by dopaminergic (DA) neuron loss and currently lacks disease-modifying treatments. We found that the long intergenic non-coding RNA LINC-EPS was markedly reduced in peripheral blood of PD patients, correlating with greater clinical severity. Similar downregulation was observed in 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) PD mice and 1-methyl-4-phenylpyridinium-treated DA neurons. Knockout of LINC-EPS, either systemically or specifically in DA neurons, aggravated motor deficits and DA neurodegeneration, whereas AAV-mediated overexpression rescued these phenotypes. LINC-EPS protected DA neurons by suppressing ferroptosis, acting as a scaffold that binds both PGC-1α protein and a T-box element in its promoter, thereby recruiting PGC-1α to enhance its own transcription through a positive feedback loop. This activation improved mitochondrial function, lowered reactive oxygen species, inhibited lipid peroxidation, and conferred ferroptosis resistance. Pharmacological activation of PGC-1α with ZLN005 rescued neurodegeneration in LINC-EPS-deficient PD mice. Our study identifies a novel LINC-EPS/PGC-1α axis that mitigates ferroptotic DA neuron loss and supports PGC-1α activation as a promising therapeutic strategy for PD progression.
Keywords: Parkinson's disease, LINC-EPS, PGC-1α, ferroptosis, mitochondria, neurodegeneration
Introduction
Parkinson's disease (PD) is a progressive neurodegenerative disorder characterized by the selective loss of dopaminergic (DA) neurons in the substantia nigra (SN) pars compacta (SNpc) and profound depletion of striatal dopamine (Str). Disruption of the nigrostriatal pathway culminates in cardinal motor symptoms, including bradykinesia, rigidity, resting tremors, and postural instability [1, 2]. Current therapeutic strategies, although effective in managing symptoms, are largely symptomatic and do not halt or reverse the underlying neurodegenerative processes. This therapeutic impasse underscores the critical gap in our understanding of the molecular mechanisms driving neuronal vulnerability in PD [3].
DA neurons in SNpc are particularly vulnerable to PD, which is intrinsically linked to their high metabolic demand and reliance on mitochondrial integrity. Mitochondrial dysfunction, a well-established hallmark of PD, results in a bioenergetic crisis leading to ATP depletion, impaired calcium homeostasis, and accumulation of damaged organelles, which collectively compromise neuronal survival [4-6]. Furthermore, compromised mitochondrial respiratory chain function, particularly in complex I, results in the excessive production of mitochondrial reactive oxygen species (mitoROS) [7-9]. This enhanced oxidative stress not only damages cellular macromolecules but also creates conditions that render neurons particularly susceptible to iron-dependent lipid peroxidation, thereby linking mitochondrial dysfunction to ferroptotic cell death [10, 11].
Ferroptosis, an iron-dependent form of regulated cell death driven by lipid peroxidation, emerges as a critical mechanism in PD pathogenesis. SNpc is characterized by a high iron content, and DA neurons are enriched with polyunsaturated fatty acids, rendering them extremely sensitive to ferroptotic insults [12-14]. Critically, mitochondrial dysfunction directly drives ferroptosis: overproduction of mitoROS, in combination with iron-catalyzed Fenton chemistry, triggers catastrophic lipid peroxidation [15-17]. This is further exacerbated by the intrinsic properties of dopamine metabolism, which inactivates key antioxidant defenses, such as glutathione peroxidase 4 (GPX4), dismantling the primary cellular defense against ferroptotic death [18, 19]. Thus, the mitochondrial dysfunction-ferroptosis axis represents a mechanistically integrated pathway central to DA neuron degeneration in PD.
Long non-coding RNAs (lncRNAs) serve as critical regulators of diverse cellular processes, including neuronal function and survival, and often act as molecular scaffolds or transcriptional modulators [20-22]. Specifically, long intergenic non-coding RNA EPS (LINC-EPS) is primarily known for its role in erythropoiesis and as a systemic anti-inflammatory regulator of immune cells. It acts as a protective factor for neuronal cells against cerebral ischemia. LINC-EPS exerts neuroprotective effects on neurons exposed to oxygen-glucose deprivation/reoxygenation by modulating the Sir1-mediated regulation of autophagy, thereby influencing the neuroinflammatory responses and apoptotic pathways [23]. However, whether LINC-EPS is involved in the pathogenesis of chronic neurodegenerative disorders, such as PD, and the molecular mechanisms by which it exerts neuroprotective effects remain unknown.
Here, we demonstrate that LINC-EPS, preferentially expressed in the SN, is significantly reduced in PD patients and correlates with disease severity. Using both systemic and cell-type-specific genetic approaches in MPTP-induced PD models, we show that LINC-EPS deficiency exacerbates dopaminergic neurodegeneration, while its overexpression confers neuroprotection. Mechanistically, LINC-EPS maintains peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) expression to preserve mitochondrial homeostasis and suppress ferroptosis-mediated neuronal death [24, 25]. Pharmacological PGC-1α activation rescues the neurodegenerative phenotypes in LINC-EPS-deficient mice, establishing a LINC-EPS/PGC-1α axis as a potential therapeutic target for PD [25, 26].
Materials and Methods
Reagents and antibodies
Comprehensive lists of key reagents, kits, and antibodies used in this study are provided in the Supplementary Materials (Table S1-S2).
Human clinical specimens and ethical statement
Peripheral blood samples were obtained from 60 participants, including 30 patients with PD and 30 healthy controls. Clinical demographics and disease characteristics, including age, gender, disease duration, and Unified Parkinson's Disease Rating Scale III (UPDRS-III) scores, are detailed in the Supplementary Materials (Table S3). This study was approved by The Ethics Committee of Yangzhou University School of Medicine (Approval No. YXYLL-2025-106) and conducted in adherence to the Declaration of Helsinki. Written informed consent was obtained from all participants, and samples were anonymized to ensure privacy.
Animals and ethical statement
All animal experiments were conducted in compliance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) of Yangzhou University Medical College. Male C57BL/6J wild-type mice (8-10 weeks old, 25 ± 2 g) were sourced from the Comparative Medicine Centre of Yangzhou University. LINC-EPS knockout (LINC-EPS⁻/⁻) mice on a C57BL/6J background were generously provided by Dr. Feng Ma (Institute of Systems Medicine, Chinese Academy of Medical Sciences), while LINC-EPS floxed (LINC-EPSfl/fl) and DAT-Cre mice were custom-generated by Cyagen Biosciences (Suzhou, China). Animals were housed in a specific-pathogen-free (SPF) environment and maintained on a 12-hour light/dark cycle at a controlled temperature (20 ± 2°C), with ad libitum access to standard chow and water. Genotyping was performed by PCR analysis of tail DNA; the specific primers and PCR protocols are detailed in the Supplementary Materials (Table S4).
Cell lines and culture
The cell lines utilized in this study included human neuroblastoma SH-SY5Y, murine microglial BV2, murine astrocyte C8-D1A, and human embryonic kidney (HEK) 293T. The identity of the SH-SY5Y line was authenticated via short tandem repeat (STR) profiling, and all cultures were routinely confirmed to be negative for mycoplasma contamination using PCR-based assays. Cells were maintained in high-glucose Dulbecco's Modified Eagle's Medium (DMEM; Servicebio) supplemented with 10% fetal bovine serum (FBS; Gibco) and 1% penicillin-streptomycin (Beyotime). For lentiviral production, HEK293T cells were cultured in Opti-MEM Reduced Serum Medium (Gibco). All cultures were maintained in a humidified incubator at 37°C with a 5% CO₂ atmosphere.
Primary neuron and glial cultures
Primary midbrain neurons were prepared from postnatal day 0-1 (P0-P1) C57BL/6J mouse pups. Briefly, midbrain tissues were dissected, dissociated with papain (2 mg/mL; Worthington) and DNase I (0.1 mg/mL; Sigma-Aldrich), and plated on poly-D-lysine-coated plates. Neurons were cultured in Neurobasal-A medium supplemented with B27 (Gibco) and GlutaMAX (Gibco). Cytosine arabinoside (Ara-C; 5 µM; Sigma-Aldrich) was added at 24 h post-plating to inhibit glial proliferation [27].
For mixed glial cultures, cerebral cortices from P0-P1 pups were dissociated with 0.125% trypsin. Cells were plated in DMEM/F12 with 10% FBS. After 14-20 days, microglia were isolated by shaking the flasks at 200 rpm for 4 h. The adherent cells remaining in the flask were cultured as primary astrocytes [28].
Parkinson's disease models
In vivo PD models were established by administering MPTP (30 mg/kg, i.p.; Sigma-Aldrich) daily for 5 consecutive days. Control mice received equivalent volumes of sterile saline. For genetic models, WT and LINC-EPS⁻/⁻ mice, as well as LINC-EPSflox/flox and DAT-Cre; LINC-EPSflox/flox (LINC-EPSΔDat) mice, were randomly assigned to saline or MPTP treatment groups (n = 10-12 mice per group).
In vitro PD models were established by treating primary midbrain neurons or SH-SY5Y cells with 500 µM MPP⁺ (Sigma-Aldrich), the active toxic metabolite of MPTP, for 24 h [29, 30].
Behavioral analyses
Mice were habituated to the testing room for at least 30 min before each test.
Pole Test: Mice were placed head-up on top of a vertical wooden pole (50 cm height, 1 cm diameter). The time to turn and descend to the floor was recorded.
Open Field Test: Spontaneous activity was recorded for 5 min in an open field arena (40 cm × 40 cm × 40 cm). Total distance, average speed, and time immobile were analyzed using an automated tracking system (ANY-maze).
Rotarod Test: Motor coordination was assessed on an accelerating rotarod (Ugo Basile). The rod accelerated from 4 to 40 rpm over 300 s. The latency to fall was recorded across three trials per day for three consecutive days [31].
Computational modeling of molecular interactions
Protein-RNA docking
To predict the binding interface between the PGC-1α protein and LINC-EPS RNA, their respective three-dimensional structures were first generated using AlphaFold3. Molecular docking simulations were subsequently performed using the HDOCK server. PGC-1α was designated as the receptor and LINC-EPS RNA as the ligand, with docking parameters set to a grid spacing of 1.200 and an angular step of 15.000. The conformation with the optimal docking score was selected for visualization in PyMOL, where key hydrogen bond interactions between PGC-1α amino acid residues and LINC-EPS nucleotides were identified and analyzed.
RNA-DNA triplex modeling
To investigate the potential formation of an RNA-DNA triplex, a computational model was constructed to simulate the interaction between LINC-EPS RNA and the double-stranded DNA sequence of the TBE. The predicted triplex structure was visualized in PyMOL to map the specific atomic interactions, including the Hoogsteen base pairing, that stabilize the complex [31-33].
RNA extraction and sequencing library preparation
Total RNA was extracted from dissected mouse SN tissues using TRIzol® Reagent (Magen). The concentration and purity of the extracted RNA were measured using a NanoDrop ND-2000 spectrophotometer, while its integrity was validated on an Agilent Bioanalyzer 4150 system. Only high-quality RNA samples with an RNA Integrity Number (RIN) of 7.0 or greater were processed for library construction. Paired-end sequencing libraries were prepared using the VAHTS Universal V6 RNA-seq Library Prep Kit (Vazyme). Briefly, polyadenylated (poly(A)) mRNA was enriched from total RNA using oligo(dT)-conjugated magnetic beads, subjected to thermal fragmentation, and then reverse-transcribed into first-strand cDNA using random hexamer primers. Following second-strand synthesis and adapter ligation, the resulting cDNA fragments were enriched by PCR amplification. The final libraries were purified and their quality confirmed on an Agilent Bioanalyzer 4150. Qualified libraries were then sequenced on the Illumina NovaSeq 6000 platform, generating 150-bp paired-end reads.
Statistical analysis
Data are presented as mean ± SEM. Statistical analyses were performed using GraphPad Prism 10.0 (GraphPad Software). Comparisons between two groups were made using a two-tailed Student's t-test. For comparisons among multiple groups, one-way ANOVA and two-way ANOVA followed by Tukey's multiple comparison test was used. Correlations between two continuous variables were assessed using Pearson's correlation coefficient analysis. A P-value < 0.05 was considered statistically significant (*P < 0.05, **P < 0.01, ***P < 0.001).
Results
LINC-EPS expression is downregulated in PD and its deficiency exacerbates nigrostriatal neuropathology
Analysis of the Genotype-Tissue Expression (GTEx) dataset revealed LINC-EPS expression in human SN (Fig. 1A) [34]. To assess a clinically accessible biospecimen, we quantified LINC-EPS expression in peripheral blood from an independent cohort by RT-qPCR, revealing significantly lower levels in PD patients compared with healthy controls (Fig. 1B). To evaluate the clinical significance of this reduction, correlations between peripheral LINC-EPS mRNA levels and motor symptom severity were examined. A significant inverse relationship was observed between LINC-EPS expression levels and the Unified PD Rating Scale part III (UPDRS-III) motor scores (Fig. 1C), indicating that low peripheral LINC-EPS expression was associated with high motor impairment. Consistent with the findings in human peripheral blood, mRNA levels of LINC-EPS were significantly reduced in the SN and striatum of MPTP-induced PD model mice (Fig. 1D).
LINC-EPS Deficiency Exacerbates MPTP-Induced Neurodegeneration. (A) Bioinformatic analysis of LINC-EPS mRNA expression across a panel of human tissues using the GTEx database. The SN is indicated to show its notable expression level. (B) RT-qPCR analysis of LINC-EPS mRNA levels in peripheral blood samples from healthy controls (HC) (n = 30) and PD patients (n = 30). (C) Pearson correlation analysis of peripheral LINC-EPS mRNA levels and UPDRS-III motor scores in PD patients (n = 30). (D) RT-qPCR analysis of LINC-EPS mRNA levels in the Substantia Nigra (SN) and striatum (Str) of C57/BL6 mice treated with saline or MPTP (n = 4). (E-J) Motor function assessments: open-field test parameters (E-H), rotarod performance (I), and pole test (J) (n=10). (K) Representative immunofluorescence images of TH (red) in the SNpc and quantification of TH-positive (TH⁺) neurons. DAPI (blue) was used for nuclear counterstaining. Scale bar, 500 µm (n = 6). (L) Representative immunofluorescence images showing TH-positive fibers in the striatum from saline- or MPTP-treated wild-type (WT) and LINC-EPS⁻/⁻ mice, together with quantification of mean fluorescence intensity (MFI) and relative density. Scale bars, 200 µm (overview) and 50 µm (magnified view) (n = 6). (M-N) Representative immunoblots and quantification of TH protein levels in the SN (M) and striatum (N) β-actin served as a loading control. Data are presented as fold change, where the ratio of TH to β-actin in the control group was designated as 1.0 (n = 3). (O) Metabolomic alterations in LINC-EPS⁻/⁻ striatum following MPTP treatment (n=3). Data are presented as mean ± SEM. Statistical significance was determined by two-tailed unpaired Student's t-test for (B), Pearson correlation for (C), two-way ANOVA with Tukey's post-hoc test for (D, F-L), and one-way ANOVA with Tukey's post-hoc test for (M, N). *P < 0.05, **P < 0.01, ***P < 0.001. ns, not significant.
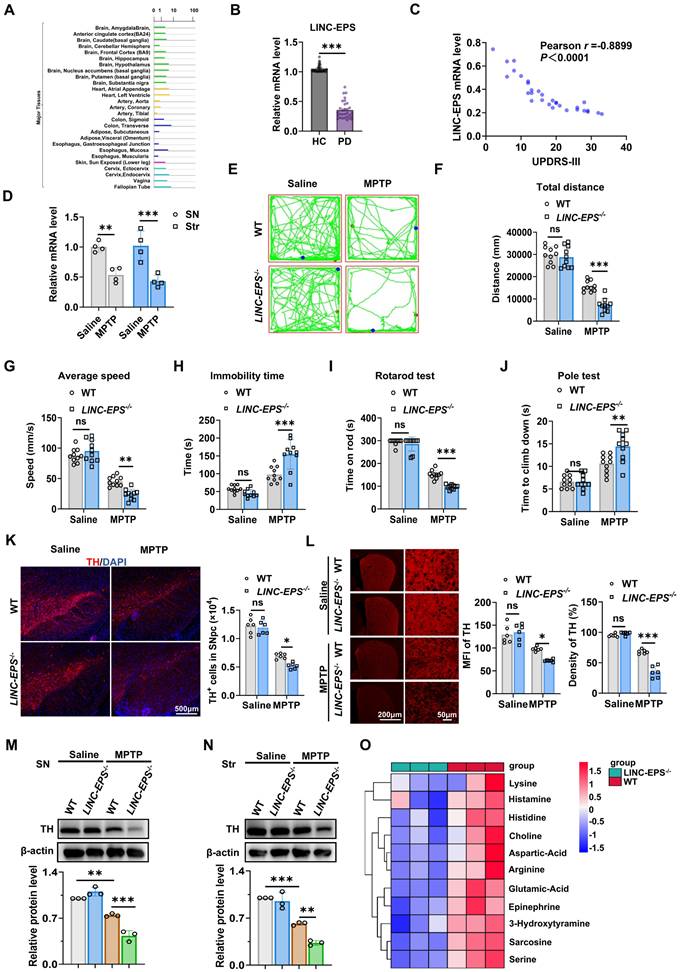
We subjected global LINC-EPS knockout (LINC-EPS⁻/⁻) mice and their wild-type (WT) littermates to the MPTP regimen to determine the role of LINC-EPS in PD-like pathology [35]. MPTP-treated LINC-EPS⁻/⁻ mice exhibited more severe motor deficits than their WT counterparts. In the open-field test, these mice traveled a shorter total distance, showed a lower average speed, and spent significantly more time immobile (Fig. 1E-H). In the accelerating rotarod test, these mice exhibited a shorter latency to fall (Fig. 1I). Furthermore, these mice showed prolonged descent time in the pole test, consistent with exacerbated bradykinesia (Fig. 1J).
Consistent with the exacerbated behavioral deficits, LINC-EPS deficiency aggravated MPTP-induced nigrostriatal neurodegeneration. Immunofluorescence analysis for tyrosine hydroxylase (TH) in SNpc revealed the significantly greater loss of TH-positive neurons in MPTP-treated LINC-EPS⁻/⁻ mice than in MPTP-treated WT controls (Fig. 1K). Quantitative analysis confirmed the significantly greater reduction in mean fluorescence intensity and TH-immunoreactive fiber density in the striatum of LINC-EPS⁻/⁻ mice than in the striatum of their WT littermates following MPTP administration (Fig. 1L). These neuropathological findings were confirmed via immunoblotting, which indicated significantly lower TH protein levels in the SN and striatum of MPTP-treated LINC-EPS⁻/⁻ mice compared to those in the SN and striatum of WT controls (Fig. 1M and N).
We performed targeted metabolomic analysis of striatal tissue to quantify the neurochemical state of the nigrostriatal system. This analysis revealed a more pronounced depletion of key neurotransmitters in LINC-EPS⁻/⁻ mice than in their WT counterparts following MPTP administration. Specifically, striatal levels of 3-hydroxytyramine (dopamine) and epinephrine were markedly reduced in LINC-EPS-deficient animals. Furthermore, a global reduction was observed across multiple neuroactive metabolites, including excitatory transmitters glutamic acid and aspartic acid, and the cholinergic metabolite choline (Fig. 1O). These data suggest that LINC-EPS deficiency causes a profound collapse of striatal neurochemical homeostasis in the Parkinsonian state. Furthermore, expression levels of key neurotrophic factors, such as brain-derived neurotrophic factor (Bdnf) and neurotrophin-3 (Nt3), were significantly lower in the midbrain of MPTP-treated LINC-EPS⁻/⁻ mice (Fig. S1A). We further examined the accumulation of α-synuclein phosphorylated at serine 129 (pS129-α-Syn), a core pathological hallmark of PD [36]. Both immunofluorescence and immunoblotting assays revealed the significantly greater accumulation of pS129-α-Syn in the nigrostriatal system of MPTP-treated LINC-EPS⁻/⁻ mice compared to that in the nigrostriatal system of MPTP-treated WT controls (Fig. S1B-E).
Collectively, these data indicate that LINC-EPS deficiency results in severe motor deficits, DA neurodegeneration, and α-synuclein pathology in MPTP induced PD model mice.
LINC-EPS deficiency in DA neurons exacerbates nigrostriatal neurodegeneration
To delineate the cellular specificity of LINC-EPS downregulation in PD brain, we performed RNA fluorescence in situ hybridization (RNA-FISH) assay in the SNpc of MPTP-treated mice. This in vivo analysis revealed significant reduction in LINC-EPS mRNA puncta in DA neurons (TH⁺), microglia (IBA1⁺), and astrocytes (GFAP⁺) following MPTP administration (Fig.2 A-D). To dissect the cell-autonomous responses to neurotoxic insult, we exposed primary murine neural cells and corresponding cell lines to MPP⁺. In stark contrast to the generalized in vivo responses, RT-qPCR analysis revealed that the MPP⁺-induced suppression of LINC-EPS was substantially more pronounced in DA neuronal cells than in glial cells (Fig. 2E). To determine the potential effects of neuron LINC-EPS on motor deficits and nigrostriatal neurodegeneration in PD model mice, we generated mice with DA neuron-specific deletion of LINC-EPS (LINC-EPSf/f; Dat-Cre, hereafter referred to as LINC-EPSΔDat) and subjected them to MPTP neurotoxicity tests. Successful generation and validation of LINC-EPSΔDat mice were confirmed by genotyping PCR (Fig. S2B). Subsequent behavioral assessments revealed that LINC-EPSΔDat mice exhibited significantly exacerbated motor deficits compared to their control (LINC-EPSf/f) littermates. These included impairments in locomotor activity (open-field test; Fig. 2F-I) and motor coordination (rotarod test; Fig. 2J) and increased bradykinesia (pole test; Fig. 2K).
Loss of LINC-EPS in dopaminergic neurons drives neurodegeneration. (A-C) Representative RNA-FISH images showing LINC-EPS mRNA puncta (red) co-localized with dopaminergic neurons (TH, green, A), microglia (IBA1, yellow, B), and astrocytes (GFAP, green, C). Nuclei were counterstained with DAPI (blue). Scale bar, 5 µm. (D) Quantification of the average number of LINC-EPS mRNA puncta per cell for each cell type, based on the analysis of images as shown in (a-c). n = 6 images per group (3 mice/group; 2 consecutive sections/mouse; 1 predefined field of view/section). (E) RT-qPCR analysis of LINC-EPS mRNA levels in primary murine neurons, microglia, and astrocytes, alongside the SH-SY5Y (neuronal), BV2 (microglial), and C8-D1A (astrocytic) cell lines, following MPP⁺ exposure (n = 4). (F-I) Representative locomotor traces from the open-field test for the indicated groups (LINC-EPSf/f control mice and LINC-EPSΔDat mice with DA neuron-specific LINC-EPS deletion, treated with saline or MPTP) (F) and quantification of total distance (G), average speed (H), and immobility time (I) (n = 10). Mouse genotype validation is shown in Supplementary Fig. S2C. (J) Motor performance on the accelerating rotarod, quantified as latency to fall (n = 10). (K) Time to descend in the pole test (n = 10). (L) Representative immunofluorescence images and stereological quantification of TH⁺ neurons in the SNpc. Scale bar, 200 µm (n = 6). (M) Representative immunohistochemistry images and quantification of TH⁺ fiber density in the striatum. Scale bar, 600 µm (overview) and 50 µm (magnified view) (n = 6). (N) Representative immunoblots and quantification of TH protein levels in the SN (n = 3). (O) Representative immunoblots and quantification of TH protein levels in the striatum (n = 3). Data are presented as mean ± SEM. Statistical significance was determined by two-way ANOVA with Tukey's post-hoc test. *P < 0.05, **P < 0.01, ***P < 0.001. ns, not significant.
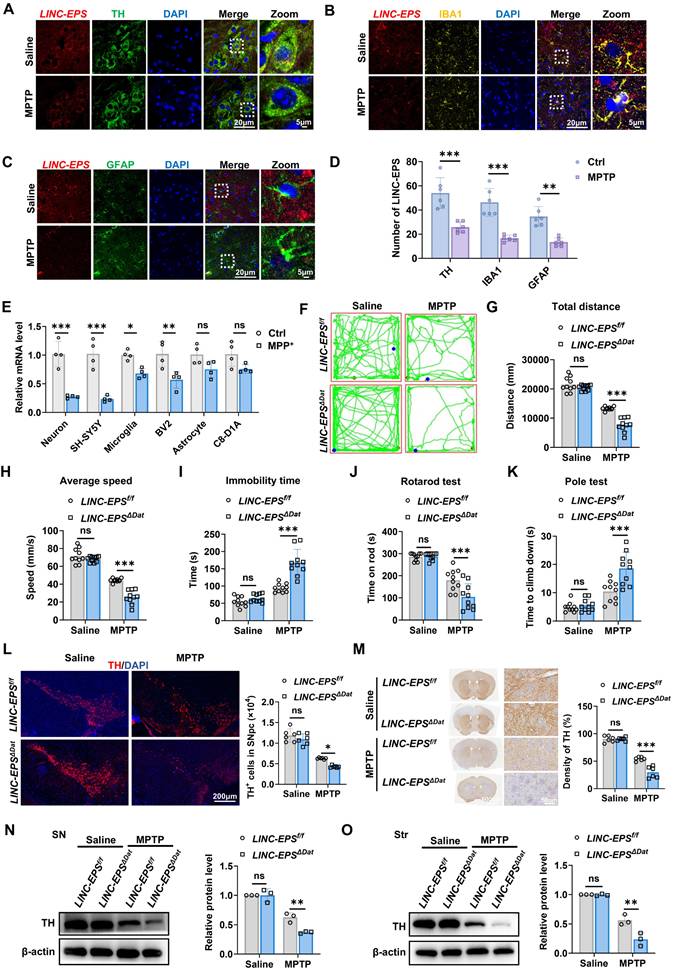
Stereological counting indicated that MPTP-induced loss of TH⁺ neurons in SNpc was significantly greater in LINC-EPSΔDat mice than in controls (Fig. 2L). This was paralleled by a more pronounced reduction in the density of TH⁺ nerve terminals in the striatum (Fig. 2M). Immunoblotting analysis further corroborated these findings, showing a greater reduction in TH protein levels in the SN and striatum of LINC-EPSΔDat mice than in the SN and striatum of controls (Fig. 2N and O).
Collectively, these results suggest that DA neuron-specific deficiency of LINC-EPS aggravates nigrostriatal neurodegeneration and motor deficits in MPTP-induced PD model mice.
LINC-EPS deficiency promotes ferroptosis in DA neurons
To elucidate the mechanisms by which LINC-EPS deficiency exacerbates nigrostriatal neurodegeneration, we performed RNA sequencing (RNA-seq) of the SN tissues of MPTP-treated WT and LINC-EPS⁻/⁻ mice, given that the SN is enriched in dopaminergic neuron somata and constitutes the primary site of selective vulnerability in Parkinson's disease. This analysis revealed the broad dysregulation of multiple cell death pathways in LINC-EPS-deficient tissues. Notably, although gene sets associated with pro-inflammatory death pathways, such as necroptosis and pyroptosis, were upregulated, ferroptosis-related genes were broadly dysregulated, exhibiting a predominant net reduction in anti-ferroptotic factors (Fig. 3A). To functionally dissect the contribution of these pathways, we treated primary neurons with specific cell death inhibitors concurrently with MPP⁺ (the active toxic metabolite of MPTP, 500 µM) for 24 hours. Pharmacological screening revealed that enhanced cell death in LINC-EPS⁻/⁻ neurons was effectively rescued by the ferroptosis inhibitor, ferrostatin-1 (Fer-1). Partial rescue was also observed with the pan-caspase inhibitor, Z-VAD-FMK, whereas inhibitors of necroptosis (Nec-1s) and pyroptosis (VX-765) conferred no significant protection against exacerbated cell loss (Fig. 3B). To probe the underlying molecular alterations, we examined the key regulators and markers of canonical cell death pathways, including ferroptosis, apoptosis, pyroptosis, and necroptosis, via immunoblotting. LINC-EPS deficiency in neurons led to the marked downregulation of core anti-ferroptotic proteins (cystine/glutamate antiporter subunit) xCT and GPX4 as well as the coenzyme Q complex (CQ10B and CQ10A) following MPP+ treatment (Fig. 3C). In contrast, expression levels of ferroptosis suppressor protein 1 (FSP1) and ferritin light chain (FTL) were not significantly altered between genotypes after MPP⁺ treatment (Fig. 3C). Moreover, no significant differences were observed in the expression levels of key proteins related to apoptosis, pyroptosis, and necroptosis between LINC-EPS⁻/⁻ and WT neurons after MPP⁺ treatment (Fig. S2A). Collectively, these convergent lines of evidence identified dysregulated ferroptosis as the primary mechanism driving heightened neuronal vulnerability associated with LINC-EPS loss.
LINC-EPS deficiency Promotes Ferroptosis in Dopaminergic Neurons In vitro and In vivo. (A) Heatmap of differentially expressed genes from regulated cell death pathways, identified by RNA-seq of SN tissue from MPTP-treated WT and LINC-EPS⁻/⁻ mice. (B)Relative cell viability of primary WT and LINC-EPS⁻/⁻ neurons treated with MPP⁺ in the presence of inhibitors for ferroptosis (Ferrostatin-1, Fer-1), apoptosis (Z-VAD-FMK), necroptosis (Necrostatin-1s, Nec-1s), or pyroptosis (VX-765). Cell viability of MPP⁺-treated WT neurons was set as 1.0 to assess the protective effects of inhibitors (n = 4). (C) Representative immunoblots of ferroptosis-related proteins (FTL, FSP1, xCT, GPX4, CQ10B, CQ10A) in primary neurons from WT and LINC-EPS⁻/⁻ mice treated with or without MPP⁺ (500 μM, 24 h). Due to limited primary neuron yield, two technical replicates from pooled neurons (six mice per pool) (D) Representative TEM images of mitochondria in primary neurons from WT and LINC-EPS⁻/⁻ mice treated with vehicle or MPP⁺. Blue arrows indicate ferroptosis-associated mitochondrial alterations, including shrinkage, increased membrane density, and outer membrane rupture. Scale bar, 500 nm. (E) Representative images of Prussian blue staining for iron in the SN of LINC-EPSf/f and LINC-EPSΔDat mice treated with saline or MPTP. Right: Quantification of the number of iron-positive cells per microscopic field (n = 12). Blue arrows highlight iron-positive cells. Scale bar, 50 µm. (F, G) Quantification of MDA content, an end-product of lipid peroxidation (F) and GSH/GSSG ratio as an indicator of the cellular redox state (G) in SN tissue from MPTP-treated LINC-EPSf/f and LINC-EPSΔDat mice (n = 3). (H) Representative immunoblots of ferroptosis-related proteins in SN tissue from LINC-EPSf/f and LINC-EPSΔDat mice treated with MPTP. Quantification is provided in Figure S2C. (I) Representative co-immunofluorescence images of GPX4 (red) and TH (green) in the SNpc of LINC-EPSf/f and LINC-EPSΔDat mice treated with MPTP. Right: Quantification of GPX4 MFI measured within the soma of individual TH-positive neurons (n = 6). Scale bar, 20 µm. Data are presented as mean ± SEM. Statistical significance was determined by two-way ANOVA with Tukey's post-hoc test or a two-tailed unpaired Student's t-test. *P < 0.05, **P < 0.01, ***P < 0.001. ns, not significant.
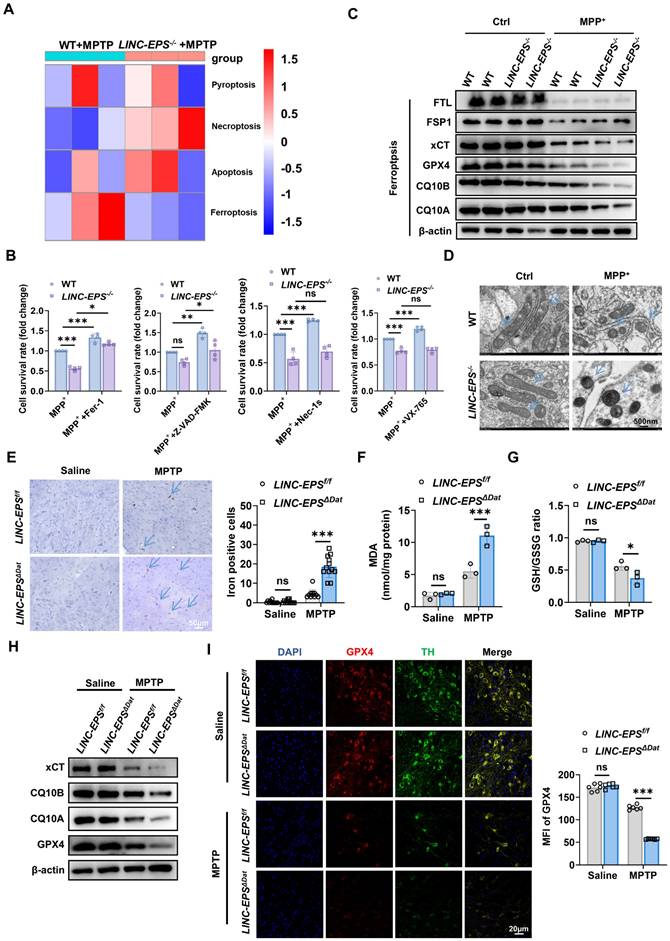
At the ultrastructural level, transmission electron microscopy (TEM) of MPP⁺-treated LINC-EPS⁻/⁻ primary neurons revealed hallmark features of ferroptosis, including shrunken mitochondria with increased membrane density and ruptured outer membranes (Fig. 3D) [38-40], confirming ferroptotic mitochondrial damage at the organellar level.
To ascertain whether LINC-EPS deficiency exacerbates ferroptosis in vivo, we assessed the SN of MPTP-treated LINC-EPSΔDat mice for key ferroptotic markers. As iron dyshomeostasis is a central feature of ferroptosis, we assessed iron accumulation in this study [41, 42]. Prussian blue staining revealed a significant increase in the number of iron-positive cells in the SN of MPTP-challenged LINC-EPSΔDat mice compared to that in the SN of controls (Fig. 3E). This finding was corroborated by the core biochemical signatures of ferroptosis in the SN tissue lysates. MPTP-treated LINC-EPSΔDat mice exhibited markedly higher levels of the lipid peroxidation marker malondialdehyde (MDA) and significantly lower ratio of reduced to oxidized glutathione (GSH/GSSG; Fig. 3F and G). Immunoblotting analysis of SN lysates revealed significantly lower protein levels of xCT, CQ10B, CQ10A, and GPX4 in MPTP-treated LINC-EPSΔDat mice than in MPTP-treated LINC-EPSf/f littermate controls (Figs. 3H and S2C). To determine whether GPX4 reduction occurred within DA neurons, we performed co-immunofluorescence analysis of SNpc. This experiment confirmed that GPX4 protein signals were significantly diminished in the TH-positive neurons of MPTP-challenged LINC-EPSΔDat mice (Fig. 3I).
Taken together, these findings showed that DA neuron-specific loss of LINC-EPS exacerbated ferroptosis in MPTP-induced PD model mice.
DA neuron-specific overexpression of LINC-EPS attenuates MPTP-induced nigrostriatal pathology
After determining that LINC-EPS deficiency exacerbated MPTP-induced nigrostriatal pathology, we examined whether the selective elevation of LINC-EPS in midbrain DA neurons mitigates these degenerative changes. To achieve DA neuron-specific overexpression, DATIRES-Cre mice were bilaterally injected with a Cre-dependent AAV vector encoding LINC-EPS (AAV-DIO-LINC-EPS) or a corresponding control vector (AAV-DIO-NC) under the hSyn promoter into SNpc using stereotaxic coordinates anteroposterior (AP) -3.0 mm, mediolateral (ML) ±1.3 mm, dorsoventral (DV) -4.4 mm relative to bregma. This strategy selectively drove LINC-EPS expression in Cre-expressing DA neurons. Thirty-four days post-injection, the mice were administered MPTP (30 mg/kg, intraperitoneally, once daily for five consecutive days) or saline. Then, motor performance, nigrostriatal DA integrity, and ferroptosis-associated indices were evaluated (Fig. 4A). FISH combined with TH immunofluorescence revealed that AAV-mediated LINC-EPS delivery resulted in the marked elevation of LINC-EPS transcript signals in TH-positive DA neurons of SN compared to those in the AAV-NC group (Fig. 4B), indicating that the AAV-LINC-EPS construct effectively enhanced LINC-EPS expression in the targeted neuronal populations.
Overexpression of LINC-EPS in Nigral Dopaminergic Neurons Mitigates MPTP-Induced Nigrostriatal Degeneration and Ferroptosis. (A) Experimental design: bilateral SNpc injection of AAV-DIO-LINC-EPS or control vector in DAT-Cre mice (3 µL/side; 2×10¹² vg/mL), followed by MPTP administration after 34 days. (B) FISH detection of LINC-EPS transcripts (red) combined with TH immunofluorescence (green) in the SN of C57BL/6J mice following injection of AAV-LINC-EPS or AAV-NC. DAPI (blue) labels cell nuclei. Insets show high-magnification views of representative TH-positive neurons co-expressing LINC-EPS. Scale bar = 25m. (C) Representative locomotor traces from the open-field test for each group. (D) Quantification of the total distance traveled by mice in the open-field test (n = 10). (E) Quantification of the average speed of mice during the open-field test (n = 10). (F) Quantification of the total immobility time during the open-field test (n = 10). (G) Assessment of motor coordination, showing the latency to fall from the accelerating rotarod (n = 10). (H) Assessment of bradykinesia, measured as the time required to descend in the pole test (n = 10). (I) Representative Immunohistochemistry images of TH positive neurons in the SNpc (n = 6). (J) Representative Immunohistochemistry images of TH-positive fibers in the striatum (n = 6). (K, L) Representative immunoblots and quantification of TH protein levels in the SN (L) and striatum (L) (n = 3). (M) Ferroptosis-related protein expression (xCT, CQ10B, CQ10A, GPX4) in SN (n=3). (N, O) Quantification of MDA levels (N) and the GSH/GSSG ratio (O) in SN lysates (n = 3). (P) GPX4-TH colocalization in SNpc with quantification of GPX4 intensity in TH-positive neurons (scale bar: 20 µm; n=6). Data are presented as mean ± SEM. Statistical significance was determined by two-way ANOVA with Tukey's post-hoc test. *P < 0.05, **P < 0.01, ***P < 0.001. ns, not significant.
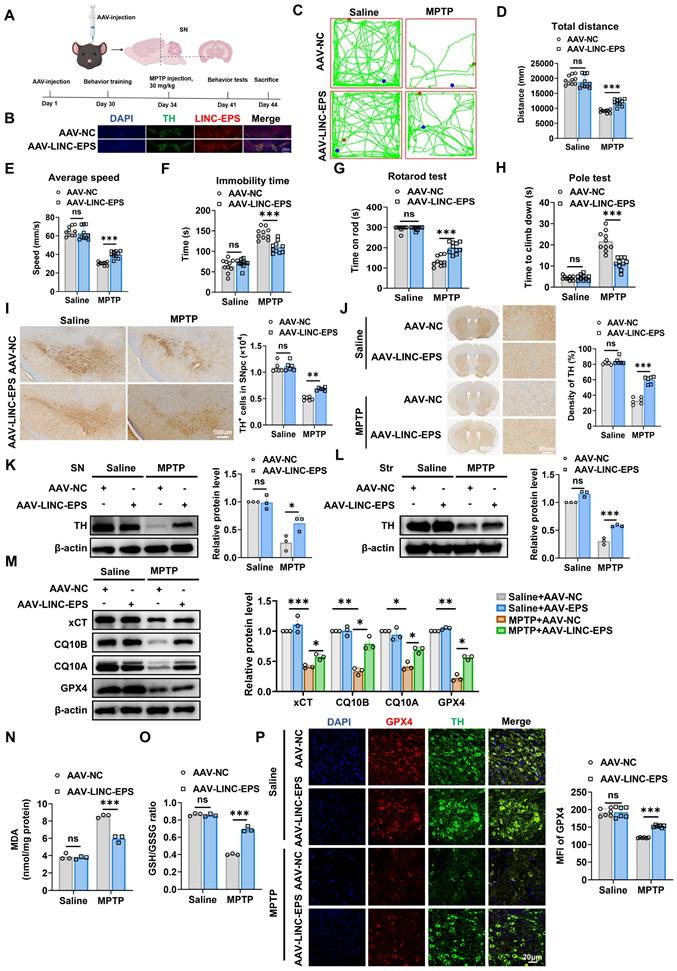
Next, we assessed the effects of the AAV-LINC-EPS construct on motor functions. In the open field test, MPTP treatment caused a marked reduction in locomotor activity in AAV-NC mice, as shown by the representative traces. However, AAV-LINC-EPS administration significantly mitigated these motor impairments, as evidenced by their greater total distance traveled, higher average speed, and lower immobility time compared to the MPTP-treated AAV-NC group (Fig. 4C-F). Furthermore, in motor coordination and bradykinesia tests, AAV-LINC-EPS group exhibited a significantly longer latency to fall in the rotarod test (Fig. 4G) and shorter descent time in the pole test (Fig. 4H). Consistent with behavioral data, LINC-EPS overexpression robustly protected against MPTP-induced DA neurodegeneration. We observed significant preservation of TH-positive neurons in the SNpc (Fig. 4I) and TH-positive nerve terminals in the striatum (Fig. 4J).
Immunoblotting revealed that the MPTP-induced loss of TH protein was significantly attenuated in both the SN (Fig. 4K) and striatum (Fig. 4L) of AAV-LINC-EPS-administered mice compared to that in the SN and striatum AAV-NC controls. We further examined whether AAV-LINC-EPS treatment attenuates ferroptosis in the MPTP-induced subacute model of PD, as indicated by changes in ferroptosis-associated biochemical and molecular markers. Immunoblotting analysis of SN tissues showed that MPTP treatment caused significant downregulation of the anti-ferroptotic regulators xCT, CQ10B, CQ10A, and GPX4 in AAV-NC mice. However, AAV-LINC-EPS delivery significantly counteracted this effect (Fig. 4M). Consistent with these protein-level observations, biochemical analysis of the SN tissues of MPTP-treated mice showed that AAV-LINC-EPS markedly suppressed the accumulation of lipid peroxidation marker MDA (Fig. 4N) and prevented the decline in the GSH/GSSG ratio (Fig. 4O). Co-immunofluorescence staining indicated that GPX4 protein levels were significantly higher in the TH-positive neurons of the AAV-LINC-EPS group than in those of the AAV-NC group (Fig. 4P).
Collectively, these data demonstrated that AAV-mediated overexpression of LINC-EPS in neurons partially protected DA neurons from MPTP-induced degeneration and ferroptosis.
LINC-EPS negatively regulates ferroptosis in a neuronal cell PD model
To elucidate the role of LINC-EPS in different cellular contexts, we used a loss-of-function strategy in the SH-SY5Y human DA neuroblastoma cell line. Among the three independent shRNA constructs targeting LINC-EPS, construct #3 showed the most robust knockdown efficiency (Fig. 5A). Therefore, this construct was selected to establish a stable LINC-EPS-knockdown (shLINC-EPS) cell line for subsequent experiments. This cell line exhibited a significant reduction in LINC-EPS transcript levels compared to the non-targeting control (shNC) cells. Upon exposure to neurotoxin MPP⁺, control shNC cells exhibited decreased protein levels of anti-ferroptotic regulators xCT, CQ10B, CQ10A, and GPX4. Notably, LINC-EPS knockdown significantly exacerbated MPP⁺-induced downregulation of all four protein levels (Fig. 5B). Consistent with the loss of these key anti-ferroptosis regulators, shLINC-EPS cells exhibited heightened sensitivity to MPP⁺-induced ferroptotic stress. This was evidenced by a more severe accumulation of total ROS, lipid ROS, intracellular ferrous iron (Fe²⁺), and MDA, along with pronounced reduction in the GSH/GSSG ratio, compared to that in the control cells (Fig. 5C-G).
LINC-EPS knockdown sensitizes SH-SY5Y cells to MPP⁺-induced ferroptosis. All experiments were performed in SH-SY5Y cells stably expressing a non-targeting control shRNA (shNC) or an shRNA against LINC-EPS (shLINC-EPS). Cells were treated with or without MPP⁺ (500 µM, 24 h).(A) qRT-PCR analysis of LINC-EPS mRNA levels confirming knockdown efficiency (n = 3). (B) Immunoblot and corresponding quantification of xCT, CQ10B, CQ10A, and GPX4 protein levels. β-actin served as a loading control (n = 3). (C) Quantification of total intracellular reactive oxygen species (ROS) using the fluorescent probe DHE (n = 4). (D) Measurement of lipid ROS accumulation using the lipid peroxidation-specific fluorescent probe Liperfluo (n = 4). (E) Determination of Fe²⁺ levels using the fluorescent probe FerroOrange (n = 4). (F) Quantification of MDA levels as an index of lipid peroxidation (n = 3). (G) Determination of the GSH/GSSG ratio as a marker of cellular redox balance (n = 3). (H-N) shLINC-EPS and shNC cells were challenged with MPP⁺ in the presence or absence of the ferroptosis inhibitor ferrostatin-1 (Fer-1; 1 µM) for 24 h. (H) Cell viability assessment using the CCK-8 assay (n = 4). (I) Representative immunoblots and corresponding quantitative analysis of the anti-ferroptosis-related proteins xCT, CQ10B, CQ10A, and GPX4, with β-actin as the loading control (n = 3). (J) Quantification of total cellular ROS levels using a fluorescent ROS probe (n = 4). (K) Quantification of lipid ROS levels using the Liperfluo probe (n = 4). (L) Quantification of Fe²⁺ levels using the FerroOrange probe (n = 4). (M) Quantification of MDA levels (n = 3). (N) Quantification of the GSH/GSSG ratio (n = 4). Data are presented as mean ± SEM. Statistical significance was determined by a one-way ANOVA followed by Tukey's post-hoc test (A) or two-way ANOVA followed by Tukey's post-hoc test (B-N). *P < 0.05, **P < 0.01, ***P < 0.001.
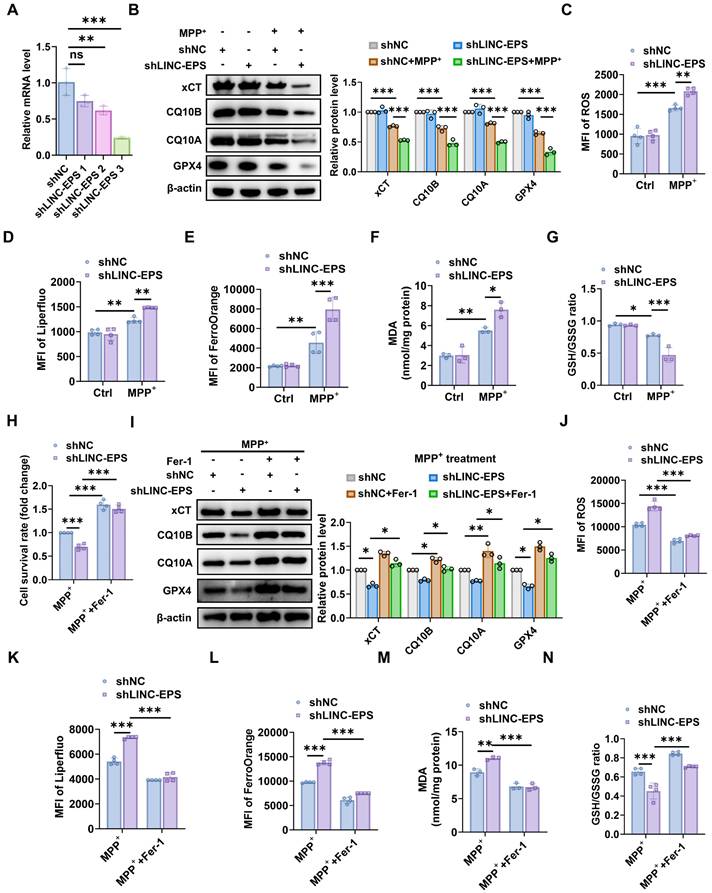
To examine whether the increased vulnerability of shLINC-EPS cells to MPP⁺ was associated with ferroptosis, a pharmacological rescue experiment was conducted using the specific ferroptosis inhibitor ferrostatin-1 (Fer-1). Specifically, shLINC-EPS cells were challenged with MPP⁺ in the presence or absence of Fer-1. Cell viability assays (CCK-8) demonstrated that Fer-1 significantly improved survival of MPP⁺-treated shLINC-EPS cells compared with MPP⁺-treated vehicle controls (Fig. 5H). Consistent with this effect, immunoblot analysis showed that Fer-1 partially restored the levels of the anti-ferroptotic proteins xCT, CQ10B, CQ10A, and GPX4, which were reduced following MPP⁺ exposure (Fig. 5I). Concurrently, Fer-1 treatment reversed the core biochemical hallmarks of ferroptosis, markedly attenuating MPP⁺-induced accumulation of total ROS, lipid ROS, Fe²⁺, and MDA, while restoring the cellular GSH/GSSG ratio (Fig. 5J-N).
In a complementary gain-of-function experiment, we generated SH-SY5Y cells stably overexpressing LINC-EPS (oeLINC-EPS; Fig. S3A). In stark contrast to LINC-EPS knockdown, its overexpression conferred significant protection against MPP⁺-induced toxicity. OeLINC-EPS cells maintained substantially higher levels of xCT, CQ10B, CQ10A, and GPX4 proteins than the control cells (empty) following MPP+ challenge (Fig. S3B). Concordantly, LINC-EPS overexpression robustly mitigated the MPP⁺-induced increase in total ROS, lipid ROS, Fe²⁺, and MDA levels and prevented the decrease in the GSH/GSSG ratio (Fig. S3C-G). To directly test whether the neuroprotective function of LINC-EPS is contingent upon the suppression of ferroptosis, we challenged oeLINC-EPS cells with ferroptosis inducer erastin. Although LINC-EPS overexpression robustly increased cell survival, this protective effect was significantly attenuated by co-treatment with erastin (Fig. S3H). At the molecular level, erastin inhibited oeLINC-EPS-mediated upregulation of the levels of anti-ferroptotic proteins xCT, CQ10B, CQ10A, and GPX4 (Fig. S3I). Concurrently, erastin treatment partially attenuated the protective effect of LINC-EPS overexpression against oxidative stress, leading to the significant elevation of total ROS, lipid ROS, Fe²⁺, and MDA levels and reduction of the GSH/GSSG ratio (Fig. S3J-N). Taken together, these findings suggest that the cytoprotective capacity of LINC-EPS is largely dependent on its ability to inhibit ferroptosis.
LINC-EPS deficiency triggers mitochondrial dysfunction and subsequent ROS-dependent ferroptosis
To determine the mechanisms by which LINC-EPS regulates ferroptosis, we analyzed the RNA-seq data of the SN tissues of MPTP-treated WT and LINC-EPS⁻/⁻ mice. Gene Ontology (GO) enrichment analysis revealed that terms such as “electron transport chain” and “oxidative phosphorylation” were among the most significantly enriched biological processes (Fig. 6A). These data strongly suggest that mitochondrial dysfunction is the primary pathological consequence of LINC-EPS deficiency. Mitochondrial membrane potential (MMP) was assessed in shLINC-EPS cells following MPP+ treatment. Upon MPP+ exposure, shLINC-EPS cells exhibited a significantly more pronounced decrease in MMP, as evidenced by the reduced percentage of cells containing JC-1 aggregates (high potential), compared to shNC cells (Fig. 6B). Decrease in MMP promoted the production of mitoROS. Accordingly, we observed a marked elevation in mitoROS levels in MPP⁺-treated shLINC-EPS cells relative to the controls (Fig. 6C). Relative to the control cells, oeLINC-EPS cells consistently exhibited preserved mitochondrial membrane potential and reduced mitoROS production following MPP+ exposure (Fig. S4A and B). As excessive mitoROS drives ferroptosis [43, 44], we explored whether ROS scavenging alleviates ferroptosis. We treated MPP⁺-challenged shLINC-EPS cells with TEMPO, a general ROS scavenger, or Visomitin, a mitochondria-targeted antioxidant. Reduced viability of shLINC-EPS cells was significantly rescued by both Mito-TEMPO and Visomitin (Fig. 6D), indicating mitochondrial oxidative stress as the primary driver of cell death. Furthermore, both Mito-TEMPO and Visomitin significantly ameliorated MPP+-induced MMP collapse (Fig. 6E) and attenuated mitoROS surge (Fig. 6F).
LINC-EPS deficiency induces mitochondrial dysfunction, leading to ROS-dependent ferroptosis. (A) Gene Ontology (GO) enrichment analysis of downregulated genes from RNA-seq of SN tissue from MPTP-treated LINC-EPS⁻/⁻ versus WT mice. The top 30 enriched biological process terms are shown (n = 3). (B) Stacked bar chart showing the distribution of JC-1 aggregates (high mitochondrial membrane potential, ΔΨm; gray) and JC-1 monomers (low ΔΨm; blue) in SH-SY5Y cells. Each bar represents one experimental group defined by the combination of MPP⁺ treatment, scrambled control (shNC), or LINC-EPS knockdown (shLINC-EPS), as indicated by the binary notation below ("+", present; "-", absent) and group labels above each bar. Data are presented as percentage of total cell count (n = 4). (C) Flow cytometric quantification of mitoROS levels in MPP⁺-treated cells, measured as the MFI of the ROS-sensitive probe MitoSOX (n = 4). (D-O) Mitochondrial-targeted antioxidant rescue experiments: cells treated with MPP⁺ (500 µM, 24h) ± Mito-TEMPO (100 µM) or Visomitin (50 nM). (D) Quantification of cell survival rate after rescue with Mito-TEMPO or Visomitin (n = 4). (E) Quantification of MMP after rescue with Mito-TEMPO or Visomitin (n = 4). (F) Quantification of mitoROS levels after rescue with Mito-TEMPO or Visomitin (n = 4). (G) Representative immunoblot of xCT, CQ10B, CQ10A, GPX4 after Mito-TEMPO rescue, quantified in Figure S4C. (H) Representative immunoblot of xCT, CQ10B, CQ10A, GPX4 after Visomitin rescue, quantified in Figure S4D. (I) Quantification of total cellular ROS levels after rescue with Mito-TEMPO or Visomitin (n = 4). (J) Quantification of lipid ROS levels after rescue with Mito-TEMPO or Visomitin using the Liperfluo probe (n = 4). (K) Quantification of Fe²⁺ levels after rescue with Mito-TEMPO or Visomitin using the FerroOrange probe (n = 4). (L) Quantification of the GSH/GSSG ratio after rescue with Mito-TEMPO (n = 3). (M) Quantification of the GSH/GSSG ratio after rescue with Visomitin (n = 3). (N) Quantification of MDA levels after rescue with Mito-TEMPO (n = 3). (O) Quantification of MDA levels after rescue with Visomitin (n = 3). Data are presented as mean ± SEM. Statistical significance was determined by two-way ANOVA with Tukey's post-hoc test. *P < 0.05, **P < 0.01, ***P < 0.001.
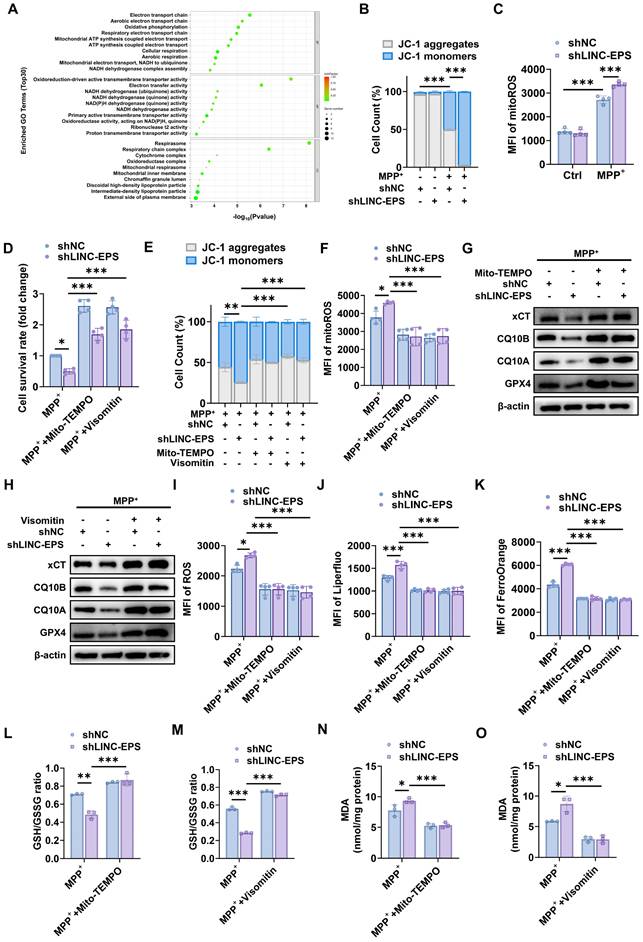
We investigated whether ROS suppression prevents ferroptosis. Immunoblotting analysis revealed that treatment with Mito-TEMPO (Figs. 6G and S4C) and Visomitin (Figs. 6H and S4D) restored the expression levels of the core ferroptotic defense proteins, xCT, CQ10B, CQ10A and GPX4, which were downregulated by MPP⁺ in shLINC-EPS cells. Both Mito-TEMPO and Visomitin significantly suppressed MPP⁺-induced elevation of total cellular ROS (Fig. 6I), lipid ROS (Fig. 6J), and Fe²⁺ (Fig. 6K) levels. Concurrently, treatment with Mito-TEMPO and Visomitin promoted the recovery of the GSH/GSSG ratio (Fig. 6L and M) and suppressed the accumulation of MDA, a key lipid peroxidation marker (Fig. 6N and O).
Collectively, these findings suggest that LINC-EPS deficiency drives the overproduction of mitochondria-derived ROS, a key proximal event to ferroptosis.
LINC-EPS sustains the PGC-1α signaling axis to preserve mitochondrial homeostasis and prevent ferroptosis
After establishing that LINC-EPS deficiency induces mitochondrial dysfunction, we aimed to identify the underlying molecular drivers. First, we screened the key mitochondrial regulators in primary neurons following MPP⁺-induced stress. This screening revealed significantly downregulated levels of PGC1A, which encodes the master transcriptional co-activator PGC-1α (Fig. 7A). As PGC-1α orchestrates mitochondrial biogenesis and antioxidant defense by regulating the activity of transcription factors, such as NRF1, NRF2, and TFAM, we hypothesized that LINC-EPS deficiency impairs the mitochondrial functions by disrupting the PGC-1α transcriptional program.
PGC-1α Restoration Rescues LINC-EPS-Deficient Cells from Mitochondrial Dysfunction and Ferroptosis under MPP⁺-Induced Stress. (A) Mitochondrial regulatory gene expression profile in primary neurons from WT and LINC-EPS⁻/⁻ mice following MPP⁺ exposure (n=3). (B) shNC or shLINC-EPS cells were treated ± MPP⁺ (500 µM, 24 h). Representative immunoblots and quantification of PGC-1α, NRF1, NRF2, and TFAM protein levels are shown. β-actin was used as a loading control (n = 3). (C) CCK-8 assay assessing cell viability in MPP⁺-treated cells after LINC-EPS knockdown, with or without concurrent PGC-1α overexpression (n = 4). (D) Western blot analysis of PGC-1α and its downstream targets in MPP+-treated cells with LINC-EPS knockdown and/or PGC-1α overexpression. All groups received MPP+ treatment (n = 3). (E) Flow cytometric assessment of MMP in MPP⁺-treated shLINC-EPS cells with or without oePGC-1α overexpression, detected using JC-1 dye (n = 4). (F) Flow cytometric quantification of mitoROS levels in MPP⁺-treated shLINC-EPS cells with or without PGC-1α overexpression, measured as the MFI using the MitoSOX Red (n = 4). (G) Mitochondrial ultrastructure by TEM (scale bar: 500 nm). (H) Quantification of total intracellular ROS levels in MPP⁺-treated shLINC-EPS cells with or without PGC-1α overexpression, detected using a general ROS detection probe (n = 4). (I) Quantification of lipid ROS levels in MPP⁺-treated shLINC-EPS cells with or without PGC-1α overexpression, measured using the Liperfluo (n = 4). (J) Quantification of Fe²⁺ levels in MPP⁺-treated shLINC-EPS cells with or without PGC-1α overexpression, assessed using FerroOrange staining (n = 4). (K) Quantification of MDA content as an indicator of lipid peroxidation in MPP⁺-treated shLINC-EPS cells with or without PGC-1α overexpression (n = 3). (L) Assessment of GSH/GSSG ratio in MPP⁺-treated shLINC-EPS cells with or without PGC-1α overexpression (n = 3). Data are presented as mean ± SEM. Statistical significance was determined using two-way ANOVA for (B, D, E) and one-way ANOVA for (C, F, H-L), with all analyses followed by Tukey's post-hoc test. *P < 0.05, **P < 0.01, ***P < 0.001.
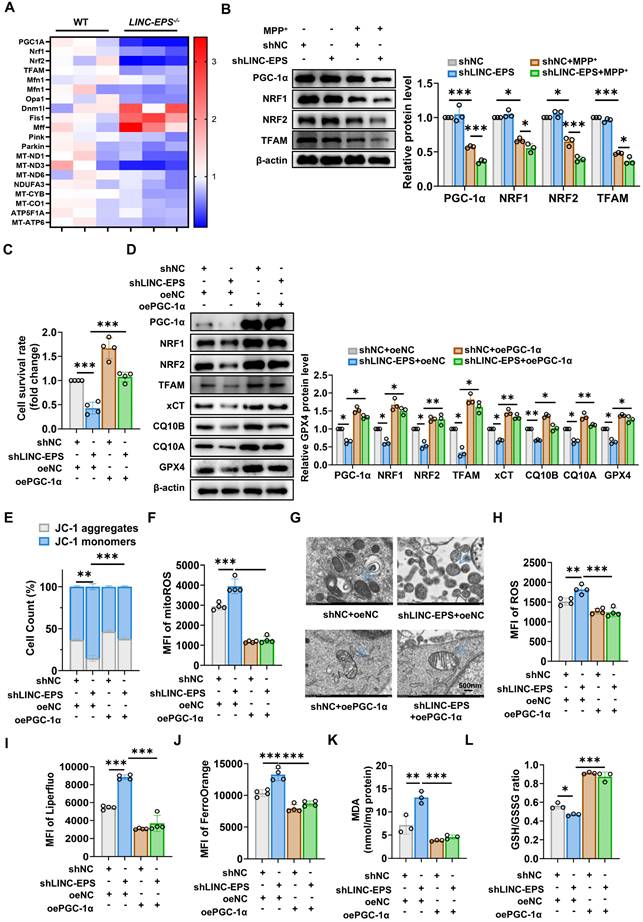
To verify this hypothesis, we first confirmed that LINC-EPS is essential to maintain the PGC-1α pathway. Challenged with MPP⁺, shLINC-EPS cells showed markedly lower protein levels of PGC-1α and its downstream transcriptional targets, NRF1, NRF2, and TFAM, than shNC cells (Fig. 7B) [45, 46]. In contrast to the exacerbation observed with LINC-EPS knockdown, MPP⁺-associated downregulation of the PGC-1α signaling pathway was alleviated in oeLINC-EPS cells (Fig. S5A).
To determine whether the detrimental phenotypes of LINC-EPS deficiency are attributed to impaired PGC-1α signaling, we performed a series of functional rescue experiments. Reduction of cell viability induced by LINC-EPS knockdown was substantially reversed by the ectopic overexpression of PGC-1α (oePGC-1α; Fig. 7C). Immunoblotting analysis confirmed that, under MPP⁺ challenge, this intervention not only restored PGC-1α levels, but also rescued the expression levels of its downstream targets (NRF1, NRF2, and TFAM) and core anti-ferroptotic proteins (xCT, CQ10B, CQ10A and GPX4; Fig. 7D). Moreover, PGC-1α overexpression effectively restored the MMP (Fig. 7E) and suppressed the excessive production of mitoROS (Fig. 7F) in MPP⁺-treated LINC-EPS knockdown cells. TEM provided key morphological evidence: Mitochondria in shLINC-EPS cells exhibited the classic hallmarks of ferroptosis, including reduced size, increased membrane density, and loss of cristae (Fig. 7G). These abnormalities were robustly reversed in the oePGC-1α rescue group.
Crucially, PGC-1α restoration was sufficient to prevent ferroptosis. This was evidenced by the significant reduction in total ROS (Fig. 7H), lipid ROS (Fig. 7I), Fe²⁺ (Fig. 7J), and lipid peroxidation product MDA (Fig. 7K) levels. Remarkably, this neuroprotective treatment not only reversed the oxidative imbalance but also elevated the GSH/GSSG ratio to a level significantly surpassing that in the controls (Fig. 7L). Consistent with this genetic rescue, pharmacological activation of PGC-1α with agonist ZLN005 also significantly mitigated MPP⁺-induced cell death in LINC-EPS-deficient neurons (Fig. S5B). ZLN005 treatment increased the protein levels of PGC-1α, its downstream targets, and ferroptosis defense proteins (Fig. S5C). This activation preserved MMP (Fig. S5D), decreased mitoROS levels (Fig. S5E), and attenuated all measured hallmarks of ferroptosis (Fig. S5F-I) and promoted the recovery of the GSH/GSSG ratio (Fig. S5J).
Collectively, these data suggest that LINC-EPS deficiency drives mitochondrial dysfunction and subsequent ferroptosis by dismantling the PGC-1α transcriptional axis.
LINC-EPS functions as a transcriptional co-activator by scaffolding PGC-1α to the PGC1A promoter
To elucidate the mechanism by which LINC-EPS regulates PGC1A expression, we assessed its potential role in post-transcriptional regulation. Alterations in LINC-EPS abundance in SH-SY5Y cells did not influence the degradation kinetics of PGC-1α protein, as determined by cycloheximide (CHX) chase assays under both shLINC-EPS and oeLINC-EPS conditions (Fig. 8A and B). Similarly, PGC1A mRNA stability, measured after actinomycin D (Act-D) treatment, remained unchanged following modulation of LINC-EPS levels (Fig. 8C and D). These observations suggest that LINC-EPS acts primarily at the transcriptional level to regulate PGC1A.
LINC-EPS Scaffolds PGC-1α for PGC1A Transcriptional Activation. (A-B) PGC-1α protein stability analysis following cycloheximide (CHX, 50 μg/mL) treatment in LINC-EPS knockdown (A) or overexpression (B) cells (n=3). (C-D) PGC1A mRNA decay kinetics following actinomycin D (5 μg/mL) treatment (n=3). (E) Proposed molecular mechanism: LINC-EPS recruits PGC-1α to the TBE regulatory element. (F) RNA-binding domain analysis of PGC-1α-LINC-EPS interaction (Pfam database). (G) ChIRP-Seq mapping of LINC-EPS occupancy at the PGC1A locus. (H) Molecular docking reveals hydrogen bonds between LINC-EPS (U780, G955) and PGC-1α (ASN344, SER8). (I) Colocalization of LINC-EPS (red) and PGC-1α (green) by FISH-immunofluorescence (scale bar: 5 μm). (J) RNA pull-down validation of LINC-EPS-PGC-1α interaction. (K) RIP-qPCR quantification of LINC-EPS enrichment by PGC-1α antibody (n=6). (L) LINC-EPS abundance-dependent PGC-1α binding (n=4). (M) Predicted RNA-DNA triplex formation between LINC-EPS and TBE element. (N) ChIRP-qPCR of LINC-EPS occupancy at TBE (n=4). (O) TBE-dependent luciferase reporter activation by LINC-EPS (n=3). (P) ChIP-qPCR demonstrating PGC-1α recruitment to PGC1A promoter (+1710-1725; CCGGCCCCCTGCCGT) modulated by LINC-EPS levels (n=4). Data are presented as mean ± SEM. Two-way ANOVA (A-D) or Student's t-test (K-P). **P<0.01, ***P<0.001.
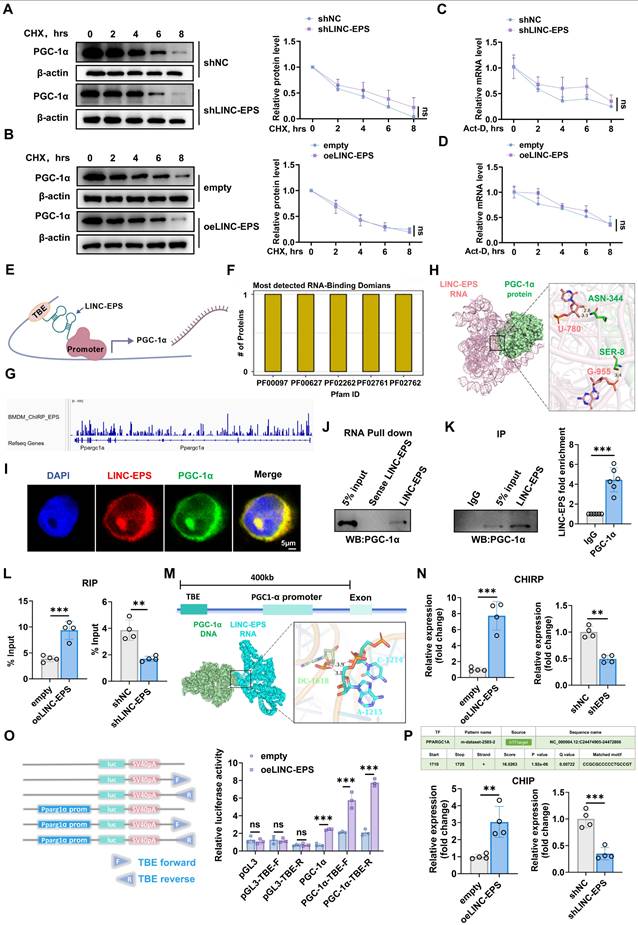
PGC-1α participates in a positive transcriptional feedback loop. LncRNA Tug1 facilitates PGC-1α protein binding to the PGC1A promoter via interaction with a T-box element (TBE) upstream of the gene. Based on this, we hypothesized that LINC-EPS functions as a scaffold to recruit PGC-1α protein to the TBE upstream of the PGC1A promoter, thereby enhancing its transcription (Fig. 8E, created with BioGDP.com) [47-49].
Bioinformatics analysis revealed several RNA-binding domains within the PGC-1α protein (Fig. 8F). To investigate whether LINC-EPS directly associates with the chromatin of its target gene, a publicly available Chromatin Isolation by RNA Purification sequencing (ChIRP-Seq) dataset (GSE115524, generated in mouse bone-marrow-derived macrophages) was analyzed [50]. The results demonstrated significant LINC-EPS enrichment at the PGC1A genomic locus (Fig. 8G), characterized by distinct peaks in both promoter-upstream and intragenic regions. The upstream peaks were consistent with potential cis-regulatory element interactions, whereas the intronic enrichment suggested involvement in chromatin-associated transcriptional modulation. The genomic distribution of these peaks was concordant with RefSeq annotation, supporting a cis-acting mechanism by which LINC-EPS may influence PGC-1α transcription. Molecular docking predicted direct RNA-protein contact, including hydrogen bonds between U-780 of LINC-EPS and ASN-344 of PGC-1α and between G-955 of LINC-EPS and SER-8 of PGC-1α (Fig. 8H). These interactions were supported by experimental evidence: RNA FISH for LINC-EPS and immunofluorescence assay for PGC-1α revealed nuclear co-localization in SH-SY5Y cells (Fig. 8I). RNA pull-down assays using biotinylated LINC-EPS transcripts specifically precipitated endogenous PGC-1α protein from nuclear lysates, whereas antisense controls showed no binding (Fig. 8J). RNA immunoprecipitation (RIP) with anti-PGC-1α antibody yielded significant enrichment of LINC-EPS compared to the IgG controls, with enrichment increasing in oeLINC-EPS cells and decreasing in shLINC-EPS cells (Fig. 8K and L).
We further evaluated whether LINC-EPS recruits PGC-1α to the PGC1A promoter. Upstream TBE, previously linked to PGC1A autoregulation, was predicted to form an RNA-DNA triplex with LINC-EPS (Fig. 8M). ChIRP-qPCR analysis revealed the enrichment of LINC-EPS at a genomic region containing the TBE located upstream of the PGC1A promoter, which was enhanced by LINC-EPS overexpression and reduced by its knockdown (Fig. 8N). Luciferase reporter assays showed that co-transfection with LINC-EPS increased the promoter activity of constructs containing the full PGC1A promoter or minimal promoter with TBE, but not of the TBE-deleted constructs, indicating a TBE-dependent effect (Fig. 8O). Bioinformatics analysis using hTFtarget identified a high-confidence transcription factor binding element for PGC-1α within the PGC1A promoter region (chr4: 24474905-24472806), positioned at nucleotides +1710 to +1725 relative to the transcription start site, with a matched core sequence of CCGGCCCCCTGCCGT. ChIP-qPCR further demonstrated that PGC-1α occupancy at the PGC1A promoter was upregulated in oeLINC-EPS cells and downregulated in shLINC-EPS cells (Fig. 8P) [51].
Together, these results support a model in which LINC-EPS serves as a molecular scaffold, recruiting PGC-1α protein to a specific TBE upstream of the PGC1A promoter, thereby amplifying a positive transcriptional feedback loop regulating PGC1A expression.
Pharmacological activation of PGC-1α ameliorates MPTP-induced neurotoxicity in LINC-EPS-deficient mice
To determine whether pharmacological activation of PGC-1α rescues the heightened pathological sensitivity caused by LINC-EPS deficiency, we administered the PGC-1α agonist ZLN005 to MPTP-treated LINC-EPSΔDat mice. ZLN005 treatment robustly ameliorated the severe motor deficits in LINC-EPSΔDat mice [25, 26]. In the open-field test, this was evidenced by the increased total distance traveled and average speed, along with reduced immobility time (Fig. 9A-D). Similarly, ZLN005 administration extended the latency to fall in the rotarod test (Fig. 9E) and shortened the descent time in the pole test (Fig. 9F). Although this intervention did not fully restore the performance of LINC-EPSΔDat mice to the level of their ZLN005-treated LINC-EPSf/f counterparts, it markedly closed the deficit gap.
Pharmacological PGC-1α Activation Protects Against Neurodegeneration in LINC-EPS Knockout PD Model Mice. Following MPTP intoxication (30 mg/kg, i.p., daily for 5 days), mice of each genotype (LINC-EPSf/f and LINC-EPSΔDat) were subsequently randomly assigned to treatment groups receiving daily administrations of either saline or the PGC-1α agonist ZLN005 (10 mg/kg, i.p.) for a duration of 14 days. Behavioral and pathological analyses were performed 7 days after the last MPTP injection. (A-D) Open field test. Representative locomotor traces (A) and quantification of total distance traveled (B), average speed (C), and immobility time (D) (n = 10). (E) Rotarod test, showing latency to fall (n = 10). (F) Pole test, showing time to descend (n = 10). (G) Representative immunofluorescence images showing TH⁺ neurons (red) in the SNpc, with corresponding stereological quantification on the right. Nuclei were counterstained with DAPI (blue). Scale bar, 200 µm (n = 6). (H) Representative immunohistochemistry images of TH-positive fibers in the striatum, including low-magnification overviews and corresponding high-magnification views. The right panel displays the quantification of TH-immunoreactive fiber density. Scale bars, 600 µm (overview) and 50 µm (magnified view) (n = 6). (I) Representative immunoblots of TH, PGC-1α pathway proteins, and ferroptosis-related proteins in midbrain lysates. β-actin was used as a loading control. Quantification is shown in Figure S6. (J) Representative immunofluorescence images and quantification of GPX4 (red) and TH (green) co-staining in the SNpc. DAPI (blue) stains nuclei (n = 6). Scale bar, 20 µm. (K) Quantification of MDA levels in midbrain tissue (n = 3). (L) Quantification of the GSH/GSSG ratio in midbrain tissue (n = 3). Data are presented as mean ± SEM. Statistical significance was determined by one-way ANOVA with Tukey's post-hoc test. *P < 0.05, **P < 0.01, ***P < 0.001.
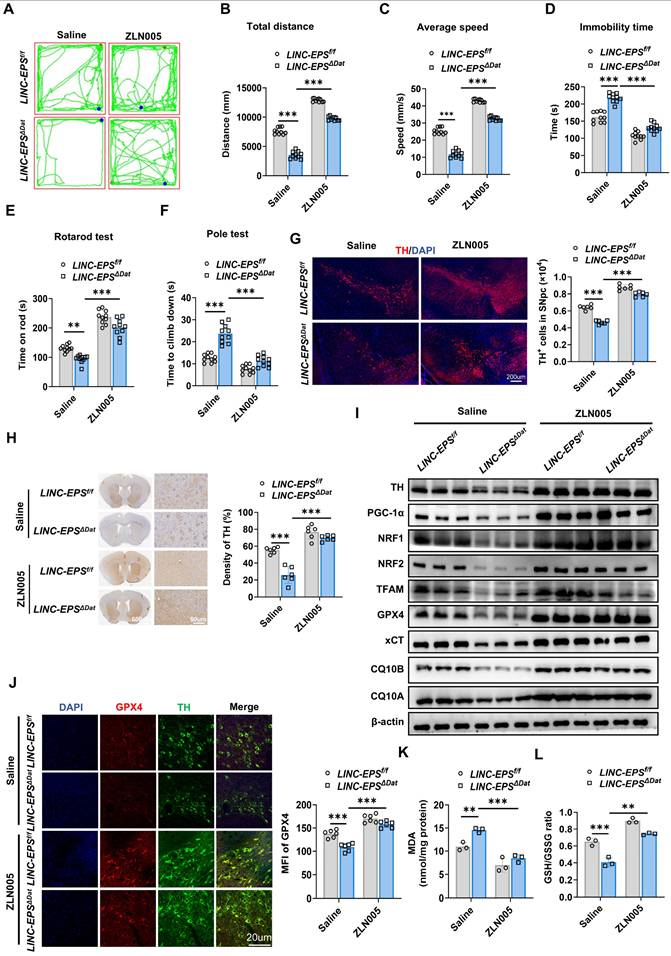
This functional recovery was accompanied by significant neuroprotective effects. ZLN005 treatment markedly attenuated the loss of TH-positive neurons in SNpc (Fig. 9G) and reduction of TH-positive fiber density in the striatum (Fig. 9H) of LINC-EPSΔDat mice. At the molecular level, ZLN005 administration increased the expression levels of TH, PGC-1α, its downstream targets (NRF1, NRF2, and TFAM), and key anti-ferroptotic proteins, including xCT, CQ10B, CQ10A and GPX4, in LINC-EPSΔDat mice (Figs. 9I and S6). Furthermore, ZLN005 treatment directly counteracted the molecular signature of ferroptosis. This reversed the decrease in GPX4 levels in the surviving TH-positive neurons (Fig. 9J), suppressed the accumulation of lipid peroxidation product MDA (Fig. 9K), and ameliorated the decrease in the GSH/GSSG ratio (Fig. 9L).
Collectively, these in vivo data suggest that pharmacological activation of PGC-1α is associated with the rescue of motor function, protection of DA neurons, restoration of the PGC-1α signaling axis and anti-ferroptosis markers in genetically-sensitized PD model mice.
Discussion
This study identified a previously unrecognized neuroprotective function of LINC-EPS in PD via suppression of ferroptosis in DA neurons. LINC-EPS served as a transcriptional co-activator of PGC-1α by directly binding to a TBE upstream of its promoter region, thereby enhancing PGC-1α expression. This molecular interaction revealed a LINC-EPS/PGC-1α axis that maintained mitochondrial homeostasis, attenuated mitoROS production, and conferred resistance to ferroptotic cell death. Both LINC-EPS overexpression and pharmacological activation of PGC-1α significantly ameliorated motor deficits and preserved DA neuronal integrity in experimental PD models [52-54]. Our findings reveal a novel LINC-EPS/PGC-1α neuroprotective axis and identify LINC-EPS as a potential therapeutic target for PD.
LINC-EPS, originally characterized as a regulator of erythroid differentiation and immune responses [55-57], exhibits broad tissue expression including substantial levels in human brain (GTEx database), suggesting neurological functions. In this study, we identified LINC-EPS as a factor implicated in the pathogenesis of PD, demonstrating its consistent downregulation across multiple disease models and patient samples. LINC-EPS expression was decreased in the SN and striatum of MPTP-intoxicated mice and also reduced in peripheral blood samples of patients with PD. Importantly, peripheral LINC-EPS levels correlated inversely with UPDRS III motor scores, linking lower expression to more severe motor impairment. In vivo RNA in situ hybridization revealed LINC-EPS downregulation across DA neurons, microglia, and astrocytes in MPTP-treated mouse SN, and in vitro studies revealed more pronounced suppression in DA neurons, suggesting neuron-specific susceptibility. Our functional validation using both global and conditional knockout strategies, combined with viral-mediated rescue experiments, established LINC-EPS as critical for DA neuron survival. Both global and neuron-specific LINC-EPS-deficient mice demonstrated cell-autonomous neuroprotective functions, with AAV-mediated LINC-EPS restoration reversing motor deficits and neurodegeneration.
Converging pathological and biochemical evidence, including prominent iron accumulation and lipid peroxidation in the SN of patients with PD, implicates ferroptosis as a contributor to DA neuronal loss [12-14]. Cellular defense against ferroptosis relies primarily on the glutathione/GPX4 axis and the coenzyme Q system [15-19]. In our models, LINC-EPS deficiency broadly dysregulated ferroptosis-related genes and resulted in a net reduction of anti-ferroptotic factors, including mitochondrial CoQ pathway genes (e.g., CQ10A/CQ10B) and core defenses such as xCT and GPX4. To address iron directly, we quantified intracellular labile Fe²⁺ using FerroOrange and flow cytometric MFI, providing a quantitative readout of bioavailable iron relevant to ferroptotic chemistry. Together with increased lipid peroxidation and GSH depletion, these findings support a coordinated collapse of anti-ferroptotic defenses under LINC-EPS loss. Furthermore, our datasets indicate that LINC-EPS deficiency preferentially engages ferroptosis, with limited changes in canonical markers of apoptosis, necroptosis, and pyroptosis. This pathway preference is plausible for DA neurons, which combine high basal oxidative burden from dopamine metabolism, high iron content in the nigrostriatal system, and PUFA-enriched membranes that are susceptible to lipid peroxidation.
Ferroptosis and mitochondrial dysfunction are tightly coupled in our PD-relevant stress context. MPP⁺-driven Complex I inhibition increases mitoROS and perturbs cellular redox balance, thereby facilitating iron-catalyzed lipid peroxidation and weakening GPX4- and CoQ10-dependent antioxidant defenses. In turn, lipid peroxidation damages mitochondrial membranes and impairs bioenergetic function, amplifying ROS production and establishing a feed-forward vicious cycle. Accordingly, ferroptosis inhibitors (e.g., ferrostatin-1 and liproxstatin-1) are expected to confer neuroprotection in part by preserving mitochondrial integrity: by arresting lipid peroxide propagation within mitochondrial membranes, these agents may limit membrane damage, maintain mitochondrial membrane potential, and prevent secondary mitochondrial collapse, thereby breaking the vicious cycle of oxidative stress and bioenergetic failure [8, 37, 57].
Our findings demonstrate that LINC-EPS functions as a molecular scaffold, facilitating a positive transcriptional feedback loop by recruiting PGC-1α to its own promoter. This mechanism sustains a transcriptional program that bolsters mitochondrial antioxidant defenses, including the glutathione/GPX4 system and coenzyme Q10 biosynthesis, thereby suppressing lipid peroxidation and conferring resistance to ferroptotic cell death. The observed downregulation of LINC-EPS in both preclinical models and PD patients suggests that disruption of this protective axis contributes to progressive neurodegeneration. Our work delineates a specific upstream regulatory cascade that controls mitochondrial dysfunction and ferroptotic execution.
Our analysis revealed a significant negative correlation between peripheral PBMC LINC-EPS levels and UPDRS-III motor scores in PD patients, indicating lower LINC-EPS expression in blood associates with greater motor impairment. From a translational perspective, peripheral blood LINC-EPS may offer several clinical utilities, including a supplementary diagnostic biomarker, a progression indicator for longitudinal monitoring, and a potential pharmacodynamic marker to assess therapeutic responses. However, several limitations warrant cautious interpretation. First, age and sex were not matched between PD patients and controls at enrollment in this study. Second, most PD patients were receiving dopaminergic therapy, which may alter PBMC genes expression independently of PD pathology. Third, the cohort size (30 PD and 30 controls) limited covariate-adjusted analyses to control for confounders such as disease duration, medication exposure, systemic inflammation, and comorbidities. Fourth, a direct link between peripheral LINC-EPS levels and nigral pathology in living patients remains unvalidated because matched brain tissue is not available. Future studies are warranted to utilize neuroimaging (e.g., DAT-SPECT or neuromelanin-sensitive MRI) as surrogate markers for nigral pathology to definitively establish this blood-brain correlation in humans. Finally, public transcriptomic resources suggest that LINC-EPS dysregulation may also occur in other neurodegenerative disorders (e.g., Alzheimer's disease and amyotrophic lateral sclerosis), underscoring the need for larger, well-matched, multicenter validation to define disease specificity and clinical utility [21-23].
Although we demonstrated efficacy of LINC-EPS overexpression in alleviating PD pathology, clinical translation of lncRNA-based therapies faces significant hurdles, particularly concerning stability and blood-brain barrier delivery. Consequently, pharmacological activation of the downstream PGC-1α axis currently offers a more accessible therapeutic avenue [24-26]. Furthermore, MPTP/MPP⁺ models, though widely used, exhibit important limitations: they do not fully recapitulate the chronic, progressive nature of idiopathic PD; they exhibit incomplete α-synuclein pathology, lacking widespread Lewy body formation; and they predominantly capture motor deficits with inadequate modeling of non-motor symptoms such as cognitive decline and autonomic dysfunction. These constraints necessitate cautious extrapolation of findings to human PD [56-58].
Critical mechanistic gaps remain regarding how MPTP/MPP⁺ triggers LINC-EPS downregulation. MPP⁺ accumulates in mitochondria via dopamine transporter (DAT) uptake and inhibits Complex I, generating excessive ROS and impairing ATP production. This mitochondrial dysfunction and oxidative stress may activate stress-responsive transcription factors (e.g., NF-κB, p53) that repress LINC-EPS transcription or recruit epigenetic modifiers (e.g., DNA methyltransferases, histone deacetylases) to silence the LINC-EPS locus. Additionally, dopamine metabolism produces reactive quinones and hydrogen peroxide, triggering ferroptosis-priming conditions that may feedback to suppress LINC-EPS expression as a maladaptive response. MPTP-induced neuroinflammation, characterized by microglial activation and the release of pro-inflammatory cytokines (TNF-α, IL-1β, IL-6), potentially modulates LINC-EPS expression via inflammation-driven transcriptional reprogramming. These hypotheses regarding the regulatory mechanisms of LINC-EPS warrant further investigation.
Conclusion
Our study establishes the LINC-EPS/PGC-1α axis as a critical regulator of dopaminergic neuron survival by controlling susceptibility to ferroptosis. The disruption of this axis contributes to neurodegeneration in Parkinson's disease. Pharmacological potentiation of this pathway reverses neurodegenerative phenotypes in preclinical models, identifying it as a promising and mechanistically defined target for developing disease-modifying therapies for Parkinson's disease.
Supplementary Material
Supplementary figures and tables.
Acknowledgements
We are grateful to Dr. Feng Ma (CAMS Key Laboratory of Synthetic Biology Regulatory Elements, Institute of Systems Medicine, Chinese Academy of Medical Sciences & Peking Union Medical College, Beijing, China) for the generous gift of LINC-EPS knockout (LINC-EPS⁻/⁻) mice. We also thank Editage (www.editage.cn) for English language editing.
Funding
This study was supported by the National Natural Science Foundation of China (81771689, 81373130, and 81001308), the Undergraduate Science and Technology Innovation Fund of Yangzhou University (Grant No. XCX20240831), and the Jiangsu Province College Students' Innovation and Entrepreneurship Training Program (Grant No. 202411117253Y).
Author contributions
ZL, WL, WG and LQ designed and conceptualized the study. ZL wrote the original manuscript draft. WL, WG and LQ revised and edited the manuscript. RW and MJ analyzed the data. RL contributed to data visualization. ZL, RW, XL, MS and JL performed the investigations. JZ developed the software. SW performed validation. QW and MS provided resources. XX administered the project. WL, WG and LQ supervised the study and acquired funding. All authors contributed to this manuscript. All authors had full access to the data and approved the final version of the manuscript.
Availability of data and materials
The datasets supporting the conclusions of this article are included within the article and its additional files.
Ethics approval and consent to participate
Human studies
The study protocol, which involved the collection of peripheral blood samples from individuals with Parkinson's disease (PD) and healthy controls, was reviewed and approved by The Ethics Committee of Yangzhou University School of Medicine (Approval No. YXYLL-2025-106). The study was conducted in strict accordance with the ethical principles of the Declaration of Helsinki. Prior to inclusion, all participants provided written informed consent, and all patient data were anonymized to protect privacy.
Animal studies
All animal experiments were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Yangzhou University (Protocol No. 202503184) and were performed in strict accordance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health (NIH). The study design and reporting adhere to the Animal Research: Reporting of In vivo Experiments (ARRIVE) guidelines. All surgical procedures were performed under appropriate anesthesia, and all efforts were made to minimize animal suffering. Methods for euthanasia were consistent with the American Veterinary Medical Association (AVMA) Guidelines for the Euthanasia of Animals (2020), and no unacceptable methods such as chloral hydrate or ether were used. Only male C57BL/6J mice were used to avoid the confounding effects of hormonal fluctuations from the estrous cycle and to maintain consistency with the established MPTP literature.
Competing Interests
The authors declare no competing financial interests.
References
1. Dong-Chen X, Yong C, Yang X. et al. Signaling pathways in Parkinson's disease: molecular mechanisms and therapeutic interventions. Signal Transduct Target Ther. 2023;8:1
2. Chen X, Zhang G, Liu M. et al. New perspectives on molecular mechanisms underlying exercise-induced benefits in Parkinson's disease. npj Parkinsons Dis. 2025;11:256
3. Alexander GE. Biology of Parkinson's disease: pathogenesis and pathophysiology of a multisystem neurodegenerative disorder. Dialogues Clin Neurosci. 2004;6:259-280
4. Gu Y, Zhao X, Zhang N. et al. Mitochondrial dysfunction as a therapeutic strategy for neurodegenerative diseases: current insights and future directions. Ageing Res Rev. 2024;102:102577
5. Brooker SM, Naylor GE, Krainc D. Cell biology of Parkinson's disease: mechanisms of synaptic, lysosomal, and mitochondrial dysfunction. Curr Opin Neurobiol. 2024;85:102841
6. Chithra Y, Dey G, Ghose V. et al. Mitochondrial Complex I Inhibition in Dopaminergic Neurons Causes Altered Protein Profile and Protein Oxidation: Implications for Parkinson's disease. Neurochem Res. 2023;48:2360-2389
7. Mansour HM, Mohamed AF, Khattab MM. et al. Heat Shock Protein 90 in Parkinson's Disease: Profile of a Serial Killer. Neuroscience. 2024;537:32-46
8. Wang W, Thomas ER, Xiao R. et al. Targeting mitochondria-regulated ferroptosis: A new frontier in Parkinson's disease therapy. Neuropharmacology. 2025;274:110439
9. He Y, Wang W, Yang T. et al. The Potential Role of Voltage-Dependent Anion Channel in the Treatment of Parkinson's Disease. Oxid Med Cell Longev. 2022;2022:4665530
10. Liu J, Wang X, Lu Y. et al. Pink1 interacts with α-synuclein and abrogates α-synuclein-induced neurotoxicity by activating autophagy. Cell Death Dis. 2017;8:e3056
11. Chong ZZ, Souayah N. Oxidative stress: pathological driver in chronic neurodegenerative diseases. Antioxidants. 2025;14:696
12. Ding X, Gao L, Han Z. et al. Ferroptosis in Parkinson's disease: molecular mechanisms and therapeutic potential. Ageing Res Rev. 2023;91:102077
13. Yang K, Zeng L, Zeng J. et al. Research progress in the molecular mechanism of ferroptosis in Parkinson's disease and regulation by natural plant products. Ageing Res Rev. 2023;91:102063
14. Alrouji M, Anwar S, Venkatesan K. et al. Iron homeostasis and neurodegeneration in the ageing brain: insight into ferroptosis pathways. Ageing Res Rev. 2024;102:102575
15. Zhang Y, Dou J, Dai S. et al. Human umbilical cord mesenchymal stem cells ameliorate liver fibrosis by inhibiting hepatocyte ferroptosis and macrophage polarization via the miR-455-3p/PLAU axis. Stem Cell Res Ther. 2025;16(1):412
16. Wang D, Pan Y, Chen W. et al. Nanodrugs targeting key factors of ferroptosis regulation for enhanced treatment of osteoarthritis. Adv Sci. 2025;12:e2412817
17. Huang W, Tian Y, Ma J. et al. Neutrophil membrane-based biomimetic metal-polyphenol self-assembled nanozyme for the targeting treatment of early brain injury following subarachnoid hemorrhage. Chem Eng J. 2024;498:155643
18. Li T, Liu J, Huang X. et al. Deficiency of PLA2G6 Induces Ferroptosis in Dopaminergic Neurons and Contributes to Parkinson's Disease Pathogenesis via Disruption of PRDX6/FTH1/GPX4 Axis. Mol Neurobiol. 2025;62:15916-15927
19. Mishima E, Nakamura T, Doll S. et al. Recommendations for robust and reproducible research on ferroptosis. Nat Rev Mol Cell Biol. 2025;26:615-630
20. Zhu Y, Wang D, Du C. et al. Ruthenium Single-Atom Nanozyme Driven Sonosensitizer with Oxygen Vacancies Enhances Electron-Hole Separation Efficacy and Remodels Tumor Microenvironment for Sonodynamic-Amplified Ferroptosis. Adv Sci. 2025;12:e2416997
21. Ilieva MS. Non-coding RNAs in neurological and neuropsychiatric disorders: unraveling the hidden players in disease pathogenesis. Cells. 2024;13:1063
22. Srinivas T, Mathias C, Oliveira-Mateos C. et al. Roles of lncRNAs in brain development and pathogenesis: Emerging therapeutic opportunities. Mol Ther. 2023;31:1550-1561
23. Li YH, Zhang S, Tang L. et al. The role of LincRNA-EPS/Sirt1/autophagy pathway in the neuroprotection process by hydrogen against OGD/R-induced hippocampal HT22 cells injury. J Pers Med. 2023;13:631
24. Huang X, Pan L, Zuo Z. et al. LINC00842 inactivates transcription co-regulator PGC-1α to promote pancreatic cancer malignancy through metabolic remodelling. Nat Commun. 2021;12:3830
25. Kumar AP, Puthussery DT. Regulation of PPAR-γ coactivator-1α and its implication in mitochondrial function and neurodegenerative diseases. Ageing Res Rev. 2025;102:102887
26. Huang J, Li Y, Ye Q. et al. PGC-1α-mediated mitochondrial homeostasis imbalance aggravated gingival lesions of diabetic periodontitis by disrupting ECM remodeling of human gingival fibroblast. Redox Biol. 2025;103:103829
27. Iannielli A, Luoni M, Giannelli SG. et al. Modeling native and seeded synuclein aggregation and related cellular dysfunctions in dopaminergic neurons derived by a new set of isogenic iPSC lines with SNCA multiplications. Cell Death Dis. 2022;13:881
28. Kakae M, Nakajima H, Tobori S. et al. The astrocytic TRPA1 channel mediates an intrinsic protective response to vascular cognitive impairment via LIF production. Sci Adv. 2023;9:eadh0102
29. Yi F, He X, Wang D. Lycopene protects against MPP+-induced cytotoxicity by maintaining mitochondrial function in SH-SY5Y cells. Neurochem Res. 2013;38:1747-1757
30. Zhong J, Yu H, Huang C. et al. Inhibition of phosphodiesterase 4 by FCPR16 protects SH-SY5Y cells against MPP+-induced decline of mitochondrial membrane potential and oxidative stress. Redox Biol. 2018;16:47-58
31. Hassani S, Esmaeili A. The neuroprotective effects of ferulic acid in toxin-induced models of Parkinson's disease: A review. Ageing Res Rev. 2024;97:102299
32. Yan Y, Tao H, He J. et al. The HDOCK server for integrated protein-protein docking. Nat Protoc. 2020;15:1829-1852
33. Yan Y, Zhang D, Zhou P. et al. HDOCK: a web server for protein-protein and protein-DNA/RNA docking based on a hybrid strategy. Nucleic Acids Res. 2017;45:W365-W373
34. Aguet F, Anand S, Ardlie KG. et al. The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science. 2020;369:1318-1330
35. Chen S, Zhu J, Sun LQ. et al. LincRNA-EPS alleviates severe acute pancreatitis by suppressing HMGB1-triggered inflammation in pancreatic macrophages. Immunology. 2021;163:201-219
36. McKinnon C, De Snoo ML, Gondard E. et al. Early-onset impairment of the ubiquitin-proteasome system in dopaminergic neurons caused by α-synuclein. Acta Neuropathol Commun. 2020;8:17
37. Chen Y, Luo X, Yin Y. et al. The interplay of iron, oxidative stress, and α-synuclein in Parkinson's disease progression. Mol Med. 2025;31:154
38. Liu J, Lu X, Zeng S. et al. ATF3-CBS signaling axis coordinates ferroptosis and tumorigenesis in colorectal cancer. Redox Biol. 2024;71:103118
39. Wu T, Wei P, Zhao P. et al. Engineering local coordination environment of manganese single-atom enzyme for amplified infected wound therapy. Chem Eng J. 2025;514:163247
40. Zhu Y, Ding C, Fang W. et al. Metal-polyphenol self-assembled nanodots for NIR-II fluorescence imaging-guided chemodynamic/photodynamic therapy-amplified ferroptosis. Acta Biomater. 2024;185:361-370
41. López-Aguirre M, Balzano T, Monje MHG. et al. Nigrostriatal iron accumulation in the progression of Parkinson's disease. npj Parkinsons Dis. 2025;11:72
42. Salcedo-Arellano MJ, Wang JY, McLennan YA. et al. Cerebral Microbleeds in Fragile X-Associated Tremor/Ataxia Syndrome. Mov Disord. 2021;36:1935-1943
43. Ying Y, Cai X, Dai P. et al. Neurological Emergency Treatment Strategy: A Neuron-Targeted Regulation System for Reactive Oxygen Species Metabolism through Ferroptosis Modulation. ACS Nano. 2025;19:8753-8772
44. Wei YY, Chen TT, Zhang DW. et al. Microplastics exacerbate ferroptosis via mitochondrial reactive oxygen species-mediated autophagy in chronic obstructive pulmonary disease. Autophagy. 2025;21(8):1717-1743
45. Wan S, Cui Z, Wu L. et al. Ginsenoside Rd promotes omentin secretion in adipose through TBK1-AMPK to improve mitochondrial biogenesis via WNT5A/Ca2+ pathways in heart failure. Redox Biol. 2023;60:102610
46. Pizcueta P, Vergara C, Emanuele M. et al. Development of PPARγ agonists for the treatment of neuroinflammatory and neurodegenerative diseases: leriglitazone as a promising candidate. Int J Mol Sci. 2023;24:3201
47. Jiang S, Li H, Zhang L. et al. Generic Diagramming Platform (GDP): a comprehensive database of high-quality biomedical graphics. Nucleic Acids Res. 2025;53:D1670-D1676
48. Zhu Y, Yang B, Chen S. et al. M6A RNA Methylation-Mediated TUG1 Stability Maintains Mitochondrial Homeostasis during Kidney Aging by Epigenetically Regulating PGC1-α Expression. Antioxid Redox Signal. 2024;41:993-1013
49. Long J, Badal SS, Ye Z. et al. Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. J Clin Invest. 2016;126:4205-4218
50. Mumbach MR, Granja JM, Flynn RA. et al. HiChIRP reveals RNA-associated chromosome conformation. Nat Methods. 2019;16:489-492
51. Rakshe PS, Dutta BJ, Chib S. et al. Unveiling the interplay of AMPK/SIRT1/PGC-1α axis in brain health: promising targets against aging and NDDs. Ageing Res Rev. 2024;96:102255
52. Modi HR, Musyaju S, Ratcliffe M. et al. Mitochondria-targeted antioxidant therapeutics for traumatic brain injury. Antioxidants. 2024;13:303
53. Panes JD, Wendt A, Ramirez-Molina O. et al. Deciphering the role of PGC-1α in neurological disorders: from mitochondrial dysfunction to synaptic failure. Neural Regen Res. 2022;17:237-245
54. Atianand MK, Hu W, Satpathy AT. et al. A long noncoding RNA lincRNA-EPS acts as a transcriptional brake to restrain inflammation. Cell. 2016;165:1672-1685
55. Paralkar VR, Weiss MJ. A new 'Linc' between noncoding RNAs and blood development. Genes Dev. 2011;25:2555-2558
56. Zhu J, Chen S, Sun LQ. et al. LincRNA-EPS impairs host antiviral immunity by antagonizing viral RNA-PKR interaction. EMBO Rep. 2022;23:e53937
57. Wang Y, Xu E, Musich PR. et al. Mitochondrial dysfunction in neurodegenerative diseases and the potential countermeasure. CNS Neurosci Ther. 2019;25:816-824
58. Yousaf S, Arshad M, Raza M. et al. Targeting mitochondrial damage and ER stress to inhibit ferroptosis in cadmium-induced nephrotoxicity. Toxicol Rep. 2025;15:102082
Author contact
![]() Corresponding authors: Li Qian, Ph.D., M.D., Faculty of Medicine, Yangzhou University, Yangzhou, 225001, Jiangsu, PR China; email: qianledu.cn; Weijuan Gong, Ph.D., M.D., Faculty of Medicine, Yangzhou University, Yangzhou, 225001, Jiangsu, PR China; email: wjgongedu.cn; Wei Li, Ph.D., Kunshan Hospital of Chinese Medicine, Affiliated Hospital of Yangzhou University, Kunshan, 215300, Jiangsu, China; email: liwei25edu.cn.
Corresponding authors: Li Qian, Ph.D., M.D., Faculty of Medicine, Yangzhou University, Yangzhou, 225001, Jiangsu, PR China; email: qianledu.cn; Weijuan Gong, Ph.D., M.D., Faculty of Medicine, Yangzhou University, Yangzhou, 225001, Jiangsu, PR China; email: wjgongedu.cn; Wei Li, Ph.D., Kunshan Hospital of Chinese Medicine, Affiliated Hospital of Yangzhou University, Kunshan, 215300, Jiangsu, China; email: liwei25edu.cn.