Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Subtypes and structure of UCPs
Biological functions of UCPs in...
Research progress on UCPs in...
Concluding remarks and prospects
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2026; 22(7):3522-3543. doi:10.7150/ijbs.129896 This issue Cite
Review
Deciphering Uncoupling Proteins in Cellular Homeostasis and Metabolic Health
Yu-Ting Liu1,2, Ya-Jia Ding1,2, Hong-Liang Qiu1,2, Yan Che1,2, Qi-Zhu Tang1,2 ![]()
1. Department of Cardiology, Renmin Hospital of Wuhan University, Wuhan 430060, RP China.
2. Hubei Key Laboratory of Metabolic and Chronic Diseases, Wuhan 430060, RP China.
Received 2025-12-13; Accepted 2026-2-23; Published 2026-3-17
Abstract

Uncoupling proteins (UCPs) are transmembrane proteins located in the inner membrane of mitochondria. They can reduce the efficiency of ATP synthesis and promote heat production by mediating the uncoupling oxidative phosphorylation process. Different subtypes of UCPs have distinct tissue distributions and functional characteristics, involving various biological processes including temperature regulation, energy balance, signal transduction, oxidative stress, and the inflammatory response. In recent years, many studies have shown the potential value of UCPs in the prevention and treatment of metabolic diseases such as obesity, diabetes, cardiovascular diseases, neurological diseases, and tumors. Importantly, multiple evidence reveals that innovative therapies targeting uncoupling proteins will show broad application prospects in the future. This review of recent research on UCPs aims to provide a direction for exploring the molecular mechanisms of cellular homeostasis and intervention strategies for metabolic diseases.
Keywords: Uncoupling proteins, Thermoregulation, Cellular homeostasis, Metabolic health, Cardiovascular disease, Tumor
Introduction
Uncoupling proteins (UCPs) are a class of transmembrane proteins located in the inner mitochondrial membrane and belong to the mitochondrial carrier protein family [1]. Previous studies have shown that electron transfer in the respiratory chain drives protons to be pumped out of the mitochondrial matrix into the membrane space, thereby forming a proton concentration gradient. Protons flow back to the matrix through ATP synthase to drive the generation of ATP in the mitochondrial oxidative phosphorylation process [2, 3]. UCPs form proton leak channels, which prevent the proton motive force (PMF) in the oxidative phosphorylation process from longer being completely used solely for ATP synthesis, but instead allowing some of the PMF to be released in the form of heat [4]. Multiple subtypes of UCPs have different localization distributions and structural functions, and are found mainly in brown adipose tissue (BAT), white adipose tissue (WAT), skeletal muscle, myocardium and the brain [5]. UCP1 is the main uncoupling protein, and found mainly in BAT, it is responsible for nonshivering thermogenesis, and is highly important for regulating energy metabolism and maintaining body temperature [6]. UCP2 and UCP3 are related to obesity, fatty acid metabolism and blood sugar regulation, while UCP4 and UCP5 are distributed mainly in brain nerve tissue and are closely related to the pathogenesis of neurodegenerative diseases [7] (Fig. 1).
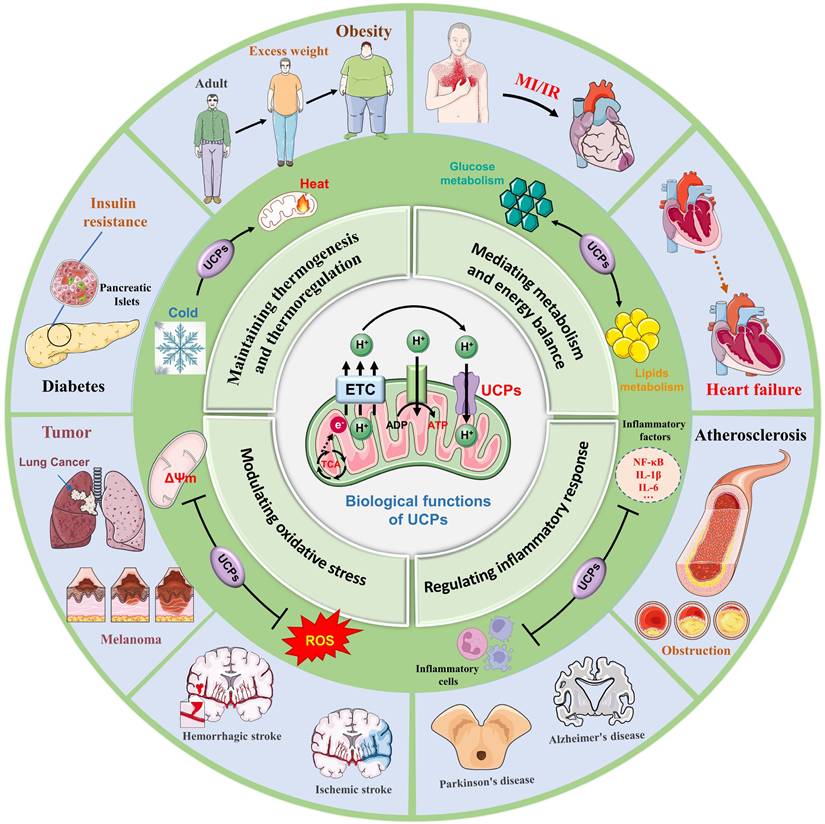
Biological processes and disease crosstalk in UCPs. UCPs are a family of transmembrane proteins located on the inner membrane of mitochondria. They are widely present in almost all cell types and play a key role in the proton leak channel transmitted by the mitochondrial TCA cycle. UCPs are widely involved in various biological processes, including (1) Maintaining thermogenesis and thermoregulation, (2) Mediating metabolism and energy balance, (3) Modulating oxidative stress, and (4) Regulating inflammatory response. Furthermore, UCPs serve a significant part in obesity and diabetes, cardiovascular disease, neurological illnesses, and tumor growth, making them an important target for disease preventive and therapy efforts. TCA, tricarboxylic acid cycle; ADP, adenosine diphosphate; ROS, reactive oxygen species; MI, myocardial infarction; IR, ischemia-reperfusion, NF-kB, nuclear factor-κB; IL-1β, nterleukin-1 beta; IL-6, nterleukin-6.
Given that UCPs are involved in mediating thermoregulation, energy balance, signal transduction, oxidative stress, and inflammatory responses, UCPs are key regulators of cellular energy metabolism and redox homeostasis, and also are core molecular switches for maintaining overall cellular homeostasis. Numerous studies have explored the interactions of UCPs with other metabolic pathways, which is conducive to clarifying the mechanism of action of uncoupling proteins in obesity, cardiovascular disease, neurodegenerative diseases, and tumors [8-10]. Recent studies have revealed that UCP1 prevents acute kidney injury (AKI) by inhibiting oxidative stress, and that endothelial UCP2 is a mechanosensitive inhibitor of atherosclerosis [11, 12]. In addition, UCP3 is closely related to myocardial susceptibility to ischaemia/reperfusion injury, and UCP4 and UCP5 are widely involved in neuroprotective and antioxidant effects, and play positive roles in the diagnosis and treatment of Parkinson's disease and Alzheimer's disease [13, 14]. With the in-depth progress of a series of studies, UCPs are expected to become innovative targets for the treatment of metabolic diseases, providing novel ideas for clinical applications [15].
Subtypes and structure of UCPs
The expression patterns of UCPs and the structural characteristics of their subtypes are specific in different tissues and physiological states [16] (Fig. 2). Current studies have revealed at least five known uncoupling protein subtypes: UCP1, UCP2, UCP3, UCP4 and UCP5, whose names and functions differ slightly across species [17]. The subtypes, distribution and functions of each group of UCPs are detailed in Table 1. All UCPs are mitochondrial inner membrane transport proteins with some common structural features. The transmembrane structure of UCP is composed of 6 α-helical transmembrane helix domains (TM1-TM6), forming a central proton channel, with both the N-terminus and the C-terminus facing the mitochondrial matrix side, which is conducive to interactions with protons and other molecules [2, 18]. UCPs have some functional binding domains such as the purine nucleotide binding domain located on the matrix side, which inhibits proton leakage by binding to ADP/GDP; the fatty acid binding site changes the protein conformation by binding to long-chain fatty acids such as palmitic acid to open the proton channel; cysteine residues are involved in redox regulation, and related studies have shown that Cys305 of UCP3 is easily modified by reactive oxygen species (ROS) [19]. In addition, UCP1 has approximately 60% homology with other subtypes, while the homology between UCP2-UCP5 is higher, reaching 70% [20].
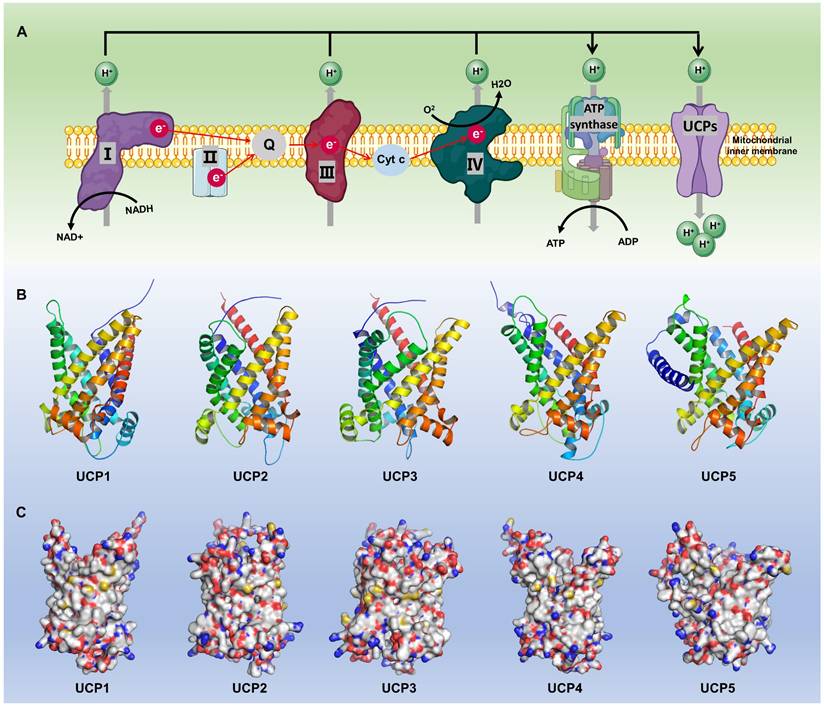
Basic biological functions and molecular structures of different subtypes of UCPs. (A) The mitochondrial respiratory chain (I-IV) pumps H+ into the mitochondrial intermembrane space, creating a concentration gradient. This electron transfer generates an electromotive force that drives ATP synthase to produce ATP. UCPs, anchored to the inner mitochondrial membrane, are activated by the H+ gradient, exerting a range of essential biological functions, including heat production. (B-C) Schematic diagrams of predicted protein structures for different members of the UCP family. UCP protein structures were predicted using AlphaFold3 and visualized using Pymol. NAD+, Nicotinamide adenine dinucleotide; NADH, Nicotinamide adenine dinucleotide; Cyt C, cytochrome complex.
Subtypes, molecular characteristics, distribution and functions of UCPs
| Subtype | Gene name | Amino acid length | Molecular weight | Main tissue distribution | Subcellular localization | Function |
|---|---|---|---|---|---|---|
| UCP1 | SLC25A7 | ~307 aa | ≈33.0 kDa | Brown adipose tissue | Mitochondrial inner membrane | Classical thermogenesis, Fatty acid metabolism |
| UCP2 | SLC25A8 | ~309 aa | ≈33.2 kDa | Pancreas, immune cells, liver, kidney, brain | Oxidative stress, energy metabolism, immune regulation | |
| UCP3 | SLC25A9 | ~312 aa | ≈33-34 kDa | Skeletal muscle, heart | Fatty acid metabolism, energy regulation, metabolic adaptation | |
| UCP4 | SLC25A27 | ~323 aa | ≈36.1 kDa | nervous system | Oxidative stress, neuronal protection, mitochondrial metabolism | |
| UCP5 | SLC25A14 / BMCP1 | ~325 aa | ≈36.2 kDa | Brain, testicles | Neuronal metabolism, lipid oxidation, mitochondrial function |
UCP1 is a classic member of the uncoupling protein family. It uncouples the mitochondrial proton gradient, preventing protons from passing through the ATP synthase complex to produce ATP, thereby exerting a thermogenic effect and increasing energy consumption [21]. UCP1 is distributed mainly in BAT and in the interscapular fat of infants. BAT contributes to heat generation and body temperature regulation through UCP1-mediated non-shivering thermogenesis. UCP1 is distributed in small amounts in WAT and skeletal muscle, but UCP1 expression in skeletal muscle is upregulated under cold adaptation conditions [22]. UCP2 is widely distributed in white adipose tissue, liver, spleen, macrophages, pancreatic β cells, heart, kidney, lung, and central nervous system. UCP2 is expressed in almost all tissues, and its expression is particularly significant in immune cells, the nervous system, and some endocrine glands [23, 24]. Current research largely suggests that the primary physiological roles of UCP2 and UCP3 are not thermogenic uncoupling like that of UCP1, but rather crucial metabolic adaptation and signal transduction proteins. Unlike UCP1, UCP2 is involved mainly in regulating the oxidative stress response and glucose metabolism [25]. Studies have confirmed that UCP2 is closely related to anti-inflammation, antioxidation and cell survival [26]. UCP3 is distributed mainly in skeletal muscle, heart and BAT, but is also expressed at low levels in WAT, placenta tissue and breast tissue. UCP3 is extremely important for fatty acid metabolism; and helps maintain energy balance by reducing heat production during fatty acid oxidation. Furthermore, the uncoupling effect of UCP3 is closely related to the metabolic function of muscle. It reduces ROS through mild uncoupling and protects muscle mitochondria from oxidative damage. UCP4 is specifically distributed in the central nervous system, especially in neuron-rich areas such as the substantia nigra, hippocampus, and cortex [27]. UCP5 is expressed mainly in brain neurons and glial cells, and in the testes, ovaries, and retina. UCP4 and UCP5 are primarily involved in the regulation of energy metabolism and neuroprotection of the nervous system, and they maintain the mitochondrial membrane potential of neurons, reduce oxidative stress damage, and delay neurodegenerative diseases [28, 29].
Different subtypes of the uncoupling protein family participate in cellular energy metabolism, oxidative stress response, and temperature regulation through their unique structural characteristics and regulatory mechanisms [30]. In-depth exploration of the location, structure, and function of UCPs is highly important for understanding the mechanisms of energy metabolism, obesity, diabetes, and other metabolic diseases.
Biological functions of UCPs in cellular homeostasis
With its diverse subtype distribution and functional differentiation, the UCP family is indispensable for thermogenesis, energy metabolism, oxidative stress, and inflammatory activation [31, 32]. Although the core mechanism involves proton leakage, the specific physiological role of each member needs to be further elucidated in combination with tissue-specific and environmental signals [33]. Next, we discuss the current studies of UCPs in terms of their physiological mechanisms.
Maintaining thermogenesis and thermoregulation
Body temperature regulation is an important physiological process for maintaining the stability of the internal environment. Nonshivering thermogenesis (NST) refers to the process of heat production mediated by mitochondrial uncoupling protein in BAT without relying on muscle contraction [34]. It is the main heat production method for long-term adaptation to cold environments [30] (Fig. 3).
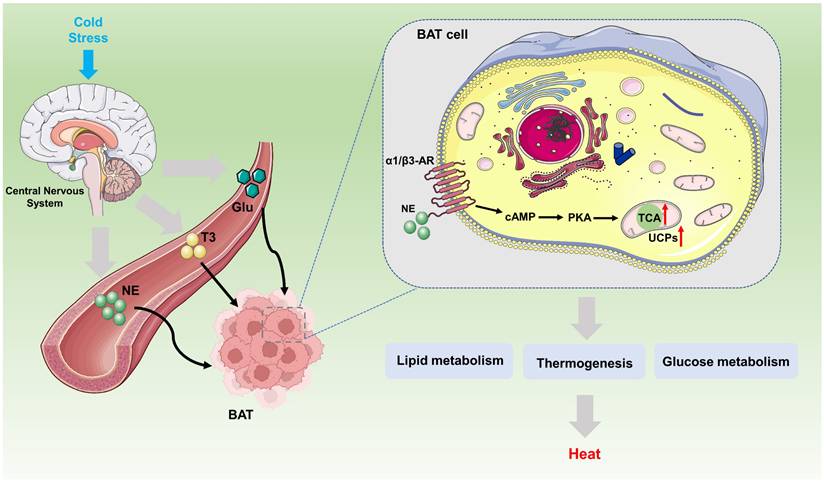
Biological mechanism pathway of UCPs regulating BAT thermogenesis. Under external stress stimuli caused by various factors, the sympathetic nervous system, under the complex regulation of the brain's central nervous system, activates the sympathetic nerves, releasing or generating NE, T3, and Glu. These molecules bind to receptors on the BAT cell membrane, such as the α1/β3-AR, activating the downstream cAMP-PKA-UCPs signaling pathway. Specifically, UCPs exert their fundamental biological function of heat production by integrating multiple metabolic pathways, such as lipid metabolism and glucose metabolism. BAT, brown adipose tissue; NE, norepinephrine; T3, triiodothyronine; Glu, glucose; CAMP, 3'-5' Cyclic adenosine monophosphate; PKA, protein kinase A.
UCP1 is the core molecule involved in nonshivering thermogenesis, and its activity can be regulated by sympathetic nerves, fatty acids and nucleotides [35]. Previous studies have shown that the activity of UCP1 is activated by free fatty acids (FFAs), ROS, and synthetic small molecules (such as 2,4-dinitrophenol (DNP)), and is inhibited by purine nucleotides or dysfunction caused by mutations in the purine binding domain [33]. Researchers have conducted detailed structural analyses of human UCP1 in the nucleotide-free state, DNP-bound state, and ATP-bound state, and revealed the structural basis of purine nucleotide inhibition of human UCP1, which is conducive to determining the binding site of the DNP activators and revealing the inhibitory mechanism of purine nucleotides [20, 22]. However, the specific molecular mechanism by which UCP1 binds to them has not yet been clarified.
Under cold conditions, postganglionic neurons release norepinephrine, which affects the activity of UCP1 via protein kinase A (PKA) through a variety of molecular mechanisms. Existing research conclusions provide evidence for a direct link between sympathetic nerves and UCP1-mediated adaptive thermogenesis. Mechanistically, AIDA, a C2-domain-containing protein located in the outer membrane of mitochondria, is phosphorylated at S161 by PKA and translocated to the membrane space, where it binds to UCP1 and promotes the cysteine oxidation of UCP1 to activate its uncoupling activity [36]. In addition, recent studies have shown that BAT activates the tension actomyosin mechanism after adrenaline stimulation. This mechanism is mediated by the mechanically sensitive transcriptional coactivator yes-associated protein 1/WW domain-containing transcription regulator protein 1 (YAP/TAZ) to regulate the thermogenic capacity of adipocytes [37]. Identifying this biomechanical signaling mechanism, which is similar to that involved in muscle tissue, is beneficial for promoting the development of brown and beige adipocytes and maintaining thermogenesis. Notably, demyristoylation of gravin-α by histone deacetylase 11 (HDAC11) inhibition was shown to promote UCP1 expression even in a catecholamine-resistant model of adipocytes in which β-adrenergic receptors(β-ARs) signaling is blocked. These findings confirm the potential for inhibiting HDAC11 to therapeutically alter thermogenesis independent of β-AR stimulation [38].
More notably, recent research has uncovered unique pathways involved in adipose tissue thermogenesis and body temperature regulation through UCPs. Creatine kinase b (CKB) as an effector of UCP1-independent thermogenesis), can regulate the energy consumption of adipocytes under cold stimulation in parallel with UCP1 [39]. Furthermore, GATA3 interacts with the transcriptional coactivator PGC1-α to increase UCP1 expression and promote energy consumption [40]. Zhang et al. demonstrated that inhibition of the PPARγ/long noncoding RNA (lncRNA) axis could increase UCP1-dependent and UCP1-independent thermogenesis [41, 42]. Interestingly, a study of two different UCP1 isoforms in mice, revealed that the long form (Ucp1L) was more efficiently translated than the short form (UCP1S), and Chen et al. reasonably posited that the cytoplasmic polyadenylation element binding protein 2 (CPEB2)-activated translation of the long 3'-UTR UCP1 mRNA might be conserved in humans [43]. With respect to mitochondria, the mitochondrial calcium uniporter (MCU) recruits UCP1 through MCU regulator (EMRE) to form the MCU-EMRE-UCP1 complex, which functions as a thermogenic uniporter. Studies have suggested that mitochondrial calcium uptake 1 (MICU1) may inhibit the formation of thermogenic uniporters to negatively regulate thermogenesis, which is essential for driving UCP1 expression and regulating metabolic disorders [44]. Additionally, data suggest that mitochondrial phosphatidylethanolamine (PE) may serve as a responder to regulate UCP1-dependent thermogenesis, modulating UCP1-dependent proton conductance across the inner mitochondrial membrane (IMM) to modulate thermogenesis [45].
Mediating metabolism and energy balance
By regulating the conversion and utilization of energy, UCPs influence the metabolism of lipids, glucose and other nutrients to ensure that the body adapts to different metabolic needs and maintains energy balance, weight control and metabolic health. Here we summarize the latest research progress on the roles of UCPs in mediating substance metabolism and energy balance.
Lipid metabolism
Previous studies have confirmed that UCPs are extremely critical in lipid metabolism, and help maintain energy balance and prevent obesity by promoting lipolysis and fatty acid oxidation, reducing fat storage, improving insulin sensitivity, and regulating adipocyte function [46, 47]. Promoting the oxidation of fatty acids is among the core functions of UCPs, especially in BAT [48]. By promoting the continued oxidation of fatty acids, allows the body to burn fat and reduce fat storage. In contrast, an obese diet impairs the oxidation of unconjugated substrates and promotes the whitening of BAT [49, 50].
With regard to UCP1, emerging evidence has shown that chronic fatty acid (FA) depletion induces UCP1 expression and that the UCP1 content can compensate for proton-lucent activity under low MMP, further suggesting a regulatory mechanism for UCP1 expression and mitochondrial energy status in human BAT cells under different nutritional conditions [51, 52]. Previous studies have confirmed that in UCP1 knockout mice, cold adaptation induced by cold stimulation induces alternative thermogenesis. Hence, the UCP1 knockout mouse model is the most commonly used animal model to determine the antiobesity effect of UCP1, but the actual effect is altered by unknown antiobesity factors in the body [53, 54]. Through high-throughput library screening of secreted peptides, two fibroblast growth factors (FGFs), FGF6 and FGF9, have been identified as potent inducers of UCP1 expression in adipocytes and preadipocytes [55]. Additionally, research has shown that UCP1 deletion increases the expression of the FGF21 gene in adipose tissue, which is an important source of endogenous FGF21. Endogenous fibroblast growth factor 21 (FGF21) can also serve as a major regulator of protection against diet-induced obesity in the absence of UCP1 [56, 57]. However, by using UCP1-FGF21 double knockout mice to further analyze the thermogenic effects of UCP1 and FGF21 in cold environments, researchers found that neither UCP1 nor FGF21 is required for maintaining body temperature or metabolic homeostasis in UCP1 knockout mice. These findings imply that more research is needed to better understand the clinical translation of UCP1 and FGF21 and their effects on cardiovascular lipid metabolism [58]. Similarly, the paradoxical effects of ambient temperature and UCP1 activation on cardiovascular disease progression have been investigated, with research revealing that thermoneutrality is associated with diminished monocyte egress from bone marrow tissue in a UCP1-dependent manner [59].
Glycerol kinase stimulates UCP1 expression through mediating fatty acid metabolism in beige adipocytes [60]. The SIRT5-mediated upregulation of C/EBPβ increases BAT browning through UCP1 [61]. As a protein phosphatase localized in mitochondria, PGAM family member 5(PGAM5) mediates molecular mechanisms including immunity, apoptosis, and metabolism. Moreover, PGAM5 has been shown to inhibit UCP1 activity and alleviate mitochondrial energy consumption by regulating its phosphatase activity and intramembrane cleavage [62]. UCP1 also exhibits diverse expression patterns in different types of adipose tissue and nonadipose tissue [63]. The expression of UCP1 in skeletal muscle fiber/adipose progenitor cells is genetically and hormonally controlled [64, 65]. Consistent with this, Wu et al. described a host gene‒microbe metabolic axis involving the regulation of intestinal lactate levels and shaping of the gut microbiota via intestinal HIF-2a, which mediates UCP1 and thereby suppresses adipose tissue thermogenesis during obesity [66]. Together, these findings reveal the indispensable status of UCP1 in fatty acid metabolism and lipid accumulation.
In addition to the crosstalk between metabolic mechanisms, research on the effects of various exogenous factors on lipid accumulation is also ongoing. Most recently, annonaceous acetogenins mimic AA005, milk fat globule membrane, whey protein and soy protein have been hypothesized to upregulate UCP1 to fight obesity [67-69]. Existing data provide evidence that dietary selenium supplementation can enhance the facultative binding of UCP1, providing a potential avenue for reducing obesity [70]. Notably, the development of corresponding tools and applications also facilitates the study of the lipid metabolism function of UCPs. For example, exploration of a functional beige adipocyte cell lineage revealed independent cell subclasses expressing UCP1 and ineffective creatine cycling [71].
Except UCP1, other UCPs can impact the activity of enzymes related to fatty acid mobilization and lipolysis. UCP2 enhances the release and mobilization of fatty acids by changing the 5' AMP-activated protein kinase (AMPK) signaling pathway [72, 73]. UCP3 was first discovered in 1997, and was shown to catalyze the exchange of C4 metabolites in a manner similar to UCP2 [74]. Similarly, emerging findings indicate that inhibiting UCP3 can affect the aggregation of mitochondrial supramolecular proteins in mouse BAT, thereby disrupting the fatty acid oxidation process [75].
Glucose metabolism and insulin resistance
The ability of UCPs to regulate metabolism is also reflected in glycolysis and insulin sensitivity, and UCPs may have a positive impact on preventing and alleviating obesity and diabetes associated with metabolic syndrome [76]. As with body thermogenesis, elevated UCP1 levels also facilitate glucose uptake in human white adipocytes [77]. As recently reported, the liver mediates preadipocyte proliferation and UCP1 expression to improve glucose metabolism [78]. Catecholamines simultaneously regulate UCP1 by mediating glycogen metabolism. Moreover, the synthesis and degradation of glycogen increase under the action of catecholamines, and glycogen metabolism is beneficial to the body's production of ROS, which leads to p38 MAPK activation, thereby driving the expression of UCP1. These results emphasize that glycogen acts as an indispensable bridge in adipocytes, linking glucose metabolism to thermogenesis [79].
In addition, the interaction between glucose metabolism and thermogenesis is being explored, and BAT ATP-citrate lyase protects against metabolic stress, tightly integrating carbohydrate utilization to thermogenesis, and providing fundamental insights into the fatty acid synthesis-oxidation paradox in BAT [80]. In addition, a prior study revealed that TUG cleavage pathway modulates both Insulin-stimulated glucose uptake in muscle and energy expenditure at the biological level by upregulating the expression of sarcolipin and UCP1. Hence, targeting this mechanism would be beneficial in promoting the thermic effect of food, and inhibiting it could lead to obesity [81].
In contrast, emerging studies have identified novel roles that are not linked to UCPs [82]. For example, WAT browning or hepatic gluconeogenesis is associated with enhanced glucose clearance when the gut microbiota is absent. However, in a mouse model of gut microbiota depletion, BAT and cecum tissue, but not white adipose tissue (WAT) or liver, contribute to glucose uptake, and this response is independent of adaptive thermogenesis [83, 84]. At the cellular level, beige fat changes energy homeostasis through Ca2+ cycling. Specifically, Ca2+ cycling stimulated by the activation α1/β3-adrenergic receptors or the SERCA2b-RyR2 pathway accelerates UCP1-independent thermogenesis. This atypical thermogenic mechanism, it demonstrated that beige fat can improve glucose tolerance independent of body weight [30]. Consistent with this effect, another study revealed that eicosapentaenoic acid profoundly alleviates weight gain and glucose intolerance in UCP1-KO mice via increased oxygen consumption and BAT PGC1α activity, independent of UCP1 [85]. Nevertheless, our understanding of metabolic and thermogenesis processes in vivo, especially in the context of obesity, remains limited, and the effects and relationships between glucose metabolism and UCPs remain to be fully explored.
Modulating redox homeostasis
UCPs significantly decrease ROS production by lowering mitochondrial membrane potential, minimizing electron leakage in the electron transport chain, and controlling calcium ion homeostasis, thereby alleviating oxidative stress. As indispensable regulators of oxidative stress, UCPs play important protective roles in various metabolic diseases and ageing [21, 86]. Now we summarize the evidence supporting the leading roles of UCPs in oxidative stress (Fig. 4).
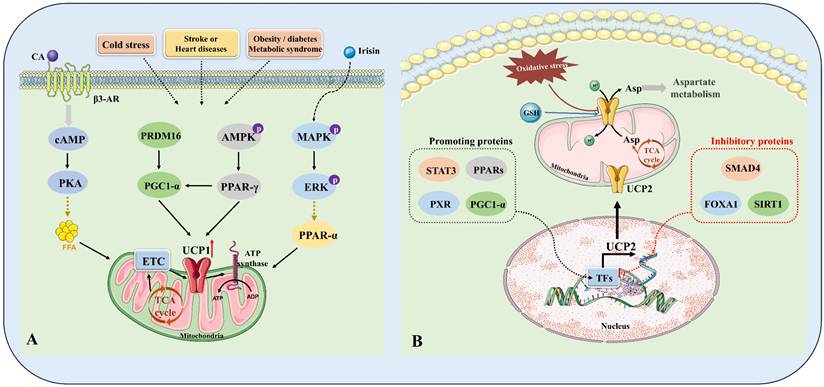
Intracellular regulatory mechanisms of the key components of UCPs. (A) Mechanisms of regulating UCP1. In the complex pathophysiological context of cold stimulation, stroke or heart disease, and obesity/diabetes, the body can release catecholamines (CA) through (1) sympathetic activation, which binds to β3-AR on the cell membrane and activates cAMP-PKA-FFA-UCP1 signaling; (2) irisin activates MAPK-ERK-PPAR-α signaling; and (3) intracellular PRDM16-PGC1-α and AMPK-PPAR-γ signaling crosstalk and regulate each other. The above signals together constitute a complex regulatory mechanism network of UCP1. (B) Mechanisms of regulating UCP2. (1) STAT3, PPARs, PXR, PGC1-α, etc. act as positive regulators of UCP2; (2) SMAD4 and FOXA1/SIRT1 act as negative regulators of UCP2. Together, these proteins constitute the regulatory network of UCP2, controlling the transcription and translation levels of UCP2, affecting the abundance of UCP2 on the mitochondrial membrane, and thereby regulating metabolic reprogramming, including aspartate metabolism, GSH metabolism, and TCA energy metabolism. FFA, free fatty acid; MAPK, mitogen-activated protein kinase; AMPK, adenosine monophosphate activated protein kinase; ERK, extracellular signal regulated kinases; PPAR-γ, peroxisome proliferator-activated receptor-γ; TFs, transcription factors; STAT3, signal transducer and activator of transcription 3; PPARs, peroxisome proliferator-activated receptors; PXR, pregnane X receptor; PGC1-α, peroxisome proliferator-activated receptor γ coactivator 1 alpha; SMAD4, SMAD family member 4; FOXA1, forkhead-box protein A1; SIRT1, NAD-dependent deacetylase sirtuin-1; GSH, Glutathione.
The role of UCPs in mitochondria
Although the mitochondrial membrane potential (ΔΨm) is essential for ATP synthesis, it also increases the probability of electron leakage in the electron transport chain, leading to increased ROS formation [87]. UCPs reduce membrane potential through proton leakage, decrease electron leakage, and inhibit ROS generation. Furthermore, complexes I and III of the electron transport chain are the main sites of ROS generation [88]. UCPs also decrease the accumulation of electrons in these complexes by lowering the membrane potential, thereby reducing the content of superoxide. By constructing an in vivo mouse AKI model and an in vitro human renal proximal tubular epithelial cell aristolochic acid I model, documented studies have confirmed that UCP1 and UCP2 respond positively to the protective effect of kidney injury by fighting oxidative stress [11, 89]. Combined with the results of skeletal muscle proteomic analysis, these findings revealed that compared with wild-type controls, mice ectopically expressing UCP1 in skeletal muscle presented lower systemic methylglyoxal-derived advanced glycation end product exposure flux, representing better aging performance, extended lifespan, and greater resistance to impaired metabolic health [90]. Similarly, it was recently reported that UCP2 alleviates mitochondrial dysfunction and oxidative damage in substantia nigra pars reticulata with hepatic encephalopathy [91]. In contrast to the above results, Sunad Rangarajan et al. have shown that silencing UCP2 in vivo induced regression of fibrosis in an aged mouse model of pulmonary fibrosis, providing evidence for the therapeutic targeting of UCP2 in age-associated organ fibrosis and impaired tissue regeneration [92]. These up-to-date studies illustrate that, UCPs also maintain Ca2+ homeostasis and decrease ROS production by indirectly affecting the uptake and release of Ca2+, thereby further protecting the integrity of mitochondrial structure and function. Aldolase B (ALDB), a glycolytic enzyme, is related to decreased calcium release from the endoplasmic reticulum (ER). It was recently discovered that UCP2 and ALDB influence insulin secretion by modulating mitochondrial activity and endoplasmic reticulum Ca2+ release, suggesting that targeting the UCP2/ALDB axis is a promising approach for restoring β-cell function [93].
With respect to the molecular mechanisms of oxidative stress, the expression and activity of UCPs are guided by multiple signaling pathways, and these processes in turn act on these patterns. As previously stated, UCPs mediate energy metabolism and ROS formation by influencing AMPK activity, regulating energy balance and antioxidant capacity. Moreover, UCPs expression is dominated by the expression of transcription factors such as PPARs, and CREB and epigenetic modifications. Previous studies have confirmed that ER stress diminishes UCP1 expression by restraining PPARγ binding activity in mouse beige adipocytes, and that procyanidin B2 targets and activates PPARγ/UCP1 signaling, exacerbating oxidative stress and thus subtly improving the developmental capacity of bovine oocytes [94, 95].
The role of UCPs in oxidative stress-related signaling pathways
To explore the relationships between UCPs and other transcription factors, Nrf2, a major transcription factor involved in the antioxidant response, was used to regulate various antioxidant enzymes, including superoxide dismutase and glutathione peroxidase. UCPs further promote antioxidant defense by reducing ROS levels and enhancing the activity of Nrf2. In addition, UCPs lower ROS generation and inhibit overactivation of the nuclear factor kappa B (NF-κB) pathway, ultimately reducing inflammatory responses and apoptosis. MAS, as a G protein-coupled receptor, is considered a new component of the renal angiotensin system (RAS). In a rat model of early brain injury in subarachnoid hemorrhage (SAH), AVE 0991, a selective agonist of Mas, has been reported to alleviate oxidative stress and neuronal apoptosis through the MAS/PKA/CREB/UCP2 pathway, which demonstrates that AVE may be a novel and effective therapeutic approach against oxidative stress in SAH patients [96]. Another study highlighted the role of UCP4 in mitochondrial homeostasis, clarifying the role of UCP4 in intermittent hypobaric hypoxia (IHH). Specifically, UCP4 overexpression alleviated oxidative stress damage in cerebellar Purkinje cells of IHH mice, enhanced mitochondrial function, and improved IHH-induced movement disorders [97].
Considering the relevance of UCPs in the biological process of oxidative stress, several novel medicines and clinical applications that rely on UCPs have been discovered. Both in vitro and in vivo experiments have shown that, dietary fish oil supplementation ameliorates adipose tissue dysfunction, oxidative stress, PPARγ, and UCP2 in insulin-resistant rats [98, 99].
Regulating the inflammatory response
There is a complex bidirectional regulatory relationship between UCPs and the inflammatory response. UCPs regulate mitochondrial function and the redox state to affect the inflammatory response, and the inflammatory response affects the expression and activity of UCPs through cytokines and signaling pathways [100]. Following that, we detail the most recent interaction mechanism between these factors.
UCPs and inflammatory cells
During the activation of inflammation, macrophages, neutrophils, and lymphocytes rapidly recruit and release a large number of inflammatory factors. At the cellular level, high expression of UCP2 decreases macrophage polarization towards a pro-inflammatory phenotype (M1 type). Furthermore, macrophages switch from oxidative phosphorylation to glycolysis under stimulation caused by inflammation, thereby regulating UCP2 through AMPK and mTOR signaling. While in T cells, UCP2 affects mitochondrial metabolism and the differentiation and functionality of T cells, ultimately affecting immune response strength. However, a recent study reported that UCP2 activity in adipose tissue during obesity-induced insulin resistance is not critical for macrophage activation [101]. In addition to this discovery, another type of immune cell, mast cells, is enriched with tryptophan hydroxylase 1 (Tph1), which is the rate-limiting enzyme for peripheral serotonin synthesis. Yabut et al. reported that Tph1 deletion in mast cells enhances UCP1 expression in BAT, further limiting the development of obesity and insulin resistance, and providing insights into unique avenues for treating obesity and diabetes [102].
Notably, inflammation also alters the methylation status of the promoter region of UCP2 and subsequently affects its transcriptional activity. Similarly, another finding revealed that the cysteine 253 residue of UCP1 is involved in sex-dependent inflammation in adipose tissue [103]. Histone deacetylase 3 (HDAC3), a transcriptional coregulator, whose activity requires interaction with nuclear receptor corepressors (NCoRs) and is closely related to the thermogenic function of UCP1. Interestingly, in the absence of HDAC3 activity, BAT lacking NCoRs activates inflammatory pathways that stimulate BAT through the sympathetic nervous system, resulting in normal heat production. This discovery provides evidence for our in-depth elucidation of the multiple physiological effects of the transcription core complex [104]. In summary, all these results emphasize that inflammation-mediated metabolic reprogramming and epigenetic regulation are also involved in influencing UCPs.
UCPs and inflammatory factors
NF-κB is a core factor in inflammation, and UCP2 has a major role in the anti-inflammatory response. As mentioned in the previous section, ROS, as crucial triggers of inflammation, can activate proinflammatory transcription factors such as NF-κB and AP-1. According to previous findings, UCPs decrease ROS levels and restrain the activation of the above transcription factors, resulting in low expression of proinflammatory cytokines, including TNF-α, IL-6, and IL-1β or fibrogenesis TGF-β [105]. In terms of the potential underlying mechanisms, several previous studies have established that UCP2 inhibits NF-κB through weakening ROS levels and increasing mitochondrial function [106]. For example, Aspergillus protease-induced mitochondrial ROS generation is associated with the downregulation of UCP2 expression through the TGF-β-SMAD4 signaling pathway, which further clarifies the mechanism of bronchial epithelial inflammation caused by allergic fungal proteases [107]. In addition, UCP2 overexpression suppresses the nuclear translocation of NF-κB, reduces the transcription of downstream proinflammatory genes, and alleviates inflammatory diseases [108].
Additionally, in both UCP1 knockout mouse and pig models, Ping Gu et al. found that UCP1 deletion promotes increased mitochondrial membrane potential and mitochondrial superoxide production, which contributes to overactivation of the NLRP3 and caspase1-mediated maturation of IL-1, and highlighted that the vascular protective effect of UCP1 is partly due to the anti-inflammatory effect of UCP1 [109]. These data shed light on the role of UCP1 in maintaining vascular health in perivascular adipose tissue. In contrast to these representative inflammatory factors, other inflammatory factors, such as IL-33, have been previously shown to trigger the early expression of proinflammatory genes in macrophages and subsequent differentiation into alternatively activated macrophages (AAMs). However, the molecular mechanisms controlling this differentiation have not been clearly elucidated. A recent study revealed that UCP2-mediated mitochondrial reprogramming promotes IL-33-induced AAM differentiation [110]. Inflammation is generally accompanied by apoptosis. In line with the findings of previous study, UCPs also inhibit apoptosis by restraining the opening of mitochondrial permeability transition pores (mPTPs), thus attenuating inflammation-induced tissue damage.
Research progress on UCPs in diseases
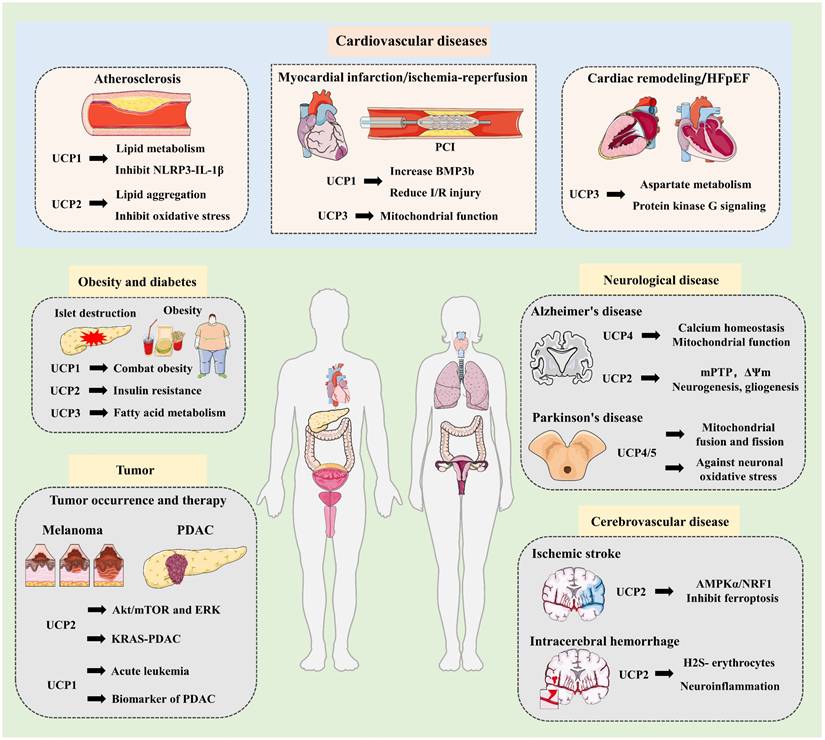
UCPs are involved in the regulation of cellular homeostasis and metabolic diseases through their diverse molecular structures and functions [71, 111]. Specifically, UCPs influence the occurrence and development of obesity, diabetes, cardiovascular diseases, neurodegenerative diseases and related inflammatory diseases [112, 113]. More importantly, UCPs exert anticancer effects by reprogramming carcinoma metabolism and improving drug resistance, and provide strategies for delaying ageing through antioxidant damage (Fig. 5).
Research progress of UCPs in multisystem diseases. UCPs are involved in the development and progression of various metabolic diseases, including obesity and diabetes, cardiovascular disease, neurological diseases, and cancer. UCP1-3 combats obesity and diabetes by improving insulin resistance and fatty acid metabolism. In cardiovascular disease, UCP1 improves atherosclerosis by mediating lipid metabolism and inhibiting the NLRP3-LI-1β inflammatory pathway. Similarly, UCP2 inhibits atherosclerosis by reducing lipid accumulation and oxidative stress. In myocardial infarction and ischemia-reperfusion injury, UCP1 promotes the cardioprotective factor BMP3b to reduce IR, while UCP3 improves mitochondrial function to exert vascular protection. Furthermore, UCP3 mediates aspartate metabolism and regulates protein kinase G signaling to alleviate cardiac hypertrophy and HFpEF. In neurological diseases, UCPs target calcium homeostasis and mitochondrial function to alleviate neurodegenerative diseases, including Alzheimer's and Parkinson's disease. UCP2 inhibits ferroptosis and inflammatory responses, effectively improving neurovascular disease. Additionally, UCPs are widely involved in tumorigenesis and progression. UCP2 promotes melanoma through Akt/mTOR and ERK and also mediates KRAS to affect PDAC. Furthermore, studies have found that UCP1-mediated thermogenesis is associated with acute leukemia, and increased UCP1 expression can serve as a biomarker for early-stage PDAC. These findings strongly suggest that UCPs-guided targeted therapies or multi-target combination therapies will become innovative treatments in the future. NLRP3, NOD-like receptor protein 3 inflammasome; PCI, percutaneous coronary intervention; HFpEF, heart failure with preserved ejection fraction; mPTP, mitochondrial permeability transition pore; AMPKα, AMP-activated protein kinase alpha; NRF1, nuclear factor E2-related factor 1; PDAC, pancreatic ductal adenocarcinoma; mTOR, mammalian target of rapamycin; ERK, Extracellular signal regulated kinases; KRAS, Kirsten rat sarcoma viral oncogene
Obesity and diabetes
In line with what is described in the above section, most of the UCPs research to date has been in the field of weight loss. Mechanistically, UCPs regulate the efficiency of energy metabolism and alter its activity to affect fat storage and burning. UCP1, UCP2, and UCP3 have considerable implications in obesity, diabetes, and metabolic syndrome [114-116]. In particular, the expression of UCP1 in BAT is closely related to thermogenic function, increasing energy expenditure to combat weight gain. Therefore, researchers are actively exploring therapeutic strategies to activate UCP1 to combat obesity [30, 36]. Contrary to the commonly held assumption, it was also discovered that increased UCP2 expression is often strongly associated with diabetes occurrence and insulin resistance [117]. Although UCP2 protects islet β cells from oxidative stress to some extent by reducing intracellular ROS levels, excessive UCP2 activity triggers ROS outbursts and impairs islet β cells and insulin secretion [106, 118]. In other words, UCP2 is widely expressed in the pancreas, liver, muscle, and adipose tissue. The active regulation of pancreatic islet β cells, has been reported to cause metabolic dysfunction, decrease ATP synthesis, and restrain insulin secretion through a reduced ATP/ADP ratio [119, 120]. In addition, in liver and adipose tissue, abnormal UCP2 content may lead to fatty acid metabolism disorders, which in turn exacerbate insulin activity [119]. However, similar to UCP1, UCP3 mainly enhances fatty acid oxidation and reduces fat accumulation to coordinate energy balance [119]. UCP3 overexpression has also been shown to help reduce fat accumulation and improve metabolic status, and UCP3 deficiency also exacerbates fatty acid accumulation, which ultimately contributes to insulin resistance and diabetes progression [121].
Obesity is a major risk factor for type 2 diabetes [122]. As outlined above, obesity alters the expression and activity of UCPs, causing a decrease in the efficiency of energy metabolism, thus exacerbating the development of obesity and diabetes [105, 123]. With respect to the critical role of UCPs in energy metabolism, modulating UCPs will be an effective strategy for the treatment of obesity and diabetes in the future. Consistent with these findings, metabolic disturbances in diabetes and obesity can be ameliorated by increasing UCP1 activity, activating thermogenic processes, or inhibiting UCP2 overexpression [24, 124]. The subsequent challenge is to clarify their various site-specific downstream targets and determine their unique signaling roles, further providing evidence for the clinical translation of UCPs.
Cardiovascular diseases
Cardiovascular disease (CVD) is the main threat to human health. Several studies have shown that UCPs participate in the development of various cardiovascular diseases by regulating energy metabolism, oxidative stress, the inflammatory response, apoptosis and other molecular pathways [10]. The expression patterns and signaling mechanisms of UCPs differ in response to different types of CVD. Herein, we provide an overview of the most recent findings concerning the role of UCPs in various forms of CVD (Table 2).
Research progress and application prospects of UCPs in cardiovascular diseases
| Cardiovascular disease | UCPs | Molecular mechanism | Application Prospects |
|---|---|---|---|
| Hypertension | UCP2 | mediating the mitochondrial activity of vascular smooth muscle cells | a biomarker for endothelium function |
| Atherosclerosis | UCP1 | regulating lipid metabolism, inhibiting the NLRP3-IL-1β inflammatory pathway | a theoretical foundation for the precise use of phenolic acid-rich food resources |
| UCP2 | reducing lipid aggregation and inhibiting oxidative stress | the function of endothelial UCP2 signaling in plaque rupture remains to be determined | |
| Myocardial infarction and ischemia-reperfusion | UCP3 | preserving the mitochondrial function, mediating fatty acid oxidation | medium-chain fatty acid supplementation might be a promising approach to improve IR injury for patients with T2DM heart disease |
| UCP1 | Mediating myocardial protective factor BMP3b | Increasing BMP3b levels or targeting Smad 1/5 could be potential therapeutic options to reducing I/R injury. | |
| Cardiac remodeling | UCP3 | downregulating the enhanced aspartate | providing a basis for targeting UCP3 to treat cardiac remodeling |
| HFpEF | UCP3 | regulating protein kinase G signaling and oxidative stress | the use of antioxidants as an adjuvant therapy for HFpEF |
| Doxorubicin-induced cardiotoxicity | UCP2 | alleviating oxidative stress and autophagy, or through the AMPKα/UCP2 pathway | Irisin and Matrine |
| Myocardial aging | UCP2 | affecting cellular proliferation and survival | Clarifying UCP2 metabolic properties that influence cardiac function |
Hypertension
Hypertension is an important indicator of a range of cardiovascular diseases, and its pathophysiology has attracted considerable attention. Previous studies have shown that UCPs can impact vascular tone and blood pressure stability by mediating the mitochondrial activity of vascular smooth muscle cells.
In an experimental analysis of clinical plasma samples, available data revealed lower circulating UCP2 levels in patients with T2DM, which were associated with endothelium-dependent vasodilation in conduit vessels. Notably, these findings imply that UCP2 can be utilized as a biomarker for endothelium function in T2DM patients, but further research should be conducted to clarify the link between UCP2 alterations and blood glucose levels and other cardiovascular event risks [125]. Consistently, in a stroke-prone spontaneously hypertensive rat model, oxidative stress and inflammatory activation were attenuated, and renal injury and stroke risk were decreased [126]. Nevertheless, another mouse experimental study examining the association between UCP2 and blood pressure (BP) yielded beneficial results. Specifically, DJ-1, also known as PARK-7, is a multifunctional oxidative stress response protein. When DJ-1 is depleted, oxidative stress induces hypertension, and NO function decreases. These findings confirmed that UCP2 overexpression in the kidney increased the blood pressure of DJ-1 knockout mice, indicating that excessive UCP2 levels may have adverse effects on BP regulation. Taken together, these findings suggest that UCP2 acts as an antioxidant and is involved in intricate mechanisms in hypertension. In terms of the expression level, activation time, and environmental variables, UCP2 can negatively impact physiological processes [127].
Atherosclerosis
Atherosclerosis (AS) is a chronic inflammatory disease involving pathological processes such as lipid deposition, smooth muscle proliferation, fibrosis and calcification. The initiation of atherosclerosis is usually multifactorial and involves the induction of endothelial cell damage. In human aortic and human umbilical vein ECs, Krüppel-like factor 2 (KLF2) has been shown to mediate fluid shear stress-dependent regulation of UCP2 expression. Both in vitro and in vivo experiments have shown that, UCP2 is a novel mechanosensitive gene in ECs that is controlled by fluid shear stress and KLF2. Moreover, RNA-seq analysis revealed that, Forkhead box protein O1 (FOXO1) is a major proinflammatory transcriptional regulator activated by UCP2 knockdown, and that the inhibition of FOXO1 induced by UCP2 expression is closely associated with the phosphorylation of AMPK. Therefore, it has become increasingly evident that UCP2 expression is critical for endothelial proinflammatory responses and atherosclerosis [128]. Apart from this, UCP2 is also found to be associated with plaque stability, especially with respect to macrophage mitochondrial function. According to existing research results, UCP2 prevents atherosclerosis by lowering lipid buildup and suppressing oxidative stress. In contrast, some studies suggest that excessive UCP2 activity can induce mitochondrial dysfunction and heart disease. Consequently, the link between UCP2 and AS is relatively complex and the function of endothelial UCP2 signaling in plaque rupture remains to be determined.
Lipid accumulation is a critical element in the process of atherosclerosis. Focusing on other UCPs, UCP1-mediated thermogenesis can alleviate lipid accumulation and offers extensive applicability for the treatment of AS. For instance, C-type natriuretic peptide (CNP) is a multidimensional protective factor in the cardiovascular system that impacts local blood flow, angiogenesis, and cardiac function. In CNP-knockout mice, researchers have reported that the thermogenic phenotype correlates with increased UCP1 levels and preferential mitochondrial utilization of lipids, implying that UCP1 regulates lipid metabolism to maintain vascular health [129]. As a consequence, an increasing number of analyses are looking into novel strategies to treat AS aiming at UCP1. AP39, a mitochondrial hydrogen sulfide donor, has been proven to stabilize atherosclerotic lesions by increasing UCP1 expression in vascular smooth muscle cells, lowering overall macrophage concentration and collagen deposition [130]. Furthermore, using UCP1 as a target, phenolic acid compounds such as ferulic acid and protocatechuic acid have been shown to have therapeutic benefits for AS through the inhibition of the NLRP3-IL-1β inflammatory pathway. This presents a theoretical foundation for the precise use of phenolic acid-rich food resources [131].
Myocardial infarction and ischemia-reperfusion injury
Myocardial infarction (MI) is typically caused by thrombosis or the rupture of atherosclerotic plaques. Ischemia-reperfusion (IR) injury is a serious consequence of MI that worsens myocardial damage, extends recovery time, and increases patient mortality and complication risk. Prior studies on the underlying molecular mechanism, revealed that UCP3 is expressed primarily in skeletal muscle and the heart; hence, it plays a significant component in the pathophysiology and prognosis of MI and IR. By suppressing the opening of mitochondrial permeability transition pores to sustain mitochondrial function through interactions with adenine nucleotide translocator and the PI3K/AKT pathway, UCP3 has been shown to eliminate the processes of cardiomyocyte apoptosis and necrosis and ultimately improve the prognosis of MI [132]. In this context, patients with insulin resistance and T2DM suffer worse cardiac endpoints following MI. Using Sprague-Dawley rats model with myocardial-specific UCP3 knockout. Edwards et al. have concluded that normal cardiac UCP3 levels are essential for the restoration of long-chain fatty acid oxidation, mitochondrial respiratory ability and myocardial contractility after I/R. Moreover, we contend that matching fatty acid metabolism to the energy needs of the heart, especially through supplementation with medium-chain fatty acids, might be a promising approach to improve IR injury for patients with T2DM heart disease [133]. With respect to other UCPs, a recent study evaluated the role of BAT in IR using UCP1-deficient mice, and revealed a cardioprotective protein that is released as bone morphogenetic protein 3b (BMP3b). Notably, BMP3b mediates cardioprotection through SMAD1/5/8, and its plasma levels are correlated with the magnitude of myocardial injury. As a consequence, increasing BMP3b levels or targeting Smad 1/5 may provide theoretical possibilities to ameliorate myocardial damage in I/R injury [134].
Heart failure
Heart failure is a common complication of several cardiovascular disorders, including cardiomyopathies, myocarditis, congenital heart disease, pericarditis, and endocarditis. Previous research has verified the presence of BAT in adults using noninvasive quantitative measurement with (18)F-FDG PET-CT, emphasizing the necessity of identifying and understanding its composition for human metabolism. A recent case‒control trial quantified the expression level of UCP1 in brown adipose tissue (BAT), and the results confirmed that UCP1 expression in BAT is negatively correlated with cardiovascular metabolic risk in adults, highlighting the potential application of targeting and activating BAT in metabolic diseases [135, 136]. In accordance with RNA sequence and STRING bioinformatics analysis, UCP2 suppression is associated with notable changes in the cellular cycle, survival signaling pathways, and mitochondrial genes [137]. Although UCP2 inhibition has been found to reduce cardiac dysfunction and heart failure in pressure overload-induced left ventricular hypertrophy, it has a distinct effect on a mouse model of pressure overload-induced right heart failure. More specifically, mouse cardiac fibroblasts express more UCP2 than myocytes do, allowing UCP2-deficient mice to maintain certain cardiac functions under right ventricular pressure overload [138]. In the treatment of heart failure with preserved ejection fraction (HFpEF), UCP3 deficiency-mediated mitochondrial oxidative stress exacerbates angiotensin II-induced hypertensive left ventricular diastolic dysfunction and exercise intolerance [139]. Moreover, recent research has demonstrated that UCP3 expression is downregulated in hypertrophic hearts and cardiomyocytes, while increasing the overexpression of UCP3 improves cardiac hypertrophy by decreasing aspartate levels [140]. Mitochondrial antioxidants have shown favorable effects on HFpEF, although research on their efficacy in animal models and their interactions with UCP3 deficiency is ongoing [139].
Cardiac dysfunction can be caused by a range of heart illnesses, genetic abnormalities, or systemic diseases. Doxorubicin-induced cardiotoxicity can also occur with heart failure and should not be overlooked during cancer chemotherapy. Current research confirms that UCP acts as a target molecule and signal bridge in various types of doxorubicin-induced cardiotoxicity therapies. For example, Irisin has been shown to alleviate DOX-induced pericardial fibrosis via decreasing ROS accumulation, autophagy impairment, NF-κB pathway activation, and the ENDMT phenotype primarily targeting UCP2. Notably, these findings provide a preliminary investigation of the potential of irisin as a cardiokine, but its regulatory mode and mechanism in the cardiac microenvironment remain to be clarified [141]. Moreover, Matrine, an alkaloid with antioxidant and anti-inflammatory properties, can also alleviate oxidative stress and cardiomyocyte apoptosis in DOX-induced cardiotoxicity via the AMPKα/UCP2 pathway [142] Further exploration of the specific site mechanisms by which traditional Chinese medicine components and known Western medicines mediate the improvement of cardiovascular diseases by UCPs may lead to more precise treatment through drug combination therapies and the development of target inhibitors or agonists.
Neurological diseases
The physiological functions of UCPs in neurological illnesses extend beyond directing mitochondrial function and oxidative stress. UCPs play a critical role in neuroprotection through numerous processes, including modulating metabolism, inflammation, and neuroplasticity. We review the latest developments in the use of UCPs in the treatment of neurodegenerative illnesses, cerebral ischemia and reperfusion injury, epilepsy, and other neuroinflammatory diseases.
Neurodegenerative diseases
Neurodegenerative diseases are characterized by the ongoing degradation of neuronal structure and function. The main pathogenic features are neuronal death, synaptic loss, and aberrant protein aggregation. Alzheimer's disease (AD), Parkinson's disease (PD) and Huntington's disease (HD) are among the most prevalent neurodegenerative diseases. Among UCPs, UCP2, UCP4, and UCP5 are highly expressed in the central nervous system and are associated with neuronal energy metabolism and antioxidant defense.
AD is characterized by the formation of β-amyloid protein (Aβ) plaques and neurofibrillary tangles (NFTs) triggered by abnormal Tau protein phosphorylation [143]. AD patients frequently experience mitochondrial dysfunction [144]. As demonstrated in previous studies, UCP4 maintains calcium ion homeostasis and mitochondrial membrane potential to protect neurons from Aβ injury, and utilizes mitophagy to eliminate damaged mitochondria, which hampers the progression of AD [145]. Similarly, UCP2 modulates the opening of the mitochondrial permeability transition pore (mPTP), suppressing cell death and substantially reducing the decrease in mitochondrial membrane potential and ROS production triggered by Aβ [146]. More crucially, UCP2 and UCP4 gene polymorphisms can increase the risk of AD [147, 148]. Recent findings revealed that endothelial UCP2 deficiency eliminates vascular diameter and affects the transition time between neurogenesis and gliogenesis [149]. These results suggest that future research should concentrate on expanding the applications of UCPs in the early detection of AD, and evaluating their potential as biomarkers, with the goal of developing AD treatment regimens that target UCPs.
In contrast to AD, which occurs mostly in the hippocampus and cortex, PD largely occurs in the substantia nigra and striatum [150]. Mechanistically, AD is caused by the loss of dopaminergic neurons, leading to motor delay or anxiety, depression, sleep disorders, etc. [151]. However, in terms of neuronal protection, UCPs particularly UCP4 and UCP5 limit ROS accumulation in dopaminergic neurons by affecting the mitochondrial membrane potential (MMP), and by regulating mitochondrial fusion and fission to maintain energy balance [13]. DJ-1, also known as PARK7, has been implicated in the pathogenesis of PD. Prior research has demonstrated that DJ-1 stimulates UCP4 overexpression to guard against neuronal oxidative stress damage by mediating autooxidation and the NF-κB pathway [152]. In addition, a study of hepatic encephalopathy-induced basal ganglia motor neuron damage, revealed that UCP2 plays a positive antioxidant role. Mechanistically, this effect is achieved by affecting the levels of K+-Cl- cotransporter-2 on GABAergic neurons, which also indicates the considerable potential of the neuroprotective effect of UCP2 in the treatment of movement disorders [153]. However, in a mouse glaucoma model, UCP2 knockout promoted mitophagy and reduced retinal ganglion cell death [154]. Therefore, these findings suggest that the regulatory effects of UCP2 on different neurological diseases involve multiple pathways and outcomes. In addition, with respect to HD aetiology, UCPs could potentially ameliorate calcium ion signaling, subsequently relieving cellular stress induced by mutant Huntingtin and improving neuronal degeneration in patients [155].
Cerebrovascular disease
In cerebral ischemia and reperfusion injury, UCPs may prevent excessive ATP consumption by lowering the mitochondrial membrane potential, and delaying energy depletion during ischemia. As previously stated, UCPs safeguard mitochondrial structure and function by limiting the rapid production of ROS. UCPs can also hinder cytochrome C leakage and initiate of apoptotic cascade processes by blocking the opening of the mPTP. For example, Lei Wang et al. reported that UCP2 expression gradually decreases with prolonged ischemic time in in vitro and in vivo ischemic stroke models. Specifically, UCP2 activates AMPKα/NRF1 signaling to protect the brain from ischemia-induced ferroptosis [156]. Moreover, a study that investigated the influence of UCP2 on the dynamic balance of mitochondrial fission and fusion in ischemic mice under normal and hyperglycaemic conditions revealed that, UCP2 deficiency enhanced ROS generation, early damage to mitochondrial ultrastructure, and disruption of mitochondrial fission dynamic equilibrium, resulting in an increase in the cerebral infarction area [157].
UCP2 is also significantly involved in intracerebral hemorrhage (ICH), and hematoma clearance is primarily accomplished through the phagocytosis of red blood cells in the hemorrhagic brain. Furthermore, spontaneous hematoma clearance after ICH requires mitochondrial complex I and UCP2. Following ICH, the gasotransmitter hydrogen sulfide (H2S), also known as the endogenous modulator of continuous phagocytosis, targets mitochondria and turns on UCP2 to stimulate microglial phagocytosis of erythrocytes [158]. Similarly, succinate has been confirmed to activate UCP2, lessen neuroinflammation, and provide protection following cerebral hemorrhage [159].
Other neuroinflammatory diseases and complications
According to recent studies, microglial UCP2 mediates inflammation and obesity caused by high-fat diets and supports the essential central regulatory mechanism directed by fuel supply-driven mitochondrial mechanisms [24]. In the adult brain, microglia participate in synaptic remodeling through the engulfment of synaptic components, a process whose mechanism of action has yet to be determined. By evaluating microglia‒neuron interactions in the ventral hippocampus, researchers have shown that the conditional ablation of UCP2 in microglia leads to the accumulation of ROS and lysosome‒lipid droplet complexes, which mediate ventral hippocampal circuit function and anxiety-like behaviors [160]. Overall, these findings reveal extensive interactions among UCP2, microglia, and neurons, which form a signaling network for anxiety behavior. In comparison to other UCPs, UCP1 plays a role in reducing anxiety-like behaviors, a function independent of its activity in BAT [161]. Compared with normal subjects, smokers have higher levels of UCP1 expression in WAT, and studies have shown that nicotine induces browning of WAT through the kappa opioid receptor, which also serves as a channel for modulating brain mechanisms to combat obesity [162].
Nevertheless, UCPs additionally govern energy supply and mitochondrial dynamics, modulating brain network stability and synaptic plasticity, which in turn influences memory and learning. Drosophila melanogaster Uncoupling Protein-4A (UCP4A) is the first biochemically described UCP4 homolog in all species, and is possibly closely related to the manufacture of β-alanine and N-acetylaspartate in the central nervous system [163]. In summary, for the purpose of developing more individualized therapies, future research must further elucidate the intricate molecular processes and regulatory networks of UCPs in the neurological system.
Tumors
Research on UCPs in tumors has focused mainly on their role in energy metabolism, oxidative stress and apoptosis [164]. Previous research has found that UCP2 is overexpressed in a variety of tumors and is associated with metabolic reprogramming and chemotherapy resistance, whereas UCP1 may play a role in BAT-related tumors [165]. The following section summarizes the current regulatory mechanisms of UCPs and their potential applications in tumor treatment.
UCPs-mediated tumorigenesis and development
In tumor research, each subtype of UCPs has distinct modes of action. Recent research has revealed that activating thermogenic adipose tissue can successfully prevent and treat acute leukemia in mice, although UCP1 is expressed at low levels in the majority of cancers. These findings suggest that further study is essential to completely understand its role in tumors involving adipose tissue [166]. In contrast to UCP1, UCP2 is overexpressed in a variety of tumors, including lung, breast, and colon cancer. Numerous studies have shown that UCP2 shields tumor cells from oxidative damage and stimulates their survival and growth by eliminating the production of ROS and mitochondrial membrane potential. For instance, UCP2 expression is increased in melanoma cells, stimulating tumor expansion through the activation of the Akt/mTOR and ERK signaling pathways [167]. Additionally, elevated UCP2 expression is related to heightened tumor cell resistance to chemotherapeutic medications [168, 169]. Considering the complex mechanism of UCP2 in cancer metabolism, it is considered as a marker of tumor malignant potential [170]. A whole metabolome analysis of 20 human primary acute myeloid leukemia (AML) cases revealed that ROS increased UCP2 levels and AMPK phosphorylation, upregulated the expression of the glycolytic enzyme 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB3), and consequently exacerbated AML [171]. Similarly, oncogenic KRAS mutations are strongly associated with human pancreatic ductal adenocarcinoma (PDAC) and regulate the redox equilibrium by regulating glutamine metabolism. Recent studies have indicated that UCP2-mediated aspartate transport is essential for the ability of KRAS to regulate glutamine metabolism and support pancreatic cancer progression, suggesting that UCP2 could be a critical metabolic target for treating refractory pancreatic cancer [172]. In general, these findings reveal that UCP2 is broadly distributed in numerous organs and affects the evolution of several malignant tumors. In-depth study of the molecular mechanism of UCP2 is critical for future quasi-medical tumor treatment.
With respect to other UCPs, UCP3 is mostly expressed in skeletal muscle and heart, and its role in malignancies has received less attention. However, studies have shown that UCP3 modulates fatty acid oxidation, which affects tumor cell metabolism and survival. Furthermore, UCP4 and UCP5 influence tumor development and apoptosis in nervous system cancers such as gliomas by controlling mitochondrial function and lowering ROS levels. Furthermore, UCP4 and UCP5 levels affect the growth and death of nervous system tumors, including gliomas [173, 174]. Comprehensive studies into the specific activities of each UCP subtype in various types of tumors will assist with the development of innovative tumor therapy techniques.
Prospects of UCPs in tumor treatment and prognosis
Tumor escape and immunosuppression are challenging issues in tumor treatment. Myeloid-derived suppressor cells in the tumor microenvironment (TEM) reduce uncoupling protein 1 expression, thereby enhancing immunosuppressive activity, emphasizing the critical function of UCP1 in immune evasion [175]. The increased expression of UCP1 in abdominal subcutaneous fat could serve as a biomarker of early-stage pancreatic ductal adenocarcinoma, although more in-depth studies need to be conducted [176]. Nevertheless, a novel concept known as tumor cell “slimming” provides an emerging treatment strategy for tumor formation. Specifically, phospholipase C-like 1(PLCL1)/UCP1-mediated lipid browning decreases aberrant lipid accumulation, and therefore decreases the progression of clear cell renal cell carcinoma (ccRCC) [177]. Consistently, melatonin activated peroxisome proliferator-activated receptor γ coactivator 1A (PGC1A) and UCP1-dependent lipophagy and lipid browning programs, mediating “tumor slimming” to inhibit tumor progression, and revealing the therapeutic potential of monitoring and manipulating UCP1 in ccRCC [178]. Undoubtedly, this therapeutic method is based on the fundamental role of UCP1 and expands the new scope of UCP1 application. In another analysis of colon cancer prognosis, UCP1 was discovered to be an independent predictive factor, and its expression was identified as an important prognostic biomarker for colorectal cancer, highlighting that the brown fat-like phenotype is strongly associated to colorectal cancer development [179].
Although UCP2 has been shown to increase the emergence and development of numerous tumors, it also depicts the boundless possibilities of UCP2-targeted treatment options. Beikbaghban et al. reported that UCP2 levels varied dramatically among different types of cancer. Moreover, the abundance of UCP2 is altered by nutritional availability in multiple kinds of cancerous cells, highlighting the complexity and diversity of UCP2 in the regulation of cancer formation [170]. With genetic or pharmacological techniques, UCP2-reprogrammed TEM sensitizes melanoma to programmed cell death protein-1 blocking therapy, which leads to beneficial anti-tumor effects [9]. Additionally, the UCP2 inhibitor genipin, a natural cross-linker, also exhibits positive antiinflammatory, antitumor and neuroprotective effects and plays an active role in glioblastoma by inducing ferroptosis [180]. By restoring oxidative stress equilibrium, disrupting tumor energy metabolism, and enhancing the immune response, UCPs generally show considerable potential for the treatment of malignancies and the prevention of cancer growth. UCP-guided targeted medications or multitarget combination treatment techniques are predicted to emerge as innovative treatments in the future.
Concluding remarks and prospects
Agonists and inhibitors targeting UCPs have potential therapeutic value in the fields of cardiovascular disease, tumors and other metabolic diseases. The known UCPs regulators are included in Table 3. Additionally, through the modulation of UCPs, numerous medicinal compounds and preparations have been shown to be highly effective in preventing metabolic disorders and preserving physiological homeostasis. Current research has indicated that dolutegravir suppresses thermogenesis through modifying the expression of UCP1 in brown/beige adipocytes, contributing to weight gain [181]. Similarly, Cyclopia intermedia (Honeybush), Baicalein and Cordycepin have been shown to balance energy expenditure and prevent obesity through UCP1-mediated thermogenesis [182-184]. In line with these findings, Yiqi Huoxue granules, a traditional Chinese medicine formula, increase UCP2 levels, reducing hydrogen peroxide-induced apoptosis. TaoHeChengQi decotion improves chronic renal failure by regulating the PHD2/UCP1 and RIPK3/AKT/TGF-β pathways [185]. However, to determine their precise regulation and therapeutic application, more studies on the development of medications and reagents related to UCPs are needed.
Clinical trials of regulators involving UCPs
| Name | Identifier | Clinical phase | Conclusion | Reference |
|---|---|---|---|---|
| Tirzepatide | NCT04081337 | phase I | increased fat oxidation | [189] |
| Bisoprolol | NCT04823442 | a randomized crossover trial | blunted the mirabegron-stimulated thermogenesis | [190] |
| Rapamycin | CRAD001ADE12 | 2-year randomized controlled trial | centrally mediated reduced food intake and increased fat oxidation and mobilization | [191] |
| Boysenberry juice | UMIN000043476 | an open-label single-arm nonrandomized study | increase fat oxidation | [192] |
| High-dose glucocorticoid | NCT03269747 | a randomised, double-blinded cross-over trial | altering skeletal muscle calcium cycling | [193] |
| Glucose-dependent insulinotropic polypeptide | NCT03734718 | a randomized, placebo-controlled, double-blind, crossover study | affects Hepatic Fat and Brown Adipose Tissue Thermogenesis | [194] |
| Ginger | NCT02570633 | a randomized, double-blind, placebo-controlled clinical trial | not increase energy expenditure in female adults | [195] |
| Creatine | NCT04086381 | a double-blind, randomized, placebo-controlled, cross-over trial | not enhance BAT activation | [196] |
| Mirabegron | NCT03049462 | an open-label study | increases human brown fat, HDL cholesterol, and insulin sensitivity | [197] |
| Salt | - | a Randomized Placebo-Controlled Study | decreases Diet-Induced Thermogenesis | [198] |
Owing to the roles of UCPs in a variety of cellular biological processes, many innovative technologies and applications for their study have emerged [186]. For example, BML-260, a rhodanine derivative, has been identified as a JSP-1 inhibitor for the treatment of inflammatory and proliferative illnesses related to the dysregulation of JNK signaling. Recently, BML-260 was shown to have thermogenic effects independent of JSP-1 activation of UCP1, which has potential antiobesity implications [187]. More importantly, by computationally analysing the molecular structure of AAC and developing a corresponding mathematical model, the researchers found that mitochondrial uncouplers induce proton leakage by activating the ADP/ATP carrier (AAC) and UCP1, thereby paving the way for the development of novel activators that target both [33]. Moreover, UCP1 in ThermoMouse was used to track UCP1 expression. Translocator protein-18 kDa (TSPO) is located in the outer mitochondrial membrane, and TSPO-targeting PET can be used to image interscapular brown adipose tissue(iBAT). Cerenkov luminescence imaging of iBAT utilizing a TSPO-targeting PET probe in the UCP1 ThermoMouse could be a more efficient method for reflecting UCP1 expression [188].
In summary, UCP-targeting strategies for metabolic illnesses include gene regulation, medication intervention, physiological stimulation, and lifestyle changes, with the aim of regulating the expression and activity of UCPs to treat diseases such as obesity, diabetes and metabolic syndrome. Recently, intensified exploration in the UCPs field has provided new opportunities to improve cellular homeostasis and health and to further develop precision medicine targeting UCPs.
Acknowledgements
This review was supported by the Regional Innovation and Development Joint Fund of National Natural Science Foundation of China (No. U22A20269).
Author Contributions
Yu-Ting Liu and Qi-Zhu Tang contributed to the design and conception of the review. Ya-Jia Ding, Yan Che and Hong-Liang Qiu proofread and improved the content. Qi-Zhu Tang was responsible for the financial support, and all authors approved the final version.
Competing Interests
The authors have declared that no competing interests exist.
References
1. Chouchani ET, Kazak L, Spiegelman BM. New Advances in Adaptive Thermogenesis: UCP1 and Beyond. Cell Metab. 2019;29:27-37
2. Toda C, Kim JD, Impellizzeri D, Cuzzocrea S, Liu ZW, Diano S. UCP2 Regulates Mitochondrial Fission and Ventromedial Nucleus Control of Glucose Responsiveness. Cell. 2016;164:872-83
3. Ramage LE, Akyol M, Fletcher AM, Forsythe J, Nixon M, Carter RN. et al. Glucocorticoids Acutely Increase Brown Adipose Tissue Activity in Humans, Revealing Species-Specific Differences in UCP-1 Regulation. Cell Metab. 2016;24:130-41
4. Berardi MJ, Shih WM, Harrison SC, Chou JJ. Mitochondrial uncoupling protein 2 structure determined by NMR molecular fragment searching. Nature. 2011;476:109-13
5. Szabo I, Zoratti M. Now UCP(rotein), Now You Don't: UCP1 Is Not Mandatory for Thermogenesis. Cell Metab. 2017;25:761-2
6. Jastroch M. Uncoupling protein 1 controls reactive oxygen species in brown adipose tissue. Proc Natl Acad Sci U S A. 2017;114:7744-6
7. Gorgoglione R, Porcelli V, Santoro A, Daddabbo L, Vozza A, Monne M. et al. The human uncoupling proteins 5 and 6 (UCP5/SLC25A14 and UCP6/SLC25A30) transport sulfur oxyanions, phosphate and dicarboxylates. Biochim Biophys Acta Bioenerg. 2019;1860:724-33
8. Jones SA, Ruprecht JJ, Crichton PG, Kunji ERS. Structural mechanisms of mitochondrial uncoupling protein 1 regulation in thermogenesis. Trends Biochem Sci. 2024;49:506-19
9. Cheng WC, Tsui YC, Ragusa S, Koelzer VH, Mina M, Franco F. et al. Uncoupling protein 2 reprograms the tumor microenvironment to support the anti-tumor immune cycle. Nat Immunol. 2019;20:206-17
10. Pravednikova AE, Shevchenko SY, Kerchev VV, Skhirtladze MR, Larina SN, Kachaev ZM. et al. Association of uncoupling protein (Ucp) gene polymorphisms with cardiometabolic diseases. Mol Med. 2020;26:51
11. Jia P, Wu X, Pan T, Xu S, Hu J, Ding X. Uncoupling protein 1 inhibits mitochondrial reactive oxygen species generation and alleviates acute kidney injury. EBioMedicine. 2019;49:331-40
12. Blanc J, Alves-Guerra MC, Esposito B, Rousset S, Gourdy P, Ricquier D. et al. Protective role of uncoupling protein 2 in atherosclerosis. Circulation. 2003;107:388-90
13. Kwok KH, Ho PW, Chu AC, Ho JW, Liu HF, Yiu DC. et al. Mitochondrial UCP5 is neuroprotective by preserving mitochondrial membrane potential, ATP levels, and reducing oxidative stress in MPP+ and dopamine toxicity. Free Radic Biol Med. 2010;49:1023-35
14. Crivelli SM, Gaifullina A, Chatton JY. Exploring the role of mitochondrial uncoupling protein 4 in brain metabolism: implications for Alzheimer's disease. Front Neurosci. 2024;18:1483708
15. Nicholls DG, Rial E. A novel regulatory mechanism for the brown-fat uncoupling protein? Nat Struct Mol Biol. 2016;23:364-5
16. Lim JH, Kim DG, Yu DY, Kang HM, Noh KH, Kim DS. et al. Stabilization of E2-EPF UCP protein is implicated in hepatitis B virus-associated hepatocellular carcinoma progression. Cell Mol Life Sci. 2019;76:2647-62
17. Burgess L, Jones AR, Hay S, Natrajan LS. Evaluating spectral overlap with the degree of quenching in UCP luminescence energy transfer systems. Methods Appl Fluoresc. 2019;7:034003
18. Gaudry MJ, Jastroch M. Hotly awaited structures obtained for the human protein UCP1. Nature. 2023;620:42-3
19. Jia X, Luo Z, Gao Y, Liu H, Liu X, Mai W. et al. EGCG Upregulates UCP(3) Levels to Protect MIN(6) Pancreatic Islet Cells from Interleukin-1beta-Induced Apoptosis. Drug Des Devel Ther. 2020;14:4251-61
20. Kang Y, Chen L. Structural basis for the binding of DNP and purine nucleotides onto UCP1. Nature. 2023;620:226-31
21. Chouchani ET, Kazak L, Jedrychowski MP, Lu GZ, Erickson BK, Szpyt J. et al. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature. 2016;532:112-6
22. Jones SA, Gogoi P, Ruprecht JJ, King MS, Lee Y, Zogg T. et al. Structural basis of purine nucleotide inhibition of human uncoupling protein 1. Sci Adv. 2023;9:eadh4251
23. Wang L, Yu C, You T, Zhang X, Su H, Cao B. et al. Injection of ROS-Responsive Hydrogel Loaded with IL-1beta-targeted nanobody for ameliorating myocardial infarction. Bioact Mater. 2025;46:273-84
24. Kim JD, Yoon NA, Jin S, Diano S. Microglial UCP2 Mediates Inflammation and Obesity Induced by High-Fat Feeding. Cell Metab. 2019;30:952-62 e5
25. Kutsche HS, Schreckenberg R, Weber M, Hirschhauser C, Rohrbach S, Li L. et al. Alterations in Glucose Metabolism During the Transition to Heart Failure: The Contribution of UCP-2. Cells. 2020 9
26. American Diabetes A. Statement of Retraction. Zhonglin Xie, Junhua Zhang, Jiliang Wu, Benoit Viollet, and Ming-Hui Zou. Upregulation of Mitochondrial Uncoupling Protein-2 by the AMP-Activated Protein Kinase in Endothelial Cells Attenuates Oxidative Stress in Diabetes. Diabetes 2008;57:3222-3230. DOI: 10.2337/db08-0610. PMID: 18835932. PMCID: PMC2584127. Diabetes. 2023;72:1039
27. Smorodchenko A, Rupprecht A, Fuchs J, Gross J, Pohl EE. Role of mitochondrial uncoupling protein 4 in rat inner ear. Mol Cell Neurosci. 2011;47:244-53
28. Alves-Bezerra M, Cosentino-Gomes D, Vieira LP, Rocco-Machado N, Gondim KC, Meyer-Fernandes JR. Identification of uncoupling protein 4 from the blood-sucking insect Rhodnius prolixus and its possible role on protection against oxidative stress. Insect Biochem Mol Biol. 2014;50:24-33
29. Perreten Lambert H, Zenger M, Azarias G, Chatton JY, Magistretti PJ, Lengacher S. Control of mitochondrial pH by uncoupling protein 4 in astrocytes promotes neuronal survival. J Biol Chem. 2014;289:31014-28
30. Ikeda K, Kang Q, Yoneshiro T, Camporez JP, Maki H, Homma M. et al. UCP1-independent signaling involving SERCA2b-mediated calcium cycling regulates beige fat thermogenesis and systemic glucose homeostasis. Nat Med. 2017;23:1454-65
31. Reyad-Ul-Ferdous M, Gul I, Raheem MA, Pandey V, Qin P. Mitochondrial UCP1: Potential thermogenic mechanistic switch for the treatment of obesity and neurodegenerative diseases using natural and epigenetic drug candidates. Phytomedicine. 2024;130:155672
32. Hass DT, Barnstable CJ. Uncoupling proteins in the mitochondrial defense against oxidative stress. Prog Retin Eye Res. 2021;83:100941
33. Bertholet AM, Natale AM, Bisignano P, Suzuki J, Fedorenko A, Hamilton J. et al. Mitochondrial uncouplers induce proton leak by activating AAC and UCP1. Nature. 2022;606:180-7
34. Ikeda K, Maretich P, Kajimura S. The Common and Distinct Features of Brown and Beige Adipocytes. Trends Endocrinol Metab. 2018;29:191-200
35. Fromme T, Kleigrewe K, Dunkel A, Retzler A, Li Y, Maurer S. et al. Degradation of brown adipocyte purine nucleotides regulates uncoupling protein 1 activity. Mol Metab. 2018;8:77-85
36. Shi M, Huang XY, Ren XY, Wei XY, Ma Y, Lin ZZ. et al. AIDA directly connects sympathetic innervation to adaptive thermogenesis by UCP1. Nat Cell Biol. 2021;23:268-77
37. Tharp KM, Kang MS, Timblin GA, Dempersmier J, Dempsey GE, Zushin PH. et al. Actomyosin-Mediated Tension Orchestrates Uncoupled Respiration in Adipose Tissues. Cell Metab. 2018;27:602-15 e4
38. Robinson EL, Bagchi RA, Major JL, Bergman BC, Matsuda JL, McKinsey TA. HDAC11 inhibition triggers bimodal thermogenic pathways to circumvent adipocyte catecholamine resistance. J Clin Invest. 2023 133
39. Rahbani JF, Bunk J, Lagarde D, Samborska B, Roesler A, Xiao H. et al. Parallel control of cold-triggered adipocyte thermogenesis by UCP1 and CKB. Cell Metab. 2024;36:526-40 e7
40. Son MJ, Oh KJ, Park A, Kwon MG, Suh JM, Kim IC. et al. GATA3 induces the upregulation of UCP-1 by directly binding to PGC-1alpha during adipose tissue browning. Metabolism. 2020;109:154280
41. Zhang Z, Cui Y, Su V, Wang D, Tol MJ, Cheng L. et al. A PPARgamma/long noncoding RNA axis regulates adipose thermoneutral remodeling in mice. J Clin Invest. 2023 133
42. Kovacova Z, Tharp WG, Liu D, Wei W, Xie H, Collins S. et al. Adipose tissue natriuretic peptide receptor expression is related to insulin sensitivity in obesity and diabetes. Obesity (Silver Spring). 2016;24:820-8
43. Chen HF, Hsu CM, Huang YS. CPEB2-dependent translation of long 3'-UTR Ucp1 mRNA promotes thermogenesis in brown adipose tissue. EMBO J. 2018 37
44. Xue K, Wu D, Wang Y, Zhao Y, Shen H, Yao J. et al. The mitochondrial calcium uniporter engages UCP1 to form a thermoporter that promotes thermogenesis. Cell Metab. 2022;34:1325-41 e6
45. Johnson JM, Peterlin AD, Balderas E, Sustarsic EG, Maschek JA, Lang MJ. et al. Mitochondrial phosphatidylethanolamine modulates UCP1 to promote brown adipose thermogenesis. Sci Adv. 2023;9:eade7864
46. Cohen P, Kajimura S. The cellular and functional complexity of thermogenic fat. Nat Rev Mol Cell Biol. 2021;22:393-409
47. Betz MJ, Enerback S. Targeting thermogenesis in brown fat and muscle to treat obesity and metabolic disease. Nat Rev Endocrinol. 2018;14:77-87
48. Lu WH, Chen HF, King PC, Peng C, Huang YS. CPEB2-activated Prdm16 translation promotes brown adipocyte function and prevents obesity. Mol Metab. 2024;89:102034
49. Da Eira D, Jani S, Ceddia RB. An obesogenic diet impairs uncoupled substrate oxidation and promotes whitening of the brown adipose tissue in rats. J Physiol. 2023;601:69-82
50. Herrnhold M, Hamp I, Plettenburg O, Jastroch M, Keuper M. Adverse bioenergetic effects of N-acyl amino acids in human adipocytes overshadow beneficial mitochondrial uncoupling. Redox Biol. 2023;66:102874
51. Takeda Y, Dai P. Chronic Fatty Acid Depletion Induces Uncoupling Protein 1 (UCP1) Expression to Coordinate Mitochondrial Inducible Proton Leak in a Human-Brown-Adipocyte Model. Cells. 2022 11
52. Gagelin A, Largeau C, Masscheleyn S, Piel MS, Calderon-Mora D, Bouillaud F. et al. Molecular determinants of inhibition of UCP1-mediated respiratory uncoupling. Nat Commun. 2023;14:2594
53. Musiol E, Fromme T, Hau J, Di Pizio A, Klingenspor M. Comparative functional analysis reveals differential nucleotide sensitivity between human and mouse UCP1. Acta Physiol (Oxf). 2024;240:e14209
54. Ukropec J, Anunciado RP, Ravussin Y, Hulver MW, Kozak LP. UCP1-independent thermogenesis in white adipose tissue of cold-acclimated Ucp1-/- mice. J Biol Chem. 2006;281:31894-908
55. Shamsi F, Xue R, Huang TL, Lundh M, Liu Y, Leiria LO. et al. FGF6 and FGF9 regulate UCP1 expression independent of brown adipogenesis. Nat Commun. 2020;11:1421
56. Keipert S, Lutter D, Schroeder BO, Brandt D, Stahlman M, Schwarzmayr T. et al. Endogenous FGF21-signaling controls paradoxical obesity resistance of UCP1-deficient mice. Nat Commun. 2020;11:624
57. Keipert S, Kutschke M, Lamp D, Brachthauser L, Neff F, Meyer CW. et al. Genetic disruption of uncoupling protein 1 in mice renders brown adipose tissue a significant source of FGF21 secretion. Mol Metab. 2015;4:537-42
58. Keipert S, Kutschke M, Ost M, Schwarzmayr T, van Schothorst EM, Lamp D. et al. Long-Term Cold Adaptation Does Not Require FGF21 or UCP1. Cell Metab. 2017;26:437-46 e5
59. Williams JW, Elvington A, Ivanov S, Kessler S, Luehmann H, Baba O. et al. Thermoneutrality but Not UCP1 Deficiency Suppresses Monocyte Mobilization Into Blood. Circ Res. 2017;121:662-76
60. Iwase M, Tokiwa S, Seno S, Mukai T, Yeh YS, Takahashi H. et al. Glycerol kinase stimulates uncoupling protein 1 expression by regulating fatty acid metabolism in beige adipocytes. J Biol Chem. 2020;295:7033-45
61. Zhai X, Dang L, Wang S, Sun C. The SIRT5-Mediated Upregulation of C/EBPbeta Promotes White Adipose Tissue Browning by Enhancing UCP1 Signaling. Int J Mol Sci. 2024 25
62. Sugawara S, Kanamaru Y, Sekine S, Maekawa L, Takahashi A, Yamamoto T. et al. The mitochondrial protein PGAM5 suppresses energy consumption in brown adipocytes by repressing expression of uncoupling protein 1. J Biol Chem. 2020;295:5588-601
63. Kim K, Wann J, Kim HG, So J, Rosen ED, Roh HC. Uncoupling protein 1-driven Cre (Ucp1-Cre) is expressed in the epithelial cells of mammary glands and various non-adipose tissues. Mol Metab. 2024;84:101948
64. Gorski T, Mathes S, Krutzfeldt J. Uncoupling protein 1 expression in adipocytes derived from skeletal muscle fibro/adipogenic progenitors is under genetic and hormonal control. J Cachexia Sarcopenia Muscle. 2018;9:384-99
65. Yuliana A, Jheng HF, Kawarasaki S, Nomura W, Takahashi H, Ara T. et al. beta-adrenergic Receptor Stimulation Revealed a Novel Regulatory Pathway via Suppressing Histone Deacetylase 3 to Induce Uncoupling Protein 1 Expression in Mice Beige Adipocyte. Int J Mol Sci. 2018 19
66. Wu Q, Liang X, Wang K, Lin J, Wang X, Wang P. et al. Intestinal hypoxia-inducible factor 2alpha regulates lactate levels to shape the gut microbiome and alter thermogenesis. Cell Metab. 2021;33:1988-2003 e7
67. Han B, Li ZM, Zhao XY, Liang K, Mao YQ, Zhang SL. et al. Annonaceous acetogenins mimic AA005 targets mitochondrial trifunctional enzyme alpha subunit to treat obesity in male mice. Nat Commun. 2024;15:9100
68. Ji A, Chen W, Zhang T, Shi R, Wang X, Wang Y. et al. Whey protein and soy protein prevent obesity by upregulating uncoupling protein 1 to activate brown adipose tissue and promote white adipose tissue browning in high-fat diet-fed mice. Food Funct. 2022;13:12836-51
69. Li T, Gao J, Du M, Song J, Mao X. Milk Fat Globule Membrane Attenuates High-Fat Diet-Induced Obesity by Inhibiting Adipogenesis and Increasing Uncoupling Protein 1 Expression in White Adipose Tissue of Mice. Nutrients. 2018 10
70. Jedrychowski MP, Lu GZ, Szpyt J, Mariotti M, Garrity R, Paulo JA. et al. Facultative protein selenation regulates redox sensitivity, adipose tissue thermogenesis, and obesity. Proc Natl Acad Sci U S A. 2020;117:10789-96
71. Vargas-Castillo A, Sun Y, Smythers AL, Grauvogel L, Dumesic PA, Emont MP. et al. Development of a functional beige fat cell line uncovers independent subclasses of cells expressing UCP1 and the futile creatine cycle. Cell Metab. 2024;36:2146-55 e5
72. Zhao P, Li X, Yang Q, Lu Y, Wang G, Yang H. et al. Malvidin alleviates mitochondrial dysfunction and ROS accumulation through activating AMPK-alpha/UCP2 axis, thereby resisting inflammation and apoptosis in SAE mice. Front Pharmacol. 2022;13:1038802
73. Tripathi R, Banerjee SK, Nirala JP, Mathur R. Exposure to Electromagnetic Fields from Mobile Phones and Fructose consumption Coalesce to Perturb Metabolic Regulators AMPK/SIRT1-UCP2/FOXO1 in Growing Rats. Biomed Environ Sci. 2023;36:1045-58
74. Kreiter J, Tyschuk T, Pohl EE. Uncoupling Protein 3 Catalyzes the Exchange of C4 Metabolites Similar to UCP2. Biomolecules. 2023 14
75. Silvestri E, Senese R, De Matteis R, Cioffi F, Moreno M, Lanni A. et al. Absence of uncoupling protein 3 at thermoneutrality influences brown adipose tissue mitochondrial functionality in mice. FASEB J. 2020;34:15146-63
76. Lomax TM, Ashraf S, Yilmaz G, Harmancey R. Loss of Uncoupling Protein 3 Attenuates Western Diet-Induced Obesity, Systemic Inflammation, and Insulin Resistance in Rats. Obesity (Silver Spring). 2020;28:1687-97
77. Tews D, Pula T, Funcke JB, Jastroch M, Keuper M, Debatin KM. et al. Elevated UCP1 levels are sufficient to improve glucose uptake in human white adipocytes. Redox Biol. 2019;26:101286
78. Okagawa S, Sakaguchi M, Okubo Y, Takekuma Y, Igata M, Kondo T. et al. Hepatic SerpinA1 improves energy and glucose metabolism through regulation of preadipocyte proliferation and UCP1 expression. Nat Commun. 2024;15:9585
79. Keinan O, Valentine JM, Xiao H, Mahata SK, Reilly SM, Abu-Odeh M. et al. Glycogen metabolism links glucose homeostasis to thermogenesis in adipocytes. Nature. 2021;599:296-301
80. Korobkina ED, Calejman CM, Haley JA, Kelly ME, Li H, Gaughan M. et al. Brown fat ATP-citrate lyase links carbohydrate availability to thermogenesis and guards against metabolic stress. Nat Metab. 2024;6:2187-202
81. Habtemichael EN, Li DT, Camporez JP, Westergaard XO, Sales CI, Liu X. et al. Insulin-stimulated endoproteolytic TUG cleavage links energy expenditure with glucose uptake. Nat Metab. 2021;3:378-93
82. Karlina R, Flexeder C, Musiol S, Bhattacharyya M, Schneider E, Altun I. et al. Differential effects of lung inflammation on insulin resistance in humans and mice. Allergy. 2022;77:2482-97
83. Li M, Li L, Li B, Hambly C, Wang G, Wu Y. et al. Brown adipose tissue is the key depot for glucose clearance in microbiota depleted mice. Nat Commun. 2021;12:4725
84. Chen B, Bai Y, Tong F, Yan J, Zhang R, Zhong Y. et al. Glycoursodeoxycholic acid regulates bile acids level and alters gut microbiota and glycolipid metabolism to attenuate diabetes. Gut Microbes. 2023;15:2192155
85. Pahlavani M, Ramalingam L, Miller EK, Scoggin S, Menikdiwela KR, Kalupahana NS. et al. Eicosapentaenoic Acid Reduces Adiposity, Glucose Intolerance and Increases Oxygen Consumption Independently of Uncoupling Protein 1. Mol Nutr Food Res. 2019;63:e1800821
86. Mo J, Enkhjargal B, Travis ZD, Zhou K, Wu P, Zhang G. et al. Corrigendum to "AVE 0991 attenuates oxidative stress and neuronal apoptosis via Mas/PKA/CREB/UCP-2 pathway after subarachnoid hemorrhage in rats" [Redox Biol. 20 (2019) 75-86]. Redox Biol. 2024;73:103177
87. Kitamura H, Fujimoto M, Hashimoto M, Yasui H, Inanami O. USP2 Mitigates Reactive Oxygen Species-Induced Mitochondrial Damage via UCP2 Expression in Myoblasts. Int J Mol Sci. 2024 25
88. Barreto P, Dambire C, Sharma G, Vicente J, Osborne R, Yassitepe J. et al. Mitochondrial retrograde signaling through UCP1-mediated inhibition of the plant oxygen-sensing pathway. Curr Biol. 2022;32:1403-11 e4
89. Feng C, Anger EE, Zhang X, Su S, Su C, Zhao S. et al. Protective Effects of Mitochondrial Uncoupling Protein 2 against Aristolochic Acid I-Induced Toxicity in HK-2 Cells. Int J Mol Sci. 2022 23
90. Masania J, Wijten P, Keipert S, Ost M, Klaus S, Rabbani N. et al. Decreased methylglyoxal-mediated protein glycation in the healthy aging mouse model of ectopic expression of UCP1 in skeletal muscle. Redox Biol. 2023;59:102574
91. Bai Y, Li K, Li X, Chen X, Zheng J, Wu F. et al. Effects of oxidative stress on hepatic encephalopathy pathogenesis in mice. Nat Commun. 2023;14:4456
92. Rangarajan S, Locy ML, Chanda D, Kurundkar A, Kurundkar D, Larson-Casey JL. et al. Mitochondrial uncoupling protein-2 reprograms metabolism to induce oxidative stress and myofibroblast senescence in age-associated lung fibrosis. Aging Cell. 2022;21:e13674
93. Inoue R, Tsuno T, Togashi Y, Okuyama T, Sato A, Nishiyama K. et al. Uncoupling protein 2 and aldolase B impact insulin release by modulating mitochondrial function and Ca(2+) release from the ER. iScience. 2022;25:104603
94. Luo Y, Li J, Zheng L, Reyimjan Y, Ma Y, Huang S. et al. Procyanidin B2 improves developmental capacity of bovine oocytes via promoting PPARgamma/UCP1-mediated uncoupling lipid catabolism during in vitro maturation. Cell Prolif. 2024;57:e13687
95. Yuliana A, Daijo A, Jheng HF, Kwon J, Nomura W, Takahashi H. et al. Endoplasmic Reticulum Stress Impaired Uncoupling Protein 1 Expression via the Suppression of Peroxisome Proliferator-Activated Receptor gamma Binding Activity in Mice Beige Adipocytes. Int J Mol Sci. 2019 20
96. Mo J, Enkhjargal B, Travis ZD, Zhou K, Wu P, Zhang G. et al. AVE 0991 attenuates oxidative stress and neuronal apoptosis via Mas/PKA/CREB/UCP-2 pathway after subarachnoid hemorrhage in rats. Redox Biol. 2019;20:75-86
97. Wu FF, Liu BZ, Wang RQ, Huang YQ, Liu H, Ni ZW. et al. Mitochondrial Ucp4 Ameliorates Motor Disorders by Protecting Cerebellar Purkinje Cells from Oxidative Stress in Intermittent Hypobaric Hypoxia Mice. Antioxid Redox Signal. 2025
98. Selenscig D, Ferreira MDR, Chicco A, Lombardo YB. Dietary fish oil ameliorates adipose tissue dysfunction in insulin-resistant rats fed a sucrose-rich diet improving oxidative stress, peroxisome proliferator-activated receptor gamma and uncoupling protein 2. Food Funct. 2018;9:2496-507
99. Oliveira TE, Castro E, Belchior T, Andrade ML, Chaves-Filho AB, Peixoto AS. et al. Fish Oil Protects Wild Type and Uncoupling Protein 1-Deficient Mice from Obesity and Glucose Intolerance by Increasing Energy Expenditure. Mol Nutr Food Res. 2019;63:e1800813
100. Shen H, Jiang L, Lin JD, Omary MB, Rui L. Brown fat activation mitigates alcohol-induced liver steatosis and injury in mice. J Clin Invest. 2019;129:2305-17
101. van Dierendonck X, Sancerni T, Alves-Guerra MC, Stienstra R. The role of uncoupling protein 2 in macrophages and its impact on obesity-induced adipose tissue inflammation and insulin resistance. J Biol Chem. 2020;295:17535-48
102. Yabut JM, Desjardins EM, Chan EJ, Day EA, Leroux JM, Wang B. et al. Genetic deletion of mast cell serotonin synthesis prevents the development of obesity and insulin resistance. Nat Commun. 2020;11:463
103. Mills EL, Harmon C, Jedrychowski MP, Xiao H, Gruszczyk AV, Bradshaw GA. et al. Cysteine 253 of UCP1 regulates energy expenditure and sex-dependent adipose tissue inflammation. Cell Metab. 2022;34:140-57 e8
104. Richter HJ, Hauck AK, Batmanov K, Inoue SI, So BN, Kim M. et al. Balanced control of thermogenesis by nuclear receptor corepressors in brown adipose tissue. Proc Natl Acad Sci U S A. 2022;119:e2205276119
105. Luo J, Chen M, Ji H, Su W, Song W, Zhang D. et al. Hypolipidemic and Anti-Obesity Effect of Anserine on Mice Orally Administered with High-Fat Diet via Regulating SREBP-1, NLRP3, and UCP-1. Mol Nutr Food Res. 2024;68:e2300471
106. Yu J, Shi L, Lin W, Lu B, Zhao Y. UCP2 promotes proliferation and chemoresistance through regulating the NF-kappaB/beta-catenin axis and mitochondrial ROS in gallbladder cancer. Biochem Pharmacol. 2020;172:113745
107. Kim YH, Lee SH. TGF-beta/SMAD4 mediated UCP2 downregulation contributes to Aspergillus protease-induced inflammation in primary bronchial epithelial cells. Redox Biol. 2018;18:104-13
108. Gupta AK, Ghosh K, Palit S, Barua J, Das PK, Ukil A. Leishmania donovani inhibits inflammasome-dependent macrophage activation by exploiting the negative regulatory proteins A20 and UCP2. FASEB J. 2017;31:5087-101
109. Gu P, Hui X, Zheng Q, Gao Y, Jin L, Jiang W. et al. Mitochondrial uncoupling protein 1 antagonizes atherosclerosis by blocking NLRP3 inflammasome-dependent interleukin-1beta production. Sci Adv. 2021;7:eabl4024
110. Faas M, Ipseiz N, Ackermann J, Culemann S, Gruneboom A, Schroder F. et al. IL-33-induced metabolic reprogramming controls the differentiation of alternatively activated macrophages and the resolution of inflammation. Immunity. 2021;54:2531-46 e5
111. Mills EL, Harmon C, Jedrychowski MP, Xiao H, Garrity R, Tran NV. et al. UCP1 governs liver extracellular succinate and inflammatory pathogenesis. Nat Metab. 2021;3:604-17
112. Xiong W, Xiong Z, Song A, Lei C, Ye C, Zhang C. Relieving lipid accumulation through UCP1 suppresses the progression of acute kidney injury by promoting the AMPK/ULK1/autophagy pathway. Theranostics. 2021;11:4637-54
113. Yang Q, Yang S, Liang Y, Sun Q, Fang Y, Jiang L. et al. UCP2 deficiency impairs podocyte autophagy in diabetic nephropathy. Biochim Biophys Acta Mol Basis Dis. 2023;1869:166705
114. Moon JS, Lee S, Park MA, Siempos II, Haslip M, Lee PJ. et al. UCP2-induced fatty acid synthase promotes NLRP3 inflammasome activation during sepsis. J Clin Invest. 2023 133
115. Ikeda K, Yamada T. UCP1 Dependent and Independent Thermogenesis in Brown and Beige Adipocytes. Front Endocrinol (Lausanne). 2020;11:498
116. Della Guardia L, Luzi L, Codella R. Muscle-UCP3 in the regulation of energy metabolism. Mitochondrion. 2024;76:101872
117. Gomez-Hernandez A, Perdomo L, de las Heras N, Beneit N, Escribano O, Otero YF. et al. Antagonistic effect of TNF-alpha and insulin on uncoupling protein 2 (UCP-2) expression and vascular damage. Cardiovasc Diabetol. 2014;13:108
118. Forte M, Bianchi F, Cotugno M, Marchitti S, Stanzione R, Maglione V. et al. An interplay between UCP2 and ROS protects cells from high-salt-induced injury through autophagy stimulation. Cell Death Dis. 2021;12:919
119. Schiffer TA, Lof L, Gallini R, Kamali-Moghaddam M, Carlstrom M, Palm F. Mitochondrial Respiration-Dependent ANT2-UCP2 Interaction. Front Physiol. 2022;13:866590
120. Zhao C, Yang Z, Chen Z, Liang W, Gong S, Du Z. AAV-ie-mediated UCP2 overexpression accelerates inner hair cell loss during aging in vivo. Mol Med. 2022;28:124
121. Kim S, Yazawa T, Koide A, Yoneda E, Aoki R, Okazaki T. et al. Potential Role of Pig UCP3 in Modulating Adipocyte Browning via the Beta-Adrenergic Receptor Signaling Pathway. Biology (Basel). 2024 13
122. Gonzalez-Muniesa P, Martinez-Gonzalez MA, Hu FB, Despres JP, Matsuzawa Y, Loos RJF. et al. Obesity. Nat Rev Dis Primers. 2017;3:17034
123. da Fonseca ACP, Assis I, Salum KCR, Palhinha L, Abreu GM, Zembrzuski VM. et al. Genetic variants in DBC1, SIRT1, UCP2 and ADRB2 as potential biomarkers for severe obesity and metabolic complications. Front Genet. 2024;15:1363417
124. Wang T, Sharma AK, Wu C, Maushart CI, Ghosh A, Yang W. et al. Single-nucleus transcriptomics identifies separate classes of UCP1 and futile cycle adipocytes. Cell Metab. 2024;36:2130-45 e7
125. Kakarla M, Puppala VK, Tyagi S, Anger A, Repp K, Wang J. et al. Circulating levels of mitochondrial uncoupling protein 2, but not prohibitin, are lower in humans with type 2 diabetes and correlate with brachial artery flow-mediated dilation. Cardiovasc Diabetol. 2019;18:148
126. Busceti CL, Cotugno M, Bianchi F, Forte M, Stanzione R, Marchitti S. et al. Brain Overexpression of Uncoupling Protein-2 (UCP2) Delays Renal Damage and Stroke Occurrence in Stroke-Prone Spontaneously Hypertensive Rats. Int J Mol Sci. 2020 21
127. De Miguel C, Hamrick WC, Sedaka R, Jagarlamudi S, Asico LD, Jose PA. et al. Uncoupling Protein 2 Increases Blood Pressure in DJ -1 Knockout Mice. J Am Heart Assoc. 2019;8:e011856
128. Luo JY, Cheng CK, He L, Pu Y, Zhang Y, Lin X. et al. Endothelial UCP2 Is a Mechanosensitive Suppressor of Atherosclerosis. Circ Res. 2022;131:424-41
129. Perez-Ternero C, Aubdool AA, Makwana R, Sanger GJ, Stimson RH, Chan LF. et al. C-type natriuretic peptide is a pivotal regulator of metabolic homeostasis. Proc Natl Acad Sci U S A. 2022;119:e2116470119
130. Stachowicz A, Wisniewska A, Czepiel K, Pomierny B, Skorkowska A, Kusnierz-Cabala B. et al. Mitochondria-targeted hydrogen sulfide donor reduces atherogenesis by changing macrophage phenotypes and increasing UCP1 expression in vascular smooth muscle cells. Biomed Pharmacother. 2024;180:117527
131. Hong K, Wang J, Kang X, Xue H, Gao Y, Liang H. et al. Ferulic acid and protocatechuic acid alleviate atherosclerosis by promoting UCP1 expression to inhibit the NLRP3-IL-1beta signaling pathway. Food Funct. 2025;16:40-53
132. Chen Y, Liu J, Zheng Y, Wang J, Wang Z, Gu S. et al. Uncoupling protein 3 mediates H(2)O(2) preconditioning-afforded cardioprotection through the inhibition of MPTP opening. Cardiovasc Res. 2015;105:192-202
133. Edwards KS, Ashraf S, Lomax TM, Wiseman JM, Hall ME, Gava FN. et al. Uncoupling protein 3 deficiency impairs myocardial fatty acid oxidation and contractile recovery following ischemia/reperfusion. Basic Res Cardiol. 2018;113:47
134. Marti-Pamies I, Thoonen R, Morley M, Graves L, Tamez J, Caplan A. et al. Brown Adipose Tissue and BMP3b Decrease Injury in Cardiac Ischemia-Reperfusion. Circ Res. 2023;133:353-65
135. Kwok TC, Ramage LE, Kelman A, Suchacki KJ, Gray C, Boyle LD. et al. UCP1 expression in human brown adipose tissue is inversely associated with cardiometabolic risk factors. Eur J Endocrinol. 2024;191:106-15
136. Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB. et al. Identification and importance of brown adipose tissue in adult humans. N Engl J Med. 2009;360:1509-17
137. Kurian J, Yuko AE, Kasatkin N, Rigaud VOC, Busch K, Harlamova D. et al. Uncoupling protein 2-mediated metabolic adaptations define cardiac cell function in the heart during transition from young to old age. Stem Cells Transl Med. 2021;10:144-56
138. Esfandiary A, Kutsche HS, Schreckenberg R, Weber M, Pak O, Kojonazarov B. et al. Protection against pressure overload-induced right heart failure by uncoupling protein 2 silencing. Cardiovasc Res. 2019;115:1217-27
139. Chen X, Ashraf S, Ashraf N, Harmancey R. UCP3 (Uncoupling Protein 3) Insufficiency Exacerbates Left Ventricular Diastolic Dysfunction During Angiotensin II-Induced Hypertension. J Am Heart Assoc. 2021;10:e022556
140. Wang Y, Tan J, Li L, Liu S, Li X, Shan H. et al. Uncoupling protein 3 protects against pathological cardiac hypertrophy via downregulation of aspartate. J Mol Cell Cardiol. 2025;202:1-12
141. Pan JA, Zhang H, Lin H, Gao L, Zhang HL, Zhang JF. et al. Irisin ameliorates doxorubicin-induced cardiac perivascular fibrosis through inhibiting endothelial-to-mesenchymal transition by regulating ROS accumulation and autophagy disorder in endothelial cells. Redox Biol. 2021;46:102120
142. Hu C, Zhang X, Wei W, Zhang N, Wu H, Ma Z. et al. Matrine attenuates oxidative stress and cardiomyocyte apoptosis in doxorubicin-induced cardiotoxicity via maintaining AMPKalpha/UCP2 pathway. Acta Pharm Sin B. 2019;9:690-701
143. Scheltens P, De Strooper B, Kivipelto M, Holstege H, Chetelat G, Teunissen CE. et al. Alzheimer's disease. Lancet. 2021;397:1577-90
144. Jucker M, Walker LC. Alzheimer's disease: From immunotherapy to immunoprevention. Cell. 2023;186:4260-70
145. He Y, Yang X, Jiao M, Anoopkumar-Dukie S, Zeng Y, Mei H. Housefly (Musca domestica) larvae powder, preventing oxidative stress injury via regulation of UCP4 and CyclinD1 and modulation of JNK and P38 signaling in APP/PS1 mice. Food Funct. 2019;10:235-43
146. Jun Z, Ibrahim MM, Dezheng G, Bo Y, Qiong W, Yuan Z. UCP2 protects against amyloid beta toxicity and oxidative stress in primary neuronal culture. Biomed Pharmacother. 2015;74:211-4
147. Galmes S, Palou A, Serra F. Increased Risk of High Body Fat and Altered Lipid Metabolism Associated to Suboptimal Consumption of Vitamin A Is Modulated by Genetic Variants rs5888 (SCARB1), rs1800629 (UCP1) and rs659366 (UCP2). Nutrients. 2020 12
148. Rosenberg N, Reva M, Binda F, Restivo L, Depierre P, Puyal J. et al. Overexpression of UCP4 in astrocytic mitochondria prevents multilevel dysfunctions in a mouse model of Alzheimer's disease. Glia. 2023;71:957-73
149. Wang W, Su L, Wang Y, Li C, Ji F, Jiao J. Endothelial Cells Mediated by UCP2 Control the Neurogenic-to-Astrogenic Neural Stem Cells Fate Switch During Brain Development. Adv Sci (Weinh). 2022;9:e2105208
150. Bloem BR, Okun MS, Klein C. Parkinson's disease. Lancet. 2021;397:2284-303
151. Reich SG, Savitt JM. Parkinson's Disease. Med Clin North Am. 2019;103:337-50
152. Xu S, Yang X, Qian Y, Xiao Q. Parkinson's disease-related DJ-1 modulates the expression of uncoupling protein 4 against oxidative stress. J Neurochem. 2018;145:312-22
153. Bai Y, Bai Y, Wang S, Wu F, Wang DH, Chen J. et al. Targeted upregulation of uncoupling protein 2 within the basal ganglia output structure ameliorates dyskinesia after severe liver failure. Free Radic Biol Med. 2018;124:40-50
154. Hass DT, Barnstable CJ. Mitochondrial Uncoupling Protein 2 Knock-out Promotes Mitophagy to Decrease Retinal Ganglion Cell Death in a Mouse Model of Glaucoma. J Neurosci. 2019;39:3582-96
155. Naia L, Ly P, Mota SI, Lopes C, Maranga C, Coelho P. et al. The Sigma-1 Receptor Mediates Pridopidine Rescue of Mitochondrial Function in Huntington Disease Models. Neurotherapeutics. 2021;18:1017-38
156. Wang L, Li X, Chen L, Mei S, Shen Q, Liu L. et al. Mitochondrial Uncoupling Protein-2 Ameliorates Ischemic Stroke by Inhibiting Ferroptosis-Induced Brain Injury and Neuroinflammation. Mol Neurobiol. 2025;62:501-17
157. He M, Ma Y, Wang R, Zhang J, Jing L, Li PA. Deletion of Mitochondrial Uncoupling Protein 2 Exacerbates Mitochondrial Damage in Mice Subjected to Cerebral Ischemia and Reperfusion Injury under both Normo- and Hyperglycemic Conditions. Int J Biol Sci. 2020;16:2788-802
158. Yan X, He M, Huang H, Wang Q, Hu Y, Wang X. et al. Endogenous H(2)S targets mitochondria to promote continual phagocytosis of erythrocytes by microglia after intracerebral hemorrhage. Redox Biol. 2022;56:102442
159. Wang Y, Huang C, Wang X, Cheng R, Li X, Wang J. et al. Succinate Activates Uncoupling Protein 2 to Suppress Neuroinflammation and Confer Protection Following Intracerebral Hemorrhage. Antioxid Redox Signal. 2024
160. Yasumoto Y, Stoiljkovic M, Kim JD, Sestan-Pesa M, Gao XB, Diano S. et al. Ucp2-dependent microglia-neuronal coupling controls ventral hippocampal circuit function and anxiety-like behavior. Mol Psychiatry. 2021;26:2740-52
161. Sideromenos S, Gundacker A, Nikou M, Oberle R, Horvath O, Stoehrmann P. et al. Uncoupling Protein-1 Modulates Anxiety-Like Behavior in a Temperature-Dependent Manner. J Neurosci. 2022;42:7659-72
162. Seoane-Collazo P, Linares-Pose L, Rial-Pensado E, Romero-Pico A, Moreno-Navarrete JM, Martinez-Sanchez N. et al. Central nicotine induces browning through hypothalamic kappa opioid receptor. Nat Commun. 2019;10:4037
163. Lunetti P, Gorgoglione R, Curcio R, Marra F, Pignataro A, Vozza A. et al. Drosophila melanogaster Uncoupling Protein-4A (UCP4A) Catalyzes a Unidirectional Transport of Aspartate. Int J Mol Sci. 2022 23
164. Caggiano EG, Taniguchi CM. UCP2 and pancreatic cancer: conscious uncoupling for therapeutic effect. Cancer Metastasis Rev. 2024;43:777-94
165. Peixoto AS, Moreno MF, Castro E, Perandini LA, Belchior T, Oliveira TE. et al. Hepatocellular carcinoma induced by hepatocyte Pten deletion reduces BAT UCP-1 and thermogenic capacity in mice, despite increasing serum FGF-21 and iWAT browning. J Physiol Biochem. 2023;79:731-43
166. Chen R, Cheng T, Xie S, Sun X, Chen M, Zhao S. et al. Effective Prevention and Treatment of Acute Leukemias in Mice by Activation of Thermogenic Adipose Tissues. Adv Sci (Weinh). 2024;11:e2402332
167. Li J, Jia Y, An L, Niu C, Cong X, Zhao Y. Uncoupling protein 2 is upregulated in melanoma cells and contributes to the activation of Akt/mTOR and ERK signaling. Int J Oncol. 2020;56:1252-61
168. Park HK, Choi YD, Shim HJ, Choi Y, Chung IJ, Yun SJ. Comparative Whole-Genome Sequencing Analysis of In-situ and Invasive Acral Lentiginous Melanoma: Markedly Increased Copy Number Gains of GAB2, PAK1, UCP2, and CCND1 are Associated with Melanoma Invasion. Am J Surg Pathol. 2024;48:1061-71
169. Yu J, Shi L, Shen X, Zhao Y. UCP2 regulates cholangiocarcinoma cell plasticity via mitochondria-to-AMPK signals. Biochem Pharmacol. 2019;166:174-84
170. Beikbaghban T, Proietti L, Ebner J, Sango R, Rattei T, Weichhart T. et al. Differential regulation of mitochondrial uncoupling protein 2 in cancer cells. Biochim Biophys Acta Bioenerg. 2024;1865:149486
171. Robinson AJ, Hopkins GL, Rastogi N, Hodges M, Doyle M, Davies S. et al. Reactive Oxygen Species Drive Proliferation in Acute Myeloid Leukemia via the Glycolytic Regulator PFKFB3. Cancer Res. 2020;80:937-49
172. Raho S, Capobianco L, Malivindi R, Vozza A, Piazzolla C, De Leonardis F. et al. KRAS-regulated glutamine metabolism requires UCP2-mediated aspartate transport to support pancreatic cancer growth. Nat Metab. 2020;2:1373-81
173. Zhang J, Pan L, Zhang Q, Zhao Y, Wang W, Lin N. et al. MFN2 deficiency affects calcium homeostasis in lung adenocarcinoma cells via downregulation of UCP4. FEBS Open Bio. 2023;13:1107-24
174. Yamada H, Munetsuna E, Yamazaki M, Mizuno G, Sadamoto N, Ando Y. et al. Maternal fructose-induced oxidative stress occurs viaTfam and Ucp5 epigenetic regulation in offspring hippocampi. FASEB J. 2019;33:11431-42
175. Zhao J, Gu M, Zhang Y, Jia X, Xiao W, Lu G. et al. Myeloid-derived suppressor cells in the tumor microenvironment reduce uncoupling protein 1 expression to boost immunosuppressive activity. Biochem Biophys Res Commun. 2024;732:150408
176. Sah RP, Sharma A, Nagpal S, Patlolla SH, Sharma A, Kandlakunta H. et al. Phases of Metabolic and Soft Tissue Changes in Months Preceding a Diagnosis of Pancreatic Ductal Adenocarcinoma. Gastroenterology. 2019;156:1742-52
177. Xiong Z, Xiao W, Bao L, Xiong W, Xiao H, Qu Y. et al. Tumor Cell "Slimming" Regulates Tumor Progression through PLCL1/UCP1-Mediated Lipid Browning. Adv Sci (Weinh). 2019;6:1801862
178. Xiao W, Xiong Z, Xiong W, Yuan C, Xiao H, Ruan H. et al. Melatonin/PGC1A/UCP1 promotes tumor slimming and represses tumor progression by initiating autophagy and lipid browning. J Pineal Res. 2019;67:e12607
179. Alnabulsi A, Cash B, Hu Y, Silina L, Alnabulsi A, Murray GI. The expression of brown fat-associated proteins in colorectal cancer and the relationship of uncoupling protein 1 with prognosis. Int J Cancer. 2019;145:1138-47
180. Dong H, Sun K, Wang X, Cui M, Ma Y, Li K. et al. Repurposed genipin targeting UCP2 exhibits antitumor activity through inducing ferroptosis in glioblastoma. Acta Biochim Biophys Sin (Shanghai). 2024
181. Jung I, Tu-Sekine B, Jin S, Anokye-Danso F, Ahima RS, Brown TT. et al. Dolutegravir Suppresses Thermogenesis via Disrupting Uncoupling Protein 1 Expression and Mitochondrial Function in Brown/Beige Adipocytes in Preclinical Models. J Infect Dis. 2022;226:1626-36
182. Qi G, Zhou Y, Zhang X, Yu J, Li X, Cao X. et al. Cordycepin promotes browning of white adipose tissue through an AMP-activated protein kinase (AMPK)-dependent pathway. Acta Pharm Sin B. 2019;9:135-43
183. Reyad-Ul-Ferdous M, Song Y. Baicalein modulates mitochondrial function by upregulating mitochondrial uncoupling protein-1 (UCP1) expression in brown adipocytes, cytotoxicity, and computational studies. Int J Biol Macromol. 2022;222:1963-73
184. Jack BU, Ramharack P, Malherbe C, Gabuza K, Joubert E, Pheiffer C. Cyclopia intermedia (Honeybush) Induces Uncoupling Protein 1 and Peroxisome Proliferator-Activated Receptor Alpha Expression in Obese Diabetic Female db/db Mice. Int J Mol Sci. 2023 24
185. Liu W, Hu C, Qian X, He C, Gu R, Meng Z. et al. TaoHeChengQi Decotion alleviate chronic renal failure via regulation of PHD2/UCP1 and RIPK3/AKT/TGF-beta pathway. Phytomedicine. 2025;141:156548
186. Chen X, He X, Gao R, Lan X, Zhu L, Chen K. et al. Aptamer-Functionalized Binary-Drug Delivery System for Synergetic Obesity Therapy. ACS Nano. 2022;16:1036-50
187. Feng Z, Wei Y, Zhang Y, Qiu Y, Liu X, Su L. et al. Identification of a rhodanine derivative BML-260 as a potent stimulator of UCP1 expression. Theranostics. 2019;9:3501-14
188. Lee SY, Oh HR, Kim YH, Bae SH, Lee Y, Lee YS. et al. Cerenkov luminescence imaging of interscapular brown adipose tissue using a TSPO-targeting PET probe in the UCP1 ThermoMouse. Theranostics. 2022;12:6380-94
189. Ravussin E, Sanchez-Delgado G, Martin CK, Beyl RA, Greenway FL, O'Farrell LS. et al. Tirzepatide did not impact metabolic adaptation in people with obesity, but increased fat oxidation. Cell Metab. 2025;37:1060-74 e4
190. Dumont L, Caron A, Richard G, Croteau E, Fortin M, Frisch F. et al. The effects of the beta(1)-adrenergic receptor antagonist bisoprolol administration on mirabegron-stimulated human brown adipose tissue thermogenesis. Acta Physiol (Oxf). 2024;240:e14127
191. Mannaa M, Pfennigwerth P, Fielitz J, Gollasch M, Boschmann M. Mammalian target of rapamycin inhibition impacts energy homeostasis and induces sex-specific body weight loss in humans. J Cachexia Sarcopenia Muscle. 2023;14:2757-67
192. Furuuchi R, Kato S, Maejima D, Amano T, Fujiki S, Shimizu I. et al. Preliminary study on the effects of boysenberry juice intake on brown adipose tissue activity in healthy adults. Sci Rep. 2024;14:25259
193. Maushart CI, Sun W, Othman A, Ghosh A, Senn JR, Fischer JGW. et al. Effect of high-dose glucocorticoid treatment on human brown adipose tissue activity: a randomised, double-blinded, placebo-controlled cross-over trial in healthy men. EBioMedicine. 2023;96:104771
194. Heimburger SMN, Hoe B, Nielsen CN, Bergman NC, Skov-Jeppesen K, Hartmann B. et al. GIP Affects Hepatic Fat and Brown Adipose Tissue Thermogenesis but Not White Adipose Tissue Transcriptome in Type 1 Diabetes. J Clin Endocrinol Metab. 2022;107:3261-74
195. Braga Tibaes JR, Martins LB, Rodrigues A, Amaral MHA, Teixeira AL, Ferreira AVM. Ginger supplementation does not increase energy expenditure in female adults. Nutrition. 2022;103-104:111803
196. Connell NJ, Doligkeit D, Andriessen C, Kornips-Moonen E, Bruls YMH, Schrauwen-Hinderling VB. et al. No evidence for brown adipose tissue activation after creatine supplementation in adult vegetarians. Nat Metab. 2021;3:107-17
197. O'Mara AE, Johnson JW, Linderman JD, Brychta RJ, McGehee S, Fletcher LA. et al. Chronic mirabegron treatment increases human brown fat, HDL cholesterol, and insulin sensitivity. J Clin Invest. 2020;130:2209-19
198. Mahler A, Klamer S, Maifeld A, Bartolomaeus H, Marko L, Chen CY. et al. Increased Salt Intake Decreases Diet-Induced Thermogenesis in Healthy Volunteers: A Randomized Placebo-Controlled Study. Nutrients. 2022 14
Author contact
![]() Corresponding author: Qi-Zhu Tang, Department of Cardiology, Renmin Hospital of Wuhan University, Hubei Key Laboratory of Metabolic and Chronic Diseases, Wuhan University at Jiefang Road 238, Wuhan 430060, PR China; Tel.: +86 27 88073385; Fax: +86 27 88042292, E-mail: qztangedu.cn.
Corresponding author: Qi-Zhu Tang, Department of Cardiology, Renmin Hospital of Wuhan University, Hubei Key Laboratory of Metabolic and Chronic Diseases, Wuhan University at Jiefang Road 238, Wuhan 430060, PR China; Tel.: +86 27 88073385; Fax: +86 27 88042292, E-mail: qztangedu.cn.