Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2026; 22(7):3886-3908. doi:10.7150/ijbs.129636 This issue Cite
Research Paper
DHCR24 Deficiency Drives Age-Related Meibomian Gland Dysfunction by Regulating Lipid Metabolic Imbalance and Cytosolic mtDNA-Induced cGAS-STING Activation
Yuchen Cai1,2*, Tianyi Zhou1,2*, Jiaming Sun3*, Xueyao Cai3*, Wenjun Shi3, Junzhao Chen1,2, Christos C. Zouboulis4, Hao Sun1,2 ![]() , Yao Fu1,2
, Yao Fu1,2 ![]() , Liangbo Chen1,2
, Liangbo Chen1,2 ![]()
1. Department of Ophthalmology, Shanghai Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China.
2. Shanghai Key Laboratory of Orbital Diseases and Ocular Oncology, Shanghai, China.
3. Department of Plastic and Reconstructive Surgery, Shanghai Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai, China.
4. Departments of Dermatology, Venereology, Allergology and Immunology, Dessau Medical Center, Brandenburg Medical School Theodor Fontane and Faculty of Health Sciences Brandenburg, Dessau, Germany.
* These authors contributed equally to this work.
Received 2025-12-8; Accepted 2026-3-12; Published 2026-3-28
Abstract

Dysregulated lipid metabolism and chronic inflammation are hallmarks of aging, yet their interplay in age-related tissue disorders remains poorly defined. In the ocular surface, age-related meibomian gland dysfunction (ARMGD) is highly prevalent but mechanistically unclear, leading to significant visual impairment without targeted therapies. To identify key molecular drivers of ARMGD, we performed integrated multi-omics screening of aging mouse meibomian glands (MGs) and identified DHCR24, a key cholesterol metabolism enzyme, as a critical regulator of gland homeostasis. Single-cell sequencing identified age-associated downregulation of Dhcr24 predominantly in meibocytes. Based on this finding, we generated a meibocyte-specific Dhcr24 knockout (cKO) model, which exhibited typical ARMGD pathology including glandular atrophy, disrupted lipid homeostasis, and inflammatory activation. Further in vitro studies using SZ95 sebocytes demonstrated that DHCR24 deficiency induces mitochondrial dysfunction and cytosolic mitochondrial DNA (mtDNA) leakage, triggering cGAS/STING-dependent inflammatory senescence. Notably, AAV-mediated restoration of DHCR24 in mice reversed age-related gland pathology. Our findings establish DHCR24 as a dual-target regulator that maintains cholesterol metabolic homeostasis while suppressing mtDNA-driven inflammation via the cGAS-STING pathway, highlighting its therapeutic potential for ARMGD and related disorders characterized by lipid-inflammatory imbalance.
Keywords: DHCR24, aging, lipid dysregulation, meibomian glands, cGAS-STING, mtDNA
Introduction
Aging represents one of the most significant health challenges of the 21st century, driven by a progressive decline in cellular function and tissue homeostasis [1]. This process is characterized by typical features, including genomic instability, mitochondrial dysfunction, metabolic abnormalities, and chronic low-grade inflammation, which contribute to the pathogenesis of age-related diseases [2,3]. Among these, ocular conditions such as age-related macular degeneration (AMD), cataracts, and dry eye disease have emerged as major causes of visual impairment [4]. In particular, meibomian gland dysfunction (MGD) has gained increasing recognition as a highly prevalent yet mechanistically elusive disorder [5]. With aging being the primary risk factor for this condition, age-related MGD (ARMGD) now affects approximately 70% of the population aged over 60 years in the US, highlighting its clinical and public health significance [6,7].
ARMGD is a chronic and progressive condition characterized by abnormal lipid secretion and inflammation in the meibomian glands (MGs), significantly impacting patients' vision and quality of life [7]. As specialized sebaceous glands of the eyelid margin, MGs provide essential lipids via holocrine secretion to the tear film, which are crucial for preventing tear evaporation and maintaining ocular surface homeostasis [8]. Recent studies have identified significant age-dependent alterations in the lipid profiles of MGs, including imbalanced wax ester (WE) to cholesteryl ester (ChE) ratios, reduced phospholipid levels, and altered fatty acid profiles [9,10]. In parallel, aged MGs exhibit increased inflammation, marked by elevated proinflammatory cytokine release and nuclear factor-κB (NF-κB) activation, which drives acinar atrophy and gland dropout [11]. Current treatment modalities for ARMGD, such as warm compresses, eyelid hygiene, artificial tears, and anti-inflammatory agents, provide only symptomatic relief and fail to address the underlying pathogenic mechanisms [12,13]. While advanced therapies such as thermal pulsation and intense pulsed light (IPL) have shown some efficacy in improving meibum delivery, they cannot reverse glandular atrophy or restore normal meibum composition [14]. However, the intricate nature of the association between age-related lipid dysregulation and inflammatory activation in MGs remains poorly understood, posing a challenge for targeted therapeutic development.
The interplay between inflammation and lipid metabolism is increasingly recognized as a hallmark of aging across multiple organ systems. In conditions such as atherosclerosis, Alzheimer's disease, and metabolic syndrome, age-dependent activation of innate immune pathways, including the cyclic GMP-AMP synthase-stimulator of interferon genes (cGAS-STING) axis and NF-κB signaling, has been shown to disrupt lipid homeostasis, leading to cytotoxic lipid species accumulation, organelle stress, and tissue dysfunction [15-17]. Pro-inflammatory cytokines such as interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) not only amplify local inflammatory responses but also interfere with lipid synthesis and trafficking, resulting in altered lipid composition and impaired cellular function [18,19]. Central to this immunometabolic interplay are regulatory factors such as peroxisome proliferator activated receptors (PPARs), whose age-related decline compromises both lipid metabolism and inflammatory resolution [20]. Emerging evidence further underscores the vital role of cholesterol homeostasis in maintaining mitochondrial integrity. As a key component of mitochondrial membranes, cholesterol regulates membrane fluidity, permeability, and cristae architecture. Dysregulation of cholesterol metabolism can therefore compromise mitochondrial structure and function, potentially leading to the leakage of pro-inflammatory mitochondrial components, including mitochondrial DNA (mtDNA) [21-23]. Notably, mitochondrial dysfunction and subsequent mtDNA release have been identified as critical triggers of cGAS-STING activation in age-related diseases, establishing a link between metabolic stress and inflammation [24,25]. While these mechanisms are increasingly well-characterized in systemic and neurological disorders, their roles in ocular aging, particularly in ARMGD, remain largely unexplored. Given that MGs are highly active lipid-secreting organs that exhibit both inflammatory infiltration and lipid abnormalities with aging, it is plausible that similar immunometabolic crosstalk underlies their age-related decline. Therefore, a systematic investigation into how lipid dysregulation and inflammatory signaling impact MG function in ARMGD holds promise for identifying novel disease mechanisms for therapeutic intervention.
In this study, we employed integrative multi-omics screening of aging MGs and identified 24-dehydrocholesterol reductase (DHCR24), a key enzyme in cholesterol biosynthesis, as a pivotal regulator of ARMGD. To functionally characterize its role, we constructed a meibocyte-specific Dhcr24 conditional knockout (cKO) mouse model using a meibocyte-enriched promoter identified from our single-cell sequencing data. Genetic ablation of Dhcr24 recapitulated ARMGD pathology, including disrupted acinar architecture, aberrant lipid metabolism, and marked inflammatory activation. Mechanistically, DHCR24 deficiency induced mitochondrial dysfunction and cytosolic mtDNA leakage, leading to activation of the cGAS-STING signaling and subsequent NF-κB-mediated inflammation. Strikingly, AAV-mediated restoration of DHCR24 in mice effectively reversed age-related glandular pathology. Together, these results establish DHCR24 as a critical node linking cholesterol homeostasis, mitochondrial integrity, and inflammatory signaling in ARMGD, offering a mechanistic foundation for targeted therapies to restore MG function and potentially mitigate related age-associated disorders.
Materials and Methods
Animals models
Two-month and two-year-old female C57/BL6J mice were purchased from the Cyagen Biosciences Inc., China. We used female mice to maintain homogeneity in our model and to avoid potential confounding factors associated with male aggression or fluctuations in androgen levels. Ocular surface injury was induced using 0.2% benzalkonium chloride (BAC) eye drops, administered twice daily for 14 days [26]. Dhcr24fl/fl mice (Strain S-CKO-16104; Cyagen Biosciences) were engineered via the CRISPR/Cas9 system, which introduced two loxP sites flanking exon 3. Krt14-CreERT mice (C001297; Cyagen Biosciences) were crossed with Dhcr24fl/fl mice to produce Krt14-CreERT Dhcr24fl/fl (conditional knockout, cKO; Dhcr24-cKO) offspring, lacking Dhcr24 expression in K14 expressing meibocytes, with Dhcr24fl/fl mice serving as controls. Gene deletion was induced by administering tamoxifen (TAM) intraperitoneally at 20 mg/kg/day for 7 days.
All mice were maintained in pathogen-free environment and provided with a standard chow diet while subjected to a 12-hour light/dark cycle. Animals were randomly allocated to various experimental groups, ensuring that each cage housed mice from different groups. The procedures were conducted in accordance with the guidelines established by the Association for Research in Vision and Ophthalmology (ARVO) regarding the ethical use of animals in ophthalmic and vision research. Approval for all animal experiments was obtained from the Animal Ethics Committee of Shanghai Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine (SH9H-2024-A1387-1).
Immunohistochemistry (IHC) of patient samples
Human tarsal plate specimens were surgically obtained from a 3-year-old with congenital eyelid deformity and a 40-year-old with traumatic injury and were fixed in 4% paraformaldehyde. Informed consent was obtained from all patients or their legal representatives, and the study received ethical approval from the Institutional Review Board of Shanghai Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine (SH9H-2021-T171-1).
Paraffin-embedded human tarsal plates were sectioned at 4 µm thickness and deparaffinized. Antigen retrieval was performed using a 10 mM citrate buffer at 95°C for 20 minutes. Sections were treated with 3% hydrogen peroxide to block endogenous peroxidase activity and were incubated overnight at 4 °C with anti-DHCR24 (Bioss; #bs-5390R). An HRP-conjugated secondary antibody was then applied, followed by DAB for visualization. Images were processed using ImageJ (IHC Profiler plugin). The H-score was calculated using the formula: H-score = (% of cells at low positive × 1) + (% at positive × 2) + (% at high positive × 3).
Mouse ocular surface and MG clinical examinations
Corneal opacity and fluorescein test
A single ophthalmologist performed all examinations using a slit-lamp microscope to image mouse eyelid margins and corneas. Corneal opacity severity was evaluated following the Draize method, which involves multiplying the observed degree of opacity by the affected corneal area [9]. To assess corneal epithelial integrity, 1 µL of a 1% sodium fluorescein solution (Jingming, China) was applied to the conjunctival sac. After 1 minute, fluorescein staining was observed and photographed under cobalt blue light. Staining intensity was scored according to the following criteria: 0 (no staining), 0.5 (slight punctate staining), 1 (widespread punctate staining), 2 (staining covering less than one-third of the cornea), 3 (staining covering more than one-third), or 4 (staining covering more than two-thirds) [27].
Rose Bengal staining
To evaluate subtle corneal epithelial damage, 5 μL of a 0.25% Rose Bengal solution (Aladdin, China) was applied into the conjunctival sac, followed by gentle blinking to distribute the dye. After rinsing with 0.9% saline, residual staining was photographed under a slit-lamp microscope. Staining intensity was scored per quadrant: 0 (no staining), 1 (punctate staining), 2 (punctate staining covering < 50%), 3 (non-confluent staining covering > 50%), or 4 (confluent staining). Total scores were calculated as the sum of all quadrants.
Tear secretion test
Tear production was assessed using the non-anesthetic Schirmer I test [28]. Initially, excess tears were absorbed, and a phenol red cotton thread (Jingming, China) was placed in the lateral conjunctival sac for one minute. The wetted length (mm) was recorded three times per eye, and the average was calculated for analysis. Mice were gently held to minimize blinking during the measurements.
MG dissection and scoring
Following euthanasia, the upper and lower tarsal plates of the mice were dissected bilaterally and rinsed in phosphate-buffered saline (PBS). Under a microscope, micro-scissors were used to carefully separate the skin, subcutaneous tissue, and conjunctiva of the eyelids to obtain a pure sample of mouse MG tissue. MG morphology was photographed using a Keratograph (Oculus, Germany) and clinically scored as follows: 0 points for no irregularities or absence of MG; 1 point for irregular MGs without absence; 2 points for irregularities with less than one-third absence; and 3 points for irregularities with one-third or more absence. MG scores were assessed based on the photographs and presented as mean ± SD.
Intra-MG adeno-associated virus (AAV) injection
Recombinant adeno-associated virus rAAV2/8 designed for Dhcr24 overexpression was purchased from Genomeditech, China. Under anesthesia, 5 μL of rAAV2/8-DHCR24 (5 × 1012 VG/mL) or control AAV-vehicle was microinjected into the mouse MGs using a 34-gauge needle attached to a 10 μL microsyringe (Hamilton, Switzerland) under microscope. Two injections were performed at different points within each gland to ensure broader coverage. To analyze AAV tropism, mice were microinjected AAV-DHCR24 or saline solution, and MG sections were evaluated via direct fluorescence imaging two weeks post-injection.
Histological examinations
Mouse eyelid tissues were paraffin-embedded or cryopreserved in tissue-freezing medium. Sagittal sections (6 μm thick) were prepared from both tissue types. Paraffin-embedded sections were subjected to H&E staining. Cryopreserved eyelid sections underwent Oil Red O (ORO) analysis. Briefly, sections were fixed in 4% paraformaldehyde (15 min), rinsed with PBS, incubated with fresh ORO solution (Servicebio, China) for 15 min, counterstained with hematoxylin (2 min), and finally mounted using an aqueous medium. To evaluate potential systemic effects, major organs including skin, heart, liver, spleen, lung, and kidney were collected, paraffin-embedded, sectioned, and stained with H&E.
Transmission electron microscopy (TEM)
Freshly dissected MG tissues were immersed in 2.5% glutaraldehyde/0.1 M phosphate buffer (pH 7.4) for 24 hours at 4°C, followed by post-fixation with 1% osmium tetroxide for 2 hours. The samples were then dehydrated through a graded series of ethanol and embedded in Epon 812 epoxy resin. Ultrathin sections, measuring 70 nm, were cut using a Leica UC7 ultramicrotome, placed onto copper grids, and contrasted with uranyl acetate and lead citrate. Imaging was examined on a Hitachi HT7800 TEM.
Multi-omics analyses of mouse MGs
Transcriptomic profiling
Total RNA was isolated from mouse MGs using the RNeasy Micro Kit (Qiagen), with integrity verified on an Agilent 2100 Bioanalyzer. Sequencing libraries were constructed with the TruSeq Stranded mRNA Library Prep Kit (Illumina) and sequenced on an Illumina NovaSeq 6000 platform (Shanghai Newcore Biotech). The raw sequencing reads were mapped to the mouse reference genome (GRCm38) and subsequently analyzed using edgeR (v3.28.1) and limma (v3.42.2) in R (v4.0.2). Differentially expressed genes (DEGs) were identified using thresholds of |fold change (FC)| > 1.2 and p < 0.05. Functional enrichment analyses of Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) were performed using clusterProfiler (v4.0.5). Principal component analysis (PCA), volcano plots, and heatmaps were visualized with ggplot2 (v3.3.5). The correlation between Dhcr24 expression and lipid metabolism was assessed via Mantel test and Pearson correlation coefficient using the R package ggcor (v0.9.6).
Lipidomic profiling
Lipids were isolated from MG tissues using the methyl tert-butyl ether (MTBE) method. Briefly, tissue homogenization was performed in 400 μL ice-cold methanol spiked with internal standards, mixed with MTBE and centrifuged (14,000 × g, 15 min, 10°C). Lipid separation was achieved using an UltiMate 3000 UHPLC system (Thermo Fisher). The mobile phase gradient was optimized for effective lipid separation. Lipids were analyzed in both positive and negative ionization modes on a Q-Exactive Plus mass spectrometer (Thermo Fisher) by Shanghai Applied Protein Technology. LipidSearch (v4.2.3) software was utilized for lipid identification and quantification. Variable importance in projection (VIP) scores and orthogonal partial least squares discriminant analysis (OPLS-DA) and were calculated using MetaX (v1.6.0). Lipids meeting VIP > 1 and p < 0.05 were classified as differentially expressed. Lipid saturation was quantified via LipidSearch's fragmentation pattern analysis. GO, KEGG, and Gene Set Enrichment Analysis (GSEA) were performed for functional annotations. Sankey diagrams were created using the R package ggalluvial (v0.12.5), integrating lipid differential expression results with KEGG enrichment outcomes.
Proteomic profiling
MG tissues were lysed in RIPA buffer (Beyotime, China) containing PMSF, followed by ultrasonication. Protein concentration was quantified via the BCA assay. Proteins were subjected to tryptic digestion, further analyzed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) using a Thermo Scientific Orbitrap mass spectrometer (analysis performed by Shanghai OE Biotech). For TMT labeling, samples were resuspended in TEAB, mixed with acetonitrile, and incubated. Labeled peptides underwent reverse-phase separation via HPLC and were processed on a Q-Exactive mass spectrometer. Raw MS data were analyzed via Proteome Discoverer (v2.4). Search parameters included trypsin digestion, carbamidomethylation (static), and oxidation (M)/acetylation (N-term) (dynamic). Protein quantification required ≥ 2 unique peptides and a false discovery rate (FDR) < 1%. Proteins exhibiting |fold change (FC)| > 1.2 and p < 0.05 were designated as differentially expressed proteins (DEPs). PCA and functional enrichment (GO/KEGG) were performed as described for transcriptomics.
Single-cell RNA sequencing
Freshly dissected MGs from young (Y; 2-month-old, n = 1) and aged (O; 2-year-old, n = 1) mice were minced and digested. Dissociated cells were harvested and treated with Red Blood Cell Lysis Solution (Thermo Fisher) to remove erythrocytes. Cell viability (>85%) was confirmed via Trypan Blue staining using an automated cell counter. Single-cell suspensions were partitioned into Gel Beads-in-Emulsion (GEMs) using the Chromium Controller (10× Genomics) and Chromium Next GEM Single Cell 3′ Reagent Kits v3.1. cDNA synthesis and amplification were performed according to the manufacturer's protocol. Amplified cDNA libraries were quantified using a Bioanalyzer 2100 (Agilent) and Qubit 4.0 Fluorometer (Thermo Fisher). Paired-end sequencing (2 × 150 bp) was carried out on an Illumina NovaSeq 6000 platform. Raw sequencing reads (FASTQ files) were aligned to the mouse reference genome (mm10) using the STAR aligner. Raw data were processed and filtered using Cell Ranger v7.1.0 (10× Genomics) to remove empty droplets (criteria: nGene > 200, nUMI > 500, mitochondrial gene ratio < 20%). Unsupervised clustering based on transcriptional similarity partitioned cells into distinct subpopulations. Cluster-specific DEGs were identified via Seurat's FindAllMarkers. Dimensionality reduction was achieved through t-distributed stochastic neighbor embedding (t-SNE) and uniform manifold approximation and projection (UMAP) methods. Cell clusters were annotated using characteristic marker genes for each subpopulation, cross-referenced with the Cell Taxonomy database and established literature [29,30]. Expression levels of Krt14 and Dhcr24 in meibocyte were extracted, and Pearson correlation coefficients were calculated using ggpubr (v0.6.0) in R.
Cell culture and RNA interference
Immortalized human SZ95 sebaceous gland cells were maintained in DMEM (Gibco) containing 10% FBS (Gibco), 5 ng/mL recombinant human EGF (Peprotech), and 100 U/mL penicillin/streptomycin (Gibco) at 37 °C/5% CO2 [31]. The medium was replaced every other day. For DHCR24 knockdown, SZ95 sebocytes were transfected with 50 nM DHCR24-targeting siRNA (Genomeditech). Nontargeting siRNA (siNC) was used as control. The siDHCR24 sequence was as follows: 5′-CGCUUAUCUUCGAUAUCUAdTdT-3′ (forward), 5′-UAGAUAUCGAAGAUAAGCGdTdT-3′ (reverse). For the treatment of STING inhibitor H-151 (Targetmol, USA; #T5674), SZ95 cells were cultured with 1 µM H-151 for 12 h.
Immunofluorescence (IF)
Mouse MG cryosections were permeabilized (0.1% Triton X-100), blocked with 5% BSA, and incubated with primary antibodies against KRT14 (Bioss; #bsm-52054R), DHCR24 (Bioss; #bs-5390R), IL-6 (Proteintech; 21865-1-AP), IL-17 (Bioss; #bs-1183R), TNF-α (Proteintech; #17590-1-AP), NF-κB p65 (Abcam; #ab32536), phospho-NF-kB p65 (S536) (Abcam; #ab76302), and STING (Proteintech; #19851-1-AP) overnight. Alexa Fluor 488/594 secondary antibodies (Invitrogen) were applied for 1 hour, and nuclei were counterstained with DAPI. Images were acquired using a Nikon A1R confocal microscope, and fluorescence intensity was analyzed with ImageJ software.
For mitochondrial double-stranded DNA (dsDNA) detection, siRNA-transfected SZ95 cells were fixed and permeabilized by utilizing 0.1% Triton X-100 for 20 min. Following the blocking of non-specific binding with 2% BSA, the cells were incubated with Tom20 (Proteintech; #66777-1-Ig) and dsDNA (Progen; #AC-30-10) antibodies overnight at 4 °C. After applying an Alexa Fluor 488/594-conjugated secondary antibody for 1 hour, immunofluorescence images were captured using a Leica Stellaris 5 confocal microscope.
Quantitative reverse transcription-polymerase chain reaction (RT-qPCR)
Total RNA was extracted from MGs or cells using RNA extraction kit (TianGen). Subsequently, cDNA synthesis was performed with the PrimeScript RT Kit (Takara). For quantitative PCR, the ABI Prism 7000 (Applied Biosystems) was utilized, employing Hieff UNICON SYBR Green master mix (Yeasen) and conducting technical triplicates for accuracy. Normalization was achieved using the 2-ΔΔCt method relative to GAPDH, with primers sourced from Sangon Biotech (see Table S1).
Cytosolic fraction and mitochondrial DNA extraction
Cytosolic fraction was performed as previously described [32]. Briefly, SZ95 cells were harvested and centrifuged at 900g for 5 minutes. The supernatant was removed, and the cells were resuspended in PBS before being split into two aliquots of equal volume. After a second centrifugation at 600g for 5 minutes, the pellet from one tube was designated as the whole-cell fraction for subsequent measurement of nuclear DNA (nDNA) and mtDNA. The pellet from the other tube was incubated in 500 μL of buffer containing 25 mg/mL digitonin, 150 mM NaCl, and 50 mM HEPES (pH 7.4) for 10 minutes at room temperature. Following this, the cells were centrifuged at 150g at 4 °C. The supernatant was then centrifuged twice more at 150g at 4 °C and once at 17,000g for 10 minutes to obtain the cytosolic fraction. DNA Extraction was performed using DNA isolation kit (Tiangen) according to the maufacturer's instructions. DNA concentration was measured using the Nanodrop ND-1000 Spectrophotometer. qPCR was conducted on 10 ng of DNA using the SYBR qPCR master mix (Yeasen) on the ABI Prism 7000 instrument. The qPCR targets included mtDNA regions ND1 and DLOOP, as well as nDNA regions 18S and TERT. The cytosolic mtDNA to whole-cell nDNA ratio [33] and the cytosolic mtDNA to whole-cell mtDNA ratio were calculated [32,34].
Enzyme-linked immunosorbent assay (ELISA)
MG lysates were used to assess the levels of murine IL-6, IL-17, and TNF-α using commercial ELISA kits (ELK Biotechnology, USA; #ELK1157 for IL-6, #ELK1147 for IL-17, and #ELK1387 for TNF-α) following the manufacturer's instructions. Absorbance was measured with a microplate reader (ELX800, BioTek), and protein concentrations were calculated from a standard curve.
Western blot (WB)
Total tissue or cellular protein was extracted using RIPA lysis, followed by quantification using a BCA kit (Beyotime). The samples were then denatured by adding loading buffer and applying heat treatment. Electrophoresis was performed using sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), after which the proteins were transferred to a PVDF membrane (Millipore). Immunodetection involved blocking with 5% skim milk, followed by incubation with primary antibodies at 4℃ overnight. Primary antibodies included DHCR24 (Proteintech; #10471-1-AP), NF-κB p65 (Abcam; #ab32536), phospho-NF-kB p65 (S536) (Abcam; #ab76302), cGAS (Proteintech; #26416-1-AP), STING (Proteintech; #19851-1-AP), phospho-STING (Ser366) (CST; #85735), GAPDH (ABclonal; #AC002), and β-Actin (ABclonal; #AC004). Afterward, HRP-conjugated secondary antibodies were applied at room temperature for 1 h. Visualization was achieved using an ECL chemiluminescence (Beyotime), and images were captured with the ChemiDoc System (Bio-Rad).
Statistical analysis
Statistical analysis was performed using GraphPad Prism 7.0 and R version 4.0.4, with data presented as mean ± standard deviation (SD). Comparisons between two groups were conducted using Student's t-test, while comparisons among three or more groups utilized one-way analysis of variance (ANOVA). To account for multiple testing, the Benjamini-Hochberg method was applied. Significance thresholds were set at p < 0.05 (*); p < 0.01 (**); p < 0.001 (***), p < 0.0001 (****).
Results
Multi-omics profiling identified DHCR24 as a central regulator in ARMGD
To systematically identify molecular drivers of ARMGD, we conducted an integrated multi-omics analysis of MGs from young (2-month-old) and aged (2-year-old) mice. MGs from each age group (n = 15 per group) were subjected to lipidomic (6 vs. 6), proteomic (3 vs. 3), and transcriptomic (6 vs. 6) profiling, followed by systematic bioinformatics analysis (Fig. 1A). Before molecular analysis, ocular morphological examinations revealed age-related morphological degeneration characterized by glandular atrophy, ductal dropout, elevated MG pathology scores (Fig. S1A-C), and a significant reduction in lipid content as shown by ORO staining (Fig. S1D), consistent with clinical ARMGD features. Multi-omics datasets revealed clear molecular separation between young and aged MG samples (Fig. S2A-C). Lipidomic identified 46 significantly altered lipid species across major classes, including ChE, triglycerides (TG), sphingomyelin (SM), diglycerides (DG), and phospholipids (phosphatidylcholine [PC], phosphatidylethanolamine [PE], phosphatidylserine [PS]) (Fig. 1B). To visually integrate these findings, a Sankey diagram illustrates the flow from these dysregulated lipids to significantly enriched metabolic and inflammatory pathways, including cholesterol metabolism, glycerophospholipid metabolism, sphingolipid signaling, adipocytokine signaling, the NF-κB signaling pathway, autophagy, and efferocytosis (Fig. S3).
Multi-omics analyses identified dehydrocholesterol reductase 24 (DHCR24) as a biomarker of age-related meibomian gland dysfunction (ARMGD). (A) Multi-omics study design for biomarker screening. In general, meibomian glands (MGs) of two-month-old (n = 15) and two-year-old (n = 15) mice were dissected for lipidomics (6 vs. 6), proteomics (3 vs. 3), and transcriptomics (6 vs. 6) profiling, followed by systematic bioinformatics analyses for biomarker identification. (B) Heatmap visualization of 46 differentially expressed lipids in 7 classes (PE, SM, PS, PC, DG, ChE, and TG) between the two-month-old (Y) and two-year-old (O) groups. ChE, cholesteryl esters; DG, diglyceride; PE, phosphatidylethanolamine; PC, phosphatidylcholine; PS, phosphatidylserine; SM, sphingomyelin; TG, triglyceride. (C) Integrated proteomics and transcriptomics analyses identified the shared significant pathways (*p < 0.05; dashed line) in three categories, including human diseases, organismal systems, and metabolism. (D) Combined transcriptomics and proteomics analyses of steroid biosynthesis pathway identified DHCR24 as the only biomarker differentially expressed in both mRNA and protein levels between the two-month-old and two-year-old groups. (E) Schematic diagram for subsequent DHCR24 verification, including immunohistochemistry (IHC) for paraffin-embedded human tarsal plate samples, and Western blot (WB) and immunofluorescence (IF) for mouse MGs. (F, G) IHC staining and H-score for DHCR24 in 3-year-old and 40-year-old human tarsal plate samples. Scale bars: 50 μm. (H, I) IF and relative fluorescence intensity for DHCR24 in two-month-old and two-year-old mouse MGs. Scale bars: 50 μm. (J, K) DHCR24 protein expression in two-month-old and two-year-old mouse MGs.
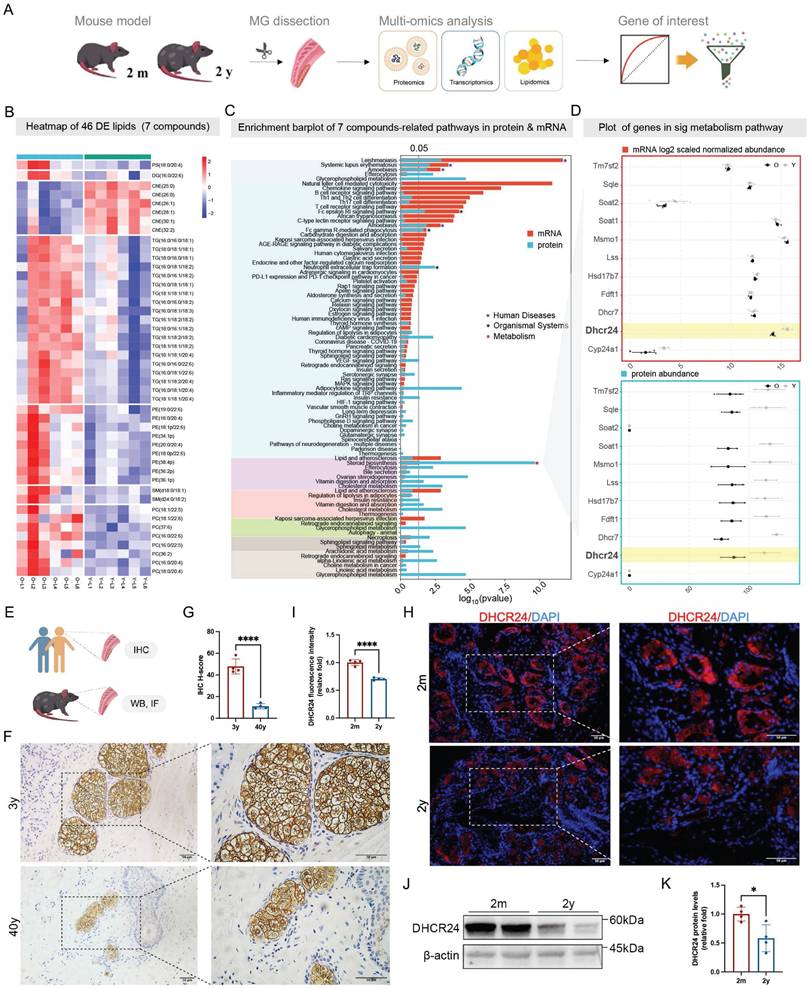
Moreover, integrated pathway analysis of transcriptomic and proteomic data highlighted significant dysregulation in immune-inflammation networks, such as chemokine signaling, T cell receptor signaling pathway, alongside lipid metabolic pathways including cholesterol metabolism, steroid biosynthesis, and glycerophospholipid metabolism (Fig. 1C). Notably, ChE levels were markedly reduced in aged mice (Fig. S4A), and pathway enrichment analyses highlighted disruptions in cholesterol metabolism and steroid biosynthesis at both the proteomic and transcriptomic levels (Fig. 1C and Fig. S4A). Strikingly, DHCR24, a key enzyme in cholesterol and steroid biosynthesis, emerged as the only gene/protein consistently downregulated at both the mRNA and protein levels in aged MGs (Fig. 1D), underscoring its potential role as a key regulator of age-related lipid dysfunction. In addition, the analysis of downregulated lipid classes, including DG, PC, PE, PS, SM, and TG, revealed their association with enriched metabolic and inflammatory signaling, such as glycerophospholipid metabolism, chemokine signaling pathway, necroptosis, and cAMP signaling pathway, which were further supported by transcriptomic and proteomic evidence (Fig. S4B).
Validation studies in human and murine tarsal plates confirmed age-dependent decline in DHCR24 (Fig. 1E). IHC of human samples revealed significantly lower DHCR24 levels in MGs from a 40-year-old donor compared to a 3-year-old child, as quantified by H-score (Fig. 1F, G). Consistent with this, IF and WB analyses in mice demonstrated a pronounced reduction of DHCR24 in aged (two-year-old) MGs relative to young (two-month-old) controls, evidenced by decreased fluorescence intensity (Fig. 1H, I) and diminished protein expression (Fig. 1J, K). Collectively, these results suggest DHCR24 as a pivotal factor in MG homeostasis during aging, with its loss linked to MGD pathogenesis.
Single-cell sequencing indicated meibocyte-specific Dhcr24 downregulation in aging MGs and the construction of meibocyte-specific Dhcr24-cKO mouse model
To elucidate the cellular origins and functional implications of DHCR24, we performed single-cell RNA sequencing (scRNA-seq) on MGs from young and aged mice and generated meibocyte-specific Dhcr24-cKO mice using the Cre-LoxP system (Fig. 2A). Dimensional reduction of the scRNA-seq data was performed using PCA (Fig. S5A). UMAP and t-SNE analysis identified 37 distinct cellular clusters (Fig. 2B, Fig. S5B), further annotated into 14 subpopulations according to distinct marker genes (Fig. S5C), including meibocytes, ductal epithelial cells, conjunctival cells, dermal papillae and sheath cells, endothelial cells (including lymphatic subtypes), immune cells (macrophages/dendritic cells, T cells, neutrophils), fibroblasts, melanocytes, pericytes, and hair follicle cells (Fig. 2C and Fig. S5D). Notably, meibocytes, the lipid-secreting epithelial population critical for MG function, were markedly reduced in aged MGs, while conjunctival cells, T cells, and melanocytes exhibited age-dependent increase (Fig. 2D). Dhcr24 expression was highly enriched in meibocyte-associated clusters (clusters 8, 16, and 22; Fig. 2E) but significantly downregulated in meibocytes of aged mice (Fig. 2F and Fig. S5E), indicating its essential role in meibocyte function during aging. Further functional analysis revealed significant dysregulation of multiple pathways in aged meibocytes, including cholesterol homeostasis, sterol biosynthesis, gland development, response to oxidative stress, mitochondrial organization, regulation of inflammatory responses, cellular aging, and key pathways such as NIK/NF-κB, PPAR, Wnt, and Hippo signaling pathways (Fig. 2G, H). These findings highlight the complex molecular landscape affecting meibocyte function and MG health in aging.
Construction and validation of Dhcr24 conditional knockout (cKO) mice. (A) Study design of the single-cell RNA sequencing (scRNA-seq) of mouse MGs and construction of meibocyte-specific cKO mice. In brief, meibomian glands (MGs) of two-month (n = 1) and two-year-old (n = 1) female mice were dissected for scRNA-seq to identify the Dhcr24-expressing cells, followed by the construction of Dhcr24-cKO mice based on Cre-LoxP system. (B, C) Uniform manifold approximation and projection (UMAP) of young (Y) and aged (O) mouse MGs (B) classified MG cells into 37 distinct clusters, (C) annotated into 14 major cell types, with Dhcr24 predominantly expressed in meibocytes. (D) Cell proportions for 14 cell types in MGs. (E) Scatter plot of Dhcr24 expression in each cell cluster. (F) Volcano plot of differentially expressed genes (DEGs) in meibocytes comparing O vs. Y mice, showing significantly reduced Dhcr24 expression. (G, H) Functional annotation of (G) Gene ontology (GO) and (H) Kyoto encyclopedia of genes and genomes (KEGG) for the DEGs in the meibocyte populations. (I) Pearson correlation analysis for the correlation between Dhcr24 and Krt14 expression in meibocyte. (J) Immunofluorescence (IF) staining confirmed co-localization (white arrows) of DHCR24 and KRT14 in two-month-old MG sections. Scale bars: 50 μm. (K) IF and (N) relative fluorescence intensity for DHCR24 in Dhcr24-cKO and Dhcr24fl/fl mouse MGs. Scale bars: 50 μm. (L) Western blot comparing DHCR24 expression in MGs of Dhcr24-cKO mice with wild type (WT) and Dhcr24fl/fl mice. (M) Normalized Dhcr24 gene expression in Dhcr24-cKO vs. Dhcr24fl/fl MGs from transcriptome analysis.
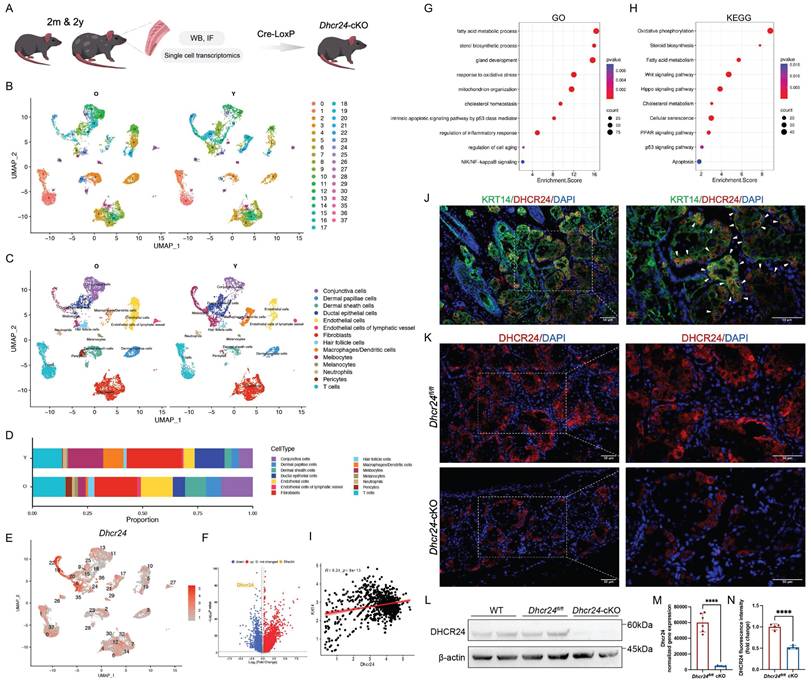
Keratin 14 (KRT14), a structural component of intermediate filaments essential for ocular surface integrity, is broadly expressed during MG development and critical for gland homeostasis [35]. Further UMAP visualization showed significant Krt14 expression in meibocytes (Fig. S5F). Given the strong transcriptional correlation between Dhcr24 and Krt14 in meibocytes (Fig. 2I), we validated their co-localization in young murine MGs via IF co-staining (Fig. 2J). This finding provided evidence for employing the Krt14 promoter to drive Cre recombinase expression and construct meibocyte-specific Dhcr24-cKO model (Krt14-CreERT Dhcr24fl/fl; Schematic diagram of the conditional deletion shown in Fig. S6). Efficient Dhcr24 knockout was confirmed through IF, WB, and transcriptomics analyses compared to Dhcr24fl/fl or wild type (WT) controls (Fig. 2K-N).
Meibocyte-specific Dhcr24-cKO mice exhibited marked MGD phenotypes and exacerbated BAC-induced ocular damage
Two-month-old Dhcr24-cKO (n = 10) and Dhcr24fl/fl (n = 10) mice were treated with TAM and raised for an additional 3 months to allow for phenotype development. Throughout this period, Dhcr24-cKO mice exhibited normal growth comparable to their Dhcr24fl/fl littermates without premature mortality or obvious signs of systemic distress (Fig. S7A). Body weight monitoring revealed no significant differences between the genotypes (Fig. S7B). H&E staining of tissues, including the liver, heart, kidney, spleen, lung, and skin, confirmed normal tissue architecture without inflammation, degeneration, or neoplasia (Fig. S7C), confirming the absence of systemic toxicity or health decline in Dhcr24-cKO mice. Comprehensive ocular surface assessments were then conducted using slit-lamp biomicroscopy, corneal fluorescein/Rose Bengal staining, MG morphological analysis, and lipid metabolic/inflammatory profiling (Fig. 3A). Both groups exhibited clear corneas with comparable opacity scores (Fig. 3B, D), but fluorescein staining revealed mild corneal epithelial defects in Dhcr24-cKO mice (Fig. 3B), confirmed by significantly elevated fluorescein scores (Fig. 3E). Minimal Rose Bengal staining was observed in Dhcr24fl/fl mice, while Dhcr24-cKO mice showed prominent staining (Fig. 3B, F), reflecting tear film instability and impaired epithelial barrier of the ocular surface. Notably, Schirmer's test demonstrated no differences in basal tear secretion between groups (Fig. 3G), suggesting that epithelial damage in Dhcr24-cKO mice arises primarily from abnormal lipid secretion and ocular surface integrity rather than reduced aqueous tear production.
Phenotypes of meibomian gland dysfunction (MGD) in meibocyte-specific Dhcr24 conditional knockout (cKO) mice. (A) Experimental timeline for the ocular surface assessments in Dhcr24-cKO mice. Two-month-old Dhcr24-cKO and Dhcr24fl/fl mice were injected with tamoxifen (TAM) for 7 days and raised for 3 months before evaluation of ocular surface phenotype and analysis of lipid metabolic and inflammatory alterations. (B) Slit lamp images showing representative white light photos, fluorescein staining, and Rose Bengal staining of Dhcr24fl/fl and Dhcr24-cKO mouse corneas. (C) Representative images of the upper tarsal plate for Dhcr24fl/fl and Dhcr24-cKO mice, with arrows indicating truncated meibomian glands (MGs). (D-H) Ocular surface and MG scoring comparisons between Dhcr24fl/fl and Dhcr24-cKO mouse, including (D) corneal opacity score (n = 10), (E) corneal fluorescein staining score (n = 10), (F) Rose Bengal staining score (n = 10), (G) relative tear secretion (n = 10), and (H) clinical MG score (n = 6). (I) Upper tarsal plate weight comparison between Dhcr24fl/fl and Dhcr24-cKO mice (n = 10). (J) Hematoxylin-eosin (H&E) staining of the upper tarsal plate, with a dotted circle indicating a single MG acinus (ac) and arrows pointing to disrupted basement membrane and gland structures. (K) Oil Red O staining of upper tarsal plate sections, with arrows highlighting aberrant lipid accumulation. (L) Experiment timeline for BAC treatment and ocular surface assessments. Two-month-old female Dhcr24-cKO and Dhcr24fl/fl mice were injected with TAM for 7 days and raised for 2 weeks before administered with BAC or phosphate-buffered saline (PBS) eye drops, followed by comprehensive phenotype evaluation. (M) Slit lamp images showing representative white light photos, fluorescein staining, and Rose Bengal staining of the Dhcr24fl/fl with PBS, Dhcr24-cKO with PBS, Dhcr24fl/fl with BAC, and Dhcr24-cKO with BAC. (N-Q) Ocular surface and MG scoring for the treatment groups, including (N) corneal opacity score (n = 5), (O) corneal fluorescein staining score (n = 5), (P) Rose Bengal staining score (n = 5), and (Q) relative tear secretion (n = 5).
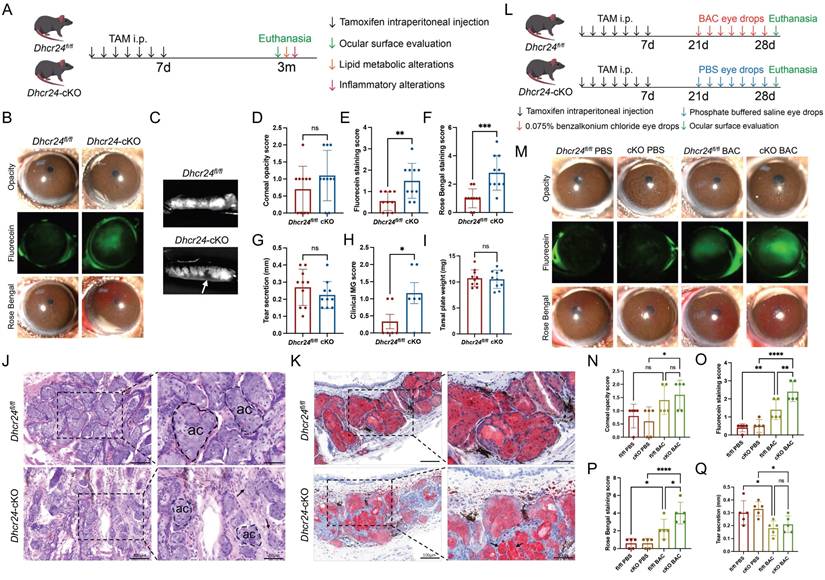
Morphological analysis of upper eyelids revealed clear contrasts in MG structure. Dhcr24fl/fl mice exhibited well-organized acinar structures, whereas Dhcr24-cKO mice demonstrated focal glandular atrophy and ductal dropout (Fig. 3C). These structural abnormalities correlated with significantly elevated MG pathology scores in Dhcr24-cKO mice (Fig. 3H), despite comparable total gland weights between groups (Fig. 3I), implying localized rather than systemic gland atrophy. Histological H&E evaluation further confirmed these findings, showing disrupted basement membranes and reduced, irregularly shaped acini in Dhcr24-cKO MGs (Fig. 3J). ORO staining highlighted diminished lipid content within Dhcr24-cKO acini and aberrant lipid droplet accumulation in ducts (Fig. 3K). Collectively, these results establish that meibocyte-specific Dhcr24 deletion leads to MGD features, including glandular atrophy, lipid secretion defects, and ocular surface compromise. This phenotype closely resembles ARMGD, emphasizing DHCR24's essential role in preserving MG functional integrity and ocular surface homeostasis.
To further investigate the implications of DHCR24 deficiency, we employed a BAC-induced ocular surface injury model, a well-established approach for studying dry eye disease pathogenesis. Following TAM induction, two-month-old Dhcr24-cKO (n = 5) and Dhcr24fl/fl mice (n = 5) were administered BAC eye drops twice daily for two weeks, while an equal number of Dhcr24-cKO and Dhcr24fl/fl mice received PBS as the control group (Fig. 3L). After BAC treatment, Dhcr24-cKO mice exhibited a relatively opalescent cornea, contrasting with the clear and homogeneous corneas observed in PBS-treated counterparts (Fig. 3M). Quantitative scores confirmed significantly greater corneal opacity in Dhcr24-cKO mice following BAC treatment (Fig. 3N), implicating DHCR24 deficiency in worsening chemical stress-induced ocular surface inflammation. Furthermore, fluorescein and Rose Bengal staining revealed severe corneal epithelial defects in BAC-treated Dhcr24-cKO mice compared to BAC-treated Dhcr24fl/fl group (Fig. 3M), with increased scorings reflecting compromised barrier integrity (Fig. 3O, P). Schirmer's test demonstrated comparable basal tear secretion between Dhcr24fl/fl and Dhcr24-cKO mice post-BAC treatment (Fig. 3Q), supporting previous evidence that Dhcr24 knockout primarily disrupts lipid secretion and ocular surface balance rather than tear production. Notably, the ocular surface pathology in Dhcr24-cKO mice was substantially less severe at the early 2-week time point post-TAM induction (Fig. 3N-Q) than after an extended 3-month period (Fig. 3D-G), suggesting that the pathological process requires longer duration to fully develop. Overall, these findings highlight DHCR24's essential role in mitigating chemical stress-induced ocular damage, further linking its deficiency to pathological lipid imbalance and barrier dysfunction.
DHCR24 deficiency disrupted lipid metabolism and cholesterol homeostasis
Given DHCR24's pivotal role in cholesterol biosynthesis, we hypothesized that its deficiency would disrupt lipid metabolism in MGs. Untargeted lipidomic profiling of MGs from Dhcr24-cKO (n = 5) and Dhcr24fl/fl mice (n = 5) revealed significant metabolic divergence between groups via OPLS-DA (Fig. 4A, B). Differential analysis identified 25 significantly altered lipid metabolites (16 upregulated, 9 downregulated; p < 0.05, VIP > 1) spanning nine subclasses: PE, ChE, phosphatidylinositol (PI), ceramides (Cer), lysophosphatidylcholine (LPC), phosphatidic acid (PA), SM, DG, and TG (Fig. 4C, D). Notably, a comparison of significantly altered lipid species revealed overlap between aged and Dhcr24-cKO conditions, with common dysregulation observed in ChE, DG, PC, PE, SM, and TG classes (Fig. S8A), indicating that DHCR24 deficiency mirrors age-related lipid remodeling. Among the most differentially expressed lipid species, several ChE species (e.g., ChE(18:1), ChE(22:1), and ChE(24:1)) were significantly downregulated in Dhcr24-cKO mice, while PI, SM, PC species (e.g., PI(18:0/20:4), SM(d18:1/24:0), and PC(16:0/16:1)) were markedly elevated (Fig. 4E and Fig. S8B). ChE exhibited a negative correlation with other lipid species, suggesting a potential relationship involving their synthesis and transformation (Fig. S8C). These changes suggest a shift away from cholesterol esterification toward altered phospholipid and sphingolipid metabolism, potentially impacting MG lipid secretion and ocular surface stability.
Comprehensive lipidomics profiling revealing alterations in steroid and cholesterol metabolic process in Dhcr24 conditional knockout (cKO) mice. (A) Orthogonal partial least squares discriminant analysis (OPLS-DA) and (B) OPLS-DA-spot indicating significant trends distinguishing the lipidomics profiling of Dhcr24fl/fl and Dhcr24-cKO groups. (C) Volcano plot showing 582 lipid metabolites, in which 16 were significantly upregulated and 9 were downregulated in the Dhcr24-cKO vs. Dhcr24fl/fl group (p < 0.05 & VIP > 1). (D) Sub class identification of the differentially expressed lipid metabolites between the Dhcr24-cKO and Dhcr24fl/fl groups. (E) Heatmap of the 25 differentially expressed lipids. (F, G) Kyoto encyclopedia of genes and genomes (KEGG) pathway analysis identifying significantly (F) upregulated and (G) downregulated pathways between the Dhcr24-cKO and Dhcr24fl/fl groups. (H-J) Gene set enrichment analysis (GSEA) based on lipid metabolites identifying significantly enriched pathways between the Dhcr24-cKO and Dhcr24fl/fl groups, including (H) cholesterol metabolism, (I) steroid biosynthesis, and (J) glycerophospholipid metabolism. (K, L) Heatmap based on lipid metabolites for significantly enriched metabolic pathways of (K) cholesterol metabolism and (L) glycerophospholipid metabolism. (M, N) Comparison of the (M) ratio of saturation for ChE, TG, PS, PI, and LPC, and (N) total unsaturation patterns between the Dhcr24-cKO and Dhcr24fl/fl groups. ChE, cholesteryl esters; LPC, lysophosphatidylcholine; PI, phosphatidylinositol; PS, phosphatidylserine; TG, triglyceride. (O) Significantly enriched signaling pathways identified through integrated transcriptomics and lipidomics of the Dhcr24-cKO vs. Dhcr24fl/fl mice, including cholesterol metabolic process, steroid biosynthesis, glycerophospholipid metabolism, and sphingolipid metabolism. (P) Correlation networks between DHCR24 and four significant lipid metabolic pathways in both Dhcr24-cKO and Dhcr24fl/fl mice.
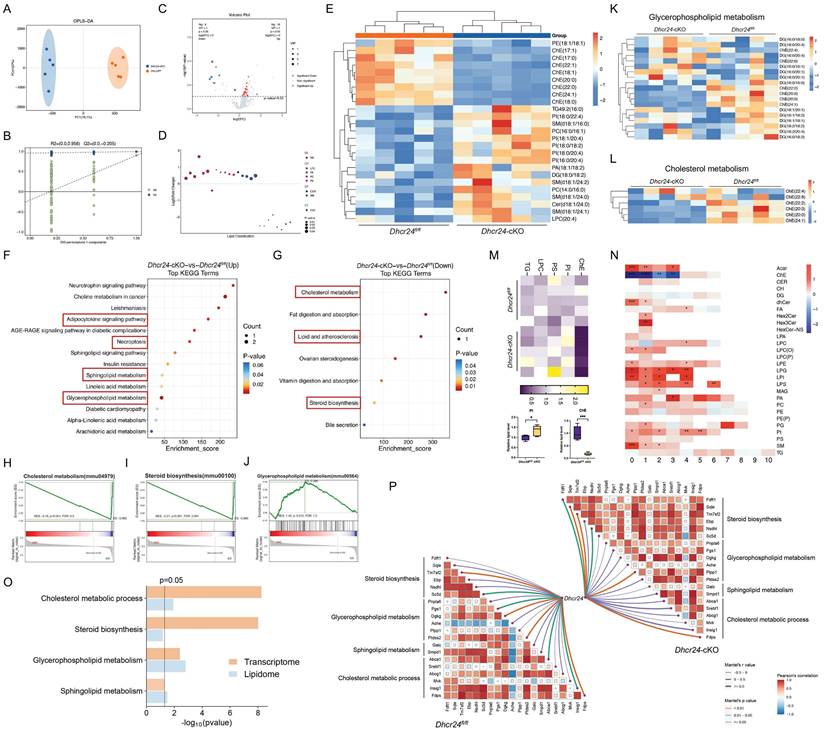
Pathway enrichment analysis substantiated these findings. KEGG mapping highlighted upregulated pathways such as adipocytokine signaling, necroptosis, and sphingolipid/glycerophospholipid metabolism (Fig. 4F), alongside downregulated cholesterol metabolism and steroid biosynthesis (Fig. 4G). Complementary GSEA of transcriptomic data confirmed downregulation in cholesterol biosynthesis, steroid biosynthesis, and glycerophospholipid metabolism (Fig. 4H-J), with heatmaps further visualizing these dysregulated pathways (Fig. 4K, L). To visually integrate the lipidomic alterations observed in Dhcr24-cKO mice, a Sankey diagram illustrates the flow from the dysregulated lipid classes (ChE, DG, PA, LPC, PC, PE, PI, SM, TG, Cer) to significantly enriched metabolic and inflammatory pathways, including cholesterol metabolism, glycerophospholipid metabolism, sphingolipid signaling, adipocytokine signaling, the NF-κB signaling pathway, autophagy, efferocytosis, Fc gamma R-mediated phagocytosis, and cAMP signaling (Fig. S8D). Additionally, analysis of lipid saturation revealed significantly reduced levels of saturated ChE species and increased levels of saturated PI species in Dhcr24-cKO mice (Fig. 4M), which paralleled lipid compositional shifts observed in aged mice [9]. Furthermore, the unsaturation of total ChE and PI was significantly altered (Fig. 4N). Integrated transcriptomic and lipidomic analysis confirmed enrichment of cholesterol/steroid biosynthesis pathways (Fig. 4O), and correlation networks linked Dhcr24 levels to dysregulated metabolic pathways, including steroid biosynthesis, glycerophospholipid metabolism, and sphingolipid metabolism (Fig. 4P). Collectively, these findings position DHCR24 as a crucial regulator of lipid homeostasis in MGs. Its deficiency disrupts cholesterol metabolism and drives pathogenic lipid redistribution, providing mechanistic insights into lipid-driven pathology in ARMGD.
Activation of cytokines and inflammatory pathways in Dhcr24-cKO mice
Transcriptomic profiling of MGs from Dhcr24-cKO (n = 6) and Dhcr24fl/fl mice (n = 6) three month after TAM treatment revealed significant enrichment in pathways associated with dysregulated lipid metabolism and inflammation. GO analysis highlighted disruptions lipid metabolism, including sterol and steroid biosynthesis and cholesterol metabolic processes, in addition to the activation of the NIK/NF-κB signaling pathway, intrinsic apoptotic signaling in response to oxidative stress, interferon (IFN)-γ production, and regulation of mitochondrial organization (Fig. 5A). Cellular component (CC) and molecular function (MF) analyses further identified enrichment in focal adhesion complexes, organelle membrane components, NF-κB binding activity, and cholesterol/sterol transporter functions (Fig. 5B, C). KEGG confirmed these findings, demonstrating enrichment in steroid biosynthesis, glycerophospholipid metabolism, FoxO signaling, and sphingolipid metabolism (Fig. 5D), highlighting the interplay between lipid dysregulation and inflammatory cascades in MGD pathogenesis.
Activation of cytokines and inflammatory pathways in meibocyte-specific Dhcr24 conditional knockout (cKO) mice. (A-C) Gene ontology (GO) analysis of differentially expressed genes (DEGs) revealing significantly enriched pathways in three categories, including (A) biological process (BP), (B) cellular component (CC), and (C) molecular function (MF). (D) Kyoto encyclopedia of genes and genomes (KEGG) enrichment analysis of DEGs between Dhcr24fl/fl and Dhcr24-cKO mice. (E) Representative immunofluorescence (IF) images of interleukin-6 (IL-6), interleukin-17 (IL-17), and tumor necrosis factor-α (TNF-α) in Dhcr24-cKO vs. Dhcr24fl/fl mice. Scale bars: 50 μm. (F-H) Enzyme-linked immunosorbent assay (ELISA) measurement of meibomian gland IL-6, IL-17, and TNF-α concentrations from Dhcr24-cKO and Dhcr24fl/fl mice. (I) Representative IF images of nuclear factor κB (NF-κB) p65 and phospho-NF-κB p65 in Dhcr24-cKO vs. Dhcr24fl/fl group. Scale bars: 50 μm. (J, K) Heatmap based on differentially expressed genes for significantly enriched pathways of (J) cytokines and inflammatory reponse and (K) NF-κB signaling. (L) Western blot of phospho-NF-κB p65 and NF-κB p65 protein levels. (M) Relative fluorescence intensity and (N) protein levels of phospho-NF-κB p65 and NF-κB p65.
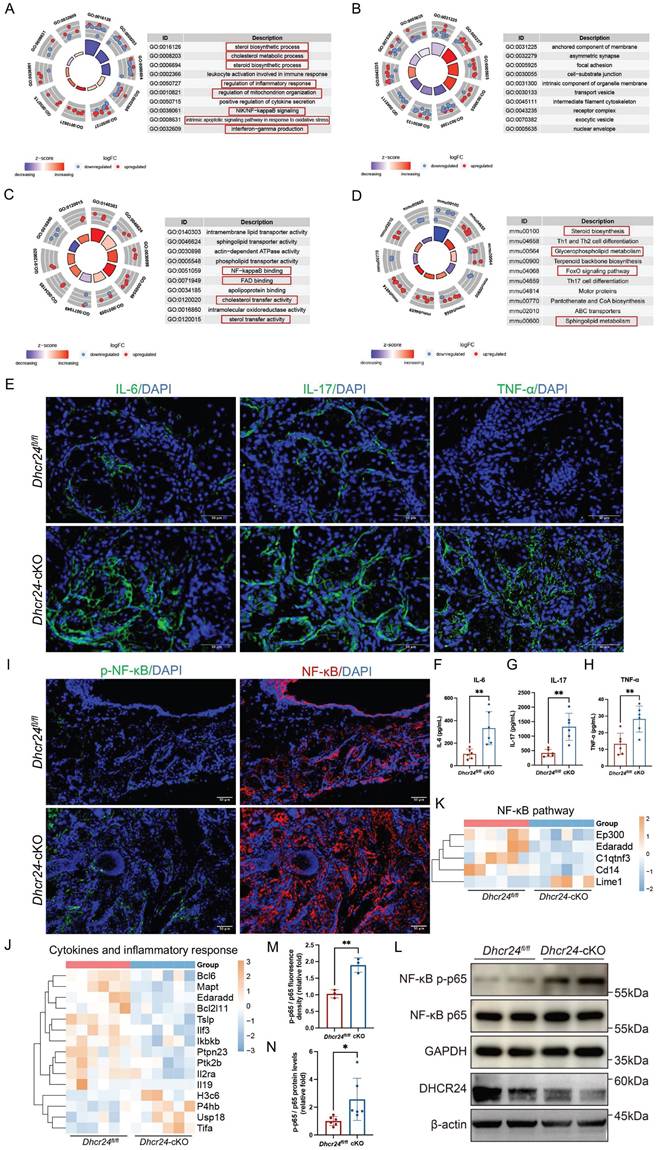
IF confirmed increased expression of proinflammatory cytokines in Dhcr24-cKO MGs, specifically IL-6, IL-17, and TNF-α (Fig. 5E). To quantitatively measure these cytokines, we conducted ELISA on MG tissue lysates, which revealed a significant increase in the protein concentrations of IL-6, IL-17, and TNF-α in Dhcr24-cKO mice compared to Dhcr24fl/fl controls (Fig. 5F-H). Additionally, activation of NF-κB was evidenced by elevated phosphorylation levels of NF-κB p65 and increased p-NF-κB/NF-κB ratios (Fig. 5I, M). Transcriptomic analysis revealed significant alterations in genes related to cytokines and inflammatory response (Fig. 5J), along with notable changes in the expression of NF-κB-associated genes (Lime 1, Cd14, C1atnf3, Edaradd, and Ep300) in Dhcr24-cKO mice (Fig. 5K). WB analysis validated these findings, showing markedly increased p-NF-κB protein levels in Dhcr24-cKO mice (Fig. 5L, N). Overall, these data suggest that DHCR24 deficiency promotes MGD through chronic inflammation and activation of the NF-κB pathway, linking lipid metabolic dysfunction to immune dysregulation in MG pathology.
Mitochondrial dysfunction and cGAS-STING activation underlie DHCR24-deficient MG senescence
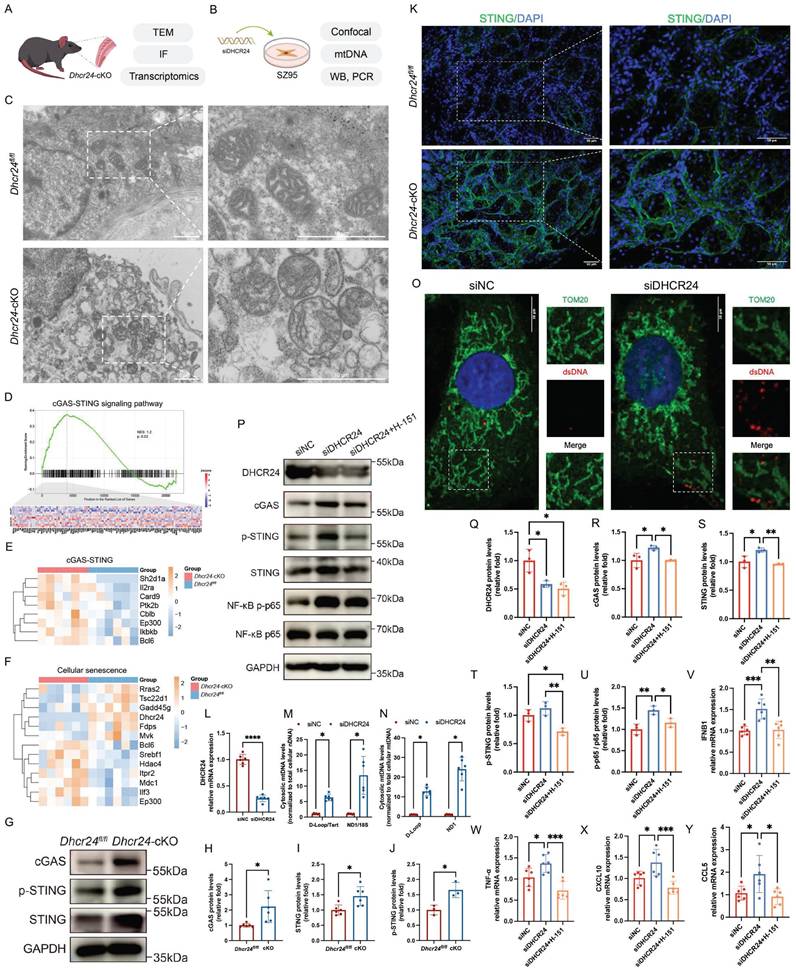
While mitochondrial morphological and functional alterations have been observed in MGD, the regulatory mechanisms driving these changes remain elusive [36,37]. Notably, the release of mtDNA into the cytoplasm has been implicated in triggering inflammation through the cGAS-STING signaling pathway in dry eye disease [33]. However, whether mtDNA release and subsequent activation of cGAS-STING similarly contribute to MGD pathogenesis remains unclear and warrants further investigation.
To address this gap, we investigated mitochondrial dysfunction in DHCR24-deficient MGD models, combining in vivo studies in Dhcr24-cKO mice (Fig. 6A) with in vitro experiments in human sebaceous gland SZ95 cells subjected to DHCR24 siRNA knockdown (Fig. 6B). TEM of Dhcr24-cKO MGs revealed swollen mitochondria with fragmented cristae, indicative of severe mitochondrial stress (Fig. 6C). GSEA identified significant dysregulation of the cGAS-STING (Fig. 6D, E) and cellular senescence pathways (Fig. 6F), supported by elevated cGAS, STING, and its activated form p-STING in Dhcr24-cKO MGs (Fig. 6G; quantification shown in Fig. 6H-J). IF further demonstrate upregulated STING in Dhcr24-cKO MGs (Fig. 6K). Using cytosolic fractionation, we quantified the release of cytosolic mtDNA in DHCR24-knockdown SZ95 sebocytes (Fig. 6L). The cytosolic mtDNA to total cellular nDNA ratio was significantly increased upon DHCR24 knockdown (Fig. 6M). To obtain a direct measurement of mtDNA leakage, we further normalized cytosolic mtDNA to the levels of total cellular mtDNA, which also demonstrated a marked increase (Fig. 6N). Additional IF for Tom20 (a mitochondrial protein) and dsDNA demonstrated that DHCR24 deficiency led to significant accumulation of cytosolic dsDNA (Fig. 6O), providing evidence of mitochondrial membrane destabilization and mtDNA leakage. WB confirmed the activation of both the cGAS-STING and NF-κB pathways upon DHCR24 knockdown, as evidenced by upregulated levels of cGAS, STING, and p-STING in SZ95 sebocytes (Fig. 6P, quantification shown in Fig. 6Q-T). Notably, introduction of the STING inhibitor H-151 in DHCR24-knockdown cells attenuated this activation and reduced downstream NF-κB pathway, indicating that DHCR24 regulates NF-κB signals through the cGAS-STING pathway (Fig. 6P, quantification shown in Fig. 6U). To determine whether cGAS-STING activation led to functional transcription of inflammatory genes, we measured the mRNA levels of key STING-dependent inflammatory and interferon-stimulated genes [38,39]. In DHCR24-knockdown SZ95 sebocytes, we observed a significant upregulation of IFNB1, CXCL10, CCL5, and TNF-α (Fig. 6V-Y). Crucially, treatment with H-151 suppressed the induction of all these downstream genes (Fig. 6V-Y). Taken together, these results establish that DHCR24 deficiency induces mitochondrial stress, leading to cytosolic mtDNA release and cGAS-STING-driven inflammation—a mechanism central to MGD-associated senescence and glandular dysfunction.
DHCR24 knockdown induces mitochondrial dysfunction and activates senescence-related cGAS-STING pathway. (A, B) Experimental workflow for (A) studies in Dhcr24-cKO mice and (B) in vitro experiments in DHCR24-knockdown SZ95 sebocytes. (C) Transmission electron microscopy (TEM) revealed mitochondrial swelling and altered cristae structure in meibomian glands (MGs) of Dhcr24-cKO mice compared to Dhcr24fl/fl controls. Scale bars: 10 μm. (D, E) Gene set enrichment analysis (GSEA) revealed significant enrichment of cGAS-STING signaling pathway in Dhcr24-cKO mice, with (E) differentially expressed genes (DEGs) shown by a heatmap. (F) Heatmap revealed significant enrichment of DEGs associated with cellular senescence. (G) Western blot (WB) and relative protein levels of (H) cGAS, (I) STING, and (J) p-STING in Dhcr24-cKO vs. Dhcr24fl/fl MGs. (K) Representative immunofluorescence (IF) images of STING in Dhcr24-cKO vs. Dhcr24fl/fl MGs. Scale bars: 50 μm. (L) Relative DHCR24 mRNA expression in SZ95 sebocytes treated with control siRNA (siNC) and siDHCR24. (M, N) qPCR measurement of (M) cytosolic mitochondrial DNA (mtDNA) normalized to total cellular nuclear DNA (nDNA) and (M) cytosolic mtDNA normalized to total cellular mtDNA in SZ95 sebocytes treated with siNC and siDHCR24. (O) IF for Tom20 and double-stranded DNA (dsDNA) in SZ95 sebocytes treated with siNC and siDHCR24. Scale bars: 10 μm. (P) WB for SZ95 sebocytes treated with siNC, siDHCR24, or siDHCR24 plus the STING inhibitor H-151, and relative protein levels were quantified, including (Q) DHCR24, (R) cGAS, (S) STING, (T) p-STING, and (U) phospho-NF-κB p65 and NF-κB p65. (V-Y) Relative mRNA expression of (V) IFNB1, (W) TNF-α, (X) CXCL10, and (Y) CCL5 in SZ95 sebocytes treated with siNC, siDHCR24, or siDHCR24 plus H-151.
AAV-mediated DHCR24 overexpression rescued MG function in aged Dhcr24-cKO mice
To investigate whether restoring Dhcr24 expression can rescue MG function, we performed intra-MG injections of AAV for Dhcr24 overexpression in Dhcr24-cKO mice. Following TAM induction, two-month-old Dhcr24-cKO and Dhcr24fl/fl mice received either AAV-DHCR24 or AAV-vehicle, followed by a 3-month observation period prior to phenotypic evaluation. Additionally, a separate group of two-month-old Dhcr24-cKO mice was raised for 6 months post-TAM before undergoing the same AAV treatment and evaluation procedures (Fig. 7A).
Adeno-associated virus (AAV)-mediated DHCR24 overexpression reverses ocular surface pathology in Dhcr24 conditional knockout (cKO) mice. (A) Experiment timeline for AAV treatment and ocular surface assessments. Two-month-old Dhcr24-cKO and Dhcr24fl/fl mice were injected with tamoxifen (TAM) for 7 days and raised for 2 weeks before intra-meibomian gland (MG) injection of AAV-vehicle or AAV-DHCR24, followed by a 3-month period before ocular phenotype evaluation. A separate group of two-month-old Dhcr24-cKO mice was raised for 6 months post-TAM before undergoing the same AAV treatment and evaluation procedures. (B) Representative white light photos, fluorescein staining, Rose Bengal staining, and upper tarsal plate images for Dhcr24fl/fl, young Dhcr24-cKO (3m), and aged Dhcr24-cKO (9m) with AAV-vehicle or AAV-DHCR24 treatment. Arrows pointing to truncated or absent MGs. (C-F) Ocular surface and MG scoring in the treatment groups, including (C) corneal opacity score (n = 5), (D) corneal fluorescein staining score (n = 5), (E) Rose Bengal staining score (n = 5), and (F) clinical MG score (n = 5). (G) Oil Red O staining of upper tarsal plate sections in the different treatment groups, with circles indicating absent MGs. Scale bars: 200 μm. (H) Hematoxylin-eosin (H&E) staining comparing the Dhcr24-cKO mice treated with AAV-vehicle and AAV-DHCR24. Scale bars: 50 μm. (I) Immunofluorescence (IF) staining and (J) relative fluorescence intensity of STING for young Dhcr24-cKO (3m) and aged Dhcr24-cKO (9m) treated with either AAV-vehicle or AAV-DHCR24. Scale bars: 50 μm. (K-M) Relative mRNA expression of (K) interleukin-6 (IL-6), (L) interleukin-17 (IL-17), and (M) tumor necrosis factor-α (TNF-α) in mouse MGs of Dhcr24-cKO (3m) and Dhcr24-cKO (9m) following AAV-vehicle or AAV-DHCR24 treatment.
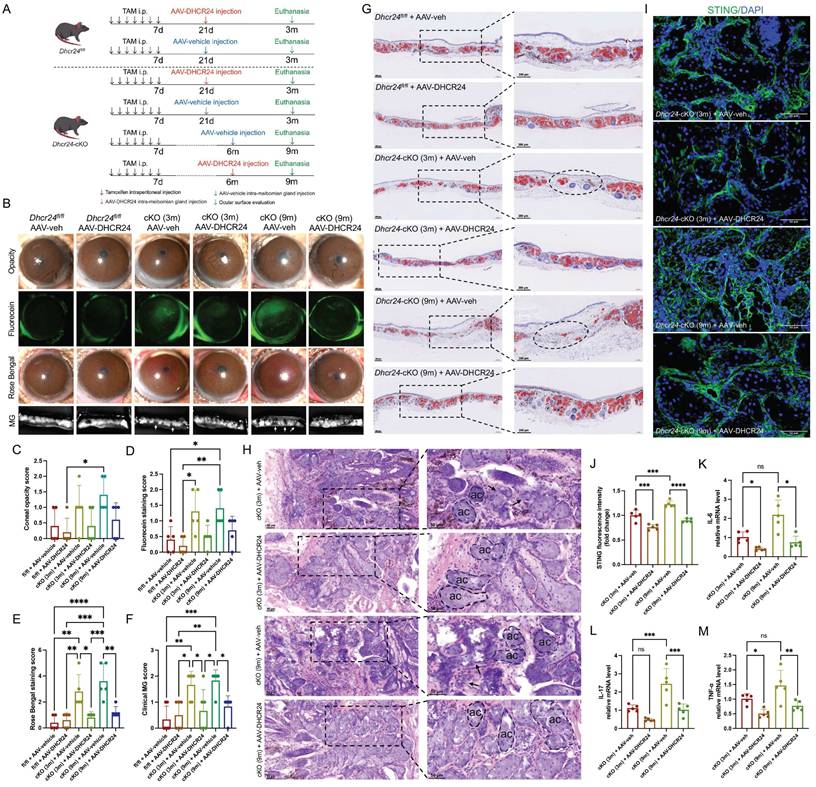
AAV-mediated DHCR24 overexpression in MGs was validated by IF (Fig. S9). Therapeutic intervention via AAV-mediated DHCR24 overexpression demonstrated significant rescue effects in Dhcr24-cKO mice. No significant differences in corneal opacity were observed between Dhcr24-cKO and Dhcr24fl/fl mice following AAV injection (Fig. 7B, C). Specifically, intra-MG delivery of AAV-DHCR24 resulted in a substantial reduction of corneal epithelial defects in both young (3m) and aged (9m) Dhcr24-cKO mice, as evidenced by diminished fluorescein staining (Fig. 7B, D) and Rose Bengal staining intensities (Fig. 7B, E), suggesting restored corneal epithelial integrity and improved tear film stability. Morphological analyses revealed near-normal MG architecture in AAV-DHCR24-treated Dhcr24-cKO mice, characterized by reduced focal dropout and normalized acinar structure (Fig. 7B), confirmed by significantly lowered MG clinical scores (Fig. 7F). Notably, aged (9m) Dhcr24-cKO mice exhibited more severe histological features compared to young (3m) counterparts, indicating greater disruption and dropout of acinar morphology (Fig. 7G, H). AAV-mediated DHCR24 overexpression demonstrated significant therapeutic improvements. ORO staining indicated recovery of acinar morphology and lipid droplet content in previously atrophic Dhcr24-cKO MGs post-treatment (Fig. 7G). H&E staining further highlighted well-defined basement membranes and a restoration of normal acinar shape in the Dhcr24-cKO group after treatment (Fig. 7H).
Importantly, IF analysis revealed a marked reduction in STING expression in both young and aged Dhcr24-cKO mice following AAV-DHCR24 treatment (Fig. 7I, J), indicating effective suppression of the STING signaling pathway. Quantitative PCR analysis demonstrated substantial downregulation of pro-inflammatory cytokines, including IL-6, IL-17, and TNF-α, in MGs of AAV-DHCR24-treated mice compared to vehicle-treated controls (Fig. 7K-M). These results suggest that DHCR24 overexpression attenuates MG inflammation and mitigates ocular surface damage by inhibiting the STING pathway and subsequent cytokine production. Overall, our findings highlight the potential of targeted DHCR24 delivery to reverse both structural and functional deficits in aging MGs.
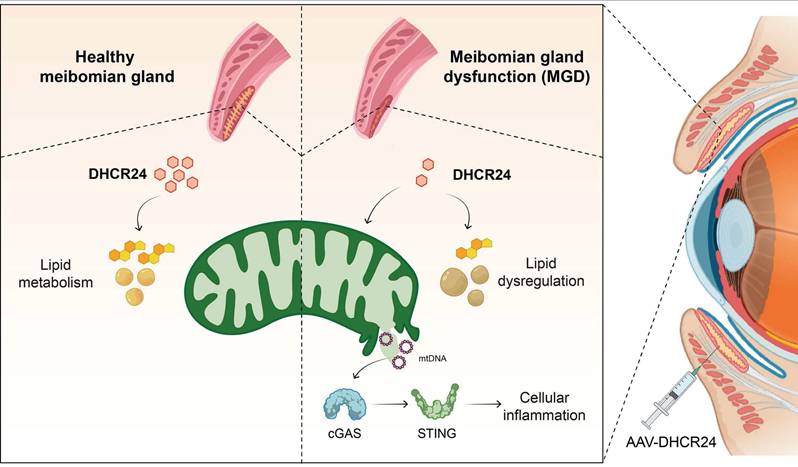
As summarized in the graphical mechanism (Fig. 8), deficiency in DHCR24 contributes to MGD through lipid dysregulation and cGAS-STING-dependent cellular inflammation in MGs. Notably, AAV-mediated delivery of DHCR24 restores lipid homeostasis, suppresses inflammatory senescence, and effectively reverses age-related pathological changes, highlighting its potential as a therapeutic strategy for age-related disorders associated with lipid-inflammatory imbalance.
Graphical mechanism illustrating the role of DHCR24 deficiency in age-related meibomian gland dysfunction (ARMGD). Deficiency in DHCR24, a key enzyme in cholesterol metabolism, contributes to ARMGD through lipid dysregulation and activation of the cGAS-STING pathway, resulting in chronic cellular inflammation and gland atrophy. Notably, AAV-mediated delivery of DHCR24 restores lipid homeostasis, suppresses STING-dependent inflammatory senescence, and reverses age-related pathological changes. This highlights its potential as a therapeutic strategy for age-related disorders associated with lipid-inflammatory imbalances.
Discussion
This study identified the cholesterol metabolism enzyme DHCR24 as a critical regulator of ARMGD through multi-omics screening. Using a meibocyte-specific Dhcr24 cKO mouse model, we demonstrated that DHCR24 deficiency mimics the pathological features of ARMGD, including ocular surface damage, gland atrophy, lipid metabolic abnormalities, and sustained inflammatory activation. Additional in vitro experiments indicated that loss of DHCR24 triggers mitochondrial dysfunction and cytosolic mtDNA leakage, leading to activation of the cGAS-STING signaling pathway, which was further validated in mouse MG tissue. Most significantly, AAV-mediated restoration of DHCR24 reversed age-related MG pathology. These findings establish DHCR24 as a central regulator of lipid metabolism and inflammation in MGs, highlighting its potential as a therapeutic target for ARMGD and other age-related disorders characterized by metabolic-inflammatory imbalance.
Our findings expand the understanding of DHCR24's functional repertoire by revealing its essential role in coordinating lipid metabolism and inflammatory responses during MG aging. DHCR24, a key enzyme in cholesterol biosynthesis, catalyzes desmosterol conversion to cholesterol and regulates cholesterol trafficking and homeostasis [40]. Beyond its core lipid metabolic function, its roles span cellular redox regulation, senescence suppression, and inflammatory control across tissues [41]. By regulating cholesterol-derived oxidative byproducts, DHCR24 helps maintain redox balance and mitigates oxidative damage, underpinning its therapeutic potential in metabolic disorders such as non-alcoholic fatty liver disease, obesity, and diabetes [42-44], as well as aging-related pathologies like Alzheimer's disease and age-related hearing loss [45]. In the brain, age-related DHCR24 decline disrupts cholesterol metabolism, impairing neuronal membrane integrity and synaptic formation [46]. Furthermore, its anti-inflammatory and antioxidant properties suppress pro-inflammatory signaling in neurons and microglia, reducing neuroinflammation and oxidative stress [47,48]. DHCR24 also intersects with apoptosis pathways, such as Akt/mTOR and Wnt/β-catenin, influencing cell proliferation and stem cell maintenance [49]. The pathological changes observed in Dhcr24-cKO mice, including disrupted cholesterol metabolism, mitochondrial cristae disorganization, and cytosolic mtDNA leakage, demonstrate that DHCR24 deficiency impairs the metabolic and structural foundations of meibocyte function. The subsequent activation of cGAS-STING signaling and NF-κB-driven inflammation suggests a mechanistic link between cholesterol metabolic disruption and inflammation in ARMGD, representing a previously unrecognized crosstalk in ocular surface pathogenesis. By identifying DHCR24 as a central orchestrator of this lipid-inflammatory axis, our work not only addresses a significant knowledge gap in ocular surface biology but also redefines ARMGD as a disorder of integrated metabolic-inflammatory dysfunction rather than merely an obstructive gland disease. These findings provide a new perspective for understanding and MG aging, positioning DHCR24-mediated pathways as strategic frontiers for therapeutic development in ocular diseases.
The age-related decrease in Dhcr24 expression reflects a dual mechanism. Consistent with established literature [7,50], our single-cell analysis confirms an age-related decrease in the proportion of meibocytes, implicating mechanisms such as diminished basal cell proliferation and progenitor pool exhaustion in overall cellular loss. Notably, our analysis moves beyond cellular proportion to assess expression at the cluster level. Even after accounting for the reduced number of meibocytes, the cluster-level expression of Dhcr24 is significantly downregulated in aged MGs compared to young ones. This statistical comparison is based on the mean expression values from all cells within the meibocyte cluster for each group, and its validity is supported by a sufficient cell count in both age groups. Thus, the age-related decline in DHCR24 is not merely a passive consequence of having fewer meibocytes but involves active transcriptional downregulation within the surviving meibocyte population. This downregulation appears to initiate a pathogenic cascade, as evidenced by the close recapitulation of age-associated lipid remodeling in Dhcr24-cKO mice. These animals showed a marked reduction in ChE together with alterations in phospholipids, TG, and SM profiles that mirror those observed in aged mice. Furthermore, the model reproduced characteristic shifts in lipid saturation states, including decreased saturated ChE and increased saturated PI species. This finding aligns with published studies demonstrating that the saturation state of meibum lipids shifts with age, particularly in ChE, TG, and PI subclasses [9,51], which are known to impact meibum viscosity and ocular surface stability. In line with clinical observations that ChE unsaturation levels are reduced in MGD and correlate with disease severity [52], our lipidomic analysis revealed significant alterations in total ChE unsaturation patterns between Dhcr24-cKO and Dhcr24fl/fl mice, further supporting the notion that the Dhcr24-cKO model captures a key physiochemical alteration relevant to ARMGD. At a systems level, both aging and DHCR24 deficiency converge on common dysregulated pathways, including suppression of cholesterol and steroid biosynthesis, as well as activation of glycerophospholipid metabolism and NF-κB signaling. Collectively, these data identify DHCR24 deficiency as a central driver of the lipid metabolic dysfunction underlying ARMGD.
The cGAS-STING pathway has emerged as a crucial mediator of chronic inflammation in aging tissues [53], yet its specific connection with lipid metabolic dysregulation in ARMGD remains poorly defined. Aging contributes to MG decline through interconnected mechanisms, including lipid dysregulation, oxidative stress, and cellular senescence. Reduced PPAR-γ activity in meibocytes impairs lipid synthesis and secretion, leading to changes in meibum composition, such as shorter fatty acid chains and increased oxidized lipids, which contribute to acinar atrophy [54]. Oxidative stress, driven by aging or environmental factors, results in excessive reactive oxygen species (ROS) accumulation, leading to lipid peroxidation, DNA damage, and accelerated MG aging [55,56]. Senescent meibocytes release senescence-associated secretory phenotype (SASP) factors, disrupting the balance of proliferation, differentiation, and secretion, and causing structural abnormalities in acinar and ductal systems [57,58]. A key factor in these processes is mitochondrial dysfunction, particularly critical in energy-intensive meibomian epithelial cells, though its regulatory mechanisms have remained elusive [59,60]. In dry eye disease, corneal epithelial mitochondrial dysfunction correlates with impaired antioxidant defenses and NF-κB-driven inflammation [61,62]. Additionally, ROS-induced mitochondrial permeability transition pore (mPTP) may also trigger cGAS-STING pathway activation via mtDNA leakage, amplifying SASP-driven senescence [33]. Our data establish that DHCR24 deficiency in ARMGD models leads to concurrent dysregulation of cholesterol metabolism, mitochondrial impairment, and cGAS-STING-driven inflammation. We observed mitochondrial damage with cristae disorganization in Dhcr24-cKO mice, while in vitro DHCR24 knockdown promoted cytosolic mtDNA leakage and activated cGAS-STING signaling. Further STING inhibition with H-151 in DHCR24-knockdowned SZ95 cells suppressed both cGAS-STING pathway activity and downstream NF-κB-mediated inflammation. Notably, the subsequent reduction in STING levels following AAV-mediated restoration of DHCR24 aligns with the dynamic regulation of this pathway. In the Dhcr24-cKO model, constitutive mtDNA leakage likely serves as a persistent ligand driving cGAS-STING activation, which can lead to the transcriptional upregulation and stabilization of STING protein [63]. We hypothesize that the restoration of DHCR24 mitigates upstream mitochondrial damage and mtDNA leakage, thereby removing the chronic activating signal. In the absence of this signal, STING may then be subject to normal protein turnover and degradation mechanisms, such as autophagy-lysosomal clearance, allowing its expression to return to baseline [64]. While these results position DHCR24 deficiency as a critical node associated with this interconnected pathology, the precise mechanistic link between disrupted cholesterol metabolism and the loss of mitochondrial integrity requires further clarification.
The structural and functional compromise of mitochondria observed in Dhcr24-cKO models points to a critical mechanistic link in meibocyte senescence. We propose that DHCR24 deficiency disrupts cholesterol homeostasis, serving as a primary driver of mitochondrial membrane instability. This "lipid-mitochondria-inflammation" axis is supported by recent evidence characterizing mitochondria as pivotal signal relays that translate metabolic dysregulation into cGAS-STING activation [23]. We hypothesize that DHCR24 deficiency alters the sterol-to-phospholipid ratio, increasing membrane fragility and facilitating mitochondrial permeabilization or aberrant opening of the mPTP. Notably, inhibition of the cholesterol biosynthetic enzyme ACLY has been shown to induce polyunsaturated fatty acid (PUFA) peroxidation and mitochondrial damage, subsequently triggering mtDNA-mediated cGAS-STING activation [65]. Furthermore, enzymes in the cholesterol pathway, such as squalene epoxidase (SQLE), can localize to mitochondria to stabilize mitochondrial transcription factor A (TFAM) and preserve organelle integrity [66]. Moreover, the interplay between lipid metabolism and innate immunity involves complex molecular crosstalk that extends beyond mitochondrial stress. Recent studies demonstrate that LXR-mediated lipid metabolism induces enzymes such as SMPDL3A, which acts as a cGAMP-degrading enzyme to restrict cGAS-STING DNA sensing [22,67]. This highlights the presence of endogenous feedback mechanisms that may be compromised in aging or DHCR24-deficient states, further promoting a shift toward chronic inflammation.
Beyond elucidating disease mechanisms, our findings provide a translational framework consistent with the urgent clinical need for targeted therapies in ARMGD. Current management strategies are mainly palliative and unable to prevent glandular atrophy or address the underlying metabolic-inflammation imbalance. Our findings establish a foundation for a more targeted therapeutic approach: restoring DHCR24 activity to correct the metabolic disruption and preserve mitochondrial integrity, combined with cGAS-STING inhibitors to attenuate the downstream chronic inflammatory cascade. Preclinical studies support the effectiveness of STING inhibitors in treating ocular surface inflammation [33,68]. A combined regimen that simultaneously normalizes lipid metabolism and reduces innate immune activation may exhibit synergistic benefits. This strategy is consistent with the emerging framework of "metabolic-immune" therapies in age-related disorders. However, several challenges must be addressed for clinical translation. Current research on DHCR24 has primarily focused on inhibitors [69,70], while the development of agonists remains underexplored due to the enzyme's complex transmembrane structure [71]. Future work should prioritize the development of tissue-specific delivery systems, such as CRISPR activation (CRISPRa) [72], to safely upregulate DHCR24 expression. Additionally, the upstream mechanisms driving age-related DHCR24 downregulation, whether driven by epigenetic modifications, transcriptional silencing, or age-related shifts in the cellular microenvironment, require systematic investigation. Further validation in human MG samples correlating DHCR24 expression with clinical severity will be essential for translational applications. These findings not only enhance our understanding of ARMGD but also create a foundation for addressing metabolic-inflammatory interactions in other age-related conditions, opening new avenues for therapeutic intervention within the broader scope of aging research.
It is noteworthy that this study utilized female mice. Although human epidemiological studies suggest a higher prevalence of dry eye disease in women, of which MGD is a major component, sex-specific prevalence for MGD remains inconsistently reported and is confounded by factors such as age and diagnostic criteria [73,74]. In C57BL/6J mice, young adults show no significant sex-based differences in baseline meibomian lipid profiles or gene expression, supporting the use of either sex for fundamental MG research [75]. We selected female mice for several reasons: (1) to reduce biological variation introduced by sex as a variable, thereby enhancing the sensitivity of our integrated multi-omics screen; (2) to avoid ocular surface trauma resulting from male-male aggression, which could confound MGD phenotyping; and (3) to prevent potential complications from androgen fluctuations that may influence lipid secretion and gland homeostasis.
This study has several limitations. First, the human tissue analysis was restricted to two samples from non-MGD donors (a 3-year-old and a 40-year-old), due to the significant practical challenge of obtaining human tarsal plate specimens. While this preliminary validation shows an age-associated decline in DHCR24 expression that aligns with our murine findings, the small sample size and lack of tissues from clinically diagnosed ARMGD patients prevent a definitive correlation between DHCR24 loss and human disease pathology. Future studies that include a larger cohort of well-characterized ARMGD patient samples will be essential to establish the clinical relevance and translational potential of DHCR24 as a therapeutic target. Second, while we hypothesize that disrupted cholesterol homeostasis serves as an upstream trigger for mitochondrial stress, the intricate link between these processes remains to be fully elucidated. Future investigations utilizing targeted lipidomics, Seahorse bioenergetic assays, and super-resolution imaging are necessary to define the specific ChE species or cholesterol metabolites and biophysical mechanisms responsible for compromising mitochondrial integrity in ARMGD. These candidate metabolites should then be functionally validated through rescue experiments, such as supplementation in DHCR24-deficient SZ95 sebocytes, to determine whether they can prevent or reverse mitochondrial damage and mtDNA leakage. Defining the functional consequences of these lipid mediators will be crucial in establishing a causal pathway from cholesterol dysregulation to mitochondrial dysfunction. Third, while our data support the hypothesis that AAV-DHCR24 restoration alleviates inflammation by mitigating mitochondrial damage and mtDNA-driven cGAS-STING activation, experimental confirmation that this intervention reduces cytosolic mtDNA leakage in vivo is still lacking. Future studies employing in vivo imaging or direct quantification of cytosolic mtDNA in MG tissue before and after AAV-DHCR24 treatment would provide crucial mechanistic validation of this proposed sequence of events.
Conclusion
In summary, this study identifies the cholesterol metabolism enzyme DHCR24 as a critical regulator of ARMGD through multi-omics screening. Utilizing meibocyte-specific Dhcr24-cKO mice, we observed MGD phenotypes, including glandular atrophy and aberrant lipid secretion. Further experiments demonstrated that DHCR24 deficiency disrupts lipid metabolism and cholesterol homeostasis and leads to the activation of cytokines and inflammatory pathways in Dhcr24-cKO mice. In vitro studies with DHCR24-knockdown SZ95 sebocytes demonstrated mtDNA leakage and subsequent activation of the cGAS-STING pathway. Remarkably, AAV-mediated restoration of DHCR24 effectively reversed age-related ocular surface pathology and restored MG integrity. Collectively, these findings establish DHCR24 as a central therapeutic target for lipid dysregulation to inflammation in MGD, highlighting its potential for treating related age-related conditions.
Supplementary Material
Supplementary figures and table.
Acknowledgements
We express our sincere gratitude to the Shanghai Newcore Biotech for their assistance with the bioinformatics analyses. We also acknowledge that the Graphic Abstract and Figure 8 was created using BioRender (Zhou, T. 2026, https://BioRender.com/920qszj), with all elements authorized for use.
Funding
This work was supported by the funding grants including the National Natural Science Foundation of China (82501245, 82471039, 82201136, 82271041), China Postdoctoral Science Foundation (2025M771943), Shanghai Post-doctoral Excellence Program (2024409), Shanghai “Rising Stars of Medical Talents” Youth Development Program (SHWSRS (2025)_71), Program of Shanghai Academic Research Leader (22XD1401800), and Biomaterials and Regenerative Medicine Institute Cooperative Research Project, Shanghai Jiao Tong University School of Medicine (2022LHA06).
Data availability
The datasets used and analyzed during the current study are available from the corresponding authors upon reasonable request.
Ethics committee approval and patient consent
All experimental procedures involving human participants were conducted in compliance with the Declaration of Helsinki. Human tarsal plate specimens were procured from Shanghai Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine under protocols approved by the Institutional Review Board (SH9H-2021-T171-1). Informed consent was obtained from all patients or their legal representatives. Approval for all animal experiments was obtained from the Animal Ethics Committee of Shanghai Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine (SH9H-2024-A1387-1).
Author contributions
YC: Conceptualization, Methodology, Investigation, Formal analysis, Funding acquisition, Writing - original draft. TZ: Methodology, Investigation, Formal analysis, Visualization. JS: Methodology, Investigation, Formal analysis, Visualization. XC: Investigation, Software, Visualization, Writing - review & editing. WS: Formal analysis, Writing - review & editing. JC: Methodology. CZ: Resources, Writing - review & editing. HS: Methodology, Supervision, Resources, Writing - review & editing. YF: Conceptualization, Funding acquisition, Supervision, Project administration. LC: Conceptualization, Funding acquisition, Supervision, Project administration, Writing - review & editing.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Fried LP, Rowe JW. Health in Aging - Past, Present, and Future. N Engl J Med. 2020;383:1293-6
2. Tian YE, Cropley V, Maier AB, Lautenschlager NT, Breakspear M, Zalesky A. Heterogeneous aging across multiple organ systems and prediction of chronic disease and mortality. Nat Med. 2023;29:1221-31
3. Uyar B, Palmer D, Kowald A, Murua Escobar H, Barrantes I, Möller S. et al. Single-cell analyses of aging, inflammation and senescence. Ageing Res Rev. 2020;64:101156
4. Cvekl A, Vijg J. Aging of the eye: Lessons from cataracts and age-related macular degeneration. Ageing Res Rev. 2024;99:102407
5. Ding J, Sullivan DA. Aging and dry eye disease. Exp Gerontol. 2012;47:483-90
6. Chader GJ, Taylor A. Preface: The aging eye: normal changes, age-related diseases, and sight-saving approaches. Invest Ophthalmol Vis Sci. 2013;54:ORSF1-4
7. Moreno I, Verma S, Gesteira TF, Coulson- Thomas VJ. Recent advances in age-related meibomian gland dysfunction (ARMGD). Ocul Surf. 2023;30:298-306
8. Arita R, Fukuoka S, Morishige N. New insights into the morphology and function of meibomian glands. Exp Eye Res. 2017;163:64-71
9. Cai Y, Zhang S, Chen L, Fu Y. Integrated multi-omics and machine learning approach reveals lipid metabolic biomarkers and signaling in age-related meibomian gland dysfunction. Comput Struct Biotechnol J. 2023;21:4215-27
10. Borchman D, Ramasubramanian A, Foulks GN. Human Meibum Cholesteryl and Wax Ester Variability With Age, Sex, and Meibomian Gland Dysfunction. Invest Ophthalmol Vis Sci. 2019;60:2286
11. Kitazawa K, Inomata T, Shih K, Hughes J-WB, Bozza N, Tomioka Y. et al. Impact of aging on the pathophysiology of dry eye disease: A systematic review and meta-analysis. Ocul Surf. 2022;25:108-18
12. Arita R, Fukuoka S. Non-pharmaceutical treatment options for meibomian gland dysfunction. Clin Exp Optom. 2020;103:742-55
13. Sabeti S, Kheirkhah A, Yin J, Dana R. Management of meibomian gland dysfunction: a review. Surv Ophthalmol. 2020;65:205-17
14. Tashbayev B, Yazdani M, Arita R, Fineide F, Utheim TP. Intense pulsed light treatment in meibomian gland dysfunction: A concise review. Ocul Surf. 2020;18:583-94
15. Xin Y, Yuan Z, Wang J, Li S. Complex Interplay Between Estrogen and Aging via Lipid Metabolism and Inflammation Forms the Novel Treatment Strategies for Atherosclerosis. FASEB J. 2025;39:e70877
16. Parhizkar S, Holtzman DM. APOE mediated neuroinflammation and neurodegeneration in Alzheimer's disease. Semin Immunol. 2022;59:101594
17. Denisenko YK, Kytikova OY, Novgorodtseva TP, Antonyuk MV, Gvozdenko TA, Kantur TA. Lipid-Induced Mechanisms of Metabolic Syndrome. J Obes. 2020;2020:5762395
18. Li Q, Xiao N, Zhang H, Liang G, Lin Y, Qian Z. et al. Systemic aging and aging-related diseases. FASEB J. 2025;39:e70430
19. Tighanimine K. Lipid remodeling in context of cellular senescence. Biochimie. 2024;227:47-52
20. Christofides A, Konstantinidou E, Jani C, Boussiotis VA. The role of peroxisome proliferator-activated receptors (PPAR) in immune responses. Metabolism. 2021;114:154338
21. Mao H, Angelini A, Li S, Wang G, Li L, Patterson C. et al. CRAT links cholesterol metabolism to innate immune responses in the heart. Nat Metab. 2023;5:1382-94
22. Shin H, Chung H. SMPDL3A links cholesterol metabolism to the cGAS-STING pathway. Immunity. 2023;56:2459-61
23. Ma Y, Pang Y, Liu C, Tian Y, Zheng K, Yao M. et al. Mitochondria relay cholesterol signal exacerbates osteoarthritis in mice. Nat Commun. 2025;16:10123
24. Liu Z, Wang M, Wang X, Bu Q, Wang Q, Su W. et al. XBP1 deficiency promotes hepatocyte pyroptosis by impairing mitophagy to activate mtDNA-cGAS-STING signaling in macrophages during acute liver injury. Redox Biol. 2022;52:102305
25. Oduro PK, Zheng X, Wei J, Yang Y, Wang Y, Zhang H. et al. The cGAS-STING signaling in cardiovascular and metabolic diseases: Future novel target option for pharmacotherapy. Acta Pharm Sin B. 2022;12:50-75
26. Li Q, Wu X, Xin S, Wu X, Lan J. Preparation and characterization of a naringenin solubilizing glycyrrhizin nanomicelle ophthalmic solution for experimental dry eye disease. Eur J Pharm Sci. 2021;167:106020
27. Pauly A, Brignole-Baudouin F, Labbe´ A, Liang H, Warnet J-M, Baudouin C. New Tools for the Evaluation of Toxic Ocular Surface Changes in the Rat. Invest Ophthalmol Vis Sci. 2007;48:5473
28. Wang X, Fan X, Wu Y, Mou Y, Min J, Jin X. Rear 4-min Schirmer test, a modified indicator of Schirmer test in diagnosing dry eye. Sci Rep. 2022;12:6272
29. Wiedemann J, Kashgari G, Lane S, Leonard BC, Knickelbein KE, Andersen B. et al. The effects of age and dysfunction on meibomian gland population dynamics. Ocul Surf. 2024;34:194-209
30. Butovich IA, Wilkerson A. Dynamic Changes in the Gene Expression Patterns and Lipid Profiles in the Developing and Maturing Meibomian Glands. Int J Mol Sci. 2022;23:7884
31. Zouboulis CC, Seltmann H, Neitzel H, Orfanos CE. Establishment and characterization of an immortalized human sebaceous gland cell line (SZ95). J Invest Dermatol. 1999;113:1011-20
32. Victorelli S, Salmonowicz H, Chapman J, Martini H, Vizioli MG, Riley JS. et al. Apoptotic stress causes mtDNA release during senescence and drives the SASP. Nature. 2023;622:627-36
33. Ouyang W, Wang S, Yan D, Wu J, Zhang Y, Li W. et al. The cGAS-STING pathway-dependent sensing of mitochondrial DNA mediates ocular surface inflammation. Sig Transduct Target Ther. 2023;8:371
34. Maekawa H, Inoue T, Ouchi H, Jao T-M, Inoue R, Nishi H. et al. Mitochondrial Damage Causes Inflammation via cGAS-STING Signaling in Acute Kidney Injury. Cell Rep. 2019;29:1261-1273.e6
35. Call M, Fischesser K, Lunn MO, Kao WW-Y. A unique lineage gives rise to the meibomian gland. Mol Vis. 2016;22:168-176
36. Ji C, Guo Y, Liu Y, Xu S, Zhao S, Luo X. et al. Inhibition of ceramide de novo synthesis ameliorates meibomian gland dysfunction induced by SCD1 deficiency. Ocul Surf. 2021;22:230-41
37. Ibrahim OMA, Dogru M, Matsumoto Y, Igarashi A, Kojima T, Wakamatsu TH. et al. Oxidative stress induced age dependent meibomian gland dysfunction in Cu, Zn-superoxide dismutase-1 (Sod1) knockout mice. PLoS One. 2014;9:e99328
38. Zhang S, Liu M, Tan D, Lv K, Li W, Huang X. et al. p57 increases immunotherapy efficacy by promoting cGAS-STING-mediated innate sensing in hepatocellular carcinoma. Cell Rep. 2026;45:116769
39. Liu J, Liu C, Ma Y, Pan X, Chu R, Yao S. et al. STING inhibitors sensitize platinum chemotherapy in ovarian cancer by inhibiting the CGAS-STING pathway in cancer-associated fibroblasts (CAFs). Cancer Lett. 2024;588:216700
40. Daimiel LA, Fernández-Suárez ME, Rodríguez-Acebes S, Crespo L, Lasunción MA, Gómez-Coronado D. et al. Promoter analysis of the DHCR24 (3β-hydroxysterol Δ24-reductase) gene: characterization of SREBP (sterol-regulatoryelement-binding protein)-mediated activation. Biosci Rep. 2012;33:57-69
41. Zerenturk EJ, Sharpe LJ, Ikonen E, Brown AJ. Desmosterol and DHCR24: Unexpected new directions for a terminal step in cholesterol synthesis. Prog Lipid Res. 2013;52:666-80
42. Dhana K, Braun KVE, Nano J, Voortman T, Demerath EW, Guan W. et al. An Epigenome-Wide Association Study of Obesity-Related Traits. Am J Epidemiol. 2018;187:1662-9
43. Zhou E, Ge X, Nakashima H, Li R, Van Der Zande HJP, Liu C. et al. Inhibition of DHCR24 activates LXRa to ameliorate hepatic steatosis and inflammation. EMBO Mol Med. 2023;15:e16845
44. Juvinao-Quintero DL, Sharp GC, Sanderson ECM, Relton CL, Elliott HR. Investigating causality in the association between DNA methylation and type 2 diabetes using bidirectional two-sample Mendelian randomisation. Diabetologia. 2023;66:1247-59
45. Tian K, Chang H-M, Wang J, Qi M, Wang W, Qiu Y. et al. Inhibition of DHCR24 increases the cisplatin-induced damage to cochlear hair cells in vitro. Neurosci Lett. 2019;706:99-104
46. Vanmierlo T, Bloks VW, Van Vark-van Der Zee LC, Rutten K, Kerksiek A, Friedrichs S. et al. Alterations in Brain Cholesterol Metabolism in the APPSLxPS1mut mouse, a Model for Alzheimer's Disease. J Alzheimers Dis. 2010;19:117-27
47. Martiskainen H, Paldanius KMA, Natunen T, Takalo M, Marttinen M, Leskelä S. et al. DHCR24 exerts neuroprotection upon inflammation-induced neuronal death. J Neuroinflammation. 2017;14:215
48. Kuehnle K, Crameri A, Kälin RE, Luciani P, Benvenuti S, Peri A. et al. Prosurvival Effect of DHCR24/Seladin-1 in Acute and Chronic Responses to Oxidative Stress. Mol Cell Biol. 2008;28:539-50
49. Yang X, Zu H, Zhao Y, Yao K. Agomelatine Prevents Amyloid Plaque Deposition, Tau Phosphorylation, and Neuroinflammation in APP/PS1 Mice. Front Aging Neurosci. 2022;13:766410
50. Yang X, Reneker LW, Zhong X, Huang AJW, Jester JV. Meibomian gland stem/progenitor cells: The hunt for gland renewal. Ocul Surf. 2023;29:497-507
51. Sledge S, Henry C, Borchman D, Yappert MC, Bhola R, Ramasubramanian A. et al. Human Meibum Age, Lipid-Lipid Interactions and Lipid Saturation in Meibum from Infants. Int J Mol Sci. 2017;18:1862
52. Nguyen A, Naidoo KK, Ajouz L, Xu X, Zhao C, Robinson MR. et al. Changes in Human Meibum Lipid Composition Related to the Presence and Severity of Meibomian Gland Dysfunction. J Ocul Pharmacol Ther. 2024;40:562-70
53. Paul BD, Snyder SH, Bohr VA. Signaling by cGAS-STING in Neurodegeneration, Neuroinflammation, and Aging. Trends Neurosci. 2021;44:83-96
54. Hwang HS, Parfitt GJ, Brown DJ, Jester JV. Meibocyte differentiation and renewal: Insights into novel mechanisms of meibomian gland dysfunction (MGD). Exp Eye Res. 2017;163:37-45
55. Nezzar H, Mbekeani JN, Noblanc A, Chiambaretta F, Drevet JR, Kocer A. Investigation of antioxidant systems in human meibomian gland and conjunctival tissues. Exp Eye Res. 2017;165:99-104
56. Böhm EW, Buonfiglio F, Voigt AM, Bachmann P, Safi T, Pfeiffer N. et al. Oxidative stress in the eye and its role in the pathophysiology of ocular diseases. Redox Biol. 2023;68:102967
57. Sato S, Ogawa Y, Shimizu E, Asai K, Okazaki T, Rusch R. et al. Cellular senescence promotes meibomian gland dysfunction in a chronic graft-versus-host disease mouse model. Ocul Surf. 2024;32:198-210
58. Zhu X, Xu M, Portal C, Lin Y, Ferdinand A, Peng T. et al. Identification of Meibomian gland stem cell populations and mechanisms of aging. Nat Commun. 2025;16:1663
59. Guo Y, Luo S, Li W. et al. Ferroptosis in Meibomian Gland Progenitor Cells Contributes to Pathogenesis of Meibomian Gland Dysfunction. Invest Ophthalmol Vis Sci. 2026;67(1):9 doi:10.1167/iovs.67.1.9
60. Xie L, Song W, Dong W, Li Y, Chen S, Sun X. et al. Indirect Application of Intense Pulsed Light Induces Therapeutic Effects on Experimental Murine Meibomian Gland Dysfunction. Front Med. 2022;9:923280
61. Zhang Y, An Y, He X, Zhang D, He W. Esculetin protects human corneal epithelial cells from oxidative stress through Nrf-2 signaling pathway. Exp Eye Res. 2021;202:108360
62. Yu D, Cai W, Shen T, Wu Y, Ren C, Li T. et al. PM2.5 exposure increases dry eye disease risks through corneal epithelial inflammation and mitochondrial dysfunctions. Cell Biol Toxicol. 2023;39:2615-30
63. Zhang Q, Wei J, Liu Z, Huang X, Sun M, Lai W. et al. STING signaling sensing of DRP1-dependent mtDNA release in kupffer cells contributes to lipopolysaccharide-induced liver injury in mice. Redox Biol. 2022;54:102367
64. Xu Y, Wan W. Lysosomal control of the cGAS-STING signaling. Trends Cell Biol. 2024;34:622-5
65. Xiang W, Lv H, Xing F, Sun X, Ma Y, Wu L. et al. Inhibition of ACLY overcomes cancer immunotherapy resistance via polyunsaturated fatty acids peroxidation and cGAS-STING activation. Sci Adv. 2023;9:eadi2465
66. Dong Y, Jiang X, Yang X, Zhang J, Fu Q, Zhou Y. et al. SQLE drives bladder cancer progression by boosting mitochondrial oxidative phosphorylation. Oncogene. 2025;44:4796-813
67. Hou Y, Wang Z, Liu P, Wei X, Zhang Z, Fan S. et al. SMPDL3A is a cGAMP-degrading enzyme induced by LXR-mediated lipid metabolism to restrict cGAS-STING DNA sensing. Immunity. 2023;56:2492-2507.e10
68. Sun X, Zhang J, Li X, Li Y, Zhao X, Sun X. et al. Fenofibrate inhibits activation of cGAS-STING pathway by alleviating mitochondrial damage to attenuate inflammatory response in diabetic dry eye. Free Radic Biol Med. 2025;235:364-78
69. Müller C, Hemmers S, Bartl N, Plodek A, Körner A, Mirakaj V. et al. New chemotype of selective and potent inhibitors of human delta 24-dehydrocholesterol reductase. Eur J Med Chem. 2017;140:305-20
70. Allen LB, Genaro-Mattos TC, Anderson A, Porter NA, Mirnics K, Korade Z. Amiodarone Alters Cholesterol Biosynthesis through Tissue-Dependent Inhibition of Emopamil Binding Protein and Dehydrocholesterol Reductase 24. ACS Chem Neurosci. 2020;11:1413-23
71. Müller C, Hank E, Giera M, Bracher F. Dehydrocholesterol Reductase 24 (DHCR24): Medicinal Chemistry, Pharmacology and Novel Therapeutic Options. Curr Med Chem. 2022;29:4005-25
72. Bendixen L, Jensen TI, Bak RO. CRISPR-Cas-mediated transcriptional modulation: The therapeutic promises of CRISPRa and CRISPRi. Mol Ther. 2023;31:1920-37
73. Stapleton F, Alves M, Bunya VY, Jalbert I, Lekhanont K, Malet F. et al. TFOS DEWS II Epidemiology Report. Ocul Surf. 2017;15:334-65
74. Stapleton F, Argüeso P, Asbell P, Azar D, Bosworth C, Chen W. et al. TFOS DEWS III: Digest. Am J Ophthalmol. 2025;279:451-553
75. Butovich IA, McMahon A, Wojtowicz JC, Bhat N, Wilkerson A. Effects of sex (or lack thereof) on meibogenesis in mice (Mus musculus): Comparative evaluation of lipidomes and transcriptomes of male and female tarsal plates. Ocul Surf. 2019;17:793-808
Author contact
![]() Corresponding authors: Hao Sun, Yao Fu, and Liangbo Chen. Department of Ophthalmology, Shanghai Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine, 639 Zhi-Zao-Ju Road, Huangpu District, Shanghai 200011, China. E-mail: sunhao6666com (HS), fuyaoedu.cn (YF), and chenliangboedu.cn (LC).
Corresponding authors: Hao Sun, Yao Fu, and Liangbo Chen. Department of Ophthalmology, Shanghai Ninth People's Hospital, Shanghai Jiao Tong University School of Medicine, 639 Zhi-Zao-Ju Road, Huangpu District, Shanghai 200011, China. E-mail: sunhao6666com (HS), fuyaoedu.cn (YF), and chenliangboedu.cn (LC).