Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2008; 4(1):15-22. doi:10.7150/ijbs.4.15 This issue Cite
Research Paper
Quantitative analysis on the characteristics of targets with FDA approved drugs
Meena K. Sakharkar1, 2, Peng Li1, Zhaowei Zhong1, Kishore R. Sakharkar2 ![]()
1. ADAMs Lab, Mechanical and Aerospace Engineering, Nanyang Technological University, Singapore
2. Biopharmaceutical Engineering Cluster, Nanyang Technological University, Singapore
Received 2007-11-19; Accepted 2007-9-12; Published 2007-12-10
Abstract
Accumulated knowledge of genomic information, systems biology, and disease mechanisms provide an unprecedented opportunity to elucidate the genetic basis of diseases, and to discover new and novel therapeutic targets from the wealth of genomic data. With hundreds to a few thousand potential targets available in the human genome alone, target selection and validation has become a critical component of drug discovery process. The explorations on quantitative characteristics of the currently explored targets (those without any marketed drug) and successful targets (targeted by at least one marketed drug) could help discern simple rules for selecting a putative successful target. Here we use integrative in silico (computational) approaches to quantitatively analyze the characteristics of 133 targets with FDA approved drugs and 3120 human disease genes (therapeutic targets) not targeted by FDA approved drugs. This is the first attempt to comparatively analyze targets with FDA approved drugs and targets with no FDA approved drug or no drugs available for them. Our results show that proteins with 5 or fewer number of homologs outside their own family, proteins with single-exon gene architecture and proteins interacting with more than 3 partners are more likely to be targetable. These quantitative characteristics could serve as criteria to search for promising targetable disease genes.
Keywords: Target, drug, disease, homologs, Pfam, tissue, protein-protein interactions, FDA
1. Introduction
The genomics revolution and advances in disease mechanisms and systems biology has provided a deluge of new potential targets for drug discovery [1]. Technological advances continue to be a central driving force in the acceleration of the drug discovery process. High-throughput gene sequencing has revolutionized the process used to identify novel targets. Thousands of new gene sequences have been generated but only a limited number of these can be converted into validated targets likely to be involved in disease. The increased number of potential targets and the decreased amount of information is generating a bottleneck in the target validation process [2].
Several new and improved methods [3], and integrated and systems-based approaches [4-6], are being explored for identifying targets and druggable proteins. The commonly used computational methods have primarily been based on the detection of sequence and functional similarity to known targets [7-8], drug-binding domain family affiliation [9, 8], and structural analysis of geometric and energetic features [10-11]. These methods are less effective in finding targets that exhibit no or low homology to known targets, disease proteins and proteins with available 3D structures. As such non-homologous and structurally unknown proteins constitute a substantial percentage, ~20–100%, of the open reading frames in many of the completed genomes and therefore, they are an untapped source of novel drug targets [12]. Hence, methods independent of sequence and functional similarity, and structural availability, are highly desirable.
Han et al. described on the use of Support Vector Machine (SVM) algorithms and their potential applications for facilitating the discovery of innovative targets and reported that the prediction accuracy for non-druggable proteins is better than that of druggable proteins. This probably results from the more diverse set of non-druggable proteins compared with that of druggable proteins, enabling SVMs to better recognize non-druggable proteins [3]. Recently, Sakharkar et al. reported on the use of integrative analyses approaches and highlighted on the utility of large genomic databases for in silico (computational) systematic drug target identification in the post-genomic era [13]. However, the two main bottlenecks in drug discovery and development are in identifying which protein targets may respond to drugs and which targets are relevant in disease. Also, there are a number of critical issues that must be considered as strategies are developed to elucidate the inherited determinants of targetability of a disease protein. In light of the above, there is a need to identify and quantify the characteristics of commercially available therapeutic targets, particularly with respect to those of the non-targeted disease proteins. Here, we quantitatively analyze the characteristics of 133 therapeutic targets (human disease genes) of FDA approved drugs and compare them with those of 3120 therapeutic targets that have no-FDA approved drugs or no drugs available for them. The possible common features of these targets are presented and discussed.
2. Methods
The human disease genes list was downloaded from the GeneCards database [14]. GeneCards is an automated and integrated database of human genes, genomic maps, proteins, and diseases. 3253 genes were identified that are reported to be involved in human diseases (Dataset-1). We manually extracted the drugs available for the disease genes from the DrugBank database. DrugBank is a unique bioinformatics/cheminformatics resource that combines detailed drug (i.e. chemical) data with comprehensive drug target (i.e. protein) information. DrugBank combines the strengths of, PharmGKB, PubChem and Swiss-Prot to create a single, fully searchable in silico drug resource that links sequence, structure and mechanistic data about drug molecules (including biotech drugs) with sequence, structure and mechanistic data about their drug targets [15].
Information on protein-protein interaction data was from Biogrid database [16]. This information could be derived for 1554 disease gene products; Tissues in which the genes are expressed from TissueDistributionDB (http://genome.dkfz-heidelberg.de/menu/tissue_db/index.html), level-4, tissue distribution data. A target is assumed to be primarily distributed in a tissue if no less than 8% of the total protein contents are distributed in that tissue. This information could be derived for 1924 therapetic targets; Pathways in which the disease gene products are involved is derived from the SwissProt knowledgebase [17]. Pathway information could be derived for 1159 therapeutic targets. We further performed Protein family assignments using SwissProt to the proteins for the disease genes and their BLASTP homologs (at a cutoff value of 0.001) [18]. 2276 therapeutic targets (human disease gene products) could be assigned to a protein family. Further categorization of homologs was performed to identify the number of unique protein families a protein was homologous to. Gene architecture information pertaining to the number of exons was extracted from CCDS (Consensus CDS) database. This information could be extracted for 2087 disease genes. We further divided the 3253 human disease genes (therapeutic targets) into two datasets one with FDA approved drugs (FDA) and the other set with no FDA approved drugs or no drugs available for them (here on referred to as 'no-FDA').
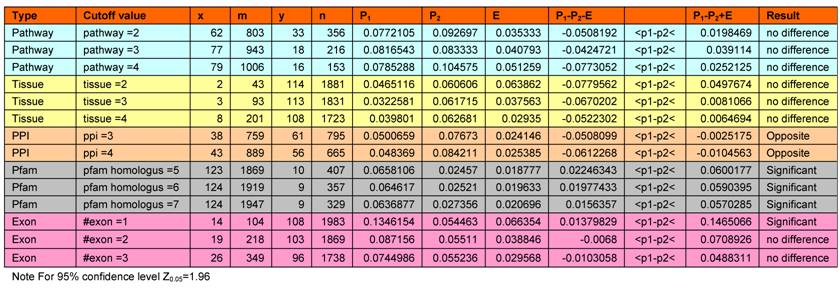
Statistical analyses were performed as described below to provide clues to the differences in the properties for disease genes with FDA approved drugs and disease genes with no-FDA approved drugs (Table 2). A test of determining the confidence interval for the difference of population proportions was performed on these two sets to check for differences at 95% confidence level, whereby:
x is the number of proteins/genes with FDA approved drugs within the cut-off region (for example pathway <=1).
m is the total number of genes (both FDA and no-FDA) within the cut-off region.
y is the number of genes with FDA approved outside the cut-off region (for example pathway >1).
n is the total number of genes (both FDA and no-FDA) outside the cut-off region.
P1=x/m; and P2=y/n
For 95% confidence level: Z=1.96; Then the sampling error E is given by
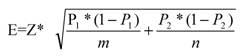
The upper and lower limits of a 95% confidence interval for P1- P2 are between
x/m - y/n -E and x/m - y/n +E
Note: This formula is based on the following assumptions:
- The population proportions P1 and P2 are not too close to 0 or 1.
- Two random samples are taken, one for each population, and the two samples are independent.
- Sample sizes m and n are large.
The results of our analyses are presented.
3. Results
Mapping drugs to targets
The mapping of FDA approved drugs to genes involved in diseases (targets) identified 133 unique targets with 289 distinct (non-duplicate) FDA approved drugs (Table 1). These results clearly suggest that one target may have multiple drugs that are reported as binding to it. An analyses on targets available for no-FDA approved drugs shows that, there are 385 disease genes targeted by 684 distinct no-FDA approved drugs (investigational agents). It should be noted that based on data available in Drugbank 2735 disease genes are not targeted by any drug or investigational agent (Table 1).
Number of targets with and without drugs
| Characteristic | Genes with drugs | # of Drugs |
|---|---|---|
| FDA approved | 133 | 289 |
| Not-FDA approved | 385 | 684 |
| No drugs | 2735 | 0 |
| Total | 3253 |
Mapping targets to pathways, and tissue information
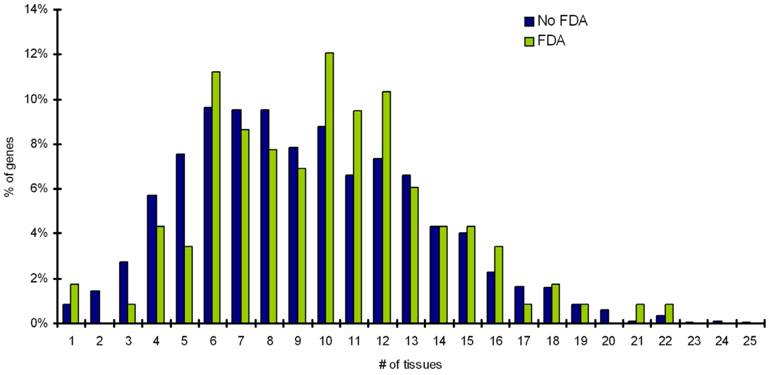
The mapping of targets with FDA approved drugs and targets with no-FDA approved drugs onto SwissProt knowledgebase and TissueDB was performed, to extract information on the number of pathways a target is involved in and the number of tissues a target is expressed. The distribution of pathway frequency for percentage of targets with FDA drugs and proteins with no-FDA drugs and number of tissues a target is expressed in, is shown in Figure 1 and Figure 2, respectively. Our results shows that, the targets with FDA approved drugs and targets with no-FDA approved drugs, when compared, show no significant bias in the number of pathways involved and the number of tissues a target is expressed (p value = 0.05, implies 95% confidence level) (Table 2).
Distribution of pathway frequency for percentage of targets with FDA drugs and proteins with noFDA drugs. The number of pathways is shown along X axis and Y axis represents the % of genes involved in diseases (targets) with FDA approved drugs and noFDA approved drugs. It is interesting to see that more than 40% of targets are involved in only 1 pathway.
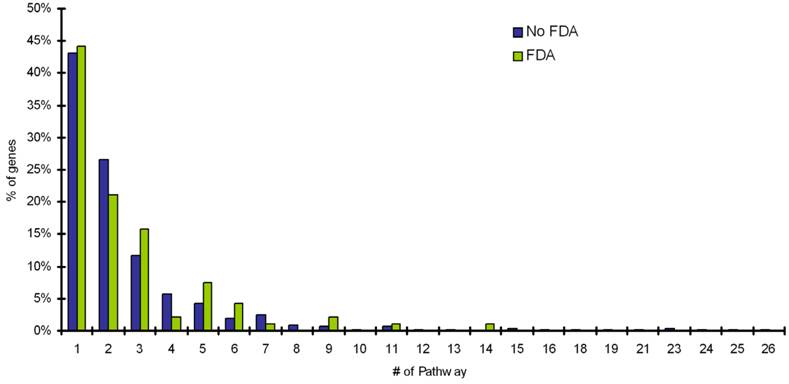
Distribution of number of tissues a target is expressed in for percentage of targets with FDA drugs and targets with noFDA approved drugs.
Statistical analyses on significance of characteristics in targets with FDA and targets with noFDA drugs. This table shows that targets with single exonic gene architectures and more than 3 interacting partners are significantly more likely to have an FDA approved drug. Targets with >5 homologs outside their own protein family are significant less likely to have an FDA approved drug. Please see text for details.
Mapping of pathways to homologs, protein-protein interaction data and gene architecture information
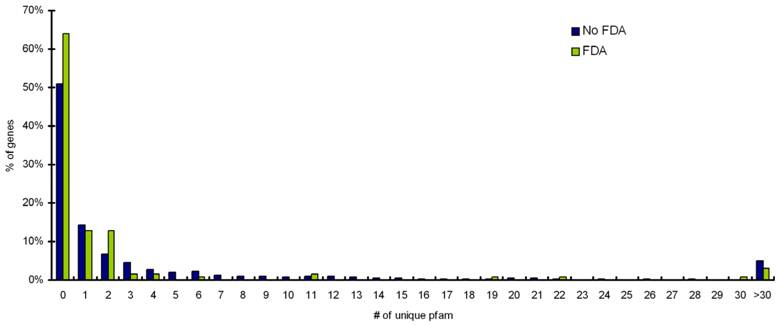
Pfam assignments were performed for targets with FDA approved, and targets with no-FDA approved drugs. A distribution of homologs outside the target's protein family for percentage targets with FDA drugs and targets with no-FDA drugs is shown in Figure 3. We observe that ~60% of targets with FDA approved drugs have no homologs outside their own Pfam family. Our statistical calculation confirms that targets having ≤ 5 BLAST homologs outside its own Pfam family (at an e-value cutoff of 0.001) are more likely (p value=0.05) to be targetable (i.e. have FDA approved drugs available for them) than targets proteins having >5 BLAST homologs outside its own Pfam family (Table 2).
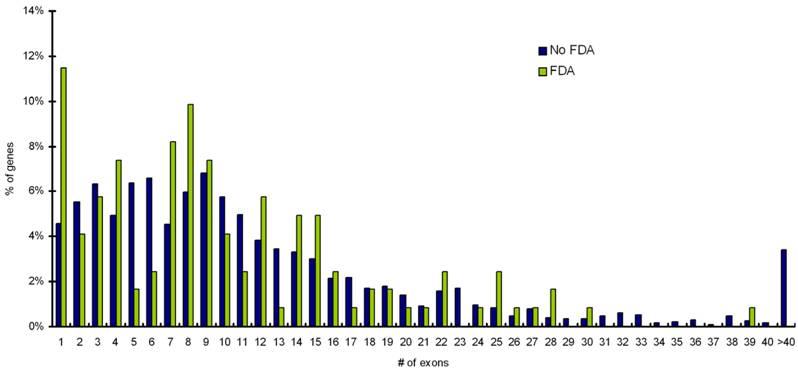
Mapping of targets to CCDS database shows that targets with single exon gene architectures are more likely to have FDA approved drugs available for them than targets with multi-exon gene architecture (p value = 0.05). Distribution for exon numbers for percentage of disease genes (targets) with FDA approved drugs and with no-FDA approved drugs is shown in Figure 4. Statistical analysis confirms that single exon genes are more likely to have FDA approved drugs (p value = 0.05).
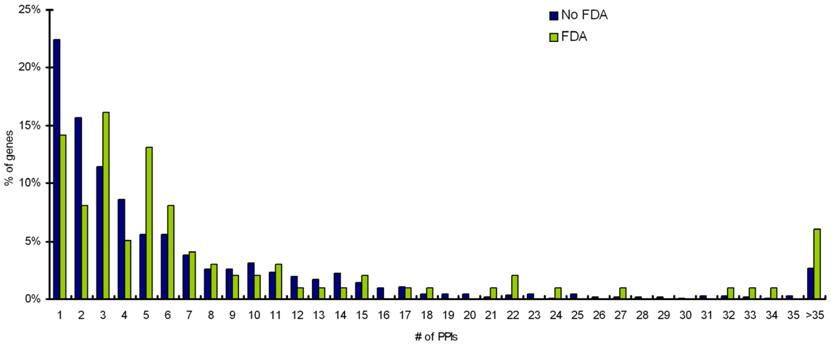
Statistical analyses on protein-protein interaction information from Biogrid database reveals that targets interacting with more than 3 partners are more likely to be targetable than proteins that do not interact with other proteins or interact with 3 or less number of partners (Table 2). Distribution of Interacting partners for percentage of targets with FDA approved drugs and percentage of targets with no-FDA drugs is shown in Figure 5.
Distribution of homologs outside the target's protein family for percentage targets with FDA drugs and targets with noFDA drugs. Targets with less than 5 homlogs outside their own protein family are more likely to have FDA approved drugs available for them (p value =0.05).
Distribution for exon numbers for percentage of disease genes (targets) with FDA approved drugs and with noFDA approved drugs. Targets with single exonic gene architectures are more likely to have FDA approved drugs available for them (p value = 0.05).
Distribution of Interacting partners for percentage of targets with FDA approved drugs and percentage of targets with noFDA drugs.
4. Discussion
It is well established that incorrect target selection accounts for the failures of some drug candidates [19]. Experience from the biopharmaceutical industry indicates that currently only 5% of newly explored targets eventually lead to FDA-approved products [19]. Thus, innovative approaches to identify a “promising” target are highly helpful in boosting productivity. Despite the clear need for better therapies for several disorders, novel drugs — particularly those that could revolutionize treatment — have been rare in recent years. Furthermore, biological complexity can often reveal unexpected and untoward effects of various treatment regimens [20-21]. However, the pharmaceutical industry must provide innovative medications to treat disease. In the quest to find pharmacologic treatments for human diseases, many targets are screened, but in silico identification of efficacy remains essential towards shortening the path from target identification to verification of its efficacy as a target. Once a set of candidate proteins have been identified, suitability of a target for small-molecule or biological drug design is a key decision making criterion. The vast varieties of in silico resources that are available in life sciences research hold much promise towards aiding the drug discovery process. Here, we perform quantitative analyses to discern the salient features of the targets with FDA approved drugs. The principles learned from this exercise would serve as technical guidelines for better choices for the identification of putative successful targets leading to cost and time savings.
Pathway affiliation
Collective actions of protein pathways are responsible for regulating disease processes and for response to drug actions. Therefore, the extent and specificity of target affiliation of pathways is likely to have statistically significant impact on drug actions [3]. In both therapeutic targets with FDA approved drugs and therapeutic targets no-FDA categories the number of proteins decrease with an increase in the number of pathways (Figure 1). This data suggests that involvement of a single protein in multiple pathways is not a preferred situation in a cell as this may lead to greater interference of a protein in related/non-related pathways and may be a reason or cause for cross-reactivity.
Comparison of therapeutic targets with FDA approved drugs and targets in no-FDA category (no FDA approved drugs or targets with no drugs available) suggests that the number of pathways a protein is involved in does not appear to be a factor determining its success as a target.
Number of tissues
Although, highly selective tissue expression of a drug target, is attractive, as the potential for unwanted side effects may be more restricted, many effective drugs have been developed against targets that are widely expressed in the body (e.g. the angiotensin converting enzyme). Our analyses shows that as of today, based on targets with FDA approved drugs, the number of tissues a gene is expressed in does not affect the targetability of the protein (Table 2, Figure 2). This is also complemented by the fact that localization of a gene in a particular tissue does not necessarily shed light on all the functions of that gene and at this stage in post-genomic era does not give sufficient details to infer any information in this direction. Since, in diseased tissue, gene expression levels often differ from those observed in normal tissues, with certain genes being over/under-expressed, or new genes being expressed or completely absent. Perhaps the most promising aspect is the information on differential expression in disease, since, the up or down-regulation of a gene may be the cause or result of the disease.
Protein homologs outside its own family
In the present day drug development processes, drug candidates have frequently been intentionally designed to bind to their target specifically and to avoid strong interactions with other human protein members of the same protein family to which the target belongs [22-25]. However, their possible interactions with human proteins outside the family are not intentionally avoided at the design stage, and the potential unwanted effects associated with some of these interactions can only be detected at the later testing stages. Our analysis showed that proteins having more than 5 BLAST homologs outside its own family are significantly less targetable than proteins that have 5 or less number of homologs outside their own family (P=0.05) (Table 2, Figure 3). This can be attributed to the fact that they can accommodate less target specific drugs that minimally interact with other pathways. Interactions with more number of targets may lead to secondary target effects, which may lead to cross-reactivity and unwanted interference. It is noteworthy that only 10 out of 133 targets with FDA approved drugs have more than 5 homologs outside their own protein family (Table 3). Therefore, it tends to be easier to find successful drugs for those targets that have fewer human similarity proteins outside of their family.
List of genes with maximum (Top10) homologs outside their protein family and their characteristics. PPI=number of protein-protein interactions or # of interacting partners. # pathway = number of pathways a target is involved, # tissue = number of tissues a target is expressed, # of exons = gene architecture information for the target. FDA approved drug shows the list of FDA approved drugs available for the target. Targets may have more than one FDA approved drug available against them. Drugs are separated by ***.
| Gene name | # Pathway | # PPI | # Tissue | # Homologs | # Exons | FDA drugs |
|---|---|---|---|---|---|---|
| ABL1 | 6 | 50 | 5 | 45 | 11 | Imatinib |
| EGFR | 11 | 95 | 9 | 40 | 28 | Erlotinib***Gefitinib |
| PDGFRB | 9 | 32 | 9 | 39 | 22 | Imatinib |
| KIT | 3 | 27 | 6 | 39 | 21 | Imatinib |
| NPR1 | 3 | 3 | 18 | 30 | 22 | Nitroglycerin |
| LDLR | 0 | 9 | 6 | 22 | 18 | Porfimer***Methyl aminolevulinate |
| F10 | 1 | 12 | 8 | 19 | 8 | Enoxaparin***Heparin |
| PLG | 2 | 33 | 14 | 11 | 19 | Aminocaproic Acid |
| F2 | 3 | 24 | 0 | 11 | 14 | Argatroban***Enoxaparin***Heparin |
| FCGR1A | 1 | 10 | 9 | 6 | 6 | Porfimer***Methyl aminolevulinate |
Exon number
Data reveals an over-representation of single-exonic genes, among genes that have FDA approved drugs available for them (P=0.05). This can be explained based on the fact that single-exon genes do not undergo alternative splicing and hence can be used as drug targets with less caution [26]. These results also corroborate with the fact that a major proportion of druggable genes have been reported as G-protein coupled receptors (GPCRs), and a major proportion of which (GPCRs) have been reported to be single exonic [13, 27]. These data support the fact that integration of data on gene annotation and gene architecture for genes involved in diseases has the potential to contribute to drug discovery and will be a step towards designing of safe, efficacious and promising drug targets. Accurate information on gene architecture and gene annotation allows us to at least be informed on the issue of splice variants. Besides, alternative splicing information is also useful at many stages of the drug discovery process including anti-sense mediated silencing and RNA interference (RNAi) for knock-down or down-regulation of specific genes products or designing of knock-out mice [28]. It must be however, noted that many of the computationally derived annotations in the databases are either minimal or incorrect (apart from a carefully manually-curated database such as Swiss-Prot). Also, as annotation of genes is provided by multiple public resources, using different methods, it results in information that is similar but not always identical. However, the database used in this study, the CCDS database overcomes these issues as it is a collaborative effort to identify a core set of human protein coding regions that are consistently annotated and of high quality (Table 2, Figure 4).
Number of interacting proteins
It is becoming increasingly clear that genes and their products interact in complex biological networks with local and global properties and perturbations of these networks contribute to the disease state. Understanding of interacting proteins is of importance in cell physiology and for developing novel treatments against disease. Small molecules that occlude crucial binding site(s) may be sufficient for modulating protein interactions that occur over large surface area and can thus act as drugs. It is known that these versatile protein-protein interactions are central to many key biological pathways and thus are attractive targets for drug discovery. Our data suggests that proteins interacting with more than 3 partners are preferred drug targets (P=0.05) (Table 2, Figure 5). However, for the drug discovery process, it is important to determine the dynamics of interactions involving proteins having multiple interacting partners as well as identifying interaction surfaces for each partner. Moreover, research to discover small-molecule drugs that target protein-protein interactions is still at an early stage.
The top 10 targets (with FDA approved drugs) based on number of interaction proteins are listed in Table 4. It is noteworthy that 5 out of these have more than 3 homologs outside their own family. These results hint on the fact that the above described characteristics do not work collectively/together to determine the success of a target.
List of genes with maximum (Top10) interaction partners and their characteristics. PPI=number of protein-protein interactions or # of interacting partners, # pathway = number of pathways a target is involved, # tissue = number of tissues a target is expressed, # of exons = gene architecture information for the target. FDA approved drug shows the list of FDA approved drugs available for the target. Targets may have more than one FDA approved drug available against them. Drugs are separated by ***.
| Gene name | # Pathway | # PPI | # Tissue | # Homologs | # Exons | FDA approved drugs |
|---|---|---|---|---|---|---|
| EGFR | 11 | 95 | 9 | 40 | 28 | Erlotinib***Gefitinib |
| AR | 0 | 81 | 12 | 2 | 8 | Testosterone***Bicalutamide***Flutamide***Oxandrolone |
| ESR1 | 0 | 81 | 11 | 2 | 8 | Fulvestrant***Raloxifene***Medroxyprogesterone***Progesterone***Estradiol***Ethinyl Estradiol***Estramustine***Tamoxifen***Conjugated Estrogens |
| NR3C1 | 1 | 71 | 4 | 2 | 8 | Hydrocortisone***Methylprednisolone***Budesonide***Mometasone***Betamethasone***Loteprednol Etabonate***Amcinonide***Dexamethasone |
| BCL2 | 7 | 55 | 4 | 0 | 2 | Paclitaxel***Docetaxel |
| ABL1 | 6 | 50 | 5 | 45 | 11 | Imatinib |
| RARA | 0 | 34 | 0 | 2 | 8 | Tazarotene***Adapalene***Alitretinoin***Isotretinoin***Tretinoin***Acitretin |
| PLG | 2 | 33 | 14 | 11 | 19 | Aminocaproic Acid |
| PDGFRB | 9 | 32 | 9 | 39 | 22 | Imatinib |
| KIT | 3 | 27 | 6 | 39 | 21 | Imatinib |
5. Conclusion
We have to keep in mind that the drug discovery and development process is extremely difficult due to our poor understanding of biology of the disease and biology of the host (i.e., Homo sapiens). We are making steady progress, but there is still a long way to go. Knowledge on characteristics of targets could be helpful for predicting features and if possible deriving rules that guide new drug design and the search for new targets from genomic data. Our analyses hint that proteins with 5 or fewer homologs outside their own family, proteins with single-exon gene architectures and proteins with more than 3 interacting partners are promising targets. For targets with a higher number of similarity proteins outside their own family, or multiple exons or interacting with less than 3 partners, it is still possible to find drugs. The characteristics defined above, merely make the tasks for finding successful drugs against these targets easier. As of today there is only 1 successful target NPR1 that is targeted by Nitroglycerin (interacts with 3 proteins, has 22 exons and has 30 homologs outside its own protein family) that does not satisfy all the three characteristics defined above. Moreover, in the FDA list there are 10 targets with more than 5 homologs outside their own protein family, 72 targets with 3 or lesser interacting partners and 108 targets with multi-exon gene architectures. These results suggest that the above quantitative characteristics selectively function in a combined, collective and differential mode and may help define and determine the targetability of a protein.
Acknowledgements
MKS and PL wishes to acknowledge funding support from A*STAR-BMRC, Singapore (Grant # 03/1/22/19/242). The authors wish to thank Prof. Chen Yu Zong, National University of Singapore, Singapore for helpful comments on the manuscript.
Conflict of interest
The authors have declared that no conflict of interest exists.
References
1. Loging W, Harland L, Williams-Jones B. High-throughput electronic biology: mining information for drug discovery. Nat Rev Drug Discov. 2007;6:220-30
2. Ofran Y, Punta M, Schneider R, Rost B. Beyond annotation transfer by homology: novel protein-function prediction methods to assist drug discovery. Drug Discov. Today. 2005;21:1475- 82
3. Zheng C. et al. Progress and problems in the exploration of therapeutic targets. Drug Discov. Today. 2006;11:412- 20
4. Lindsay MA. Finding new drug targets in the 21st century. Drug Discov. Today. 2005;10:1683-87
5. Sams-Dodd F. Target-based drug discovery: is something wrong? Drug Discov. Today. 2005;10:139-47
6. Hardy LW, Peet NP. The multiple orthogonal tools approach to define molecular causation in the validation of druggable targets. Drug Discov. Today. 2004;9:117-26
7. Hopkins AL, Groom CR. The druggable genome. Nat. Rev. Drug Discov. 2002;1:727-30
8. Wang S. et al. Tools for target identification and validation. Curr. Opin. Chem. Biol. 2004;8:371-77
9. Kramer R, Cohen D. Functional genomics to new drug targets. Nat. Rev. Drug Discov. 2004;3:965-72
10. Hajduk PJ, Huth JR, Fesik SW. Druggability indices for protein targets derived from NMRbased screening data. J. Med. Chem. 2005;48:2518-25
11. Hajduk PJ, Huth JR, Tse C. Predicting protein druggability. Drug Discov. Today. 2005;10:1675-82
12. Han LY, Cai CZ, Ji ZL, Cao ZW, Chen YZ. Predicting functional family of novel enzymes irrespective of sequence similarity: a statistical learning approach. Nucleic Acids Res. 2004;32:6437-44
13. Sakharkar MK, Sakharkar KR, Pervaiz S. Druggability of human disease genes. Int J Biochem Cell Biol. 2007;39:1156-64
14. Safran M, Solomon I, Shmueli O, Lapidot M, Shen-Orr S, Adato A. et al. GeneCards 2002: towards a complete, object-oriented, human gene compendium. Bioinformatics. 2002;18:1542-43
15. Wishart DS, Knox C, Guo AC, Shrivastava S, Hassanali M, Stothard P. et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:D668-72
16. Stark C, Breitkreutz BJ, Reguly T, Boucher L, Breitkreutz A, Tyers M. BioGRID: A General Repository for Interaction Datasets. Nucleic Acids Res. 2006;34:D535-39
17. Boeckmann B, Bairoch A, Apweiler R, Blatter M, Estreicher A, Gasteiger E, Martin MJ. et al. The SWISS-PROT protein knowledgebase and its supplement TrEMBL in 2003. Nucleic Acids Res. 2003;31:365-70
18. Finn RD, Mistry J, Schuster-Bockler B, Griffiths-Jones S, Hollich V, Lassmann T. et al. Pfam: clans, web tools and services. Nucleic Acids Res. 2006;34:D247-51
19. Caskey TC. The Drug Development Crisis: Efficiency and Safety. Annual Review of Medicine. 2007;58:1-16
20. Mulder BJ, van der Doef RM, van der Wall EE, Tijssen JG, Piek JJ, van der Meer J. et al. Effect of various antithrombotic regimens (aspirin, aspirin plus dipyridamole, anticoagulants) on the functional status of patients and grafts one year after coronary artery bypass grafting. Eur Heart J. 1994;15:1129-34
21. Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457-71
22. Drews J. Proceedings of the Roche Symposium “The Genetic Basis of Human Disease”. In: (ed.) Drews J, Ryser S. Human Disease—From Genetic Causes to Biochemical Effects. Berlin: Blackwell. 1997:5-9
23. Drews J. Strategic choices facing the pharmaceutical industry: a case for innovation. Drug Discov Today. 1997;2:72-8
24. Ohlstein EH, Ruffolo RR Jr, Elliott JD. Drug discovery in the next millennium. Annu Rev Pharmacol Toxicol. 2000;40:177-91
25. Terstappen GC, Reggiani A. In silico research in drug discovery. Trends Pharmacol Sci. 2001;22:23-6
26. Sakharkar MK, Kangueane P, Petrov DA, Kolaskar AS, Subbiah S. SEGE: A database on 'intron less/single exonic' genes from eukaryotes. Bioinformatics. 2002;18:1266-7
27. Gentles AJ, Karlin S. Why are human G-protein coupled receptors predominantly intronless? Trends Genet. 1999;15:47-9
28. Levanon EY, Sorek R. The importance of alternative splicing in the drug discovery process. Targets. 2003;2:109-14
Author contact
![]() Correspondence to: Meena K. Sakharkar, Ph.D., Advanced Design and Modeling Lab, MAE and BioPharmaceutical Engineering Cluster, Nanyang Technological University, Singapore. Tel: +65-67905836. E-mail: mmeenaedu.sg or ksakharkarcom
Correspondence to: Meena K. Sakharkar, Ph.D., Advanced Design and Modeling Lab, MAE and BioPharmaceutical Engineering Cluster, Nanyang Technological University, Singapore. Tel: +65-67905836. E-mail: mmeenaedu.sg or ksakharkarcom