Impact Factor ISSN: 1449-2288
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Natural Mutagenesis and...
Physical Mutagenesis and...
Chemical Mutagenesis and Tilling...
Agrobacterium Transferred DNA...
Transposon Mutagenesis and Its...
Gene Trap, Promoter Trap and...
Tissue Culture Mutagenesis and...
Gene Silencing Mutagenesis and...
Natural and Artificial Mutants...
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2010; 6(3):228-251. doi:10.7150/ijbs.6.228 This issue Cite
Review
Natural and artificial mutants as valuable resources for functional genomics and molecular breeding
Shu-Ye Jiang, Srinivasan Ramachandran ![]()
Rice Functional Genomics Group, Temasek Life Sciences Laboratory, 1 Research Link, Singapore 117604
Received 2010-3-12; Accepted 2010-4-20; Published 2010-4-28
Abstract
With the completion of rice genome sequencing, large collection of expression data and the great efforts in annotating rice genomes, the next challenge is to systematically assign functions to all predicted genes in the genome. The generations and collections of mutants at the genome-wide level form technological platform of functional genomics. In this study, we have reviewed currently employed tools to generate such mutant populations. These tools include natural, physical, chemical, tissue culture, T-DNA, transposon or gene silencing based mutagenesis. We also reviewed how these tools were used to generate a large collection of mutants and how these mutants can be screened and detected for functional analysis of a gene. The data suggested that the current population of mutants might be large enough to tag all predicted genes. However, the collection of flanking sequencing tags (FSTs) is limited due to the relatively higher cost. Thus, we have proposed a new strategy to generate gene-silencing mutants at the genome-wide level. Due to the large collection of insertion mutants, the next step to rice functional genomics should be focusing on functional characterization of tagged genes by detailed survey of corresponding mutants. Additionally, we also evaluated the utilization of these mutants as valuable resources for molecular breeding.
Keywords: Functional Genomics, Molecular Breeding, Mutagenesis, Mutants, Rice
Introduction
Functional genomics is the branch of genomics that determines the biological function of genes and their products. Both Arabidopsis and rice plants have been regarded as model organisms for dicots and monocots, respectively [1]. Now both japonica and indica rice genomes have been completely sequenced [2-4]. With the sequencing of rice genome, gene prediction /annotation has been carried out. Various annotation databases were set up and were freely available for public researchers. One of these databases is RiceGAAS (http://ricegaas.dna.affrc.go.jp/; [5]). The second one is the Rice Annotation Project Database RAP-DB (http://rapdb.dna.affrc.go.jp/; [6]). The third is the TIGR rice genome annotation database (now moved to Michigan State University (MSU); http://rice.plantbiology.msu.edu/; [7, 8]). The releasing of these databases has been significantly contributing to the research of rice functional genomics. As a result of ongoing annotation efforts, predicted gene numbers continue to be changed [9]. More than 50,000 genes were predicted upon publication of its draft sequence [2, 3]. Subsequently, 40,612 non-transposable element-related genes were predicted by the MSU rice genome annotation project. However, 37,544 genes were predicted to be protein-coding genes [4]. Now only 30,000 or less protein-coding genes were estimated [10]. The large differences in the annotated gene numbers suggest the necessary to further validate these annotated genes by various experimental approaches. Such necessity was strengthened by the fact that at least 40% of predicted Arabidopsis genes were wrongly annotated based on subsequent validation by experiments [11]. Besides the efforts in genome-wide gene prediction, many rice gene families were also annotated by individual researchers. For example, we have identified and characterized members from 5 gene families at the genome-wide level. These included 14 rice myosin gene family members [12], 114 pollen-allergen-like genes [13], 49 rice cyclin genes [14], 111 small GTPase genes and 85 genes encoding small GTPase activating proteins [15] and 17 GRAM domain containing proteins [16]. These works together with other community annotation of rice gene families (http://rice.plantbiology.msu.edu/ca/rice_ca.shtml) significantly contributed to and complemented genome-wide gene annotations.
Additionally, large amount of rice Expressed Sequence Tags (EST) data are available in public databases including the MSU (http://rice.plantbiology.msu.edu/), NCBI (http://www.ncbi.nlm.nih.gov/dbEST/index.html), Gramene (http://www.gramene.org/Oryza_sativa_japonica/index.html) databases and so on. For example, until October 29, 2009, total of 1,249,001 EST sequences have been released into NCBI database (http://www.ncbi.nlm.nih.gov/dbEST/dbEST_summary.html). In addition to these, more than 32,000 full-length cDNA clones from japonica rice have been sequenced [17] (http://cdna01.dna.affrc.go.jp/cDNA/) and 10,096 indica full-length cDNA clones were also released [18] (http://www.ncgr.Ac.cn/cDNA/indexe.html).
With the completion of rice genome sequencing, large collection of expression data and great efforts in annotating rice genomes, the next challenge is to systematically assign functions to all predicted genes in the genome. To broadly assign functions to unknown genes, various old approaches are improved and new methods are developed. The different methodologies have been developed to form their own fields within the functional genomics technological platform and are termed transcriptomics, proteomics, metabolomics and phenomics [19]. However, all tools to identify functions of genes are based on the analyses in phenotypic variations between wild type and its mutant. Therefore, the generations and collections of mutants at the genome-wide level form the technological platform of functional genomics.
On the other hand, during long breeding history, farmers and breeders have been selecting new rice varieties with better agronomic traits such as higher yield, improved resistance to various diseases and better quality of grains and so on. These varieties were developed by altering the genetic makeup of the crop. Therefore, genetic variation is the basis of breeding selection. The variation may be produced by natural and artificial mutations as well as sexual crossing. Among the hundreds and thousands of variations, elite germplasms may be developed, which form the important resource for rice breeding. The evidence has shown that a breakthrough might be achieved when such germplasms have been found and used for rice breeding practice. For example, the rice yield has been greatly improved by the utilization of dwarf germplasm [20]. Similarly, other important germplasms such as cytoplasmic male sterile lines and photoperiod/temperature-sensitive male sterile lines have led to the development of various hybrid rice combinations, which have further improved crop yields by 20-30% when compared with the conventional varieties [21]. Therefore, it is very important for us to collect, to generate, and to evaluate rice germplasms for better serving rice breeding.
In this review, we will focus on the collections and characterizations of large rice mutants generated from various methods of mutagenesis such as maize two-element Ac/Ds system and T-DNA insertion mutagenesis and so on. We also review the applications of these tools and mutants in identifying gene functions and in rice breeding.
Natural Mutagenesis and Map-based Cloning
Natural mutants were generated during species evolution. Generally, the ratio of natural mutation is very low at only 10-5-10-8 in higher plants. However, a large collection is still available during long evolutionary history. Some of such mutants were harmful or neutral and might be lost during evolution. Others might exhibit higher resistance to various abiotic / biotic stresses or have some specific agricultural traits, which were valuable germplasm resources for rice breeding. One example is the utilization of dwarf germplasm Dee-geo-woo-gen from China and release of rice variety IR8, which was developed from the dwarf line [22]. Another example is the application of cytoplasmic male sterile (CMS) and photoperiod-sensitive genic male sterile rice lines, which are widely utilized to develop hybrid rice seeds for commercial release [23].
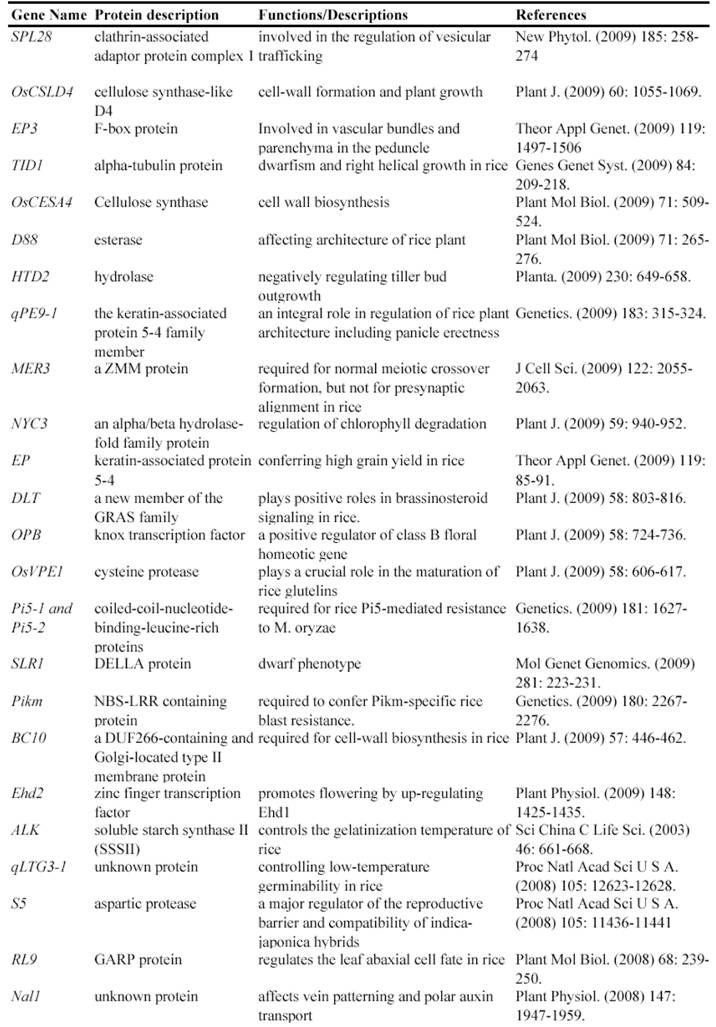
Map-based cloning is a widely-used method to isolate genes using such mutants. Genetic analysis is the first step to use these mutants for identifying gene functions, by which we know that how many genetic loci control the mutated phenotype. The next step is to finely map these loci to rice genome and then to clone the mutated gene confirmed by genetic complementation experiments. In fact, many genes have been isolated and functionally characterized by using natural mutant lines. For example, the rice Xa21 and Xa27 gene, which confers resistance to Xanthomonas oryzae pv. oryzae race 6, was isolated by map-based cloning from a natural mutated rice variety [24, 25]. Another such example is the isolation of rice semidwarf gene sd-1, which encodes a gibberellin 20-oxidase [26]. Up to now, at least 67 rice genes have been isolated and functionally characterized by the map-based cloning (Table 1).
Some of rice genes functionally characterized through mapped-based cloning.
Physical Mutagenesis and Deleteagene Detecting System
In 1930, Muller observed that mutation could be induced by X-rays [27]. Subsequent researches found that the most efficient mutagenesis was mediated by fast neutron bombardment [28]. A short deletion of DNA fragment was usually observed following the bombardment. Thus, a truncated gene might be detected by genomic subtraction and its functions could be identified by corresponding mutant phenotype. One example is the isolation and characterization of Arabidopsis ga1-3 gene [29]. Currently, a new reverse genetics method has been developed to identify and isolate such mutants [30, 31]. This method was named as Deleteagene. In this system, DNA samples were extracted from the fast neutron-treated plants and were used to screen for deletion mutants by polymerase chain reaction (PCR) using specific primers flanking the targeted genes. Li et al. (2001) has generated an Arabidopsis population of 51,840 lines by fast neutron mutagenesis [30]. This library was then used for screening deletion mutants of 25 gene loci, among which deletion mutants were obtained for 21 (84%) gene loci. Similarly, they also generated a rice fast neutron mutant pool with 24,660 lines and similar method was successfully used for identification and isolation of targeted genes. Evidence showed that this method can be efficiently used for the identification of small genes or tandemly arrayed genes [30]. Wu et al. (2005) reported the generation of around 10,000 rice mutant lines by the fast neutron bombardment and around 20,000 lines by γ-ray [32]. Since the establishment of the method, many genes have been isolated and functionally characterized including the phytochrome family gene PHYC [33] and phytochrome-interacting transcription factor PIF3 [34], 3 genes encoding TGA transcription factors TGA2, TGA5, TGA6 [35] and so on.
Chemical Mutagenesis and Tilling Detecting System
Chemical mutagenesis is mediated by certain chemical reagents. One of the most frequently used reagents is ethyl methane sulfonic acid (EMS). This alkylating agent can efficiently induce chemical modification of nucleotides, which results in various point mutations including nonsense, missense and silent mutations, among which silent mutations can not generate any modification in phenotype and thus can not be used for mutagenesis. In Arabidopsis, EMS mainly induces C to T changes resulting in C/G to T/A substitutions and at a low frequency, EMS generates G/C to C/G or G/C to T/A transversions by 7-ethylguanine hydrolysis or A/T to G/C transition by 3-ethyladenine pairing errors [36]. Based on codon usage in Arabidopsis, the frequency of EMS-induced stop codon and missense mutations has been calculated to be ~5% and ~65%, respectively [37].
In Arabidopsis, at least 125,000 M1 lines should be generated in order to achieve saturation of EMS mutagenesis [38]. However, it is not difficult to produce such a population since viable seeds can be used for EMS treatment. The difficulty is how to detect single-nucleotide polymorphisms or substitutions in these mutation lines in a large scale. Based on the technology in detecting single-nucleotide polymorphisms [39, 40], McCallum et al (2000) established a new detecting method named as TILLING (Targeting Induced Local Lessions In Genomes) [37, 41] complemented with denaturing high-performance liquid chromatography (DHPLC). These technologies allow chemically induced mutant pools to be used for reverse genetics. With help of automation, robust and rapid detection makes it possible to screen a wide range of mutant pools in a short time and to avoid the laborious process of forward genetic screening [42, 43]. Now the technology has been used in various species including animals and plants and some improved methods were also provided [44-51].
In rice, around 18,000 and 9,000 mutants were generated from diepoxybutane and EMS mutagenesis, respectively [32]. Total of 10 genes were screened using TILLING and independent mutations were detected in two genes: pp2A4 encoding serine/threonine protein phosphatase catalytic subunit and cal7 encoding callose synthase, suggesting the feasibility of this screen method in chemical mutagenesis. In another report, they screened 10 genes including Os1433 (LOC_Os02g36974), OsBZIP (LOC_Os01g64000), OsCALS8R (LOC_Os01g55040), OsDREB (LOC_Os01g07120), OsEXTE (LOC_Os10g33970), OsMAPK (LOC_Os07g38530) OsPITA (LOC_Os12g18360), OsR1A (LOC_Os05g41290), OsRPLD1 (LOC_Os01g07760) and OsTPS1 (LOC_Os02g44230). Independent mutants were detected for all 10 genes [52]. They also found that multiple nucleotide changes can be detected in each gene [52], suggesting that they have developed a useful method for more reliable and exact functional identification of a gene.
Agrobacterium Transferred DNA (T-DNA) Mutagenesis and Functional Characterization of Rice Genes
With the development of high efficient Agrobacterium-mediated transformation of rice [53], T-DNA mutagenesis has become a major method to generate a large collection of insertion mutants. Generally, T-DNA can be randomly and stably inserted into plant genome, which made it possible to generate a population saturated with insertions, i.e. having at least one insertion in each gene [54]. In Arabidopsis, at least 225,000 independent T-DNA insertion lines have been created that represent near saturation of the gene space; the precise locations were determined for more than 88,000 T-DNA insertions, which resulted in the identification of mutations in more than 21,700 of the approximately 29,454 predicted Arabidopsis genes [55].
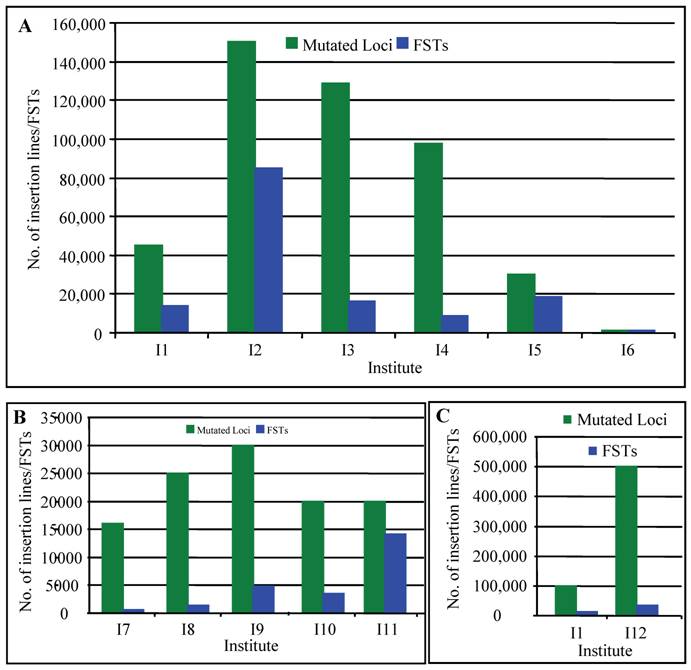
In rice, several research groups have contributed to the generation of T-DNA insertion lines. For example, An's group has generated approximately 100,000 insertion lines [56, 57]. Around 42,000 T-DNA insertion lines have been generated by Zhang and Wu's group [58, 59]. Recently, Hsing et al. (2007) have reported the generation of 55,000 T-DNA insertion lines [60]. Several other groups also independently produced T-DNA insertional mutant lines in rice [61-63]. According to the previous reports, the average copy number of T-DNA inserts per line is 1.4-2.0 [64]. Thus, more than 450,000 T-DNA tags have now been generated in rice (Fig. 1A). Recent progresses on the generation of T-DNA insertion lines have been reviewed by several researchers [65-67]. If there are only 30,000 or less protein-coding genes in rice genome [10], these populations are large enough to find a knockout in a given gene, assuming that T-DNA is randomly inserted into a chromosome. This suggestion was strengthened by the fact that T-DNA have been observed to insert preferentially in gene-rich regions [58-59, 68-70].
Collection of insertion mutants in rice. This figure summarizes the collection of T-DNA (A), Ac/Ds/Spm/dSPM transposon (B) and retrotransposon Tos17 (C) insertion lines in rice. Green columns indicate the numbers of mutated loci carried out in each institute and blue columns indicate the numbers of insertion lines with FSTs. I1, including Centre de Coopération Internationale en Recherche Agronomique pour le Développement, Institut National de la Recherche Agronomique and Centre National de la Recherche Scientifique; I2, Pohang University of Science and Technology; I3, Huazhong Agricultural University, China; I4, Shanghai Institute of Plant Physiology and Ecology, China; I5, Institute of Plant and Microbial Biology, Academia Sinica, Taiwan; I6, Zhejiang University, China; I7, CSIRO Plant Industry, Australia; I8, Centre de Coopération Internationale en Recherche Agronomique pour le Développement; I9, Gyeongsang National University, Korea; I10, Temasek Life Sciences Laboratory, Singapore; I11, University of California, Davis; I12, National Institute of Agrobiological Sciences, Japan. The data are based on the following references: [56-60, 62, 63, 65-67, 76-83, 105, 107, 111, 121-124].
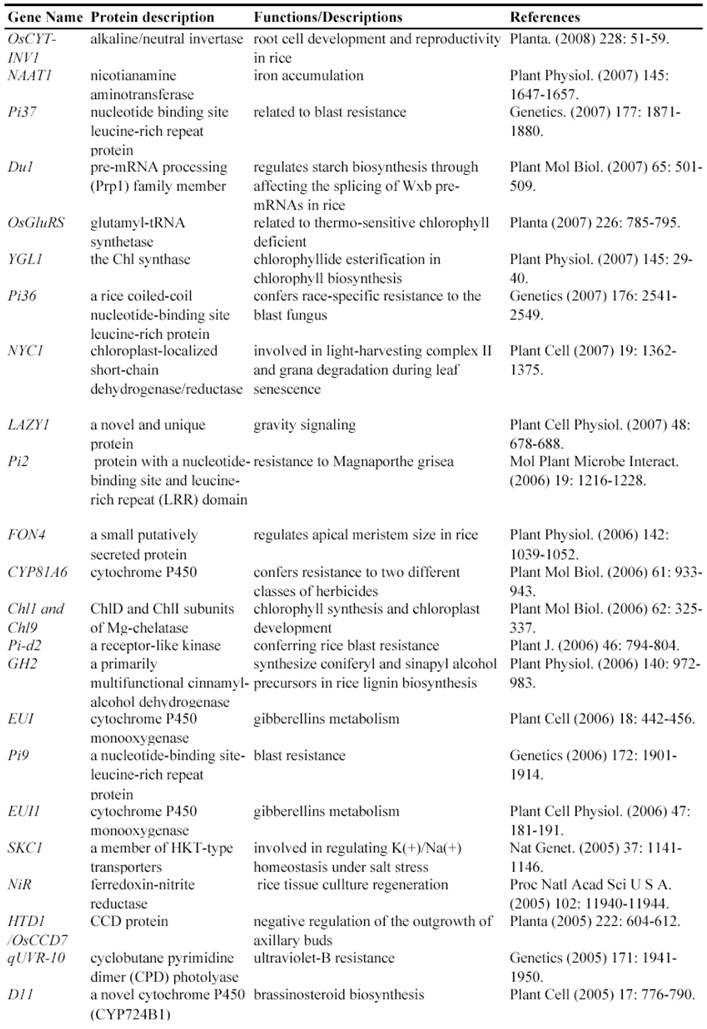
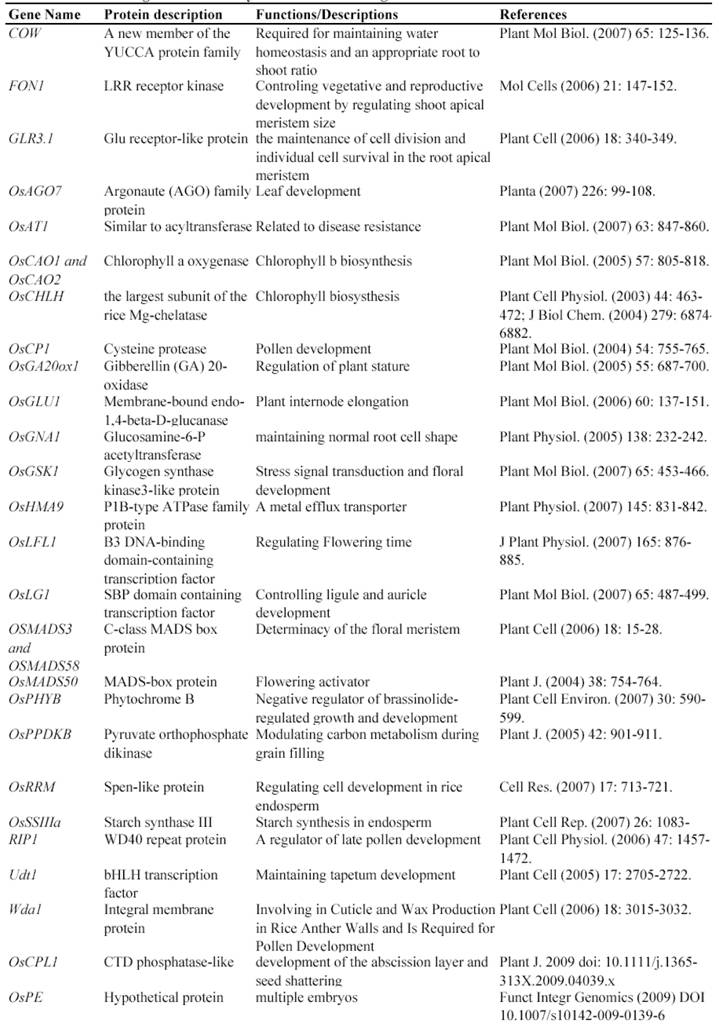
After T-DNA insertion, various phenotypes have been observed including changed growth rates, different plant statues, pollen and seed fertility and so on [67, 71-74]. Those visible differences in phenotypes could significantly contribute to the identification of gene functions. Since the establishment of T-DNA insertion populations, at least 43 genes have been functionally characterized by T-DNA insertion mutants (Table 2). For example, a knockout line of OSMADS3 by T-DNA insertion shows homeotic transformation of stamens into lodicules and ectopic development of lodicules in the second whorl near the palea where lodicules do not form in the wild type but carpels develop almost normally [75]. Their data show that this gene plays a crucial role in regulating stamen identity.
Some of rice genes functionally characterized through T-DNA insertion mutants
Transposon Mutagenesis and Its Application on Functional Identification of Rice Genes
Maize transposon Ac and Ds elements have been successfully used as insertion mutagens for rice insertion mutagenesis. For obtaining stable insertion lines, a two-element Ac/Ds tagging system has been established. In this system, two different transgenic parental lines were used for sexual crossing. One parental line contains Ac element, in which Ac is immobilized and provides only Ac transposase under the control of 35S promoter. Another parental line is transgenic Ds plant, in which Ds is also non-autonomous element and provides only two wings of Ds element (5' Ds and 3' Ds). Thus, in both Ac and Ds parental lines transposon Ac or Ds can not mobilize by themselves. However, after crossing between Ac and Ds plants, Ds element will be mobilized and inserted into different genome positions under the presence of Ac transposase. In the next generation, these lines containing only Ds element and without Ac transposase were selected. Therefore these Ds insertional lines will be stable since the plants contained no Ac transposase. Besides Ac and Ds, other transposons such as En and Spm were also used to generate transposon insertion mutants [76].
Currently, multiple research groups have been greatly contributed to the large collection of transposon insertion mutants and various databases have been set up for better utilization of these resources (Fig. 1B; [65-67, 76-84]). Totally, more than 153,000 transposon insertion lines have been generated, providing valuable resources for the survey of functional genomics.
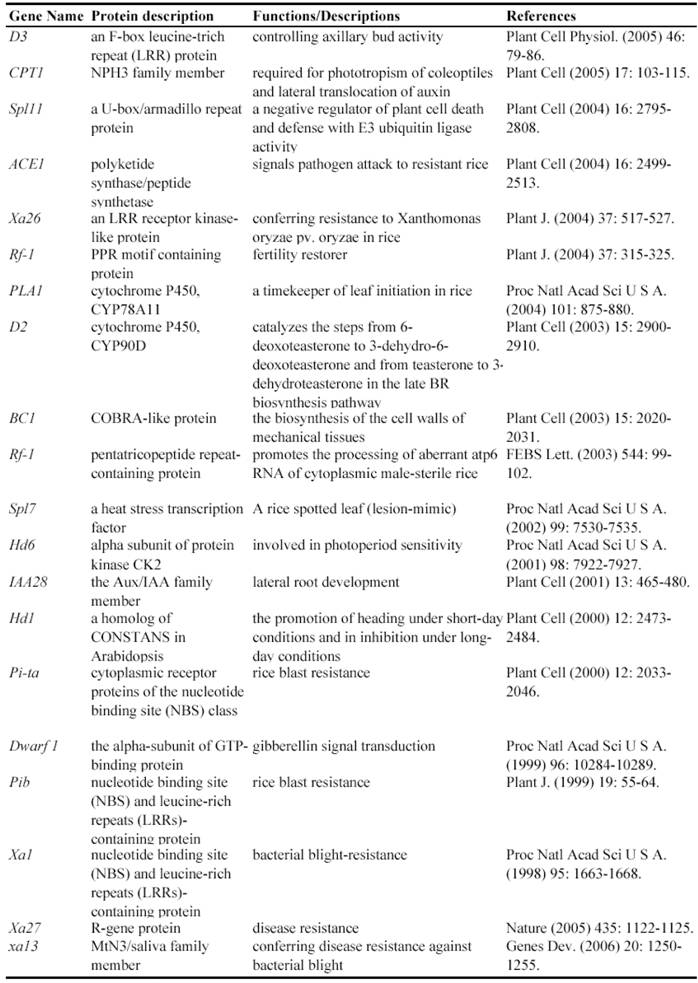
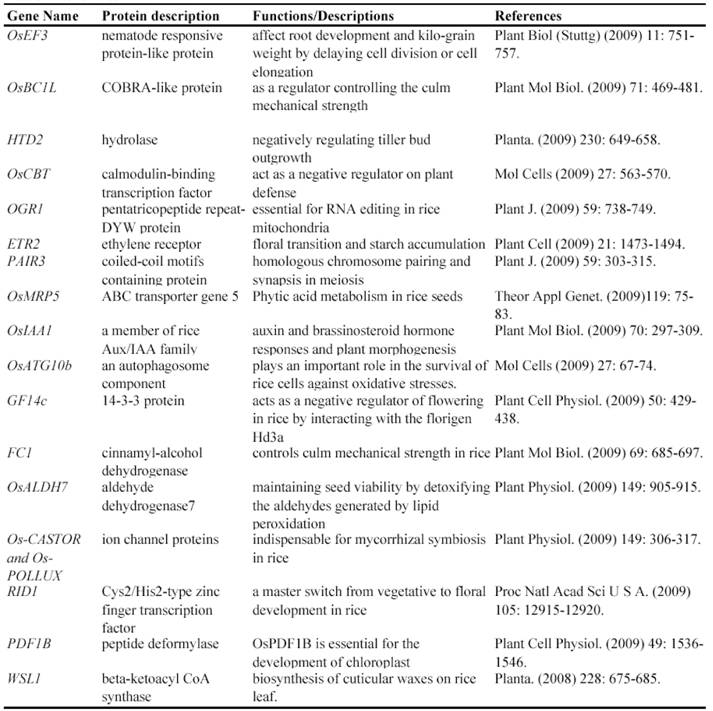
Since a large collection of transposon insertion lines were generated during 2000's, not so many genes have been functionally identified by such insertion mutants. Currently, at least 9 genes have been functionally characterized by Ds insertion mutants including ANTHER INDEHISCENCE1, BRANCHED FLORETLESS 1, CSL1, OSH6, OsKS1, OSMYOXIB, OsNAC2, OSNOP and OsPE (Table 3). These data suggested the feasibility and potential of transposon insertion lines as a tool to decipher gene functions.
Some of rice genes functionally characterized through transposon insertion mutants
In our lab, we have used a two-element Ac/Ds gene trap system to tag rice genes. In this system, an immobilized version of Ac, in which the transposase gene is under the control of cauliflower mosaic virus (CaMV) 35S promoter was used. The non-autonomous Ds element carries the bar gene encoding phosphinothricin acetyltransferase conferring resistance to phosphinothricin (herbicide Basta), which serves as a positive selection marker and a modified promoterless gusA gene encoding β-glucuronidase as a reporter gene. The gusA gene used in the Ds construct has the intron and triple splice acceptor sequences upstream of the ATG codon to trap the expression of tagged genes at 3' Ds. The synthetic green fluorescence protein (sGFP) was used under maize ubiquitin promoter as negative selection markers within both the Ac and the Ds T-DNA constructs as a negative selection marker.
These two constructs were then introduced into rice genome by Agrobacterium-mediated transformation. Transgenic Ac and Ds rice plants were used as parent lines for sexual crossing. In next generation, stable Ds insertion lines were obtained by selecting Basta positive and GFP negative plants. Homozygous Ds insertion lines were obtained after the fifth/sixth generations by self-crossing. These homozygous lines were used for phenotype investigation.
In this system, the germinal transposition frequency of Ds was estimated as an average of 51% by analyzing 4413 families. Study of Ds transposition pattern in siblings revealed that 79% had at least two different insertions, suggesting late transposition during rice development. Analysis of 2057 Ds flanking sequences showed that 88% of them were unique, whereas the rest within T-DNA. The insertions were distributed randomly throughout the genome; however, there was a bias toward chromosomes 4 and 7, which had two times as many insertions as that expected. A hot spot for Ds insertions was identified on chromosome 7 within a 40-kbp region. One-third of Ds flanking sequences was homologous to either proteins or rice ESTs, confirming a preference for Ds transposition into coding regions. Analysis of 200 Ds lines on chromosome 1 revealed that 72% insertions were found in genic region. Anchoring of more than 800 insertions to yeast artificial chromosome (YAC)-based EST map showed that Ds transposes preferentially into regions rich in expressed sequences. High germinal transposition frequency and independent transpositions among siblings show that the efficiency of this system is suitable for large-scale transposon mutagenesis in rice [78].
Additionally, we have performed a systematic analysis to survey the transposition activities of Ac/Ds parent lines in the following generations. We found that high somatic and germinal transposition frequencies were maintained as late as T4 and T5 generations; thus the propagation of parental lines did not induce transposon silencing. Moreover, the stably transposed Ds element was active even at the F5 generation, since Ac could remobilize the Ds element as indicated by the footprint analysis of several revertants. Strikingly, substantial transgenic silencing was not observed in any of the generations tested. We analyzed the timing of transposition during rice development and provide evidence that Ds was transposed late after tiller formation. Our study validates the Ac/Ds system as a tool for large-scale mutagenesis in rice, since the Ds elements were active in the starter and insertion lines even in the later generations [85].
Gene Trap, Promoter Trap and Enhancer Trap in T-DNA or Transposon Mutagenesis
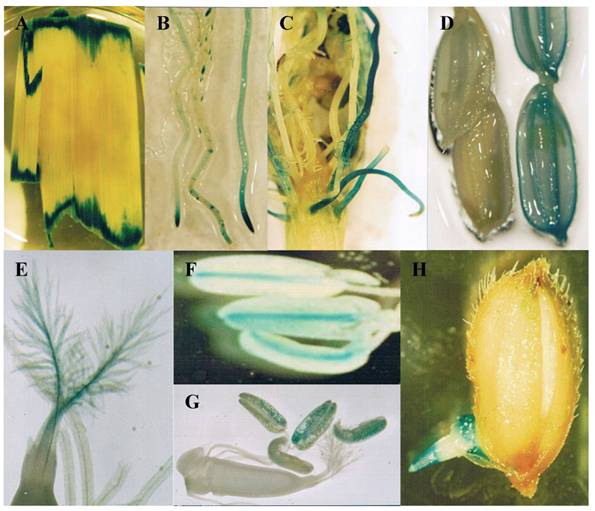
Not all genes can be functionally identified by mutagenesis. One of the reasons is that many genes are functionally redundant and mutation of such genes may not lead to an easily recognizable phenotype. Another reason is that many genes function at multiple stages of development and mutations of these genes may lead to early lethality. Therefore, it is necessary to develop a system to monitor gene expression patterns to better understand functions of these genes. Gene trap, promoter trap or enhancer trap is a system that allows gene activity to be monitored by creating gene fusions with a reporter gene. In an enhancer trap, a reporter gene has a minimal promoter that is only expressed when inserted near cis-acting chromosomal enhancers. Reporter genes in gene trap and promoter trap have no promoter, so that reporter genes expression can occur only when the reporter gene inserts within a transcribed chromosomal gene, creating a transcriptional fusion. Expression of a promoter trap reporter gene requires that it be inserted into an exon, leading to a transcriptional fusion. In contrast, gene trap constructs contain one or more splice acceptor sequences preceding the reporter gene. Thus reporter genes can be detected even if insertion occurs in an intron since splicing from the splice donor sites to the splice acceptor sites in the reporter gene results in fusion of upstream exon sequences to the reporter gene [86]. Currently, this system has been widely used for T-DNA or transposon mutagenesis. The GUS reporter gene is the mostly used gene for various trap systems in rice. In an enhancer trap Ds insertion population, around 8% of the lines were detected with GUS expression in panicles [81]. For T-DNA promoter trap lines, histochemical GUS assays were carried out in the leaves and roots from 5353 lines, mature flowers from 7026 lines, and developing seeds from 1948 lines. The data revealed that 1.6-2.1% of tested organs were GUS-positive and that their GUS expression patterns were organ- or tissue-specific or ubiquitous in all parts of the plant [56]. In our lab, 2852 Ds lines were subjected to GUS assay and the result showed that around 8.1% of the lines were with GUS activities [87]. Some of the examples are shown in Figure 2.
Expression of GUS in gene trapped Ds insertion lines. These images show various GUS expression patterns. (A) Expressed in wounded leaves. (B) Expressed in root tips. (C) Expressed in lateral roots. (D) Expressed in grain hulls (left image is WT control). (E) Expressed in stigma. (F) Expressed in connective tissues of anthers. (G) Expressed in pollens. (H) Expressed in geminated seeds.
These analyses suggested that Ds-tagged genes exhibited different expression patterns due to the Ds insertion into different genomic positions. Furthermore, the multimerized transcriptional enhancers from the cauliflower mosaic virus 35S promoter were positioned next to the left border of the T-DNA to make activation tagged lines [57]. Histochemical GUS assays have revealed that the GUS-staining frequency from those lines is about twice as high as that from lines without the enhancer element. This result suggests that the enhancer sequence presented in the T-DNA improves the GUS-tagging efficiency [57]. In another report, a CaMV35S enhancer was cloned in eight tandem repeats. This octamer configuration may serve as more potent activator than the traditional tetramer, as gene distances as far as 12.5 kb from the ATG start codon led to gene activation [60]. Thus, most insertions of the CaMV35S enhancers into the rice genome (excluding insertions in exons and introns that lead to gene knockout) have the potential to activate at least one native gene based on the average gene density of one gene per 9.9 kb in the rice genome [4]. Recently, a versatile transposon-based activation tag vector system was used for functional genomics in cereals and other monocot plants to further enhance rice gene expression [88]. All these data showed that gene trap, promoter trap and enhancer trap in T-DNA or transposon mutagenesis can be used as efficient tools to trap gene expression and to analyze their functions.
Besides the activation tagging system, gain-of-function type mutants may also be obtained from over-expression of individual rice genes. Both over-expression and gene silencing (see below) have been widely used for the annotation of gene functions. Currently, many rice genes have been functionally characterized by the over-expression of their genes. For example, the biological function of ETHYLENE RESPONSE2 (ETR2) was annotated by comparing the difference between gain-of-function and knockout mutants [89]. To investigate the over-expression mutants at the genome-wide level, special binary vectors have been designed to globally over-express all genes in an organism [90-92]. Up to now, at least 45,000 FOX hunting rice lines have been generated [93, 94].
Tissue Culture Mutagenesis and Retrotransposon Tos17
Tissue culture is also an efficient tool to induce various mutations, which is called somaclonal variations [95]. Tissue culture mutagenesis formed the important resources for rice breeding [96, 97]. However, little is known about the application of this technique in functional genomics until that some transposon elements in maize can be activated during tissue culture, indicating that some tissue culture-derived genetic variability may be the result of insertion or excision of transposable elements, or both [98]. Subsequent studies showed that active DNA transposon elements were also observed during rice tissue culture [99-101]. In addition to transposons, active retrotransposons were also detected during rice tissue culture [102]. Differentiated from transposons, retrotransposons are mobile genetic elements that transpose through reverse transcription of an RNA intermediate. One of these retrotransposons was named as Tos17, a widely utilized retrotransposon in rice [102]. One to five copies of Tos17 elements can be detected in normal growth conditions, varying with different rice varieties. For example, two copies of Tos17 were detected in japonica variety Nipponbare. These Tos17 elements have usually no activity in normal growth conditions. However, Tos17 will be activated during tissue culture and its copy number will increase to 5-30 [102]. For example, at least 5 new insertions of Tos17 were induced during 3- to 9- month tissue culture. Although Tos17 is actively transcribed during tissue culture, no transcript of Tos17 was detected in plants regenerated from tissue culture [102-104], suggesting that transposition is active only under tissue culture conditions. This result indicated that Tos17 could be used for mutagenesis to generate stabilized insertion lines. Subsequent studies showed that insertion sites were mostly found in genic regions and preferably in coding sequences [105, 106]. In 2001, 32,000 rice lines were generated from Tos17, containing 256,000 insertions [103]. Now they have produced around 50,000 insertion lines (Fig. 1C; [107]). Phenotypic investigation of these insertion lines indicated that nearly half of the lines showed more than one mutant phenotype; the most frequently observed phenotype was low fertility, followed by dwarfism [107]. These phenotype data have been submitted into Tos17 mutant database with a dataset of sequences flanking Tos17 insertion points in rice genome (http://tos.nias.affrc.go.jp/).
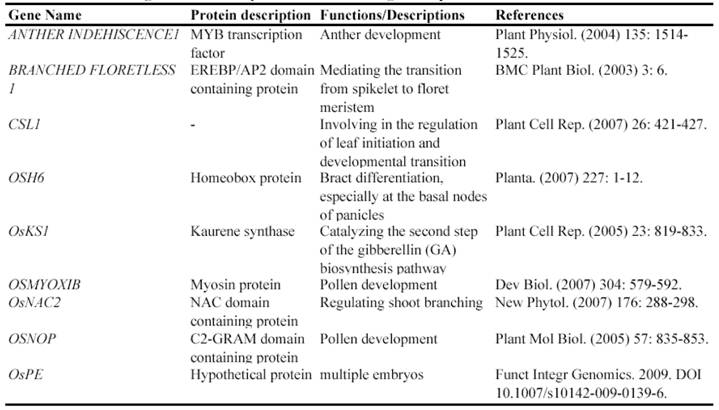
Since the identification and characterization of Tos17, many genes have been isolated and functionally characterized through Tos17 insertion lines. Currently, at least 24 genes have been characterized by Tos17 insertion (Table 4). For example, oshkt2;1 is the first mutant that greatly diminishes sodium influx into plant roots. Further investigator showed that OsHKT2;1 is the central transporter for nutritional Na+ uptake into K+ starved rice roots [108].
Some of rice genes functionally characterized through retrotransposon Tos17

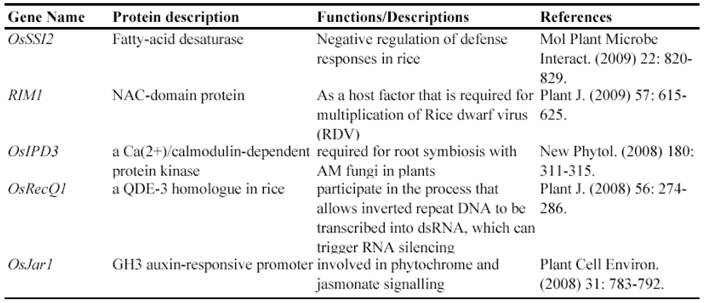
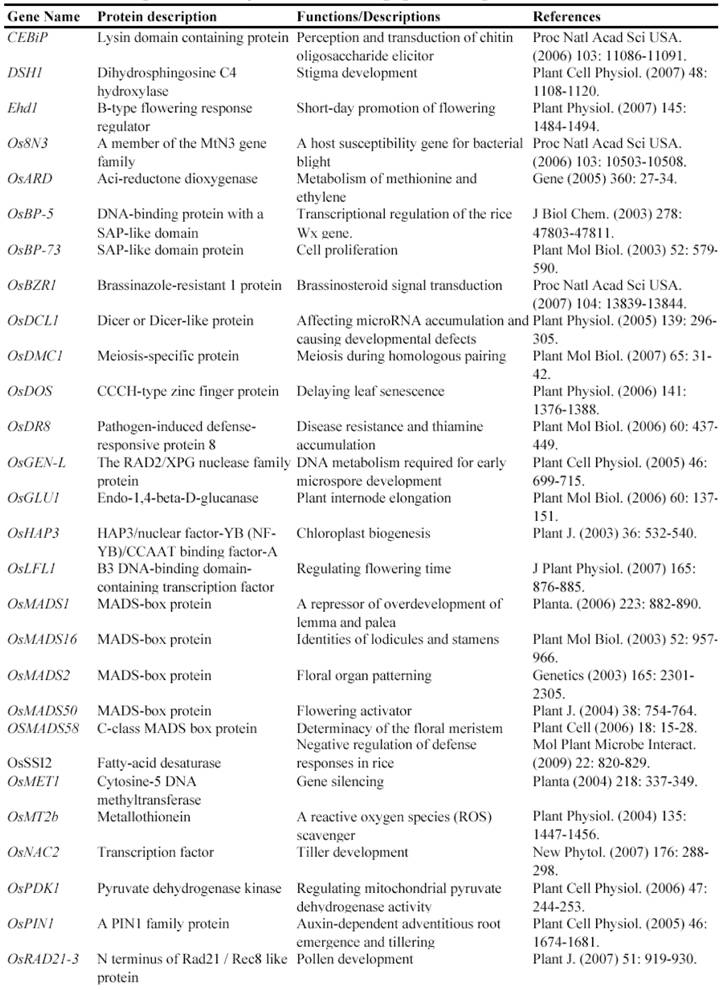
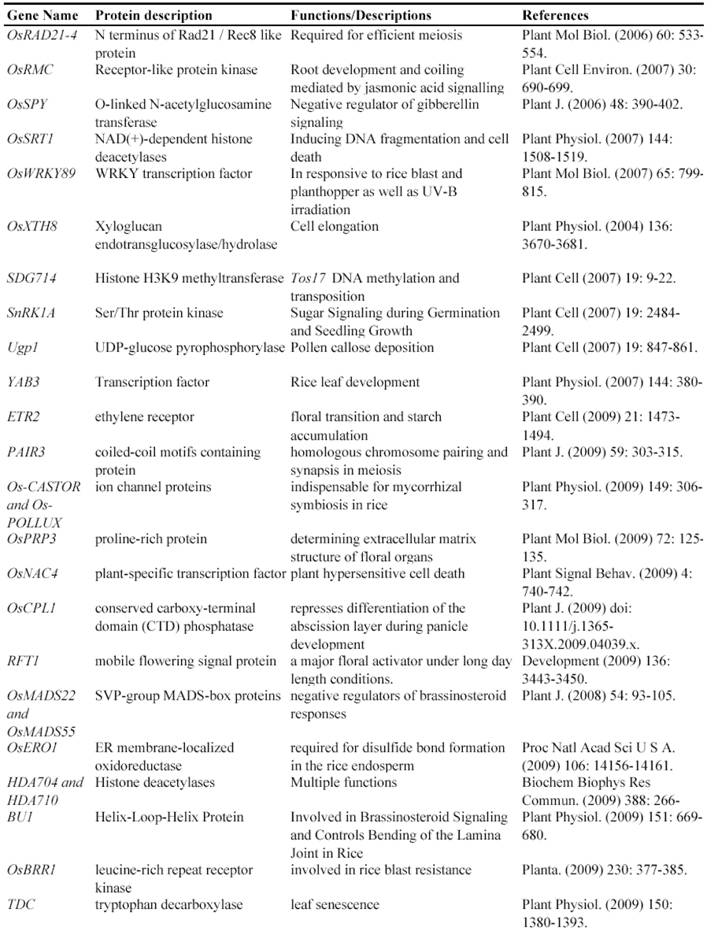
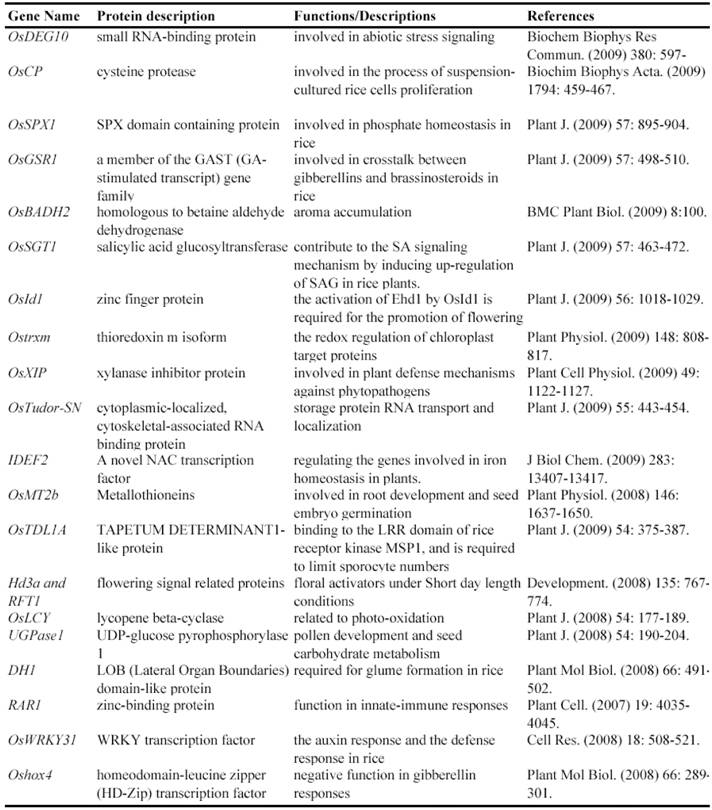
Gene Silencing Mutagenesis and its Application on Functional Identification of Rice Genes
Silencing a gene is also an efficient tool to determine its functions. Several methods can be used to silence a gene. For example, anti-sense or co-suppression was frequently observed in transgenic plants [109, 110]. However, in some cases, only partial functions can be suppressed by anti-sense or co-suppression. Among RNA silencing methods, RNA interference (RNAi) is now widely used for gene silencing. During silencing, double-stranded RNA (dsRNA) is processed into 20- to 25-nt short interfering RNA (siRNA) and microRNA (miRNA) by RNaseIII-like enzymes called Dicers1. siRNAs and miRNAs guide RNA-induced silencing complexes (RISCs) to suppress gene expression at the level of transcription, RNA stability or translation. siRNAs are 21-23 nucleotide double-stranded RNA molecules. Once incorporated into RISC they facilitate the cleavage and degradation of their recognized mRNA. MicroRNAs are single-stranded RNAs of 22-nucleotides that are processed from ~70-nucleotide hairpin RNA precursors by Dicers. Similar to siRNAs, miRNAs can silence gene activity through destruction of homologous mRNA or blocking its translation in plants.
The key to utilize the RNAi silencing in plants is how to transfer double strand RNAs into cells. In animals, RNAi can be initiated by injecting or feeding dsRNA into cells [111]. However, these methods can not be used in plants. Currently, at least two types of constructs have been used for RNAi-mediated silencing in plants. One is by virus-based vectors to transfer dsRNAs into plant cells. However, the virus-based silencing method can not be genetically inherited. Another one is by hairpin RNA (hpRNA)-mediated gene silencing. In this method, the target gene is cloned as an inverted repeat spaced with an unrelated sequence and is driven by a strong promoter, such as the 35S CaMV promoter for dicots or the maize ubiquitin 1 promoter for monocots. When an intron is used as the spacer, the efficiency becomes very high: almost 100% of transgenic plants show gene silencing [112]. However, this technique cannot be applied to genes whose silencing may block plant regeneration or result in embryo lethality. To obviate these potential problems, a chemical-inducible Cre/loxP (CLX) recombination system was used to trigger the expression of an intron-containing inverted-repeat RNA (RNAi) in plants [113]. In addition, a vector for high-throughput cloning of target genes as inverted repeats has been constructed for genome-wide analysis of gene functions [114]. Another such RNAi vector is based on the spreading of RNA targeting (transitive RNAi) from an inverted repeat of a heterologous 3'UTR [115]. Thus, the RNAi-mediated gene silencing can be used as a tool not only to analyze gene functions of a single gene or a gene family but also for genome-wide analysis of gene functions.
Now, this RNAi-mediated gene silencing has been successfully used for identification of gene functions in rice plants. In less than 7 years from 2003, at least 71 genes were functionally characterized by the RNAi silencing (Table 5). These genes included genes encoding various transcription factors, flowering-related proteins, pathogen/membrane-related proteins, various protein kinases and cell division-related proteins and so on. These results suggested that various genes can be silenced by RNAi and this method may be universally employed for characterizing various rice genes.
Some of rice genes functionally characterized through gene silencing mutants
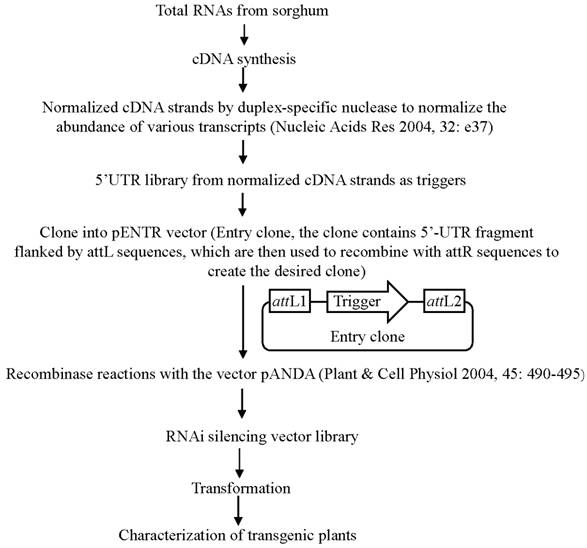
In order to use this RNAi-mediated gene silencing at the genome-wide level, we have designed a binary vector to globally silence rice genes (Fig. 3). Currently, in plants, genome-wide RNAi technology has been developed only in Arabidopsis [116]. However, this method was based on whole genome sequence information and was costly. The new system is based on the construction of a cDNA library and subsequent RNAi constructs followed by transformation; thus, it is relatively cheap and can be used for other plants since no sequence information is required to generate such system. On the other hand, gene silence-based phenotypes can be investigated during the first or second generation; thus, it is a time-saved system. In this system, 300-500 bp 5'-UTR fragments are generated from a normalized cDNA library and are then cloned into the Gateway pENTR vector, which carry two recombination sites (attL1 and attL2) for LR clonase reactions (Fig. 3). These cloned UTR fragments are then transferred into a pANDA destination vector, which was developed by Miki and Shimamoto (2004) [117], through recombinase reactions. Thus, a RNAi silencing vector library is constructed, where UTR fragments are inserted into two regions flanked by two inverted repeats. This library can be used for transformation to develop a collection of transgenic rice plants integrated with different RNAi silencing T-DNAs.
Binary vector construction for genome-wide gene silencing. This figure shows the detail of the construction of gene silencing vector.
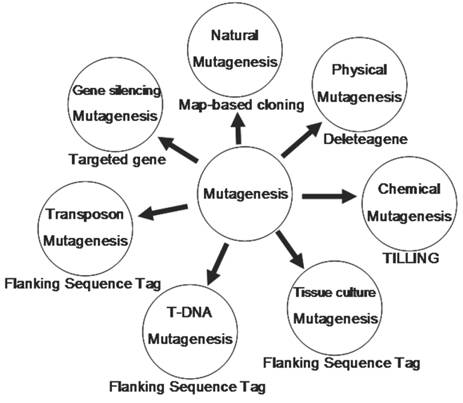
In summary, we have reviewed 7 different methods for mutagenesis including natural, physical, chemical, tissue culture, T-DNA, transposon and gene silencing mutagenesis (Fig. 4). Natural mutagenesis has been widely used to identify gene functions but this method can not be used for genome-wide analysis due to its low frequency of mutation and its difficulty in identifying mutated genes by map-based cloning. Chemical mutagenesis can be efficiently used to produce a large number of point mutants in a short period and the induced mutants can be detected by TILLING. However, multiple point mutations were sometimes observed in one mutant. Thus, it is necessary to genetically segregate these point mutants. Similarly, Physical mutagenesis can be applied for producing a large number of deletion-based mutants in a short period, and the deletion mutants can be screened by Deleteagene. However, it is also very difficult to identify a deletion mutant and its phenotype when the induced deletion occurs covering multiple genes or within an intron. Insertional mutagenesis based on T-DNAs, transposons and retrotransposons is becoming a major approach to produce a saturated mutant pool. A large number of T-DNA insertion lines have been produced in rice; but T-DNA insertional mutagenesis can be used only for those rice varieties with highly efficient transformation. The retrotransposon Tos17 has been successfully applied for rice functional genomics. But it is also difficult to identify a mutant related to Tos17 because there are multiple copies of Tos17 in a mutant and only about 10 percent of mutants are tagged by Tos17 insertion. Theoretically, Ac/Ds two-element system is regarded as a best approach for rice insertional mutagenesis because more than 95% of Ds insertion lines have only single copy of Ds insertion. An additional advantage is that Ds can be remobilized from a tagged gene in the presence of Ac transposase, resulting in phenotypic reversion to the wild type or giving rise to alleles with weaker phenotypes. However, it is also difficult to identify a mutant when there are Ds excision footprints in the mutant caused by multiple Ds excision-insertion events in the presence of Ac transposase. RNAi can efficiently silence a gene, but not all genes can be silenced. In addition, RNAi can interfere in genes with redundant and overlapping functions or gene families with high homolog in sequence, making it difficult to identify a silenced gene. So it is obvious that each method has its advantage and disadvantage and different methods should be combined to produce a saturated mutant population.
The technological platform for rice functional genomics. This figure shows various tools used to generate mutants and the strategies to screen these mutants.
Natural and Artificial Mutants as Valuable Resources for Molecular Breeding
Large-scale phenotype investigation has been carried out in rice using several mutant resources. Chern et al (2007) reported 11 categories of the visible phenotypes including growth condition, leaf color, leaf morphology, plant morphology, mimic response, tiller, heading date, flower, panicle, seed fertility and seed morphology, which were subdivided into 65 subcategories [71]. Miyao et al (2007) also investigated around 50,000 Tos17 insertion lines in their phenotype variation including germination, growth, leaf color, leaf shape, culm shape, spotted leaf/lesion mimic, tillering, heading date, spikelet, panicle, sterility and seed [107]. Park et al (2009) have analyzed 115,000 Ds insertion lines in their phenotype variation and 437 mutants from 12,162 Ds-tagged lines were catalogued in their agronomic traits including tillers, panicles, leaves, flowers, seed, chlorophyll content, and plant height [83]. Furthermore, several rice mutant phenotype databases are now established including Tos17 insertion lines [107], T-DNA-tagged lines [72], and chemical- and irradiation-induced lines [32]. Kuromori et al (2009) have reviewed a detail phenome analysis [67]. However, they have not discussed their application in rice breeding. Recently, Mochida and Shinozaki (2010) have summarized the genomics and bioinformatics resources for crop improvement [118].
We have subjected around 20,000 Ds insertion lines to phenotypic and abiotic stress screens and evaluated these lines with respect to their seed yields and other agronomic traits as well as their tolerance to drought, salinity and cold. Based on this evaluation, we observed that random Ds insertions into rice genome have led to diverse variations including a range of morphological phenotypes. We have observed various variations in these Ds insertion lines including the differences in plant height, growth vigor, growth period of duration and stigma and so on (Fig. 5). Among the various phenotypes identified, some Ds lines showed significantly higher grain yield compared to wild-type plants under normal growth conditions indicating that rice could be improved in grain yield by disrupting certain endogenous genes [87]. In addition, several thousands of Ds lines were subjected to abiotic stresses to identify conditional mutants. Subsequent to these screens, over 800 lines responsive to drought, salinity or cold stress were obtained, suggesting that rice has the genetic potential to survive under abiotic stresses when appropriate endogenous genes were suppressed. The mutant lines that have higher seed yielding potential or display higher tolerance to abiotic stresses may be used for rice breeding by conventional backcrossing combining with molecular marker-assisted selection. In addition, by exploiting the behavior of Ds to leave footprints upon remobilization, we have shown an alternative strategy to develop new rice varieties without foreign DNA sequences in their genome [87].
Phenotype investigation of Ds insertion lines. Left image is wild type (WT) control and right image is mutant line in A to D. (A) Dwarf phenotype. (B) Taller mutant. (C) weak-growth mutant, (D) Late-flowering mutant. E, Yellow-leaf mutant. F, Stigma in WT. G and H, Stigmas with brown color.

Phenotype screens of Ds insertion lines have identified two male sterile mutants. One is Orysa sativa no pollen (Osnop) mutant with a pollen-less phenotype at the flowering stage. The mutant phenotype showed linkage to Ds insertion into OSNOP gene region. This mutant contained a deletion of 65 kb chromosomal region at the site of Ds insertion containing 14 predicted genes. Among them, delegen 14 was expressed only in late stage of pollen development with the highest expression at the stage of pollen release and germination by RT-PCR, Northern blotting, in situ hybridization, and promoter-GUS transgenic plants. Thus, this gene is the best candidate for OSNOP. Since this gene encoded C2 and GRAM domains, it can be assumed that this gene cross-links both calcium and phosphoinositide signaling pathways. This is the first report to suggest possible functions for this gene in plant development [119].
Another one is the myosin mutant osmyoXIB. This mutant showed male sterility under short day length (SD) conditions and fertility under long day length (LD) conditions. Under both SD and LD conditions, the OSMYOXIB transcript was detected in whole anthers. However, under SD conditions, the OSMYOXIB-GUS fusion protein was localized only in the epidermal layer of anthers due to the lack of 3'-untranslated region (3'-UTR) and to dilute (DIL) domain sequences following the Ds insertion. As a result, mutant pollen development was affected, leading to male sterility. By contrast, under LD conditions, the fusion protein was localized normally in anthers. Despite normal localization, the protein was only partially functional due to the lack of DIL domain sequences, resulting in limited recovery of pollen fertility [120]. Since this mutant is a photoperiod sensitive male sterile line, it can be a candidate line to develop new male sterile lines for producing two-line hybrid rice.
Acknowledgements
We thank Drs. Ildiko Szeverenyi and Tatiana Kolesnik for their help in generation of transposant lines and Dr. Doris Bachmann for her help in phenotyping and GUS staining. We take this opportunity to thank Zhigang Ma and Hongfen Luan for their technical assistance.
Conflict of Interest
The authors have declared that no conflict of interest exists.
References
1. Rensink WA, Buell CR. Arabidopsis to rice. Applying knowledge from a weed to enhance our understanding of a crop species. Plant Physiol. 2004;135:622-9
2. Goff SA, Ricke D, Lan TH. et al. A draft sequence of the rice genome (Oryza sativa L. ssp. japonica). Science. 2002;296:92-100
3. Yu J, Hu S, Wang J. et al. A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science. 2002;296:79-92
4. International Rice Genome Sequencing Project. The map-based sequence of the rice genome. Nature. 2005;436:793-800
5. Sakata K, Nagamura Y, Numa H. et al. RiceGAAS: an automated annotation system and database for rice genome sequence. Nucleic Acids Res. 2002;30:98-102
6. Ohyanagi H, Tanaka T, Sakai H. et al. The rice annotation project database (RAP-DB): hub for Oryza sativa ssp. japonica genome information. Nucleic Acids Res. 2006;34(Database issue):D741-4
7. Yuan Q, Ouyang S, Wang A. et al. The institute for genomic research Osa1 rice genome annotation database. Plant Physiol. 2005;138:18-26
8. Ouyang S, Zhu W, Hamilton J. et al. The TIGR Rice Genome Annotation Resource: improvements and new features. Nucleic Acids Res. 2007;35(Database Issue):D846-51
9. Sterck L, Rombauts S, Vandepoele K. et al. How many genes are there in plants (.. and why are they there)? Curr Opin Plant Biol. 2007;10:199-203
10. Itoh T, Tanaka T, Barrero RA. et al. Curated genome annotation of Oryza sativa ssp. japonica and comparative genome analysis with Arabidopsis thaliana. Genome Res. 2007;17:175-83
11. Haas BJ, Wortman JR, Ronning CM. et al. Complete reannotation of the Arabidopsis genome: methods, tools, protocols and the final release. BMC Biol. 2005;3:7
12. Jiang SY, Ramachandran S. Identification and molecular characterization of myosin gene family in Oryza sativa genome. Plant Cell Physiol. 2004;45:590-9
13. Jiang SY, Phua JXH, Yeo YT. et al. Genome-wide Identification and Molecular Characterization of Ole_e_I, Allerg_1 and Allerg_2 Domain-containing Pollen-Allergen-like Genes in Oryza sativa. DNA Res. 2005;12:167-179
14. La H, Li J, Ji Z. et al. Genome-wide analysis of cyclin family in rice. Mol. Genet. Genomics. 2006;275:374-86
15. Jiang SY, Ramachandran S. Comparative and evolutionary analysis of genes encoding small GTPases and their activating proteins in eukaryotic genomes. Physiol Genomics. 2006;24:235-51
16. Jiang SY, Ramamoorthy R, Ramachandran S. Comparative transcriptional profiling and evolutionary analysis of the GRAM domain family in eukaryotes. Dev Biol. 2008;314:418-32
17. Kikuchi S, Satoh K, Nagata T. et al. Collection, mapping, and annotation of over 28,000 cDNA clones from japonica rice. Science. 2003;301:376-9
18. Liu X, Lu T, Yu S. et al. A collection of 10,096 indica rice full-length cDNAs reveals highly expressed sequence divergence between Oryza sativa indica and japonica subspecies. Plant Mol Biol. 2007;65:403-15
19. Holtorf H, Guitton MC, Reski R. Plant functional genomics. Naturwissenschaften. 2002;89:235-49
20. Evenson RE, Gollin D. Assessing the impact of the green revolution, 1960 to 2000. Science. 2003;300:758-62
21. Yuan L. Hybrid rice technology for food security in the world. FAO Rice Conference, Rome, Italy. 2004
22. International Rice Research Institute. Annual Report for 1966. 1967: 59-82.
23. Virmani SS, Sun ZX, Mou TM. et al. Two-line hybrid rice breeding manual. Los Banos, Philippines: International Rice Research Institute. 2003
24. Song WY, Wang GL, Chen LL. et al. A receptor kinase-like protein encoded by the rice disease resistance gene, Xa21. Science. 1995;270:1804-6
25. Gu K, Yang B, Tian D. et al. R gene expression induced by a type-III effector triggers disease resistance in rice. Nature. 2005;435:1122-5
26. Spielmeyer W, Ellis MH, Chandler PM. Semidwarf (sd-1), green revolution rice, contains a defective gibberellin 20-oxidase gene. Proc Natl Acad Sci USA. 2002;99:9043-8
27. Muller HJ. Types of visible variations induced by X-rays in Drosophila. J Genet. 1930;22:299-334
28. Koorneef M, Dellaert LWM, Vab der Veen JH. EMS- and radiation-induced mutation frequencies at individual loci in Arabidopsis thaliana (L.), Heynh. Mutation Res. 1982;93:109-23
29. Sun TP, Goodman HM, Ausubel FM. Cloning the Arabidopsis GA1 locus by genomic subtraction. Plant Cell. 1992;4:119-28
30. Li X, Song Y, Century K. et al. A fast neutron deletion mutagenesis-based reverse genetics system for plants. Plant J. 2001;27:235-42
31. Li X, Lassner M, Zhang Y. Deleteagene: a fast neutron deletion mutagenesis-based knockout system for plants. Comp Funct Genom. 2002;3:158-60
32. Wu JL, Wu C, Lei C. et al. Chemical- and irradiation-induced mutants of indica rice IR64 for forward and reverse genetics. Plant Mol Biol. 2005;59:85-97
33. Monte E, Alonso JM, Ecker JR. et al. Isolation and characterization of phyC mutants in Arabidopsis reveals complex crosstalk between phytochrome signaling pathways. Plant Cell. 2003;15:1962-80
34. Monte E, Tepperman JM, Al-Sady B. et al. The phytochrome-interacting transcription factor, PIF3, acts early, selectively, and positively in light-induced chloroplast development. Proc Natl Acad Sci USA. 2004;101:16091-8
35. Zhang Y, Tessaro MJ, Lassner M. et al. Knockout analysis of Arabidopsis transcription factors TGA2, TGA5, and TGA6 reveals their redundant and essential roles in systemic acquired resistance. Plant Cell. 2003;15:2647-53
36. Krieg DR. Ethyl methanesulfonate-induced reversion of bacteriophage T4rII mutants. Genetics. 1963;48:561-80
37. McCallum CM, Comai L, Greene EA. et al. Targeting induced local lessions in genomes (TILLING) for plant functional genomics. Plant Physiol. 2000;123:439-42
38. Jander G, Baerson SR, Hudak JA. et al. Ethyl methanesulfonate saturation mutagenesis in Arabidopsis to determine frequency of herbicide resistance. Plant Physiol. 2003;131:139-146
39. Gross E, Arnold N, Goette J. et al. A comparison of BRCA1 mutant analysis by direct sequencing, SSCP and DHPLC. Human Genet. 1999;105:72-8
40. Li-Sucholeiki XC, khrapko K, Andre PC. et al. Applications of constant denaturant capillary electrophoresis/high-fidelity polymerase chain reaction to human genetic analysis. Electrophoresis. 1999;20:1224-32
41. McCallum CM, Comai L, Greene EA. et al. Targeted screening for induced mutations. Nat Biotechnol. 2000;18:455-7
42. Colbert T, Till BJ, Tompa R. et al. High-throughput screening for induced point mutations. Plant Physiol. 2001;126:480-4
43. Henikoff S, Comai L. Single-nucleotide mutations for plant functional genomics. Ann Rev Plant Biol. 2003;54:375-401
44. Triques K, Sturbois B, Gallais S. et al. Characterization of Arabidopsis thaliana mismatch specific endonucleases: application to mutation discovery by TILLING in pea. Plant J. 2007;51:1116-25
45. Triques K, Piednoir E, Dalmais M. et al. Mutation detection using ENDO1: application to disease diagnostics in humans and TILLING and Eco-TILLING in plants. BMC Mol Biol. 2008;9:42
46. Cooper JL, Till BJ, Laport RG. et al. TILLING to detect induced mutations in soybean. BMC Plant Biol. 2008;8:9
47. Moens CB, Donn TM, Wolf-Saxon ER. et al. Reverse genetics in zebrafish by TILLING. Brief Funct Genomic Proteomic. 2008;7:454-9
48. Suzuki T, Eiguchi M, Kumamaru T. et al. MNU-induced mutant pools and high performance TILLING enable finding of any gene mutation in rice. Mol Genet Genomics. 2008;279:213-23
49. Wang N, Wang Y, Tian F. et al. A functional genomics resource for Brassica napus: development of an EMS mutagenized population and discovery of FAE1 point mutations by TILLING. New Phytol. 2008;180:751-65
50. Xin Z, Wang ML, Barkley NA. et al. Applying genotyping (TILLING) and phenotyping analyses to elucidate gene function in a chemically induced sorghum mutant population. BMC Plant Biol. 2008;8:103
51. Weil CF. TILLING in grass species. Plant Physiol. 2009;149:158-64
52. Till BJ, Cooper J, Tai TH. et al. Discovery of chemically induced mutations in rice by TILLING. BMC Plant Biol. 2007;7:19
53. Hiei Y, Ohta S, Komari T. et al. An efficient transformation of rice (Oryza sativa L.) mediated by Agrobaterium and sequence analysis of boundaries of the T-DNA. Plant J. 1994;6:271-82
54. Azpiroz-Leehan R, Feldmann KA. T-DNA insertion mutagenesis in Arabidopsis: going back and forth. Trends Genet. 1997;13:152-6
55. Alonso JM, Stepanova AN, Leisse TJ. et al. Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science. 2003;301:653-7
56. Jeon JS, Lee S, Jung KH. et al. T-DNA insertional mutagenesis for functional genomics in rice. Plant J. 2000;22:561-70
57. Jeong DH, An S, Kang HG. et al. T-DNA insertional mutagenesis for activation tagging in rice. Plant Physiol. 2002;130:1636-44
58. Wu C, Li X, Yuan W. et al. Development of enhancer trap lines for functional analysis of the rice genome. Plant J. 2003;35:418-27
59. Chen S, Jin W, Wang M. et al. Distribution and characterization of over 1000 T-DNA tags in rice genome. Plant J. 2003;36:105-13
60. Hsing YI, Chern CG, Fan MJ. et al. A rice gene activation/knockout mutant resource for high throughput functional genomics. Plant Mol Biol. 2007;63:351-64
61. Yin Z, Wang GL. Evidence of multiple complex patterns of T-DNA integration into rice genome. Theor Appl Genet. 2000;100:461-70
62. Sallaud C, Meynard D, van Boxtel J. et al. Highly e.cient production and characterization of T-DNA plants for rice (Oryza sativa L.) functional genomics. Theor Appl Genet. 2003;106:1396-408
63. Sha Y, Li S, Pei Z. et al. Generation and.anking sequence analysis of a rice T-DNA tagged population. Theor Appl Genet. 2004;108:306-14
64. An G, Jeong DH, Jung KH. et al. Reverse genetic approaches for functional genomics of rice. Plant Mol Biol. 2005;59:111-23
65. Droc G, Périn C, Fromentin S. et al. OryGenesDB 2008 update: database interoperability for functional genomics of rice. Nucleic Acids Res. 2009;37(Database issue):D992-995
66. Krishnan A, Guiderdoni E, An G. et al. Mutant Resources in Rice for Functional Genomics of the Grasses. Plant Physiol. 2009;149:165-70
67. Kuromori T, Takahashi S, Kondou Y. et al. Phenome analysis in plant species using loss-of-function and gain-of-function mutants. Plant Cell Physiol. 2009;50:1215-31
68. Barakat A, Gallois P, Raynal M. et al. The distribution of T-DNA in the genomes of transgenic Arabidopsis and rice. FEBS Lett. 2000;471:161-4
69. An S, Park S, Jeong DH. et al. Generation and analysis of end sequence database for T-DNA tagging lines in rice. Plant Physiol. 2003;133:2040-7
70. Zhang J, Guo D, Chang Y. et al. Non-random distribution of T-DNA insertions at various levels of the genome hierarchy as revealed by analyzing 13,804 T-DNA flanking sequences from an enhancer-trap mutant library. Plant J. 2007;49:947-59
71. Chern CG, Fan MJ, Yu SM. et al. A rice phenomics study-phenotype scoring and seed propagation of a T-DNA insertion-induced rice mutant population. Plant Mol Biol. 2007;65:427-38
72. Larmande P, Gay C, Lorieux M. et al. Oryza Tag Line, a phenotypic mutant database for the Genoplante rice insertion line library. Nucleic Acids Res. 2008;36(Database issue):D1022-7
73. Fu FF, Ye R, Xu SP. et al. Studies on rice seed quality through analysis of a large-scale T-DNA insertion population. Cell Res. 2009;19:380-91
74. Ma Y, Liu L, Zhu C. et al. Molecular analysis of rice plants harboring a multi-functional T-DNA tagging system. J Genet Genomics. 2009;36:267-76
75. Yamaguchi T, Lee DY, Miyao A. et al. Functional diversification of the two C-class MADS box genes OSMADS3 and OSMADS58 in Oryza sativa. Plant Cell. 2006;18:15-28
76. Kumar CS, Wing RA, Sundaresan V. Efficient insertional mutagenesis in rice using the maize En/Spm elements. Plant J. 2005;44:879-92
77. Upadhyaya NM, Zhou XR, Zhu QH. et al. An iAc/Ds gene and enhancer trapping system for insertional mutagenesis in rice. Funct Plant Biol. 2002;29:547-59
78. Kolesnik T, Szeverenyi I, Bachmann D. et al. Establishing an efficient Ac/Ds tagging system in rice: large-scale analysis of Ds flanking sequences. Plant J. 2004;37:301-14
79. Greco R, Ouwerkerk PB, De Kam RJ. et al. Transpositional behaviour of an Ac/Ds system for reverse genetics in rice. Theor Appl Genet. 2003;108:10-24
80. van Enckevort LJ, Droc G, Piffanelli P. et al. EU-OSTID: a collection of transposon insertional mutants for functional genomics in rice. Plant Mol Biol. 2005;59:99-110
81. Chin HG, Choe MS, Lee SH. et al. Molecular analysis of rice plants harboring an Ac/Ds transposable element-mediated gene trapping system. Plant J. 1999;19:615-23
82. Park SH, Jun NS, Kim CM. et al. Analysis of gene-trap Ds rice populations in Korea. Plant Mol Biol. 2007;65:373-84
83. Park DS, Park SK, Han SI. et al. Genetic variation through Dissociation (Ds) insertional mutagenesis system for rice in Korea: progress and current status. Mol Breeding. 2009;24:1-15
84. Jung KH, An G, Ronald PC. Towards a better bowl of rice: assigning function to tens of thousands of rice genes. Nat Rev Genet. 2008;9:91-101
85. Szeverenyi I, Ramamoorthy R, Teo ZW. et al. Large scale systematic study on stability of Ds element and timing of transposition in rice. Plant Cell Physiol. 2006;47:84-95
86. Springer PS. Gene traps: tools for plant development and genomics. Plant Cell. 2000;12:1007-20
87. Jiang SY, Bachmann D, La H. et al. Ds insertion mutagenesis as an efficient tool to produce diverse variations for rice breeding. Plant Mol Biol. 2007;65:385-402
88. Qu S, Desai A, Wing R. et al. A versatile transposon-based activation tag vector system for functional genomics in cereals and other monocot plants. Plant Physiol. 2008;146:189-99
89. Wuriyanghan H, Zhang B, Cao WH. et al. The ethylene receptor ETR2 delays floral transition and affects starch accumulation in rice. Plant Cell. 2009;21:1473-94
90. Ichikawa T, Nakazawa M, Kawashima M. et al. The FOX hunting system: an alternative gain-of-function gene hunting technique. Plant J. 2006;48:974-985
91. Weiste C, Iven T, Fischer U. et al. In planta ORFeome analysis by large-scale over-expression of GATEWAY-compatible cDNA clones: screening of ERF transcription factors involved in abiotic stress defense. Plant J. 2007;52:382-90
92. Pogorelko GV, Fursova OV, Ogarkova OA. et al. A new technique for activation tagging in Arabidopsis. Gene. 2008;414:67-75
93. Nakamura H, Hakata M, Amano K. et al. A genome-wide gain-of function analysis of rice genes using the FOX-hunting system. Plant Mol Biol. 2007;65:357-71
94. Kondou Y, Higuchi M, Takahashi S. et al. Systematic approaches to using the FOX hunting system to identify useful rice genes. Plant J. 2009;57:883-94
95. Larkin PJ, Scowcroft P. Somaclonal variation-----a novel source of variability from cell culture for plant improvement. Theor Appl Genet. 1981;60:197-214
96. Adkins SW, Kunauvatchaidach R, godwin ID. Somaclonal variation in rice-drought tolerance and other agronomic characters. Aust J Bot. 1995;43:201-9
97. Xie Q, linscombe R, Rush MC. et al. Registration of LSBR-5 sheath blight-resistant germplasm lines in rice. Crop Sci. 1992;32:507
98. Peschke VM, Phillips RL, Gengenbach BG. Discovery of transposable element activity among progeny of tissue culture--derived maize plants. Science. 1987;238:804-7
99. Jiang N, Bao Z, Zhang X. et al. An active DNA transposon family in rice. Nature. 2003;421:163-7
100. Kikuchi K, Terauchi K, Wada M. et al. The plant MITE mPing is mobilized in anther culture. Nature. 2003;421:167-70
101. Nakazaki T, Okumoto Y, Horibata A. et al. Mobilization of a transposon in the rice genome. Nature. 2003;421:170-2
102. Hirochika H, Sugimoto K, Otsuki Y. et al. Retrotransposons of rice involved in mutations induced by tissue culture. Proc Natl Acad Sci USA. 1996;93:7783-8
103. Hirochika H. Contribution of the Tos17 retrotransposon to rice functional genomics. Curr Opin Plant Biol. 2001;4:118-22
104. Yamazaki M, Tsugawa H, Miyao A. et al. The rice retrotransposon Tos17 prefers low copy sequences as integration targets. Mol Gen Geneti. 2001;265:336-44
105. Miyao A, Tanaka K, Murata K. et al. Target site specificity of the Tos17 retrotransposon shows a preference for insertion within genes and against insertion in retrotransposon-rich regions of the genome. Plant Cell. 2003;15:1771-80
106. Piffanelli P, Droc G, Mieulet D. et al. Large-scale characterization of Tos17 insertion sites in a rice T-DNA mutant library. Plant Mol Biol. 2007;65:587-601
107. Miyao A, Iwasaki Y, Kitano H. et al. A large-scale collection of phenotypic data describing an insertional mutant population to facilitate functional analysis of rice genes. Plant Mol Biol. 2007;63:625-35
108. Horie T, Costa A, Kim TH. et al. Rice OsHKT2;1 transporter mediates large Na+ influx component into K+-starved roots for growth. EMBO J. 2007;26:3003-14
109. Baulcombe DC. RNA as a target and an initiator of post-transcriptional gene silencing in transgenic plants. Plant Mol Biol. 1996;32:79-82
110. Jorgensen RA, Que Q, Stam M. Do unintended antisense transcripts contribute to sense co-suppression in plants? Trends Genet. 1999;15:11-2
111. Zamore PD. RNA interference: listening to the sound of silence. Nat Struct Biol. 2001;8:746-50
112. Kusaba M. RNA interference in crop plants. Curr Opin Biotechnol. 2004;15:139-43
113. Guo HS, Fei JF, Xie Q. et al. A chemical-regulated inducible RNAi system in plants. Plant J. 2003;34:383-92
114. Wesley SV, Helliwell CA, Smith NA. et al. Construct design for efficient, effective and high-throughput gene silencing in plants. Plant J. 2001;27:581-90
115. Brummell DA, Balint-Kurti P, Harpster MH. et al. Inverted repeat of a heterologous 3'-untranslated region for high-efficiency, high-throughput gene silencing. Plant J. 2003;33:798-800
116. Hilson P, Allemeersch J, Altmann T. et al. Versatile gene-specific sequence tags for Arabidopsis functional genomics: transcript profiling and reverse genetics applications. Genome Res. 2004;14:2176-89
117. Miki D, Shimamoto K. Simple RNAi vectors for stable and transient suppression of gene function in rice. Plant Cell Physiol. 2004;45:490-5
118. Mochida K, Shinozaki K. Genomics and bioinformatics resources for crop improvement. Plant Cell Physiol. 2010;51:497-523
119. Jiang SY, Cai M, Ramachandran S. The Oryza sativa no pollen (Osnop) gene plays a role in male gametophyte development and most likely encodes a C2-GRAM domain-containing protein. Plant Mol Biol. 2005;57:835-53
120. Jiang SY, Cai M, Ramachandran S. ORYZA SATIVA MYOSIN XI B controls pollen development by photoperiod-sensitive protein localizations. Dev Biol. 2007;304:579-592
121. Sallaud C, Gay C, Larmande P. et al. High throughput T-DNA insertion mutagenesis in rice: a first step towards in silico reverse genetics. Plant J. 2004;39:450-64
122. Han B, Xue Y, Li J. et al. Rice functional genomics research in China. Philos Trans R Soc Lond B Biol Sci. 2007;362:1009-21
123. Wan S, Wu J, Zhang Z. et al. Activation tagging, an efficient tool for functional analysis of the rice genome. Plant Mol Biol. 2009;69:69-80
124. Zhang J, Li C, Wu C. et al. RMD: a rice mutant database for functional analysis of the rice genome. Nucleic Acids Res. 2006;34:D745-8
Author contact
![]() Corresponding author: E-mail: sriorg.sg, Telephone: 65-68727480, Fax: 65-68727007
Corresponding author: E-mail: sriorg.sg, Telephone: 65-68727480, Fax: 65-68727007