Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Innate Immunity and the...
Inflammatory diseases in the...
Recent developments
Discussion
Conclusions
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(3):261-268. doi:10.7150/ijbs.7.261 This issue Cite
Review
Innate Immunity and the Pathogenicity of Inhaled Microbial Particles
C. Henrik J. Wolff ![]()
Finnish Institute of Occupational Health (FIOH), Biological Mechanisms and the Prevention of Work related Diseases
Received 2011-1-19; Accepted 2011-3-9; Published 2011-3-15
Abstract
Non-infectious inhaled microbial particles can cause illness by triggering an inappropriate immunological response. From the pathogenic point of view these illnesses can be seen to be related to on one hand autoimmune diseases and on the other infectious diseases.
In this review three such illnesses are discussed in some detail. Hypersensitivity pneumonitis (HP) is the best known of these illnesses and it has also been widely studied in animal models and clinically. In contrast to HP Pulmonary mycotoxicosis (PM) is not considered to involve immunological memory, it is an acute self-limiting condition is caused by an immediate "toxic" effect. Damp building related illness (DBRI) is a controversial and from a diagnostic point poorly defined entity that is however causing, or attributed to cause, much more morbidity than the two other diseases.
In the recent decade there has been a shift in the focus of immunology from the lymphocyte centered, adaptive immunity towards innate immunity. The archetypal cell in innate immunity is the macrophage although many other cell types participate. Innate immunity relies on a limited number of germline coded receptors for the recognition of pathogens and signs of cellular damage. The focus on innate immunity has opened new paths for the understanding of many chronic inflammatory diseases. The purpose of this review is to discuss the impact of some recent studies, that include aspects concerning innate immunity, on our understanding of the pathogenesis of inflammatory diseases associated with exposure to inhaled microbial matter.
Keywords: innate immunity, inflammasome, hypersensitivity pneumonitis, pulmonary mycotoxicosis, mold, damp building related inflammatory disease, sick building syndrome
Innate Immunity and the inflammasome
Vertebrates have a two-tiered system to defend themselves against pathogen. The innate immunity system is the first one to be activated by pathogens and it is for the most part sufficient on its own, however when the strength of the challenge from pathogens reaches an critical threshold the innate immunity triggers the adaptive immunity system. The adaptive immunity relies on receptors on the B and T cells generated through somatic gene rearrangements and hypermutation allowing the adaptive immunity to specifically recognize any type of microorganisms. In the innate system microbes are recognized by a limited number of germline coded receptors called pattern recognition receptors (PRRs) [1-3]. The PRRs recognize structures called pathogen-associated molecular patterns (PAMPs) that are common to many pathogens. The PAMPs are usually essential for the viability of the microbes, thus minimizing the possibilities of the pathogens to avoid detection by simply altering the PAMP. Substances recognized by PAMPs include cell wall components peptidoglycan, lipopolysaccharide and bacterial DNA. The PRRs include Toll-like receptors (TLRs), NOD- of like receptors (NLRs), C-type lectin receptors, and RIG-I like receptors [4, 5]. The model based on PAMP recognition was proposed by Charles Janeway Jr [6]. It was soon realized that an additional element was required to explain why non- pathogenic bacteria did not cause activation of the innate immunity. Polly Matzinger suggested that the innate immunity system in addition relies on the recognition of cell injury by danger associated molecular patterns (DAMPs) [7]. Subsequently it has indeed been shown that that among others ATP, mammalian double stranded DNA and uric acid crystals activate an inflammatory response [8].
The receptors engaged then induce the synthesis of a spectrum of inflammatory mediators. Central for most inflammatory reactions is the production of IL-1β and a related protein IL-18, both are produced in the cytosol as inactive precursor forms. IL-18 is present in resting cells while IL-1β precursor is only produced in activated cells [9]. The formation of bioactive secreted 17- or 18kDa forms of IL-1β and IL-18 is dependent on the proteolytic activity of a caspase which is activated in molecular platforms named inflammasomes [10]. Eleven caspases are presently known in humans, these are divided into pro-inflammatory caspases (caspases 1, 4, and 5) and apoptotic caspases (caspases 2, 3, and 6-10) [11, 12]. Inflammasomes can be described as multiprotein complexes that link the recognition of cellular damage by members of the Nod-like receptor (NLR) to the processing and release of IL-1β and IL-18. NLR:s are cytosolic intracellular PRRs that recognize PAMPS and endogenous ligands. In addition to activating the inflammasome NLR:s can activate the NFκB pathway [13]. In general there is a surprising tendency for DAMP:s and PAMPS to use similar molecular pathways. This is apparent with HSP:s (heat shock proteins) that are endogenous damage associated proteins recognized by the typically PAMP related TLR-MyD88-NFκB pathway [14]. Another DAMP released by damaged cells is HMGB-1 which is also extensively recognized by TLR:s [15].
Central components of the inflammasome are NLRs, the pyrin domain (PYD) and caspase recruitment domain (CARD) containing adaptor protein ASC (apoptosis-associated speck-like protein containing a CARD) and caspase-1.
The activation of the inflammasome is initiated by the recognition of intracellular PAMPS or signs of stress or danger from the hosts, DAMPS, by a cytosolic NLR. The NLRS undergo ATP-dependent oligomerization and recruit ASC by PYD-PYD interaction. Subsequently, caspase 1 is recruited through the CARD of ASC.
NLRP3 (NLR pyrin domain containing 3,) and the inflammasome it is a part of has been linked to a number of inflammatory diseases. These include contact hypersensitivity, sunburn, essential hypertension, gout and pseudogout. NLRP3 is activated by exogenous and endogenous ligands. Exogenous ligands include microbial ligands (PAMP)s such as the bacterial product MDP (muramyl dipeptide) bacterial and viral RNA. Endogenous ligands (PAMPS )include uric acid (monosodium urate crystals), calcium pyrophosphate crystals, NLRP3 in combination with the P2X7 receptors take part in recognizing extracellular ATP[3]. Fig 1 shows a schematic representation of the activation of IL-1β production by LPS as an example of a PAMP and uric acid as an example of a DAMP.
Example of IL-1β production activation after ligation of a TLR receptor by LPS and activation of the NALP3 inflammasome by uric acid.
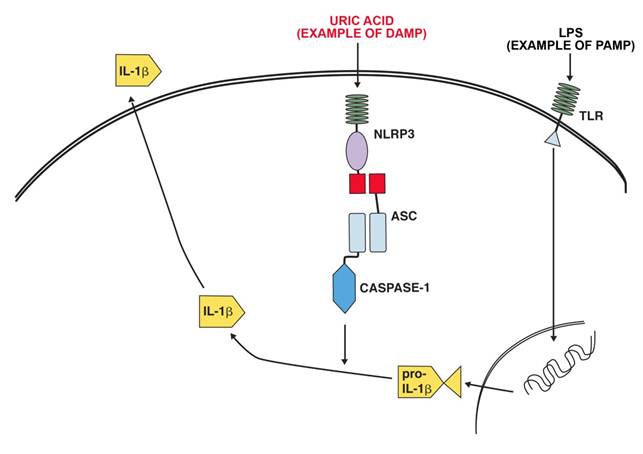
The complexity of inflammatory regulation is understandable from the point of view that there are severe risks for the organism both from a lack of sufficient response to a pathogen or an exaggerated response to a pathogen. There are thus two separate major control points for the production of IL-1β, transcription is initiated by the recognition of pathogen (and some DAMPS) by PRR:s and secretion of the active form is initiated by activation of the inflammasome by DAMPS and intracellular PAMPS [16].
Inflammatory diseases in the airways caused by exposure to microbial substances
Pulmonary mycotoxicosis (PM, organic dust syndrome) is an acute febrile respiratory illness associated with the exposure to high levels of microbial matter. Typically the exposure occurs in an agricultural context, but every occupation associated with the handling of organic material possesses some risk. PM does not depend on immunological sensitization, so it can occur during or immediately after the first exposure. Airborne bacterial and fungal spore counts are mostly higher than those associated with HP [17]. In cases of heavy exposure to organic dust most of the exposed become ill. However in conditions of lighter exposure the proportion individuals affected can be very small. The clinical picture is an influenza like reaction with 70-95 % of affected individuals experiencing shivering with fever and myalgias. Approximately half will experience chest tightness and cough, headache and nausea is also fairly common. Irritation of eyes, nose and throat has also been reported. Symptoms will occur 4-12 hours after the start of the exposure but they rarely persist beyond 1 or 2 days. Continuing the exposure will often decrease the symptoms, in contrast to the other illnesses discussed in this review. PM is characterized by macrophages and neutrophiles in the BAL fluid [18] and an interstitial and intra-alveolar neutrophilic and macrophage infiltrate associated with bronchiolitis [18], In some cases lymphocytosis has been reported at later stages[19, 20]. In general there is not much ongoing research activity in this disease, probably because it is a relatively benign and self-limiting condition. In addition the incidence of PM is probably decreasing because of changing work methods. In a study on Swedish farmers that had experienced PM it was a single event for almost half of farmers studied while the rest had two or more attacks often several years apart[21]. Some clinical features of PM as well as HP and DBRI are shown in table 1.
Some features of illnesses attributed to the exposure of microbial particles
| Pulmonary mycotoxicosis | DBRI | HP | |
|---|---|---|---|
| Exposures required | Single | Repeated (see text) | Repeated |
| Consequence of re-exposure | Activity of illness can decrease | Activity of illness increases | Activity of illness increases |
| Susceptibility of the exposed | All of the exposed can develop illness | Part of the exposed develop illness (see text) | Part of the exposed develop illness |
| Cells increased in BAL | neutrophils macrophages lymphocytes | lymphocytes, macrophages ? | lymphocytes, macrophages |
Hypersensitivity pneumonitis (HP), also known as allergic alveolitis, is caused by an exposure to a wide range of thermophilic bacteria, molds, other bacteria and bacterial products, amoebae, and some chemicals. In some cases HP is caused by a reaction to a single environmental agent while in other cases a mixture of microbes is required [22]. Only a small portion (1%-15%) [22, 23], of those exposed develop HP. The particles causing HP are generally of organic origin and their size is suitable for reaching the alveolar space in lungs. The particles usually have adjuvant activity, interestingly many of the animal models for HP is dependent on adjuvant (typically freunds complete adjuvant CFA) for costimulation [23]. Clinically HP is divided in acute, subacute and chronic forms, [18]. The symptoms in the acute form are reminiscent of pulmonary mycotoxicosis while main symptoms in the subacute and chronic forms are shortness of breath. Lymphocytosis, often accompanied by an increase in macrophages[24] is seen in BAL (bronchoalveolar lavage) [25], the earliest lymphocytes are CD4+, later the proportion of CD8+ cells increase [22]. Histologically HP is characterized by a temporally uniform inflammation of the interstitial portion of the lung tissue. Early in the course this process is distributed around respiratory bronchioles. Lymphocytes comprise the majority of infiltrating cells; scattered plump macrophages in clusters around bronchioles are also seen. Small non-necrotizing granulomas are found in two-thirds of the cases. The granulomas contain a mixture of epitheloid macrophages, multinucleated giant cells and lymphocytes [18]. Repeat or continuous exposures will cause progression of the disease with increasing deterioration of lung function and fibrotic changes in the lungs.
The patients have often IgG against antigens on the microbes they are exposed to, this is however not specific for patients with HP and these antibodies can also be seen in exposed individuals without HP [17]. Earlier the formation of immune complexes with this IgG was considered to be a central part of the pathogenesis. However the current view is that HP is caused by cytotoxic delayed hypersensitivity lymphocytes [22]. Some recent results suggest that the development of chronic HP is associated with the skewing of CD4 and CD8 cells toward a Th2 phenotype [26]. It has been suggested that the mechanisms ultimately leading to lung fibrosis are similar to those (as yet poorly known) in interstitial lung diseases in rheumatoid arthritis and idiopathic pulmonary fibrosis [22, 27, 28].
In animal models the earliest cellular event is an activation of macrophages followed by an influx of neutrophiles and lymphocytes [29]. Normal alveolar macrophages (AM) have a low expression of the co stimulatory molecule CD80/CD86 and and a poor capacity to function as antigen presenting cells (APC:s), In HP the macrophages are activated and express increased levels of CD80 and CD86 [30-33]. CD 80 and CD86 bind to CD28 and CD 152 (earlier called CTL4) present on the surface of T-cells. CD 28 is constitutively expressed on the surface of T-cells while CD152 is not expressed on resting T-cells but it is upregulated during cell cycle progression. The relationship between CD28 and CD152 is complex [34]. In short CD28 promotes T-cell activation while CD152 appears to restrict T-cell activation. Studies in mouse models have shown that a blockade of co stimulation by CTLA4 -Ig inhibits lung inflammation in the murine model for HP, both the amounts of lymphocytes and macrophages in the BAL fluid decrease [35]. In mice models for HP the mice develop tolerance [36] if the exposure is prolonged.
Sick building syndrome (SBS) was first described in 1982 [37], a major, but not the only cause, for SBS is dampness in buildings. Damp building related illness (DBRI) refers to those cases of SBS associated with water damage in dwellings or workplaces. There is a consensus for the association of dampness with health effects, [38-40]. By definition DBRI include both HP and pulmonary mycotoxicosis if they are caused by damp buildings. It is however rare for damp buildings to cause HP and still rarer for them to cause a full-fledged pulmonary mycotoxicosis. The typical DBRI is associated with a prolonged exposure to water damaged buildings, usually only a portion of the exposed develop symptoms [41, 42]. As in the case of allergic alveolitis continuation of the exposure will usually worsen the symptoms. There is currently no well-defined diagnostic criteria for the great majority of DBRI:s. The symptoms tend to be quite diverse but include fever, myalgias, cough and headaches somewhat reminiscent of PM. Recently we have shown that exposure to water damage in buildings is associated with an increase in lymphocytes in BAL, this is similar to findings in HP and some cases of pulmonary mycotoxicosis, although the there was no change in the CD4/CD8 ratio in contrast to the increase of CD8 cells usually seen in HP [43]. In this study the number of macrophages was also increased but the increase was not significant possibly because of a lag between the start of symptoms and the BAL procedure.
Recent developments
A component of fungal walls, (1-3)- β-glucans have been shown to be recognized by the C-type lectin dectin-1 [44]. In mice exposure to a (1-3)- β- glucan, curdlan has been shown to promote interleukin 17 (IL-17) producing T helper cells (Th 17 cells) [45]. In humans it has been shown that candida albicans specific memory cells have a Th17 phenotype [46, 47]. IL-1β is known to be necessary for the production of Th17 cells [48] and it can also induce Il-23 and IL-6 both of which promote the formation of Th17 cells [47]. It has also been hypothesized that curdlan might support Th17 development because of its ability to preferentially promote IL-23 production. More recently it has been shown in a mouse model of HP [49, 50] using saccharopolyspora rectivirgula (SR) that immune response is Th17 -polarized and that in absence of IL-17 receptor signaling or T cells the lung inflammation and fibrosis is significantly decreased. It should be noted that SR is a thermophilic actinomycete, not a mold, and does thus not contain 1-3β-glucan. However, production of IL-23 was increased and transcription of TLR 2,6 and dectin-1 was upregulated in the mice exposed to SR [50]. TLR-6 seems central for the IL-17 expression [51]. In humans many of the settings causing HP is associated with exposure to molds containing (1-3)-β-glucans.
In cell culture experiments we have recently found that a group of toxins present in a number of molds (trichotecene mycotoxins) are able to cause an increased production of IL-1 and IL-18 by activating the inflammasome [52]. In another work we found that (1-3)-β-glucans, mentioned above in the context of Th17, in addition to being able induce the production of IL-1 mRNA, is able to induce the activation of the inflammasome [53].
Discussion
The possible role of inflammasome activation in the pathogenesis of PM and other conditions which include a response to inhaled non-infectious particles poses an interesting question. As described earlier IL-1β production requires two distinct signals for activation. The first signal, activating the transcription, is typically caused by PAMPS, abundant in the dusts causing PM. The second signal, activation of the inflammasome is however typically caused by danger signals related to tissue damage tissue. As the dust causing PM is not infectious there is presumably no primary tissue damage and thus no DAMP to activate the inflammasome. Recent work by Kankkunen et al [52, 53] has however shown on other hand that trichotecane mycotoxins can activate the inflammasome and on the other that β-glucans can provide both signals for the activation of IL-1β production. Interestingly both the PRR recognizing β-glucan on the surface of the cells (dectin-1) and the inflammasome require the dectin-1/syk signalling pathway for activation. Thus the same molecule can activate both the signals required for IL-1β production using in part the same signal transduction pathway. This finding seems related to the findings of convergence of signalling pathways (between DAMPS and PAMPS) in the activation of IL-1β production. Together these findings seems to suggests that in the activation of Il-1β production the central question is not what (DAMP or PAMP) but where (cell surface or cytosol) the activating molecule is.
If we thus consider pulmonary mycotoxicosis in the context of innate immunity, it is clear that there is an abundance of explanations to the macrophage activation seen in this condition. High levels of various moulds and other microbes contain both pathogen related structures that PRR:s on the cells surface recognize and toxins and β -glucans that can provide a second signal activating the inflammasome.
The first crucial events in the pathogenesis of HP is the formation of efficient APC:s. This requires an adjuvant activity of the microbial components in question [23]. The mechanism for the adjuvant activity can be explained by essentially identical mechanisms as those described in conjunction with PM. Consistent with this, increases in IL-1 β level has been recorded in the BAL fluid of HP patients [23]. Studies on mice models have demonstrated that IL-1β has adjuvant properties [54], and that it is necessary for the production of antibodies against T-cell dependent antigens [55].
It is fairly easy to understand the pathogenesis of HP up to the point of production of IgG specific for an antigen present in the microbial matter in question. One of the unresolved questions in HP, is why only a small proportion of those exposed and producing specific IgG, go on to develop HP. A part of the explanation could be associated with the repeated exposures to antigen required in HP [56, 57]. IL-1 β has been demonstrated to have a direct effect on memory T Cells [54, 58]. It has also been shown in a mouse model that IL-1β acts directly on CD4 T cells to enhance their antigen driven expansion and differentiation, especially IL-17 and IL-4 producing cells.[48]. The engagement of the dectin receptor by the 1-3 β glucans can also lead to a Th17 polarization of the immunological reaction that is, at least in mouse models a crucial step for the development HP. Thus it would seem possible that IL-1β production is involved in the pathogenesis of HP both in the initial exposure and re-exposures. A further obvious requirement for HP is a lack of tolerance [36].
Key features in the development of allergic alveolitis.
| Feature | Macrophage activation | Lymphocyte activation | No developement of tolerance | Formation of: Cytotoxic delayed hypersensitivity lymhocytes? Th2 cells? | Interstitial fibrosis |
|---|---|---|---|---|---|
| Special properties of the cells | CD80/86 high macrophages | Th 17 cells |
DBRI is as already mentioned a poorly defined entity with few objective diagnostic criteria. In this context it is of interest that in cases of HP in agricultural settings, symptomless persons also exposed to the dust in question have increased levels of lymphocytes in their BAL fluid, as do people exposed to water damaged buildings [43]. To some extent DBRI can perhaps be considered to be related to HP with some cases including features of pulmonary mycotoxicosis. However the types of microbial exposure in these diseases are quite divergent. In HP of farmer's lung type the patients have often been exposed to the antigens since childhood and the exposures are usually rather intense. In DBRI the exposure is usually not of as long duration and intensity as in farmer's lung. In any case the finding that microbial components have the ability to activate the inflammasome and IL-1β production suggests a mechanism for the various phenomena associated with exposure to microbes in water damaged buildings.
Conclusions
The airways are in constant contact with non-infectious microbial particles and have evolved a tolerance for these. In PM the tolerance on the innate immunity level is breached by high concentrations of microbial particles. Interestingly in PM repeated exposure to microbial particles sometimes results in some form tolerance at the innate immunity level. The development of HP probably requires a breakdown of tolerance both at the innate and specific immunity level. It is however unquestionable that the inappropriate activation of specific immunity is causing the progress of HP. The pathogenesis of DBRI is largely unclear but the complexity of symptoms suggests a more complicated condition than that caused solely by the activation of innate immunity. The rather unstructured clinical information available suggests that the mechanisms may be to some extent similar to those seen in HP.
The recent advances innate immunity and its relationship with specific immunity suggest many mechanisms to explain the phenomena seen in illnesses caused by inhaled microbial particles. Further studies, especially clinical ones, are however badly needed in order to determine if the explanations are valid. Much of the possible explanations are centered on the regulation of Il-1β production. As there is a receptor antagonist to IL-1β used as a drug for various inflammatory diseases (Anakinra) it can be possible to plan intervention studies on patients [59]. The same can be said about CTL4 antagonists. In general the patients should avoid exposure which obviously makes studies meaningless; however there might be some groups where avoidance of exposure cannot be achieved. Another way to approach the question is studies on the immunological response of cell samples from exposed patients [60]. Perhaps the best type of sample for this purpose is bronchoalveolar lavage (BAL) fluid. Studies on HP using BAL are relatively common but much less common for pulmonary mycotoxicosis and DBRI. The yield from the standard BAL procedure is somewhat disappointing but various advances in modern research techniques would make it possible to fully utilize the potential of BAL cells and fluids.
Acknowledgements
I wish to thank Sampsa Matikainen, Helena Mussalo-Rauhamaa, Tuula Putus and Aino Nevalainen for helpful discussions about various aspects in this review. Graduate students Päivi Kankkunen and Laura Teirilä are thanked for their interest in this field. Jouko Rytkönen has helped with the illustration. This review has been supported by a grant from the Nummela foundation (Nummela sanatoriums stiftelse).
Conflict of Interests
The author has declared that no conflict of interest exists.
References
1. Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819-826
2. Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783-801
3. Medzhitov R. Origin and physiological roles of inflammation. Nature. 2008;454:428-435
4. Creagh EM, O'Neill LA. TLRs, NLRs and RLRs: a trinity of pathogen sensors that co-operate in innate immunity. Trends Immunol. 2006;27:352-357
5. Dennehy KM, Brown GD. The role of the beta-glucan receptor Dectin-1 in control of fungal infection. J Leukoc Biol. 2007;82:253-258
6. Medzhitov R, Janeway CAJr. Innate immunity: the virtues of a nonclonal system of recognition. Cell. 1997;91:295-298
7. Matzinger P. The danger model: a renewed sense of self. Science. 2002;296:301-305
8. Liu Y, Chen GY, Zheng P. CD24-Siglec G/10 discriminates danger- from pathogen-associated molecular patterns. Trends Immunol. 2009;30:557-561
9. Puren AJ, Fantuzzi G, Dinarello CA. Gene expression, synthesis, and secretion of interleukin 18 and interleukin 1beta are differentially regulated in human blood mononuclear cells and mouse spleen cells. Proc Natl Acad Sci U S A. 1999;96:2256-2261
10. Kanneganti TD, Lamkanfi M, Nunez G. Intracellular NOD-like receptors in host defense and disease. Immunity. 2007;27:549-559
11. Fuentes-Prior P, Salvesen GS. The protein structures that shape caspase activity, specificity, activation and inhibition. Biochem J. 2004;384:201-232
12. Turk B, Stoka V. Protease signalling in cell death: caspases versus cysteine cathepsins. FEBS Lett. 2007;581:2761-2767
13. Kumar H, Kawai T, Akira S. Pathogen recognition in the innate immune response. Biochem J. 2009;420:1-16
14. Vabulas RM, Ahmad-Nejad P, Ghose S, Kirschning CJ, Issels RD, Wagner H. HSP70 as endogenous stimulus of the Toll/interleukin-1 receptor signal pathway. J Biol Chem. 2002;277:15107-15112
15. Park JS, Svetkauskaite D, He Q, Kim JY, Strassheim D, Ishizaka A, Abraham E. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370-7377
16. Dinarello CA. Mutations in cryopyrin: bypassing roadblocks in the caspase 1 inflammasome for interleukin-1beta secretion and disease activity. Arthritis Rheum. 2007;56:2817-2822
17. Hendrick DJ, Burge PS, Beckett WS, Churg A. Occupational Disorders of the Lung, 1 Edn. WB Saunders. 2002
18. Katzenstein A.-L. Katzensteins and Askin's Surgical Pathology of Non-Neoplastic Lung Disease, 3 Edn. WB SAUNDERS COMPANY. 1997
19. Raymenants E, Demedts M, Nemery B. Bronchoalveolar lavage findings in a patient with the organic dust toxic syndrome. Thorax. 1990;45:713-714
20. Lecours R, Laviolette M, Cormier Y. Bronchoalveolar lavage in pulmonary mycotoxicosis (organic dust toxic syndrome). Thorax. 1986;41:924-926
21. Rask-Andersen A, Malmberg P. Organic dust toxic syndrome in Swedish farmers: symptoms, clinical findings, and exposure in 98 cases. Am J Ind Med. 1990;17:116-117
22. Patel AM, Ryu JH, Reed CE. Hypersensitivity pneumonitis: current concepts and future questions. J Allergy Clin Immunol. 2001;108:661-670
23. McSharry C, Anderson K, Bourke SJ, Boyd G. Takes your breath away--the immunology of allergic alveolitis. Clin Exp Immunol. 2002;128:3-9
24. Woda BA. Hypersensitivity pneumonitis: an immunopathology review. Arch Pathol Lab Med. 2008;132:204-205
25. Richerson HB, Bernstein IL, Fink JN, Hunninghake GW, Novey HS, Reed CE, Salvaggio JE, Schuyler MR, Schwartz HJ, Stechschulte DJ. Guidelines for the clinical evaluation of hypersensitivity pneumonitis. Report of the Subcommittee on Hypersensitivity Pneumonitis. J Allergy Clin Immunol. 1989;84:839-844
26. Barrera L, Mendoza F, Zuniga J, Estrada A, Zamora AC, Melendro EI, Ramirez R, Pardo A, Selman M. Functional diversity of T-cell subpopulations in subacute and chronic hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2008;177:44-55
27. Jinta T, Miyazaki Y, Kishi M, Akashi T, Takemura T, Inase N, Yoshizawa Y. The pathogenesis of chronic hypersensitivity pneumonitis in common with idiopathic pulmonary fibrosis: expression of apoptotic markers. Am J Clin Pathol. 2010;134:613-620
28. Churg A, Wright JL, Tazelaar HD. Acute exacerbations of fibrotic interstitial lung disease. Histopathology. 2010 [Epub ahead of print]
29. Schuyler M, Gott K, Cherne A. Mediators of hypersensitivity pneumonitis. J Lab Clin Med. 2000;136:29-38
30. Chelen CJ, Fang Y, Freeman GJ, Secrist H, Marshall JD, Hwang PT, Frankel LR, DeKruyff RH, Umetsu DT. Human alveolar macrophages present antigen ineffectively due to defective expression of B7 costimulatory cell surface molecules. J Clin Invest. 1995;95:1415-1421
31. Zissel G, Ernst M, Schlaak M, Muller-Quernheim J. Accessory function of alveolar macrophages from patients with sarcoidosis and other granulomatous and nongranulomatous lung diseases. J Investig Med. 1997;45:75-86
32. Israel-Assayag E, Dakhama A, Lavigne S, Laviolette M, Cormier Y. Expression of costimulatory molecules on alveolar macrophages in hypersensitivity pneumonitis. Am J Respir Crit Care Med. 1999;159:1830-1834
33. Dakhama A, Israel-Assayag E, Cormier Y. Altered immunosuppressive activity of alveolar macrophages in farmer's lung disease. Eur Respir J. 1996;9:1456-1462
34. Sansom DM, Manzotti CN, Zheng Y. What's the difference between CD80 and CD86? Trends Immunol. 2003;24:314-319
35. Israel-Assayag E, Fournier M, Cormier Y. Blockade of T cell costimulation by CTLA4-Ig inhibits lung inflammation in murine hypersensitivity pneumonitis. J Immunol. 1999;163:6794-6799
36. Bogaert P, Tournoy KG, Naessens T, Grooten J. Where asthma and hypersensitivity pneumonitis meet and differ: noneosinophilic severe asthma. Am J Pathol. 2009;174:3-13
37. Hodgson M. Field studies on the sick building syndrome. Ann N Y Acad Sci. 1992;641:21-36
38. Bornehag CG, Sundell J, Bonini S, Custovic A, Malmberg P, Skerfving S, Sigsgaard T, Verhoeff A. Dampness in buildings as a risk factor for health effects, EUROEXPO: a multidisciplinary review of the literature (1998-2000) on dampness and mite exposure in buildings and health effects. Indoor Air. 2004;14:243-257
39. Fisk WJ, Lei-Gomez Q, Mendell MJ. Meta-analyses of the associations of respiratory health effects with dampness and mold in homes. Indoor Air. 2007;17:284-296
40. Heseltine E, Rosen J. WHO GUIDELINES FOR INDOOR AIR QUALITY: DAMPNESS AND MOULD. WHO. 2009
41. Straus DC, Cooley JD, Wong WC, Jumper CA. Studies on the role of fungi in Sick Building Syndrome. Arch Environ Health. 2003;58:475-478
42. Akpinar-Elci M, Siegel PD, Cox-Ganser JM, Stemple KJ, White SK, Hilsbos K, Weissman DN. Respiratory inflammatory responses among occupants of a water-damaged office building. Indoor Air. 2008;18:125-130
43. Wolff H, Mussalo-Rauhamaa H, Raitio H, Elg P, Orpana A, Piilonen A, Haahtela T. Patients referred to an indoor air health clinic: exposure to water-damaged buildings causes an increase of lymphocytes in bronchoalveolar lavage and a decrease of CD19 leucocytes in peripheral blood. Scand J Clin Lab Invest. 2009;69:537-544
44. Brown GD, Gordon S. Immune recognition. A new receptor for beta-glucans. Nature. 2001;413:36-37
45. LeibundGut-Landmann S, Gross O, Robinson MJ. et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol. 2007;8:630-638
46. Acosta-Rodriguez EV, Rivino L, Geginat J, Jarrossay D, Gattorno M, Lanzavecchia A, Sallusto F, Napolitani G. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat Immunol. 2007;8:639-646
47. Palm NW, Medzhitov R. Antifungal defense turns 17. Nat Immunol. 2007;8:549-551
48. Ben-Sasson SZ, Hu-Li J, Quiel J, Cauchetaux S, Ratner M, Shapira I, Dinarello CA, Paul WE. IL-1 acts directly on CD4 T cells to enhance their antigen-driven expansion and differentiation. Proc Natl Acad Sci U S A. 2009;106:7119-7124
49. Simonian PL, Roark CL, Wehrmann F, Lanham AK. Diaz del Valle F, Born WK, O'Brien RL, and Fontenot AP, Th17-polarized immune response in a murine model of hypersensitivity pneumonitis and lung fibrosis. J Immunol. 2009;182:657-665
50. Joshi AD, Fong DJ, Oak SR, Trujillo G, Flaherty KR, Martinez FJ, Hogaboam CM. Interleukin-17-mediated immunopathogenesis in experimental hypersensitivity pneumonitis. Am J Respir Crit Care Med. 2009;179:705-716
51. Fong DJ, Hogaboam CM, Matsuno Y, Akira S, Uematsu S, Joshi AD. Toll-like receptor 6 drives interleukin-17A expression during experimental hypersensitivity pneumonitis. Immunology. 2010;130:125-136
52. Kankkunen P, Rintahaka J, Aalto A, Leino M, Majuri ML, Alenius H, Wolff H, Matikainen S. Trichothecene mycotoxins activate inflammatory response in human macrophages. J Immunol. 2009;182:6418-6425
53. Kankkunen P, Teirila L, Rintahaka J, Alenius H, Wolff H, Matikainen S. (1,3)-{beta}-Glucans Activate Both Dectin-1 and NLRP3 Inflammasome in Human Macrophages. J Immunol. 2010 epub
54. Dinarello CA. The many worlds of reducing interleukin-1. Arthritis Rheum. 2005;52:1960-1967
55. Nakae S, Asano M, Horai R, Iwakura Y. Interleukin-1 beta, but not interleukin-1 alpha, is required for T-cell-dependent antibody production. Immunology. 2001;104:402-409
56. Nance S, Cross R, Fitzpatrick E. Chemokine production during hypersensitivity pneumonitis. Eur J Immunol. 2004;34:677-685
57. Fink JN. Clinical features of hypersensitivity pneumonitis. Chest. 1986;89:193S-195S
58. Nakae S, Asano M, Horai R, Sakaguchi N, Iwakura Y. IL-1 enhances T cell-dependent antibody production through induction of CD40 ligand and OX40 on T cells. J Immunol. 2001;167:90-97
59. Pascual V, Allantaz F, Arce E, Punaro M, Banchereau J. Role of interleukin-1 (IL-1) in the pathogenesis of systemic onset juvenile idiopathic arthritis and clinical response to IL-1 blockade. J Exp Med. 2005;201:1479-1486
60. Dai H, Guzman J, Chen B, Costabel U. Production of soluble tumor necrosis factor receptors and tumor necrosis factor-alpha by alveolar macrophages in sarcoidosis and extrinsic allergic alveolitis. Chest. 2005;127:251-256
Author contact
![]() Corresponding author: Henrik Wolff, Biological Mechanisms and the Prevention of Work related Diseases, Finnish Institute of Occupational Health (FIOH), Topeliuksenkatu 41 a A, 00250 Helsinki, Finland. E-mail: Henrik.Wolfffi
Corresponding author: Henrik Wolff, Biological Mechanisms and the Prevention of Work related Diseases, Finnish Institute of Occupational Health (FIOH), Topeliuksenkatu 41 a A, 00250 Helsinki, Finland. E-mail: Henrik.Wolfffi