Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Metabolic Syndrome
Potential Causal Factors
The Insulin and IGF Systems
Interaction of causal factors...
Inflammatory Factors
Transcription factors
Other Factors
Conclusion
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(7):1003-1015. doi:10.7150/ijbs.7.1003 This issue Cite
Review
The Link between the Metabolic Syndrome and Cancer
Sandra Braun, Keren Bitton-Worms, Derek LeRoith ![]()
Diabetes and Metabolism Clinical Research Center of Excellence, Legacy Heritage Clinical Research Institute at Rambam (LHCRIR), Haifa, Israel.
Received 2011-6-16; Accepted 2011-7-24; Published 2011-8-16
Abstract
Since the incidence of the metabolic syndrome is on the rise in the western world, its coherence to cancer is becoming more apparent. In this review we discuss the different potential factors involved in the increase of cancer in the metabolic syndrome including obesity, dyslipidemia and Type 2 Diabetes Mellitus (T2DM) as well as inflammation and hypoxia. We especially focus on the insulin and IGF systems with their intracellular signaling cascades mediated by different receptor subtypes, and suggest that they may play major roles in this process. Understanding the mechanisms involved will be helpful in developing potential therapeutics.
Keywords: metabolic syndrome, cancer
Metabolic Syndrome
The metabolic syndrome and its concomitant diseases are a severe health problem world-wide and most likely will gain even more importance in the future since the prevalence of obesity is rising (1). The metabolic syndrome includes abdominal obesity, hypertension, dyslipidemia and hyperglycemia and is linked to insulin resistance and the development of diabetes mellitus as well as to nonalcoholic fatty liver disease (2). The Aerobic Center Longitudinal Study of 33 230 cancer-free men revealed an up to 56% enhanced risk of cancer mortality associated with the metabolic syndrome after 14 years of following-up (2). Also other studies support that the metabolic syndrome, or its components, might play an important role in the etiology and progression of certain cancer types and a worse prognosis for some cancers (3). Obesity and diabetes, individually, have been associated with breast, endometrial, colorectal, pancreatic, hepatic and renal cancer (4-5).
Obesity and Cancer
Worldwide there are 1.1 billion overweight people with a BMI between 25 kg/m2 and 30 kg/m2 and 312 million with a BMI > 30 kg/m2 (6). Within the last four decades the prevalence of obese people in the US increased and is currently 66% of adults with a body mass index (BMI) > 25 kg/m2 and half of those have a BMI of > 30 kg/m2 (7). It was seen that obese patients tend to manifest more localized tumors, earlier relapse and a diminished overall survival (8). The American Cancer Society calculates that currently new cancer cases are in the order of 1.5 million with half a million cancer deaths per year, nearly one in five due to obesity (4, 9-10). A projection for the year 2030 estimated that 366 million people will suffer from obesity and the accompanying Type 2 Diabetes Mellitus (T2DM) (11). A large epidemiologic study showed evidence for the association between obesity, T2DM and particular cancer types. The CPS II study revealed a significantly enhanced relative risk for colorectal cancer in obese people (4). This risk was maintained even after adjusting for factors such as BMI, family history, physical activity, smoking, red meat consumption, hormone and aspirin use (12). Interestingly, not the BMI but waist circumference seems to be a strong predictor of colorectal cancer (13). Furthermore, it has been shown that there is an association between obesity and cancer of the gastric cardia, esophageal adenocarcinoma and cholangiocarcinoma (4). The tendency to develop these tumors was ascribed to Barret´s esophagus caused by gastroesophageal reflux disease (GERD) (14), which appears to be common in obese people. Another study also linked a high BMI in both genders to an enhanced risk of colorectal, esophageal and kidney cancer as well as the non-Hodgkins lymphoma and multiple myeloma (4, 15). Multiple myeloma and large B cell lymphoma were especially linked to obesity in men (16-17). Also breast cancer has been linked to obesity in postmenopausal women (4). For obese, diabetic, postmenopausal women there exists an augmented risk especially for estrogen receptor-positive breast cancer (5). Obese women were found to display many fold higher estrogen levels than normal-weight individuals. After adjusting for known risk factors for breast cancer like family history, use of hormones or menopausal status, a positive correlation between breast cancer mortality and a BMI > 25 kg/m2 was found. Female patients with a BMI > 40 kg/m2 showed twice the risk of slim women to develop breast cancer (4). Approximately 30-50% of deaths caused by breast cancer are due to obesity and overweight (18). It is hypothesized that the increased endogenous estrogen level in obese women plays a key role in both the postmenopausal breast and endometrial cancer incidence (4). Compared to lean, non-diabetic individual, the risk of obese persons to develop endometrial cancer rises from 2-fold to 6-fold if they develop T2DM (19). Cervical adenocarcinoma has also been associated with obesity (20). Regarding the association between obesity and prostate cancer the data in the literature is inconsistent (5).
Dyslipidemia and Cancer
Dyslipidemia includes low high-density-lipoprotein cholesterol (HDL-C), high low-density-lipoprotein cholesterol (LDL-C) and high serum triglycerides (TG) levels. Low HDL-C serum levels were associated with lung cancer incidence as well as the Non-Hodgkin lymphoma (NHL) and was suggested to be a marker for increased breast cancer risk in pre-menopausal as well as post-menopausal women, since it might reflect an unfavorable hormonal profile with particularly increased estrogen levels especially in obese women (3, 21-24). Furthermore, high serum levels of total cholesterol and TG raise the risk of prostate and post-menopausal breast cancer (3, 25-26). Inconsistent with these data, low LDL-C serum levels were also linked to an 15-fold increased risk of developing hematological cancer (3).
Cancer association with obesity, diabetes and obesity or dyslipidemia.
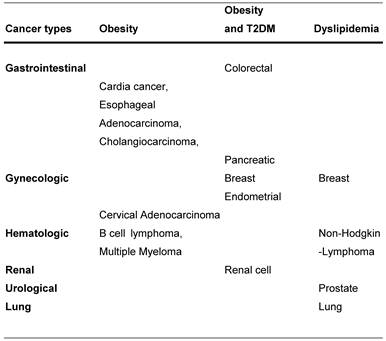
Diabetes and Cancer
Several studies analyzed the association between diabetes and cancer development. A large prospective study in the US followed a cohort of 467,922 men and 588,321 women for 16 years who had no reported history of cancer. After this long follow-up, the results showed that independent of a high body mass, T2DM acts as a predictor of mortality from cancer of the colon, pancreas, female breast, male liver and bladder (27). An increased incidence of colorectal cancer in diabetic patients independent of obesity is supported by the Physician Health Study (28). Another study from Italy observed a slight rise of cancer mortality in diabetic patients which achieved statistical significance in women but not in men. The mortality of diabetic women was mainly caused by pancreatic and breast tumors in which the mortality of breast cancer was especially evident in obese women with diabetes (29). Even though there is a strong association between pancreatic cancer and diabetes, it is still a subject of speculation if diabetes is the outcome of the pancreatic cancer or vice versa. But one percent of newly diagnosed diabetic patients >50 years will contract pancreatic cancer within three years (5). The main risk factors for pancreatic and lung cancer are considered to be cigarette smoking. Lung cancer in most studies did not show any connection to diabetes, although a Korean study revealed an enhanced risk of lung cancer in non-smoking, diabetic women (10). An augmented risk for estrogen receptor-positive breast cancer was found in postmenopausal, obese and diabetic women (5). However, diabetes was not associated with increased breast cancer in premenopausal women (30). Regarding diabetes and prostate cancer incidence and mortality, most studies suggest an inverse connection (5).
Potential Causal Factors
Some studies suggest a link between obesity, inflammation and insulin resistance in patients with the metabolic syndrome potentially caused by adipose tissue hypoxemia. The adipose tissue of obese patients show inflammation characterized by elevated inflammatory cytokines in plasma and adipose tissue as well as macrophage infiltration and activation in the adipose tissue. Inflammation by means of TNF-α is able to contribute to insulin resistance by intervening in the intracellular signalling cascade of insulin (31-32). Especially systemically elevated free fatty acids (FFA) and decreased adiponectin levels found in the blood of obese people might contribute to insulin resistance since FFA and inflammatory cytokines are capable and thus of aggravate insulin resistance. Furthermore, the transcriptional activity of the nuclear factor PPARγ plays an important role in insulin sensitivity. TNFα and IL-1, both inflammatory mediators induced by NF-κB, and may inhibit PPARγ, that in turn might promote insulin resistance (31).
Another interesting aspect is the role of the IGF-1 receptor, which is expressed by almost all normal and transformed cells. Data from several studies suggest that IGF-1 and the IGF-1R are necessary for the normal growth and development of cells (33-34). But IGF-1R and IR have been found to be overexpressed in cancer cells (35-36). The PI3K signaling cascade is a major pathway of the IGF-1R and IR (34, 37) and commenly deregulated in cancer cells (38-39). In the 1980´s the Middle T antigen of Polyoma virus was found to mediate its oncogenic activity by inducing PI3K. Thus, it is conceivable that mutations in tumor suppressor genes resulting in a dysregulation of insulin and IGF-1 signaling pathways, might lead to cancer development. It is of special interest to investigate the role of signaling through IR and IGF-1 in diabetes and obesity in regards to cancer because signaling through IR and IGF-1R is increased in hyperinsulinemia (50). It is well known that a large number of tumor types and cancer-derived cells overexpress the IGF-1 receptor, which mediates mitogenic effects. Some tumors, like squamous carcinoma and small cell lung cancer produce high levels of IGF-1 themselves. However, the main source of IGF-1 seems to be the liver since it provides 75% of the circulating IGF-1 (40). Hyperinsulinemia was shown to even increase the IGF-1 production of the liver (41). In contrast to the mentioned cancer types above, breast cancer does not generally produce IGF-1 but does express and secrete small amounts of IGF-2. Furthermore, estrogen was found to induce IGF-1 receptor expression in estrogen receptor-positive breast cancer cell lines (37). Since IGF-2 is produced by adult human tissues but not by the adult tissue of the mouse and mice with an IGF-1 null mutation in the liver did not demonstrate any reduction in somatic growth (40), it is not clear for now whether the hepatic- or the non-hepatic IGF-1 or IGF-2 plays the primary role in promoting cancer.
Recent studies have strongly suggested that the relationship between obesity and T2DM and the increased risk of cancer and cancer-related mortality maybe explained by the hyperinsulinemia, particularly but not restricted to breast cancer (42-43). Whether this effect is mediated by the IR or the IGF-1R, is as yet undefined, though both are capable of this effect (44). However, the mechanisms which actually promote cancer growth in patients with metabolic syndrome demand further investigation.
The Insulin and IGF Systems
IGF-1 and its effects
The IGF-1 system comprises three peptides, insulin, IGF-1, IGF-2 and each with its receptor (IR, IGF-1R, IGF-2R) as well as the IGF-binding proteins (IGFBPs). Both IGF-1 and IGF-2 bind the IGF-1 receptor with a high affinity. The IGF-2R is the cation-independent mannose-6-phosphate receptor and its signaling pathway, if any, is unclear (40). IGF-2 was identified as being a fetal growth factor (45) in comparison to IGF-1, which stimulates fetal as well as post-natal growth. The liver-derived IGF-1 is the growth promoting mediator of growth hormone (33, 46-47). Even though the liver is the main source of the IGF-1 production, non-hepatic IGF-1, which is produced by many cell types, also plays an important role. Thus, IGF-1 acts via endocrine, paracrine and autocrine mechanisms (40). The growth promoting effects of IGF-1 include stimulation of proliferation, differentiation and protein synthesis and are accompanied by the consistent effect on cells by reducing apoptosis (40). IGF-1 also regulates the cell cycle through modulation of cyclins, cyclin-dependent kinases and cyclin-dependent kinase inhibitors (48). Tumors often express IGF-2 which is more mitogenic than IGF-1 and signals via the IGF-1R and the mitogenic subtype of the IR, IR-A (49).
IGF-2
As with insulin and IGF-1, the overexpression of IGF-2 is also associated with cancer development. Like IGF-1 it is mainly expressed by the liver but also by other tissues in adult humans. Its regulation is carried out by genetic imprinting. Reduced methylation of the differentially methylated region (DMR) on the maternal allele leads to an overexpression of IGF-2. Thus, a loss of imprinting and overexpression of IGF-2 is observed in many tumors. Furthermore, IGF-2 initiates endocytosis of its bound IGF-2R and thus leads to a clearance of IGF-2 (50).
IGF-binding proteins (IGFBPs)
There are six IGFBPs showing different characteristics and functions. They bind IGF-1 and IGF-2 with a higher affinity than the IGF-1 receptor. 99% of circulating IGF-1 is bound to IGFBPs and 80% thereof to IGFBP-3 (40). IGFBPs are produced by the liver and most tissues and can be found in the circulation as well as the extracellular compartments. They are able to inhibit or stimulate the effects of IGFs and display IGF-independent effects (37). They enhance the half-life of the IGF-1 and IGF-2, protect them from degradation and regulate their bioavailability and their release to the target cells (40). IGFBP-2 was found to suppress the tumor suppressor gene product PTEN in MCF-7 breast cancer cell lines. The phosphatase PTEN induces reduction of protein synthesis and cell cycle progression and antagonizes the PI3K pathway and in IGF-2 signaling (50). IGFBP-3 has protective effects against cancer development. It is up-regulated by p53 and might either promote apoptosis through a p53-dependent mechanism or through binding a putative IGFBP-3 receptor, which mediates its anti-apoptotic effects through caspase-8 (51). IGFBP-3 also might have a protective impact because of its strong affinity and thus slow release of IGF-1 and IGF-2 to the receptor. Hyperinsulinemia and increased levels of IGF-1 lead to a decreased secretion of GH and thus secondarily leads to less IGFBP-3 expression. This might cause an increased IGF-1 signaling and a reduction of the apoptosis promoting effects of IGFBP-3 (50).
The insulin receptor (IR) with its two subtypes IR-A and IR-B, the insulin growth factor 1 receptor (IGF-IR) and the hybrid receptors (IGF-1R/IR-A and IGF-1R/IR-B). Structurally, IR and the IGF-1R have two extracellular α-subunits and two transmembrane β-subunits that are jointed to each other by disulfide bonds. Affinity, insulin binds with high affinity to IR-A or IR-B but has low affinity for IGF-1R, while insulin has no binding to the hybrid receptors. IGF-1 binds to the IGF-1R and to the hybrid receptor IGF-1R/IR-A or IGF-1R/IR-B. IGF-2 binds to IR-A, IGF-1R or to IGF-1R/IR-A hybrid receptor. Signaling, ligand binding to insulin receptor-A or to IGF-1 receptor mediates the mitogenic signaling pathway, while ligand binding to insulin receptor-B activates metabolic signaling. Binding to the hybrid receptors, leading to mitogenic or metabolic signaling, is determined by the IR isoform that formed the hybrid receptors. Reproduced by permission of the RMMJ (54).
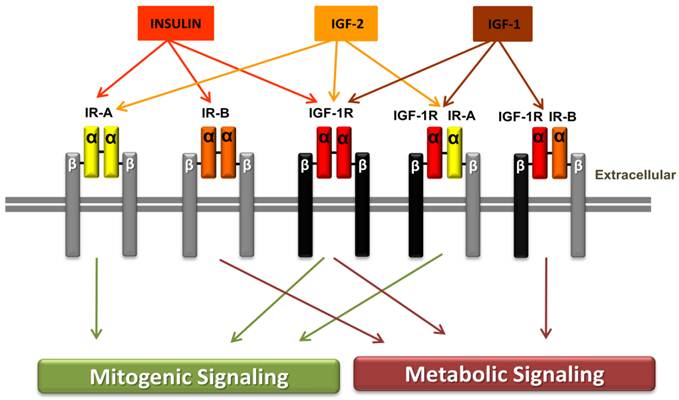
The insulin receptor and IGF-1 receptors
Both the IR and the IGF-1R are transmembrane oligomer receptors which consist of one α- and one β-subunit. The β-subunit comprises a tyrosine kinase which undergoes autophosphorylation after the ligand binds to the extracellular α-subunit of the receptor. Subsequently insulin receptor substrates and adaptor proteins are recruited, activated and induce two major signaling pathways. On one hand the mitogenic MAPK pathway and on the other hand the metabolic and anti-apoptotic PI3K pathway. The MAPK pathway plays in important role in cell growth and proliferation. The anti-apoptotic influence of the PI3K pathway is mediated through the activation of protein kinase B (AKT) which controls among other things apoptosis-regulating transcriptions factors (40). Cells that express the IR and the IGF-1R may also form a hybrid receptor from two subunits of these two receptors. Insulin manifests a decreased affinity to the IGF-1R and a very low affinity to the hybrid receptors. However IGF-1 retains its high affinity to the IGF-1R as well as to the hybrid receptors. Since there are two subtypes of the IR, the IR-A and the IR-B, there also exist two different hybrid receptors of IGF-1 and IR (IGF-1/IR-A, IGF-1/IR-B). As the IR-A homoreceptor, the IGF-1R/IR-A hybrid receptor mainly results in mitogenic signalling. Comparatively the IGF-1R/IR-B hybrid receptor results in metabolic signaling (37, 52). The IR-A receptor has been seen to be aberrantly expressed especially in fetal cells and many tumor cells and it additionally has a high affinity to IGF-2, compared to IR-B. This may explain why hyperinsulinemia has a cancer-promoting effect in diabetic and obese patients. The IR-B receptor is predominantly expressed by the liver, muscle and adipocytes, which generally causes metabolic signaling in adult, well-differentiated tissues (53). Furthermore, studies found an up-regulation of IR splicing in insulin target tissues of patients with insulin resistance. However, its role in type 2 diabetes is not as yet well understood (53).
IGF-1 receptor signaling and its potential link to cancer development
The IR and the IGF-1R are able to mediate their effects through recruitment, phosphorylation and thus activation of IRS-1, Shc, Grb2, Ras and subsequent of PI3K, AKT and mTOR or Raf-1, MAPK/ERK. In the following we try to clarify some potential linking points of the IGF-1R signaling cascades to cancer development.
mTOR and PI3K
PI3K signaling activates Akt resulting in activation of mTOR and thus, mediating its effects on cell growth by increasing ribosomal protein synthesis and preparation of mitosis through S6k1 and 4E-BP-1. Activation of mTOR results in protein synthesis, cell growth and the preparation of cells for mitosis, all mechanisms that favor tumor growth (50). Dysregulated signaling of mTOR has been linked to numerous human cancers (56). Apart from that, mTOR plays an important role in mediating the signaling of insulin, growth factors, nutrients and energy. A homozygous deletion of the mTOR target S6K1 (ribosomal protein S6 kinase 1) in mice was found to lead to hyperinsulinemia and glucose intolerance (58).
mTOR and TSC
mTOR regulation is controlled not just by PTEN but also by the tumor suppressor gene products tuberous sclerosis (TSC) 1 and TSC-2. These two proteins together form the Tuberous Sclerosis Complex (TSC) and incorporate and transfer cellular growth factor and stress signals to negatively regulate TOR activity (56). TSC-1 and TSC-2 receive input from several signaling pathways like the PI3K, LKB1-AMPK, MAPK and in response to hypoxia. Activated AKT leads to a phosphorylation of TSC-2 renouncing its inhibitory influence on mTOR through the small GTPase Ras-homolog-enriched-in-brain (Rheb) (50). mTOR stimulating activity of Rheb is regulated by TSC-1 and TSC-2. Whether TSC-1 or TSC-2 mediates cell growth promoting or inhibiting effects, depends on its upstream signaling cascade and which of the four binding sites of TSC gets phosphorylated. AKT results in an inhibition of TSC-2 and thus, relieves the inhibition on mTOR, in which AMPK seems to mediate stimulating effects on TSC. AMPK interacts through phosphorylation with TSC-2 as well as with mTOR and thus is capable of inhibiting the activation of mTOR by a direct and indirect way. The detailed mechanism of the AMPK-TSC interaction is not clear (59).
mTOR regulation
Insulin resistance leads to a decreased entry of glucose into cells and consequently results consequently in a deprivation of energy. Therefore the concentration of the high-energy compound ATP drops and AMP rises. An enhanced concentration of AMP shows a low energy level of the cell and leads to activation of AMPK, which is controlled by the tumor suppressor gene LKB1. Basically, AMPK activation results in inactivation of mTOR, thereby preventing protein production, cell growth and proliferation (50). mTOR was found not just to be inhibited by energy depletion but also by hypoxia. The TSC-1/TSC-2 tumor suppressor complex inhibits mTOR and also regulates accumulation of HIF-α. Tsc-2-deficient cells did not influence up-regulation of HIF-α in response to hypoxia, whereas Tsc2+/+ cells down-regulated HIF-α with prolonged hypoxia. LKB1 and AMPK are not involved in inhibition of mTOR in hypoxia but this is achieved by REDD1 (regulated in development and DNA damage responses 1) (55). The stress response gene REDD1 is induced by hypoxia and energy stress. REDD1-deficient cells manifest a highly defective TOR regulation in response to either of these stress signals (56). REDD1 mRNA was found to be induced by hypoxia. However, how mechanistically REDD1 interacts with TSC is not known yet but its significance in inhibiting mTOR in hypoxia is clear (55). Since it is known that hypoxic TSC-2-deficient cells demonstrate high levels of cell proliferation, it seems to be important in hypoxia to inhibit mTOR to prevent tumor development.
Insulin-like growth factor 1 receptor (IGF-1R) signaling pathway. ( "->" : activation, "-●": inhibition). Binding of IGF-1 or IGF-2 or insulin to the IGF-1R α-subunit leads to autophosphorylation of β-subunit residues, which then act as docking site to insulin receptor substrates (IRS-1 to 4). Bound IRS-1 results in PI3K activation, which in turn activates Akt. The tumor suppressor phosphates and tensin homolog deleted on chromosome 10 (PTEN) inhibits PI3K. Activated Akt has many substrates; in one pathway Akt inhibits apoptosis by inactivating BCL-2 antagonist of cell death (BAD), and in the second pathway Akt regulates protein synthesis by phosphorylating tuberous sclerosis complex (TSC1/2). This phosphorylation removes the inhibition of TSC from mammalian target of rapamycin (mTOR). mTOR activates the ribosomal S6 kinase (S6K) and eukaryotic initiation factor 4E-binding protein-1 (4E-BP-1), leading to protein synthesis. In energy depletion expression of the suppressor gene LKB1 and AMP raise. AMPK is activated by both mechanisms. AMPK inhibits protein synthesis through direct inhibition of mTOR or indirectly by activating the TSC complex. Hypoxia induces REDD1. The detailed interaction between REDD1 and TSC is not clear yet. We conclude that the inhibition of HIF-α by TSC might be independent of REDD1. The mitogen-activated protein kinase (MAPK) pathway can also be activated by IGF-1R activation. In this pathway IGF-1R activates the adaptor proteins, Shc and Grb2, leading to activation of Ras, Raf, MEK1/2, and ERK1/2, which results in cell proliferation.
The detailed interaction between REDD1 and TSC is not clear yet. We conclude that the inhibition of HIF-α by TSC might be independent of REDD1. The mitogen-activated protein kinase (MAPK) pathway can also be activated by IGF-1R activation. In this pathway IGF-1R activates the adaptor proteins, Shc and Grb2, leading to activation of Ras, Raf, MEK1/2, and ERK1/2, which results in cell proliferation.
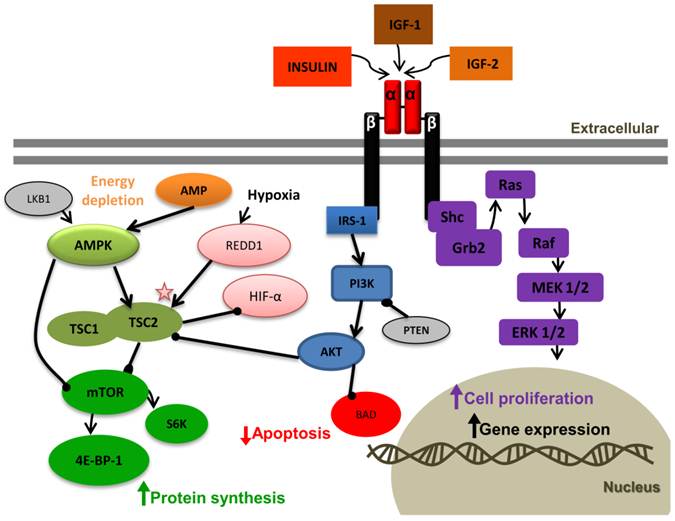
PTEN, MAPK and PI3K
PTEN is, after p53, the most commonly mutated tumor suppressor gene in human cancer. A mutation or inactivation of at least one copy of PTEN emerges in more than 50% of women breast cancer. In MCF-7 breast cancer cells it has been shown that a loss of PTEN results in an increased signaling of IGF-2 mediating through the IGF-1R and IR-A (57). Another mechanism whereby PTEN counters cell growth and cell cycle progression is to inactivate PI3K by dephosphorylation.
PI3K and MAPK
One example for a cancer cell line which hosts a mutation of a kinase of the IGF-1R signaling cascade is the MET-1 breast cancer cells overexpressing polyoma virus middle T antigen (PyVmT). When stimulated by insulin or IGF-1, MET-1 breast cancer cells overexpressing PyVmT and thus actually inducing PI3K, have been observed to demonstrate an increased interaction with Src and PLCγ1 (50, 60). Src initiates MAPK pathway resulting in cell proliferation and is known, like phospholipase Cγ1 (PLCγ1), to induce tumor growth (50).
It is conceivable that a mutation of the tumor suppressor genes PTEN and/or TSC, combined with hyperinsulinemia and an increased signaling of insulin and/or IGF-1 might promote tumor growth. Furthermore, non-functional and dysfunctional IGF-1Rs were shown to inhibit growth of lung cancer and Ewing sarcoma cell lines and to decrease proliferation in mammary glands in mice. Additionally, breast and prostate cancer cell lines were found to display an enhanced level of IGF-1R and decreased level of IR (50). This data provide strong support for the significance of the IGF-1R and/or IR signaling for cancer development and growth.
IGF-1R and regulation of apoptosis
IGF-1 through the IGF-1R affects the apoptotic machinery at several levels. It is capable of mediating both anti-apoptotic and pro-apoptotic effects but acts mainly anti-apoptotic. By activation of PI3K-AKT pathway, IGF-1 may indirectly inactivate caspase-9 through phosphorylation and thus realize its anti-apoptotic effects. IGF-1 inhibits the intrinsic pathway by initiating phosphorylation of Bcl-2 family proteins like BAD. IGF-1 also controls the extrinsic apoptotic pathway via regulation of the death-inducing receptor. IGF-1 regulates multiple transcription factors like CREB, FKHR, NF-κB, mdm2 and p53, which are involved in the regulation of apoptotic proteins. It is known that IGF-1 mediates its effects through different signaling cascades such as MEK-ERK, 14.4.4-raf, PI3K-AKT and p38-MAPKAP-K3. It is conceivable that in some cell types it might be necessary to activate multiple signaling pathways at once to reach full protection against apoptosis. By being capable of mediating through many different signaling pathways the IGF-1R may be able to protect cells against multiple apoptotic factors. However, which pathway is used by the IGF-1R to inhibit apoptosis depends on the specific cell type (40).
Interaction of causal factors involved in the connection between Diabetes and Obesity and Cancer
Diabetes and the increased resistance against therapeutic agents
It has been observed that patients with diabetes tend to show impaired response to cancer treatment (10). By a variety of mechanisms an aberrant mitogenic IR-A expression may favor cancer resistance to both conventional and targeted therapies (53). A resistance to chemotherapeutic agents like trastuzumab and tamoxifen in breast cancer cell lines was associated with an activation of mTOR. One activator of mTOR and thus promotor of cell growth and proliferation is displayed by AKT. Interestingly, Metformin was found to activate AMPK and decrease AKT and insulin levels in mice (61). Both mechanisms result in a decreased signaling of cell growth. This might explain why Metformin is associated with a decreased risk of cancer development and a better response to chemotherapy in patients with breast cancer (50).
Connection between obesity, insulin, IGF-1 and cancer development
Compared to normal-weight individuals the augmented adipose tissue in obesity produces an increased amount of FFAs, triglycerides, leptin and inflammatory cytokines. These metabolic changes, combined with reduced physical activity raise the secretion of insulin. This mechanism results in hyperinsulinemia and insulin resistance common in the pre-diabetic condition.
The liver seems to be the main source providing 75% of the circulating IGF-1 (40). The hepatic IGF-1 production is dependent on the signaling of growth hormone (GH) mediated by the growth hormone receptor (GHR). Obesity and hyperinsulinemia are known to influence the level of GH and as a result of IGF-1. Hyperinsulinemia was also found to stimulate the expression of the growth hormone receptor (GHR) in the liver tissue. Thus it is comprehensible that hyperinsulinemia and obesity cause an increased production of hepatic IGF-1 by a enhanced signaling of the GHR in the liver. This in turn, raises the amount of circulating IGF-1 and therefore results in cell growth and proliferation (62-63).
Estrogen, obesity and breast cancer
The enhanced percentage of fat tissue in obesity delivers an augmented level of aromatase, which results in an increased synthesis of estrogen. Furthermore, obesity as well as hyperinsulinemia and elevated IGF-1 levels were shown to reduce the production of sex hormone-binding globulin (SHBG). This also leads to an increased bioavailability of estrogen (63). It was shown that the pathways of the IGF-1R and the estrogen receptor (ER) synergize in the activation of the mitogen-activated protein kinase (MAPK). Estrogen was demonstrated to induce the expression of the IGF-1R as well as the insulin receptor substrates IRS-1 and IRS-2. These effects of estrogen led to an enhanced IRS-1 phosphorylation and hence an increased activation of MAPK after IGF-1 stimulation of MCF-7 breast tumor cells (64). Leptin and insulin as well as TNF-α and IL-6 are known to induce aromatase and thus stimulate estrogen biosynthesis (65). These results might explain the connection of the enhanced growth of breast cancer in patients with T2DM and obesity.
Additional factors
Leptin
The major source of leptin is the white adipose tissue but it can also be secreted by cells of the placenta, ovaries, mammary epithelial, brown adipose tissue, skeletal muscle, the fundal glands of the stomach, bone marrow, pituitary and the liver (66). Leptin mediates the feeling of satiety through its receptors in the hypothalamus. The absence of leptin as well as a dysfunction of the leptin receptor results in an uncontrolled food intake and obesity (67). Leptin as well as insulin levels in the plasma were positively correlated with body weight and especially with the adipose mass (68). However, obese people were found to manifest high leptin levels in the plasma and demonstrate leptin resistance (69). Leptin was found to improve insulin resistance and hyperglycemia by increasing the hepatic responsiveness to insulin and thus most likely decreasing the gluconeogenesis of the liver. In this study, they further observed an improvement of the lipotoxic condition through an increased fatty acid oxidation and inhibition of the liponeogenesis that led to a reduction of lipid stores in liver and muscles (70). Leptin might be associated with cancer cell proliferation in colorectal cancer as well as acute myeloid leukemia and was found to reduce apoptosis by mediating cytokines in MO7E and TF-1 cells. Similarly in cancer cell lines of esophageal, breast and prostate cancer leptin was seen to stimulate cellular proliferation (71-72). A breast cancer study showed that leptin up-regulates VEGF and found that leptin mainly requires activation of HIF-1α and NF-κB for its regulation of VEGF (73). Other studies suggest that hypoxia might lead to a secretion of leptin by adipocytes which is induced by HIF-1α (31). However, it still requires further investigation to determine the specific role of leptin in cancer development.
Adiponectin
Adiponectin is a protein, which is produced of the adipocytes and regulates energy homeostasis, glucose and lipid metabolism (74-75). It has anti-inflammatory characteristics and was found to be decreased in obese individuals and elevated in normal-weight people (5). In adults the level of adiponectin is inversely associated to the percentage of body fat even though it was shown to be the most abundantly expressed protein in the adipose tissue (76). Loss of weight in turn results in a significant rise of adiponectin concentration in the blood circulation (77). Some studies suggest a decreased adiponectin production secondary to adipose tissue hypoxia. They found that hypoxia induces expression of inflammatory cytokines and concurrently reduces the mRNA of adiponectin. Furthermore it was shown that the gene promotor activity of adiponectin was down-regulated by hypoxia and TNFα. Since it is known, that TNFα might reduce adiponectin mRNA levels, it is not clear if hypoxia realizes its effects in a direct manner or through TNFα (31, 78-80). Furthermore, women demonstrate a higher level of adiponectin than men (77). Adiponectin also correlates with systemic insulin sensitivity (81) and was found to be reduced in diabetic patients compared to healthy individuals. Its insulin-sensitizing effects are mediated through its two receptors Adipo R1 and Adipo R2 (82). By activation of AMP-activated protein kinase (AMPK) and thus indirectly suppressing mTOR, adiponectin was found to inhibit colorectal cancer cell growth (83). By using the AMPK pathway, adiponectin also affects the regulation of glucose utilization and fatty-acid oxidation (5, 75, 84-85). Adiponectin is considered to have anticancer effects because of its anti-inflammatory character and further was found to be a negative regulator of angiogenesis (86). It was shown that addition of adiponectin to gastric cancer cell lines inhibited their proliferation. A continuous intraperitoneal infusion of adiponectin succeeded in suppressing the formation of peritoneal metastasis (87).
Inflammatory Factors
Hyperinsulinemia, obesity and hypoxia are linked to inflammation and cancer development. The elevated levels inflammatory cytokines produced by macrophages and adipocytes within the adipose tissue include IL-6 and TNF-α (5, 84, 88). Cytokines are considered to form one link between inflammation and cancer. Cancer causing mechanisms like a loss of tumor suppressor function, increase of cell cycling and stimulation of oncogene expression were found to be related to cytokines, reactive oxygen species (ROS) and mediators of the inflammatory pathways (TNF-α, COX-2) (3).
IL-6
IL-6 and TNF-α are known to promote angiogenesis (65). The secretion of IL-6 by human adipocytes rises significantly with the BMI (89). Enhanced levels of IL-6 were found in breast cancer patients. Patients with insulin resistance showed even higher levels. However, highest levels of IL-6 were detected in patients with ER positive breast cancer. In prostate cancer the IL-6 levels were significantly higher in hormone-resistant tumors compared to hormone-dependent cancer (5). IL-6 is correlated with obesity and was additionally shown to be necessary for the differentiation of immature plasmablasts into mature antibody producing plasma cells (5). IL-6 may explain the association of B cell lymphoma and multiple myeloma which are increased in obesity.
TNF-α
TNF-α is also causally related to cancer, acute sepsis and chronic inflammation. In obesity, it was shown that adipocytes and infiltrated macrophages express an increased level of TNF-α, which had been positively correlated with insulin resistance and waist circumference (90). TNF-α demonstrates apoptotic effects by inhibiting mTOR and protein synthesis through IκB kinase (IKK) and MAPK pathways. Also necrotic cell death might be induced by TNF-α. Furthermore, TNF-α was found to phosphorylate IRS-1 and IRS-2 and therefore interfere in the signaling of the tyrosine kinase of the IR which might contribute to insulin resistance. TNF-α function lacking, obese mice even were shown to be protected from developing insulin resistance (5, 32, 84). However, one study analyzing the effects of TNF-α on myoblasts observed an anti-apoptotic effect of TNF-α. Treatment of C2C12 myotubes with TNF-α for 24 hours led to an increase in protein synthesis and increased activity of cellular dehydrogenase up to 26%. The PI3K-Akt and MEK-ERK signaling cascades were detected to be the activated pathways mediated by the TNF-α receptor 1 (TNF-R1) (91). This was ascribed by an enhanced protein synthesis also mediated through the PI3K / AKT / NF-κB and the MAPK / ERK pathway (5). However, TNF-α mediates the transcription of a variety of proteins involved in inflammation, cell survival, proliferation and prevention of apoptosis through its activation of NF-κB pathway and MAPK pathway, we can conclude from this data that TNF-α also might have a protective impact by improving insulin resistance and thus attenuating its cancer promoting influence.
CRP (C-reactive protein)
C-reactive protein (CRP) is an acute phase protein produced and secreted mainly by the liver and a sensitive unspecific marker for inflammation, infection and tissue injury. It binds to exogenous and autologous molecules containing phosphocholine (PC) which are released from damaged cells and expressed on the surface of some types of bacteria. After binding, the concentration of CRP in the blood rises rapidly and extensively and activates the complement system (92).
In 35% of obese men and 60% of obese women with a BMI > 30 kg/m2, increased levels of CRP were found (93). There is a close correlation between the proportion of CRP levels and the amount of adipose tissue. Given that adipose tissue secretes a remarkable amount of pro-inflammatory mediators, including IL-6, TNF-α and leptin and that IL-6 is the main regulatory cytokine of hepatic CRP synthesis, it is comprehensible that the enhanced amount of adipose tissue in obesity leads to an increased concentration of CRP. Moreover, expression of CRP was found in human, mouse and rat adipose tissue showing a twofold increase in obese animals, compared to lean controls (94). CRP and IL-6 levels were reported to predict the development of diabetes in both obese men and women what identifies CRP as the potential link between obesity and diabetes (95-96). Furthermore, CRP is associated with an increased risk to develop colorectal, cervical and ovarian cancer (97). These data may clarify an association between inflammation, T2DM, obesity and cancer.
Transcription factors
NF-κB (nuclear factor 'kappa-light-chain-enhancer' of activated B-cells)
NF-κB is a transcription factor which is present in the cytoplasm of almost all cell types in an inactive state. This allows a fast response and immediate expression change to any harmful cellular stimuli. Thus, NF-κB is activated by many factors such as several cytokines, reactive oxygen species (ROS), bacterial or viral antigens like bacterial lipopolysaccharides (LPS) and ionizing radiation. NF-κB activates the expression of genes which promote cell proliferation and inhibit apoptosis and therefore enhances cell survival. Thus, it is not remarkable that several different types of human tumors display dysregulated NF-κB function (98).
HIF-1α (hypoxia inducible factor 1alpha)
HIF-1α is a transcriptional factor, which is induced by hypoxia and activates the expression of certain genes. Activation of HIF-1α relies on the oxygen-dependent hydroxylation of prolyl residues and leads to an elevation of vascularization in tumors. In normoxia the HIF-1α level is regulated by ubiquitination and subsequently degradation in the proteasome so that the half-life of this protein is less than five minutes. An enhanced HIF-1α level results from decreased ubiquitination which is induced by EGF, insulin and IGFs through their PI3K-AKT and MAPK pathways (31). The bacterial lipopolisaccharide is able to activate HIF-1α even in normoxic conditions which lead to the initiation of an inflammatory response. The HIF-1α pathway interacts with the NF-κB pathway and hence links hypoxia to inflammation. Solid tumors were found to contain enhanced levels of HIF-1α. Also oncogenes and the loss of function of tumor suppressor genes are capable of stabilizing HIF-1α. These observations have been associated with an increased risk of metastasis and tumor invasiveness. Furthermore, HIF-1α inhibition might improve sensitivity of tumors to radiation (88).
PPARs (Peroxisome proliferator-activated receptors)
PPARs are transcription factors which belong to the nuclear hormone receptor superfamily and are activated by ligands. One of three different PPAR-subtypes, the PPARα, is mostly found in the liver and plays an important role in activation of fatty acid catabolism. The ubiquitously expressed PPARβ (also known as δ) induces fatty acid oxidation and differentiation of keratinocytes. PPARγ is able to improve insulin resistance through its interactions in glucose metabolism whereas PPARγ2 was found to be important for the differentiation of adipose tissue. However, several studies suggested activated PPARγ as a anti-tumorigenic and pro-differentiation factor in which PPARβ seems to act in a tumorigenic manner (3).
Other Factors
COX-2 (Cyclooxygenase-2)
Many cell types produce the inducible enzyme COX-2. It was found to be overexpressed in several cancer types and linked to promotion of carcinogenesis by increasing the production of prostaglandins, conversion of pro-carcinogens to carcinogens, inhibition of apoptosis, promotion of angiogenesis, modulation of inflammation and immune function and the increase of tumor cell invasiveness (3).
MIF (macrophage migration inhibition factor)
MIF is a target gene of HIF-1α thereby activated by hypoxia. MIF is secreted by macrophages, adipocytes as well as lymphocytes and its effect results in a decreased departure of macrophages out of hypoxic areas in tissues. MIF was negatively correlated with insulin sensitivity and its level rises with the BMI. The detailed role of MIF is unclear but there might be a connection between the MIF-induced inflammation of adipocytes and insulin resistance and thus with cancer (31).
Conclusion
In this article we describe the effects of the metabolic syndrome (obesity and Type 2 diabetes) on cancer development and progression. We stress that the insulin and IGF systems are important causal factors in this connection. However, there are a number of other factors that may play important roles as well. Clearly the tumor promoting potential of the IR and IGF-1R might be a promising answer to identify major cancer promoting mechanisms, with therapeutic possibilities, in such a high-prevalent disease like the metabolic syndrome.
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Zimmet PZ, Alberti KG. Introduction: Globalization and the non-communicable disease epidemic. Obesity (Silver Spring). 2006;14:1-3
2. Jaggers JR, Sui X, Hooker SP, LaMonte MJ, Matthews CE, Hand GA, Blair SN. Metabolic syndrome and risk of cancer mortality in men. Eur J Cancer. 2009;45:1831-8
3. Pothiwala P, Jain SK, Yaturu S. Metabolic syndrome and cancer. Metab Syndr Relat Disord. 2009;7:279-88
4. Calle EE, Rodriguez C, Walker-Thurmond K, Thun MJ. Overweight, obesity, and mortality from cancer in a prospectively studied cohort of U.S. adults. N Engl J Med. 2003;348:1625-38
5. Gallagher EJ NR, Yakar S. The Increased Risk of Cancer in Obesity and Type 2 Diabetes: Potential Mechanisms; Principles of Diabetes Mellitus, 2nd ed. New York, USA: Springer. 2010:579-99
6. Haslam DW, James WP. Obesity. Lancet. 2005;366:1197-209
7. Ogden CL, Carroll MD, Curtin LR, McDowell MA, Tabak CJ, Flegal KM. Prevalence of overweight and obesity in the United States, 1999-2004. JAMA. 2006;295:1549-55
8. Pavelka JC, Brown RS, Karlan BY, Cass I, Leuchter RS, Lagasse LD, Li AJ. Effect of obesity on survival in epithelial ovarian cancer. Cancer. 2006;107:1520-4
9. Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43-66
10. Gallagher EJ, LeRoith D. Insulin, insulin resistance, obesity, and cancer. Curr Diab Rep. 2010;10:93-100
11. Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047-53
12. Hu FB, Manson JE, Liu S, Hunter D, Colditz GA, Michels KB, Speizer FE, Giovannucci E. Prospective study of adult onset diabetes mellitus (type 2) and risk of colorectal cancer in women. J Natl Cancer Inst. 1999;91:542-7
13. Larsson SC, Wolk A. Obesity and colon and rectal cancer risk: a meta-analysis of prospective studies. Am J Clin Nutr. 2007;86:556-65
14. Hampel H, Abraham NS, El-Serag HB. Meta-analysis: obesity and the risk for gastroesophageal reflux disease and its complications. Ann Intern Med. 2005;143:199-211
15. Gallagher EJ, Fierz Y, Ferguson RD, Leroith D. The pathway from diabetes and obesity to cancer, on the route to targeted therapy. Endocr Pract. 2010;16:864-73
16. Larsson SC, Wolk A. Obesity and risk of non-Hodgkin's lymphoma: a meta-analysis. Int J Cancer. 2007;121:1564-70
17. Birmann BM, Giovannucci E, Rosner B, Anderson KC, Colditz GA. Body mass index, physical activity, and risk of multiple myeloma. Cancer Epidemiol Biomarkers Prev. 2007;16:1474-8
18. Petrelli JM, Calle EE, Rodriguez C, Thun MJ. Body mass index, height, and postmenopausal breast cancer mortality in a prospective cohort of US women. Cancer Causes Control. 2002;13:325-32
19. Friberg E, Mantzoros CS, Wolk A. Diabetes and risk of endometrial cancer: a population-based prospective cohort study. Cancer Epidemiol Biomarkers Prev. 2007;16:276-80
20. Smith HO, Tiffany MF, Qualls CR, Key CR. The rising incidence of adenocarcinoma relative to squamous cell carcinoma of the uterine cervix in the United States--a 24-year population-based study. Gynecol Oncol. 2000;78:97-105
21. Kucharska-Newton AM, Rosamond WD, Mink PJ, Alberg AJ, Shahar E, Folsom AR. HDL-cholesterol and incidence of breast cancer in the ARIC cohort study. Ann Epidemiol. 2008;18:671-7
22. Furberg AS, Veierod MB, Wilsgaard T, Bernstein L, Thune I. Serum high-density lipoprotein cholesterol, metabolic profile, and breast cancer risk. J Natl Cancer Inst. 2004;96:1152-60
23. Furberg AS, Jasienska G, Bjurstam N, Torjesen PA, Emaus A, Lipson SF, Ellison PT, Thune I. Metabolic and hormonal profiles: HDL cholesterol as a plausible biomarker of breast cancer risk. The Norwegian EBBA Study. Cancer Epidemiol Biomarkers Prev. 2005;14:33-40
24. Lim U, Gayles T, Katki HA, Stolzenberg-Solomon R, Weinstein SJ, Pietinen P, Taylor PR, Virtamo J, Albanes D. Serum high-density lipoprotein cholesterol and risk of non-hodgkin lymphoma. Cancer Res. 2007;67:5569-74
25. Magura L, Blanchard R, Hope B, Beal JR, Schwartz GG, Sahmoun AE. Hypercholesterolemia and prostate cancer: a hospital-based case-control study. Cancer Causes Control. 2008;19:1259-66
26. Manjer J, Berglund G, Bondesson L, Garne JP, Janzon L, Lindgren A, Malina J, Matson S. Intra-urban differences in breast cancer mortality: a study from the city of Malmo in Sweden. J Epidemiol Community Health. 2000;54:279-85
27. Coughlin SS, Calle EE, Teras LR, Petrelli J, Thun MJ. Diabetes mellitus as a predictor of cancer mortality in a large cohort of US adults. Am J Epidemiol. 2004;159:1160-7
28. Sturmer T, Buring JE, Lee IM, Gaziano JM, Glynn RJ. Metabolic abnormalities and risk for colorectal cancer in the physicians' health study. Cancer Epidemiol Biomarkers Prev. 2006;15:2391-7
29. Verlato G, Zoppini G, Bonora E, Muggeo M. Mortality from site-specific malignancies in type 2 diabetic patients from Verona. Diabetes Care. 2003;26:1047-51
30. Michels KB, Solomon CG, Hu FB, Rosner BA, Hankinson SE, Colditz GA, Manson JE. Type 2 diabetes and subsequent incidence of breast cancer in the Nurses' Health Study. Diabetes Care. 2003;26:1752-8
31. Ye J. Emerging role of adipose tissue hypoxia in obesity and insulin resistance. Int J Obes (Lond). 2009;33:54-66
32. Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature. 1997;389:610-4
33. Baker J, Liu JP, Robertson EJ, Efstratiadis A. Role of insulin-like growth factors in embryonic and postnatal growth. Cell. 1993;75:73-82
34. Liu JP, Baker J, Perkins AS, Robertson EJ, Efstratiadis A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell. 1993;75:59-72
35. Papa V, Pezzino V, Costantino A, Belfiore A, Giuffrida D, Frittitta L, Vannelli GB, Brand R, Goldfine ID, Vigneri R. Elevated insulin receptor content in human breast cancer. J Clin Invest. 1990;86:1503-10
36. Papa V, Gliozzo B, Clark GM, McGuire WL, Moore D, Fujita-Yamaguchi Y, Vigneri R, Goldfine ID, Pezzino V. Insulin-like growth factor-I receptors are overexpressed and predict a low risk in human breast cancer. Cancer Res. 1993;53:3736-40
37. LeRoith D, Werner H, Beitner-Johnson D, Roberts CTJr. Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr Rev. 1995;16:143-63
38. Schatz JH. Targeting the PI3K/AKT/mTOR Pathway in Non-Hodgkin's Lymphoma: Results, Biology, and Development Strategies. Curr Oncol Rep. 2011 Epub
39. Neal CL, Xu J, Li P, Mori S, Yang J, Neal NN, Zhou X, Wyszomierski SL, Yu D. Overexpression of 14-3-3zeta in cancer cells activates PI3K via binding the p85 regulatory subunit. Oncogene. 2011 Epub
40. Kooijman R. Regulation of apoptosis by insulin-like growth factor (IGF)-I. Cytokine Growth Factor Rev. 2006;17:305-23
41. Baxter RC, Bryson JM, Turtle JR. Somatogenic receptors of rat liver: regulation by insulin. Endocrinology. 1980;107:1176-81
42. LeRoith D. Can endogenous hyperinsulinaemia explain the increased risk of cancer development and mortality in type 2 diabetes: evidence from mouse models. Diabetes Metab Res Rev. 2010;26:599-601
43. Cannata D, Fierz Y, Vijayakumar A, LeRoith D. Type 2 diabetes and cancer: what is the connection? Mt Sinai J Med. 2010;77:197-213
44. Novosyadlyy R, Lann DE, Vijayakumar A, Rowzee A, Lazzarino DA, Fierz Y, Carboni JM, Gottardis MM, Pennisi PA, Molinolo AA, Kurshan N, Mejia W, Santopietro S, Yakar S, Wood TL, LeRoith D. Insulin-mediated acceleration of breast cancer development and progression in a nonobese model of type 2 diabetes. Cancer Res. 2010;70:741-51
45. DeChiara TM, Efstratiadis A, Robertson EJ. A growth-deficiency phenotype in heterozygous mice carrying an insulin-like growth factor II gene disrupted by targeting. Nature. 1990;345:78-80
46. Liu JL, LeRoith D. Insulin-like growth factor I is essential for postnatal growth in response to growth hormone. Endocrinology. 1999;140:5178-84
47. Salmon WDJr, Daughaday WH. A hormonally controlled serum factor which stimulates sulfate incorporation by cartilage in vitro. J Lab Clin Med. 1957;49:825-36
48. Dupont J, Le Roith D. Insulin-like growth factor 1 and oestradiol promote cell proliferation of MCF-7 breast cancer cells: new insights into their synergistic effects. Mol Pathol. 2001;54:149-54
49. LeRoith D, Helman L. The new kid on the block(ade) of the IGF-1 receptor. Cancer Cell. 2004;5:201-2
50. Gallagher EJ, LeRoith D. The proliferating role of insulin and insulin-like growth factors in cancer. Trends Endocrinol Metab. 2010;21:610-8
51. Ingermann AR, Yang YF, Han J, Mikami A, Garza AE, Mohanraj L, Fan L, Idowu M, Ware JL, Kim HS, Lee DY, Oh Y. Identification of a novel cell death receptor mediating IGFBP-3-induced anti-tumor effects in breast and prostate cancer. J Biol Chem. 2010;285:30233-46
52. Yakar S, Pennisi P, Kim CH, Zhao H, Toyoshima Y, Gavrilova O, LeRoith D. Studies involving the GH-IGF axis: Lessons from IGF-I and IGF-I receptor gene targeting mouse models. J Endocrinol Invest. 2005;28:19-22
53. Belfiore A, Frasca F, Pandini G, Sciacca L, Vigneri R. Insulin receptor isoforms and insulin receptor/insulin-like growth factor receptor hybrids in physiology and disease. Endocr Rev. 2009;30:586-623
54. LeRoith D. et al. The role of insulin and insulin-like growth factors in the increased risk of cancer in diabetes. RMMJ. 2011;2(2):e0043
55. Brugarolas J, Lei K, Hurley RL, Manning BD, Reiling JH, Hafen E, Witters LA, Ellisen LW, Kaelin WGJr. Regulation of mTOR function in response to hypoxia by REDD1 and the TSC1/TSC2 tumor suppressor complex. Genes Dev. 2004;18:2893-904
56. Ellisen LW. Growth control under stress: mTOR regulation through the REDD1-TSC pathway. Cell Cycle. 2005;4:1500-02
57. Perks CM, Vernon EG, Rosendahl AH, Tonge D, Holly JM. IGF-II and IGFBP-2 differentially regulate PTEN in human breast cancer cells. Oncogene. 2007;26:5966-72
58. Soliman GA. The mammalian target of rapamycin signaling network and gene regulation. Curr Opin Lipidol. 2005;16:317-23
59. Nellist M, Burgers PC, van den Ouweland AM, Halley DJ, Luider TM. Phosphorylation and binding partner analysis of the TSC1-TSC2 complex. Biochem Biophys Res Commun. 2005;333:818-26
60. Novosyadlyy R, Vijayakumar A, Lann D, Fierz Y, Kurshan N, LeRoith D. Physical and functional interaction between polyoma virus middle T antigen and insulin and IGF-I receptors is required for oncogene activation and tumour initiation. Oncogene. 2009;28:3477-86
61. Algire C, Amrein L, Zakikhani M, Panasci L, Pollak M. Metformin blocks the stimulative effect of a high-energy diet on colon carcinoma growth in vivo and is associated with reduced expression of fatty acid synthase. Endocr Relat Cancer. 2010;17:351-60
62. LeRoith D, Roberts CTJr. The insulin-like growth factor system and cancer. Cancer Lett. 2003;195:127-37
63. Gallagher EJ. et al. The increased risk of cancer in obesity and type 2 diabetes: potential mechanisms. In: (ed.) Poretsky L. Principles of Diabetes Mellitus. US: Springer. 2010:583
64. Lee AV, Jackson JG, Gooch JL, Hilsenbeck SG, Coronado-Heinsohn E, Osborne CK, Yee D. Enhancement of insulin-like growth factor signaling in human breast cancer: estrogen regulation of insulin receptor substrate-1 expression in vitro and in vivo. Mol Endocrinol. 1999;13:787-96
65. Rose DP. et al. Obesity, adipocytokines, and insulin resistance in breast cancer. Obes. Rev. 2004;5:153-165
66. Margetic S, Gazzola C, Pegg GG, Hill RA. Leptin: a review of its peripheral actions and interactions. Int J Obes Relat Metab Disord. 2002;26:1407-33
67. Zhang Y, Proenca R, Maffei M, Barone M, Leopold L, Friedman JM. Positional cloning of the mouse obese gene and its human homologue. Nature. 1994;372:425-32
68. Benoit SC, Clegg DJ, Seeley RJ, Woods SC. Insulin and leptin as adiposity signals. Recent Prog Horm Res. 2004;59:267-85
69. Maffei M, Fei H, Lee GH, Dani C, Leroy P, Zhang Y, Proenca R, Negrel R, Ailhaud G, Friedman JM. Increased expression in adipocytes of ob RNA in mice with lesions of the hypothalamus and with mutations at the db locus. Proc Natl Acad Sci U S A. 1995;92:6957-60
70. Toyoshima Y, Gavrilova O, Yakar S, Jou W, Pack S, Asghar Z, Wheeler MB, LeRoith D. Leptin improves insulin resistance and hyperglycemia in a mouse model of type 2 diabetes. Endocrinology. 2005;146:4024-35
71. Konopleva M, Mikhail A, Estrov Z, Zhao S, Harris D, Sanchez-Williams G, Kornblau SM, Dong J, Kliche KO, Jiang S, Snodgrass HR, Estey EH, Andreeff M. Expression and function of leptin receptor isoforms in myeloid leukemia and myelodysplastic syndromes: proliferative and anti-apoptotic activities. Blood. 1999;93:1668-76
72. Endo H, Hosono K, Uchiyama T, Sakai E, Sugiyama M, Takahashi H, Nakajima N, Wada K, Takeda K, Nakagama H, Nakajima A. Leptin acts as a growth factor for colorectal tumours at stages subsequent to tumour initiation in murine colon carcinogenesis. Gut. 2011 epub
73. Gonzalez-Perez RR, Xu Y, Guo S, Watters A, Zhou W, Leibovich SJ. Leptin upregulates VEGF in breast cancer via canonic and non-canonical signalling pathways and NFkappaB/HIF-1alpha activation. Cell Signal. 2010;22:1350-62
74. Scherer PE, Williams S, Fogliano M, Baldini G, Lodish HF. A novel serum protein similar to C1q, produced exclusively in adipocytes. J Biol Chem. 1995;270:26746-9
75. Yamauchi T, Kamon J, Minokoshi Y, Ito Y, Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, Eto K, Akanuma Y, Froguel P, Foufelle F, Ferre P, Carling D, Kimura S, Nagai R, Kahn BB, Kadowaki T. Adiponectin stimulates glucose utilization and fatty-acid oxidation by activating AMP-activated protein kinase. Nat Med. 2002;8:1288-95
76. Ukkola O, Santaniemi M. Adiponectin: a link between excess adiposity and associated comorbidities? J Mol Med. 2002;80:696-702
77. Coppola A, Marfella R, Coppola L, Tagliamonte E, Fontana D, Liguori E, Cirillo T, Cafiero M, Natale S, Astarita C. Effect of weight loss on coronary circulation and adiponectin levels in obese women. Int J Cardiol. 2009;134:414-6
78. Ye J, Gao Z, Yin J, He Q. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am J Physiol Endocrinol Metab. 2007;293:E1118-28
79. Chen B, Lam KS, Wang Y, Wu D, Lam MC, Shen J, Wong L, Hoo RL, Zhang J, Xu A. Hypoxia dysregulates the production of adiponectin and plasminogen activator inhibitor-1 independent of reactive oxygen species in adipocytes. Biochem Biophys Res Commun. 2006;341:549-56
80. Fasshauer M, Klein J, Neumann S, Eszlinger M, Paschke R. Hormonal regulation of adiponectin gene expression in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 2002;290:1084-9
81. Berg AH, Combs TP, Scherer PE. ACRP30/adiponectin: an adipokine regulating glucose and lipid metabolism. Trends Endocrinol Metab. 2002;13:84-9
82. Tsatsanis C, Zacharioudaki V, Androulidaki A, Dermitzaki E, Charalampopoulos I, Minas V, Gravanis A, Margioris AN. Peripheral factors in the metabolic syndrome: the pivotal role of adiponectin. Ann N Y Acad Sci. 2006;1083:185-95
83. Sugiyama M, Takahashi H, Hosono K, Endo H, Kato S, Yoneda K, Nozaki Y, Fujita K, Yoneda M, Wada K, Nakagama H, Nakajima A. Adiponectin inhibits colorectal cancer cell growth through the AMPK/mTOR pathway. Int J Oncol. 2009;34:339-44
84. Poretsky. the increased risk of cancer in obesity and type 2 diabetes: potential mechanisms. Principles of Diabetes Mellitus Chapter. 2010;36:589-91
85. Diez JJ, Iglesias P. The role of the novel adipocyte-derived hormone adiponectin in human disease. Eur J Endocrinol. 2003;148:293-300
86. Brakenhielm E, Veitonmaki N, Cao R, Kihara S, Matsuzawa Y, Zhivotovsky B, Funahashi T, Cao Y. Adiponectin-induced antiangiogenesis and antitumor activity involve caspase-mediated endothelial cell apoptosis. Proc Natl Acad Sci U S A. 2004;101:2476-81
87. Ishikawa M, Kitayama J, Yamauchi T, Kadowaki T, Maki T, Miyato H, Yamashita H, Nagawa H. Adiponectin inhibits the growth and peritoneal metastasis of gastric cancer through its specific membrane receptors AdipoR1 and AdipoR2. Cancer Sci. 2007;98:1120-7
88. Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364:656-65
89. Onuma M BJ, Rummel TL. Prostate cancer cell-adipocyte interaction:leptin mediates androgen-independent prostate cancer cell proliferation through c-Jun NH2-terminal kinase. J Biol Chem. 2003;278:42660-42667
90. Zinman B, Hanley AJ, Harris SB, Kwan J, Fantus IG. Circulating tumor necrosis factor-alpha concentrations in a native Canadian population with high rates of type 2 diabetes mellitus. J Clin Endocrinol Metab. 1999;84:272-8
91. Plaisance I, Morandi C, Murigande C, Brink M. TNF-alpha increases protein content in C2C12 and primary myotubes by enhancing protein translation via the TNF-R1, PI3K, and MEK. Am J Physiol Endocrinol Metab. 2008;294:E241-50
92. Thompson D, Pepys MB, Wood SP. The physiological structure of human C-reactive protein and its complex with phosphocholine. Structure. 1999;7:169-77
93. Visser M, Bouter LM, McQuillan GM, Wener MH, Harris TB. Elevated C-reactive protein levels in overweight and obese adults. JAMA. 1999;282:2131-5
94. Lau DC, Dhillon B, Yan H, Szmitko PE, Verma S. Adipokines: molecular links between obesity and atheroslcerosis. Am J Physiol Heart Circ Physiol. 2005;288:H2031-41
95. Festa A, D'Agostino RJr, Tracy RP, Haffner SM. Elevated levels of acute-phase proteins and plasminogen activator inhibitor-1 predict the development of type 2 diabetes: the insulin resistance atherosclerosis study. Diabetes. 2002;51:1131-7
96. Freeman DJ, Norrie J, Caslake MJ, Gaw A, Ford I, Lowe GD, O'Reilly DS, Packard CJ, Sattar N. C-reactive protein is an independent predictor of risk for the development of diabetes in the West of Scotland Coronary Prevention Study. Diabetes. 2002;51:1596-600
97. Erlinger TP, Platz EA, Rifai N, Helzlsouer KJ. C-reactive protein and the risk of incident colorectal cancer. JAMA. 2004;291:585-90
98. Puszynski K, Bertolusso R, Lipniacki T. Crosstalk between p53 and nuclear factor-B systems: pro- and anti-apoptotic functions of NF-B. IET Syst Biol. 2009;3:356-67
Author contact
![]() Corresponding author: Derek LeRoith, M.D., Ph.D., Director, Diabetes and Metabolism Clinical Research Center of Excellence, Legacy Heritage Clinical Research Institute at Rambam (LHCRIR). Tel: 972-4-854-1260. d_leroithhealth.gov.il
Corresponding author: Derek LeRoith, M.D., Ph.D., Director, Diabetes and Metabolism Clinical Research Center of Excellence, Legacy Heritage Clinical Research Institute at Rambam (LHCRIR). Tel: 972-4-854-1260. d_leroithhealth.gov.il