Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(7):1016-1026. doi:10.7150/ijbs.7.1016 This issue Cite
Research Paper
Solexa Sequencing of Novel and Differentially Expressed MicroRNAs in Testicular and Ovarian Tissues in Holstein Cattle
Jinming Huang ![]() , Zhihua Ju, Qiuling Li, Qinlei Hou, Changfa Wang, Jianbin Li, Rongling Li, Lingling Wang, Tao Sun, Suqin Hang, Yundong Gao, Minghai Hou, Jifeng Zhong
, Zhihua Ju, Qiuling Li, Qinlei Hou, Changfa Wang, Jianbin Li, Rongling Li, Lingling Wang, Tao Sun, Suqin Hang, Yundong Gao, Minghai Hou, Jifeng Zhong ![]()
Laboratory of Molecular Breeding, Center of Dairy Cattle Research, Shandong Academy of Agricultural Sciences, China.
Received 2011-1-30; Accepted 2011-7-1; Published 2011-8-18
Abstract
The posttranscriptional gene regulation mediated by microRNA plays an important role in the development and function of male and female reproductive organs and germ cells in mammals, including cattle. In the present study, we identified novel and differentially expressed miRNAs in the testis and ovary in Holstein cattle by combining the Solexa sequencing with bioinformatics. In total 100 and 104 novel pre-miRNAs were identified in testicular and ovarian tissues, encoding 122 and 136 mature miRNAs, respectively. Of these, 6 miRNAs appear to be bovine-specific. A total of 246 known miRNAs were co-expressed in the testicular and ovarian tissues. Of the known miRNAs, twenty-one testis-specific and nine ovary-specific (1-23 reads) were found. Approximately 30.5% of the known bovine miRNAs in this study were found to have >2-fold differential expression within the two respective reproductive organ systems. The putative miRNA target genes of miRNAs were involved in pathways associated with reproductive physiology. Both known and novel tissue-specific miRNAs are expressed by Real-time quantitative PCR analysis in dairy cattle. This study expands the number of miRNAs known to be expressed in cattle. The patterns of miRNAs expression differed significantly between the bovine testicular and ovarian tissues, which provide important information on sex differences in miRNA expression. Diverse miRNAs may play an important regulatory role in the development of the reproductive organs in Holstein cattle.
Keywords: MicroRNA, testis, ovary, dairy cattle
Introduction
MicroRNAs (miRNAs) are a class of small (18-24 nucleotides), endogenous and non-coding RNAs which bind to target mRNAs so as to regulate protein expression by repressing translation or by promoting degradation of the target mRNA [1, 2], or by enhancing translation at the posttranscriptional level in RNA-induced silencing complex (RISC) [3]. It is estimated that 1 to 5% of all mammalian genes encode for miRNA, which regulate the expression of approximately 30% of all of the protein-coding genes [4, 5], and that each miRNA on average regulates 200 target genes through an interaction between the seed sequence and the complementary target sites [6]. Several reviews have summarized the diverse miRNA biological roles developmental timing, including embryogenesis, organ differentiation, organogenesis, growth and programmed cell death as well as stem and germline cell maintenance, disease and evolution [1, 7-9]. The number of the known miRNAs has rapidly increased in recent years [10-12], and currently there are 662 Bos taurus miRNA sequences deposited in the Sanger miRNA database release 16.0 (miRBase) [13-15], with potentially many more yet to be.
The insights which have been obtained in the field of reproductive biology are of help in efforts to enhance the reproductive and economic efficiencies in dairy cattle. The testes are among the most important reproductive organs of the bull since they produce sperm and the hormone testosterone. The ovaries are the corresponding primary reproductive organs in the dairy cow. The ovaries are comprised of a unique organ system produce ova as well as the hormones involved in regulating the estrous cycle and pregnancy. There are many important events in these processes, including the recruitment of growing follicles, ovulation, and luteal tissue formation and regression, which are dynamically regulated in the bovine ovary. Lack of regulatory control can lead to ovarian failure due to disruption of folliculogenesis, blockage of ovulation, or corpus luteum (CL) insufficiency. Considerable light has been shed on transcriptional gene regulation and cellular signaling pathways within the reproductive organs and tissues over the past 40 years. However, much less is known about posttranscriptional gene regulation in these crucially important reproductive organs.
Recent reports have shown that certain reproductive processes are strictly regulated at the transcriptional and posttranscriptional levels [16-17]. A novel mechanism of miRNA mediated post-transcriptional control has lately come to be regarded as an important regulator of reproductive processes [16-18]. The aim of the present study was to identify novel miRNAs and differentially expressed miRNAs which may play a regulatory role in the development of the testis and ovary in Holstein cattle. The data obtained will help provide a better understanding of the role of miRNA in reproductive biological processes and the mechanisms of target gene expression and regulation.
Materials and Methods
Sample preparation and RNA extraction
Four bulls and four cows were sacrificed at each of the two stages: 3d after birth (Pre-sex-maturation) and 2.5 years old (Post-sex maturation). The testis and ovary tissues were collected at the above sacrificed time points. And the same kind of tissue samples were homogenized and polled with equal amounts of each tissue type for the Solexa sequencing. The tissue samples were immediately frozen in liquid nitrogen and separately stored at -80℃. Then, total RNA was extracted from pooled samples with Trizol (Invitrogen, USA) according to the manufacturer's instructions. Quantity and integrality of RNA was detected by Agilent2100 (Agilent, USA).
Solexa sequencing
The overall flow of the sequencing and bioinformatics analysis for small RNA was shown in Additional File 1: Figure S1 (BGI, China). Briefly, after polyacrylamide gel electrophoresis purification of small RNA molecules less than 30 bases and ligation of a pair of Solexa adaptors to their 3' and 5' ends, the small RNA molecules were amplified using the adaptor primers for 17 cycles and the fragments around 90 bp (small RNA and adaptors) were isolated from agarose gel. The purified DNA was used directly for cluster generation and sequencing analysis using the Illumina's Solexa Sequencer according to the manufacturer's instructions. Then the image files generated by the sequencer were processed to produce digital-quality data. The subsequent procedures performed with Solexa were summarizing data production, evaluating sequencing quality, calculating length distribution of small RNA reads and filtrating reads contaminated by rRNA, tRNA, mRNA, snRNA, and snoRNA. Finally, clean reads were compared with a miRBase database (release 16.0).
Bioinformatics analysis for small RNAs
Firstly, Solexa reads were aligned against the Bos.taurus genome (Btau 4.0) [19] using SOAP [20]. Sequences with perfect match or one mismatch were retained for further analysis. To further analyze the RNA secondary structures comprising matched Solexa reads, 100 nucleotides of genomic sequence flanking each side of these sequences were extracted, and the secondary structures were predicted using RNAfold software [21] and analyzed by MIREAP software under these parameter settings [22]: miRNA sequence length (18-26nt); miRNA reference sequence length (20-24nt); Minimal depth of Drosha/Dicer cutting site (3) ; Maximal copy number of miRNAs on reference (20); Maximal free energy allowed for a miRNA precursor (-18 kcal/mol) ; Maximal space between miRNA and miRNA* (35) ; Minimal base pairs of miRNA and miRNA* (14) ; Maximal bulge of miRNA and miRNA* (4) ; Maximal asymmetry of miRNA/miRNA* duplex (5); Flank sequence length of miRNA precursor (10). Stem-loop hairpins were considered typical only when they accord with following three criteria: mature miRNAs are present in one arm of the hairpin precursors, which lack large internal loops or bulges; the secondary structures of the hairpins are steady, with the free energy of hybridization lower than -18kcal/mol; and hairpins are located in intergenic regions or introns. Those genes whose sequences and structures fulfilled above three criteria were considered as candidate miRNA genes. Subsequently, new miRNA genes that are paralogs or orthologs to known miRNAs were predicted by the miRAlign [23]. Finally, all remaining candidates were subjected to MiPred to filter out pseudo-pre-miRNAs with the following settings: Minimum of free energy >-20 kcal/mol or P-value > 0.05 [24]. All the novel miRNAs candidates were also analyzed to search against the most similar miRNA sequences in the miRNA database (Release16.0). Each identified miRNA was located in the genome using the search function in the Ensembl genome browser (http://www.ensembl.org/Bos_taurus). Prediction of target genes was performed using MIREAP and TargetScan (http://www.targetscan.org/). The roles of prediction for MIREAP were as follows: (1) No more than four mismatches between sRNA & target (G-U bases count as 0.5 mismatches); (2)No more than two adjacent mismatches in the miRNA/target duplex; (3) No adjacent mismatches in in positions 2-12 of the miRNA/target duplex (5' of miRNA); (4) No mismatches in positions 10-11 of miRNA/target duplex; (5) No more than 2.5 mismatches in positions 1-12 of the of the miRNA/target duplex (5' of miRNA); (6) Minimum free energy (MFE) of the miRNA/target duplex should be >= 75% of the MFE of the miRNA bound to it's perfect com plement. Blastx search was performed using the target sequence and the NCBI database to predict functions of potential targets.
Differential expression analysis of miRNAs
To compare the differential miRNA expression between two libraries (ovary and testis), first, the expression of miRNAs in two samples were normalized to obtain the expression of transcripts per million. If the normalized expression value of a given miRNA is zero, its expression value was be modified to 0.01. If the normalized expression (NE) of a given miRNA is less than 1 in both library, the miRNA was removed in future differential expression analysis. Then, the fold-change and P-value were calculated from the normalized expression using the following formulas.
Normalized expression = (Actual miRNA sequencing reads count / Total clean reads count) ×1,000,000.
Fold change = Log2 ( Ovary-NE /Testis-NE)
P-value:
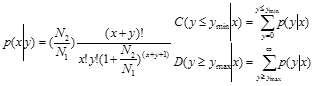
The N1 and x represent total count of clean reads and normalized expression level of a given miRNA in testis sRNA library, respectively. The N2 and y represent total count of clean reads and normalized expression level of a given miRNA in ovary sRNA library, respectively.
Hierarchical clustering of miRNAs expression
Hierarchical clustering of miRNA expression was performed using the PermutMatrix software with Pearson distance [25]. The relative cloning frequency for each miRNA was calculated as the number of sequences for each miRNA in a library divided by the total of number of sequence reads for that library.
Real time quantitative PCR (Q-PCR)
Q-PCR was performed using the mirVana™ miRNA isolation kit (Ambion) according to the manufacturer's protocol. Bta-let-7g was used as an endogenous control. The primers for miRNAs have the same sequences as bovine miRNAs. The ABI PRISM 7900 Sequence Detector System (Applied Biosystem, Foster City, CA) was used to perform the quantitative analysis using SYBRGreen PCR master mix (Applied Biosystems). The protocol referenced to the previous report [26]. The relative quantifications of miRNAs expression were calculated using the standard curve-based method for relative real time PCR [27].
Ethics statement
The Chinese Holstein bulls and cows used in this study were obtained from the Shandong OX Biotechnology Co., Ltd. Experiments were carried out according to the Regulations for the Administration of Affairs Concerning Experimental Animals published by Ministry of Science and Technology, China, 2004) and approved by the Animal Care and Use Committee in Shandong Academy of Agricultural Sciences, Shandong, P. R. China.
Results
Construction of two small RNA libraries by Solexa sequencing
In order to identify novel and differentially expressed miRNAs in the bovine testis and ovary, two small RNA libraries were constructed by Solexa sequencing. The overall flow of the sequencing and bioinformatics analysis is shown schematically in Additional File 1: Figure S1. Solexa sequencing provided a total of 17,292,838 and 17,440,637 reads of 3nt-30nt from the testis and ovary tissue libraries, respectively. After removing the low quality, adaptor, insufficiently tagged and sequences, a total of 9,122,461 and 8,454,797 reads of 18nt-30nt were ultimately obtained. The kind and the total number of the common and tissue-specific small RNA sequences in the testis and ovary are shown in Additional File 1: Figure S2. The percentages of the testis-specific and ovary-specific sequences were 0.78% and 1.62% of the total small RNAs.
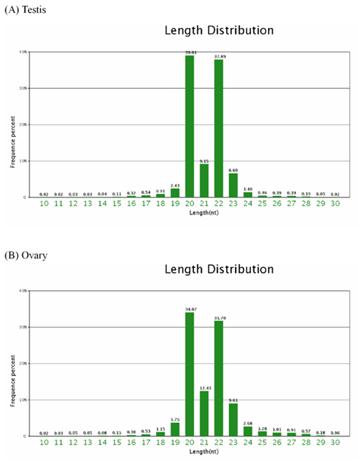
Of the total of the small sequences in two libraries, the lengths of the majority of small RNAs 19nt-24nt, which is typical of the small RNA of Dicer-processed processed product. The size distribution (10nt-30nt) of the small RNA from the testis and ovary in Chinese Holstein cattle is similar (Figure 1). For example, in the bovine testis, the 20nt and 22nt sequences are the dominant small RNAs, which account for 39.01% and 37.89% of the total sequences, respectively (Figure 1A).
Length distribution and frequency percent of sequences in testis and ovary libraries.
Next, all of the Solexa sequencing reads were aligned against the Bos taurus genome (Btau_4.0) using the SOAP Program [20]. Additional File 2: Table S1 shows that 6,973,964 reads were matched to the bovine genome in the testis library (9,122,461 reads in total). A total of 6,156,692 reads were matched to the bovine genome in the ovary library (8,454,797 reads in total).
Subsequently, the unique bovine small RNAs (sRNA) in the two libraries were annotated into different categories (Additional File 1: Figure S3). Among these genome-matched reads, 6,637,871 and 5,236,997 reads belonging to miRNA were obtained from the testis and ovary libraries, respectively.
Identification of novel cattle miRNAs
To determinate whether these small RNA sequences are genuine cattle miRNA, we scanned the bovine genome for hairpin structures comprising the candidate miRNA with MIREAP software (http://sourceforge.net/projects/mireap/), which can be used to identify both known and novel miRNAs from sRNA libraries deeply sequenced using the Solexa/454/SOLid technology. In total, 278 and 266 loci possessed the typical stem-loop structures matching the known miRNA hairpins (676 miRNA, 32 miRNAs* and 662 hairpins miRbase16.0) in the testis (Additional File 2: Table S2) and ovary (Additional File 2: Table S3). A total of 260 and 248 known miRNAs were expressed in the testicular and ovarian tissues, respectively (Additional File 2: Table S2 and S3). After the removal of loci that overlapped with protein-coding gene exons and loci with a free energy less than -18 kcal/mol, the remaining 100 and 104 loci with novel hairpins were considered candidate miRNA genes (Additional File 2: Table S4 and S5). Precursors of the novel miRNAs were identified formed proper secondary hairpin structures, with the free energy ranging from -19.3 kcal/mol to -52.74 kcal/mol (an average of -36.02 kcal/mol) (Additional File 2: Table S4 and S5). More importantly, the identification of anti-sense miRNA (miRNA*) from 35 novel miRNA candidates in the two tissues provided further evidence supporting them as novel miRNAs. The novel miRNAs are shown in Additional File 2: Table S4 and S5. Of these 258 novel miRNAs in the testis and ovary, 6 are considered to be cattle-specific, because no homologs were found in the miRBase 16.0 data. In general, most of the identified miRNAs are homologous to the known miRNAs in this species, which supports the view that miRNAs are evolutionarily conserved [2].
Analyses of the first nucleotide bias of the 18-30nt miRNAs candidates revealed that uridine (U) was the most common (97.71%) at the 5' end of the 20nt novel miRNAs, whereas adenine (A) is the most common first nucleotide of the novel 22nt (57.89%), 23nt (83.79%) and 24nt (100%) miRNAs in the testis (Additional File 2: Table S6). Interestingly, in the ovary, U was the dominant first nucleotide of the novel 19nt (100%), 20nt (96.41%) and 22nt (45.10%) miRNAs, while A was the dominant first nucleotide of the 23nt (75.92%) nucleotide miRNAs (Additional File 2: Table S7).
We investigated the chromosome locations (BTAU4.0) for the known miRNAs in the MiRBase, searching for the known and novel pre-miRNAs via BLAST analysis. The result is shown in Additional File 2: Table S8. Most of the novel miRNAs were found to be located on the autosomes and X chromosome of the cattle. However, fourteen and ten novel miRNAs from the respective testis and ovary, respectively, failed to match the Bos.taurus genomic sequences, which might be due to the incomplete sequencing of the Bos.taurus or differences between the different cattle breeds (beef and dairy cattle). Among the matching sequences, approximately 50% (43/86) and 52.13% (49/94) novel pre-miRNAs in the testis and ovary were found within the intron regions of the genes, respectively (Additional File 2: Table S4 and S5). Approximate 65.74% (165/251) and 57.68% (154/267) of the known pre-miRNAs which were successfully located in chromosomes in the testicular and ovarian tissues, respectively, were located in the intergenic regions (Additional File 2: Table S9). We also analyzed the genomic density of the pre-miRNAs (Additional File 2: Table S8). The average density distribution of the known miRNAs located in the autochromosome and X chromosome ranged from 0.09 to 1.03 miRNAs per 1Mbp. The shortest chromosome, 25, and the longest chromosome, 1, encode 24 and 18 miRNAs, respectively, corresponding to 0.55 and 0.11 miRNAs per 1Mbp genomic sequences. Chromosome 21 has the greatest number of the known miRNAs distribution in the cattle genome and also the expressed miRNAs in the testis and ovary (Additional File 2: Table S8). The X chromosome has the fourth greatest number of the known miRNAs, accounting for 5.82% (37/636) of the total miRNAs. It is noteworthy that the expressed miRNAs in the X chromosome and chromosome 21 are dominant in the testis and ovary tissues. The chromosomal densities of the expressed miRNAs in the X chromosomes were 2.36 (0.33/0.14) and 2.07 (0.29/0.14) fold the average of the autosomes in the bovine testis and ovary, respectively.
Expression of miRNAs in the testis and ovary
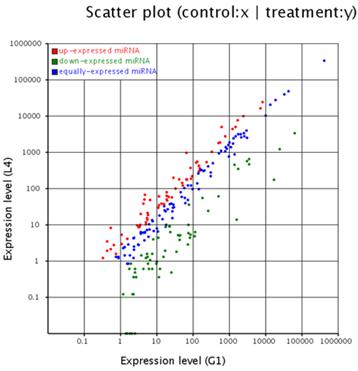
The solexa sequencing method provides a useful means to estimate the expression profiles of the miRNA genes by measuring the sequencing frequencies. The sequencing reads of the miRNAs in the testis and ovary libraries are presented in Additional File 2: Table S5 and Figure 2. The hierarchical clustering of the two tissues based on the relative cloning frequencies of the miRNAs suggested that miRNA expression is different in the testis and ovary (Figure 3). In the testis, bta-miR-143, bta- miR-378, bta-let-7f, bta-let-7a, bta-miR-1, bta-let-7c, bta-miR-206 and bta-let-7b were the dominant expressed miRNAs, with more than 100,000 reads. They constituted 63.5% of the total sequencing reads, suggesting that they are abundant expression in testis tissue. The sequencing frequencies of the 57 miRNAs (e.g. bta-miR-324, bta-miR-541, bta-miR-653) were much lower (1≤sequencing reads ≤10). However, in ovary, seven miRNAs (bta-miR-143, bta-let-7f, bta-let-7a, bta-let-7c, bta-miR-10b, bta-let-7b and bta-miR-26a), each with more than 100,000 reads, were the most abundant. Fifty-nine miRNAs (e.g., bta-miR-380-5p, bta-miR-495 and bta-miR-345) displayed the lowest sequencing frequencies (less than 10 reads). In the testis and ovary libraries, except for bta-miR-193a* and bta-miR-142*, the expression levels of the three miRNA*s (bta-miR-21*, bta-miR-126* and bta-let-7a*) were much lower than their corresponding miRNAs (Additional File 2: Table S9). This result indicates that some of the miRNA* strands are degraded rapidly during the biogenesis of the mature miRNAs.
Comparison of expression levels of miRNAs in testis and ovary. The X and Y axis show expression level of miRNAs in two samples respectively. Red points represent miRNA with ratio>2; Blue points represent miRNAs with 1/2<ratio<2; Green points represent miRNAs with ratio<1/2. Ratio=normalized expression of the treatment / normalized expression of the control.
Hierarchical clustering of miRNAs in two tissues using Peason correlation. Heatmap was constructed base on the relative cloning frequencies of miRNAs. Green and red indicate low frequency and high frequency miRNA cloned in the library.

In the testicular and ovarian tissues, a total of 246 known miRNAs are co-expressed. Among the known miRNAs, twenty-one testis-specific and nine ovary-specific were found (Additional File 2: Table S9). The expression levels of the tissue-specific expressed miRNAs were low (1-23 reads). In comparison with miRNA expression in the testis, 65 miRNAs in the ovary were significantly up-regulated, while 63 miRNAs in the ovary were significantly down-regulated (Additional File 2: Table S10).
The expression levels of novel miRNAs in the testis and ovary are shown in Additional File 2: Table S4 and S5. The analysis of the differential miRNA expression patterns in different tissues may provide valuable information on the role that miRNAs play in the testis and ovary in terms of growth and development. The Solexa sequencing technology is also a useful method for distinguishing and measuring miRNA sequences with only a small number of nucleotide changes. Based on the Solexa sequencing results, different family members exhibited drastically different expression levels. For example, the abundance of the bta-let-7 family varied from 4 reads (bta-let-7a*) to 400170 reads (bta-let-7f) in the deep sequencing results (Additional File 2: Table S9). The existence of a dominant member in a given miRNA family may suggest that the regulatory function is for the most part performed by the dominant member.
Validation of miRNAs expression with Q-PCR
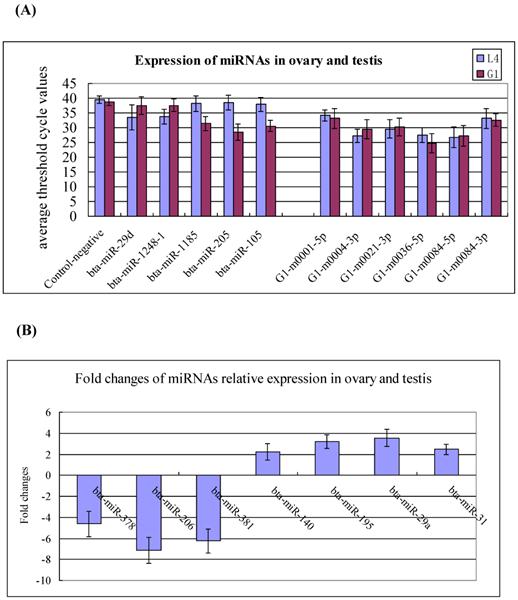
To validate the existence of the known and newly identified bovine miRNAs, the same RNA preparations used for the Solexa sequencing were subjected to Q-PCR assay. The expression levels of the 18 miRNAs were determined in the testis and ovary. The selected miRNAs including to 5 testis- and ovary-specific known miRNAs (bta-miR-29d, miR-1248-1, miR-1185, miR-205, and miR-105a), 7 testis-predominant and ovary-predominant known miRNAs (miR-381, miR-206, miR-378, miR-29a, miR-195, miR-31 and miR-140) as well as the 6 candidate novel bovine miRNAs (G1-m0001-5p, G1-m0004-3p, G1-m0021-3p, G1-m0036-5p, G1-m0084-5p and G1-m0084-3p). The expression levels of testis-specific,ovary-specific and three novel miRNAs are much lower in the testis and ovary, therefore, their relative expression represent with the average threshold cycle (CT) values (Figure 4A), while the highly expressed miRNAs represent using the fold changes (Figure 4B). This pattern of expression is consistent with the Solexa sequencing results (Figure 4, Additional File 2: Table S4, S5 and S9).
Q-PCR validation of the identified miRNAs using solexa sequencing technology. (A) Expression of the testis- and ovary-specific miRNAs and novel miRNAs. The expression of miRNAs was shown in the CT value. The novel identified miRNA in testis correspond to the novel miRNAs in ovary: G1-m0001-5p=L4-m0001-5p; G1-m0004-3p=L4-m0006-3p; G1-m0021-3p =L4-m0022-3p; G1-m0036-5p =L4-m0038-5p; G1-m0084-5p=L4-m0094-5p; G1-m0084-3p=L4-m0094-3p. (B) Expression of the testis- and ovary-predominat known miRNAs. Error bars represent one standard deviation of three different biological replicates.
Prediction of target genes and pathways analysis
To identify the potential target genes of the novel miRNAs, target prediction was performed using MIREAP software. A total of 331234 putative target sites for 100 miRNAs in the testis and 354702 target sites for 99 miRNAs in the ovary were identified (data not shown). The predicted target genes for the 20 differential expression with highly abundant miRNAs (Additional File 2: Table S10) were classfied according to KEGG functional annotations to identify pathways that were actively regulated by miRNA in testis and ovary tissues (Additional File 2: Table S11). Interestingly, the most 57 miRNA targets belonged to the MAPK signaling pathway, which plays a part in virtually every step of spermatogenesis in the testis, including spermatogonial stem cell renewal, germ cell cycle and meiosis, spermiogenesis, and spermiation. MAPK is also involved in the subsequent sperm maturation and motility, hyperactivation, capacitation and acrosome reaction in the female reproductive tract before fertilization with the ovum [28]. Another important pathway is Wnt signaling pathway, which is known to be closely involved in the mammalian reproduction, including follcilular development, ovulation, luteinization, Müllerian duct formation and regression, etc [29]. Another important pathway targeted by the highly expressed miRNAs is the TGF-beta signaling pathway, which is known to be involved in reproductive aging [30]. TGF-beta signaling pathway can also regulate the duration of germ cell quiescence and is necessary to maintain adult spermatogenesis [31]. It is worth noting that the bta-miR-143 miRNA is the one most highly expressed in the testis and ovary, a total of 266 putative conserved target genes were identified by a search using Targetscan5.1. Interestingly, the DAVID functional annotation (http://david.abcc.ncifcrf.gov/) showed that 10 putative target genes were involved in the GnRH-signaling and insulin-signaling pathways which are associated with endocrine system function. Bta-miR-206 and bta-miR-1, with respective 6.62 and 4.36 fold differences between the testis and ovary, target 584 genes sites, some of which are involved in 35 KEGG pathways, such as aldosterone-regulated sodium reabsorption, progesterone-mediated oocyte maturation, wnt signaling pathway. Seven putative target genes (Activin, ActivinRI, ActivinRII, BMPRI, Smurf1/2, p107 and SP1) of G1-m0084-3p participate in the TGF beta signaling pathway that is involved in gonadal growth, embryo differentiation and placenta formation. Although the predicted target genes are needed to be validated experimentally in the next experiments, collectively, these pathway and biological process analyses illustrate some of the possible roles of the differential highly expressed miRNAs in reproductive organ biologies.
Discussion
Mapping and location of the miRNAs sequences in genome
Of the mappable sequences, the majority of the small RNAs are 19-24 nt in size, which is typical of the small RNA of Dicer-processed products. In the present study, the 20nt and 22nt sequences in the bovine testis and ovary (including the 3d after birth and adult samples) are the dominant small RNAs. Interestingly, it was recently reported that the majority of 679 miRNAs sequences representing 58 distinct bovine miRNAs were 20-23nt in size, and the 21-22nt size was predominant in the bovine fetal ovary (230 and 250d of gestation) [32]. Our data show that the pattern of variation in the miRNA length may not remain constant during the course of bovine ovary development. Our finding is not consistent with the previous finding in the mouse testis and ovary [33]. We speculated that there are three reasons: First, different miRNA identification methods producted different throghput data. Such as, our data is based on the high throughput experiment. Second, the discrepancy may result from the isomiRs with different lengths (e.g. 22nt size) in ovary and testis (Additional File 2: Table S2 and S3). Third, the length differentce of mature miRNAs with the mouse may hint the function difference in different species. Further data collection and analysis are needed to adequately account for this discrepancy. The variations in sequence length are attributed to have certain functional roles, such as RNA editing in miRNA-mediated gene silencing [34], 3'-editing [35] and degradation of microRNAs by a family of exoribonucleases [36]. However, only the variations in the relatively low abundance miRNAs are accounted for by RNA-editing events. The frequency of these modifications was low for the vast majority of miRNAs, suggesting that these modifications play a limited role in miRNA function [34]. Currently there is little understanding of the functional role of the homologs of the highly abundant miRNAs.
miRNAs possess a diverse pattern of genomic organization. Some miRNAs contain their own independent promoters and enhancers, for example, miRNA-1-1 and miRNA-133a-2 in humans and mice. In contrast, approximately 40% of the miRNAs are located within the introns of either non-protein-coding or protein-coding transcriptional units, such as miR-208, and 10% are within exons, such as miRNA-198 [37, 38].
The XX/XY chromosomes are a biological system that determines the development of sexual characteristics in cattle. We calculated the densities of the miRNAs on the chromosomes using the miRNA data from the miRBase and found there was no available data on the Y chromosome. In addition, some sequences were not mappable to the chromosomes. Therefore, the autosomes and X chromosome were used for the subsequent density analysis. We note that 37 known bovine miRNAs (5.59%) are located in the X chromosome, on the current version of miRBase 16.0. In other animals, for example, 94 (8.97%, Homo sapiens), 26 (12.32%, Sus scrofa), 41 (12.69%, Canis familiaris) and 78 (11.61%, Mus musculus) miRNAs were located on the X chromosome. We also found a higher density of the bovine-expressed to be located on the X chromosome than the autosomes on average. It is possible that a bias in miRNA density analysis may be the reason that miRNAs from species such as the cow, pig and dog have not been fully identified. As more comprehensive miRNA databases are made available for the species analyzed here, and species as well, it will become feasible to determine whether this is a widespread phenomenon. On the basis of the current data, the analysis of the chromosomal distribution of the miRNAs showed a higher miRNA density on the X chromosome compared to the autosomes, and the miRNAs are evolving faster on the X chromosome than on the autosomes, which implies alterations in male reproductive function may be involved in mammalian speciation [39].
miRNAs expression patterns in different tissues
The miRNA expression patterns in different tissues have been investigated in dairy cattle and beef cattle [32, 40]. In beef cattle, miR-9 and miR-124 in the brain, miR-122 in the liver, and miR-1, miR-133a and miR-206 in muscle are all tissue-specific [40]. In the present study, these miRNAs were also identified in the testis and ovary. The results suggest that some of the previously reported tissue-specific miRNAs maybe have certain limits in terms of distribution in tissues. The bta-miR-424 miRNA was not identified in the bovine testis or ovary using the Solexa sequencing and Q-PCR methods in the present study. However, the bta-miR-424 expression was highly abundant in GV and MII stage oocytes as well as early stage embryos with a tendency to decrease in morula and blastocyst stage embryos [41]. Because our samples belong to two distinct stages i.e. pre- and post-sex maturation, this may explain why miR-424 was not identified. Such an expression pattern indicates that bta-miR-424 is maternally inherited and may potentially be involved in the turnover of the maternal transcript during zygotic gene activation [42].
miRNAs may have important roles in male and female reproductive physiology
Analyses of miRNAs in the field of reproduction are of considerable interest. Murchison et al. first reported that the oocytes of mice lacking Dicer activity exhibited arrested meiosis and suggested that miR function is important to the very earliest stages of development [42]. Another study also reported that maternal miRNAs contribute to zygotic development in the mouse, as deletion of Dicer from the oocyte blocked cell division [43]. Together, these data suggest that miRNAs are critical for the progression of oogenesis. On the contrary, another report showed that miRNAs are not essential for oocyte growth, meiotic maturation and early stages of preimplantation development in the mouse [44]. The expression of certain miRNA, for example miR-514 in the testis of infant (1-2 year, sexually immature) and adult rhesus monkeys (8-10 year, sexually mature ) were development-stage specific, which implies that the X-linked miRNA cluster most likely plays a regulatory role in primate testis development and spermatogenesis [45].
In the present study, bta-miR-21 exhibited a 1.97-fold significant increase in ovary compared with testis tissue. The results suggest that bta-miR-21 may play a regulatory role in female physiology. MicroRNA-21 has been widely studied due to its upregulation in almost all forms of cancer. It was shown to rapidly increase following in vivo LH/hCG administration [46]. Our finding showed that a total of 182 putative conserved target genes were identified for the bta-miR-21/590-5p. The target genes belong to 9 KEGG pathways, including the MAPK, TGF-beta, B-cell receptor, T-cell receptor pathways. The result indicates that miR-21 is involved in immune reactions. There is evidence suggesting that the targets of bta-miR-21 are cell specific [47]. A recent publication indicated that bta-miR-21 plays a role in preventing apoptosis in periovulatory granulosa cells as they transition to luteal cells, although the regulatory mechanisms exerted by miR-21 remain undetermined [48]. In the present study, the high expression levels of bta-let-7b in the testis and ovary were demonstrated by 123195 and 170290 reads, respectively. However, miR-17-5p exhibited lower expressed levels (420 and 665 reads). It is reported that the impaired CL angiogenesis in Dicerd/d mice is partly due to a lack of miR-17-5p and let-7b, 2 miRNAs that participate in the endothelial function of angiogenesis via regulation of the expression of the anti-angiogenic tissue inhibitor of metalloproteinase 1 (TIMP1) [49]. The expression levels of the two miRNAs in the ovary were higher than the testis, which indicates a role for the two miRNAs in the ovary. Bta-miR-143, the most highly expressed miRNA in the bovine testis and ovary, targets genes which participate in pathways associated with reproduction. The expression of bta-miR-143 in the mouse ovary was very low in primordial follicles, but became readily detectable in the granulosa cells of primary, secondary and antral follicles. Furthermore, the expression of bta-mir-143 is under the negative control of FSH [50].
While miRNAs have been evaluated in the mouse ovary and oocyte [42-44], there is no current literature on miRNAs in production livestock, leading us to evaluate the relative miRNAs abundance in the bovine testis and ovary, highlighting the need for research in actual livestock to compliment that performed in model species. Cattle are regarded as an animal model for the study of human reproduction, biochemistry, enzymology, endocrinology and nutrition research because of the organizational similarity at the genomic level between human and cattle, as compared to the mouse [51]. Therefore, this study should ultimately help facilitate a better understanding human reproduction physiology as well as cattle. Insights into testicular and ovarian miRNA expression and function are just beginning to be obtained. Further studies on the miRNAs function with regard to sex differences in cattle are needed in order to identify individual miRNAs playing key roles during male and female reproductive physiological processes.
Supplementary Material
Additional File 1Figure S1: The overall flow of the sequencing and informatic analysis for small RNA. Figure S2: Summary of common and specific sequences between bovine testis (G1) and ovary (L4).Figure S3: Distribution of small RNAs among different categories in testis (G1) and ovary (L4)
Table S1: Mapping statistics of testis and ovary samples. Table S2: Mireap output in Testis (The Summary of small RNAs matching known miRNA hairpins). Table S3: Mireap output in ovary (The Summary of small RNAs matching known miRNA hairpins). Table S4: Summary of novel miRNAs in testis (G1). Table S5: Summary of novel miRNAs in ovary (L4). Table S6: First nucleotide bias of 18~30nt novel miRNA candidates (G1). Table S7: First nucleotide bias of 18-30nt novel miRNA candidates (L4). Table S8: Distribution of the miRNAs in chromosomes. Table S9: Expression profiles and chromosomal positions of the total known mature miRNAs in the testis and ovary. Table S10: Comparison of the expression profiles of miRNAs in the testis and ovary. Table S11: KEGG pathways enriched for targets of the 20 differential highly expressed miRNAs in the ovary and testis in cattle.
Acknowledgements
We thank Boru K, PhD for his revision of our manuscript.
This study was supported by grants from the National Natural Science Foundation of China (no. 31000543), Major Project of National Transgene in China (2011ZX08007-001), Support Program of the Ministry of Science and Technology, P. R. China (2011BAD19B02, 2011BAD19B04), Program of National Cow Industrial Technology System (CARS-37), Youth Science Foundation from Shandong Academy of Agriculture Science (No.2006YQN030), Project of Agriclutual Fine Breed from the Department of Science and Technology of Shandong Province (2010LZ10-02) and the Natural Science Foundation of Shandong Province (Y2007D72).
Conflict of Interests
The authors have declared that no conflict of interest exists.
References
1. Carrington JC, Ambros V. Role of microRNAs in plant and animal development. Science. 2003;301:336-8
2. Bartel DP. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell. 2004;116:281-97
3. Vasudevan S, Tong YC, Steitz JA. Switching from repression to activation: MicroRNAs can up-regulate translation. Science. 2007;318:1931-4
4. Lim LP, Glasner ME, Yekta S. et al. Vertebrate microRNA genes. Science. 2003;299:1540
5. Lewis BP, Shih IH, Jones-Rhoades MW. et al. Prediction of mammalian microRNA targets. Cell. 2003;115:787-98
6. Krek A, Grun D, Poy MN. et al. Combinatorial microRNA target predictions. Nat Genet. 2005;37:495-500
7. Ambros V. The functions of animal microRNAs. Nature. 2004;431:350-5
8. Alvarez-Garcia I, Miska EA. MicroRNA functions in animal development and human disease. Development. 2005;132:4653-62
9. Kloosterman WP, Plasterk RHA. The diverse functions of MicroRNAs in animal development and disease. Developmental Cell. 2006;11:441-50
10. Coutinho LL, Matukumalli LK, Sonstegard TS. et al. Discovery and profiling of bovine microRNAs from immune-related and embryonic tissues. Physiol Genomics. 2007;29:35-43
11. Strozzi F, Mazza R, Malinverni R. et al. Annotation of 390 bovine miRNA genes by sequence similarity with other species. Anim Genet. 2009;40:125
12. Jin W, Grant JR, Stothard P. et al. Characterization of bovine miRNAs by sequencing and bioinformatics analysis. BMC Mol Biol. 2009;10:90
13. Griffiths-Jones S, Saini HK, van Dongen S. et al. miRBase: tools for microRNA genomics. Nucleic Acids Res. 2008;36:D154-D158
14. Griffiths-Jones S, Grocock RJ, van Dongen S. et al. miRBase: microRNA sequences, targets and gene nomenclature. Nucleic Acids Res. 2006;34:D140-D144
15. The miRBase Database. http://microrna.sanger.ac.uk
16. Papaioannou MD, Nef S. MicroRNAs in the Testis: Building up male fertility. J Andro. 2010;31:26-33
17. Carletti M Z, Christenson LK. MicroRNA in the ovary and female reproductive tract. J Anim Sci. 2009;87:E29-E38
18. Ohlsson Teague EMC, Print CG, Hull ML. The role of microRNAs in endometriosis and associated reproductive conditions. Human Reproduction Update. 2010;16:142-65
19. Bovine Genome Sequencing, Analysis Consortium, Elsik CG, Tellam RL, Worley KC. et al. The genome sequence of taurine cattle: a window to ruminant biology and evolution. Science. 2009;324:522-8
20. Li R, Li Y, Kristiansen K. et al. SOAP: Short Oligonucleotide Alignment Program. Bioinformatics. 2008;24:713-4
21. RNAfold. http://www.tbi.univie.ac.at/~ivo/RNA/RNAfold.html
22. MIREAP. http://https//sourceforge.net/projects/mireap/
23. Wang X, Zhang J, Li F. et al. MicroRNA identification based on sequence and structure alignment. Bioinformatics. 2005;21:3610-4
24. Jiang P, Wu H, Wang W. et al. MiPred: classification of real and pseudo microRNA precursors using random forest prediction model with combined features. Nucleic Acids Res. 2007;35:W339-W344
25. Caraux G, Pinlonche S. PermutMatrix: a graphical environment to arrange gene expression profiles in optimal linear order. Bioinformatic. 2005;21:1280-1
26. Ro S, Park C, Jin J. et al. A PCR-based method for detection and quantification of small RNAs. Biochem Biophys Res Commun. 2006;351:756-63
27. Larionov A, Krauseand A, Miller W. A standard curve based method for relative real time PCR data processing. BMC Bioinform. 2005;6:62-78
28. Li MWM, Mruk DD, Cheng CY. Mitogen-activated protein kinases in male reproductive function. Trends Mol Med. 2009;15:159-68
29. Kobayashi A, Stewart CA, Wang Y. et al. β-Catenin is essential for Müllerian duct regression during male sexual differentiation. Development. 2011 epub
30. Luo S, Shaw WM, Ashraf J. et al. TGF-ß Sma/Mab Signaling Mutations Uncouple Reproductive Aging from Somatic Aging. PLoS Genet. 2009;5:e1000789
31. Moreno SG, Attali M, Allemand I. et al. TGFbeta signaling in male germ cells regulates gonocyte quiescence and fertility in mice. Dev Biol. 2010;342:74-84
32. Tripurani SK. et al. Cloning and analysis of fetal ovary microRNAs in cattle. Anim Reprod Sci. 2010;120:16-22
33. Mishima T, Takizawa T, Luo SS. et al. MicroRNA (miRNA) cloning analysis reveals sex differences in miRNA expression profiles between adult mouse testis and ovary. Reproduction. 2008;36:811-22
34. Kawahara Y, Zinshteyn B, Sethupathy P. et al. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science. 2007;315:1137-40
35. Landgraf P, Rusu M, Sheridan R. et al. A mammalian microRNA expression atlas based on small RNA library sequencing. Cell. 2007;129:1401-14
36. Ramachandran V, Chen X. Degradation of microRNAs by a family of exoribonucleases in Arabidopsis. Science. 2008;321:1490-2
37. Zhao Y, Srivastava DA. Developmental view of microRNA function. Trends Biochem Sci. 2007;32:189-97
38. Guo XJ, Su B, Zhou ZM. et al. Rapid evolution of mammalian X-linked testis microRNAs. BMC Genomics. 2009;10:97
39. Zhang R, Peng Y, Wang W. et al. Rapid evolution of an X-linked microRNA cluster in primates. Genome Res. 2007;17:612-7
40. Jin W, Grant J, Stothard P. et al. Characterization of bovine miRNAs by sequencing and bioinformatics analysis. BMC Mol Bio. 2009;10:90
41. Giraldez AJ, Mishima Y, Rihe J. et al. Zebrafish MiR-430 promotes deadenylation and clearance of maternal mRNAs. Science. 2006;312:75-9
42. Murchison EP, Stein P, Xuan Z. et al. Critical roles for Dicer in the female germline. Genes Dev. 2007;21:682-93
43. Tang F, Kaneda M, O'Carroll D. et al. Maternal microRNAs are essential for mouse zygotic development. Genes Dev. 2007;21:644-8
44. Suh N, Baehner L, Moltzahn F. et al. MicroRNA function is globally suppressed in mouse oocytes and early embryos. Curr Biol. 2010;20:271-7
45. Ro S, Song R, Park C. et al. Cloning and expression profiling of small RNAs expressed in the mouse ovary. RNA. 2007;13:2366-80
46. Carletti MZ, Fiedler SD, Christenson LK. MicroRNA 21 blocks apoptosis in mouse periovulatory granulosa cells. Biol Reprod. 2010;83:286-95
47. Sood P, Krek A, Zavolan M. et al. Cell type-specific signatures of microRNAs on target mRNA expression. Proc Natl Acad Sci USA. 2006;103:2746-51
48. Christenson LK. MicroRNA control of ovarian function. Anim Reprod. 2010;7:129-33
49. Otsuka M, Zheng M, Hayashi M. et al. Impaired microRNA processing causes corpus luteum insufficiency and infertility in mice. J Clin Invest. 2008;118:1944-54
50. Yao N, Lu CL, Zhao JJ. et al. A network of miRNAs expressed in the ovary are regulated by FSH. Front Biosci. 2009;14:3239-45
51. Gibs R, Weinstock G, Kappes S, et al. Bovine genomic sequencing initiative. www.genome.gov/Pages/Research/Sequencing/SeqProposals/BovineSEQ.pdf
Author contact
![]() Corresponding author: Jinming Huang (huangjinmcom); Jifeng Zhong (Zhongjifengcom). Laboratory of Molecular Breeding, Center of Dairy Cattle Research, Shandong Academy of Agricultural Sciences, Industry North Road 159, Jinan, Shandong 250131, China. Telephone/Fax: 086-531-88604132.
Corresponding author: Jinming Huang (huangjinmcom); Jifeng Zhong (Zhongjifengcom). Laboratory of Molecular Breeding, Center of Dairy Cattle Research, Shandong Academy of Agricultural Sciences, Industry North Road 159, Jinan, Shandong 250131, China. Telephone/Fax: 086-531-88604132.