Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
TGF-β1 is a key mediator in...
Distinct role of active and...
TGF-β-dependent and...
Diverse roles of Smad2 and Smad3...
Diverse roles of Smad4 in renal...
Inhibitory role of Smad7 in...
Regulation of...
Concluding Remarks
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2011; 7(7):1056-1067. doi:10.7150/ijbs.7.1056 This issue Cite
Review
Diverse Roles of TGF-β/Smads in Renal Fibrosis and Inflammation
Hui Yao Lan ![]()
Li Ka Shing Institute of Health Sciences and Department of Medicine & Therapeutics, The Chinese University of Hong Kong, Hong Kong SAR, China
Received 2011-8-2; Accepted 2011-8-19; Published 2011-9-2
Abstract
TGF-β1 has been long considered as a key mediator in renal fibrosis and induces renal scarring largely by activating its downstream Smad signaling pathway. Interestingly, while mice overexpressing active TGF-β1 develop progressive renal injury, latent TGF-β1 plays a protective role in renal fibrosis and inflammation. Under disease conditions, Smad2 and Smad3 are highly activated, while Smad7 is degraded through the ubiquitin proteasome degradation mechanism. In addition to TGF-β1, many pathogenic mediators such as angiotensin II and advanced glycation end products can also activate the Smad pathway via both TGF-β-dependent and independent mechanisms. Smads interact with other signaling pathways, such as the MAPK and NF-κB pathways, to positively or negatively regulate renal inflammation and fibrosis. Studies from gene knockout mice demonstrate that TGF-β1 acts by stimulating its downstream Smads to diversely regulate kidney injury. In the context of renal fibrosis and inflammation, Smad3 is pathogenic, while Smad2 and Smad7 are protective. Smad4 exerts its diverse roles by transcriptionally enhancing Smad3-mediated renal fibrosis while inhibiting NF-κB-driven renal inflammation via a Smad7-dependent mechanism. Furthermore, we also demonstrated that TGF-β1 acts by stimulating Smad3 to positively or negatively regulate microRNAs to exert its fibrotic role in kidney disease. In conclusion, TGF-β/Smad signaling is a major pathway leading to kidney disease. Smad3 is a key mediator in renal fibrosis and inflammation, whereas Smad2 and Smad7 are renoprotective. Smad4 exerts its diverse role in promoting renal fibrosis while inhibiting inflammation. Thus, targeting the downstream TGF-β/Smad3 signaling pathway by gene transfer of either Smad7 or Smad3-dependent microRNAs may represent a specific and effective therapeutic strategy for kidney disease.
Keywords: TGF-β/Smads, fibrosis, inflammation, anti-TGF-β therapy, microRNAs.
Introduction
Renal fibrosis characterized by accumulation of fibroblasts and excessive matrix proteins along with a loss of functioning nephrons are a major pathological feature of progressive kidney disease [1]. Many studies have shown that progressive renal fibrosis is mediated by multiple mediators including growth factors, cytokines, metabolic toxins, and stress molecules via multiple mechanisms and pathways. Among them, transforming growth factor-β1 (TGF-β1) has been recognized as a key mediator in the pathogenesis of renal fibrosis [1-3].
TGF-β is a founding member of the TGF-β superfamily including activins, inhibins, growth and differentiation factors, and bone morphogenetic proteins. TGF-β1 and its isoforms (TGF-β2 and TGF-β3) are synthesized by a variety of cells including all cell types of the kidney and secreted as latent precursors (latent TGF-β1) complexed with latent TGF-β binding proteins (LTBP) [3, 4]. TGF-β1 becomes active when TGF-β1 is liberated from the latency-associated peptide (LAP) and dissociated from LTBP via proteolytic cleavage by plasmin, reactive oxygen species, thrombospondin-1, and acid [3, 4]. Active TGF-β then binds its receptors and functions as autocrine and paracrine manners to exert its biological and pathological activities via Smad-dependent and independent signaling pathways [5]. Of them, the Smad-dependent mechanism has been well studied and considered to be a major pathway in many pathophysiological processes of kidney disease [2, 3, 6].
It is now well established that the binding of TGF-β1 to its receptor II (TβRII) can activate the TGF-β receptor type I (TβRI)-kinase, resulting in phosphorylation of Smad2 and Smad3, two receptor-associated Smads (R-Smads). Subsequently, phosphorylated Smad2 and Smad3 bind to the common Smad4 and form the Smad complex, which translocates into the nucleus to regulate the target gene transcription, including Smad7 (Figure 1). Smad7 is an inhibitory Smad that negatively regulates Smad2 and Smad3 activation and functions by targeting the TβRI and Smads for degradation via the ubiquitin proteasome degradation mechanisms [7, 8].
TGF-β/Smads and crosstalk pathways in renal fibrosis and inflammation. After binding to TβRII, TGF-β1 activates the TβRI-kinase which phosphorylates Smad2 and Smad3. The phosphorylated Smad2 and Smad3 then bind to Smad4 and form the Smad complex, which translocates into the nucleus and regulates the target gene transcription, including Smad7. Smad7 is an inhibitory Smad that functions to block Smad2/3 activation by degrading the TβRI and Smads and to inhibit NF-κB-driven inflammatory response by inducing IκBα, an inhibitor of NF-κB. Note that Ang II and AGEs can activate Smads independent of TGF-β1 via the ERK/p38/MAPK crosstalk pathway. Blue lines (symbols) indicate protective or negative regulation pathways, while red arrows (symbols) represent pathogenic or positive regulation pathways.
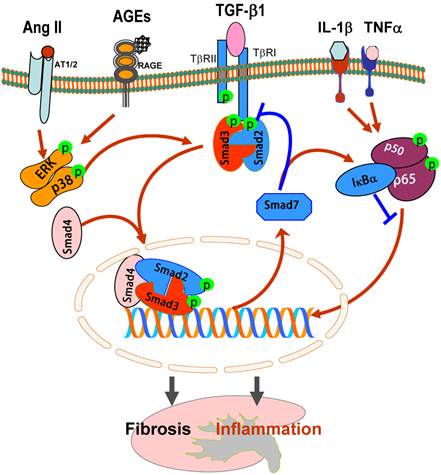
In this review, the major task is to focus on the molecular mechanisms of TGF-β/Smads in diversely regulating renal fibrosis and inflammation during progressive kidney injury. In addition, the new therapeutic approaches for kidney disease by targeting the downstream TGF-β/Smad signaling with gene transfer of Smad7 and TGF-β/Smad3-dependent microRNAs are also described.
TGF-β1 is a key mediator in kidney disease
Increasing evidence shows that TGF-β1 is a key mediator in the pathogenesis of renal fibrosis in both experimental and human kidney diseases because TGF-β1 is highly upregulated in the diseased kidney with severe renal fibrosis [2, 3]. TGF-β1 mediates progressive renal fibrosis by stimulating extracellular matrix (ECM) production while inhibiting its degradation. In addition, TGF-β1 also mediates renal fibrosis by inducing the transformation of tubular epithelial cells (TECs) to myofibroblasts through epithelial-mesenchymal transition (EMT) [9]. The functional role of TGF-β1in EMT and renal fibrosis is demonstrated by the ability of blocking TGF-β1 with neutralizing TGF-β antibodies, decorin, and antisense oligonucleotides to prevent or ameliorate renal fibrosis in vivo and in vitro [10]. Direct evidence for a role of TGF-β1 in renal fibrosis comes from studies that mice overexpressing an active form of TGF-β1 in liver develop progressive liver and renal fibrosis [11, 12].
While the critical role of TGF-β1 in renal fibrosis has been well recognized, little attention has been paid to the role of TGF-β1 in renal inflammation. TGF-β1 is also known as an anti-inflammation cytokines [13]. Mice deficient for TGF-β1 results in the development of a lethal inflammation and death at 3 weeks of age [14]. Similarly, conditional deletion of TβRII or TGF-β1 gene from T cells develops autoimmune diseases [15, 16]. In contrast, mice overexpressing human latent TGF-β1 are protected against progressive renal inflammation and fibrosis in obstructive and immunologically-induced crescentic glomerulonephritis [17-19]. These findings suggest a vital role for TGF-β1 in anti-inflammation. However, signaling mechanisms by which TGF-β1 exerts its anti-inflammatory properties remain unclear, although TGF-β-induced inhibition of NF-κB-mediated renal inflammation via induction of Smad7-dependent IκBα expression has been recently considered [17, 19].
Distinct role of active and latent TGF-β1 in renal fibrosis and inflammation
While it is well understood that active TGF-β1 acts as a critical fibrogenic growth factor mediating progressive renal fibrosis [11, 12], our recent studies showed that mice overexpressing latent TGF-β1 in the skin are protected against progressive renal inflammation and fibrosis in obstructive and immunologically-induced crescentic glomerulonephritis [17-19]. This is associated with an up-regulation of endogenous renal Smad7, an inhibitory Smad [17-19]. These remarkable findings are completely opposite to the findings from the previous reports that mice overexpressing the active TGF-β1 in the liver result in progressive liver and renal fibrosis [11, 12]. Thus, upregulation of renal Smad7 may be an important mechanism through which latent TGF-β1 protects kidney from inflammation and fibrosis. This may be associated with prevention of renal Smad7 from Smurf-mediated ubiquitination and degradation in response to higher levels of latent TGF-β1. It may be also true that latent TGF-β1 may bind its own receptor, GARP, to exert its anti-inflammatory effects via induction of Smad7 or Foxp3-dependent regulatory mechanisms [20, 21]. However, mechanisms underlying the distinct role of active versus latent TGF-β1 in renal fibrosis and inflammation remain largely unknown. In addition, mechanisms whereby Smad7 is upregulated by latent TGF-β1, but degraded by the active form of TGF-β1 are also to be determined. Nevertheless, results from these studies implicate the complexity of TGF-β1 in the pathogenesis of kidney disease and the necessity to understand the diverse roles and mechanisms of active versus latent TGF-β1under disease conditions.
TGF-β-dependent and independent Smad signaling in renal fibrosis
Increasing evidence shows that in chronic kidney diseases, TGF-β is not a sole molecule to activate Smads [3, 22]. Many mediators can activate Smad2 and Smad3 independent from TGF-β1 because Smads act as signal integrators and interact with other signaling pathways such as the mitogen-activated protein kinase (MAPK) signaling pathway and play a role in the pathophysiological processes of kidney diseases [3, 22]. As shown in Figure 1, under the diabetic conditions, advanced glycation end-products (AGEs), an critical mediator in diabetic complications, are able to activate Smad2 and Smad3 independently from TGF-β1 via the ERK/p38 MAP kinase-dependent mechanism [23, 24]. This is supported by the findings that deletion of TGF-β1 or TGF-β receptor II is unable to prevent AGE-induced Smad2 and Smad3 from activation and fibrosis response [24]. In contrast, blockade of the engagement of AGE to its receptor (RAGE) with the soluble RAGE or ERK/p38 MAP kinases with the specific inhibitors or dominant negative ERK1/2 or p38 is able to prevent AGE-induced Smad2/3 phosphorylation and nuclear translocation [23, 24]. Similarly, under the hypertensive conditions, Ang II can activate the Smad signaling pathway to stimulate ECM production via the AT1 receptor-mediated, ERK/p38 MAP kinase-Smad cross talk pathway (Figure 1), in addition to the TGF-β-dependent mechanism [25, 26]. The important role for the MAPK-Smad crosstalk pathway in renal fibrosis is further demonstrated by the ability of Ang II and AGE to activate Smad2/3 to stimulate connective tissue growth factor (CTGF) expression in kidney cells lacking TGF-β1 gene or TβRII, but not in those with blockade of MAPK signaling by overexpressing dominant negative ERK1/2 and p38 or pre-treating cells with their specific inhibitors [24, 26]. All these studies reveal that activation of Smads under disease conditions is complicated and that targeting the TGF-β signaling at the receptor levels may not be an optimal therapeutic approach due to the existing intracellular crosstalk pathways.
It should be pointed out that within the TGF-β super family, TGF-β/Smads also interact with the BMP/Smads to counter-regulate each other to maintain the balance between two pathways in the pathophysiological process. It is well known that Smad1, Smad5 and Smad8 transduce BMP action, whereas Smad2 and Smad3 mediate TGF-β1 activities and the interactions between these two pathways can be at multiple levels including receptors and individual Smads [27]. It has been reported that TGF-β-activated Smad2/3 to mediate epithelial-mesenchymal transition (EMT) is reversed by addition of human recombinant BPM-7 via the Smad1-depdnent mechanism [28]. However, a subsequent study fails to show this counter-regulating activity [29], suggesting the complexity between the TGF-β/Smad and BMP/Smad pathways under disease conditions.
Diverse roles of Smad2 and Smad3 in extracellular matrix synthesis, EMT, and angiogenesis in kidney disease
It is now well accepted that Smad2 and Smad3 are two critical downstream mediators responsible for the biological effects of TGF-β1. In the context of renal fibrosis, Smad2 and Smad3 are strongly activated in both experimental and human kidney diseases, including diabetic nephropathy [23, 24, 30-32], obstructive kidney diseases [18, 33-36], remnant kidney disease [37, 38], hypertensive nephropathy [25], drug-associated nephropathy [39], and immunologically-mediated glomerulonephritis [19, 40]. Many fibrogenic genes, such as (ColIa1, ColIa2, ColIIIa1, ColVa2, ColVIa1, and ColVIa3) and tissue inhibitor of MMP-1 (TIMP-1), are the downstream targets of TGF-β/Smad3 signaling [41], suggesting that Smad3 may be a critical mediator of TGF-β/Smad signaling in fibrosis. An essential role for Smad3 in fibrogenesis is confirmed by the findings that deletion of Smad3 from mice suppresses fibrogenesis in a number of rodent models, including diabetic nephropathy [30], obstructive nephropathy [33], and drug toxicity-related nephropathy [39]. Furthermore, the use of a Smad3 inhibitor to inhibit endothelial-myofibroblast transition and renal fibrosis in a type-1 diabetic kidney disease demonstrates a therapeutic potential for kidney disease by targeting Smad3 signaling [42].
While it is clear that TGF-β1 acts by stimulating Smad3 to mediate renal fibrosis, role of Smad2 in the kidney disease remains largely unknown. This is largely due to the unavailability of Smad2 knockout (KO) mice because of the embryonic lethality [43]. To explore the role of Smad2 in renal fibrosis, we generated conditional Smad2 KO mice by crossing the Smad2 floxed mouse with kidney specific promoter (Cadherin 16)-driven Cre transgenic mouse [44], which results in a conditional deletion of Smad2 from kidney TECs [45]. Unexpectedly, we found that specific deletion of Smad2 from the kidney significantly enhances renal fibrosis in a mouse model of UUO [45]. This is associated with enhanced Smad3 signaling. The in vivo finding is also confirmed in vitro in mouse embryonic fibroblasts (MEF) in which MEFs lacking Smad2 substantially enhance TGF-induced Smad3 signaling including phosphorylation, nuclear translocation, Smad3 promoter activities, the binding of Smad3 to the collagen I promoter (COL1A2), and collagen matrix production [45]. Thus, although it is commonly believed that Smad2 and Smad3 bind together physically and work in a nonredundant manner in embryonic development, Smad2 may function to competitively inhibit the phosphorylation of Smad3 in response to TGF-β1 (Figure 2). It is also possible that the physical interaction of Smad2 and Smad3, together with Smad4, may affect phospho-Smad3 nuclear translocation and the subsequent binding to its target genes (Figure 2). Thus, loss of Smad2 enhances Smad3 signaling, thereby promoting Smad3-mediated collagen matrix expression in response to TGF-β1 and other fibrotic mediators including angiotensin II, AGE, and drugs [24-26, 37, 39].
Protective role of Smad2 in renal fibrosis. Smad2 physically binds Smad3 but prevents Smad3 from activation through two possible mechanisms: 1) competitively inhibit Smad3 binding to the TβRI for phosphorylation; 2) block phosphorylated Smad3 nuclear translocation and binding to the DNA sequences, therefore inhibiting Smad3-mediated renal fibrosis. Blue lines (symbols) indicate protective or negative regulation pathways, while red arrows (symbols) represent pathogenic or positive regulation pathways.
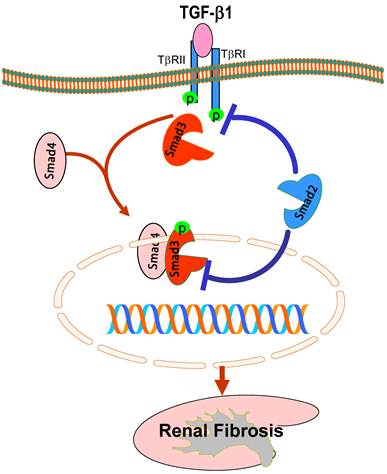
Epithelial-mesenchymal transition (EMT) has been long considered as a process leading to renal fibrosis [9]. Many studies have demonstrated that Smad3 plays a critical role in the EMT process in the kidney as well as in other disease conditions [37, 39, 46-50]. To dissect the functional role of Smad2 and Smad3 in EMT, we conditionally knocked down Smad2 or Smad3 from renal tubular epithelial cells. Interestingly, disruption of Smad3, but not Smad2, attenuates Ang II-induced EMT as identified by a loss of an epithelial marker E-cadherin and a gain of mesenchymal phenotype α-SMA [37]. Similarly, knockdown of Smad3, but not Smad2, also blocks AGE and angiotensin II-induced CTGF expression and renal fibrosis [24, 26]. It is now well known that many fibrogenic genes (collagens) and EMT markers (α-SMA and E-cadherin) are Smad3-dependent [41, 51, 52] and Smad3, but not Smad2, directly binding to the DNA sequences to regulate these target genes [52, 53]. Thus, findings that knockdown of Smad3, but not Smad2, blocks CTGF expression and EMT provide a new evidence for an essential role of Smad3 in the EMT process.
TGF-β1 has been known as a key mediator in controlling angiogenesis. Indeed, impaired angiogenesis leads to the progressive renal fibrosis [54], whereas excessive angiogenesis has been associated with diabetic nephropathy [55]. It is now understood that TGF-β activates Smad2 and Smad3 to control the process of angiogenesis by diversely regulating expression of vascular endothelial growth factor (VEGF), a proangiogenic factor, and thrombospondin-1 (TSP-1), an antiangiogenic factor [56]. By using of Smad2 and Smad3 knockout mouse embryonic fibroblasts, TGF-β1-induced VEGF mRNA and protein expression in both Smad2 and Smad3 wild-type cells is blocked in cells null for Smad3, but not in Smad2. Whereas Smad2, but not Smad3, is critical for TSP-1 expression, demonstrating a critical role for Smad3 in angiogenesis while Smad2 as antiangiogenic factor in response to TGF-β1 [56]. Consistent with the antiangiogenic role of Smad2, we also demonstrated that Smad2, but not Smad3, mediated the expression of VEGF-A antagonist, a soluble VEGF-A receptor sFlt-1, in response to TGF-beta1 [56]. Thus, Smad2 and Smad3 have distinct roles in mediating the expression of pro- and antiangiogenic growth factors in response to TGF-β1.
Diverse roles of Smad4 in renal fibrosis and inflammation
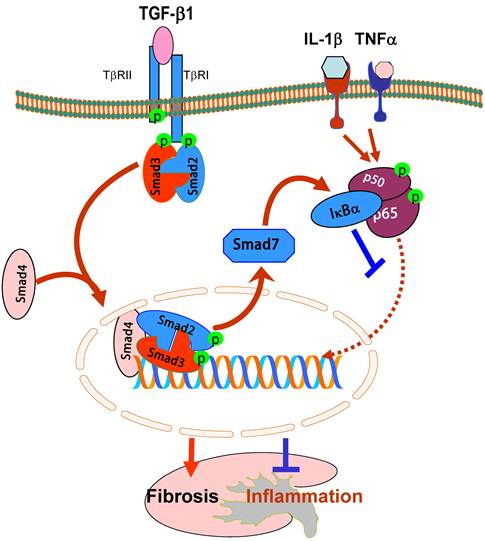
It is now well accepted that TGF-β1 mediates fibrosis by causing the phosphorylation of Smad2 and Smad3, which forms a complex with the common Smad4 and then translocates to nuclei to bind and regulate the target genes [57]. Although Smad4 has been known as the common Smad in the signal transduction pathway of the TGF-β family, its functional role in TGF-β1-regulated fibrosis and inflammatory responses remains largely unclear. This may be largely attributed to the lethality of Smad4 KO mice [58]. Recently, we generated conditional Smad4 KO mice by crossing the Smad4 floxed mouse to the kidney specific promoter-driven Cre transgenic mouse in which Smad4 is deleted from most tubular epithelial cells upon Cre recombination [59]. We found that disruption of Smad4 from the kidney enhanced renal inflammation as evidenced by a greater CD45+ leukocyte and F4/80+ macrophage infiltration and up-regulation of IL-1β, TNF-α, MCP-1, and ICAM-1 in the obstructed kidney and in IL-1β-stimulated macrophages [59]. Further studies showed that the loss of Smad4 repressed Smad7 transcription, therefore leading to a loss of Smad7 functional protein. This, in turn, inhibited IκBα expression but enhanced NF-κB activation, thereby promoting renal inflammation. In contrast, deletion of Smad4 inhibited progressive renal fibrosis such as collagen matrix expression in the obstructive nephropathy and TGF-β1-induced collagen I expression by fibroblasts [59]. Interestingly, the mechanism of deletion of Smad4 to suppress renal fibrosis is not associated with inhibition of Smad2/3 activation because disruption of Smad4 does not alter phosphorylation levels of Smad2/3 nor phosphorylated Smad2/3 nuclear translocation. This is consistent with the previous finding in Smad4 null cancer cell lines [60]. However, deletion of Smad4 influences Smad3-mediated promoter activities and the binding of Smad3 to the COL1A2 promoter [59]. It has been reported that Smad3 binding sequences are located in the promoter regions of COL1A2, COL2A1, COL3A1, COL5A1, COL6A1, and COL6A3 [61-63], therefore, disruption of Smad4 may influence the binding activity of Smad3 to the collagen promoter, thereby inhibiting the fibrotic response.
Moreover, we also found that deletion of Smad4 impairs Smad7 transcriptional regulation in inhibition of NF-κB signaling [59]. It has been shown that Smad7 transcription is regulated by TGF-β1 through the direct binding of Smad3 and Smad4 to the Smad7 promoter [64, 65]. Thus, disrupted Smad4 results in a loss of Smad7 expression transcriptionally and impairs Smad7 promoter activities functionally [59]. Because Smad7 is capable of inducing IκBα expression, an inhibitor of NF-κB [17], disruption of Smad4 reduces renal Smad7, thereby promoting NF-κB-dependent renal inflammation and the inhibitory effect of TGF-β1 on interleukin-1β-induced inflammatory response in macrophages in vivo and in vitro [59]. Taken together, Smad4 may be a key regulator for the diverse roles of TGF-β1 in inflammation and fibrogenesis by interacting with Smad7 and Smad3 to influence their transcriptional activities in renal inflammation and fibrosis. (Figure 3).
Diverse roles of Smad4 in renal fibrosis and inflammation. Smad4 binds phosphorylated Smad2/Smad3 to form the Smad complex that translocates into the nucleus to regulate target genes related to fibrogenesis including Smad7. Upregulation of Smad7 prevents NF-κB/p50/p65 from phosphorylation and nuclear translocation by inducing IκBα expression. Therefore, Smad4 acts as a fine turner to promote Smad3-mediated fibrosis while inhibiting NF-κB-driven inflammation. Blue lines (symbols) indicate protective or negative regulation pathways, while red arrows (symbols) represent pathogenic or positive regulation pathways.
Inhibitory role of Smad7 in renal fibrosis and inflammation
Smad7 is an inhibitory Smad and negatively regulates Smad2 and Smad3 activation by its negative feed-back mechanism. Expression of Smad7 is induced by TGF-β1, which, in turn, exerts its negative feedback mechanism by causing degradation of TβRI and Smads [7, 8, 66, 67]. In chronic kidney diseases, TGF-β1 and angiotensin II are able to induce Smad7 mRNA expression and also activate the Smurfs and arkadia-dependent ubiquitin-proteasome pathways that, in turn, degrade Smad7 protein via a post-transcriptional modification mechanism [7, 8, 37, 68, 69]. Smurf1, Smurf2, and arkadia are E3 ubiquitin ligases for Smad7 [7, 8, 68] and have been shown to physically interact with Smad7 [69, 70]. As shown in Figure 4, Smad7 acts as an adaptor protein to recruit E3 ubiquitin ligases such as Smurf2 and arkadia to the TGF-β receptor complex to promote its degradation through proteasomal-ubiquitin degradation pathways [7, 8, 68-70]. Once Smad7 is degraded, activation of Smad2/3 and renal fibrosis is enhanced. This is clearly demonstrated by the recent finding that up-regulation of renal Smurf2 causes an ubiquitin-dependent degradation of renal Smad7, resulting in enhanced TGF-β/Smad signaling and progressive renal fibrosis [69]. Furthermore, Smurf2 can interact with Smad2 and selectively target Smad2 and Smad transcriptional corepressors Ski, SnoN, and TG-interacting factor (TGIF) for degradation in tubular epithelial cells, leading to progressive renal fibrosis and EMT in a mouse model of obstructive kidney disease [71-73]. Thus, ubiquitin-mediated degradation of Smad7 and Smad transcriptional corepressors Ski, SnoN, and TGIF promotes further activation of TGF-β signaling and progressive renal fibrosis as evidenced in a number of animal models [36, 37, 69, 72, 73]. This is further supported by the findings that Smad7 KO mice develop more severe renal fibrosis in both obstructive nephropathy and diabetic kidney disease [32, 36].
Inhibitory role of Smad7 in renal fibrosis and inflammation. Overexpression of Smad7 prevents Smad2/3 from phosphorylation by degrading the TβRI as well as Smads via the ubiquitin degradation pathway (Ub), thereby inhibiting Smad3-dependent renal fibrosis in response to TGF-β1, AGEs, and angiotensin II (Ang II). In addition, overexpression of Smad7 can induce IκBα, an inhibitor of KF-κB, therefore inhibiting NF-κB-driven renal inflammation. Thus, Smad7 acts as a therapeutic agent for treatment of kidney diseases. Blue lines (symbols) indicate protective or negative regulation pathways, while red arrows (symbols) represent pathogenic or positive regulation pathways.
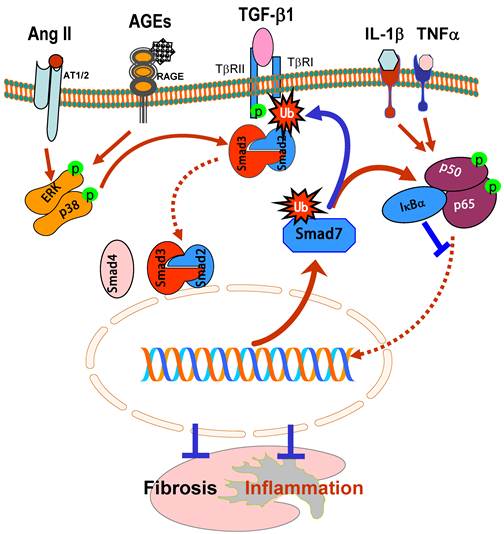
Loss of renal Smad7 not only enhances TGF-β/Smad3-mediated renal fibrosis, but also enhances renal inflammation by activating the NF-κB-dependent inflammatory pathway [17, 32, 36, 40]. It is well recognized that activation of NF-κB/p65 is associated with the renal inflammation in crescentic glomerulonephritis, diabetic nephropathy, and obstructive nephropathy [17, 32, 36, 40]. Overexpression of Smad7 substantially inhibits DNA binding activity, nuclear translocation, transcriptional activity of NF-κB/p65, as wells as NF-κB-dependent inflammatory responses induced by IL-1β and TNFα [17], implying a functional link between the Smad7 and NF-кB. As shown in Figure 4, Smad7 is able to induce IκBα expression, an inhibitor of NF-κB, suggesting that TGF-β may act by stimulating Smad7 to induce IκBα expression to suppress NF-κB activation [17].
The ability of overexpression of renal Smad7 to inhibit NF-κB activation and renal inflammation in remnant kidney disease [74], autoimmune crescentic glomerulonephritis [40], and diabetic nephropathy [32] confirms the potential role of Smad7-NF-κB crosstalk pathway in renal inflammation. Evidence for the anti-inflammatory role of Smad7 also comes from the findings that Smad7 KO mice develop more severe kidney inflammation in diabetic kidney disease and UUO models because these mice have significantly higher phosphorylation levels of NF-κB/p65 along with higher levels of renal inflammation including upregulation of IL-1β, TNFα, MCP-1, ICAM-1, and the development of more macrophage infiltration whine the kidney when compared to wild-type mice [32, 36]. In contrast, mice that transgenically express Smad7 on T cells are protected against crescentic glomerulonephritis [75]. Therefore, inhibition of the NF-κB signaling pathway by inducing IκBα expression may be the major mechanism by which Smad 7 inhibits renal inflammation (Figure 4).
The therapeutic potential for Smad7 on renal inflammation and fibrosis have been examined in rat models of obstructive nephropathy [34, 35], remnant kidney disease [38, 74], diabetic nephropathy [32], and in a mouse model of autoimmune crescentic glomerulonephritis [40]. In these studies, Smad7 was transferred into the kidney using the ultrasound-microbubble-mediated gene therapy technique. Overexpression of Smad7 not only inhibits Smad3-mediated renal fibrosis such as collagen matrix expression including epithelial-mesenchymal transition, but also blocks NF-κB-driven renal inflammation including accumulation of macrophages and T cells in both glomeruli and tubulointerstitium and upregulation of renal IL-1, TNFα, ICAM-1, and iNOS. Thus, restored the renal Smad7 rebalances the TGF-β/Smad and NF-κB signaling pathways and, therefore, inhibits progressive renal functional injury (Figure 4). All these studies demonstrate that Smad7 not only plays a negatively regulating role in renal fibrosis and inflammation, but also acts as a therapeutic agent and has therapeutic effect on renal fibrosis and inflammation [76].
Regulation of TGF-β/Smad3-dependent microRNAs in renal fibrosis
Recent studies in microRNAs have demonstrated that TGF-β regulates specific microRNAs to influence renal fibrosis in kidney diseases. Recent reports from our laboratory and others describe several microRNAs to be regulated by TGF-β1 during kidney diseases as detailed in a recent review article [77]. Indeed, TGF-β1 is able to up-regulate miR-21, miR-93, miR-192, miR-216a, miR-377, but down-regulates the miR-29 and miR-200 families [77]. We have recently identified that, among these TGF-β-dependent miRNAs, miR-21, miR-192, and the miR-29 family expression during renal fibrosis is tightly regulated by TGF-β1 via the Smad3, but not Smad2, dependent mechanism [78-80], which is also illustrated in Figure 5. Indeed, in vitro studies reveal that deletion of Smad3, not Smad2, inhibits expression of miR-21 and miR-192, but enhances the miR-29 family expression in response to TGF-β1 in both mouse embryonic fibroblasts and kidney tubular epithelial cells [78-80]. Evidence supporting the interaction of Smad3 with miR-21, miR-192, and miR-29 also came from the findings that there are conserved Smad3-binding sites in the promoter region of all three miRNAs and that Smad3 is able to interact with their individual promoter region as detected by the Chip assay [78-80]. Interestingly, expression of miRNAs can also interact with Smad3 to influence the Smad3 activity and functions. It is reported that overexpression of miR-200a could decrease Smad3 activity and attenuate TGF-β1-induced fibrosis [81]. However, although it remains undetermined if this interaction is direct or indirect, such an observation indicates the complexity between TGF-β/Smads and miRNAs under pathophysiological conditions.
Smad3-dependent miRNAs in renal fibrosis. TGF-β1 acts by stimulating Smad3 to positively regulate miR-21 and miR-192, but negatively regulate the miR-29 or miR-200 families, to mediate renal fibrosis. Blue lines (symbols) indicate protective or negative regulation pathways, while red arrows (symbols) represent pathogenic or positive regulation pathways.
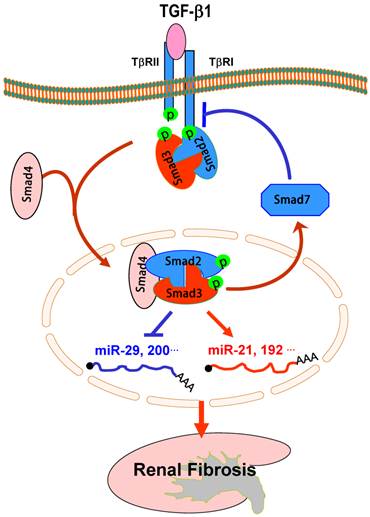
The regulating role of TGF-β/Smad3 in expression of miRNAs during renal fibrosis is also detected in an experimental mouse model of obstructive nephropathy by miRNA microarray and real-time PCR. Mice null for Smad3 are protected against renal fibrosis along with the inhibition of miR-21 and miR-192 expression [78, 79]. In contrast, while severe renal fibrosis in the obstructive nephropathy in Smad3 wild-type mice is associated with a loss of miR-29, prevention of renal fibrosis in Smad3 knockout mice is largely attributed to an increase in expression of renal miR-29 [80].
Smad7 also plays a role in regulating Smad-dependent miRNA expression in response to TGF-β1. For example, In vitro, overexpression of Smad7 in tubular epithelial cells abolished TGF-β1-induced miR-192 expression [78]. In vivo, deletion of Smad7 enhanced Smad3 signaling, thereby promoting miR-192 expression and fibrosis in obstructive kidney disease. In contrast, overexpression of Smad7 blocks TGF-β/Smad signaling and thus inhibits miR-192 expression and renal fibrosis in the rat 5/6 nephrectomy model.
The important role of TGF-β/Smad3-dependent miRNAs in renal fibrosis is demonstrated by the in vitro findings that overexpression of miR-21 and miR-192 enhances, but knockdown of miR-21 or miR-192 inhibits, collagen matrix expression in response to TGF-β1 [78, 79]. In contrast, knockdown of miR-29 enhances fibrosis, whereas overexpression of miR-29 blocks collagen I expression in response to TGF-β1 [80].
More excitingly, we recently demonstrated that the ultrasound-microbubble-mediated gene transfer technique can also be used to deliver the miRNAs into the kidney and have proved that the ultrasound-mediated miRNA therapy is a novel and effective therapeutic approach for kidney disease. Indeed, ultrasound-mediated overexpression of miR-29b or knockdown of miR-21before or after the established mouse model of obstructive nephropathy is capable of preventing or halting the progression of renal fibrosis [79, 80]. These findings suggest that specific targeting the Smad3-dependent microRNAs related to fibrogenesis such as miR-21 and miR-29 may represent a novel and specific anti-fibrosis therapy for renal fibrosis.
Concluding Remarks
The current advances in research into the TGF-β/Smad signaling pathway improve our understanding of the molecular mechanisms of renal fibrosis and inflammation in chronic kidney diseases. In general, Smad3 is a downstream key mediator of TGF-β/Smad signaling and plays a pathogenic role in both renal inflammation and fibrosis (Figure 1). Smad3 may also positively or negatively regulate its downstream specific miRNAs such as miR-21, mir-192, and miR-29 to mediate renal fibrosis (Figure 5). In contrast, Smad2 is renoprotective and suppresses renal fibrosis by competitively inhibiting Smad3 signaling including phosphorylation and nuclear translocation (Figure 2). Smad4 is the common Smad and plays a diverse role in promoting Smad3-mediated renal fibrosis but suppressing NF-κB-driven renal inflammation by stimulating Smad7 expression transcriptionally (Figure 3). Most importantly, Smad7 is a negative regulator of both renal inflammation and fibrosis. Smad7 inhibits TGF-β/Smad signaling by recruiting the E3 ligases such as Smurf2 and arkadia to the TGF-β receptor complex or Smads to promote their degradation through the proteasomal-ubiquitin degradation pathway. In addition, Smad7 can induce IκBα expression, thereby preventing NF-κB subunits from phosphorylation. However, Smad7 is lost in the diseased kidney, which causes an imbalance within and between the TGF-β/Smad and NF-κB signaling pathways, resulting in the development of renal fibrosis and inflammation. Therefore, restored renal Smad7 rebalances both Smad and NF-κB signaling pathways and inhibits renal fibrosis and inflammation (Figure 4). Thus, understanding of the specific role of individual Smads and the crosstalk pathways is the first step towards the development of novel and effective therapeutic strategies for kidney diseases.
Acknowledgements
This work is supported by grants from the Research Grant Council of Hong Kong (RGC GRF 767508 and 469110, and CUHK5/CRF/09) and Focused Investment Scheme B grant from The Chinese University of Hong Kong (1902061).
Conflict of Interests
The author has declared that no conflict of interest exists.
References
1. Eddy AA, Neilson EG. Chronic kidney disease progression. J Am Soc Nephrol. 2006;17:2964-66
2. Bottinger EP. TGF-beta in renal injury and disease. Semin Nephrol. 2007;27:309-20
3. Wang W, Koka V, Lan HY. Transforming growth factor-beta and Smad signalling in kidney diseases. Nephrology (Carlton). 2005Feb;10(1):48-56
4. Roberts AB. Molecular and cell biology of TGF-beta. Miner Electrolyte Metab. 1998;24:111-9
5. Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGFbeta family signalling. Nature. 2003;425:577-84
6. Schnaper HW, Hayashida T, Poncelet AC. It's a Smad world: regulation of TGF-beta signaling in the kidney. J Am Soc Nephrol. 2002;13:1126-8
7. Kavsak P, Rasmussen RK, Causing CG, Bonni S, Zhu H, Thomsen GH, Wrana JL. Smad7 binds to Smurf2 to form an E3 ubiquitin ligase that targets the TGF beta receptor for degradation. Mol Cell. 2000;6:1365-75
8. Ebisawa T, Fukuchi M, Murakami G, Chiba T, Tanaka K, Imamura T, Miyazono K. Smurf1 interacts with transforming growth factor-beta type I receptor through Smad7 and induces receptor degradation. J Biol Chem. 2001;276:12477-80
9. Lan HY. Tubular epithelial-myofibroblast transdifferentiation mechanisms in proximal tubule cells. Curr Opin Nephrol Hypertens. 2003;12:25-9
10. Border WA, Noble NA. Evidence that TGF-beta should be a therapeutic target in diabetic nephropathy. Kidney Int. 1998;54:1390-1
11. Kopp JB, Factor VM, Mozes M, Nagy P, Sanderson N, Bottinger EP, Klotman PE, Thorgeirsson SS. Transgenic mice with increased plasma levels of TGF-beta 1 develop progressive renal disease. Lab Invest. 1996;74:991-1003
12. Sanderson N, Factor V, Nagy P, Kopp J, Kondaiah P, Wakefield L, Roberts AB, Sporn MB, Thorgeirsson SS. Hepatic expression of mature transforming growth factor beta 1 in transgenic mice results in multiple tissue lesions. Proc Natl Acad Sci U S A. 1995;92:2572-6
13. Letterio JJ, Roberts AB. Regulation of immune responses by TGF-beta. Annu Rev Immunol. 1998;16:137-61
14. Yaswen L, Kulkarni AB, Fredrickson T, Mittleman B, Schiffman R, Payne S, Longenecker G, Mozes E, Karlsson S. Autoimmune manifestations in the transforming growth factor-beta 1 knockout mouse. Blood. 1996;87:1439-45
15. Li MO, Wan YY, Flavell RA. T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity. 2007;26:579-91
16. Li MO, Sanjabi S, Flavell RA. Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity. 2006;25:455-71
17. Wang W, Huang XR, Li AG, Liu F, Li JH, Truong LD, Wang XJ, Lan HY. Signaling mechanism of TGF-beta1 in prevention of renal inflammation: role of Smad7. J Am Soc Nephrol. 2005;16:1371-83
18. Huang XR, Chung AC, Wang XJ, Lai KN, Lan HY. Mice overexpressing latent TGF-beta1 are protected against renal fibrosis in obstructive kidney disease. Am J Physiol Renal Physiol. 2008;295:F118-27
19. Huang XR, Chung AC, Zhou L, Wang XJ, Lan HY. Latent TGF-beta1 protects against crescentic glomerulonephritis. J Am Soc Nephrol. 2008;19:233-42
20. Stockis J, Colau D, Coulie PG, Lucas S. Membrane protein GARP is a receptor for latent TGF-beta on the surface of activated human Treg. Eur J Immunol. 2009;39:3315-22
21. Wang R, Kozhaya L, Mercer F, Khaitan A, Fujii H, Unutmaz D. Expression of GARP selectively identifies activated human FOXP3+ regulatory T cells. Proc Natl Acad Sci U S A. 2009;106:13439-44
22. Lan HY, Chung AC. Transforming growth factor-β and Smads. Contrib Nephrol. 2011;170:75-82
23. Li JH, Huang XR, Zhu HJ, Oldfield M, Cooper M, Truong LD, Johnson RJ, Lan HY. Advanced glycation end products activate Smad signaling via TGF-beta-dependent and independent mechanisms: implications for diabetic renal and vascular disease. Faseb J. 2004;18:176-8
24. Chung AC, Zhang H, Kong YZ, Tan JJ, Huang XR, Kopp JB, Lan HY. Advanced glycation end-products induce tubular CTGF via TGF-beta-independent Smad3 signaling. J Am Soc Nephrol. 2010;21:249-60
25. Wang W, Huang XR, Canlas E, Oka K, Truong LD, Deng C, Bhowmick NA, Ju W, Bottinger EP, Lan HY. Essential role of Smad3 in angiotensin II-induced vascular fibrosis. Circ Res. 2006;98:1032-9
26. Yang F, Chung AC, Huang XR, Lan HY. Angiotensin II induces connective tissue growth factor and collagen I expression via transforming growth factor-beta-dependent and -independent Smad pathways: the role of Smad3. Hypertension. 2009;54:877-84
27. Wrana JL. Regulation of Smad activity. Cell. 2000;100:189-192
28. Zeisberg M, Hanai J, Sugimoto H, Mammoto T, Charytan D, Strutz F, Kalluri R. BMP-7 counteracts TGF-beta1-induced epithelial-to-mesenchymal transition and reverses chronic renal injury. Nat Med. 2003;9(7):964-8
29. Dudas PL, Argentieri RL, Farrell FX. BMP-7 fails to attenuate TGF-beta1-induced epithelial-to-mesenchymal transition in human proximal tubule epithelial cells. Nephrol Dial Transplant. 2009;24(5):1406-16
30. Fujimoto M, Maezawa Y, Yokote K, Joh K, Kobayashi K, Kawamura H, Nishimura M, Roberts AB, Saito Y, Mori S. Mice lacking Smad3 are protected against streptozotocin-induced diabetic glomerulopathy. Biochem Biophys Res Commun. 2003;305:1002-7
31. Isono M, Chen S, Hong SW, Iglesias-de la Cruz MC, Ziyadeh FN. Smad pathway is activated in the diabetic mouse kidney and Smad3 mediates TGF-beta-induced fibronectin in mesangial cells. Biochem Biophys Res Commun. 2002;296:1356-65
32. Chen H, Huang XR, Wang W, Li J, Heuchel RL, Chung AC, Lan HY. The protective role of Smad7 in diabetic kidney disease: Mechanism and therapeutic potential. Diabetes. 2010;60:590-601
33. Sato M, Muragaki Y, Saika S, Roberts AB, Ooshima A. Targeted disruption of TGF-beta1/Smad3 signaling protects against renal tubulointerstitial fibrosis induced by unilateral ureteral obstruction. J Clin Invest. 2003;112:1486-94
34. Lan HY, Mu W, Tomita N, Huang XR, Li JH, Zhu HJ, Morishita R, Johnson RJ. Inhibition of renal fibrosis by gene transfer of inducible Smad7 using ultrasound-microbubble system in rat UUO model. J Am Soc Nephrol. 2003;14:1535-48
35. Terada Y, Hanada S, Nakao A, Kuwahara M, Sasaki S, Marumo F. Gene transfer of Smad7 using electroporation of adenovirus prevents renal fibrosis in post-obstructed kidney. Kidney Int. 2002;61:94-8
36. Chung AC, Huang XR, Zhou L, Heuchel R, Lai KN, Lan HY. Disruption of the Smad7 gene promotes renal fibrosis and inflammation in unilateral ureteral obstruction (UUO) in mice. Nephrol Dial Transplant. 2009;24:1443-54
37. Yang F, Huang XR, Chung AC, Hou CC, Lai KN, Lan HY. Essential role for Smad3 in angiotensin II-induced tubular epithelial-mesenchymal transition. J Pathol. 2010;221:390-401
38. Hou CC, Wang W, Huang XR, Fu P, Chen TH, Sheikh-Hamad D, Lan HY. Ultrasound-microbubble-mediated gene transfer of inducible Smad7 blocks transforming growth factor-beta signaling and fibrosis in rat remnant kidney. Am J Pathol. 2005;166:761-71
39. Zhou L, Fu P, Huang XR, Liu F, Chung AC, Lai KN, Lan HY. Mechanism of chronic aristolochic acid nephropathy: role of Smad3. Am J Physiol Renal Physiol. 2010;298:F1006-17
40. Ka SM, Huang XR, Lan HY, Tsai PY, Yang SM, Shui HA, Chen A. Smad7 gene therapy ameliorates an autoimmune crescentic glomerulonephritis in mice. J Am Soc Nephrol. 2007;18:1777-88
41. Verrecchia F, Chu ML, Mauviel A. Identification of novel TGF-beta /Smad gene targets in dermal fibroblasts using a combined cDNA microarray/promoter transactivation approach. J Biol Chem. 2001;276:17058-62
42. Li J, Qu X, Yao J, Caruana G, Ricardo SD, Yamamoto Y, Yamamoto H, Bertram JF. Blockade of endothelial-mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes. 2010;59:2612-24
43. Ju W, Ogawa A, Heyer J, Nierhof D, Yu L, Kucherlapati R, Shafritz DA, Bottinger EP. Deletion of Smad2 in mouse liver reveals novel functions in hepatocyte growth and differentiation. Mol Cell Biol. 2006;26:654-67
44. Shao X, Somlo S, Igarashi P. Epithelial-specific Cre/lox recombination in the developing kidney and genitourinary tract. J Am Soc Nephrol. 2002;13:1837-46
45. Meng XM, Huang XR, Chung AC, Qin W, Shao X, Igarashi P, Ju W, Bottinger EP, Lan HY. Smad2 Protects against TGF-{beta}/Smad3-Mediated Renal Fibrosis. J Am Soc Nephrol. 2010;21:1477-87
46. Zhou Y, Mao H, Li S, Cao S, Li Z, Zhuang S, Fan J, Dong X, Borkan SC, Wang Y, Yu X. HSP72 inhibits Smad3 activation and nuclear translocation in renal epithelial-to-mesenchymal transition. J Am Soc Nephrol. 2010;21:598-609
47. Ng YY, Chen YM, Tsai TJ, Lan XR, Yang WC, Lan HY. Pentoxifylline inhibits transforming growth factor-β signaling and renal fibrosis in experimental crescentic glomerulonephritis in rats. Am J Nephrol. 2009;29:43-53
48. Saika S, Kono-Saika S, Ohnishi Y, Sato M, Muragaki Y, Ooshima A, Flanders KC, Yoo J, Anzano M, Liu CY, Kao WW, Roberts AB. Smad3 signaling is required for epithelial-mesenchymal transition of lens epithelium after injury. Am J Pathol. 2004;164:651-663
49. Roberts AB, Tian F, Byfield SD, Stuelten C, Ooshima A, Saika S, Flanders KC. Smad3 is key to TGFβ-mediated epithelial-to-mesenchymal transition, fibrosis, tumor suppression and metastasis. Cytokine Growth Factor Rev. 2006;17:19-27
50. Phan TT, Lim IJ, Aalami O, Lorget F, Khoo A, Tan EK, Mukhopadhyay A, Longaker MT. Smad3 signalling plays an important role in keloid pathogenesis via epithelial-mesenchymal interactions. J Pathol. 2005;207:232-42
51. Dennler S, Itoh S, Vivien D ten Dijke P, Huet S, Gauthier JM. Direct binding of Smad3 and Smad4 to critical TGF beta-inducible elements in the promoter of human plasminogen activator inhibitor-type 1 gene. Embo J. 1998;17:3091-100
52. Piek E, Ju WJ, Heyer J, Escalante-Alcalde D, Stewart CL, Weinstein M, Deng C, Kucherlapati R, Bottinger EP, Roberts AB. Functional characterization of transforming growth factor-β signaling in Smad2- and Smad3-deficient fibroblasts. J Biol Chem. 2001;276:19945-53
53. Dennler S, Huet S, Gauthier JM. A short amino acid sequence in MH1 domain is responsible for functional differences between Smad2 and Smad3. Oncogene. 1999;18:1643-48
54. Kang DH, Joly AH, Oh SW, Hugo C, Kerjaschki D, Gordon KL, Mazzali M, Jefferson JA, Hughes J, Madsen KM, Schreiner GF, Johnson RJ. Impaired angiogenesis in the remnant kidney model: I. Potential role of vascular endothelial growth factor and thrombospondin-1. J Am Soc Nephrol. 2001;12:1434-47
55. Nakagawa T, Kosugi T, Haneda M, Rivard CJ, Long DA. Abnormal angiogenesis in diabetic nephropathy. Diabetes. 2009;58:1471-8
56. Nakagawa T, Li JH, Garcia G, Mu W, Piek E, Böttinger EP, Chen Y, Zhu HJ, Kang DH, Schreiner GF, Lan HY, Johnson RJ. TGF-beta induces proangiogenic and antiangiogenic factors via parallel but distinct Smad pathways. Kidney Int. 2004;66:605-13
57. Lagna G, Hata A, Hemmati-Brivanlou A, Massagué J. Partnership between DPC4 and SMAD proteins in TGF-β signalling pathways. Nature. 1996;383:832-6
58. Yang X, Li C, Herrera PL, Deng CX. Generation of Smad4/Dpc4 conditional knockout mice. Genesis. 2002;32:80-81
59. Meng XM, Huang XR, Xiao J, Chung ACK, Qin W, Chen HY, Lan HY. Disrupted Smad4 impairs TGF-β/Smad3 and Smad7 transcriptional regulation in renal inflammation and fibrosis in vivo and in vitro. Kidney Int. in press
60. Fink SP, Mikkola D, Willson JK, Markowitz S. TGF-beta-induced nuclear localization of Smad2 and Smad3 in Smad4 null cancer cell lines. Oncogene. 2003;22:1317-23
61. Verrecchia F, Vindevoghel L, Lechleider RJ, Uitto J, Roberts AB, Mauviel A. Smad3/AP-1 interactions control transcriptional responses to TGF-beta in a promoter-specific manner. Oncogene. 2001;20:3332-40
62. Vindevoghel L, Lechleider RJ, Kon A, de Caestecker MP, Uitto J, Roberts AB, Mauviel A. SMAD3/4-dependent transcriptional activation of the human type VII collagen gene (COL7A1) promoter by transforming growth factor beta. Proc Natl Acad Sci U S A. 1998;95:14769-14774
63. Chen SJ, Yuan W, Mori Y, Levenson A, Trojanowska M, Varga J. Stimulation of type I collagen transcription in human skin fibroblasts by TGF-beta: involvement of Smad 3. J Invest Dermatol. 1999;112:49-57
64. Nagarajan RP, Zhang J, Li W, Chen Y. Regulation of Smad7 promoter by direct association with Smad3 and Smad4. J Biol Chem. 1999;274:33412-18
65. von Gersdorff G, Susztak K, Rezvani F, Bitzer M, Liang D, Böttinger EP. Smad3 and Smad4 mediate transcriptional activation of the human Smad7 promoter by transforming growth factor beta. J Biol Chem. 2000;275:11320-26
66. Afrakhte M, Moren A, Jossan S, Itoh S, Sampath K, Westermark B, Heldin CH, Heldin NE, ten Dijke P. Induction of inhibitory Smad6 and Smad7 mRNA by TGF-beta family members. Biochem Biophys Res Commun. 1998;249:505-11
67. Zhu HJ, Iaria J, Sizeland AM. Smad7 differentially regulates transforming growth factor beta-mediated signaling pathways. J Biol Chem. 1999;274:32258-64
68. Liu FY, Li XZ, Peng YM, Liu H, Liu YH. Arkadia regulates TGFβ signaling during renal tubular epithelial to mesenchymal cell transition. Kidney Int. 2008;73:588-94
69. Fukasawa H, Yamamoto T, Togawa A, Ohashi N, Fujigaki Y, Oda T, Uchida C, Kitagawa K, Hattori T, Suzuki S, Kitagawa M, Hishida A. Down-regulation of Smad7 expression by ubiquitin-dependent degradation contributes to renal fibrosis in obstructive nephropathy in mice. Proc Natl Acad Sci USA. 2004;101:8687-92
70. Inoue Y, Imamura T. Regulation of TGF-beta family signaling by E3 ubiquitin ligases. Cancer Sci. 2008;99:2107-12
71. Lin X, Liang M, Feng XH. Smurf2 is a ubiquitin E3 ligase mediating proteasome-dependent degradation of Smad2 in transforming growth factor-beta signaling. J Biol Chem. 2000;275:36818-36822
72. Yang J, Zhang X, Li Y, Liu Y. Downregulation of Smad transcriptional corepressors SnoN and Ski in the fibrotic kidney: an amplification mechanism for TGF-beta1 signaling. J Am Soc Nephrol. 2003;14:3167-77
73. Tan R, He W, Lin X, Kiss LP, Liu Y. Smad ubiquitination regulatory factor-2 in the fibrotic kidney: regulation, target specificity, and functional implication. Am J Physiol Renal Physiol. 2008;294:F1076-83
74. Ng YY, Hou CC, Wang W, Huang XR, Lan HY. Blockade of NFkappaB activation and renal inflammation by ultrasound-mediated gene transfer of Smad7 in rat remnant kidney. Kidney Int. 2005;94(Suppl):S83-91
75. Kanamaru Y, Nakao A, Mamura M, Suzuki Y, Shirato I, Okumura K, Tomino Y, Ra C. Blockade of TGF-beta signaling in T cells prevents the development of experimental glomerulonephritis. J Immunol. 2001;166:2818-23
76. Lan HY. Smad7 as a therapeutic agent for chronic kidney diseases. Front Biosci. 2008;13:4984-92
77. Kantharidis P, Wang B, Carew RM, Lan HY. Diabetes Complications: The MicroRNA Perspective. Diabetes. 2011;60:1832-7
78. Chung AC, Huang XR, Meng X, Lan HY. miR-192 mediates TGF-beta/Smad3-driven renal fibrosis. J Am Soc Nephrol. 2010;21:1317-25
79. Zhong X, Chung AC, Chen HY, Meng X, Lan HY. Smad3 mediated upregulation of miR-21 promotes renal fibrosis. J Am Soc Nephrol. 2011;22:1668-81
80. Qin W, Chung AC, Huang XR, Meng XM, Hui DS, Yu CM, Sung JJ, Lan HY. TGF-{beta}/Smad3 Signaling Promotes Renal Fibrosis by Inhibiting miR-29. J Am Soc Nephrol. 2011;22:1462-74
81. Wang B, Koh P, Winbanks C, Coughlan MT, McClelland A, Watson A, Jandeleit-Dahm K, Burns WC, Thomas MC, Cooper ME, Kantharidis P. miR-200a Prevents renal fibrogenesis through repression of TGF-b2 expression. Diabetes. 2011;60:280-87
Author contact
![]() Corresponding author: Hui Y. Lan, M.D., Ph.D., Li Ka Shing Institute of Health Sciences and Department of Medicine & Therapeutics, the Chinese University of Hong Kong, Shatin, New Territories, Hong Kong, China. Tel: 852-3763 6077; Fax: 852-2145 7190; email: hylanedu.hk
Corresponding author: Hui Y. Lan, M.D., Ph.D., Li Ka Shing Institute of Health Sciences and Department of Medicine & Therapeutics, the Chinese University of Hong Kong, Shatin, New Territories, Hong Kong, China. Tel: 852-3763 6077; Fax: 852-2145 7190; email: hylanedu.hk