Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(4):498-511. doi:10.7150/ijbs.3723 This issue Cite
Research Paper
Characterization of Bovine Induced Pluripotent Stem Cells by Lentiviral Transduction of Reprogramming Factor Fusion Proteins
Hongguo Cao ![]() , Pan Yang, Yong Pu, Xueping Sun, Huiqun Yin, Yu Zhang, Yunhai Zhang, Yunsheng Li, Ya Liu, Fugui Fang, Zijun Zhang, Yong Tao, Xiaorong Zhang
, Pan Yang, Yong Pu, Xueping Sun, Huiqun Yin, Yu Zhang, Yunhai Zhang, Yunsheng Li, Ya Liu, Fugui Fang, Zijun Zhang, Yong Tao, Xiaorong Zhang ![]()
Anhui Local Livestock Genetic Resources Conservation and Breeding Laboratory, College of Animal Science and Technology, Anhui Agricultural University, Hefei 230036, China
Received 2011-10-28; Accepted 2012-3-12; Published 2012-3-21
Abstract
Pluripotent stem cells from domesticated animals have potential applications in transgenic breeding. Here, we describe induced pluripotent stem (iPS) cells derived from bovine fetal fibroblasts by lentiviral transduction of Oct4, Sox2, Klf4 and c-Myc defined-factor fusion proteins. Bovine iPS cells showed typical colony morphology, normal karyotypes, stained positively for alkaline phosphatase (AP) and expressed Oct4, Nanog and SSEA1. The CpG in the promoter regions of Oct4 and Nanog were highly unmethylated in bovine iPS cells compared to the fibroblasts. The cells were able to differentiate into cell types of all three germ layers in vitro and in vivo. In addition, these cells were induced into female germ cells under defined culture conditions and expressed early and late female germ cell-specific genes Vasa, Dazl, Gdf9, Nobox, Zp2, and Zp3. Our data suggest that bovine iPS cells were generated from bovine fetal fibroblasts with defined-factor fusion proteins mediated by lentivirus and have potential applications in bovine transgenic breeding and gene-modified animals.
Keywords: bovine, fetal fibroblasts, iPS cells, defined-factor fusion proteins
Introduction
As a new type of pluripotent stem cells, reprogrammed by ectopic expression of defined factors Oct4, Sox2, Klf4 and c-Myc from differentiated somatic cells (1), iPS cells have unlimited self-renewal capacity, maintain pluripotency, and are very similar to embryonic stem (ES) cells in morphology, proliferation, pluripotent gene expression, promoter methylation, teratoma formation and the capacity to differentiate into all of the cell types in vivo or in vitro under appropriate induction conditions (1-5). Similar to ES cells, mouse iPS cells possess the ability to form germline chimera upon injection into early blastocysts (6) and can produce viable offspring using a tetraploid complementation strategy (7, 8). Considering the abundant resources, identical self-renewal and unlimited proliferation abilities, iPS cells are regarded as a potential source that can be easily genetically manipulated to produce transgenic, chimeric and knock-out domestic animals. iPS cells may provide an invaluable tool not only for research aimed at drug development and regenerative medicine in human beings but also for transgenic breeding and genetically modified domestic animals. Additionally, mouse ES cells can efficiently produce both presumptive oocytes and sperm cells under defined culture conditions (9-11).
Cattle are the most common type of large domesticated ungulates and are raised as livestock for meat, as dairy animals for milk and other products. Bovine iPS cells are artificially derived from adult somatic cell without the use of rare and excellent embryos, so bovine iPS cells and their derivatives especially germ cells will enable the precise genetic engineering of livestock for improved production traits, are also powerful reproductive tools and could significantly speed up the breeding process. In the current study, we report the characterization of bovine iPS cells by lentiviral transduction of Oct4, Sox2, Klf4 and c-Myc reprogramming factor fusion proteins with enhanced green fluorescent protein (EGFP) and demonstrate the differentiation capacity of bovine iPS cells, including oocytes, in vitro under defined induction conditions.
Materials and Methods
Cell culture and lentivirus infection
Bovine primary fibroblasts from the skin of individual 2.5 and 4-month-old Holstein breed fetuses were established by culturing tissue explants in high glucose DMEM medium (Hyclone) with 10% fetal bovine serum (FBS, Hyclone) and were expanded for several passages before virus transduction. Lentiviral expression vectors (pLentilox 3.7) for human Oct4, porcine Sox2, c-Myc and Klf4 fused with EGFP were constructed as described in ref (12). Lentiviruses were packaged and produced in 293T cells, collected, filtered, concentrated by ultrafiltration and added onto bovine fibroblasts (1.0 × 104 cells/cm2) with high glucose DMEM containing 10% FBS, 10 μg/ml polybrene (Sigma). After 2 days, the cells were cultured with stem cell medium consisting of 1000 U/ml LIF (Chemicon), 4 ng/ml bFGF (Sigma), 15% FBS and high glucose DMEM. After the cells formed colonies, the colonies were relocated from culture dishes using 1 mg/ml dispase (Sigma) and seeded onto mouse embryonic fibroblasts (MEFs) for subsequent culture.
Teratoma formation and in vitro differentiation
After digesting from culture dishes with trypsin solution, cells were pelleted, resuspended at 1×107 cells/ml in serum-free DMEM, and 200 μl was injected subcutaneously into the dorsal flank of 6-week-old severe combined immunodeficient BALB/C nude mice. At 9 weeks after injection, tumor tissues were generated and fixed in 4% paraformaldehyde and stained with hematoxylin/eosin.
In vitro differentiation of cells was performed via spontaneous differentiation of embryoid body (EB) formation. The cells were detached from culture dishes using 1 mg/ml dispase, collected after centrifugation, resuspended in culture medium without LIF and bFGF, and seeded in low-adhesive dishes (Qingdao Alpha). After 7 days in suspension culture, EBs were transferred to gelatin-coated dishes and cultured for another 7 days.
For the derivation of female gametes, cells were cultured and passaged without feeders after digestion with trypsin solution. After 4 days, cells were resuspended in culture medium without LIF and bFGF and formed EBs by hanging drop culture. The resulting EBs were transferred to low-adhesive dishes. After 4 days in suspension culture, EBs were transferred to gelatin-coated dishes and cultured in DMEM/F12 medium containing 10% FBS, 10% porcine follicular fluid and 0.5 μM retinoic acid (RA) for another 4 days.
Karyotype analysis and AP staining
For karyotyping, cells were treated for 2.5 hours with 0.1 μg/ml colchicines (Sigma), digested with trypsin solution, resuspended in 0.075 mol/L KCl and incubated at 37°C for 25 min before fixation with methanol and acetic acid (3:1) for 10 min. After centrifugation, cells were resuspended, spread on slides, stained with giemsa dye, air-dried, mounted and examined.
Cellular AP activity was displayed with AP staining solution after fixation with 4% paraformaldehyde using standard protocols. Cells exhibiting brown stain in cytoplasm were identified as positive.
RT-PCR and quantitative real-time PCR
Purified RNA samples were isolated from the different cells and treated with an RNase-free DNase kit to effectively remove contaminating genomic DNA. After genomic DNA elimination, the RNA samples were ready for reverse transcription to synthesize first-strand DNA according to the manufacturer's instructions (Qiagen). The primers used are described in Table 1. PCR products were examined by 1% agarose gel electrophoresis.
The primers used for RT-PCR
| Genes | Sequences of primers | Productions (bp) | Tm (°C ) |
|---|---|---|---|
| ExoOct4 | F: 5′-GCTCTCCCATGCATTCAAAC-3′ R: 5′- TCGTCCTTGAAGAAGATGGTG -3′ | 350 | 55 |
| EndoOct4 | F: 5′- CAGACACCACCGCCACCAGC -3′ R: 5′- TCCCTCCACACAAGTCATAG -3 | 416 | 55 |
| ExoKlf4 | F: 5′- TCCAGTGCCAGAAGTGCGAC -3′ R: 5′- TCGTCCTTGAAGAAGATGGTG -3′ | 380 | 55 |
| EndoKlf4 | F: 5′-ACTGTCATCCTGCCCTGCC -3′ R: 5′-CCTTGTGTCTCCTGATTATC -3′ | 412 | 55 |
| Nanog | F: 5′- ACCAGA GAATGAAATGTAAG -3′ R: 5′- ATTTTCCCCAGCAGTTCC -3′ | 382 | 55 |
| GDF9 | F: 5′- TTTGCCTGGCTCTGTTT-3′ R: 5′- GTGGCTTCTGTTGGATTTA -3′ | 288 | 53 |
| ZP2 | F: 5′- AGAATGACGGTGAGGTGC -3′ R: 5′- AAGGTGGTTCTGTGGTTGTC -3′ | 353 | 53 |
| ZP3 | F: 5′- CTTCAGCAAGTCCTCCAACA-3′ R: 5′- CAGCCAGAGTCAAGGTCATC-3′ | 293 | 57 |
| VASA | F: 5′- GGTAGTTTCCGAGGTTGC -3′ R: 5′- ATGCCTGTTTGATAATGTGC-3′ | 347 | 52 |
| DAZL | F: 5′- TCCTCCACCACAATTTCA-3′ R: 5′- CACCGTCTGTATGCTTCT-3′ | 364 | 50 |
| GAPDH | F: 5′- TTGGTATCGTGGAAGGACTCTA -3′ R: 5′- TGTCATATTTGGCAGGTT -3′ | 270 | 55 |
Real-time PCR was performed in an ABI Step-One Plus real-time PCR system using a Power SYBR Green PCR Master mix (ABI 4367659) using the standard curve method with β-actin as the internal control. The primers used to amplify EndoOct4, Nanog andβ-actin (13) are described in Table 2. Amplification was performed in 20 μl reaction volume according to the manufacturer's instructions.
The primers used for quantitative real-time RT-PCR△ and bisulphite genomic sequencing◇
| Genes | Sequences of primers | Productions(bp) | Tm (°C ) |
|---|---|---|---|
| △EndoOct4 | F: 5′- GAACCCTGAGGAGTCCCAGGACATCA -3′ R: 5′- ACACCGGCTACTCTTCCCAGAGAA -3′ | 306 | 60 |
| △Nanog | F: 5′- CGACACGGACACTGTCTCTCCTCTTC -3′ R: 5′- ACCAGGTCTTCACCTGCTTGTAGCT -3′ | 287 | 60 |
| △β-Actin | F: 5′- GAGCGGGAAATCGTCCGTGAC -3′ R:5′- GTGTTGGCGTAGAGGTCCTTGC -3′ | 278 | 60 |
| ◇Oct4 | F:5′- GTTTGGAGAGGGGTTTTGAAGAATGTGTAG -3′ R:5′- ATCCCACCCACTAACCTTAACCTCTAAC-3′ | 260 | 60 |
| ◇Nanog | F:5′- TAGGTGGTTATAGGAGATGTATTTTTGATT - 3′ R:5′- ACCTATAAAATAAAAACCATCCAATCCAAT -3′ | 260 | 60 |
Bisulphite genomic sequencing
Genomic DNA was prepared from cell colonies using a DNA isolation kit (Takara) according to the manufacturer's instructions. The isolated DNA was treated with sodium bisulfite using a MethylampTM DNA modification kit (Epigentek) according to the manufacturer's instructions. The primers used to amplify Oct4 and Nanog are described in Table 2. Amplified fragments were cloned into the T-vector and at least 10 randomly selected positive clones were chosen and sequenced for each sample.
Flow cytometry
Cells were washed with PBS and digested from culture dishes with trypsin solution and then incubated with a 1:200 dilution of mouse-anti SSEA1 antibody (Chemicon, MAB4301) for 60 minutes on ice. Cells were washed with PBS prior to staining with fluorescein isothiocyanate (FITC)-conjugated donkey anti-mouse IgM secondary antibody (Jackson) for 60 min in a 1:200 dilution on ice and washed again with PBS prior to flow cytometry analysis. Cells were analyzed using BD FACS Calibur flow cytometer and cell quest software. Next, cells were probed with an appropriate isotype antibody as a negative control. All treatments were performed in duplicate.
Fluorescent immunochemistry and western blotting
For immunocytochemistry, cells were fixed with 4% paraformaldehyde for 15 min, permeablized with 0.5% Triton X-100 in PBS for 10 min, and blocked with 1% BSA in PBS for 30 min. The primary antibodies anti-Oct4 in rabbit (Abcam, ab19875), anti-Nanog in goat (R&D, BAF1997), anti-SSEA1 in mouse (Chemicon, MAB4301), anti-SSEA3 in mouse (Chemicon, MAB4303), anti-SSEA4 in mouse (Chemicon, MAB4304), anti-TRA-1-60 (Chemicon, MAB4360) and TRA-1-80 in mouse (Chemicon, MAB4381), anti-α-Actinin (Sarcomeric) in mouse (Sigma, A7811), Anti-α-Fetoprotein (AFP) in mouse (Sigma, A8452), Anti-Neurofilament 200 in rabbit (Sigma, N4142), anti-Nobox (Abcam, ab41521) in rabbit and anti-Vasa (Abcam, ab13840) in rabbit were diluted 1:200, added onto cells and incubated at 4°C overnight. Cells were washed with PBS, incubated with CY3-labeled secondary antibody (1:200 dilution) at room temperature for 1 h and then washed with PBS. The cells were stained with DAPI (Roche) and then examined.
Cells were washed with PBS, collected in sample buffer, and then heated to 100℃ for 10 min. After centrifuging at 12 000 rpm for 4 min, whole-cell lysates were separated using SDS-PAGE and then transferred to polyvinylidene difluoride (PVDF) membranes. The membranes were blocked for 1 h at room temperature in TBST buffer (10 mM Tris, 100 mM NaCl, 0.2% Tween-20, pH 7.4) containing 5% low-fat milk, incubated in fresh buffer containing primary antibodies anti-Oct4 in rabbit (Abcam, ab19875) and anti-β-actin in rabbit and then diluted 1: 1000 for 1 h at room temperature. After five 5-min washes in TBST, the membranes were incubated with AP-conjugated anti- rabbit (1:1000) for 1 hour at room temperature. Immunoreactive proteins were displayed with AP staining solution.
Results
Derivation of bovine iPS cells
After infection with lentiviruses 2-4 times, a small portion of bovine fibroblasts gradually displayed sphere morphology, proliferated and formed cell colonies that consist of two cell types: one with EGFP in the nucleus and the other without EGFP expression 2 days after the 4th lentivirus infection. The spherical cells were able to form colonies with clear boundaries, displayed morphological uniformity, grew rapidly after digestion with dispase, and passaged at a 1:4 ratio every 2-3 days (Fig.1A, B, C, D). In these experiments, under the culture conditions of medium containing 1000 U/ml LIF, 4 ng/ml bFGF and feeders, the bovine iPS cells morphologically resembled human ES cells rather than mouse ES cells. After the 4th lentivirus infection, 45-150 AP+ cell colonies from 2.15×105 bovine fetal fibroblasts were obtained, and the induction efficiency was approximately 0.0002-0.0007%. However, after only 1 round of lentivirus infection, 5-10 AP+ cell colonies from 2.15×105 bovine fetal fibroblasts were obtained. Repeated lentivirus infections could clearly improve the induction efficiency and shorten the induction period. As the number of passages increased, the proportion of cells expressing EGFP in the nucleus decreased and the fluorescence intensity in the nucleus of cells attenuated gradually. After passaging more than 20 generations, the fluorescence in cell colonies was no longer visible. Through continuous culture and passage, 4 bovine iPS cell lines were established and have been stably passaged for over 40 passages.
Generation of bovine iPS cells from Holstein fetal fibroblasts by lentiviral transduction. (A) Time schedule of bovine iPS cell generation after the 4th lentivivus infection. (B)Round cells and colonies (1: normal light, 2: under fluorescence) appeared after the 4th lentivivus infection. (C) The 10th passage bovine iPS cell colonies at the 2nd day after passage are shown (1: normal light; 2: under fluorescence). (D) The 10th passage bovine iPS cell colonies at the 3rd day after passage are shown (1: normal light; 2: under fluorescence), scale bar: 200 μm. (E) Positive AP staining of the 9th passage bovine iPS cells is shown. (F) Normal karyotype (30 pairs) of the 10th passage bovine iPS cells is shown.

AP staining and karyotype analysis of bovine iPS cells
AP is highly expressed in ES cells, but expression is low or even absent in differentiated cells. Therefore, AP expression is a key criterion for ES cells. Bovine iPS cells produced in the current study displayed strong AP expression. After staining with AP solution, colonies displayed a reddish-brown or reddish-purple color. The intensity of color varied among colonies; for example, some colonies were strongly positive along the boundary but weak in the center, while others were strong in the center but weak along the boundary. In contrast, mouse feeder cells were negative (Fig.1E). When cultured with feeders and stem cell medium, iPS cells generated in this study exhibited normal karyotype (2n = 60) (Fig.1F).
Stem cell gene expression of bovine iPS cells
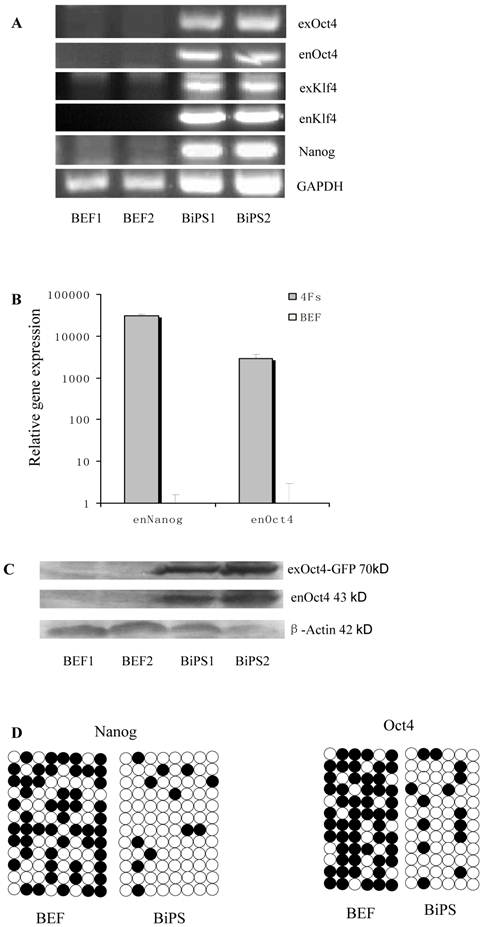
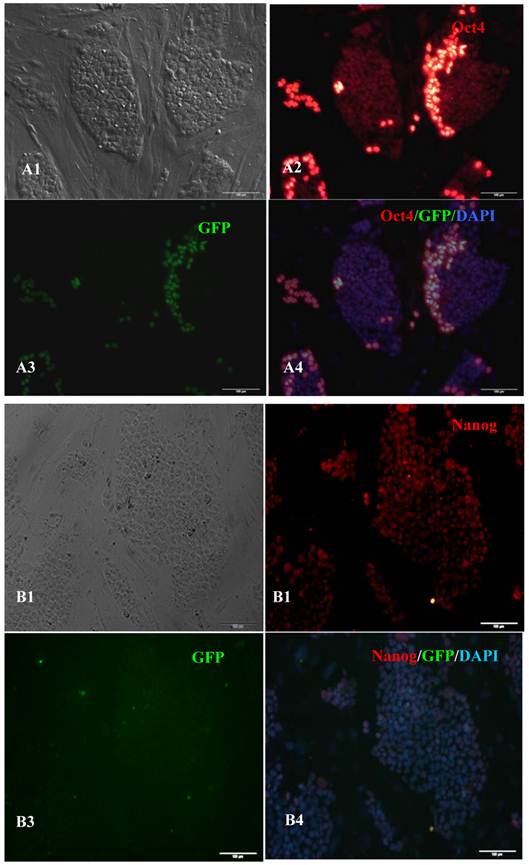
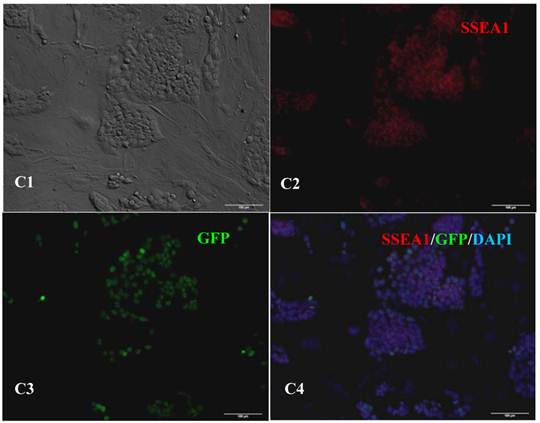
RT-PCR analysis indicated that the bovine iPS cells that were generated in the current study expressed not only endoOct4, endoKlf4 and Nanog but also exoOct4 and exoKlf4. In contrast, the bovine fibroblasts did not express these genes (Fig.2A). The expression of the endogenous pluripotent genes Oct4 and Nanog in bovine iPS cells were highly evaluated by quantitative real-time PCR (Fig.2B). In addition, bovine iPS cells expressed not only endoOct4 but also exoOct4 protein by western blotting (Fig.2C). Examination of stem cell antigens indicated the presence of Oct4, Nanog and SSEA1 antigens in bovine iPS cells (Fig.3). Under the same condition, feeders or bovine fibroblasts were negative for these antigens. The cells with strong EGFP expression were also strongly positive for Oct4 antigen expression, while those with weak or negative EGFP were moderate in Oct4 antigen expression. However, SSEA1 and Nanog antigen expression were not different in colonies with or without EGFP expression. In addition, the stem cell markers SSEA3, SSEA4, TRA-1-60 and TRA-1-81 were negative in bovine iPS cells.
Expression of stem cell marker genes and methylation status in the promoter regions of Oct4 and Nanog. BEF1 and BEF2 represented different Un-infected bovine embryonic fibroblast lines; BiPS1 and BiPS2 represented different 10th passage bovine iPS cell lines. (A) The exogenous and endogenous gene expression was analyzed by RT-PCR. (B) The endogenous Oct4 and Nanog expression was analyzed by quantitative RT-PCR. (C) The exogenous and endogenous Oct4 expression was analyzed by western blotting. Exogenous Oct4 protein (70KDa) was composed of Oct4 protein (43KDa) and EGFP (27KDa). (D) Bisulfite sequencing analysis of the Nanog and Oct4 promoters is shown. White and black circles indicate unmethylated and methylated CpG, respectively.
Expression of pluripotent markers in the 10th passage bovine iPS cells. scale bar: 100 μm. (A1) Bovine iPS cells were used as pluripotent marker Oct4 analysis. (A2) The immunofluorescence staining of marker Oct4 is shown. (A3) EGFP expression is shown. (A4) The merge is shown. (B1) Bovine iPS cells were used as pluripotent marker Nanog analysis. (B2) The immunofluorescence staining of marker Nanog is shown. (B3) EGFP expression is shown. (B4) The merge is shown. (C1) Bovine iPS cells were used as pluripotent marker SSEA1 analysis. (C2) The immunofluorescence staining of marker SSEA1 is shown. (C3) EGFP expression is shown. (C4) The merge is shown.
Methylation status of bovine iPS cells
Bisulfite genomic sequencing analyses showed that the cytosine guanine dinucleotides (CpG) in the promoter regions of Oct4 and Nanog were highly unmethylated in bovine iPS cells compared to the fetal fibroblasts (Fig.2D). These results reveal that the promoters were reactivated in bovine iPS cells and the fetal fibroblasts were reprogrammed by lentiviral transduction of Oct4, Sox2, Klf4 and c-Myc reprogramming factor fusion proteins with EGFP.
In vitro differentiation and in vivo teratoma formation of bovine iPS cells
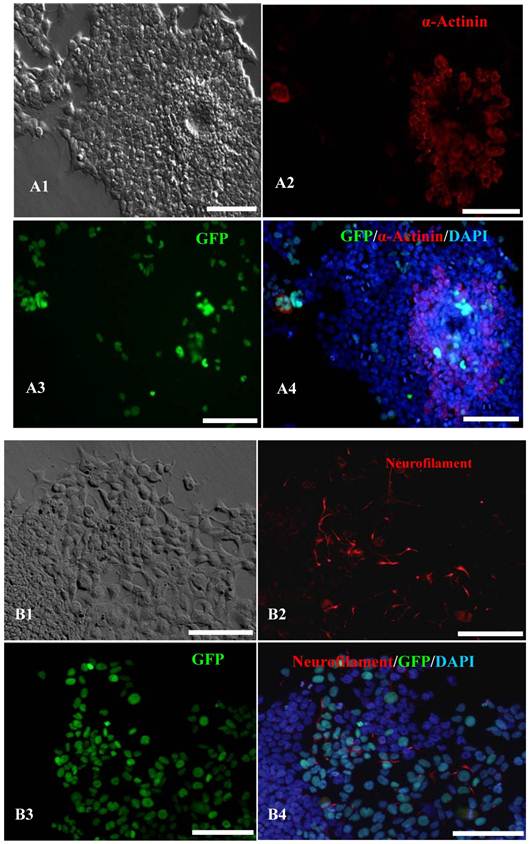
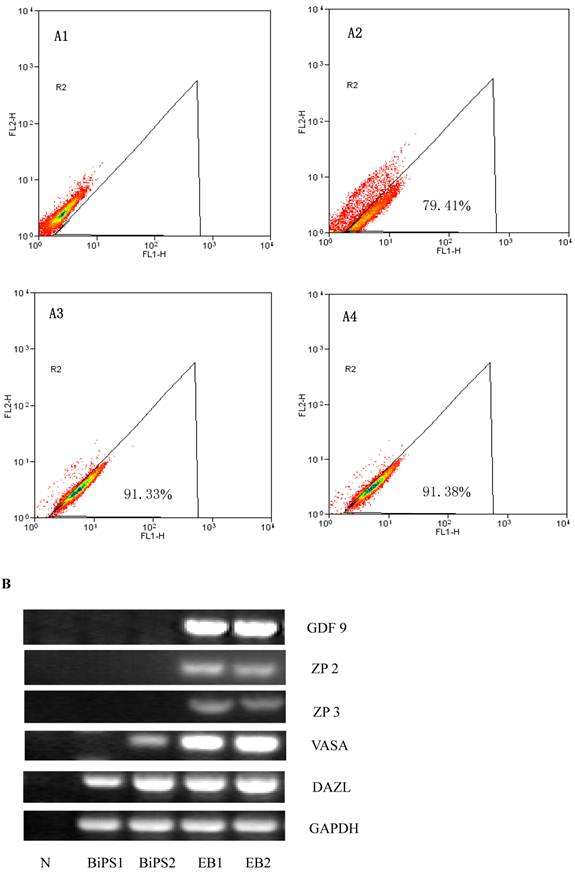
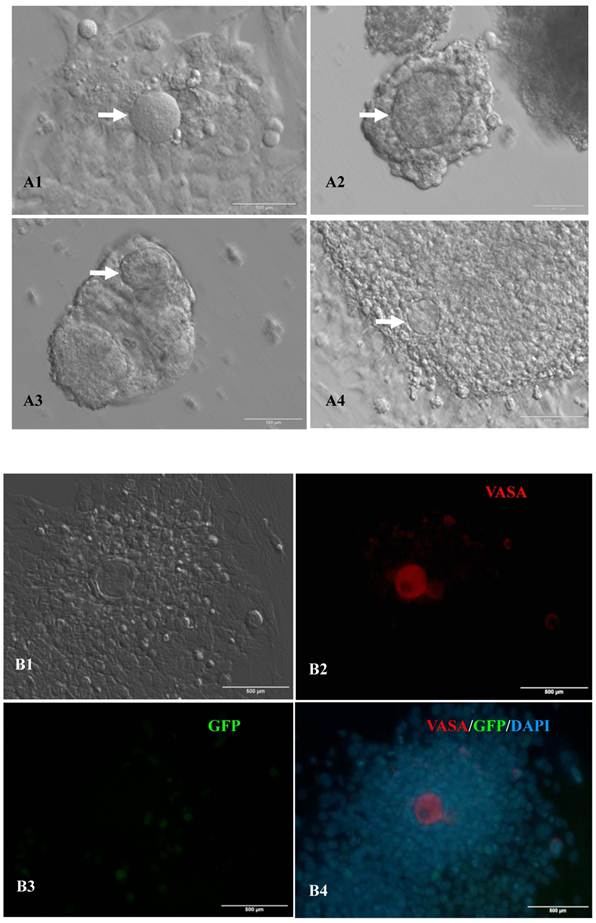
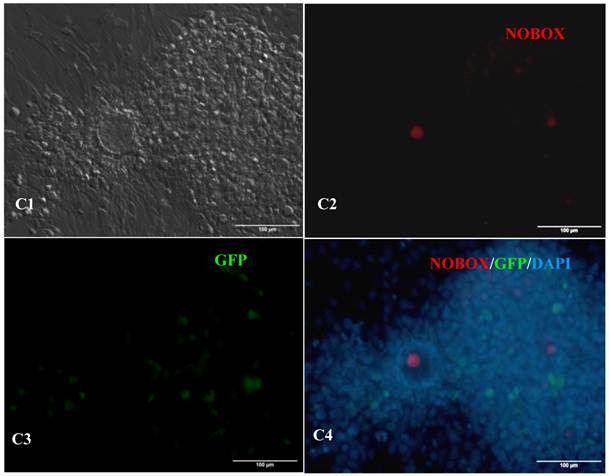
In vitro differentiation of 10th passage bovine iPS cells was performed through the spontaneous and defined differentiation of EB formation. Immunocytochemistry confirmed that 10th passage bovine iPS cells were able to differentiate into all three germ layers in vitro, as assessed by positive expression of Neurofilament (Ectoderm), α-Actinin (Sarcomeric, mesoderm) and a-Fetoprotein (AFP, endoderm). In addition, the cells with EGFP expression in the colonies had pluripotency and were able to differentiate into all three germ layers in vitro (Fig.4). When injected into subcutaneous abdominal tissue of BALB/C nude mice, the 10th passage bovine iPS cells formed teratomas in 9 weeks, and the teratomas displayed hair follicles as well as muscle and gastrointestinal tissues from ectoderm, mesoderm and endoderm germ layers (Fig.5). Flow cytometeric analysis indicated that endogenous stem cell marker SSEA1 in the colonies was stably expressed and positive 91.33% and 91.38% in 20th and 30th passage bovine iPS cells, but 79.41% in 10th passage bovine iPS cells (Fig.6A). Under defined culture conditions using porcine follicular fluid and RA to induce differentiation, the 12th passage bovine iPS cells could be induced into female germ cells and expressed early and late female germ cell specific genes Vasa, Dazl, Gdf9, Zp2, and Zp3 (Fig.6B). Multiple oocyte-like cells and follicular-like structures were observed, and the diameter of some oocyte-like complexes was measured and varied from 30μm to 150μm. Immunofluorescence staining showed that germ cell specific markers Vasa and Nobox were positive in oocyte-like cells (Fig.7).
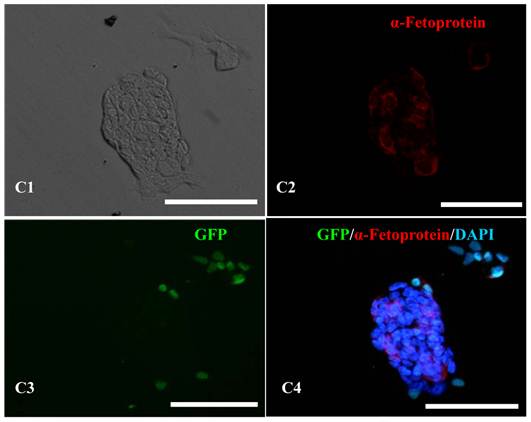
In vitro differentiation analyses for the three germ layers in the 12th passage bovine iPS cells. scale bar: 100 μm. (A1) EBs expressing marker α-Actinin (Sarcomeric, mesoderm) are shown. (A2) The immunofluorescence staining of mesoderm marker α-Actinin is shown. (A3) EGFP expression in EBs is shown. (A4)The merge is shown. (B1) EBs expressing marker Neurofilament (ectoderm) are shown. (B2)The immunofluorescence staining of ectoderm marker Neurofilament is shown. (B3) EGFP expression in EBs is shown. (B4)The merge is shown. (C1) EBs expressing marker a-Fetoprotein(AFP, endoderm) are shown. (C2) The immunofluorescence staining of endoderm marker a-Fetoprotein is shown. (C3) EGFP expression in EBs is shown. (C4)The merge is shown.
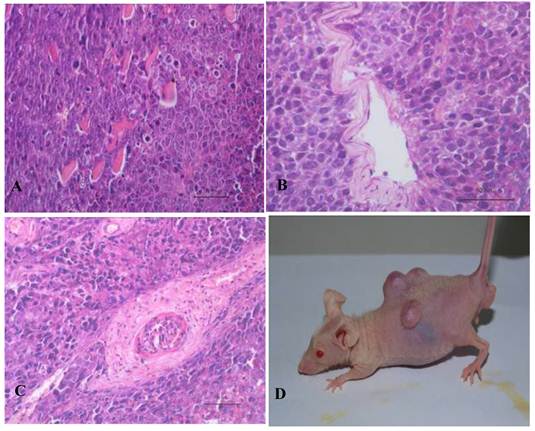
Teratoma formation in vivo including three germ layer with the 10th passage bovine iPS cells. Three germ layers including muscle tissue (mesoderm) (A), gut-like gland tissue (endoderm) (B), hair follicle (ectoderm) (C) in the teratomas (D) are shown.
SSEA1 stable expression in bovine iPS cells by flow cytometer and the expression of female gamete specific genes in the EBs by RT-PCR. (A1) Negative control as quantitative flow cytometric analysis of bovine iPS cell surface antigen SSEA1 is shown. (A2) Quantitative flow cytometric analysis of the 10th passage bovine iPS cell surface antigen SSEA1 is shown. (A3) Quantitative flow cytometric analysis of the 20th passage bovine iPS cell surface antigen SSEA1 is shown. (A4) Quantitative flow cytometric analysis of the 30th passage bovine iPS cell surface antigen SSEA1 is shown. (B) the expression of female gamete specific genes in the EBs from 10th passage bovine iPS cells is shown.
Expression of female gamete specific markers in the EBs from the 12th passage bovine iPS cells. scale bar: 100 μm. (A1) Arrow indicated oocyte-like cell. (A2) Arrow indicated oocyte-like complex in suspended EBs. (A3) Arrow indicated oocyte-like cell in suspended EBs. (A4) Arrow indicated oocyte-like cell in adherent EBs. (B1) Bovine iPS cells were used as marker Vasa analysis. (B2) The immunofluorescence staining of pluripotent marker Vasa is shown. (B3) EGFP expression is shown. (B4) The merge is shown. (C1) Bovine iPS cells were used as marker Nobox analysis. (C2) The immunofluorescence staining of marker Nobox is shown. (C3) EGFP expression is shown. (C4) The merge is shown.
Discussion
The four defined factors Oct4, Sox2, Klf4 and c-Myc have been successfully used in the induction of pluripotent stem cells from somatic cells. Previous studies have shown that the four defined factors from human or mouse are able to transform somatic cells of other species into pluripotent stem cells (14-16), demonstrating the commonality of these highly conserved factors. In our previous study, human Oct4 and the other three porcine factors were successfully used to generate porcine pluripotent stem cells (12). In the current study, the combination of these factors was also effective in the derivation of bovine iPS cells. The fusion proteins between EGFP and exogenous functional genes have been applied widely in the study of gene functions and protein properties. In the current study, EGFP fusion proteins were effective in the induction of bovine iPS cells when introduced by lentivirus, demonstrating that the EGFP portion does not interfere with the functioning of exogenous defined factors in the fusion proteins during somatic cell reprogramming.
In the current study, as the number of passages increased, the proportion of cells expressing EGFP decreased and the fluorescence intensity attenuated gradually. Additionally, after more than 20 passages, the fluorescence in cell colonies was no longer visible. During somatic cell reprogramming, partially reprogrammed cells still express high levels of exogenous factors, and the incomplete silencing of exogenous factors was also observed in iPS cells of other species, such as human (17), rat (5) and pig (16). Chin et al (18) discovered that upon comparing gene expression profiles among different passages, human iPS cells at 54-61 passages more closely resemble ES cells than those at 5-9 passages. Therefore, iPS cells may gradually establish reprogramming ability during continuous culture. In addition, through fusion proteins, the cells expressed exogenous defined factors in the colonies also exhibited pluripotency and were able to differentiate into all three germ layers in vitro. The results indicate that somatic cell reprogramming is complex and tracking the exogenous defined factors through fusion proteins is very effective.
Therefore, EGFP fusion proteins of exogenous defined factors mediated by lentivirus have been successfully applied in the generation of bovine iPS cells. Accordingly, this study is of value for further tracking the reprogramming mechanism and the application of bovine iPS cells.
Acknowledgements
This work is supported by grants from by National Natural Science Foundation of China (30800784), National Basic Research Program (2009CB941004), National High-Tech R&D Program (2008AA101003, 2008AA101010, 2011AA100307) and National Transgenic Breeding Program (2009ZX08008-007B).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663-676
2. Aasen T, Raya A, Barrero MJ. et al. Efficient and rapid generation of induced pluripotent stem cells from human keratinocytes. Nat Biotechnol. 2008;26:1276-1284
3. Takahashi K, Tanabe K, Ohnuki M. et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861-872
4. Liao J, Cui C, Chen S. et al. Generation of induced pluripotent stem cell lines from adult rat cells. Cell Stem Cell. 2009;4:11-15
5. Maherali N, Sridharan R, Xie W. et al. Directly reprogrammed fibroblasts show global epigenetic remodeling and widespread tissue contribution. Cell Stem Cell. 2007;1:55-70
6. Okita K, Ichisaka T, Yamanaka S. Generation of germline-competent induced pluripotent stem cells. Nature. 2007;448:313-317
7. Zhao XY, Li W, Lv Z. et al. iPS cells produce viable mice through tetraploid complementation. Nature. 2009;461:86-90
8. Kang L, Wang J, Zhang Y. et al. iPS cells can support full-term development of tetraploid blastocyst-complemented embryos. Cell Stem Cell. 2009;5:135-138
9. Geijsen N, Horoschak M, Kim K. et al. Derivation of embryonic germ cells and male gametes from embryonic stem cells. Nature. 2004;427:148-154
10. Hübner K, Fuhrmann G, Christenson LK. et al. Derivation of oocytes from mouse embryonic stem cells. Science. 2003;300:1251-1256
11. Kerkis A, Fonseca SA, Serafim RC. et al. In vitro differentiation of male mouse embryonic stem cells into both presumptive sperm cells and oocytes. Cloning Stem Cells. 2007;9:535-548
12. Yin HQ, Cao HG, Sun XP. et al. Generation of Induced Pluripotent Stem Cells From Porcine Fibroblasts. Prog in Biochem and Biophy. 2010;37:607-612
13. Pérez R, Tupac-Yupanqui I, Dunner S. Evaluation of suitable reference genes for gene expression studies in bovine muscular tissue. BMC Mol Biol. 2008;9:79
14. Esteban MA, Xu J, Yang J. et al. Generation of induced pluripotent stem cell lines from Tibetan miniature pig. J Biol Chem. 2009;284:17634-17640
15. Ezashi T, Telugu BP, Alexenko AP. et al. Derivation of induced pluripotent stem cells from pig somatic cells. Proc Natl Acad Sci USA. 2009;106:10993-10998
16. Wu Z, Chen J, Ren J. et al. Generation of pig induced pluripotent stem cells with a drug-inducible system. J Mol Cell Biol. 2009;1:46-54
17. Yu J, Vodyanik MA, Smuga-Otto K. et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917-1920
18. Chin MH, Mason MJ, Xie W. et al. Induced pluripotent stem cells and embryonic stem cells are distinguished by gene expression signatures. Cell Stem Cell. 2009;5:111-123
Author contact
![]() Corresponding author: Hongguo Cao; Anhui Local Livestock Genetic Resources Conservation and Breeding Laboratory, College of Animal Science and Technology, Anhui Agricultural University, Hefei 230036, China; Tel: +86 551-5786357; Fax: +86 551-5786357; Email: caohongguo1com.cn. Xiaorong Zhang; Anhui Local Livestock Genetic Resources Conservation and Breeding Laboratory, College of Animal Science and Technology, Anhui Agricultural University, Hefei 230036, China; Tel: +86 551-5785543; Fax: +86 551-5785543; Email: zxredu.cn.
Corresponding author: Hongguo Cao; Anhui Local Livestock Genetic Resources Conservation and Breeding Laboratory, College of Animal Science and Technology, Anhui Agricultural University, Hefei 230036, China; Tel: +86 551-5786357; Fax: +86 551-5786357; Email: caohongguo1com.cn. Xiaorong Zhang; Anhui Local Livestock Genetic Resources Conservation and Breeding Laboratory, College of Animal Science and Technology, Anhui Agricultural University, Hefei 230036, China; Tel: +86 551-5785543; Fax: +86 551-5785543; Email: zxredu.cn.