Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Roles of TNSALP, NPP1, and ANK...
Roles of PHOSPHO1 and PiT-1/2 in...
Discoveries from double-knockout...
Interplay between PPi and OPN in...
Conclusions and perspectives
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(6):778-790. doi:10.7150/ijbs.4538 This issue Cite
Review
Phosphate/Pyrophosphate and MV-related Proteins in Mineralisation: Discoveries from Mouse Models
Xiaoying Zhou, Yazhou Cui, Xiaoyan Zhou, Jinxiang Han ![]()
Shandong Academy of Medical Sciences, Shandong Medical Biotechnological Center, Key Laboratory for Rare Disease Research of Shandong Province, and Key Laboratory for Biotech Drugs of the Ministry of Health, Shandong, China.
Received 2012-4-30; Accepted 2012-5-22; Published 2012-6-1
Abstract
During the process of matrix vesicle (MV)-mediated initiation of mineralisation, chondrocytes and osteoblasts mineralise the extracellular matrix by promoting the seeding of basic calcium phosphate crystals of hydroxyapatite (HA) along the collagen fibrils. This orchestrated process is carefully regulated by the balanced action of propagators and inhibitors of calcification. The primary antagonistic regulators of extracellular matrix mineralisation are phosphate (Pi) and inorganic pyrophosphate (PPi). Studies in mouse models and in humans have established critical roles for Pi/PPi homeostasis in biomineralisation. In this review, we present the regulators of Pi/PPi, as derived from animal models, and discuss their clinical relevance to physiological and pathological mineralisation.
Keywords: Mineralisation, Matrix vesicles, PPi, Pi, MV-related proteins, OPN.
Introduction
During skeletogenesis and bone development, osteoblasts from bones, as well as hypertrophic chondrocytes from embryonic or growth plate cartilages, mineralise the extracellular matrix (ECM) at least in part by promoting the deposition of HA crystals in the sheltered interior of the membrane-bounded matrix vesicles (MVs). The MVs are submicroscopic, extracellular, membrane-invested bodies containing calcium and phosphate (Pi) and are released from the apical microvilli of hypertrophic chondrocytes [1-2] or osteoblasts [3-4]. Once released, the MVs serve as nucleation sites and continue to accumulate calcium and Pi, which stimulate the formation of HA crystals from the immature minerals present in the lumen and initiate mineralisation [1, 3, 5]. In a second step, the MV membranes subsequently rupture and/or break down, and the HA crystals are exposed to the extracellular fluid and continue to propagate along the extracellular collagen fibrils [6]. This MV-mediated mineralisation is coordinated by the balanced action of propagators and inhibitors of calcification. Studies of the mechanisms involved in the regulation of physiological and pathological mineralisation have indicated that the Pi/PPi homeostasis is the main determinant of the rate of HA crystal formation in bone tissue [7]. The extracellular PPi (ePPi) adsorbs tightly to HA and potently antagonises the ability of Pi to crystallise with calcium to form HA, thereby inhibiting HA crystal propagation [8]. For normal mineral deposition to proceed, a tight balance between the Pi and PPi levels must be maintained.
The MVs perform specialised roles in initiating matrix mineralisation. These roles include regulating the Pi/PPi ratio in the intra- and extracellular fluid, managing mineral nucleation, controlling calcium and Pi ion homeostasis, and interacting with the surrounding ECM to direct HA localisation and growth [1, 9-11]. The MVs possess protein and lipid machinery that is essential to carry out these functions, and they are highly enriched in certain mineralisation-relevant proteins, especially tissue-nonspecific alkaline phosphatase (TNSALP/ALPL/Akp2), ATPase, AMPase, inorganic pyrophosphatase, ectonucleotide pyrophosphatase phosphodiesterase 1 (NPP1/PC-1/Enpp1), phosphatase orphan 1 (PHOSPHO1), sodium-dependent Pi symporters (Pit1/2), and annexins [12-14]. An emerging consensus now emphasises the central role of these MV proteins, in conjunction with the cell-associated ankylosis protein (ANK), in the physiological manipulation of Pi/PPi homeostasis and in the control of osteopontin (OPN) [7, 15-16].
In this article, we discuss the regulators of Pi/PPi homeostasis, as determined from various mouse models, and how they relate to pathological or ectopic mineralisation. The models presented in this context will enable us to investigate and clarify the functional involvement of the highlighted MV-related proteins in skeletal mineralisation and soft tissue ossification abnormalities. This discussion will help to elucidate the mechanism of action for diseases such as hypomineralisation, hypermineralisation, and ectopic ossification.
Roles of TNSALP, NPP1, and ANK in PPi metabolism and mineralisation
PPi is a major inhibitor of physiologic and pathologic calcification, bone mineralisation, and bone resorption [17]. The maintenance of physiologic ePPi levels by mineralisation-competent cells suppresses spontaneous calcification, and abnormal ePPi metabolism has been implicated in abnormal calcification [18]. Decreased PPi concentrations can generate basic calcium phosphate (BCP) deposition, while an excess of PPi can lead to calcium pyrophosphate dihydrate (CPPD) formation, a marker of pathological calcification [18]. Given that MV-associated proteins are primarily responsible for producing and hydrolysing ePPi, the functional disruption of MV-associated enzymes and PPi transporters in mice and the observation of MV-related gene deficiencies in humans are anticipated to cause phenotypic changes associated with defective skeletal mineralisation.
TNSALP
TNSALP, which is encoded by the Akp2 gene, is a membrane-bound phosphomonoesterase localised to the surface of osteoblasts and chondrocytes, including the membranes of their shed MVs, via a glycosylphosphatidylinositol (GPI) anchor [19-20]. The absence of TNSALP activity results in the extracellular accumulation of its natural substrates, such as pyri-doxal-5'-phosphate (PLP), phosphoethanolamine (PEA), and PPi [20-21]. Mutations in the Akp2 gene cause the inherited skeletal disease known as hypophosphatasia (HPP), which is characterised by hypomineralisation that causes rickets in infants and children, osteomalacia in adults, spontaneous fractures, deficiencies in serum and bone alkaline phosphatase (ALP) activity, and elevated extracellular concentrations of PPi [22-23]. The severity of the six clinical forms of HPP varies widely in patients and is modulated by the nature of the Akp2 mutation. Patients with the infantile form of HPP may appear normal before the onset of failure to thrive and the associated development of rickets before the first 6 months, and severe infantile HPP is often fatal [23]. To date, cell therapy with bone marrow cells [24-25] and mesenchymal cells [26] have been showed very limited or no clinical effects to HPP. Very recently, enzyme replacement, using ENB-0040, a bone-targeted form of human TNALP, can improve skeletal manifestations and pulmonary and physical function in life-threatening HPP [27].
TNSALP knockout mice, which serve as good models for the infantile form of HPP, have been established in two independent laboratories [28-29]. Biochemical, radiographic, and histological investigations have shown that Akp2−/− mice are born with a normal skeletal phenotype but develop rachitic changes, osteopenia, and growth plate chondrocyte differentiation arrest. Subsequently, the animals manifest secondary hyperparathyroidism and epileptic seizures, and they die prematurely due to the multiple metabolic abnormalities caused by the lack of TNSALP and the resulting elevated concentrations of its substrates, including PLP, PEA, and PPi [29-30]. A subsequent study of Akp2−/− mice revealed that, in addition to the hypomineralisation of the skeleton, a severe disorder of the mineral crystal alignment pattern existed in the corticalis of growing long bones. This defect was also associated with a disordered matrix architecture, which was presumably caused by disturbed bone remodelling [31]. Recently, two strategies for therapy in Akp2−/− mice have been reported. Sustained delivery of bone-targeted form of TNSALP prevented infantile HPP in Akp2−/− mice; and lentiviral gene therapy during the neonatal period prolonged survival and corrected the phenotype of severe infantile HPP in Akp2−/− mice [21, 32]. Interestingly, there were no differences observed between the TNSALP-knockout mice and wild-type littermates immediately after birth [7-9]; perhaps maternal TNSALP or other Mn2+-dependent isoenzyme of ALP expressed in utero can compensate for the lack of TNSALP during embryogenesis in Akp2−/− mice [30, 33]. Primary osteoblasts from the calvaria of TNALP-knockout mice differentiate normally in vitro and display a normal level of mRNA expression and protein synthesis of osteocalcin (OC) and collagen type I, but they fail to form mineralising nodules in vitro [34]. Electron microscopy revealed that, in both humans and mice, TNSALP-deficient MVs contain apatite crystals but are characterised by increased levels of unhydrolysed PPi and an absence of mineral crystal proliferation and growth in the matrix surrounding the MVs. These findings indicate that TNSALP is involved but not essential in the initial step of mineralisation within MVs [19].
In addition, transgenic overexpression of TNSALP can cause a decrease in ePPi concentrations that induce spontaneous HA crystal formation within a fibrillar/collagenous scaffolding [7, 15, 35]. These findings appear to support the hypothesis that high PPi levels at the perimeter of TNSALP-deficient MVs prevent crystal growth in the localised region of the matrix. Although the TNSALP-deficient MVs may be able to initiate intravesicular mineralisation through the compensatory activity of other phosphatases hydrolysing other substrates to generate Pi, still incapable to remove excess PPi. Thus, despite TNSALP deficiency, initial mineralisation could begin within MVs, while its propagation at the perimeter of MVs would be hindered by a local excess of PPi. The main cause of the hypomineralisation observed in TNSALP knockout mice is likely due to the excessive accumulation of PPi in the extracellular matrix [7], suggesting that TNSALP is required to promote calcification by removing PPi, which inhibits mineralisation [8, 12].
Recently, a novel mouse model of autosomal semi-dominant adult HPP has been developed [36]. In contrast to Akp2−/− mice, the Akp2Hpp/Hpp mice were characterised by a normal lifespan, absence of seizures and normal initial skeletal development and growth, but developed late-onset skeletal diseases [36]. Compared with wild type control cells, osteoblasts from Akp2Hpp/Hpp mice displayed ∼10% of normal ALP activity, and significant reductions in the levels of iPi, but normal ePPi levels, yet were capable of normal mineralisation in short term in vitro [36]. These data indicated that the action of residual TNSALP activity contributed to the normal phenotype of young Akp2Hpp/Hpp mice, but fell below a threshold needed to maintain normal bone turnover with age.
NPP1
PPi is primarily generated by the members of the nucleotide pyrophosphatase/phosphodiesterase (NPP) family of isozymes, including NPP1, NPP2/autotaxin, and NPP3/B10 [8]. However, NPP1 appears to be the only NPP present in MVs [37]. Similar to the skeletal expression of TNSALP, NPP1, which is encoded by the Enpp1 gene, is highly abundant on the surfaces of osteoblasts and chondrocytes, as well as on the membranes of their MVs [38]. NPP1 potently inhibits the nucleation and propagation of HA and other BCP crystals through its PPi-generating properties [8]. In humans, loss-of-function NPP1 mutations cause ectopic ossification diseases, such as autosomal recessive hypophosphatemic rickets (ARHR2; OMIM No. 613312), ossification of the posterior longitudinal ligament (OPLL; OMIM No.602475), and generalised arterial calcification of infancy (OMIM No. 208000) [39-41].
In naturally occurring mouse models, the link between defective NPP1 expression and altered mineralisation was initially demonstrated in tiptoe-walking ttw/ttw mice, which are caused by a nonsense mutation (glycine 568 to stop) in the NPP1 gene [42]. The truncated NPP1 protein loses a vital calcium binding domain and two putative glycosylation sites. The ttw/ttw mice display marked ectopic ossification, including the postnatal development of progressive ankylosing intervertebral and peripheral joint hyperostosis, as well as spontaneous arterial and articular cartilage calcification and increased vertebral cortical bone formation [42-46]. Excess axial skeleton ligamentous calcification results in myelopathy and abnormal gait in ttw/ttw mice, reminiscent of the human disorder OPLL [47]. Additionally, ttw/ttw mice exhibit abundant calcified matrix vesicle-like chain granules in the pericellular matrix, cytoplasm, and nucleus of the articular and spinal disc chondrocytes, suggesting that NPP1 deficiency may dysregulate matrix vesicle formation and/or calcification [48]. The ttw/ttw mice also share features with idiopathic infantile arterial calcification, including spontaneous periarticular and aortic calcifications in early life and the systemic lowering of NPP1 activity and PPi levels [42, 49-50].
Consistent with the findings from ttw/ttw mice, chondrogenesis mediated by PPi depletion promotes spontaneous aortic calcification in Enpp1−/− mice [51]. The multipotential bone marrow stromal cells from Enpp1−/− mice can spontaneously undergo chondrogenesis; and the cartilage-specific expression of genes, such as ALP and OPN, was altered in aortic smooth muscle cells, which showed increased calcification in Enpp1−/− mice [51]. Enpp1−/− mice are characterised by spontaneous mineralisation of soft tissue, hypermineralisation in the talocrural joint and the cortical thickness of both the tibia and femur, which becomes increasingly more severe with age [52]. These serious disruptions to the architecture and mineralisation of the long bones were consistent with alterations in the markers of bone formation and resorption and explained their reduced mechanical properties [52]. These findings suggest that NPP1 is essential for normal bone development and for the control of physiological bone mineralisation with age through PPi production. Interestingly, the Enpp1−/− mice have also been documented to display trabecular bone loss [52]. Furthermore, circulating phosphate and calcium levels were reduced, and the concentration of the phosphaturic hormone FGF-23, which is elevated in ARHR2, was also significantly increased in the Enpp1−/− mice [40, 52]. It should be noted that NPP1 also plays important roles in calcium and phosphate regulation and the repression of soft tissue mineralisation, as well as in maintaining skeletal structure and function [52]. Moreover, it has recently been demonstrated that NPP1 regulates osteoblastic gene expression and controls cellular differentiation in calvarial osteoblasts independent of its catalytic activity [53].
ANK
ANK, which is encoded by the mouse progressive ankylosis (ank) gene, appears to function as a multiple-pass, transmembrane PPi-channelling protein, allowing PPi molecules to pass through the plasma membrane from the cytoplasm to the outside of the cell [54-55]. Unlike TNSALP and NPP1, the ANK protein is detectable on the surfaces of the osteoblasts and chondrocytes but not on the surfaces of osteoblast-derived MVs [35]. Mutations in the human ANK gene (ANKH), specifically the point mutations that cluster mostly in cytoplasmic domains close to the C terminus, have previously been linked to craniometaphyseal dysplasia (CMD; OMIM No.123000) [56-57]. CMD is a childhood disease characterised by several craniofacial anomalies, including hyperostosis and sclerosis of the skull and symmetric thickening of all facial bones. In severe cases, hyperostosis and sclerosis of the skull may lead to cranial nerve compressions, resulting in hearing loss and facial palsy [57]. The results from two studies analysing CMD mutations indicated that, other than acting as a transporter, ANK may perform one or more additional functions in the regulation of bone formation and remodelling [56-58]. Mutations in the N terminus of ANKH result in another human skeletal disorder, familial calcium pyrophosphate dihydrate deposition disease (CPPDD), also known as familial chondrocalcinosis (OMIM No.118600), which is characterised by the formation of CPPD crystals in articular cartilage, but unlike CMD, does not present with apparent defects in the skull or long bones [59-61].
A spontaneous mutation in the murine ank gene is a loss-of-function mutation [62]. These ank mutation mice display a generalised, progressive form of arthritis accompanied by mineral deposition, the formation of bony outgrowths, and joint destruction [54]. Fibroblasts from the ank/ank mutant mice show a three- to five-fold decrease in ePPi levels and an increase in intracellular PPi (iPPi) accumulation compared with wild type cells. These results indicate that the mouse ank gene regulates tissue calcification and the development of arthritis in higher animals through the control of PPi levels [54]. A recent biochemical study used a sensitive radioflux method to test the transport activity of mutant ANK proteins carrying CMD mutations (C331R and C389R) in frog oocytes and showed greatly reduced PPi transport activity [55]. However, the pathophysiological mechanisms in human CPPDD and in ank/ank mice are distinct. In human CPPDD, mutant ANK proteins have been characterised as gain-of-function mutations that result in increased ePPi levels [59, 63]. The high concentration of ePPi in joint fluid inhibits the formation of HA crystals and promotes the deposition of CPPD crystals within joint tissues, ultimately leading to cartilage destruction. However, another study reported that the number of mature osteoblasts and osteoclasts was reduced, and the bone formation, as well as bone resorption was suppressed in Ankank/ank mice. This finding indicated that ANK can also regulate bone remodelling through directly affecting osteoblast and osteoclast differentiation [62].
Mice with complete and joint-specific disruption of Ank gene function (Anknull/null and Ankank/ank) were generated simultaneously to verify whether Ank mutations could completely eliminate the function of the endogenous gene [64]. Two Ank loss-of-function models had joint and skeletal defects of similar severity, with increased HA deposition eventually leading to complete rigidity and death at approximately 6 months of age [64-66]. The study indicated that both complete and joint-specific loss of ANK function can cause mineral formation in joints and confirms a key role for Ank in joint maintenance and local regulation of mineralisation in articular cartilage [64]. In addition, Anknull/null mice shared some characteristics with CMD patients but did not fully replicate the symptoms of the human condition. The phenotype differences between Anknull/null mice and CMD patients indicate that the pathological mechanism in human CMD is not merely due to a loss of PPi transport function by ANK [55].
Recently, the first knock-in (KI) mouse model (AnkKI/KI) of CMD was generated [67]. The animals express one of the most common Ank mutations (i.e., the Phe 377 deletion) identified in the human disease. Homozygous AnkKI/KI mice share many features with human CMD patients, including hyperostosis of craniofacial bones, massive jawbones, decreased diameter of the cranial foramina, the obstruction of the nasal sinuses, the fusion of the middle ear bones, and club-shaped femurs [67]. Surprisingly, despite the hyperostotic phenotype, the bone matrix in AnkKI/KI mice is hypomineralised and less mature. Although the number of osteoclasts was increased in AnkKI/KI mice, as determined by histomorphometry, the in vitro cultures of bone marrow-derived macrophages showed decreased osteoclastogenesis [67]. Subsequently, an elaborate study was carried to investigate the effects of the ANK Phe377del mutation on osteoblastogenesis and osteoclastogenesis at the cellular level in the AnkKI/KI mouse model [68]. AnkKI/KI osteoblast cultures showed decreased expression of genes involved in mineral deposition and bone mineralisation regulation (e.g., Mmp13, Ocn, Osx, and Phex) and increased NPP1 activity, while the Fgf23 mRNA level was elevated in calvarial and femoral bones. Similar to the bone marrow-derived macrophage cultures from AnkKI/KI mice, peripheral blood cultures from CMD patients exhibited reduced osteoclastogenesis. Cell-autonomous effects in the AnkKI/KI osteoclasts caused disrupted actin ring formation and cell fusion [68]. Moreover, the osteoblasts of AnkKI/KI mice failed to adequately support osteoclastogenesis. The authors concluded that the hypomineralisation and high bone mass phenotypes of AnkKI/KI mice were due to impaired osteoblastogenesis and osteoclastogenesis resulting from the ANK Phe377del mutation [68].
Roles of PHOSPHO1 and PiT-1/2 in Pi metabolism and mineralisation
There are two extracellular mineral ions, Pi and Ca2+, that play a critical role in the regulation of ECM mineralisation. Mutations in genes regulating Pi/Ca2+ homeostasis have resulted in severe skeletal mineralisation abnormalities in mouse models [13]. Interestingly, the ECM mineralisation defects were invariably associated with low Pi levels but not always with low serum calcium levels, suggesting that the regulation of the systemic Pi levels is indispensable for proper bone formation, particularly for the osteoid mineralisation processes. In the MVs, the presence of Pi, generated from PHOSPHO1 and PiT1/2 as well as from ATPase and ADPase, allows for the formation of HA crystals when Ca2+ is available [18].
PHOSPHO1
The phosphatase PHOSPHO1, first identified in the chick, exhibits high specific activities for PEA and phosphocholine (PChol) [69-71]. PEA and PChol are the two most abundant phosphomonoesters in cartilage [72], and PHOSPHO1 is expressed at levels approximately 100-fold higher in mineralising chondrocytes than in the non-skeletal tissues [71]. These facts led us to hypothesise that PHOSPHO1 is responsible for generating Pi for skeletal mineralisation through its highly specific phosphohydrolase activity. The presence of PHOSPHO1 within the lumen of the MVs has been proven [73]. The data from an expression analysis of an embryonic chick skeleton suggested that PHOSPHO1 expression occurs in the diaphysis of long bones [73]. Inhibition of PHOSPHO1 through the use of novel small molecule inhibitors decreased the mineralisation capacity of the MVs, which further indicated that PHOSPHO1 plays an important role in controlling the first step of HA crystal deposition inside the MVs by generating Pi inside the MVs [73-74]. In addition, PHOSPHO1 also has a functional role in mineralisation during limb development. Inhibition of PHOSPHO1 activity results in impaired skeletal endochondral mineralisation during limb development in the chick [75]. To date, no human disease has been associated with a Phospho1 mutation.
Very recently, Phospho1−/− mouse models have been generated to study the role of PHOSPHO1 during endochondral ossification [76]. The Phospho1−/− mice exhibited growth plate abnormalities, spontaneous fractures, bowed long bones, osteomalacia, and scoliosis in early life. Chondrocytes and chondrocyte-derived MVs from Phospho1−/− tibial growth plate displayed a reduced ability to form minerals, which is consistent with the reduced mineralisation of their skeletons. These findings clearly demonstrate that PHOSPHO1 is required for normal endochondral ossification [76].
PiT1/2
Type III sodium/phosphate cotransporters (i.e., PiT1 and PiT2) utilise the free energy provided by the Na+ concentration gradient to mediate the influx of Pi across the cell membrane. The cotransporters share similar gene structures and functional characteristics [18, 77-78]. PiT1 and PiT2 are expressed in most organs, suggesting a “housekeeping” function for these genes [78-79].
Recent reports have suggested that PiT1 plays an important role in vascular and bone physiology [80-81], whereas PiT2 has a role in renal Pi reabsorption [82-83]. Using in situ hybridisation to study murine metatarsals, PiT-1 mRNA was found to be expressed in early hypertrophic chondrocytes, which is consistent with a potential role for the PiT-1 cotransporter in matrix calcification [84]. PiT1 plays a dominant role in the transdifferentiation of the vascular smooth muscle cells (VSMCs) to osteoblast-like cells during the process of vascular calcification [80, 85]. The knockdown of PiT-1 in human VSMCs suppressed phosphate-induced mineralisation and osteochondrogenic changes, which were rescued by overexpression of mouse PiT-1 using PiT-1-specific small interfering RNAs (siRNAs) [86]. PiT-1 was found to be required for the phosphate-induced calcification of human VSMCs in vitro [80]. A recent study of Werner Syndrome patients with subcutaneous calcification found that PiT-1 was overexpressed in the patients' skin, thereby supporting a role for PiT-1 in diseases characterised by ectopic calcification [87]. However, the transgenic overexpression of PiT-1 in rats affected bone and calcium phosphate metabolism and ALP activity, but did not influence bone matrix mineralisation or skeletal development, potentially due to the effects of an unidentified compensatory mechanism [88].
PiT-1 knockout mice have been recently generated by two different groups to understand the physiological role of these transporters. The complete deletion of PiT1 resulted in embryonic lethality. PiT-1 null embryos arrested between embryonic days 11.5 and 13.5 and displayed severe anaemia [89-90]. The anaemia may result from the decreased proliferation and massive apoptosis of the fetal liver, but a fundamental defect in hematopoiesis has not yet been ruled out [90]. However, observations of the vascular development of the yolk sac yielded contradictory results in these two studies. Although PiT1 is essential to embryonic development, the protein does not appear to be essential for skeleton and vasculature development in PiT1-deficient mice [90]. The role of PiT-1 in the bone development or calcification processes cannot be addressed in these null embryos, but the approach used to generate conditional alleles for PiT-1 would be valuable in the creation of tissue-specific knockouts. In addition, the development of a model with a hypomorphic PiT-1 allele would allow for the analysis of subtle phenotypes that are determined by the gene expression level [80]. To date, no PiT-2 knockout mice have been reported.
Discoveries from double-knockout mouse models
The restriction of mineral deposition to specific sites in the skeletal tissues and the regulation of the mineralisation process are crucial in maintaining a healthy skeleton [12-13]. Normal mineralisation is dependent on a balance between the intra- and extracellular levels of Pi and PPi [9]. The ePPi pool has been hypothesised to determine the rate of HA crystal formation in the bone tissue, whereas the action of TNSALP, NPP1 and ANK are proposed to regulate the PPi concentration. Akp2−/−, Enpp1−/−, and ank/ank mice all suffer from severe mineralisation defects due to abnormal PPi levels. The concerted action of TNSALP, NPP1, and ANK ensures that the optimal concentration of PPi is achieved under physiologic conditions, and double-knockout mice have been valuable in furthering our understanding of the roles that these proteins play in the MV-mediated process of bone mineralisation.
[Akp2−/−; Enpp1−/−]
TNSALP is proposed to be required to hydrolyse PPi at the site of mineral crystal proliferation, thus facilitating mineral precipitation and growth, and is also required to contribute to the ePi pool for the formation of HA crystals [8, 12, 19]. Mutations in the TNSALP gene in humans and mice result in a metabolic disease known as hypophosphatasia, which is characterised by hypomineralisation and elevated ePPi levels. In NPP1 deficiencies, the ePPi pool decreases because of reduced PPi production, and hypermineralisation occurs in the absence of the HA deposition inhibitor, PPi. Interestingly, the double knockout of both Akp2 and Enpp1 [Akp2−/−; Enpp1−/−] rescued the bone mineralisation abnormalities of the individual mutations, including the hypo- and hypermineralisation defects in the calvaria and spine [7]. The MVs derived from the double-knockout mouse osteoblasts showed normalisation of the ePPi content and mineral deposition. These findings suggest that TNSALP and NPP1 are antagonistic regulators that are directly involved in fine-tuning the PPi concentration to maintain steady-state levels of PPi adequate for controlled mineralisation. Further, this evidence points to both NPP1 and TNSALP as potential therapeutic targets in the treatment of mineralisation diseases such as hypophosphatasia and osteoarthritis [7].
However, when considering the trabecular bone loss of Enpp1-deficient mice, in addition to the hypermineralisation of soft tissue [42, 45, 52], the extent of mineral deposition in the bones of [Akp2−/−; Enpp1−/−] needed to be examined to ascertain whether correction of the mineralisation abnormalities in the [Akp2−/−; Enpp1−/−] mice extend to the entire skeleton [91]. The effects of the Enpp1 ablation on an Akp2−/− background were site-specific and extends only to the calvaria, spine, and incompletely in the femur and tibia in which the [Akp2−/−; Enpp1−/−] mice were much more worse than in the Enpp1−/− mice [91]. In contrast to the rescue of the phenotype in the calvaria, vertebrae, and soft tissues as a consequence of the simultaneous disruption of NPP1 and TNSALP function, the long bones of the [Akp2−/−; Enpp1−/−] mice appeared to have sustained osteomalacia. The hypomineralisation observed in the long bones of Enpp1−/− mice may be related to the different levels of NPP1 expression in these skeletal environments; that is, endogenous NPP1 expression is relatively low surrounding the long bones in comparison with the calvaria and vertebrae [52, 91]. Thus, with the complete disruption of NPP1 function, the long bones could show further reductions in ePPi to abnormally low levels, resulting in insufficient PPi substrate levels for TNSALP to generate Pi for normal mineral formation. Because inadequate quantities of PPi are generated from NPP1 or hydrolysed by TNSALP, the [Akp2−/−; Enpp1−/−] mice displayed sustained more severe osteomalacia of the long bones compared to the Enpp1−/− mice [91].
[Akp2−/−; Ank/Ank]
Given that the ANK protein functions similarly to NPP1, albeit in a different manner, [Akp2−/−; Ank/Ank] mice were generated to investigate whether the simultaneous disruption of TNSALP and ANK functions would also rescue the mineralisation defects of the mice with single gene ablations [35]. The [Akp2−/−; Ank/Ank] mice displayed a partial correction of the hypo- and hypermineralisation phenotypes, as measured by the extent of mineralisation in their vertebral apophyses. This partial rescue of the mineralisation defects observed in the single mutants was associated with a partial correction of the PPi levels. The ePPi concentration was normal in these double mutants, but the iPPi levels remained abnormal [35]. This residual phenotypic abnormality may be due to the abnormal iPPi concentration, suggesting the importance of a balanced ePPi/iPPi ratio in the regulation of HA deposition.
In addition, the ectopic calcification and soft tissue ossification phenotypes were more severe in the Enpp1−/− mice than in the ank/ank mice. The degree of soft-tissue ossification was further increased in the [Enpp1−/−; Ank/Ank] double-mutants compared to the mice with single gene ablations [35]. Together, these observations and the absence of ANK in the MVs suggest that NPP1 is essential for the second phase of mineral deposition and that NPP1 plays a more crucial role in PPi production than ANK. Presumably, there is still a sufficient amount of PPi provided by NPP1 in the ank/ank mice [35]. This hypothesis could explain the observed differences in the phenotypic abnormalities of the [Akp2−/−; Enpp1−/−] and [Akp2−/−; Ank/Ank] mice.
[Phospho1−/−; Akp2−/−]
It is well established that the first HA crystals are formed within the MVs, and TNSALP is not essential for the initiation of mineralisation in the MVs because the MVs from hypophosphatasia patients and Akp2−/− mice contain minerals but fail to extend the mineral propagation beyond the MVs and into the surrounding collagenous matrix [30]. PHOSPHO1, which is sequestered within the MV lumen, has been proposed to be responsible for the first step of the MV-mediated initiation of mineralisation by increasing the intravesicular Pi concentration [74]. Transgenic overexpression TNSALP does not rescue the skeletal abnormalities in Phospho1−/− mice, despite correcting their plasma PPi levels. The disruption of both TNSALP and PHOSPHO1 in mouse embryos showed an almost complete absence of skeletal mineralisation (i.e., both bone and cartilage), suggesting that TNSALP and PHOSPHO1 have independent and non-redundant roles for skeletal mineralisation in the mouse [76]. It is also worth noting that Phospho1−/− mice showed small increases in NPP1 activity and ANK expression, as well as a slight reduction in plasma TNSALP levels [76]. However, in a single stillborn [Phospho1−/−; Akp2−/−] pup, some calcification was observed in the axial skeleton [76].
Recently, research was carried out to analyse the kinetic behaviour of isolated wild-type (WT) osteoblast-derived MVs, as well as TNSALP-, NPP1-, and PHOSPHO1-deficient MVs, when confronted with physiologic substrates (e.g., ATP, ADP, and PPi) at the physiologic pH [92]. At the MV level, the data indicated that TNSALP was not only an efficient PPiase, but it also functioned as a potent ATPase and ADPase. In addition to its PPi-generating activity, NPP1 can also act as an efficient phosphatase, generating Pi from ATP, ADP, and PPi. However, this activity was evident only in the absence of TNSALP [92]. In contrast, when confronted with ATP, ADP, and PPi, PHOSPHO1 is not an efficient phosphatase, as it requires cooperation with PiT1/2 to achieve a sufficient Pi concentration for crystallisation in the MVs [92]. This research study first proposed that TNSALP has major roles as a pyrophosphatase, ATPase, and ADPase, thus participating in the calcification process not only by the restricting the concentration of ePPi, but also by simultaneously contributing to the Pi pool available for calcification via its ATPase, ADPase, and PPiase functions. This new data helped to explain why Akp2−/− mice, which model infantile HPP, displayed a normal phenotype immediately after birth and had a phenotype that was less severe than the lethal and perinatal HPP [23, 30]. Despite the absence of TNSALP, NPP1 can act as a backup pyrophosphatase to control the ePPi concentration, allowing the Akp2−/− mice to carry out normal mineralisation. However, this compensatory effect was temporary; after few days, the Akp2−/− mice developed progressive hypomineralisation with age [30]. The partial and temporary compensatory pyrophosphatase activity of NPP1 also explains the partial mineralisation of the axial skeleton in the single stillborn [Phospho1−/−; Akp2−/−] pup. The deletion of PHOSPHO1 would depress intravesicular Pi levels, but a sufficient intravesicular Pi concentration can also be achieved by the activity of PiT1/2. The influx of ePi could be produced primarily by the ATPase activity of TNSALP or, in the absence of TNSALP, by the ATPase activity of NPP1. This compensatory mechanism could explain why the Phospho1−/− MVs only displayed a decrease rather than a complete loss of calcification [76].
Interplay between PPi and OPN in the inhibition of mineralisation
OPN, which was first identified in an osteosarcoma as a matrix protein with strong mineral binding properties [30, 93-94], has been studied as a mineral inhibitor that is upregulated by inflammation, atherosclerosis, and vascular calcification [95-96]. Recently, a link between the proteins regulating ePPi and OPN has been widely investigated. The ank/ank and Enpp1−/− mice showed a decrease in ePPi and OPN levels, and the subsequent hypermineralisation was particularly associated with the joints and ligaments [42, 54, 97]. Extracellular PPi downregulation resulted in a concomitant OPN deficiency, and the correction of the OPN deficiency prevents hypercalcification, suggesting the synergistic inhibition of HA deposition by PPi and OPN [97]. In addition to being downregulated in Enpp1−/− and ank/ank mice, OPN expression was upregulated in Akp2−/− mice and was normalised in [Akp2−/−; Enpp1−/−] and [Akp2−/−; ank/ank] mice, which corresponded with a full or partial normalisation of mineralisation defects and PPi levels and suggests that PPi can regulate OPN expression [35]. Furthermore, PPi modulates Opn expression in osteoblasts, as the addition of exogenous PPi to WT calvarial osteoblasts caused an increase in Opn mRNA level, and down-regulated expression of both Enpp1 and Ank[35]. Thus, it was proposed that the hypo- and hypermineralisation phenotypes observed individually in the Akp2−/− mice, Enpp1−/−, and ank/ank mice were due to the combined effects of at least two mineralisation inhibitors, including PPi and OPN. It is interesting to note that the majority of studies reported that Opn−/− mice display normal development and bone structure, with no histological differences from WT mice [98], whereas other studies reported mild mineralisation abnormalities and more minerals in the Opn−/− mice than in WT controls [99-101]. The primary osteoblasts from Opn−/− mice showed markedly elevated levels of ePPi, which were even higher than in the Akp2−/− mice, attributable to an increase in Enpp1 and Ank expression and a concomitant downregulation of Akp2 expression [101]. The addition of exogenous OPN to Opn−/− mice showed the opposite result compared to the Opn−/− mice [101]. Thus, OPN likely plays a role in ePPi's production, transport, and degradation through the modulation of Akp2, Ank, and Enpp1 expression. Together with the exogenous PPi can upregulate Opn expression in WT osteoblasts [35], these datas indicated that there was a feedback mechanism between PPi and OPN. Recently, an in vitro study proposed that PPi inhibits the mineralisation of osteoblast cultures using at least three distinct mechanisms, including direct binding to minerals, increased Opn expression, and decreased ALP activity, which is consistent with the observations of the OPN−/− mice [102].
Given that both Akp2−/− and Opn−/− single knockout mice displayed elevated levels of ePPi, it was surprising that the ePPi levels of [Opn−/−; Akp2−/−] mice were midway between the values of these single knockout mice. As previously reported, the Akp2−/− osteoblast-derived MVs contained only half the amount of NPP1 compared with the WT MVs, and the transfection of Akp2 up-regulated Enpp1 expression [97], suggesting that lower NPP1 levels in the Akp2−/− mice may contribute the intermediate ePPi levels in the [Opn−/−; Akp2−/−] mice [101]. In addition, [Opn−/−; Akp2−/−] mice showed a partial correction of the bone hypomineralisation phenotype observed in the Akp2−/− mice. These findings, along with the observation that the [Akp2−/−; Enpp1−/−] and [Akp2−/−; ank/ank] mice exhibit corrected PPi and OPN levels, support the conclusion that, for a complete rescue of the hypomineralisation phenotype in the Akp2−/− mice, the concentrations of both PPi and OPN must be at normal levels [101].
Conclusions and perspectives
After decades of investigation, the MV-related proteins regulating Pi/PPi homeostasis have been shown to play a prominent role in mineral deposition and skeletal development. Studies of animal models with pathologic calcification phenotypes similar to those of their human counterparts (Table 1) strongly support the hypothesis that ePPi concentration regulates HA crystal deposition and that the Pi/PPi ratio is a key factor in the development of either physiological or pathological mineralisation. From a metabolic perspective, ePPi are derived from the TNSALP- and NPP1-catalysed production of eATP as well as by intracellular export via ANK transporters. Further, ePPi is likely to be the source of Pi necessary to sustain HA formation after being cleaved by TNSALP and NPP1. The intravesicular mineralisation propagator Pi is generated by the activity of PHOSPHO1, as well as by the transport function of PiT1/2 (Figure 1). PPi, together with OPN, inhibits HA crystal propagation and growth. Mutations of the MV-related proteins can affect Pi/PPi homeostasis, HA crystal initiation in the MVs, or crystal propagation along the extracellular collagen fibrils, resulting in hypomineralisation, hypermineralisation, or ectopic ossification at various ages. Notwithstanding our extensive knowledge of the involvement of these MV-related proteins in skeletal growth, our understanding of the interplay between TNSALP, NPP1, ANK, PHOSPHO1, and OPN in the regulation of skeletal mineralisation during growth and aging is incomplete. Other open questions regarding the functions of the MV-related proteins include their roles in the function of mineral-competent cells and in osteoclastogenesis. The last, and perhaps the most important question is how and when should intervention occur to manipulate Pi/PPi homeostasis with the goal of obtaining a healthy and more robust skeleton.
Mouse Models of Regulators of PPi/Pi and Their Human Counterparts.
| Target | Mutant name | Skeletal phenotype | PPi/Pi | OPN | Human counterpart (ref) | Ref |
|---|---|---|---|---|---|---|
| Akp2 | Akp2−/− | Born with normal, Skeletal defective first appeared radiographically at ~10 days, hypomineralization, progressive rachitic, osteopenia, and fracture | ePPi↑ | OPN↑ | Infantile hypophosphatasia (OMIM No. 241500) | [19, 29-30, 35] |
| Akp2Hpp/Hpp (862 + 5G>A) | Normal initial skeletal development and growth, late onset skeletal diseases, arthropathies | ePPi N; iPi↓ | Autosomal Semidominant Adult Hypophosphatasia (OMIM No. 146300) | [36] | ||
| Enpp1 | Enpp1 (ttw/ttw) (1813G>T) | ankylosing intervertebral, peripheral joint hyperostosis, arterial and articular cartilage calcification | ePPi↓ | Ossification of the posterior longitudinal ligament of the spine (OPLL) (OMIM No.602475) | [42, 47] | |
| Enpp1−/− | ectopic ossification, disruption to the structural and mechanical properties of long bones, severity increases with age; trabecular bone loss | ePPi↓ | OPN↓ | Not applicable | [51] | |
| Ank | AnkKI/KI | hyperostotic phenotype, significantly in craniofacial anomalies, but bone matrix is hypomineralization and less mature | ePPi N | craniometaphyseal dysplasia (CMD) (OMIM No.123000) | [67] | |
| Anknull/null and Ankank/ank | Arthritis, bony and joint fusion, loss of mobility | ePPi↓; iPPi↑ | OPN↓ | Not applicable | [35, 64] | |
| Phospho1 | Phospho1−/− | poor weight gain, growth plate and skeletal abnormalities, and thoracic scoliosis | Intravesicular Pi↓; ePPi↑ | Not applicable | [76] | |
| PiT1 | PiT1Δ5/Δ5 | embryonic lethality, no evidence defects in early skeleton formation | Not applicable | [90] | ||
| Opn | Opn−/− | Normal bone development, but mild mineral deposition | ePPi↑ | Not applicable | [98-99, 101] | |
| Double- knockouts | [Akp2−/−; Enpp1−/−] | Correct hypo- and hypermineralization in calvaria and spine, long bone sustain osteomalacia | ePPi N | OPN N | [7, 91] | |
| [Akp2−/−; Ank/Ank] | partial correction in the hypo- and hypermineralization | ePPi N; iPPi↑ | OPN N | [35] | ||
| [Akp2−/−; Phospho1−/−] | complete absence of skeletal mineralization (bone and cartilage) and and perinatal lethality | Intravesicular Pi ↓; ePi ↓ | [76] | |||
| [Akp2−/−; Opn−/−] | partial correction of hypomineralization | ePPi↑ | [101] |
↑= increase; ↓= decrease; N = normal; OMIM: http://www.ncbi.nlm.nih.gov/omim
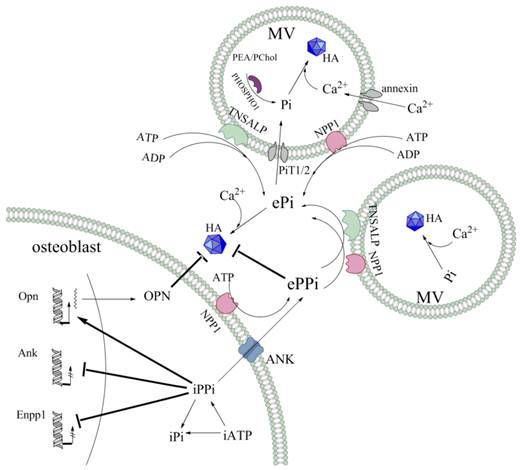
Schematic representation of Pi/PPi regulators involved in MV-mediated mineralisation. The HA crystals are composed of Pi and Ca2+. In the MVs, Pi is generated by the function of PiT1/2 phosphate transporters, as well as by the hydrolysis of the phosphomonoesters PEA and PChol by the phosphatase PHOSPHO1. Ca2+ transport is mediated by annexin channels that allow influx into the vesicles. The ePPi pool is derived both by NPP1 catalysing production from eATP and by intracellular export via the ANK transporter at the level of the chondrocyte and osteoblast membranes. For both TNSALP and NPP1, ATPase, ADPase and PPiase contribute to the ePi pool, though TNSALP is more efficient than NPP1 at the level of the MVs. Moreover, a negative feedback loop exists in intracellular. PPi, which is produced by NPP1 and transported by ANK, inhibits the expression of Enpp1 and Ank. PPi, together with its induction of OPN expression, inhibit HA crystal propagation and growth.
Abbreviations
MV: matrix vesicle; HA: hydroxyapatite; ECM: extracellular matrix; ePPi: extracellular inorganic pyrophosphate; Pi: phosphate; ALP: alkaline phosphatase; TNSALP: tissue-nonspecific alkaline phosphatase; NPP1: ectonucleotide pyrophosphatase phosphodiesterase 1; ANK: ankylosis protein; PHOSPHO1: phosphatase orphan 1; PiT1/2: sodium-dependent Pi symporters 1/2; OPN: osteopontin; BCP: basic calcium phosphate; CPPD: calcium pyrophosphate dehydrate; CPPDD: familial calcium pyrophosphate dihydrate deposition disease; HPP: hypophosphatasia; PLP: pyri-doxal-5'-phosphate; PEA: phosphoethanolamine; ARHR2: autosomal recessive hypophosphatemic rickets; OPLL: ossification of the posterior longitudinal ligament; CMD: craniometaphyseal dysplasia; PChol: phosphocholine; VSMCs: vascular smooth muscle cells.
Acknowledgements
This work was supported by the Key Project for Drug Research and Development from the Ministry of Science and Technology of China (Grant No. 2010ZX09401-302-5-07).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Anderson HC. Molecular biology of matrix vesicles. Clin Orthop Relat Res. 1995:266-80
2. Hale JE, Wuthier RE. The mechanism of matrix vesicle formation. Studies on the composition of chondrocyte microvilli and on the effects of microfilament-perturbing agents on cellular vesiculation. J Biol Chem. 1987;262:1916-25
3. Thouverey C, Strzelecka-Kiliszek A, Balcerzak M. et al. Matrix vesicles originate from apical membrane microvilli of mineralizing osteoblast-like Saos-2 cells. J Cell Biochem. 2009;106:127-38
4. Thouverey C, Malinowska A, Balcerzak M. et al. Proteomic characterization of biogenesis and functions of matrix vesicles released from mineralizing human osteoblast-like cells. J Proteomics. 2011;74:1123-34
5. Anderson HC. Matrix vesicles and calcification. Curr Rheumatol Rep. 2003;5:222-6
6. Anderson HC, Stechschulte DJ Jr, Collins DE. et al. Matrix vesicle biogenesis in vitro by rachitic and normal rat chondrocytes. Am J Pathol. 1990;136:391-8
7. Hessle L, Johnson KA, Anderson HC. et al. Tissue-nonspecific alkaline phosphatase and plasma cell membrane glycoprotein-1 are central antagonistic regulators of bone mineralization. Proc Natl Acad Sci U S A. 2002;99:9445-9
8. Terkeltaub RA. Inorganic pyrophosphate generation and disposition in pathophysiology. Am J Physiol Cell Physiol. 2001;281:C1-C11
9. Golub EE. Biomineralization and matrix vesicles in biology and pathology. Semin Immunopathol. 2011;33:409-17
10. Golub EE. Role of matrix vesicles in biomineralization. Biochim Biophys Acta. 2009;1790:1592-8
11. Xiao Z, Blonder J, Zhou M. et al. Proteomic analysis of extracellular matrix and vesicles. J Proteomics. 2009;72:34-45
12. Kirsch T. Determinants of pathological mineralization. Curr Opin Rheumatol. 2006;18:174-80
13. Murshed M, McKee MD. Molecular determinants of extracellular matrix mineralization in bone and blood vessels. Curr Opin Nephrol Hypertens. 2010;19:359-65
14. Wuthier RE, Lipscomb GF. Matrix vesicles: structure, composition, formation and function in calcification. Front Biosci. 2012;17:2812-902
15. Murshed M, Harmey D, Millan JL. et al. Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev. 2005;19:1093-104
16. Wang W, Xu J, Du B. et al. Role of the progressive ankylosis gene (ank) in cartilage mineralization. Mol Cell Biol. 2005;25:312-23
17. Fleisch H. Diphosphonates: history and mechanisms of action. Metab Bone Dis Relat Res. 1981;3:279-87
18. Kirsch T. Determinants of pathologic mineralization. Crit Rev Eukaryot Gene Expr. 2008;18:1-9
19. Anderson HC, Garimella R, Tague SE. The role of matrix vesicles in growth plate development and biomineralization. Front Biosci. 2005;10:822-37
20. Orimo H. The mechanism of mineralization and the role of alkaline phosphatase in health and disease. J Nihon Med Sch. 2010;77:4-12
21. Yamamoto S, Orimo H, Matsumoto T. et al. Prolonged survival and phenotypic correction of Akp2(-/-) hypophosphatasia mice by lentiviral gene therapy. J Bone Miner Res. 2011;26:135-42
22. Rodrigues TL, Foster BL, Silverio KG. et al. Correction of Hypophosphatasia (Hpp) Associated Mineralization Deficiencies In vitro by Phosphate/Pyrophosphate Modulation in Periodontal Ligament Cells. J Periodontol. 2012May;83(5):653-63
23. Mornet E. Hypophosphatasia. Orphanet J Rare Dis. 2007;2:40
24. Whyte MP, Kurtzberg J, McAlister WH. et al. Marrow cell transplantation for infantile hypophosphatasia. J Bone Miner Res. 2003;18:624-36
25. Cahill RA, Wenkert D, Perlman SA. et al. Infantile hypophosphatasia: transplantation therapy trial using bone fragments and cultured osteoblasts. J Clin Endocrinol Metab. 2007;92:2923-30
26. Tadokoro M, Kanai R, Taketani T. et al. New bone formation by allogeneic mesenchymal stem cell transplantation in a patient with perinatal hypophosphatasia. J Pediatr. 2009;154:924-30
27. Whyte MP, Greenberg CR, Salman NJ. et al. Enzyme-replacement therapy in life-threatening hypophosphatasia. N Engl J Med. 2012;366:904-13
28. Waymire KG, Mahuren JD, Jaje JM. et al. Mice lacking tissue non-specific alkaline phosphatase die from seizures due to defective metabolism of vitamin B-6. Nat Genet. 1995;11:45-51
29. Narisawa S, Frohlander N, Millan JL. Inactivation of two mouse alkaline phosphatase genes and establishment of a model of infantile hypophosphatasia. Dev Dyn. 1997;208:432-46
30. Fedde KN, Blair L, Silverstein J. et al. Alkaline phosphatase knock-out mice recapitulate the metabolic and skeletal defects of infantile hypophosphatasia. J Bone Miner Res. 1999;14:2015-26
31. Tesch W, Vandenbos T, Roschgr P. et al. Orientation of mineral crystallites and mineral density during skeletal development in mice deficient in tissue nonspecific alkaline phosphatase. J Bone Miner Res. 2003;18:117-25
32. Millan JL, Narisawa S, Lemire I. et al. Enzyme replacement therapy for murine hypophosphatasia. J Bone Miner Res. 2008;23:777-87
33. Kovacs CS, Kronenberg HM. Maternal-fetal calcium and bone metabolism during pregnancy, puerperium, and lactation. Endocr Rev. 1997;18:832-72
34. Wennberg C, Hessle L, Lundberg P. et al. Functional characterization of osteoblasts and osteoclasts from alkaline phosphatase knockout mice. J Bone Miner Res. 2000;15:1879-88
35. Harmey D, Hessle L, Narisawa S. et al. Concerted regulation of inorganic pyrophosphate and osteopontin by akp2, enpp1, and ank: an integrated model of the pathogenesis of mineralization disorders. Am J Pathol. 2004;164:1199-209
36. Hough TA, Polewski M, Johnson K. et al. Novel mouse model of autosomal semidominant adult hypophosphatasia has a splice site mutation in the tissue nonspecific alkaline phosphatase gene Akp2. J Bone Miner Res. 2007;22:1397-407
37. Johnson KA, Hessle L, Vaingankar S. et al. Osteoblast tissue-nonspecific alkaline phosphatase antagonizes and regulates PC-1. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1365-77
38. Terkeltaub R. Physiologic and pathologic functions of the NPP nucleotide pyrophosphatase/phosphodiesterase family focusing on NPP1 in calcification. Purinergic Signal. 2006;2:371-7
39. Saito T, Shimizu Y, Hori M. et al. A patient with hypophosphatemic rickets and ossification of posterior longitudinal ligament caused by a novel homozygous mutation in ENPP1 gene. Bone. 2011;49:913-6
40. Lorenz-Depiereux B, Schnabel D, Tiosano D. et al. Loss-of-function ENPP1 mutations cause both generalized arterial calcification of infancy and autosomal-recessive hypophosphatemic rickets. Am J Hum Genet. 2010;86:267-72
41. Levy-Litan V, Hershkovitz E, Avizov L. et al. Autosomal-recessive hypophosphatemic rickets is associated with an inactivation mutation in the ENPP1 gene. Am J Hum Genet. 2010;86:273-8
42. Okawa A, Nakamura I, Goto S. et al. Mutation in Npps in a mouse model of ossification of the posterior longitudinal ligament of the spine. Nat Genet. 1998;19:271-3
43. Baba H, Furusawa N, Fukuda M. et al. Potential role of streptozotocin in enhancing ossification of the posterior longitudinal ligament of the cervical spine in the hereditary spinal hyperostotic mouse (twy/twy). Eur J Histochem. 1997;41:191-202
44. Furusawa N, Baba H, Imura S. et al. Characteristics and mechanism of the ossification of posterior longitudinal ligament in the tip-toe walking Yoshimura (twy) mouse. Eur J Histochem. 1996;40:199-210
45. Okawa A, Goto S, Moriya H. Calcitonin simultaneously regulates both periosteal hyperostosis and trabecular osteopenia in the spinal hyperostotic mouse (twy/twy) in vivo. Calcif Tissue Int. 1999;64:239-47
46. Sakamoto M, Hosoda Y, Kojimahara K. et al. Arthritis and ankylosis in twy mice with hereditary multiple osteochondral lesions: with special reference to calcium deposition. Pathol Int. 1994;44:420-7
47. Nakamura I, Ikegawa S, Okawa A. et al. Association of the human NPPS gene with ossification of the posterior longitudinal ligament of the spine (OPLL). Hum Genet. 1999;104:492-7
48. Furuya S, Ohtsuki T, Yabe Y. et al. Ultrastructural study on calcification of cartilage: comparing ICR and twy mice. J Bone Miner Metab. 2000;18:140-7
49. Rutsch F, Vaingankar S, Johnson K. et al. PC-1 nucleoside triphosphate pyrophosphohydrolase deficiency in idiopathic infantile arterial calcification. Am J Pathol. 2001;158:543-54
50. Rutsch F, Ruf N, Vaingankar S. et al. Mutations in ENPP1 are associated with 'idiopathic' infantile arterial calcification. Nat Genet. 2003;34:379-81
51. Johnson K, Polewski M, van Etten D. et al. Chondrogenesis mediated by PPi depletion promotes spontaneous aortic calcification in NPP1-/- mice. Arterioscler Thromb Vasc Biol. 2005;25:686-91
52. Mackenzie NC, Zhu D, Milne EM. et al. Altered bone development and an increase in FGF-23 expression in Enpp1(-/-) mice. PLoS One. 2012;7:e32177
53. Nam HK, Liu J, Li Y. et al. Ectonucleotide pyrophosphatase/phosphodiesterase-1 (ENPP1) protein regulates osteoblast differentiation. J Biol Chem. 2011;286:39059-71
54. Ho AM, Johnson MD, Kingsley DM. Role of the mouse ank gene in control of tissue calcification and arthritis. Science. 2000;289:265-70
55. Gurley KA, Reimer RJ, Kingsley DM. Biochemical and genetic analysis of ANK in arthritis and bone disease. Am J Hum Genet. 2006;79:1017-29
56. Reichenberger E, Tiziani V, Watanabe S. et al. Autosomal dominant craniometaphyseal dysplasia is caused by mutations in the transmembrane protein ANK. Am J Hum Genet. 2001;68:1321-6
57. Nurnberg P, Thiele H, Chandler D. et al. Heterozygous mutations in ANKH, the human ortholog of the mouse progressive ankylosis gene, result in craniometaphyseal dysplasia. Nat Genet. 2001;28:37-41
58. Zaka R, Williams CJ. Role of the progressive ankylosis gene in cartilage mineralization. Curr Opin Rheumatol. 2006;18:181-6
59. Pendleton A, Johnson MD, Hughes A. et al. Mutations in ANKH cause chondrocalcinosis. Am J Hum Genet. 2002;71:933-40
60. Williams CJ, Zhang Y, Timms A. et al. Autosomal dominant familial calcium pyrophosphate dihydrate deposition disease is caused by mutation in the transmembrane protein ANKH. Am J Hum Genet. 2002;71:985-91
61. Zaka R, Stokes D, Dion AS. et al. P5L mutation in Ank results in an increase in extracellular inorganic pyrophosphate during proliferation and nonmineralizing hypertrophy in stably transduced ATDC5 cells. Arthritis Res Ther. 2006;8:R164
62. Kim HJ, Minashima T, McCarthy EF. et al. Progressive ankylosis protein (ANK) in osteoblasts and osteoclasts controls bone formation and bone remodeling. J Bone Miner Res. 2010;25:1771-83
63. Williams CJ. Familial calcium pyrophosphate dihydrate deposition disease and the ANKH gene. Curr Opin Rheumatol. 2003;15:326-31
64. Gurley KA, Chen H, Guenther C. et al. Mineral formation in joints caused by complete or joint-specific loss of ANK function. J Bone Miner Res. 2006;21:1238-47
65. Sweet HO, Green MC. Progressive ankylosis, a new skeletal mutation in the mouse. J Hered. 1981;72:87-93
66. Hakim FT, Cranley R, Brown KS. et al. Hereditary joint disorder in progressive ankylosis (ank/ank) mice. I. Association of calcium hydroxyapatite deposition with inflammatory arthropathy. Arthritis Rheum. 1984;27:1411-20
67. Chen IP, Wang CJ, Strecker S. et al. Introduction of a Phe377del mutation in ANK creates a mouse model for craniometaphyseal dysplasia. J Bone Miner Res. 2009;24:1206-15
68. Chen IP, Wang L, Jiang X. et al. A Phe377del mutation in ANK leads to impaired osteoblastogenesis and osteoclastogenesis in a mouse model for craniometaphyseal dysplasia (CMD). Hum Mol Genet. 2011;20:948-61
69. Houston B, Seawright E, Jefferies D. et al. Identification and cloning of a novel phosphatase expressed at high levels in differentiating growth plate chondrocytes. Biochim Biophys Acta. 1999;1448:500-6
70. Stewart AJ, Schmid R, Blindauer CA. et al. Comparative modelling of human PHOSPHO1 reveals a new group of phosphatases within the haloacid dehalogenase superfamily. Protein Eng. 2003;16:889-95
71. Roberts SJ, Stewart AJ, Sadler PJ. et al. Human PHOSPHO1 exhibits high specific phosphoethanolamine and phosphocholine phosphatase activities. Biochem J. 2004;382:59-65
72. Kvam BJ, Pollesello P, Vittur F. et al. 31P NMR studies of resting zone cartilage from growth plate. Magn Reson Med. 1992;25:355-61
73. Stewart AJ, Roberts SJ, Seawright E. et al. The presence of PHOSPHO1 in matrix vesicles and its developmental expression prior to skeletal mineralization. Bone. 2006;39:1000-7
74. Roberts S, Narisawa S, Harmey D. et al. Functional involvement of PHOSPHO1 in matrix vesicle-mediated skeletal mineralization. J Bone Miner Res. 2007;22:617-27
75. Macrae VE, Davey MG, McTeir L. et al. Inhibition of PHOSPHO1 activity results in impaired skeletal mineralization during limb development of the chick. Bone. 2010;46:1146-55
76. Yadav MC, Simao AM, Narisawa S. et al. Loss of skeletal mineralization by the simultaneous ablation of PHOSPHO1 and alkaline phosphatase function: a unified model of the mechanisms of initiation of skeletal calcification. J Bone Miner Res. 2011;26:286-97
77. Bottger P, Pedersen L. Mapping of the minimal inorganic phosphate transporting unit of human PiT2 suggests a structure universal to PiT-related proteins from all kingdoms of life. BMC Biochem. 2011;12:21
78. Collins JF, Bai L, Ghishan FK. The SLC20 family of proteins: dual functions as sodium-phosphate cotransporters and viral receptors. Pflugers Arch. 2004;447:647-52
79. Kavanaugh MP, Miller DG, Zhang W. et al. Cell-surface receptors for gibbon ape leukemia virus and amphotropic murine retrovirus are inducible sodium-dependent phosphate symporters. Proc Natl Acad Sci U S A. 1994;91:7071-5
80. Lau WL, Festing MH, Giachelli CM. Phosphate and vascular calcification: Emerging role of the sodium-dependent phosphate co-transporter PiT-1. Thromb Haemost. 2010;104:464-70
81. Yoshiko Y, Candeliere GA, Maeda N. et al. Osteoblast autonomous Pi regulation via Pit1 plays a role in bone mineralization. Mol Cell Biol. 2007;27:4465-74
82. Villa-Bellosta R, Ravera S, Sorribas V. et al. The Na+-Pi cotransporter PiT-2 (SLC20A2) is expressed in the apical membrane of rat renal proximal tubules and regulated by dietary Pi. Am J Physiol Renal Physiol. 2009;296:F691-9
83. Breusegem SY, Takahashi H, Giral-Arnal H. et al. Differential regulation of the renal sodium-phosphate cotransporters NaPi-IIa, NaPi-IIc, and PiT-2 in dietary potassium deficiency. Am J Physiol Renal Physiol. 2009;297:F350-61
84. Palmer G, Zhao J, Bonjour J. et al. In vivo expression of transcripts encoding the Glvr-1 phosphate transporter/retrovirus receptor during bone development. Bone. 1999;24:1-7
85. Jono S, McKee MD, Murry CE. et al. Phosphate regulation of vascular smooth muscle cell calcification. Circ Res. 2000;87:E10-7
86. Li X, Yang HY, Giachelli CM. Role of the sodium-dependent phosphate cotransporter, Pit-1, in vascular smooth muscle cell calcification. Circ Res. 2006;98:905-12
87. Honjo S, Yokote K, Fujimoto M. et al. Clinical outcome and mechanism of soft tissue calcification in Werner syndrome. Rejuvenation Res. 2008;11:809-19
88. Suzuki A, Ammann P, Nishiwaki-Yasuda K. et al. Effects of transgenic Pit-1 overexpression on calcium phosphate and bone metabolism. J Bone Miner Metab. 2010;28:139-48
89. Festing MH, Speer MY, Yang HY. et al. Generation of mouse conditional and null alleles of the type III sodium-dependent phosphate cotransporter PiT-1. Genesis. 2009;47:858-63
90. Beck L, Leroy C, Beck-Cormier S. et al. The phosphate transporter PiT1 (Slc20a1) revealed as a new essential gene for mouse liver development. PLoS One. 2010;5:e9148
91. Anderson HC, Harmey D, Camacho NP. et al. Sustained osteomalacia of long bones despite major improvement in other hypophosphatasia-related mineral deficits in tissue nonspecific alkaline phosphatase/nucleotide pyrophosphatase phosphodiesterase 1 double-deficient mice. Am J Pathol. 2005;166:1711-20
92. Ciancaglini P, Yadav MC, Simao AM. et al. Kinetic analysis of substrate utilization by native and TNAP-, NPP1-, or PHOSPHO1-deficient matrix vesicles. J Bone Miner Res. 2010;25:716-23
93. Sodek J, Ganss B, McKee MD. Osteopontin. Crit Rev Oral Biol Med. 2000;11:279-303
94. Oldberg A, Franzen A, Heinegard D. Cloning and sequence analysis of rat bone sialoprotein (osteopontin) cDNA reveals an Arg-Gly-Asp cell-binding sequence. Proc Natl Acad Sci U S A. 1986;83:8819-23
95. Cho HJ, Kim HS. Osteopontin: a multifunctional protein at the crossroads of inflammation, atherosclerosis, and vascular calcification. Curr Atheroscler Rep. 2009;11:206-13
96. Hunter GK, Kyle CL, Goldberg HA. Modulation of crystal formation by bone phosphoproteins: structural specificity of the osteopontin-mediated inhibition of hydroxyapatite formation. Biochem J. 1994;300( Pt 3):723-8
97. Johnson K, Goding J, Van Etten D. et al. Linked deficiencies in extracellular PP(i) and osteopontin mediate pathologic calcification associated with defective PC-1 and ANK expression. J Bone Miner Res. 2003;18:994-1004
98. Rittling SR, Matsumoto HN, McKee MD. et al. Mice lacking osteopontin show normal development and bone structure but display altered osteoclast formation in vitro. J Bone Miner Res. 1998;13:1101-11
99. Boskey AL, Spevak L, Paschalis E. et al. Osteopontin deficiency increases mineral content and mineral crystallinity in mouse bone. Calcif Tissue Int. 2002;71:145-54
100. Shapses SA, Cifuentes M, Spevak L. et al. Osteopontin facilitates bone resorption, decreasing bone mineral crystallinity and content during calcium deficiency. Calcif Tissue Int. 2003;73:86-92
101. Harmey D, Johnson KA, Zelken J. et al. Elevated skeletal osteopontin levels contribute to the hypophosphatasia phenotype in Akp2(-/-) mice. J Bone Miner Res. 2006;21:1377-86
102. Addison WN, Azari F, Sorensen ES. et al. Pyrophosphate inhibits mineralization of osteoblast cultures by binding to mineral, up-regulating osteopontin, and inhibiting alkaline phosphatase activity. J Biol Chem. 2007;282:15872-83
Author contact
![]() Corresponding author: Jinxiang Han, Ph.D., Shandong Academy of Medical Sciences, Shandong Medical Biotechnological Center, Key Laboratory for Rare Disease Research of Shandong Province, China, and Key Laboratory for Biotech Drugs of the Ministry of Health, China, Ji'nan 250062, Shandong, China. Tel: 86-531-82919888/ Fax: 86-531-82951586; E-mail: samshjxcom
Corresponding author: Jinxiang Han, Ph.D., Shandong Academy of Medical Sciences, Shandong Medical Biotechnological Center, Key Laboratory for Rare Disease Research of Shandong Province, China, and Key Laboratory for Biotech Drugs of the Ministry of Health, China, Ji'nan 250062, Shandong, China. Tel: 86-531-82919888/ Fax: 86-531-82951586; E-mail: samshjxcom