Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2012; 8(6):901-912. doi:10.7150/ijbs.4554 This issue Cite
Research Paper
Oridonin Up-regulates Expression of P21 and Induces Autophagy and Apoptosis in Human Prostate Cancer Cells
Xiang Li1, Xiang Li2, Jiaxiong Wang1, Zaiyuan Ye2, Ji-Cheng Li1 ![]()
1. Institute of Cell Biology, Zhejiang University, Hangzhou 310058;
2. Key Laboratory of Oncology, Zhejiang Province People's Hospital, Hangzhou 310012, China.
Received 2012-5-10; Accepted 2012-6-11; Published 2012-6-22
Abstract
Background: Oridonin (ORI) could inhibit the proliferation and induce apoptosis in various cancer cell lines. However, the mechanism is not fully understood.
Methods: Human prostate cancer (HPC) cells were cultured in vitro and cell viability was detected by the CCK-8 assay. The ultrastructure changes were observed under transmission electron microscope (TEM). Chemical staining with acridine orange (AO), MDC or DAPI was used to detect acidic vesicular organelles (AVOs) and alternation of DNA. Expression of LC3 and P21 was detected by Western Blot. Apoptotic rates and cell cycle arrest were detected by FACS.
Results: Our study demonstrated that after ORI treatment, the proliferations of human prostate cancer (HPC) cell lines PC-3 and LNCaP were inhibited in a concentration and time-dependent manner. ORI induced cell cycle arrest at the G2/M phase. A large number of autophagosomes with double-membrane structure and acidic vesicular organelles (AVOs) were detected in the cytoplasm of HPC cells treated with ORI for 24 hours. ORI resulted in the conversion of LC3-I to LC3-II and recruitment of LC3-II to the autophagosomal membranes. Autophagy inhibitor 3-methyladenine (3-MA) reduced AVOs formation and inhibited LC3-I to LC3-II conversion. At 48 h, DNA fragmentation, chromatin condensation and disappearance of surface microvilli were detected in ORI-treated cells. ORI induced a significant increase in the number of apoptotic cells (PC-3: 5.4% to 27.0%, LNCaP: 5.3% to 31.0%). Promoting autophagy by nutrient starvation increased cell viability, while inhibition of autophagy by 3-MA promoted cell death. The expression of P21 was increased by ORI, which could be completely reversed by the inhibition of autophagy.
Conclusions: Our findings indicated that autophagy occurred before the onset of apoptosis and protected cancer cells in ORI-treated HPC cells. P21 was involved in ORI-induced autophagy and apoptosis. Our results provide an experimental basis for understand the anti-tumor mechanism of ORI as treatment for prostate cancer.
Keywords: oridonin, autophagy, apoptosis, prostate cancer, P21.
Introduction
Prostate cancer is one of the most common cancers and the second most common cause of cancer-related deaths in men. Orchiectomy and hormone therapy are used to treat prostate cancer in current clinical practice. But, to date, there is no effective therapy for hormone-independent prostate cancer[1]. PC-SPES, a herbal mixture, has been widely used for the treatment of hormone-responsive and hormone-refractory prostate cancer[2]. Oridonin (ORI), one of the active ingredients in PC-SPES, has various pharmacological and physiological effects. It could inhibit the proliferation of several tumor cell lines by inducing cell cycle arrest and apoptosis. Thus, ORI is emerging as a promising anti-tumor agent.
In recent years, ORI has been reported that ORI could induce autophagy in HeLa [3], MCF-7 [4], A431 [5], L929 [6] and several other cancer cell lines. Autophagy is characterized by sequestration of bulk cytoplasm and organelles in autophagosomes, and their delivery to and subsequent degradation by the cell's own lysosomal system[7]. Microtubule-associated protein light chain 3 (MAP-LC3) is a critical marker for autophagic process. The amount of LC3-II or the ratio of LC3-II to LC3-I can reflect the autophagic activity.
Autophagy plays an important role in many physiological processes, including the response to starvation, cell growth control, and anti-aging mechanisms[8]. Excessive autophagy may, however, lead to cell death referred to as type II programmed cell death[9]. So, autophagy has possible roles in cell survival and death. The activation of autophagy to inhibit cell proliferation would be an important molecular mechanism for many anti-tumor drugs. Previous studies have reported that the autophagic capacity of cancer cell lines is lower than their normal counterparts. In fact, studies of carcinogen-induced pancreatic cancer in rats have shown that pancreatic adenocarcinoma cells have lower autophagic activity during tumor progression[10]. On the other hand, autophagy can act as a mechanism of cell survival during starvation. In addition, autophagy is activated as a defense mechanism in response to many cancer therapeutics[11].
The P21 gene has been widely studied as an anti-tumor gene. It is regulated directly by the P53 gene, and involved in the P53-mediated DNA-damaging response. In addition, P21 is also known as a cyclin-dependent kinase (CDK) inhibitor, and can arrest cell cycle by inhibiting the formation of cyclin-CDK complex. Tasdemir et al. have suggested the role of P53 in the regulation of autophagy[12]. However, the relationship between P21 and autophagy is still not understood.
In the present study, we detected the ORI-induced autophagy and the role of autophagy in cell death in PC-3 (androgen-independent) and LNCaP (androgen-dependent) prostate cancer cell lines. The expression levels of P21 were investigated to elucidate the relation of ORI-induced autophagy and apoptosis in prostate cancer.
Materials and Methods
Reagent
ORI was purchased from Wuhan Botanical Garden, Chinese Academy of Sciences. Cell Counting Kit-8 was obtained from Dojindo Laboratories (Kumamoto, Japan). Acridine orange (AO), monodansylcadervarine (MDC), 4,6-diamidino-2-phenylindole (DAPI), 3-methyladenine (3-MA) and rabbit polyclonal antibody against LC3 were obtained from Sigma (St. Louis, USA). Mouse monoclonal antibody against GAPDH and rabbit polyclonal antibody against P21 were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Lipofectamine 2000 was obtained from Invitrogen (Carlsbad, CA, USA). Annexin V-FITC Kit was purchased from Bender MedSystems (Vienna, Austria).
Cell Lines and Cell Culture
PC-3 and LNCaP cell lines were purchased from ATCC, and were cultured in F12 (GIBCO) and DMEM (HyClone) respectively, both supplemented with 10% (v/v) non-heat-inactivated fetal bovine serum (FBS, from ExCell Biology) and 100U/L penicillin-streptomycin. Each cell line was maintained at 37°C in an atmosphere of 5% CO2. Stock solution of ORI was prepared in DMSO at the concentration of 100mmol/L, and an equal volume of DMSO was added to the control.
The CCK-8 Assay
Cell suspension of 100 μl was dispensed (1×104 cells/ well) in 96-well plates. The plates were pre-incubated for 24 h, followed by the treatments of either DMSO (control) or various concentrations of ORI for 6, 12, 24, or 48 h, and added 10 μl of CCK-8 solution (Dojindo Laboratories) to each well of the plate. Incubate the plate for 1 h in the incubator. The absorbance was measured at 450 nm using a microplate spectrophotometer (BIO-RAD xMark).
DAPI Staining
ORI-treated cells in 96-well plate were fixed with 4% paraformaldehyde for 10 min at 37°C, and then stained with DAPI for 10 min, observed under the fluorescence microscope (OLYMPUS IX71).
Transmission Electron Microscopy (TEM) Analysis
TEM analysis was performed as described previously[13]. Briefly, cells were harvested after treatment of DMSO (control) or ORI, fixed in ice-cold 2.5% glutaraldehyde in PBS (pH 7.3) for 2 h, post-fixed in 1% osmium retraoxide and dehydrated in a graded series of ethanol (50-100%) and acetone, and embedded in Epon 812. Semithin sections were cut, double stained with uranyl acetate and lead citrate, and examined on Philips TECNAI 10 TEM.
FACS Analysis of Apoptosis and Cell Cycle Arrest
Cells were seeded onto 6-well plate and incubated overnight. The cells were treated with DMSO or ORI for 24 or 48 h and collected. For apoptotic analysis, cells were washed in PBS, and re-suspended in pre-diluted binding buffer in a density of 2-5×105/ml. Cell suspension of 195 μl was taken and added 5 μl Annexin V-FITC. After incubated 10 min at room temperature, cells were washed and suspended in 190 μl binding buffer. Then added 10 μl of the propidium iodide [PI] (20 μg/ml) and analyzed by FAC Scan. For cell cycle analysis, cells were washed and fixed with ice-cold 75% (v/v) ethanol at -20°C for 2 h, then stained with PI at the concentration of 50 μg/mL in the presence of RNase A (100 μg/mL). DNA content was analyzed.
Vital Staining with AO or MDC
ORI-treated cells cultured in 96-well plate were stained with AO (5 μg/mL) or MDC (0.05 mmol/L) directly for 10 min at 37°C, and then observed under the fluorescence microscope (OLYMPUS IX71).
Western Blot Analysis
Following the different treatments, cells were lysed with lysing solution containing 150 mmol/L NaCl, 50 mmol/L Tris-HCl (pH 8.0), 0.5% deoxycholic acid, 1%NP-40, 0.1% SDS and protease inhibitor cocktail (0.1 mmol/L PMSF, 1 mmol/L Na3VO4, 100 μmol/L lepeptin) on ice for 30 min. Cell lysates was cleared by centrifugation at 12000 rpm for 15 min at 4°C. The protein concentration was determined by Bicinchoninic acid protein assay kit (Pierce). Equal proteins were separated by 12% SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membrane (Millipore). After blocked with a solution containing 10 mmol/L Tris, 150 mmol/L NaCl, 0.1% Tween 20 (TBS-T) and 5% non-fat dry milk for 4 h at room temperature, the membrane was incubated with the respective primary antibody at 4°C overnight. Then it was treated with appropriate secondary antibody for 2 h at room temperature. The immunoblots were visualized by enhanced chemiluminescent substrate (Thermo).
Transient Transfection and Confocal Microscopy Analysis
The tandem fluorescent-tagged LC3 construct (tfLC3), which has been described previously, was provided by Dr. Tamotsu Yoshimori (national institute of Genetics, Shizuoka-ken, Japan). The transient transfection was performed using Lipofectamine Plus reagents (Invitrogen) according to the manufacturer's protocol. The transfected cells were examined by a confocal microscopy (Zeiss lsm-510).
Statistical Analysis
All data and results presented were confirmed in at least three independent experiments, unless otherwise indicated. The data are expressed as mean ± S.D. Student's t-distribution probability density function (Statistical Package for Social Sciences for Windows) was used for calculation of P-value. Differences were considered significant at P<0.05.
Results
Cytotoxicity of ORI in HPC Cells
After treatment with different concentrations (10, 25, 40, 60, 100 μmol/L) at different times (12, 24, 48 and 72 h), the anti-tumor activity of ORI was assessed in PC-3 and LNCaP HPC cell lines by CCK-8 kit. ORI induced cell growth inhibition in a concentration and time-dependent manner in both cell lines, while relative concentration of DMSO (≤ 0.1%) has little influence on cell proliferation. The IC50 of ORI treatment at 48 h for PC-3 and LNCAP cell lines were approximately 25μmol/L (Fig.1A).
ORI inhibited the proliferation of HPC cells. A: Cell viability assay. HPC cells in 96-well plates were treated with different concentrations of ORI or DMSO for different time, and cell viability was determined using the CCK-8 assay (10 μl/well CCK-8 solution for 1 h). B: ORI reduced cell density. Cells in 96-well plate were treated by ORI or DMSO for 24 or 48 h, and observed under phase-contrast microscope (×200). C: Cell cycle was arrested at G2/M phase by ORI. Cells treated with 25 μmol/L ORI or DMSO for 24 h were fixed with ice-cold 75% ethanol (-20°C for 2 h), and stained with PI solution of 50 μg/ml. DNA content was analyzed by FACS. The data in A and C are presented as means ± S.D. from three independent experiments.
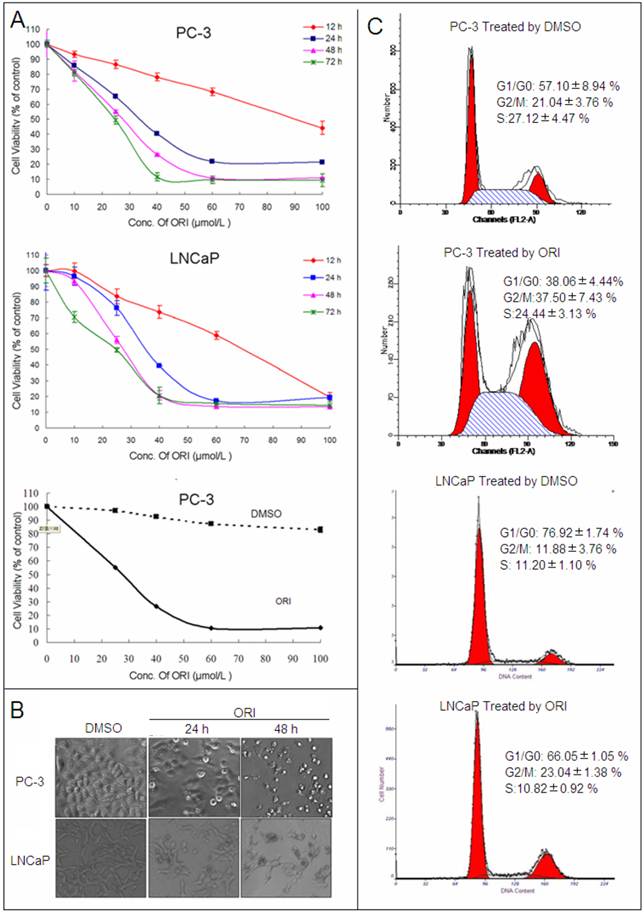
Under phase-contrast microscope, the number of ORI-treated HPC cells decreased at 24 h. At 48 h, it decreased significantly and the majority of cells became rounded and smaller in size (Fig. 1B). The cell cycle was analyzed by PI staining and FACS. In PC-3 cells, ORI treatment for 24 h induced an increase in the cell numbers at G2/M phase (21.04±3.76% to 37.50±7.43%). Similar cell cycle arrest was also detected in LNCaP cells (11.88±0.64% to 23.04±1.38%). Meanwhile, sub-G1 peak was detected neither in PC-3 nor in LNCaP cells (Fig. 1C, Table 1).
Cell cycle was arrested at G2/M phase by ORI.
| PC-3 Cells | LNCap Cells | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DMSO | ORI | DMSO | ORI | ||||||||||
| Mean | S.D. | Mean | S.D. | p-value | Mean | S.D. | Mean | S.D. | p-value | ||||
| G1/G0 | 57.10 | 8.94 | 38.06 | 4.44 | 0.088 | 76.92 | 1.74 | 66.05 | 1.05 | 0.001** | |||
| G2/M | 21.04 | 3.76 | 37.50 | 7.43 | 0.200 | 11.88 | 3.76 | 23.04 | 1.38 | 0.000** | |||
| S | 27.12 | 4.47 | 24.44 | 3.13 | 0.696 | 11.20 | 1.10 | 10.82 | 0.92 | 0.666 | |||
Cells were treated by ORI(25μmol/L) or DMSO for 24h, stained by PI and detected by FACS. The data in Table 1 were from three independent experiments and analyzed using Student's test(*,P<0.05;**,P<0.01).
Change of LC3 Protein Expression
The tfLC3 plasmid was successfully transfected into PC-3 cells and fluorescent changes in the cells were observed by laser scanning confocal microscopy before and after ORI treatment. The results showed that GFP-LC-3 protein was evenly distributed in the cytoplasm in untreated PC-3 cells. While, treatment with ORI for 1 h produced a punctuate pattern for GFP-LC3 fluorescence. The spots were further enhanced after 4 h (Fig. 2A).
ORI treatment induced autophagy in HPC cells. A: LC3II protein was recruited to autophagosomsal membranes. After transfected with tfLC3 plasmid, PC-3 cells in 24-well plates were treated with ORI (25 μmol/L) and examined by confocal microscopy (×400). A punctuate pattern for GFP-LC3 fluorescence was observed. B: ORI increased LC3 expression. Cells were treated with ORI (25 μmol/L) or DMSO for 12, 24 or 48 h, then collected and subject to Western Blot. C: Increase of LC3 level was inhibited by 3-MA. Cells were treated by ORI (25 μmol/L) with or without 3-MA (5 mmol/L) for 24 h, then the expression of LC3 protein were detected by Western Blot. The data in B and C were from three independent experiments.
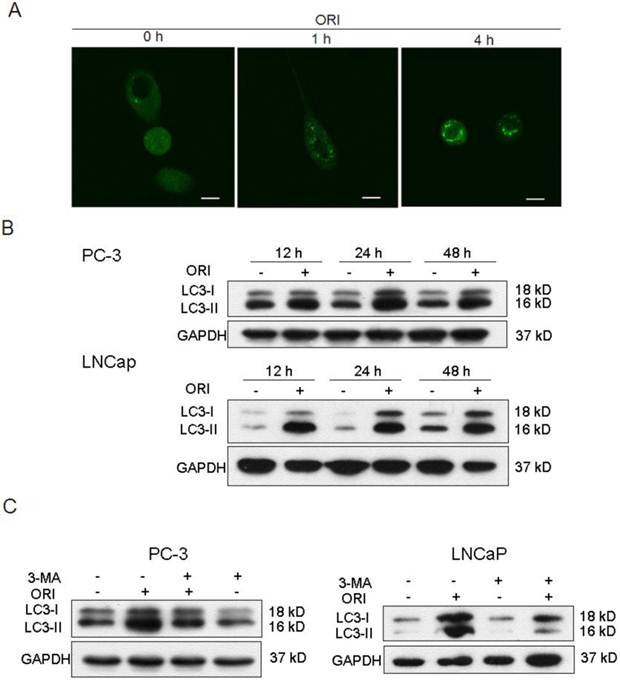
Levels of LC3-I and LC3-II in PC-3 cells were up-regulated after 12 h of ORI treatment, and reached maximum at 24 h. At 48 h, the levels of both LC3-I and LC3-II were reduced, but still remained higher than the controls. Similar changes of LC3 were detected in ORI-treated LNCaP cells (Fig.2B). These results suggested that ORI treatment for 24 h could induce high expressions of LC3-I and II.
We further examined the influence of 3-MA in ORI-induced increase of LC3 in HPC cells. The results showed that the levels of LC3 expression in ORI and 3-MA co-treated cells were lower than that of the cells treated with ORI alone, but remained higher than the controls. The treatment of 3-MA alone did not change the expression of LC3 (Fig. 2C).
Change of Cell Ultra-structure in HPC cells
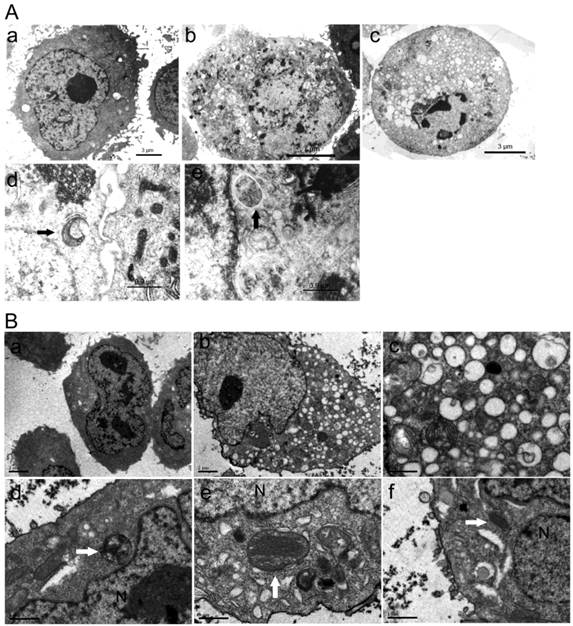
No changes in cells nuclei and membranes were observed in ORI-treated PC-3 cells at 24 h. But interestingly, the appearance of a large number of free membrane structures and double-membrane vacuoles were found in the cytoplasm. These free membrane structures resembled pre-autophagosomal structure (PAS), and some of the later vacuoles resembled autophagosomes. After 48 h, typical apoptotic changes with chromatin condensation and nuclear fragmentation were observed in ORI-treated cells (Fig.3A). The nuclei were normal and there was no chromatin condensation in the ORI-treated LNCaP cells for 24 h (Fig. 4). But, the formation of a large number of vacuoles indicated cytoplasmic vacuolization. Autophagosomes were also observed in the cytoplasm, which contained portions of cytosol and organelles, such as endoplasmic reticulum (ER) and mitochondria (Fig. 3B).
Formation of autophagosomes was induced by ORI in HPC cells. Cells were treated with ORI (25 μmol/L) or DMSO, and examined under TEM A: Changes of ultra-structure in PC-3 cells induced by ORI. (a) Controls, (b) Cells treated with ORI for 24 h, (c) Cells treated with ORI for 48 h, (d) ORI-induced PAS, (e) ORI-induced autophagosome (arrows). B: Changes of ultrastructure in LNCaP cells. Cells were treated with DMSO or ORI (25 μmol/L) for 24 h. (a) Controls, (b) ORI-treated cells, (c) Cytoplasmic vacuolization, (d) Autophagosome with cytoplasmic material, (e) Autophagosome with ER, (f) PAS with mitochondria (arrows).
Accumulation of AVOs induced by ORI treatment (×200). Cells in 96-well plate were treated by ORI (25 μmol/L) with or without 3-MA (5 mmol/L) for 24 h, stained with AO (5 μg/mL) or MDC (0.05 mmol/L) for 10 min, then observed. Red fluorescent spots of AO and prominent accumulations of MDC were observed in both ORI-treated cells (arrows).
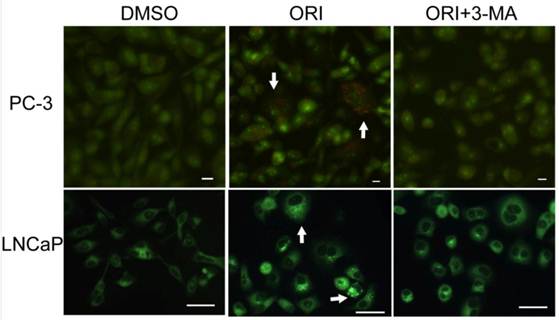
Detection of AVOs
By AO staining, red fluorescent spots appeared on ORI-treated PC-3 cells, while the control cells, and the cells co-treated with 5 mmol/L 3-MA and ORI showed mainly green cytoplasmic fluorescence. Similarly, prominent accumulation of autophagic specific staining MDC was observed around the nuclei in ORI-treated PC-3 cells (Fig.4).
Autophagy Played a Protective Role in ORI-treated PC-3 Cells
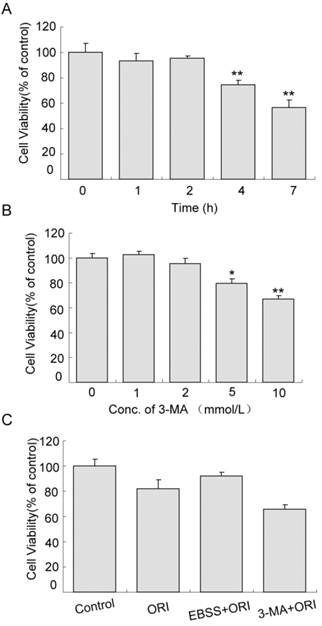
Cell proliferation was not affected in cells treated with starvation (in EBSS medium) for 1 or 2 h. When the time was extended to 4 h, cell viability reduced to 74.54% (Fig.5A, P<0.01). Similarly, the effects of 1 or 2 mmol/L 3-MA on cell proliferation were very small. As the concentration increased to 5 mmol/L, cell viability decreased to 79.57% (Fig.5B, P<0.05). The viability of cells treated with ORI alone was 80.75% compared to the control group. Pretreatment with starvation for 2 h and treatment with ORI for 24 h increased cell viability to 91.40%. Combined treatment of ORI and 3-MA (2 mmol/L) reduced the viability to 65.56%, indicating that autophagy could play a protective role in ORI-treated PC-3 cells (Fig.5C).
Protective role of autophagy in ORI-treated PC-3 cells. A: Inhibition of proliferation induced by starvation. Cells were cultured in EBSS for different times (1, 2, 4, and 7 h). B: Cytotoxicity of 3-MA. Cells in 96-well plates were treated by 3-MA in different concentrations (1, 2, 5 and 10 mmol/L) for 24 h. C: Autophagy protected cells survival. Cells in 96-well plate were treated with media containing 25 μmol/L ORI, or ORI plus 2 mmol/L 3-MA, or pre-cultured in EBSS for 2 h then treated by ORI in full media for 24 h. Cell viabilities were measured by the CCK-8 assay. The data were presented as means ± S.D. from 3 independent experiments (*P<0.05, **P<0.01).
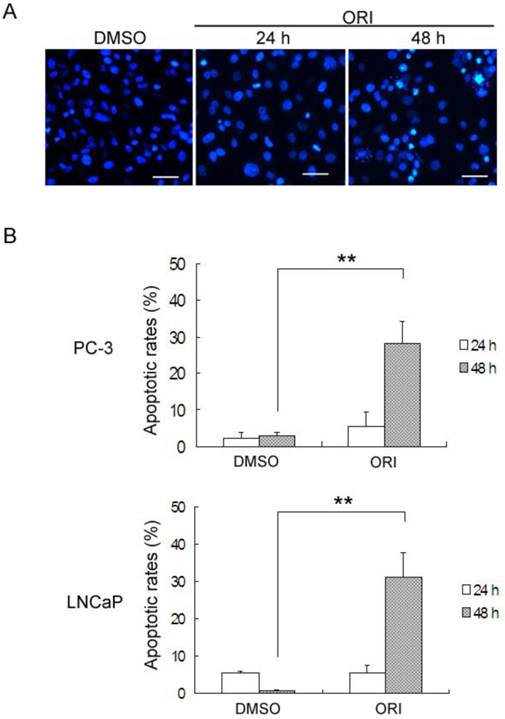
Detection of Apoptosis in ORI-treated HPC Cells
By DAPI staining, PC-3 cells treated with DMSO or ORI at 24 h exhibited normal nuclei, but typical apoptotic changes with chromatin condensation and nuclear fragmentation were observed at 48 h (Fig.6A). The levels of ORI-induced apoptosis in HPC cells were determined by Annexin V-FITC/PI staining and FACS. In PC-3 cells, treatment with 25 µmol/L ORI for 24 h caused only 5.4% of the cells to undergo apoptosis (2.25% for control). In spite of this, ORI treatment for 48 h resulted in an approximately 4.2-fold (27.0%) increase of the apoptotic rate (2.94% for controls). Similar changes of apoptotic rates were detected in LNCaP cells, and the apoptotic rate increased from 5.3% for 24 h to 31.0% for 48 h (Fig. 6B).
ORI induced apoptosis in HPC cells after longer treatment. A: Longer treatment with ORI resulted in chromatin condensation and nuclear fragmentation (arrows, ×200). PC-3 cells in 96-well plates were treated with 25 μmol/L ORI for 24 or 48 h, fixed and stained with DAPI (2 μg/mL), and observed for nuclear alterations under the fluorescence microscope. B: The apoptotic rates were evidently increased after longer treatment with ORI. Cells in 6-well plates were treated with ORI (25 μmol/L) for 24 or 48 h, stained with Annexin V-FITC for 10 min and PI for 10 min, then analyzed by FACS. (*P<0.05, **P<0.01).
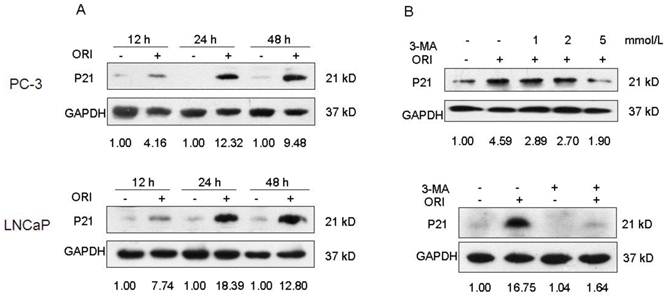
Change in P21 Protein Expression
The P21 protein expression in ORI-treated PC-3 cells increased by 4.16, 12.32, and 9.48 folds at 12 h, 24 h, and 48 h, respectively, compared to the control group. Also, the P21 protein expression in ORI-treated LNCaP cells increased by 7.74, 18.39, and 12.80 folds at 12 h, 24 h, and 48 h, respectively, compared to the control group. Co-treatment with 5 mmol/L 3-MA and ORI could reduce the increase in P21 expression (Fig. 7).
Inhibition of autophagy blocked the augment of P21 induced by ORI. A: The expression of P21 was up-regulated by ORI. Cells were treated with ORI (25 μmol/L) for 12, 24 or 48 h, and detected by Western Blot. B: ORI-induced augment of P21 protein was blocked by 3-MA. Cells were treated by ORI with or without 3-MA (5 mmol/L), and subjected to Western Blot. The data are from three independent experiments.
Discussion
ORI is an active diterpenoid compound extracted from the Chinese traditional medicine Rabdosia rubescens. It has the potential to develop into a highly effective anti-tumor chemotherapeutic drug. ORI has shown anti-tumor effects against a variety of human cancers such as, lung and breast cancers, and leukemia[14]. In the present study, we found that ORI could inhibit the proliferation of both PC-3 and LNCaP prostate cancer cell lines and induce G2/M phase cell cycle arrest. ORI treatment increased the expression of LC3 and induced the formation of AVOs in HPC cells. Meanwhile, the expression of P21 was increased, which could be blocked by 3-MA.
LC3 is recognized as the most common autophagy marker. Sometimes, because of LC3-I instability, LC3-II is typically used as a marker for autophagosome formation. We detected the intracellular localization and expression of the LC3 protein, and found that ORI significantly increased the expressions of both LC3-I and LC3-II, and promoted the conversion of water-soluble LC3-I to lipidated LC3-II, which was recruited to autophagosomsal membranes. Although ORI has been reported to induce PC-3 cells autophagy and promote LC3 expression[15], but in this study, similar results were observed in both PC-3 and LNCaP cells. Our results further suggested that the induction of autophagy in HPC cells were unlikely to be cell type-specific. Besides, it is evident that the level of ORI-induced autophagy reached its peak at 24 h after drug treatment, but began to decline thereafter.
In recent years, the autophagic process has been observed in response to various cancer-relevant stimuli, including radiation[16], ceramide[17], rapamycin[18], and arsenic trioxide[19]. In the present study, a large number of double-membrane autophagosomes and PAS were observed in both PC-3 and LNCaP cell lines treated with ORI. The mature autophagosome fuses with lysosome to become autolysosome, which is one structure of the AVOs. The fluorescent dyes AO and MDC were used as specific traces for AVOs. AO dye in acidic site fluoresced bright red and apparent accumulation of MDC in the cytoplasm indicated the accumulation of AVOs.
In addition, we co-treated HPC cells with 3-MA and ORI, and found that 3-MA could reverse the accumulation of MDC and AO in acidic organelles and reduce the expression levels of LC3-II. Considering the conserved positive role of Class III phosphatidylinositol 3-kinase (PI3K) in the autophagic process, 3-MA as a PI3K inhibitor, has been reported as a specific autophagy inhibitor. It inhibited or blocked the formation of autophagosomes by blocking the Class III PI3K/beclin 1 signal pathway[20, 21]. All these results indicated that 3-MA could specifically inhibit ORI-induced autophagy in HPC cells by inhibiting the increase of LC3. Interestingly, it has been reported that 3-MA has dual roles in the modulation of autophagy. 3-MA has been found to promote autophagy flux when treated under nutrient-rich condition with a prolonged period of treatment, whereas it is still capable of suppressing starvation-induced autophagy[22]. There are two explanations for this contradiction. First, ORI is able to inactivate Class I PI3K/Akt pathway[23, 24]. The blockade of Class I PI3K/PKB pathway weakens the positive effect of 3-MA on autophagy. Second, class III PI3K is associated with the protein Beclin 1, another marker of autophagy. Low expression of Beclin 1 in various cancer cells has been detected[21], thus, the suppressive effect of 3-MA on autophagy is weakened under nutrient-rich conditions. In oridonin-induced Hela cells, pretreatment with 3-MA has been shown to decrease the autophagic ratio accompanied with downregulation of the protein expression of Beclin 1 [25]. Further studies are still required to understand the mechanism of 3-MA as an inhibitor of ORI-induced autophagy.
Our research also showed that autophagy could play a protective role in ORI-treated HPC cells. Nutrient starvation has been widely used to induce autophagy[26]. It is evident that viability of PC-3 cells with high level of autophagy induced by co-treatment of starvation and ORI were higher than that of cells treated with ORI alone. And inhibition by 3-MA increased ORI-induced cell death. In addition, apoptotic rates of HPC cells were significantly increased after treatment with ORI for 48 h. Characteristic morphological features of apoptosis, such as chromatin condensation, nuclear fragmentation and disappearance of the surface microvilli were detected. These results indicated that apoptosis was induced by prolong treatment of ORI in HPC cells. It has been reported that ORI can induce apoptosis in A431 human epidermoid carcinoma cells[27], HeLa human cervical carcinoma cell line[24], breast cancer cells[28], hepatocelluar carcinoma cells[29], and osteosarcoma cells[23]. The apoptotic process was mainly associated with activation of death receptors, release of cytochrome c and cleavage of caspase through inhibition of the Ras/ERK and PI3K/Akt signaling pathways. It shows that the induction of apoptosis plays an essential role in the ORI anti-tumor activity. So, a better understanding of the relationship between autophagy and apoptosis will help to understand the protective mechanism of autophagy in ORI-induced cell death.
In our research, at 24 h, the level of autophagy reached its peak and a large number of autophagosomes were detected. While apoptotic cells with sub-G0-G1 DNA content were not detected, cellular nuclei and microvilli were normal, indicating that HPC cells did not undergo apoptosis at 24 h after drug treatment. As the time was extended to 48 h, apoptotic rates of HPC cells were significantly increased. Typical morphological features of apoptosis, such as chromatin condensation, nuclear fragmentation and disappearance of the surface microvilli were detected. This indicates that autophagy was induced and occurred before the onset of apoptosis in ORI-treated HPC cells. Autophagy preceding apoptosis has also been reported in sulforaphane-treated prostate cancer cells[30]. Even though the connection between autophagy and apoptotic cell death is not clear, several studies have demonstrated that both apoptosis and autophagy can occur concomitantly in the same cells under certain circumstances. In some studies, autophagy seems to promote apoptosis or be a prelude to apoptosis, and inhibition of autophagy inhibits DNA fragment[31]. However, in osteoblastic cells, a negative relationship between autophagy and apoptosis has been reported[32]. These negative relationships have also been observed in ORI-treated tumor cells. The inhibition of autophagy could increase the number of apoptotic cells[6, 33, 34]. Therefore, by delaying the onset of apoptosis, autophagy represents a defense mechanism against ORI-induced cell death. Further research in molecular biological chemistry is needed to understand the relationship between autophagy and apoptosis in prostate cancer cells.
P21 was originally identified as a universal inhibitor of cyclin-dependent kinases (CDK). But, accumulating evidence has shown that P21 has multiple functions in addition to CDK inhibition including cell cycle regulation, inhibition or mediator of apoptotic signaling pathways[35]. Previous studies have reported P21 relative to ORI-induced cell cycle arrest and apoptosis in various tumor cell lines. ORI has been shown to induce S phase cell cycle arrest in MCF-7 human breast cancer cells and G2/M phase arrest in murine fibrosarcoma L929 cells through up-regulating phosphorylated P53 and P21[28, 34]. In human laryngeal carcinoma Hep-2 cells, ORI has been shown to induce G2/M phase arrest and apoptosis in a P53-independent and P21/WAF1-dependent manner[36]. ORI has also been shown to induce apoptosis in colorectal cancer SW1116 cells by the regulation of P21[37]. In our study, increased expression of P21 was detected in ORI-treated HPC cells for 12 h and reached maximum at 24 h. At the same time, the existence of a large number of autophagosomes was observed in the cytoplasm under TEM. In addition, P21 showed a tendency for the autophagy marker, LC3-II. After 3-MA treatment, ORI-induced autophagy was inhibited and the augment of P21 was also blocked. Therefore, we speculated that there was a correlation between ORI-induced autophagy and P21 expression. However, whether P21 mediate autophagy signal molecules or is merely a result of autophagy downstream remains to be unclear. Fujiwara et al. found that autophagy was not induced in ceramide-treated P21+/+ mouse embryonic fibroblasts (MEFs). Interfere of P21 expression increased autophagy. In ceramide-treated P21-/- MEFs, there was a high autophagic activity, while exogenous expression of P21 decreased the number of autophagic cells. These results indicated that P21 played an essential role in determining the type of cell death, positively for apoptosis and negatively for autophagy[38]. So, low expression of P21 is conductive to the occurrence of autophagy. Elevated levels of P21 could be down-regulated through the reactive oxygen species/mTOR complex 1 (ROS/mTORC1) pathway[39]. The mTOR pathway is an important negative regulator of autophagy. With the activation of autophagy, the mTOR pathway is down-regulated, and the P21 expression is promoted. The feedback regulation may increase the expression of P21, which may further increase the number of apoptotic cells. Therefore, promoting endogenous P21 protein expression may improve cells sensitivity to ORI.
In summary, we demonstrated that ORI treatment could induce autophagy in HPC cells. Autophagy could protect cancer cells from apoptotic cell death by delaying the onset of apoptosis. P21 protein plays an important role in ORI-induced autophagy and apoptosis. The present study not only provides a new idea for further studying the biological activity of P21 and the anti-tumor mechanism of ORI, but also shows that autophagy is a possible mechanism for HPC cells to resist anti-tumor agent ORI. Promoting endogenous expression of P21 protein may improve cell sensitivity to ORI. This study also provides evidence for potential of ORI as a valuable component in therapeutic strategies to treat prostate cancer.
Acknowledgements
We thank Dr. Tamotsu Yoshimori from Osaka University and Dr. Hanming Shen from National University of Singapore for generous gift of tfLC3 plasmid.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Crawford ED. Epidemiology of prostate cancer. Urology. 2003;62:3-12
2. Marks LS, DiPaola RS, Nelson P. et al. PC-SPES: herbal formulation for prostate cancer. Urology. 2002;60:369-75
3. Cui Q, Tashiro S, Onodera S. et al. Augmentation of oridonin-induced apoptosis observed with reduced autophagy. J Pharmacol Sci. 2006;101:230-9
4. Cui Q, Tashiro S, Onodera S. et al. Autophagy preceded apoptosis in oridonin-treated human breast cancer MCF-7 cells. Biol Pharm Bull. 2007;30:859-64
5. Li D, Cui Q, Chen SG. et al. Inactivation of ras and changes of mitochondrial membrane potential contribute to oridonin-induced autophagy in a431 cells. J Pharmacol Sci. 2007;105:22-33
6. Cheng Y, Qiu F, Huang J. et al. Apoptosis-suppressing and autophagy-promoting effects of calpain on oridonin-induced L929 cell death. Arch Biochem Biophys. 2008;475:148-55
7. Tsujimoto Y, Shimizu S. Another way to die: autophagic programmed cell death. Cell Death Differ. 2005;12(Suppl 2):1528-34
8. Terman A, Gustafsson B, Brunk UT. Autophagy, organelles and ageing. J Pathol. 2007;211:134-43
9. Baehrecke EH. Autophagy: dual roles in life and death? Nat Rev Mol Cell Biol. 2005;6:505-10
10. Toth S, Nagy K, Palfia Z. et al. Cellular autophagic capacity changes during azaserine-induced tumour progression in the rat pancreas. Up-regulation in all premalignant stages and down-regulation with loss of cycloheximide sensitivity of segregation along with malignant transformation. Cell Tissue Res. 2002;309:409-16
11. Wu YT, Tan HL, Huang Q. et al. Autophagy plays a protective role during zVAD-induced necrotic cell death. Autophagy. 2008;4:457-66
12. Tasdemir E, Chiara Maiuri M, Morselli E. et al. A dual role of p53 in the control of autophagy. Autophagy. 2008;4:810-4
13. Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452-60
14. Ikezoe T, Chen SS, Tong XJ. et al. Oridonin induces growth inhibition and apoptosis of a variety of human cancer cells. Int J Oncol. 2003;23:1187-93
15. Ye LH, Li WJ, Jiang XQ. et al. Study on the autophagy of prostate cancer PC-3 cells induced by oridonin. Anat Rec (Hoboken). 2012;295:417-22
16. Paglin S, Hollister T, Delohery T. et al. A novel response of cancer cells to radiation involves autophagy and formation of acidic vesicles. Cancer Res. 2001;61:439-44
17. Daido S, Kanzawa T, Yamamoto A. et al. Pivotal role of the cell death factor BNIP3 in ceramide-induced autophagic cell death in malignant glioma cells. Cancer Res. 2004;64:4286-93
18. Takeuchi H, Kondo Y, Fujiwara K. et al. Synergistic augmentation of rapamycin-induced autophagy in malignant glioma cells by phosphatidylinositol 3-kinase/protein kinase B inhibitors. Cancer Res. 2005;65:3336-46
19. Kanzawa T, Kondo Y, Ito H. et al. Induction of autophagic cell death in malignant glioma cells by arsenic trioxide. Cancer Res. 2003;63:2103-8
20. Kanzawa T, Germano IM, Komata T. et al. Role of autophagy in temozolomide-induced cytotoxicity for malignant glioma cells. Cell Death Differ. 2004;11:448-57
21. Cao Y, Klionsky DJ. Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res. 2007;17:839-49
22. Wu YT, Tan HL, Shui G. et al. Dual role of 3-methyladenine in modulation of autophagy via different temporal patterns of inhibition on class I and III phosphoinositide 3-kinase. J Biol Chem. 2010;285:10850-61
23. Jin S, Shen JN, Wang J. et al. Oridonin induced apoptosis through Akt and MAPKs signaling pathways in human osteosarcoma cells. Cancer Biol Ther. 2007;6:261-8
24. Hu HZ, Yang YB, Xu XD. et al. Oridonin induces apoptosis via PI3K/Akt pathway in cervical carcinoma HeLa cell line. Acta Pharmacol Sin. 2007;28:1819-26
25. Cui Q, Tashiro S, Onodera S. et al. Oridonin induced autophagy in human cervical carcinoma HeLa cells through Ras, JNK, and P38 regulation. J Pharmacol Sci. 2007;105:317-25
26. Martinez-Borra J, Lopez-Larrea C. Autophagy and self-defense. Adv Exp Med Biol. 2012;738:169-84
27. Mei Y, Xu J, Zhao J. et al. An HPLC method for determination of oridonin in rabbits using isopsoralen as an internal standard and its application to pharmacokinetic studies for oridonin-loaded nanoparticles. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;869:138-41
28. Cui Q, Yu JH, Wu JN. et al. P53-mediated cell cycle arrest and apoptosis through a caspase-3- independent, but caspase-9-dependent pathway in oridonin-treated MCF-7 human breast cancer cells. Acta Pharmacol Sin. 2007;28:1057-66
29. Zhang JF, Chen GH, Lu MQ. et al. Change of Bcl-2 expression and telomerase during apoptosis induced by oridonin on human hepatocelluar carcinoma cells. Zhongguo Zhong Yao Za Zhi. 2006;31:1811-4
30. Herman-Antosiewicz A, Johnson DE, Singh SV. Sulforaphane causes autophagy to inhibit release of cytochrome C and apoptosis in human prostate cancer cells. Cancer Res. 2006;66:5828-35
31. Jia L, Dourmashkin RR, Allen PD. et al. Inhibition of autophagy abrogates tumour necrosis factor alpha induced apoptosis in human T-lymphoblastic leukaemic cells. Br J Haematol. 1997;98:673-85
32. Lai EH, Hong CY, Kok SH. et al. Simvastatin alleviates the progression of periapical lesions by modulating autophagy and apoptosis in osteoblasts. J Endod. 2012;38:757-63
33. Cui Q, Tashiro S, Onodera S. et al. Mechanism of downregulation of apoptosis by autophagy induced by oridonin in HeLa cells. Yao Xue Xue Bao. 2007;42:35-9
34. Cheng Y, Qiu F, Ye YC. et al. Autophagy inhibits reactive oxygen species-mediated apoptosis via activating p38-nuclear factor-kappa B survival pathways in oridonin-treated murine fibrosarcoma L929 cells. Febs J. 2009;276:1291-306
35. Gartel AL, Tyner AL. The role of the cyclin-dependent kinase inhibitor p21 in apoptosis. Mol Cancer Ther. 2002;1:639-49
36. Kang C, Avery L. To be or not to be, the level of autophagy is the question: dual roles of autophagy in the survival response to starvation. Autophagy. 2008;4:82-4
37. Gao FH, Hu XH, Li W. et al. Oridonin induces apoptosis and senescence in colorectal cancer cells by increasing histone hyperacetylation and regulation of p16, p21, p27 and c-myc. BMC Cancer. 2010;10:610
38. Fujiwara K, Daido S, Yamamoto A. et al. Pivotal role of the cyclin-dependent kinase inhibitor p21WAF1/CIP1 in apoptosis and autophagy. J Biol Chem. 2008;283:388-97
39. Ji WT, Yang SR, Chen JY. et al. Arecoline downregulates levels of p21 and p27 through the reactive oxygen species/mTOR complex 1 pathway and may contribute to oral squamous cell carcinoma. Cancer Sci. 2012 Epub ahead of print
Author contact
![]() Corresponding author: Prof. Ji-Cheng Li, Institute of Cell Biology, Zhejiang University, E-mail: lijichenedu.cn; Tel.& Fax: 0086-571-88208088.
Corresponding author: Prof. Ji-Cheng Li, Institute of Cell Biology, Zhejiang University, E-mail: lijichenedu.cn; Tel.& Fax: 0086-571-88208088.