Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(6):531-540. doi:10.7150/ijbs.5718 This issue Cite
Research Paper
A Dynein Motor Attachment Complex Regulates TGFß/Smad3 Signaling
Qunyan Jin†, Guofeng Gao†, Kathleen M. Mulder ![]()
Department of Biochemistry and Molecular Biology, Penn State Hershey College of Medicine, Hershey, PA 17033, USA.
† The first two authors contributed equally.
Received 2012-12-15; Accepted 2013-5-22; Published 2013-6-9
Abstract
Our previous results have demonstrated that km23-2 has functions in TGFß signaling that are distinct from those for km23-1. In the current report, we demonstrate that blockade of km23-2 decreased TGFß activation of the human Smad7 promoter Smad7-Luc, an endogenous Smad3-target promoter. Luminescence-based mammalian interaction mapping (LUMIER) analyses showed that TGFß stimulated the interaction of km23-2 preferentially with Smad3, relative to that with Smad2. Size exclusion chromatography experiments revealed that km23-2 and Smad3 were recruited into the same complex after TGFß treatment. Moreover, in the presence of TGFß, but not in the absence, km23-2 was present in early endosomes with the TGFß receptors (TßRs) and Smad3. Collectively, our data indicate that km23-2 is a critical signaling intermediate in a Smad3-dependent TGFß signaling pathway. We also provide evidence of the novel finding that TGFß stimulates the rapid recruitment of the km23-2 dimer to the dynein intermediate chain (DIC) of the dynein complex, whereas a kinase-deficient form of TßRII prevented this interaction. Finally, we demonstrate for the first time that TGFß stimulated not only assembly of the dynein motor attachment complex, but also triggered the tethering of the km23-2-Smad3 cargo to the other dynein components. Thus, our data demonstrate a novel function for km23-2 as a motor receptor to recruit Smad3 to the dynein complex for intracellular transport, thereby mediating Smad3-dependent TGFß signaling.
Keywords: TGFß, km23, Smad, signaling, receptor, dynein.
Introduction
TGFß is the prototype for the TGFß superfamily of highly conserved growth regulatory polypeptides, which regulate a variety of physiologic and pathologic processes, such as embryogenesis, wound healing, fibrosis, growth control, and oncogenesis [1-4]. TGFß initiates its signaling by activating two pairs of structurally similar, single-pass transmembrane receptors, TGFß receptor type II (TßRII) and type I (TßRI). The activated receptor complex stimulates various TGFß signaling events, including receptor-regulated Smads (RSmads) and non-Smad pathways [1-4]. The activated RSmads form a complex with the common partner Smad4 and translocate to the nucleus, where they regulate transcription of target genes such as Smad7 [1-4].
Although Smad2 and Smad3 are structurally similar, accumulating evidence suggests that Smad2 and Smad3 play distinct roles in mediating the cellular responses induced by TGFß [5]. For example, it has been shown that TGFß-mediated induction of matrix metalloproteinase-2 is selectively dependent upon Smad2, whereas induction of c-fos and Smad7 relies on Smad3 [6]. However, the mechanisms underlying this differential regulation of TGFß responses by Smad2 and Smad3 are still unclear. We have previously reported that differential utilization of km23 dynein light chain (DLC) isoforms by Smad2 and Smad3 could be partially responsible for Smad2-dependent versus Smad3-dependent TGFß signaling [7]. Along these lines, cytoplasmic dynein is a motor complex that transports membrane vesicles and diverse motor cargoes along microtubules in a retrograde manner [8, 9]. The cargo attachment complex is variably composed of light intermediate chains (DYNC1LI), intermediate chains (IC2; DYNC1I), and three dimeric light chain subunits [9]. The three classes of cytoplasmic DLCs that have been identified in mammals to date are LC8 (DYNLL), Tctex-1/rp3 (DYNLT), and km23/LC7/roadblock (DYNLRB) [8-10]. While the LC8 and Tctex families of DLCs have been shown to bind to diverse cargos, often independent of the dynein motor complex, the km23/LC7 family displays unique characteristics, with comparatively fewer binding partners identified to date [9].
With regard to the km23/LC7 family, we first described the km23-1 DLC as a novel TßR-interacting protein, also termed mLC7-1[11], Robl1[12], DNLC2A [13] and DYNLRB1[10]. In addition, we have demonstrated that km23-1 is required for both Smad2-dependent and Smad-independent TGFß responses [14-16]. In contrast to km23-1, we have also demonstrated that km23-2, another member of the km23/DYNLRB/LC7/robl family of DLCs, has functions in TGFß signaling that are distinct from those for km23-1 [7]. In the current report, we describe the role of km23-2 in TGFß/Smad3 signaling. Further, we demonstrate for the first time that TGFß regulates the interplay between the km23-2 dimer and other dynein motor subunits, further supporting the unique nature of the km23/LC7 family of dynein adaptors.
Materials and Methods
Reagents--The anti-Flag M2 antibody (Ab) was from Sigma. The anti-dynein intermediate chain (DIC) monoclonal Ab (M11618) was from Chemicon (Temecula, CA). The anti-HA Ab (1-583-816) was from Boehringer Manheim. The TGFß RII Ab (SC-220), rabbit IgG, dynein heavy chain (DHC) Ab, protein A/G plus agarose were from Santa Cruz Biotech. 32P- orthophosphate (NEX-053) and [3H]-thymidine (NET-027X) were from Perkin Elmer (Boston, MA). The rabbit Smad3 Ab (51-1500) was from Zymed (South San Francisco, CA). The p150 Glued Ab was from BD Transduction Laboratories. TGFß1 was purchased from R & D Systems (Minneapolis, MN). The Fugene 6 transfection reagent was from Roche Applied Science (Indianapolis, IN). The Lipofectamine™ 2000 transfection reagent was from Invitrogen (Carlsbad, CA). The Dual-Luciferase Reporter Assay System (Cat. # E1960) was purchased from Promega (Madison, MI). Gel filtration standards were purchased from Bio-Rad and Amersham.
Cell Culture--HaCaT cells and Mv1Lu cells (CCL-64) were obtained from ATCC (Rockville, MD) and were grown in DMEM supplemented with 10% fetal bovine serum. 293T cells were obtained from T-W. Wong (Bristol-Myers Squibb) and were maintained as for Mv1Lu cells. The rat IEC 4-1 cell line was cultured as described previously [17]. Cells were routinely screened for mycoplasma using Hoechst staining. For studies involving TGFß treatment, responsiveness to TGFß was assessed by thymidine assays as described previously [11].
LUMIER assays were performed as described as previously [18]. Briefly, IEC4-1 cells were transiently co-transfected with hRL-Smad2, hRL-Smad3, pRL-TK, together with Ski-Flag or km23-2-Flag as indicated. pRL-TK is renilla luciferase (RL) driven by the thymidine kinase (TK) promoter and is a negative control [18]. 28h after transfection, cells were incubated in serum-free (SF) medium for 1h prior to incubation for 5 min in the absence and presence of TGFß1 (5 ng/ml). Cells were lysed, followed by immunoprecipitation (IP) using an anti-Flag Ab, or using IgG as a control. Protein interactions on anti-Flag IP's were determined by performing a renilla luciferase enzymatic assay.
siRNAs-- Human km23-2 stealth siRNA (5'-GGACAACUCAACAACUGUUCAAUAU-3'), km23-1 stealth siRNA (5'-ACCAGAUAAAGACUAUUUCCUGAUU-3'), and negative control (NC) stealth siRNA (5'-AAUUCUCCGAACGUGUCACGUGAGA-3') were designed by BLOCK-iT™ RNAi Designer (Invitrogen) and synthesized by Invitrogen.
Luciferase reporter assays-- HaCaT cells were plated at 1×104 cells/cm2 in 24-well plates. 24h after plating, the cells were transfected with the indicated amounts of either km23-2 siRNA or NC siRNA, together with Smad7-Luc. Renilla was used to normalize transfection efficiencies. 24h after transfections, the cells were washed once with SF medium and incubated in SF medium for 1h. The cells were then cultured in the absence and presence of TGFß1 (5 ng/ml) for an additional 18 h. Luciferase activity was measured using the Dual-Luciferase Reporter Assay System following the manufacturer's instructions. All assays were performed in triplicate. Data are expressed as mean ±SEM.
Size exclusion chromatography--IEC4-1 cells (10 near 60-80% confluent 10-cm dishes) were transiently transfected with km23-2-Flag using Fugene 6 according to the manufacturer's instructions. 24h after transfections, IEC4-1 cells were washed once with SF medium, incubated in SF medium for 1h, and then incubated in the absence and presence of TGFß1 for 5 min. The cells were washed twice with ice-cold 1xPBS, scraped in lysis buffer as described [11], and centrifuged at 100,000 x g for 10 min. Clarified whole cell lysates of 500 μl (containing 1-2 mg of protein) were loaded on a fast protein liquid chromatography (FPLC)/Superose 12 HR 10/30 column (Amersham Biosciences, Piscataway) equilibrated in lysis buffer without Triton x-100 and glycerol. The column was calibrated with two sets of gel filtration standards according to the manufacturer's instructions. The flow rate was 0.5 ml/min and fractions were collected every min with a total of 60 fractions being collected.
In vivo phosphorylation assays were performed essentially as described previously [11]. Briefly, cells were washed and incubated for 30 min in phosphate-free medium prior to incubation in the presence of [32Pi]-orthophosphate (1mCi/plate) for 3h. TGFß1 was added during the last 15 min of the labeling period. Cells were lysed on ice, followed with IP with an anti-Flag Ab and SDS-PAGE.
Sucrose gradient assays-- Early-endosome-containing fractions were prepared as described previously [14], except using IEC4-1 cells transiently transfected with km23-2-Flag.
Transient transfections, IP/blotting, Westerns were performed as described previously [11].
Statistical analyses were by Student's t test. Triplicate samples were analyzed and mean ± SE plotted unless otherwise indicated.
Results
Our previous results have shown that TGFß receptor (TßR) expression and activation in 293T cells stimulated the interaction of km23-2 preferentially with Smad3 over Smad2. To further confirm that km23-2 interacts with Smad3 in a ligand-dependent manner, we performed LUMIER analyses in the absence and presence of TGFß in IEC4-1 intestinal epithelial cells. Using LUMIER, proteins of interest are fused to Renilla luciferase [18], and the association with Flag-tagged proteins co-expressed in mammalian IEC4-1 cells is assessed by performing a luciferase assay on anti-Flag immunoprecipitates collected using an automated robotics platform. IEC4-1 cells were used in these studies because of their high level of TGFß sensitivity and their well-characterized TGFß pathways [17]. As shown in Fig. 1A, in the absence of TGFß, a basal level of interaction of km23-2 with Smad3 was observed (lane 7, left panel). However, addition of TGFß1 resulted in a strong interaction between km23-2 and Smad3 (lane 8, left panel). In contrast, no interaction between Smad2 and km23-2 was observed in the absence of TGFß (lane 9, left panel). TGFß stimulation caused a baseline level of interaction of km23-2 with Smad2 (lane 10, left panel). As prescribed previously [18], the negative control pRL-TK (lanes 1-2, 5-6, left panel) was used for comparison. As expected, there was no interaction observed between pRL-TK and Ski or between pRL-TK and km23-2 in the absence or presence of TGFß. The interaction between the Ski and Smad3-Flag (lanes 3-4, left panel) is shown as a positive control for comparison [18]. The IgG control was also negative (lane 11, left panel). Equal expression and loading of Ski-Flag, km23-2-Flag and γ-tubulin were confirmed (top, right panel), as was the expression of pRL-TK, hRL-Smad2 and hRL-Smad3 (bottom, right panel). Thus, LUMIER analyses confirm that km23-2 interacts preferentially with Smad3, relative to Smad2, in a ligand-dependent manner.
TGFß stimulates km23-2 and Smad3 recruitment to the same complex. A: IEC4-1 cells were transiently co-transfected with hRL-Smad2, hRL-Smad3, pRL-TK, together with Ski-Flag or km23-2-Flag as indicated. 28h after transfection, cells were incubated in SF medium for 1h prior to incubation for 5 min in the absence and presence of TGFß1 (5 ng/ml). Cell lysates were subjected to IP using an anti-Flag Ab, or IgG (control). Protein interactions on anti-Flag immunoprecipitates were determined by performing a renilla luciferase enzymatic assay (LUMIER) as described in “Materials and Methods.” Western blot analysis with the indicated Abs demonstrates equal loading and expression of Ski-Flag, km23-2-Flag, hRL-Smad2, hRL-Smad3, pRL-TK and γ-tubulin (right panel). The results shown are representative of two similar experiments. B: IEC4-1 cells were transiently transfected with km23-2-Flag and were treated with TGFß1 as described in “Materials and Methods.” FPLC size exclusion chromatography analyses were performed as described in “Materials and Methods.” The numbers 158, 44, 29 and 17 on the top indicate the position of protein markers of molecular weights 158-, 44-, 29- and 17-kDa. The results shown are representative of two similar experiments.
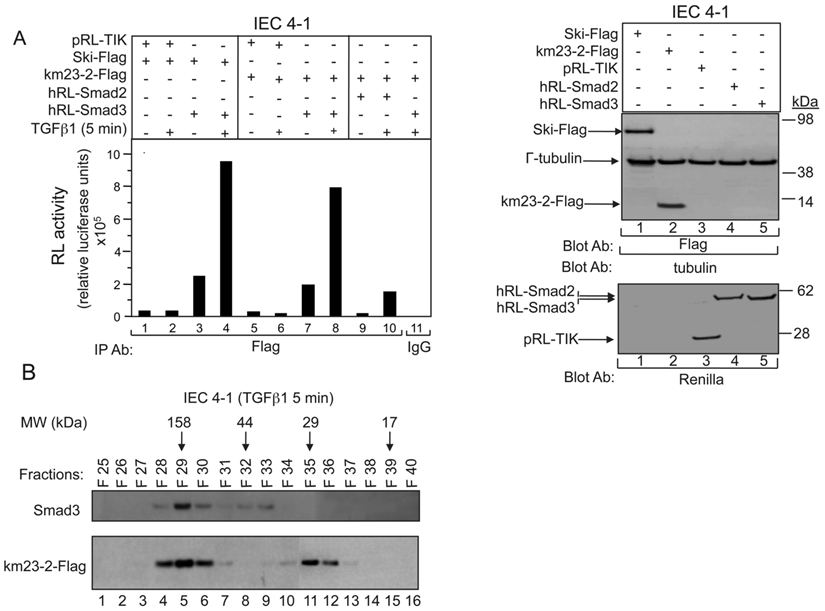
To determine whether the km23-2 DLC and Smad3 proteins were in the same complex in vivo under native conditions, lysates from TGFß-treated IEC4-1 cells were fractionated by FPLC/size exclusion chromatography [19, 20]. This strategy has been used successfully to separate various protein complexes, including Smad and dynein subunit proteins [21, 22]. The collected fractions were examined for Smad3 and km23-2 by Western blot analysis. As shown in Fig. 1B, km23-2 was detected in the fractions close to 29 kDa (lanes 35-36), suggesting that the km23-2 dimer is present after TGFß treatment in vivo, consistent with previous results indicating that km23 can form stable homodimers [23]. In addition, km23-2 co-fractionated with Smad3 in fractions 28-30 (approx. 150 kDa), which would be consistent with the km23-2 dimer bound to the Smad3/4 complex. Collectively, we demonstrate by two different approaches that TGFß induces a rapid complex between the km23-2 DLC and Smad3 proteins.
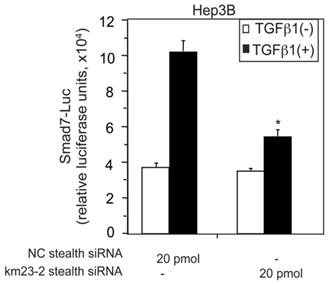
TGFß activates varied and intricate cellular responses as a result of differential transcriptional regulation, as well as non-transcriptional effects that depend upon the cell context and physiological environment [1, 3]. Among the transcriptional mediators of TGFß effects, the RSmad proteins (Smad2/3) are key regulators of TGFß signaling and downstream responses [1, 2]. In addition to R-Smads and the co-Smad Smad4, there is a third Smad protein family, namely the inhibitory Smads (Smad6 and Smad7), which have been documented to play key roles in regulating signal transduction of TGFß family cytokines [24, 25]. Since we show here that km23-2 interacts with Smad3, it is conceivable that km23-2 might play a role in TGFß regulation of the promoters of Smad3 targets. The Smad7 promoter represents the first natural promoter in vertebrates that has been shown to contain the 8-base pair (GTCTAGAC) palindromic Smad-binding element (SBE) [26], a key regulatory element of Smad3-targeted promoters [6, 27]. In order to more definitively establish whether km23-2 was required for TGFß induction of Smad7 promoter activity, we utilized a small interfering RNA (siRNA) approach to block km23-2 expression by km23-2-specific stealth siRNAs, as described previously [7, 14-16]. We examined the effects on transcriptional regulation of the human Smad7 promoter, as assessed by the Smad7-Luc reporter. As shown in Fig. 2, in the NC stealth siRNA-transfected cells, TGFß stimulated a 3-fold induction of the Smad7-Luc activity. However, in the km23-2 stealth siRNA-transfected cells, TGFβ-induction of the Smad7 promoter reporter activity was significantly decreased (to levels of only 1.5-fold), compared to that in the NC stealth siRNA-transfected cells. Thus, km23-2 inhibition attenuated TGFß-inducible transcriptional activity of the human Smad7 promoter, suggesting that km23-2 is required for Smad3-dependent TGFß signaling.
siRNA blockade of endogenous km23-2 expression inhibits TGFß-mediated transcriptional activation of the human Smad7 promoter in Hep3B cells. Hep3B cells were transfected with either NC stealth siRNA or km23-2 stealth siRNA along with Smad7-Luc, followed by treatment with TGFß1, and luciferase reporter analysis as described in “Materials and Methods.” *p< 0.001 indicates a statistically significant difference in the fold changes for +/-TGFß between NC stealth siRNA-transfected and km23-2 stealth siRNA-transfected Hep3B cells. The results shown are representative of three similar experiments.
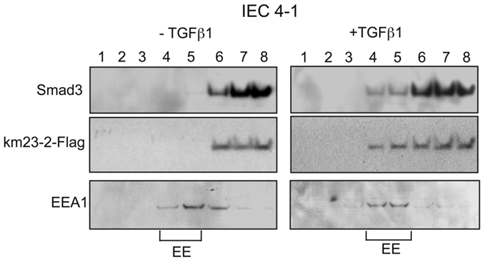
Since km23-2 appeared to preferentially modulate Smad3 signaling, we investigated the precise subcellular location for km23-2's effects on Smad3-dependent TGFß signaling. Along these lines, previous reports have shown that the TßRs and associated signaling components are localized to early endosomes (EEs) for optimal signal transduction [28, 29]. To determine whether km23-2 was co-localized with Smad3 after TGFß treatment, we performed sucrose flotation gradients to isolate the endosomal compartments enriched for early endosome antigen-1 (EEA1), as described previously [14, 30]. As shown in Fig. 3, in the absence of TGFß (left panel), the majority of Smad3 (top panel) and km23-2 (2nd panel) was present in fractions 6-8. However, upon TGFß treatment for 5 min (right panel), the localization of Smad3 and km23-2 to the EEA1-enriched fractions was increased (fractions 4-5). The bottom panel designates the EEA1-enriched EE fractions (fractions 4-5). Thus, km23-2 is present in early endosomes with Smad3 in the presence of TGFß.
km23-2 is present in EEA1-enriched early endosomes together with Smad3 after TGFß treatment. IEC4-1 cells were transiently transfected with km23-2-Flag. 24 h after transfection, cells were incubated in SF medium for 1 h, followed by incubation of the cells in the absence (left panel) or presence of TGFß1 (5 ng/ml) for 5 min. The cells were then harvested for sucrose gradient analysis, followed by Western blot analysis as described in “Materials and Methods.” EE indicates early endosome/EEA1-enriched fractions. The results shown are representative of two similar experiments.
As mentioned earlier, km23-2 is a member of the km23/LC7/robl family of light chains of the motor protein dynein in mammalian cells [7, 11]. To determine whether TGFß could stimulate the interaction between the km23-2 DLC and the DIC, we performed IP/blot analyses, in this case in HaCaT cells that express endogenous TßRs, in order to further extend our observations to TGFß-sensitive human keratinocyte cells. As shown in Fig. 4A, there was no detectable interaction between the km23-2 and DIC in the absence of TGFß (lane 2, top panel). However, TGFß induced a time-dependent increase in the recruitment of km23-2 to DIC beginning as early as 2 min after TGFß treatment (lanes 3-7, top panel). The IgG control was negative (lane 1, top panel). Western blot analysis with anti-Flag (middle panel) or anti-DIC (bottom panel) demonstrates equal protein expression and loading. Similar results were observed in IEC 4-1 cells (data not shown). Collectively, these data demonstrate for the first time that TGFß induced a rapid recruitment of km23-2 to DIC in two different TGFß-responsive cell lines expressing endogenous TßRs. Also of interest, however, was the escalating degree of complex formation, extending to at least 60 min after TGFß treatment, at a time when Smad3 would have translocated to the nucleus and become actively engaged in mediating downstream TGFß responses [31]. These findings are consistent with a novel stabilizing role for km23-2 in the cytoplasm during the transmission of TGFß/Smad3 events.
The interaction between DIC and km23-2 is induced by TGFß and regulated by TβRII kinase activity. A: HaCaT cells were transiently transfected with km23-2-Flag. 32 h after transfection, cells were incubated in SF medium for 1h prior to addition of TGFß1 (5 ng/ml) for the indicated times. Top panel: Cell lysates were subjected to IP/blot analyses using a DIC Ab or IgG (control) as the IP Ab and a Flag Ab as the blotting Ab. Middle panel: Western blot analysis to demonstrates equal loading and expression of km23-1-Flag and DIC. Bottom panel: Plot of densitometric scan of results in top. The results shown are representative of three similar experiments. B: HaCaT cells were transiently transfected with km23-2-Flag, EV, and KNRII-HA as indicated. Studies were performed as for A. The same membrane was re-blotted with anti-DIC (middle panel) to show equal protein loading. Western blot analysis (two bottom panels) with anti-Flag or anti-HA demonstrates protein expression of KNRII and km23-2. The results shown are representative of three similar experiments. C: HaCaT cells were transiently transfected with km23-2-Flag, and either RII-HA or KNRII-HA. 32 h after transfection, cells were incubated in SF medium for 1h prior to addition of TGFß1 (5 ng/ml) for the indicated times. Cell lysates were subjected to IP using a monoclonal anti-DIC Ab and were separated by 4-12% NuPAGE under non-reducing conditions (native), followed by immunoblot analysis using an anti-Flag Ab (top panel). Western blotting with anti-DIC (middle panel) or anti-Flag (bottom panel) demonstrates equal protein loading and expression. The results shown are representative of three similar experiments.
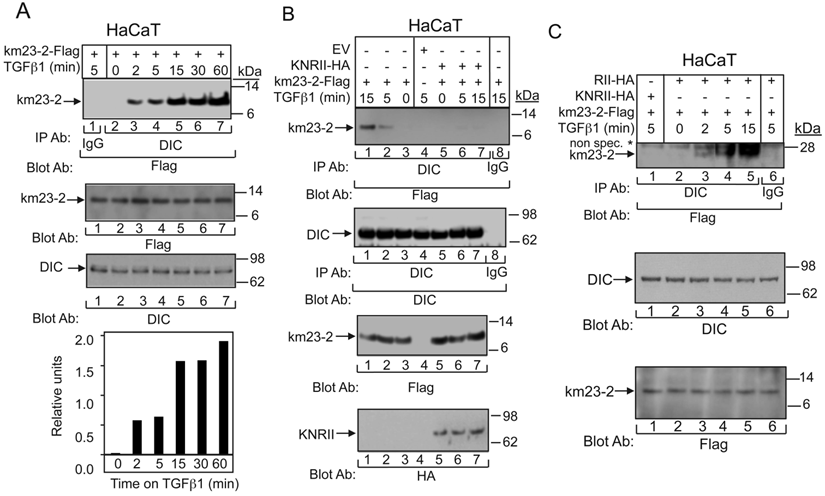
To provide definitive evidence that TßR activation is required for the km23-2-DIC interaction, we examined the interaction between km23-2 and DIC in the absence and presence of a kinase-deficient form of TßRII (KNRII). This receptor mutant can function in a dominant-negative manner to block the kinase activity of endogenous TßRII when overexpressed in cells [32]. As shown in Fig. 4B (left), TGFß stimulated the recruitment of km23-2 to DIC beginning at 5 min after treatment and increasing further at 15 min post-TGFß incubation (lanes 2, 1). In the absence of TGFß, km23-2 binding to DIC was not observed (lane 3). As shown in Fig. 4B (right), expression of KNRII completely blocked the interaction between km23-2 and DIC in the presence of TGFß treatment (lanes 6-7, top panel). IgG control and empty vector (EV) control were both negative (lanes 8, 4). Equal expression and loading of DIC and km23-2 were confirmed (two middle panels), as was the expression of KNRII (bottom panel). Thus, we show for the first time that TßRII kinase activity regulates the recruitment of km23-2 to DIC, suggesting a connection between TGFß signaling and DLC recruitment.
Since the results in Fig. 1B suggest that km23-2 forms a dimer after TGFß treatment in vivo, it was of interest to determine whether TGFß could stimulate binding of the km23-2 dimer to DIC. Hence, we performed IP/blot analyses under non-reducing conditions, after co-expression of km23-2-Flag, and either KNRII-HA or RII-HA in the absence and presence of TGFß. As shown in Fig. 4C, TGFß rapidly induced the formation of a complex between the km23-2 dimer and DIC (lanes 2-5). More importantly, expression of KNRII completely blocked this interaction between km23-2 and DIC at 5 min after TGFß treatment (lane 1). No specific band was detectable in the IgG control (lane 6). Equal expression and loading of DIC and km23-2 was also confirmed (middle and bottom panels). Thus, the kinase activity of TßRII was required for the recruitment of the km23-2 dimer to the DIC.
It is thought that DLCs may be important for specifying the nature of the cargo that will be transported by the motor [33]. Therefore, it is likely that extracellular factors (such as growth factors, cytokines, etc) might be able to select the particular DLCs to be recruited to the motor in specific cellular contexts to specify and carry the cargo. Along these lines, we had previously shown that TßR activation was associated with not only km23-1-Smad2 complex formation, but also an increase in km23-1 binding to the DIC [11, 14]. However, we had not previously examined the ability of TGFß to regulate the interplay among other components of the dynein motor complex. The cargo attachment complex is also associated with the DHC, which conveys the ATPase activity for motoring along microtubules (MTs) [9, 34]. In addition to the dynein motor itself, intracellular transport of signaling cargoes generally requires the attachment of the dynactin complex [9, 34]. This multisubunit complex contains 11 different subunits, including the largest subunit, p150Glued, and a filament of actin-related protein 1 (Arp1). It has been found to be essential for many cellular functions of cytoplasmic dynein [9, 34]. Here we show for the first time (Fig. 5A) that TGFß stimulates complex formation not only between the DHC and DIC (top panel), but also between the p150Glued subunit of dynactin and DIC (middle panel). The bottom panel confirms equal loading, supporting the presence of a TGFß-regulated dynein complex in TGFß-responsive epithelial cells. Our findings provide the first evidence that dynein motor subunit assembly can be rapidly stimulated by a multifunctional cytokine.
TGFß regulates the tethering of the specific km23-2-Smad3 cargo to the other dynein components. A: MDCK cells were incubated in SF medium for 1h prior to addition of TGFß1 (5 ng/ml) for the indicated times. Cell lysates were subjected to IP using a monoclonal anti-DIC Ab and were separated by 4-12% NuPAGE, followed by immunoblot analysis using an anti-DHC Ab (top panel) or an anti-p150 Ab (2nd panel). The same membrane was re-blotted with anti-DIC (bottom panel) to show equal protein loading. The results shown are representative of three similar experiments. B: MDCK cells were transiently transfected with either EV or km23-2-Flag as indicated. 24h after transfection, cells were incubated in SF medium for 1h prior to addition of TGFß1 (5 ng/ml) for the indicated times. Cell lysates were subjected to IP using a monoclonal anti-DIC Ab and were separated by 4-12% NuPAGE, followed by immunoblot analysis using an anti-Flag Ab (top panel) and an anti-Smad3 Ab (2nd panel). The same membrane was re-blotted with anti-DIC (3rd panel) to show equal protein loading. Western blot analysis (two bottom panels) with anti-Flag or anti-DIC demonstrates equal protein expression of km23-2 and DIC. The results shown are representative of two similar experiments. C: A schematic diagram illustrating the role of km23-1 in Smad3-dependent Smad7 transcriptional activation after TGFß stimulation.
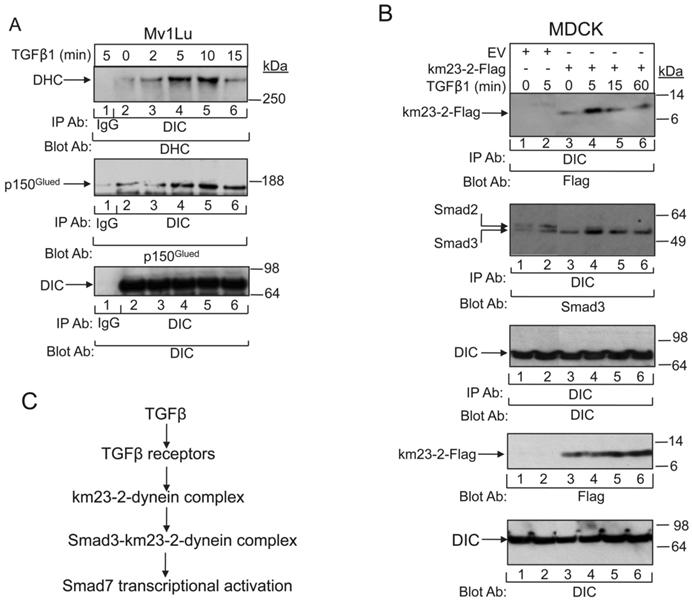
Since TGFß stimulated dynein motor complex assembly very rapidly after treatment of the TGFß-sensitive cells, it was conceivable that the km23-2-Smad3 complexes analyzed in Figs. 1 and 3 were actually signaling cargo recruited to the dynein motor. In order to investigate whether TGFß stimulated not only assembly of the dynein motor attachment complex, but also triggered the tethering of the km23-2-Smad3 cargo to the other dynein components known to be required for motoring along MTs, we performed IP/blot analyses in TGFß-responsive MDCK cells at various times after ligand stimulation. As shown in Fig. 5B, TGFß stimulated a rapid interaction of DIC with km23-2 at 5 min after TGFß treatment (lane 4, top panel). In contrast, the association between km23-2 and DIC was significantly decreased at 15 and 60 min after TGFß addition (lanes 5, 6). The EV controls were negative (lanes 1- 2, top panel). In addition, TGFß induced a rapid interaction of Smad3 with DIC in km23-2-transfected cells (lanes 3-6, 2nd panel). The kinetics were similar to those for km23-2 binding to the DIC (lanes 3-6, top panel), showing some basal interaction, but with increased association at 5 min after TGFß addition. Since the Smad3 Ab that we used does cross-react with Smad2, the two specific bands detected in the EV-transfected cells (lanes 1-2, middle panel) suggest that endogenous DIC co-precipitated with both endogenous Smad2 and Smad3. In addition, TGFß stimulated a rapid interaction of DIC with Smad2/3 (lane 2, 2nd panel). Collectively, our results indicate for the first time that TGFß regulated the tethering of km23-2-Smad3 to the other dynein components, suggesting that Smad3 is a specific cargo of the dynein motor complex.
Discussion
Our previous results have demonstrated that km23-2 has functions in TGFß signaling that are distinct from those for km23-1. In the current report, we showed that TGFß regulated the interaction of the km23-2 DLC preferentially with Smad3, relative to that with Smad2, in cells expressing endogenous TßRs. km23-2 and Smad3 proteins were recruited into the same complex upon ligand activation of the TßRs. We also show for the first time that knockdown of endogenous km23-2 expression resulted in a reduction of Smad3-dependent Smad7 transcriptional activation. In addition, km23-2 was present in early endosomal compartments with Smad3 after TGFß treatment. TGFß stimulated not only the phosphorylation of km23-2, but also the recruitment of km23-2 dimer to DIC in TGFß-sensitive cells. Kinase-active TßRII was required for both km23-2 phosphorylation and interaction with DIC. Most importantly, we demonstrate for the first time that TGFß not only stimulated assembly of the dynein motor attachment complex, but also triggered the tethering of the km23-2-Smad3 cargo to the other dynein components. Collectively, based upon our results and those of others, we propose a model for km23-2 action in Smad3-dependent TGFβ signaling. According to this model, upon TßRs activation, km23-2 may function as a motor receptor to recruit Smad3 to the dynein complex for intracellular transport, thereby mediating Smad3-dependent TGFβ signaling, as assessed by Smad7 transcriptional activation (Fig. 5C).
The TßRs are known to be internalized with Smad2/3 into early endosomes within minutes of TGFß addition to cells [29]. This is in contrast to non-Smad signaling in response to TGFß, which has been shown to occur in lipid rafts [16, 35]. Further, differential activation of Smad2 and Smad3 can occur as a result of interactions with unique subsets of proteins after TßR activation [36]. In addition, our previous results have shown that km23-1 selectively interacts with Smad2 complexes in early endosomes [14]. In contrast, herein we show that Smad3 was recruited into an early endosomal complex with km23-2 at 5 min after TGFß treatment. It is possible that the formation of the specific Smad3-km23-2 complexes described herein may help to selectively direct Smad3 to the appropriate sub-domains or sub-compartments in early endosomes.
Cytoplasmic dynein with distinct light chain content can exhibit target -binding specificity. For example, LC8 is thought of as an ordered hub protein, which regulates the ordering and dimerization of locally disordered partner proteins by binding [37]. Interestingly, although the Tctex/DYNLT light chain family is a structural homolog of the LC8/DYNLL family, with no apparent sequence similarity, there is no overlap in known targets of the two light chains [8]. More importantly, even members of the same DLC family exhibit target-binding specificity. For example, Tctex-1 binds rhodopsin, whereas rp3 does not [38]. Our previous results have shown that km23-1 interacts with the TGFß/Smad2 complex [14]. Similarly, kinesin, the motor protein that transports signaling cargoes toward the cell periphery, regulates Smad2 localization after TGFß treatment [39]. Further, a light chain of kinesin has been shown to undergo phosphorylation and binding to Smad2 after activation of a specific kinase by TGFß [40]. Here we show that km23-2 selectively interacts with Smad3 and is required for Smad3-depedent Smad7 transcriptional activation [14]. More importantly, TGFß stimulated not only assembly of the dynein motor attachment complex, but also triggered the tethering of the km23-2-Smad3 cargo to the other dynein components. Thus, our results demonstrate for the first time that the km23-2 DLC may function as a dynein cargo adaptor linking TGFß/Smad3 signaling complex to dynein.
Phosphorylation is one of the mechanisms that regulates the functions of the DLC families. For example, biochemical studies clearly showed that LC8 phosphorylation at Ser88 can disrupt dimer assembly [41]. In contrast to LC8, the phosphomimetic mutant of Tctex-1 retains its association state in the native dimer [41]. Our previous results have shown that km23-1 and zebrafish km23 (zkm23) are phosphorylated upon TGFß activation [11, 42]. Here we show that the kinase activity of TβRII is required for the regulation of km23-2 binding to DIC. In relation to these findings, a recent crystal structure and in vitro binding studies suggest that phosphorylation does not directly affect km23-1 binding to DIC [43]. Therefore, it is possible that TGFß-stimulated interactions between km23-1 or km23-2 and an additional signaling component or components are required to regulate DIC binding [43]. Specific kinases and other relevant signaling components are likely involved in these dynamic regulatory events.
In the current report, we also show that blockade of km23-2 diminished TGFß induction of Smad7 transcriptional activation. Smad7 has been shown to be a direct gene target of Smad3/4 in several cell types. For example, an absolute requirement for Smad3, but not Smad2, has been demonstrated for TGFß induction of the Smad7 promoter in human MD-MBA-468 cells [27], human Hep3B cells [44], and mouse embryonic fibroblasts [6]. Similarly, km23-2 appears to be required for TGFß induction of Smad7 promoter activity via a Smad3-specific TGFß pathway. Our findings in this regard are particularly significant in that Smad7 is an important negative feedback regulator of signaling pathways involved in development and tumorigenesis [6, 27, 35, 36]. For example, inhibitor of differentiation-1 (Id1), a dominant-negative transcriptional antagonist, was found to be rapidly induced by TGFß, dependent upon Smad3, but not Smad2, and regulated by Smad7 feedback inhibition [45, 46]. Further, TGFß regulation of this Smad3-/Smad7-dependent target plays critical roles in both the metastatic potential of pancreatic cancer and in the self-renewal capacity of human colon cancer-initiating cells, events that may involve p21, a known km23-2/Smad3 downstream target [47, 48]. Overall then, the ability of km23-2 to facilitate TGFß/Smad3 signaling to downstream events such as p21 and Smad7 provides clear evidence of the importance of km23-2 in TGFß signaling, and implicates km23-2 in cancer progression and metastasis. Altogether, our results emphasize the unique nature of the km23 family of DLCs, and stress the importance of considering the in vivo regulation of dynein subunit assembly and cargo interactions by extracellular stimuli such as cytokines or growth factors.
Abbreviations
Ab, antibody; DHC, dynein heavy chain; DIC, dynein intermediate chain; DLC, dynein light chain; EE, early endosome; EEA1, early endosome antigen 1; EV, empty vector; FPLC, fast protein liquid chromatography; IP, immunoprecipitation; LUMIER, luminescence-based mammalian interaction mapping; MT, microtubule; NC, negative control; RSmads, receptor-regulated Smads; SF, serum-free; TßR, TGFß receptor; TK, thymidine kinase.
Acknowledgements
This work was supported by National Institutes of Health Grants CA090765, CA092889 to K.M.M. We also thank Dr. J. Massague (Memorial Sloan-Kettering Cancer Center, New York, NY) for KNRII-HA; Dr. Erwin Bottinger (Mount Sinai School of Medicine, New York, NY) for the Smad7-Luc; Dr. Jeffrey L. Wrana (Samuel Lunenfeld Res. Institute, Toronto, Canada) for the pCMV5-HA-RII, hRL-Smad2, hRL-Smad3, pRL-TK, and Ski-Flag constructs.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Heldin CH, Vanlandewijck M, Moustakas A. Regulation of EMT by TGFbeta in cancer. FEBS Lett. 2012;586:1959-1970
2. Massague J. TGFbeta in Cancer. Cell. 2008;134:215-230
3. Yue J, Mulder KM. Transforming growth factor-beta signal transduction in epithelial cells. Pharmacol Ther. 2001;91:1-34
4. Roberts AB, Russo A, Felici A. et al. Smad3: a key player in pathogenetic mechanisms dependent on TGF-beta. Ann N Y Acad Sci. 2003;995:1-10
5. Uemura M, Swenson ES, Gaca MD. et al. Smad2 and Smad3 play different roles in rat hepatic stellate cell function and alpha-smooth muscle actin organization. Mol Biol Cell. 2005;16:4214-4224
6. Piek E, Ju WJ, Heyer J. et al. Functional characterization of transforming growth factor beta signaling in Smad2- and Smad3-deficient fibroblasts. J Biol Chem. 2001;276:19945-19953
7. Jin Q, Gao G, Mulder KM. Requirement of a dynein light chain in TGFbeta/Smad3 signaling. J Cell Physiol. 2009;221:707-715
8. Pfister KK, Shah PR, Hummerich H. et al. Genetic analysis of the cytoplasmic dynein subunit families. PLoS Genet. 2006;2:e1
9. Allan VJ. Cytoplasmic dynein. Biochem Soc Trans. 2011;39:1169-1178
10. Pfister KK, Fisher EM, Gibbons IR. et al. Cytoplasmic dynein nomenclature. J Cell Biol. 2005;171:411-413
11. Tang Q, Staub CM, Gao G. et al. A novel transforming growth factor-beta receptor-interacting protein that is also a light chain of the motor protein dynein. Mol Biol Cell. 2002;13:4484-4496
12. Nikulina K, Patel-King RS, Takebe S. et al. The Roadblock light chains are ubiquitous components of cytoplasmic dynein that form homo- and heterodimers. Cell Motil Cytoskeleton. 2004;57:233-245
13. Jiang J, Yu L, Huang X. et al. Identification of two novel human dynein light chain genes, DNLC2A and DNLC2B, and their expression changes in hepatocellular carcinoma tissues from 68 Chinese patients. Gene. 2001;281:103-113
14. Jin Q, Ding W, Mulder KM. Requirement for the dynein light chain km23-1 in a Smad2-dependent transforming growth factor-beta signaling pathway. J Biol Chem. 2007;282:19122-19132
15. Jin Q, Ding W, Staub CM. et al. Requirement of km23 for TGFbeta-mediated growth inhibition and induction of fibronectin expression. Cell Signal. 2005;17:1363-1372
16. Jin Q, Ding W, Mulder KM. The TGFbeta receptor-interacting protein km23-1/DYNLRB1 plays an adaptor role in TGFbeta1 autoinduction via its association with Ras. J Biol Chem. 2012;287:26453-26463
17. Liu G, Ding W, Neiman J. et al. Requirement of Smad3 and CREB-1 in mediating transforming growth factor-beta (TGF beta) induction of TGF beta 3 secretion. J Biol Chem. 2006;281:29479-29490
18. Barrios-Rodiles M, Brown KR, Ozdamar B. et al. High-throughput mapping of a dynamic signaling network in mammalian cells. Science. 2005;307:1621-1625
19. Zhang Y, Hancock WO. The two motor domains of KIF3A/B coordinate for processive motility and move at different speeds. Biophys J. 2004;87:1795-1804
20. Sharma C, Vomastek T, Tarcsafalvi A. et al. MEK partner 1 (MP1): regulation of oligomerization in MAP kinase signaling. J Cell Biochem. 2005;94:708-719
21. Kawabata M, Inoue H, Hanyu A. et al. Smad proteins exist as monomers in vivo and undergo homo- and hetero-oligomerization upon activation by serine/threonine kinase receptors. Embo J. 1998;17:4056-4065
22. King SJ, Bonilla M, Rodgers ME. et al. Subunit organization in cytoplasmic dynein subcomplexes. Protein Sci. 2002;11:1239-1250
23. Ilangovan U, Ding W, Zhong Y. et al. Structure and dynamics of the homodimeric dynein light chain km23. J Mol Biol. 2005;352:338-354
24. Yan X, Chen YG. Smad7: not only a regulator, but also a cross-talk mediator of TGF-beta signalling. Biochem J. 2011;434:1-10
25. ten Dijke P, Hill CS. New insights into TGF-beta-Smad signalling. Trends Biochem Sci. 2004;29:265-273
26. Denissova NG, Pouponnot C, Long J. et al. Transforming growth factor beta -inducible independent binding of SMAD to the Smad7 promoter. Proc Natl Acad Sci USA. 2000;97:6397-6402
27. von Gersdorff G, Susztak K, Rezvani F. et al. Smad3 and Smad4 mediate transcriptional activation of the human Smad7 promoter by transforming growth factor beta. J Biol Chem. 2000;275:11320-11326
28. Hayes S, Chawla A, Corvera S. TGF beta receptor internalization into EEA1-enriched early endosomes: role in signaling to Smad2. J Cell Biol. 2002;158:1239-1249
29. Di Guglielmo GM, Le Roy C, Goodfellow AF. et al. Distinct endocytic pathways regulate TGF-beta receptor signalling and turnover. Nat Cell Biol. 2003;5:410-421
30. Lin HK, Bergmann S, Pandolfi PP. Cytoplasmic PML function in TGF-beta signalling. Nature. 2004;431:205-211
31. Nicolas FJ, Hill CS. Attenuation of the TGF-beta-Smad signaling pathway in pancreatic tumor cells confers resistance to TGF-beta-induced growth arrest. Oncogene. 2003;22:3698-3711
32. Wieser R, Attisano L, Wrana JL. et al. Signaling activity of transforming growth factor beta type II receptors lacking specific domains in the cytoplasmic region. Mol Cell Biol. 1993;13:7239-7247
33. Mallik R, Gross SP. Molecular motors: strategies to get along. Curr Biol. 2004;14:R971-982
34. Kardon JR, Vale RD. Regulators of the cytoplasmic dynein motor. Nat Rev Mol Cell Biol. 2009;10:854-865
35. Zuo W, Chen YG. Specific activation of mitogen-activated protein kinase by transforming growth factor-beta receptors in lipid rafts is required for epithelial cell plasticity. Mol Biol Cell. 2009;20:1020-1029
36. Brown KA, Pietenpol JA, Moses HL. A tale of two proteins: differential roles and regulation of Smad2 and Smad3 in TGF-beta signaling. J Cell Biochem. 2007;101:9-33
37. Barbar E. Dynein light chain LC8 is a dimerization hub essential in diverse protein networks. Biochemistry. 2008;47:503-508
38. Tai AW, Chuang JZ, Bode C. et al. Rhodopsin's carboxy-terminal cytoplasmic tail acts as a membrane receptor for cytoplasmic dynein by binding to the dynein light chain Tctex-1. Cell. 1999;97:877-887
39. Batut J, Howell M, Hill CS. Kinesin-mediated transport of Smad2 is required for signaling in response to TGF-beta ligands. Dev Cell. 2007;12:261-274
40. Manser C, Guillot F, Vagnoni A. et al. Lemur tyrosine kinase-2 signalling regulates kinesin-1 light chain-2 phosphorylation and binding of Smad2 cargo. Oncogene. 2012;31:2773-2782
41. Song Y, Benison G, Nyarko A. et al. Potential role for phosphorylation in differential regulation of the assembly of dynein light chains. J Biol Chem. 2007;282:17272-17279
42. Jin Q, Gao G, Mulder KM. Requirement of a dynein light chain in transforming growth factor beta signaling in zebrafish ovarian follicle cells. Mol Cell Endocrinol. 2012;348:233-240
43. Hall J, Song Y, Karplus PA. et al. The crystal structure of dynein intermediate chain-light chain roadblock complex gives new insights into dynein assembly. J Biol Chem. 2010;285:22566-22575
44. Felici A, Wurthner JU, Parks WT. et al. TLP, a novel modulator of TGF-beta signaling, has opposite effects on Smad2- and Smad3-dependent signaling. Embo J. 2003;22:4465-4477
45. Li Y, Yang J, Luo JH. et al. Tubular epithelial cell dedifferentiation is driven by the helix-loop-helix transcriptional inhibitor Id1. J Am Soc Nephrol. 2007;18:449-460
46. Liang YY, Brunicardi FC, Lin X. Smad3 mediates immediate early induction of Id1 by TGF-beta. Cell Res. 2009;19:140-148
47. Shuno Y, Tsuno NH, Okaji Y. et al. Id1/Id3 knockdown inhibits metastatic potential of pancreatic cancer. J Surg Res. 2010;161:76-82
48. O'Brien CA, Kreso A, Ryan P. et al. ID1 and ID3 regulate the self-renewal capacity of human colon cancer-initiating cells through p21. Cancer cell. 2012;21:777-792
Author contact
![]() Corresponding author: Department of Biochemistry and Molecular Biology-MC H171, Penn State Hershey College of Medicine, 500 University Drive, Hershey, PA 17033. Tel: 1-717-531-6789; FAX: 1-717-531-0939.
Corresponding author: Department of Biochemistry and Molecular Biology-MC H171, Penn State Hershey College of Medicine, 500 University Drive, Hershey, PA 17033. Tel: 1-717-531-6789; FAX: 1-717-531-0939.