Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
INTRODUCTION
ENDOTHELIAL FUNCTIONS
ENDOTHELIAL DYSFUNCTION
PHARMACOLOGICAL REMEDIES
CONCLUSIONS
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2013; 9(10):1057-1069. doi:10.7150/ijbs.7502 This issue Cite
Review
The Vascular Endothelium and Human Diseases
Peramaiyan Rajendran1, Thamaraiselvan Rengarajan1, Jayakumar Thangavel2, Yutaka Nishigaki1, Dhanapal Sakthisekaran3, Gautam Sethi4, Ikuo Nishigaki1 ![]()
1. NPO-International Laboratory of Biochemistry,1-166, Uchide, Nakagawa-ku, Nagoya 454-0926, Japan;
2. Department of Pharmacology and Center of Lung and Vascular Biology, University of Illinois College of Medicine, Chicago, Illinois, United States of America;
3. Department of Medical Biochemistry, Dr. ALM PG Institute of Basic Medical Sciences, University of Madras, Taramani Campus, Chennai, India 600 113;
4. Department of Pharmacology,Yong Loo Lin School of Medicine,National University Health System, 10 Medical Drive, MD11, #05-09, Clinical Research Centre, Singapore 117597.
Received 2013-8-26; Accepted 2013-10-7; Published 2013-11-9
Abstract
Alterations of endothelial cells and the vasculature play a central role in the pathogenesis of a broad spectrum of the most dreadful of human diseases, as endothelial cells have the key function of participating in the maintenance of patent and functional capillaries. The endothelium is directly involved in peripheral vascular disease, stroke, heart disease, diabetes, insulin resistance, chronic kidney failure, tumor growth, metastasis, venous thrombosis, and severe viral infectious diseases. Dysfunction of the vascular endothelium is thus a hallmark of human diseases. In this review the main endothelial abnormalities found in various human diseases such as cancer, diabetes mellitus, atherosclerosis, and viral infections are addressed.
Keywords: Endothelium, Endothelial dysfunction, Atherosclerosis, Stroke, Cancer.
INTRODUCTION
The endothelium was once thought of as the ''cellophane wrapper'' of the vascular tree, with no other specific functions than affording selective permeability to water and electrolytes (1). However, enormous advances since the 1980's have led to an understanding of the complex functions of this large endocrine organ. Vascular endothelial cells line the entire circulatory system, from the heart to the smallest capillaries. These cells have very distinct and unique functions that are paramount to vascular biology. These functions include fluid filtration, such as in the glomeruli of the kidneys, blood vessel tone, hemostasis, neutrophil recruitment, and hormone trafficking. Researchers in vascular biology know well that the endothelium embodies a wide range of homeostatic functions (2), with the ability to act in both sensory and effector capacities. The role of the endothelium is effected through the presence of membrane-bound receptors for numerous molecules including proteins, lipid-transporting particles, metabolites, and hormones, as well as through specific junctional proteins and receptors that govern cell-cell and cell-matrix interactions (3). Endothelial cells (EC's) also play a pivotal role in regulating blood flow. In part, this role is achieved due to the capacity of quiescent EC's to generate an active antithrombotic surface that facilitates the transit of the plasma and cellular constituents throughout the vasculature. Perturbations, such as those that may occur at sites of inflammation or high hydrodynamic shear stress, disrupt these activities and induce EC's to create a prothrombotic and antifibrinolytic microenvironment. Blood flow is also regulated, in part, through secretion and uptake of vasoactive substances by the endothelium that act in a paracrine manner to constrict and dilate specific vascular beds in response to stimuli such as endotoxin (4). The endothelium is a cell layer lining the blood luminal surface of vessels. It was until recently considered to be just a lining, but it is now realized that EC's have important functions besides merely providing a lining for vessel walls. For example, the endothelium is now known to be involved in the control of thrombosis and thrombolysis, platelet and leukocyte interaction with the vessel wall, and the regulation of vascular tone and growth of blood vessels (5).
ENDOTHELIAL FUNCTIONS
Thrombosis and thrombolysis
The endothelium plays a crucial role in providing the proper haemostatic balance. The function of endothelial cells far exceeds that of providing a non-thrombogenic inner layer of the vascular wall that helps to maintain blood fluidity. Under physiological conditions, endothelial cells prevent thrombosis by means of different anticoagulant and antiplatelet mechanisms. These cells are involved in all main haemostatic pathways triggered upon vascular injury and limit clot formation to the areas where haemostasis is needed to restore vascular integrity. Breakdown of this complex balance between pro- and anticoagulant systems because of genetic or acquired disturbances may result in bleeding or thrombosis. Endothelial heterogeneity assures adequate homeostasis in the different organs and parts of the vascular tree. The local environment elicits heterogeneous endothelial cell phenotypes determined by local needs. This heterogeneity also explains the diverse pathological responses to a disturbed vascular integrity. Localised manifestation of thrombosis in spite of disturbance of systemic procoagulant systems depends on vascular bed-specific properties. Endothelial dysfunction not only precedes atherogenesis but may also predispose to arterial thrombosis. The potential role of the endothelium in venous thrombosis with and without overt vessel wall injury has been discussed. The vast majority of endothelial cells are located in the microvessels. Therefore, it is no surprise that endothelial cells play a key role in microcirculatory diseases such as thrombotic microangiopathies and diffuse intravascular coagulation. Microcirculatory endothelial cell activation is an important feature in all thrombotic microangiopathies. In diffuse intravascular coagulation, the endothelium is the interface between inflammation and inappropriate activation of the coagulation system (6).
Coagulant mechanisms
Endothelial cells form the luminal vascular surface and thus have a central role in the regulation of coagulation. One important way in which endothelial cells control the clotting system is by regulating the expression of binding sites for anticoagulant and procoagulant factors on the cell surface. In the quiescent state, endothelial cells maintain blood fluidity by promoting the activity of numerous anticoagulant pathways, including the protein C/protein S pathway. After activation, as can be brought about by cytokines, the balance of endothelial properties can be tipped in favor of clot formation through the coordinated induction of procoagulant and suppression of anticoagulant mechanisms. Tumor necrosis factor suppresses the formation of throbomodulin, an endothelial anticoagulant cofactor, and induces the expression of tissue factor, which is a t procoagulant cofactor. Working in concert, these changes can allow fibrin formation to proceed in an inflamed focus but still maintain blood fluidity in the surrounding area of normal vasculature. Recent studies suggest that similar changes in endothelial coagulant properties can be induced by advanced glycosylation end products, which are proteins modified by glucose and accumulate in the vasculature at a rapid rate in diabetic subjects, indicating the potential relevance of these mechanisms to diabetic vascular disease (7).
Platelet and leukocyte interaction
The first description of rolling of blood cells along the endothelial surface of venules was reported more than 160 years ago, when leukocytes were shown to adhere to blood vessel walls, an interaction that increased in inflammation (8). Giulio Bizzozero in 1882 first described platelets as a new blood corpuscle, playing a relevant role in thrombosis and haemostasis, and observed that “Every time when a vascular wall is damaged arrest of white blood corpuscles represents a secondary phenomenon and may, perhaps, be caused by increased stickiness of blood platelets whereby the latter react with white blood corpuscles which have been brought in contact with the former by blood circulation”(9). Many decades later, the modern version of intravital microscopy allowed the first quantitative observations of leukocyte rolling in the cheek pouch of hamsters and in mouse mesentery (10). Platelet adhesion to and leukocyte rolling on the endothelium represent the initial stage of a multistep process leading to extravasation of white blood cells to sites of inflammation or infection, to platelet-leukocyte interaction and aggregation on a thrombogenic surface, and finally to vascular occlusion. Platelets may interact with the endothelium even in the absence of any apparent morphological damage. Indeed, they may stick to an apparently intact endothelium inflamed by different stimuli, such as infection, mechanic alteration or ischemia and reperfusion, or to endothelium located at lesion-prone sites, such as the carotid artery bifurcation. The recruitment of platelets and leukocytes at sites of vascular injury is a very rapid response and is mediated by the release of preformed components elaborated by the endothelium, including Weibel-Palade bodies and their major constituents, the largest multimers of von Willebrand factor, and P-selectin, which are the most active promoters of platelet and leukocyte adhesion. P-selectin mediates both leukocyte and platelet adhesion and during secretion fuses with the endothelial plasma membrane. The process of leukocyte rolling is initiated by P-selectin secretion and is concluded by leukocyte adhesion and transmigration into inflamed tissue (11-14).
Regulation of vascular tone and growth
Endothelial cells play an important regulatory role in the circulation as a physical barrier and as a source of a variety of regulatory substances. Endothelium-derived nitric oxide and prostacyclin are released in response to physical stimuli, hormones, and platelet-derived substances and induce vascular relaxation and inhibit platelet function. Certain substances can evoke a hyperpolarization of smooth muscle cells. In addition, endothelial cells can release several contraction-inducing factors (e.g., endothelin, thromboxane A2, angiotensin II, superoxide, and unidentified endothelium-derived contraction-inducing factors), at least under certain conditions. Endothelial cells are also a source of growth inhibitors and promoters, such as heparin and heparin sulphates, platelet-derived growth factor, and thrombospondin. Several vasoactive substances produced by the endothelium, such as nitric oxide, endothelin, and angiotensin II may also play a role in the regulation of vascular growth. Thus, the endothelial layer can regulate vascular tone and growth. Dysfunction of these endothelium-dependent regulatory systems may play a role in cardiovascular diseases, such as hypertension and atherosclerosis (15).
Cell Proliferation and Angiogenesis
The endothelium also involved in blood vessel formation. The development of a functional vascular network requires a remarkable degree of coordination between different cell types undergoing complex changes and is exquisitely dependent upon signals exchanged between these cell types. Vascular endothelial growth factor (VEGF) provided the first example of a growth factor specific for the vascular endothelium. More recently, an entirely unrelated family of growth factors known as the angiopoietins (Ang) and particular members of the very large ephrin family have been identified as having unique effects on the endothelium. Recent insights have led to a model of vascular formation that attempts to incorporate the known vascular-specific growth factors. According to this model, VEGF is the most critical driver of vascular formation, as it is required to initiate the formation of immature vessels by vasculogenesis or angiogenic sprouting. Ang1 and ephrinB2 are subsequently required for further remodeling and maturation of this initially immature vasculature, notably as endothelial cells integrate with supporting cells such as smooth muscle cells and pericytes. Following vessel maturation, Ang1 seems to continue to be important for maintaining the quiescence and stability of the mature vasculature (16).
ENDOTHELIAL DYSFUNCTION
Endothelial dysfunction is characterized by a shift in the actions of the endothelium toward reduced vasodilation, a proinflammatory state, and prothrombic properties. It is associated with most forms of cardiovascular disease, such as hypertension, coronary artery disease, chronic heart failure, peripheral vascular disease, diabetes, chronic kidney failure, and severe viral infections. Free radicals can disrupt the balance of NO, damage the endothelium, and leave it overly permeable, allowing toxins to pass into body tissues (17). In most instances, the human body has an adequate supply of antioxidants obtained from various foods to neutralize these free radicals; but if the body is depleted of these antioxidants, or if there are too many coexistent factors, injury to the endothelium and a change in the balance of NO may occur. If the endothelium becomes damaged and the NO levels become imbalanced, cells that should remain in the blood can pass through blood vessels into the adjacent body tissue. Some of these proteins include C - reactive protein, which is produced by the liver and causes inflammation (18). When NO action is inhibited, endothelial signaling can become impaired, resulting in widespread disease, because the endothelium actively maintains approximately 60,000 miles of blood vessels in the human body. Several factors that can increase the number of free radicals in the body including obesity, smoking, sleep deprivation, acute microbial infections, high glucose intake, and exposure to metals and air pollutants. When the endothelium is functioning normally, it helps to regulate blood clotting, assists the body's immune response, controls the volume of fluid and the amount of electrolytes and other substances that pass from the blood into the tissues, and produces dilation or constriction of the blood vessels (Fig. 1) (19). When endothelial dysfunction is present, however, the ability to perform one or more of these functions is reduced. Actually measuring a patient's endothelial function is not routinely done; but when it is, the ability of the blood vessels to dilate and/or constrict in response to drug administration can be assessed. Endothelial dysfunction is thought to play a major role in the development of atherosclerosis, angiogenesis in cancer, vascular leakage, infectious diseases, and stroke. Endothelial dysfunction can be caused by several conditions, including diabetes or metabolic syndrome, hypertension, smoking, and physical inactivity [20].
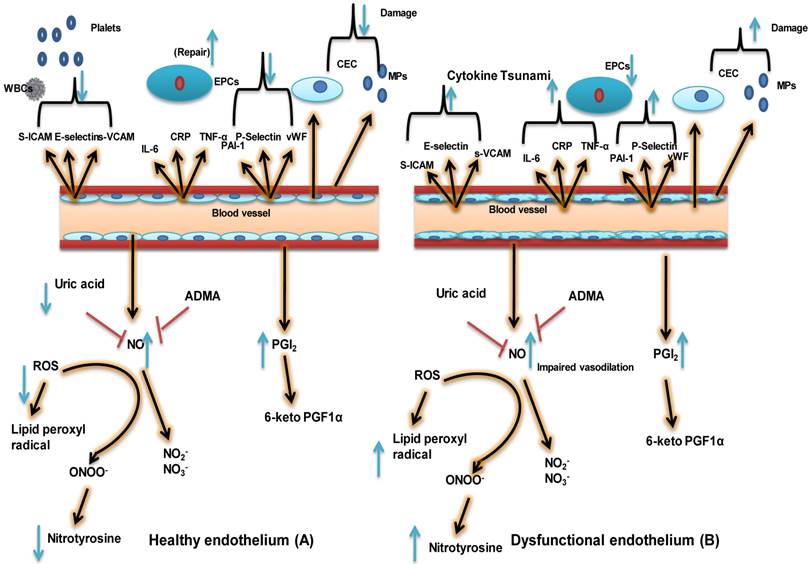
The healthy endothelium not only arbitrates endothelium-dependent vasodilation, but also actively suppresses thrombosis, vascular inflammation, and hypertrophy. This schematic depicts differences between a healthy endothelium (A) and a dysfunctional one (B). A healthy endothelium displays a vasodilatory phenotype consisting of high levels of vasodilators such as nitric oxide (NO) and prostacyclin (PGI2) and low levels of reactive oxygen species (ROS) and uric acid. A healthy endothelium also has an anticoagulative phenotype consisting of low levels of plasminogen activator inhibitor 1 (PAI-1), von Willebrand factor (vWF), and P-selectin. Very little inflammation may be present, as indicated by low levels of soluble vascular cell adhesion molecule (sVCAM.), soluble intercellular adhesion molecule (sICAM), E-selectin, C-reactive protein (CRP), tumor necrosis factor alpha (TNF-α), and interleukin-6 (IL-6). Finally, the population of endothelial progenitor cells (EPCs, indicative of vascular repair capacity) is high; whereas levels of endothelial microparticles (EMPs) and circulating endothelial cells (CECs), indicative of endothelial stress/damage, are low. In the case of a dysfunctional endothelium, the phenotypic characteristics include impaired vasodilation, increased oxidative stress/uric acid, lipid peroxide radical,Nitrotyrosine and Nitirc oxide, and a procoagulant and pro-inflammatory phenotype with decreased vascular repair capacity and increased numbers of EMPs and CECs. 6-keto PGF1α: 6 keto prostaglandin F1-alpha, a stable product of PGI2; ADMA; asymmetric dimethyl arginine, inhibitor of NO biosynthesis; EC: endothelial cell; NO2-: nitrite ion, stable degradation product of NO; NO3- nitrate ion, stable degradation product of NO; ONOO-: peroxynitrite, the product of superoxide-mediated inactivation of NO; VSMC: vascular smooth muscle cell; WBC: white blood cell. (Modified from Dylan Burger and Rhian MT 2012).
Peripheral vascular disease
Endothelial dysfunction can contribute to the clinical status in peripheral vascular disease (PAD) in a number of ways. Loss of nitric oxide in skeletal muscle microvessels blunts the hyperemic responses to exercise and ischemia, which would be expected to limit oxygen delivery under conditions of increased demand (21-22). As that in epicardial coronary arteries, the loss of nitric oxide in the lower extremities can worsen the vasoconstrictor effects of catecholamines and impair flow-mediated dilation, which might increase the severity of stenosis and resistance to blood flow during exercise (23). Importantly, such abnormalities can be reversed over weeks to months with risk-reduction therapy; even if lesion regression does not occur (24). This time course corresponds to improvement of claudication symptoms and reduced cardiovascular events during intervention studies. As described by Glagov et al. (25), outward remodelling helps to maintain a patent arterial lumen as atherosclerotic plaques increase in size. Experimental studies have shown that compensatory remodeling depends on the activity of nitric oxide synthase (26), and the authors of these studies recently demonstrated that the remodelling response is blunted in patients with local endothelial dysfunction. Chronic and recurrent limb ischemia stimulates compensatory formation of collateral vessels (27-30), and this response depends on the bioavailabity of nitric oxide (31-33). Thus, endothelial dysfunction may worsen the clinical status in PAD because of impaired formation of collateral vessels and remodelling. Endothelial activation with expression of proinflammatory and prothrombotic factors may also contribute to ischemia by promoting plaque rupture (35, 36). Studies completed by us and others have shown that endothelial dysfunction predicts acute coronary events in PAD patients, suggesting a relationship between such dysfunction and plaque rupture in the coronary circulation (23, 35, 36). Plaque rupture in the lower extremities would produce an acute reduction in blood flow, and this mechanism has been proposed to contribute to the development of critical limb ischemia (34). The observed benefits of antiplatelet therapy support this concept. Furthermore, thrombus reorganization following subacute rupture has been proposed as a mechanism for lesion progression (37-39). Consistent with these potential mechanisms, several small cross-sectional studies have demonstrated the loss of nitric oxide bioavailability in patients with PAD. For example, PAD patients have blunted vasodilator responses to acetylcholine and impaired endothelium-dependent flow-mediated dilation of their brachial artery (40-42). Flow-mediated dilation also has been reported to correlate inversely with the ankle-brachial index (43-45). Urine nitrate and cyclic GMP levels are reduced in patients with PAD, suggesting decreased total body nitric oxide production. Polymorphisms of the nitric oxide synthase gene correlate with the ankle-brachial index. Finally, PAD is associated with increased production of endothelin and plasminogen activator inhibitor-1 (46). These studies support a link between endothelial dysfunction and PAD.
Stroke
Endothelial dysfunction may not only be a vascular disease marker, but may also actually play an important pathogenetic role, leading to progression of the disease and unfavourable outcomes. Among vascular diseases, cerebrovascular accidents, namely, stroke, clearly represent a paradigmatic example of the potential role of a dysfunctional endothelium. In fact, in the world's growing elderly population few diseases are more dreaded than stroke. With an increasing incidence and a mortality of 30%, stroke carries the threat of death or long-term disability and suffering (47). Elevated blood pressure has long been recognized as one of the most important risk factors for stroke; however, other factors appear to play an important role as well. Indeed, epidemiological evidence suggests that in spite of an improved control of blood pressure, the secular trend of stroke in well-controlled populations is increasing. Moreover, although the hemorrhagic subtype of stroke has continuously decreased in incidence over the years; ischemic stroke is little affected by the current pharmacological treatment for hypertension (48). Many cardiovascular risk factors increase the production of reactive oxygen species (ROS) and promote inflammation in systemic and cerebral blood vessels. The predominant vascular sources of ROS are the superoxide-producing enzyme NADPH oxidase, xanthine oxidase, and mitochondrial enzymes, as well as the uncoupling of nitric oxide synthase (NOS), a state in which this enzyme generates superoxide instead of NO. Thus, oxidative stress and vascular inflammation are major pathways through which risk factors exert their deleterious effects on blood vessels. However, it remains to be determined how individual risk factors trigger the activation of one or both of these processes. This is a critical question for targeting preventive strategies in patients with specific risk factors (49). Finally, beyond the influence of reactive oxygen species and environmental factors, there is growing evidence that may contribute to the etiology of stroke (50). There is much evidence suggesting that endothelial dysfunction can play a role in the pathogenesis of ischemic stroke (51).
Hypertension and atherosclerosis
Endothelial cell damage occurs in many vascular beds during hypertension (52,53). However, it is not clear whether hypertension is the cause or the result of this damage. In 1978, Moncada and Vane suggested that endothelial dysfunction follows the course of a chronic increase in blood pressure and is therefore a consequence of hypertension (54). On the other hand, another study found that treatment of hypertension did not improve endothelial function (55), arguing against endothelial dysfunction as being a consequence of hypertension. An important consideration in this context is the mode of treatment of hypertension. For example, lowering blood pressure with beta-blockers does not improve endothelial function; whereas treatment with angiotensin-converting enzyme inhibitors (ACEI's) or angiotensin-receptor blockers (ARB's) significantly improves it (56). Supporting the notion that endothelial dysfunction is one of the causes of hypertension is the finding of impaired endothelial function in the normotensive offspring of patients with essential hypertension (57). A common abnormal finding in hypertensive states is a lower production of endothelial vasodilatory factors or their ineffectiveness and/or over production of or sensitivity to vasoconstrictor agents. One suggestion is that increased oxidative stress in hypertensive states leads to decreased availability of NO (58). In agreement with this suggestion is the finding that antioxidants improve endothelium-dependent relaxation (59). Higher oxidative stress in some hypertensive states could be due to increased levels of angiotensin-II, which stimulates NADPH oxidase to generate ROS, thus causing vascular inflammation (60). By preventing these effects, ACEI's and ARB's improve vasorelaxation in hypertensive patients. Other than angiotensin-II, endothelial vasoconstrictor prostanoids have also been implicated in the production of increased vascular tone in patients with essential hypertension (61). It is also likely that overproduction of endothelin-1 may play a role in hypertension. Measurement of plasma levels of ET-1 in hypertensive rats has been promising, but is confounded by opposite results in human patients. In pulmonary hypertension, however, a higher plasma level of ET-1 occurs in both human and animal disease (62). There is some success in the clinical use of bosentan, an antagonist of ET-1 receptors A and B, in patients with pulmonary hypertension (63).
Predisposing risk factors for atherosclerosis, which include hypertension, diabetes, smoking, and hypercholesterolemia, are all associated with endothelial dysfunction. In these conditions, the endothelial phenotype changes to a proinflammatory and prothrombotic state (64) by increased expression of leukocyte adhesion molecules (such as VCAM-1) and cytokines such as monocyte chemoattractant protein-1. These changes augment monocyte adhesion to and penetration through the vascular wall. A reduction in endothelium-derived NO is suggested to be one of the causes of such endothelial phenotypical changes. The antiatherogenic role of NO is supported by several studies on apo-E knockout mice and other animal models of atherosclerosis.
In these models, the inhibition of endothelial NO production accelerates lesion formation in the aorta and coronary arteries, and L-arginine treatment preserves vessel morphology. One mechanism for lower NO bioavailability in arteries predisposed to atherosclerosis could be the increased production of superoxide (65). The ROS will either degrade NO or tetrahydrobiopterin (BH4), a cofactor in its synthetic pathway (66). The source for ROS in the arterial bed is through augmented production by NADPH oxidase, xanthine oxidase (67) or reduced degradation by superoxide dismutase (68). Endothelial dysfunction also occurs in inflammatory conditions that promote atherosclerosis due to increased levels of C-reactive protein (CRP). CRP decreases eNOS-mediated NO production by decreasing the stability of eNOS mRNA (69). Moreover, in vitro experiments suggest that oxidized lipoproteins and lysophosphatidylcholine, two important mediators of atherogenesis, inhibit both NO and EDHF release (70). Oxidized LDL also decreases the expression of eNOS or its function in vitro (71), effects that are reversed by antioxidants. Lipids and oxidants also decrease the bioavailability of NO by impairing the structure of caveolae and b activating proinflammatory pathways such as those mediated by NFκB. Further, in response to inflammatory mediators (IL-1, TNF-α) that are released from activated monocytes, endothelial cells as well as SMCs secrete growth factors that enhance atherogenesis (72). Excessive endothelial secretion of factors such as ET-1 can have a significant role in atherogenesis, since both plasma and coronary vascular endothelin immunoreactivity is increased in hyperlipidemia as well as in early and advanced atherosclerosis. Endothelin enhances atherogenesis through several mechanisms. First, it is a strong chemoattractant that acts by stimulating ETB receptors on circulating monocytes. Second, ET-1 activates macrophages leading to over secretion of inflammatory mediators such as IL-6, IL-8, TNF, PGE2, and superoxide anion (73). Third, ET-1 stimulates smooth muscle cell migration and hypertrophy and the production of firoblast growth factor-2, making them hyper responsive to angiotensin-2 (74). Fourth, ET-1 increases fibroblast proliferation, chemotaxis, and matrix biosynthesis (74). Fifth, ET-1 causes PKC activation and increases platelet adherence through increased expression of P-selectin; however, little is known about the effect of ET-1 on platelets in vivo (75). Finally, endothelial dysfunction also causes instability of the atheromatous plaque, worsening the consequences of plaque rupture due to ineffective anti-aggregation, antithrombotic, and fibrinolytic functions. In fact, a dysfunctional endothelium may be prothrombotic by the overproduction of plasminogen activator inhibitor-1, an inhibitor of fibrinolysis (76).
Diabetes
Patients with diabetes invariably show impaired endothelium-dependent vasodilation. This is partly due to the frequent association of the disease with other cardiovascular risk factors, including hypertension, obesity, and dyslipidemia. Moreover, diabetic as well as obese patients usually consume a high-calorie diet rich in macronutrients that per se is able to induce vascular abnormalities (77). Indeed, protein, lipid, and glucose loads are associated with a marked production of ROS (78); and high-fat meals, with impaired endothelium-dependent vasodilation (79). A crucial negative effect of such meals is particularly attributable to high levels of circulating free fatty acids, which are able to induce ROS production and impair endothelial function (80). Mechanisms leading to endothelial damage in diabetes, independent of the damage due to other cardiovascular risk factors, include insulin resistance, hyperglycemia, and low-grade systemic inflammation (81). A large number of studies have been published on the interaction between insulin and the NO system. It was shown that, in normal subjects, insulin is able to induce a dose-dependent increase in lower limb blood flow by reducing vascular resistance in skeletal muscle (82), mainly by vasodilating the microcirculation (83). This observed vasodilatory effect of insulin is, at least partly, mediated by the enhanced production of NO through both the activation of the insulin receptor substrate- 1/phosphoinositol 3-kinase/Akt pathway (84) and increased expression of eNOS (85). Interestingly, studies on lower limb circulation showed that the magnitude of vasodilation in response to insulin appears to be linked to the rate of insulin-mediated glucose metabolism (86). However, some controversies exist on this topic, with another group failing to detect a net direct effect of insulin on vasodilation (87). The reasons for this could be related to the difference in methodology used and different vascular districts analyzed. Indeed, Taddei et al. showed no net direct effect of insulin on forearm microcirculation, but a potentiating effect of insulin on acetylcholine mediated vasodilation at this level, possibly through a hyperpolarizing effect on the endothelium (88). However, pathways downstream of insulin, whether through a direct interaction with the eNOS/NO system or some other intracellular system(s), have been implicated in the regulation of vascular tone and reactivity; since the presence of insulin resistance is associated with the presence of endothelial dysfunction not only in diabetes and obesity, but also in clearer models of insulin resistance, such as polycystic ovary syndrome (89).
Endothelium vs. Insulin resistance
Individuals with atherosclerosis exhibit both endothelial dysfunction and impaired insulin action. The endothelium plays an important role in the regulation of hemostasis, blood flow, maintenance of vascular architecture, and mononuclear cell transmigration-all of primary significance in atherogenesis. The endothelium also transports small molecules, macromolecules, and hormones such as insulin and degrades lipoprotein particles. Might endothelial dysfunction contribute to the individual components of the insulin resistance syndrome? Endothelial cells express the cognate insulin receptor (IR), which belongs to a family of membrane-bound receptors with intrinsic tyrosine kinase activity, whose ligands include growth factors such as insulin-like growth factor-1, vascular endothelial growth factor, platelet-derived growth factor, and epidermal growth factor. In addition to its crucial metabolic actions, insulin plays a critical role in the maintenance of physiological endothelial function through its ability to stimulate NO release via a cascade of signaling that involves activation of the PI3K-Akt axis and the downstream serine phosphorylation of endothelial NO synthase (eNOS) (90). In addition to its NO-dependent vasodilatory actions, insulin stimulates endothelial release of the vasoconstrictor ET-1, as suggested by increased insulin vasodilatory effects in humans under ET-1 receptor blockade. Thus, insulin has multiple opposing hemodynamic actions, the net effect of which on blood pressure is negligible in normal individuals. Insulin resistance is characterized by specific impairment in PI3K-dependent signaling pathways, whereas other insulin-signaling branches, including Ras/mitogen-activated protein kinase-dependent pathways, are unaffected (91). In addition, metabolic insulin resistance is usually paralleled by a compensatory hyperinsulinemia to maintain euglycemia. Thus, consequent hyperinsulinemia in insulin-resistant states will overdrive unaffected mitogen-activated protein kinase-dependent pathways. In the endothelium, decreased PI3K signaling and increased mitogen-activated protein kinase signaling in response to insulin may lead to decreased production of NO and increased secretion of ET-1, a characteristic of endothelial dysfunction. Indeed, insulin-resistant patients have elevated plasma ET-1 levels, and hyperinsulinemia increases ET-1 secretion in humans (91, 92). Pharmacological blockade of ET-1 receptors (ET-A isoform) improves endothelial function in obese and diabetic patients but not in lean, insulin-sensitive subjects. Endothelial dysfunction might also play a causal role in the development of insulin resistance. Insulin can relax resistance vessels and increase blood flow to skeletal muscle. Insulin acts on the vasculature in three discrete steps to enhance its own delivery to muscle/fat tissues (93): Relaxation of resistance vessels to increase total blood flow (94), relaxation of precapillary arterioles to increase the microvascular exchange surface perfused within the skeletal muscle, i.e. microvascular recruitment (95), and promotion of the transendothelial transport of insulin itself (96). Indeed, insulin resistance is associated with functional disturbances of the coronary circulation. Conversely, insulin infusion improves coronary flow, even in the setting of type 2 diabetes mellitus and coronary artery disease. Thus, such an imbalance between production of NO and secretion of ET-1 leads to decreased blood flow, which worsens insulin resistance. The reciprocal relationship of insulin resistance and endothelial dysfunction has been a subject of excellent review (97).
Endothelial dysfunction precedes the development of both the insulin resistance syndrome and atherosclerosis. Peripheral endothelial dysfunction, at the arteriolar and capillary levels, arises through a complex interplay of genetic and environmental factors and leads to a multifaceted metabolic disturbance comprising insulin resistance and the other features of the insulin resistance syndrome. Thus, this syndrome is a marker of peripheral endothelial dysfunction and plays a major role in atherogenesis but has little direct metabolic impact. The coexistence of central and peripheral endothelial dysfunction explains the observed association between atherosclerotic vascular disease and insulin resistance syndrome. This hypothesis could offer new insights into several clinical observations. First, it may be proposed that not all patients with coronary heart disease have an insulin resistance syndrome phenotype, because this syndrome is not an obligatory precursor of large-vessel atherogenesis, but rather a marker of peripheral endothelial dysfunction. However, subjects with peripheral endothelial dysfunction, such as those with diabetes or poor skeletal muscle capillarization, will be more likely to generate the insulin resistance syndrome phenotype, which accelerates endothelial dysfunction and atherogenesis in the large vessels. It is recognized, however, that additional local factors such as shear stress and rates of cholesterol deposition are likely to play important roles in restricting plaque formation to specific sites (98). In summary, we propose that peripheral endothelial dysfunction is the principal cause of insulin resistance and insulin resistance syndrome.
Chronic kidney failure
Renal dysfunction per se may be an underlying abnormality conducive to hypertension because hypertension “goes with the kidney” in cross-transplantation experiments, and a congenital reduction in the number of nephrons likely represents an important cause of glomerular endothelial dysfunction and hypertension in humans (99, 100). Endothelial cell activation was previously described as an increased adhesion between leukocytes and cultured endothelial cells when the latter are exposed to inflammatory mediators, implying that endothelium is a simple on-off device. Subsequently, other findings such as procoagulant activity, changes in vasomotor tone, and loss of barrier function were included in the definition of endothelial activation to describe a broader spectrum of phenotypic changes (101, 102). Chronic kidney disease in essential hypertension presents the expression of a medium- and small-size arteriolopathy characterized by intimal hyperplasia, hyalinosis, and smooth muscle cell hypertrophy (nephroangiosclerosis). Such arteriolopathy could be reflective of systemic dysfunction of the vascular endothelium. It seems to be the basic anatomic disturbances that may eventually lead to disastrous vascular events in the heart, brain, and kidney (103). Moreover, the small- and medium-size vessels respond inappropriately to vasodilatory stimuli such as acetylcholine, the production of which is mediated by nitric oxide. In the recent study of Raff et al. (104), the renal resistive index was disproved as a valid tool to assess renal endothelial dysfunction in humans. Following inhibition of nitric oxide synthase, the renal resistive index increases significantly but is not related to renal perfusion or renal vascular resistance. Renal vascular resistance changes to a similar extent in hypertensives, diabetics, and healthy volunteers (105). The pluripotent role of fat cells in renal dysfunction is still not completely understood. Fat tissue secretes a number of adipocytokines, including leptin, adiponectin, resistin, vaspin, visfatin, as well as proinflammatory cytokines such as TNFα and IL-6 (106). It appears likely that a reduced glomerular filtration rate (GFR) can contribute to the accumulation of adipocytokines, which could explain the marked dysmetabolism in chronic kidney disease. However, in a recent population-based study, the Hoorn study (107), which enrolled a cohort of 613 relatively healthy, elderly (50-75 years) subjects with an average GFR of 68 ml/min, endothelial dysfunction (estimated by plasma vWF and VCAM-1) but not inflammation (as measured by CRP and ICAM-1) correlated negatively with the GFR. Patients with established renal disease exhibit a consistent association between renal function and biomarkers of inflammation. In a survey based on 103 patients with established renal diseases, creatinine clearance correlated with both CRP and interleukin-6 (IL-6) levels, the IL-6-creatinine clearance link being fairly strong (R2=30%; 108). Similarly, in a sizeable group of patients with mild-to-severe renal insufficiency enrolled in a renal clinic in Birmingham (CRIB study), a lower GFR was associated with both low-grade inflammation and endothelial dysfunction, even among patients with moderate renal impairment (109). Furthermore, in another survey on patients with renal diseases performed by the Hoorn study investigators (110), creatinine clearance exhibited a fairly strong relationship (R2=28%) with an inflammation score composed of secretory phospholipase A(2) and CRP and with a complex endothelial function score (a combination of vWf, ICAM-1, VCAM-1, tissue-type plasminogen activator [tPA], plasminogen activator inhibitor-1 [PAI-1] and E-selectin [ES]) as well (R2=19%). On the other hand, in a post hoc analysis of a randomized placebo-controlled trial of a statin in persons with previous myocardial infarction, higher CRP and soluble tumor necrosis factor receptor II were independently associated with faster rates of kidney function loss; and the statin prevented loss of kidney function to a greater extent in individuals with greater evidence of inflammation (111). Thus, inflammation and perhaps endothelial dysfunction may not only signal but also be causally involved in the loss of kidney function among subjects with CKF (chronic kidney failure). Overall, inflammation seems to be a coherent, functional correlate of a reduced GFR in patients with established renal diseases; and at least in part it seems to contribute to renal and cardiovascular damage by altering endothelial function. Hemodynamic endothelial dysfunction (i.e., flow-mediated vasodilation) went in parallel with a reduced GFR in two studies on patients with moderate-to-severe renal insufficiency (112, 113) and was also fairly well associated with inflammation in the second of these studies (113,114). Some possible therapies to improve or to ameliorate endothelial dysfunction in the kidneys include non-pharmacologic approaches such as smoking cessation; weight reduction, particularly in obese patients with metabolic syndrome or type 2 diabetes mellitus; a low fat/Mediterranean diet; and regular exercise.
Cancer
Endothelial cell migration is an essential component of angiogenesis, which migration requires a tight regulation of the contractile and noncontractile states of the cell. These processes require the integration of signals elicited by chemotactic, haptotactic, and mechanotactic stimuli. In turn, this movement is associated with the activation of intracellular pathways that converge on cytoskeleton remodeling. Actin is a major cytoskeletal component of endothelial cells. It is composed of 43-kDa monomeric globular subunits (G-actin) that polymerize into helical filaments (F-actin). The assembly of F-actin is tightly associated with the hydrolysis of ATP by its intrinsic ATPase activity. Mg2+/ATP-bound monomeric G-actin is incorporated into the growing filaments at the barbed end, and ATP-actin is then converted into ADP-actin as actin monomers are shifted along the filaments toward the pointed end (115, 116). The constant remodeling of the actin cytoskeleton into filopodia, lamellipodia, and stress fibers is essential for cell migration, as briefly overviewed below and discussed in detail by K. Mizumo and colleagues in a forthcoming article within this thematic review series. Filopodia are membrane projections that contain long parallel actin filaments arranged in tight bundles. These particular structures act as sensors of motile stimuli. Classically, the formation of filopodia is regulated by activation of the small GTPase Cdc42, which associates with Wiskott-Aldrich syndrome proteins (WASP's). Lamellipodia are cytoplasmic protrusions that form at the leading edge of spreading or migrating cells (117). These protrusions are approximately 1 to 5 µm wide and approximately 2µm thick. The formation of lamellipodia is associated with important actin polymerization involving the Rac and Arp2/3 complex. Stress fibers are actin filaments of inverted polarity linked by β-actinin and myosin and distributed along contractile fibers (118). All three structures are essential to drive the several steps of actin-based endothelial cell motility: 1) Sensing of the motogenic signal by filopodia; 2) formation and protrusion of lamellipodia and pseudopodia-like forward extension; 3) attachment of the protrusions to the extracellular matrix (ECM); 4) stress fiber-mediated contraction of the cell body to allow forward progress; 5) rear release of the stress fibers; and 6) recycling of adhesive and signaling materials (114).
Endothelial cell relocation during angiogenesis
Endothelial cell migration involves three major mechanisms, namely, chemotaxis, the directional migration toward an increasing gradient of soluble chemoattractants; haptotaxis, the directional migration toward an increasing gradient of immobilized ligands; and mechanotaxis, the directional migration generated by mechanical forces (115). Endothelial cell migration during angiogenesis is the integrated result of these three mechanisms. Typically, chemotaxis of endothelial cells is driven by growth factors such as VEGF and basic fibroblast growth factor (bFGF), whereas haptotaxis is associated with increased endothelial cell migration activated in response to integrins binding to ECM components (116, 117). Because of their location at the inner surface of blood vessels, endothelial cells are constantly in contact with shear stress, which contributes to the activation of migratory pathways.
Severe infectious diseases
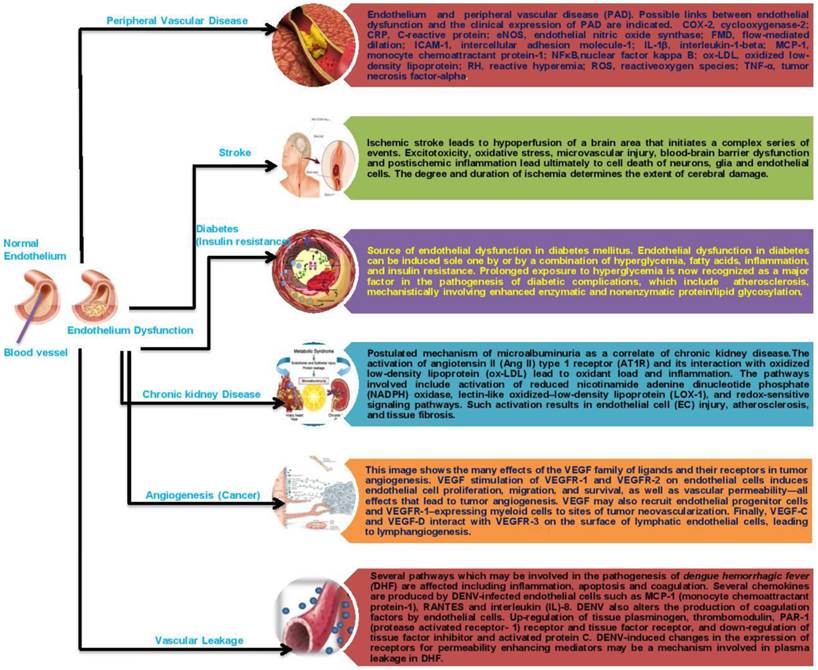
The endothelium plays a key role in the pathogenesis of coagulation disorders in infectious diseases, although the precise mechanisms are not yet clear in some cases. The endothelium is involved in both bacterial and non-bacterial infections and is important for the initiation and regulation of hemostasis. The loss of the endothelium barrier and vascular leakage play a central role in the pathogenesis of hemorrhagic fever viruses in general. This can be caused either directly by the viral infection and damage to the vascular endothelium or indirectly by a dysregulated immune response resulting in an excessive activation of the endothelium. Disruption of the vascular endothelial barrier occurs in two severe disease syndromes, dengue hemorrhagic fever and hantavirus pulmonary syndrome. Both viruses cause changes in vascular permeability without damaging the endothelium. In the leaky vascular endothelium seen in dengue severe syndrome (DSS), various mechanisms that have been considered include immune complex disease, T-cell-mediated reactions, antibodies cross-reacting with the vascular endothelium, enhancing antibodies, complement and its products, various soluble mediators including cytokines, selection of virulent strains, and viral virulence; but the most favoured are enhancing antibodies and memory T cells in a secondary infection that results in a cytokine “tsunami.” Whatever the mechanism, the vascular endothelium is ultimately targeted (making it a battlefield), leading to this severe disease syndrome. Extensive recent work has been done using in vitro endothelial cell monolayer models to understand the pathophysiology of the vascular endothelium during a dengue virus infection, and the results may help in understanding the pathogenesis of DHF[Dengue Hemorrhagic Fever] /DSS (118). Understanding the dynamics between viral infection and the dysregulation of the endothelial cell barrier will help us to define potential therapeutic targets for reducing disease severity (119). Given the above data, it is conceivable that the therapeutic correction of endothelial dysfunction may lead to an improvement of prognosis in patients with PAD, cardiovascular diseases, stroke, chronic kidney failure, cancer or infectious disease. However, scant data are available on this topic, and most of the conclusions that can be drawn are highly speculative. There is, therefore, virtually no available substance able to specifically target the endothelium (Fig. 2).
Schematic representation of endothelial dysfunction on human diseases.
PHARMACOLOGICAL REMEDIES
Endothelial dysfunction is the initial step in the pathogenesis of PAD, cardiovascular diseases, stroke, chronic kidney failure, cancer, and infectious diseases, all of which are highly complicated human diseases. Advances in our understanding of endothelial function/ physiology have been the basis for many therapeutic strategies. Many pharmacological interventions have been targeted to the endothelium with the intent of restoring it to its quiescent state (120). Various pharmacological interventions such as angiotensin-converting enzyme (ACE) inhibitors, statins, insulin sensitizers, and L-arginine, as well as agents that target endothelial nitric oxide synthase (eNOS) “coupling” such as folates or tetrahydrobiopterin (BH4) have been noted to improve the function of the endothelium. Various experimental studies have demonstrated the protective action of inhibitors of Rho-kinase, PARP, PTPase, geranyl transferase, and transketolase and of activators of Akt and PKA toward the endothelium, which would be novel future candidates for treating cardiovascular disorders (121). It is foreseeable that expanding our understanding of endothelial function further will lead to targeted therapies for a myriad of diseases, including cancer, cardiovascular disease, and inflammation.
CONCLUSIONS
The endothelium appears to play a critical role in a variety of human disorders, including peripheral vascular disease, stroke, heart disease, diabetes, insulin resistance, chronic kidney failure, tumor growth, metastasis, venous thrombosis, and severe viral infectious diseases. Because endothelial function may serve as a marker for the initiation and progression of insulin resistance, chronic kidney failure, and tumor metastasis, a more aggressive approach is warranted for early detection and treatment of abnormal endothelial function. In addition, although many agents can lessen endothelial dysfunction in the short term, more long-term reversibility studies need to be performed in humans. A better understanding of the different functions fulfilled by endothelial cells would be expected to result not only in a better comprehension of these diseases but also in their prevention and more effective therapeutic treatments. New pharmacological strategies are also being designed to target endothelial cells specifically, and their development is an exciting challenge of and goal for future research.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wilson SH, Lerman A. Function of Vascular Endothelium. Heart Physiology and Pathophysiology. 2001:27
2. Durand MJ, Gutterman DD. Diversity in Mechanisms of Endothelium-Dependent Vasodilation in Health and Disease. Microcirculation. 2013;20(3):239-247
3. Douglas B Cines, Eleanor S Pollak, Clayton A Buck. et al. Endothelial Cells in Physiology and in the Pathophysiology of Vascular Disorders. The Journal of The American Society of Hematology Blood. 1998;91(10):3527-3561
4. Patel JC. Functions of endothelium. Indian Journal of Medical Sciences. 2001;55:165-6
5. Verhamme P, Hoylaerts MF. The pivotal role of the endothelium in haemostasis and thrombosis. Acta clinica Belgica. 2006;61(5):213-9
6. Stern DM. et al. Endothelium and Regulation of Coagulation. Diabetes care. 1991Feb;14(2):160-6
7. Atherton A, Born GV. Quantitative investigations of the adhesiveness of circulating polymorphonuclear leucocytes to blood vessel walls. Journal of Physiology. 1972;222:447-74
8. Cerletti C, Evangelista V, de Gaetano G. P-selectin-β2 integrin cross-talk: a molecular mechanism for polymorphonuclear leukocyte recruitment at the site of vascular damage. The Journal of Thrombosis and Haemostasis. 1999;82:787-93
9. De Gaetano G, Donati MB, Cerletti C. Prevention of thrombosis and vascular inflammation: benefits and limitations of selective or combined COX-1, COX-2 and 5-LOX inhibitors. Trends Pharmacolology Science. 2003;24:245-52
10. Wagner DD, Frenette PS. The vessel wall and its interactions. Blood. 2008;111:5271-81
11. Smyth SS, McEver RP, Weyrich AS. et al. Platelet Colloquium Participants. Platelet functions beyond hemostasis. The Journal of Thrombosis and Haemostasis. 2009;7:1759-66
12. Wagner R. Erlauterungstalfeln zur Physiologie and Entwicklungsgeschichte. Leipzig, Germany: Leopold Voss. 1839
13. Boilard E, Nigrovic PA, Larabee K. et al. Platelets amplify inflammation in arthritis via collagen-dependent microparticle production. Science. 2010;327:580-3
14. Hara T, Shimizu K, Ogawa F. et al. Platelets control leukocyte recruitment in a murine model of cutaneous arthus reaction. American Journal of Pathology. 2010;176:259-69
15. Barton M, Baretella O, Meyer MR. Obesity and risk of vascular disease: importance of endothelium-dependent vasoconstriction. Br J Pharmacol. 2012;165(3):591-602
16. Michiels C. Endothelial Cell Functions. Journal of Cellular Physiology. 2003;196:430-443
17. Rubanyi GM, Vanhoutte PM. Superoxide anions and hyperoxia inactivate endothelium-derived relaxing factor(s). American Journal of Physiology. 1986;250:H822-H827
18. Devaraj S, Singh U, Jialal I. The Evolving Role of C-Reactive Protein in Atherothrombosis. Clinical Chemistry. 2009;55:229-238
19. Paulo Roberto BE. et al. Endothelium dysfunction classification: Why is it still an open discussion? International Journal of Cardiology. 2009;137(2):175-176
20. Hadi AR Hadi, Cornelia S Carr, Jassim Al Suwaidi. Endothelial Dysfunction: Cardiovascular Risk Factors, Therapy, and Outcome. Vascular Health Risk Management. 2005;1(3):183-198
21. Vita JA, Hamburg NM. Does endothelial dysfunction contribute to the clinical status of patients with peripheral arterial disease? Canadian Journal of Cardiology. 2010;26( A):45A-50A
22. Gordon MB, Jain R, Beckman JA. et al. The contribution of nitric oxide to exercise hyperemia in the human forearm. Vascular Medicine. 2002;7:163-8
23. Gokce N, Keaney JF, Menzoian JO. et al. Risk stratification for postoperative cardiovascular events via noninvasive assessment of endothelial function. Circulation. 2002;105:1567-72
24. Widlansky ME, Gokce N, Keaney JF. et al. The clinical implications of endothelial dysfunction. Journal of the American College of Cardiology. 2003;42:1149-60
25. Glagov S, Weisenberg E, Zarins CK. et al. Compensatory enlargement of human atherosclerotic coronary arteries. New England Journal of Medicine. 1987;316:1371-5
26. Kawashima S, Yamashita T, Ozaki M. et al. Endothelial NO synthase overexpression inhibits lesion formation in mouse model of vascular remodeling. Arteriosclerosis Thrombosis and Vascular Biology. 2001;21:201-7
27. Vita JA, Holbrook M, Palmisano J. et al. Flow-induced arterial remodeling relates to endothelial function in the human forearm. Circulation. 2008;117:3126-33
28. McGuigan MR, Bronks R, Newton RU. et al. Muscle fiber characteristics in patients with peripheral arterial disease. Medicine & Science in Sports & Exercise. 2001;33:2016-21
29. Porcu P, Emanueli C, Kapatsoris M. et al. Reversal of angiogenic growth factor upregulation by revascularization of lower limb ischemia. Circulation. 2002;105:67-72
30. Tsui JC, Baker DM, Biecker E. et al. Potential role of endothelin 1 in ischaemia-induced angiogenesis in critical leg ischaemia. British Journal of Surgery. 2002;89:741-7
31. Yu J, deMuinck ED, Zhuang Z. et al. Endothelial nitric oxide synthase is critical for ischemic remodeling, mural cell recruitment, and blood flow reserve. Proceedings of the National Academy of Sciences. 2005;102:10999-1004
32. Shiojima I, Walsh K. Role of Akt signaling in vascular homeostasis and angiogenesis. Circulation Research. 2002;90:1243-50
33. Libby P. Current concepts of the pathogenesis of the acute coronary syndromes. Circulation. 2001;104:365-72
34. Faxon DP, Fuster V, Libby P. et al. Atherosclerotic Vascular Disease Conference: Writing Group III: Pathophysiology. Circulation. 2004;109:2617-25
35. Huang AL, Silver AE, Shvenke E. et al. Predictive value of reactive hyperemia for cardiovascular events in patients with peripheral arterial disease undergoing vascular surgery. Arteriosclerosis, Thrombosis, and Vascular Biology. 2007;27:2113-9
36. Gokce N, Keaney JF, Hunter LM. et al. Predictive value of noninvasively- determined endothelial dysfunction for long-term cardiovascular events in patients with peripheral vascular disease. Journal of American College of Cardiology. 2003:1769-75
37. Burke AP, Kolodgie FD, Farb A. et al. Healed plaque ruptures and sudden coronary death: Evidence that subclinical rupture has a role in plaque progression. Circulation. 2001;103:934-40
38. Makin A, Silverman SH, Lip GY. Peripheral vascular disease and Virchow's triad for thrombogenesis. QJM. 2002;95:199-210
39. Virmani R, Kolodgie FD, Burke AP. et al. Atherosclerotic plaque progression and vulnerability to rupture: Angiogenesis as a source of intraplaque hemorrhage. Arteriosclerosis, Thrombosis, and Vascular Biology. 2005;25:2054-61
40. Fronek A, DiTomasso DG, Allison M. Noninvasive assessment of endothelial activity in patients with peripheral arterial disease and cardiovascular risk factors. Endothelium. 2007;14:199-205
41. Komai H, Higami Y, Tanaka H. et al. Impaired flow-mediated endothelium-dependent and endothelium-independent vasodilation of the brachial artery in patients with atherosclerotic peripheral vascular disease. Angiology. 2008;59:52-6
42. Poredos P, Golob M, Jensterle M. Interrelationship between peripheral arterial occlusive disease, carotid atherosclerosis and flow mediated dilation of the brachial artery. International Angiology. 2003;22:83-7
43. Brevetti G, Silvestro A, Di Giacomo S. et al. Endothelial dysfunction in peripheral arterial disease is related to increase in plasma markers of inflammation and severity of peripheral circulatory impairment but not to classic risk factors and atherosclerotic burden. Journal of vascular surgery. 2003;8:374-9
44. Kull I.J, Greene MT, Boerwinkle E. et al. Association of polymorphisms in NOS3 with the ankle-brachial index in hypertensive adults. Atherosclerosis. 2008;196:905-12
45. Silvestro A, Scopacasa F, Ruocco A. et al. Inflammatory status and endothelial function in asymptomatic and symptomatic peripheral arterial disease. Vascular Medicine. 2003;8:225-32
46. Tsui JC, Baker DM, Biecker E. et al. Evidence for the involvement of endothelin-1 but not urotensin-II in chronic lower limb ischaemia in man. European Journal of Vascular and Endovascular Surgery. 2003;25:443-50
47. Chang CC, Chen CJ. Secular trend of mortality from cerebral infarction and cerebral hemorrhage in Taiwan, 1974-1988. Stroke. 1993;24:212-18
48. Diaz JF, Hachinski VC, Pederson LL, Donald A. Aggregation of multiple risk factors for stroke in siblings of patients with brain infarction and transient ischemic attacks. Stroke. 1986;17:1239-42
49. Michael A. Moskowitz, Eng H. Lo, Costantino Iadecol. The Science of Stroke: Mechanisms in Search of Treatments. Neuron Review. 2010;67:181-198
50. Cosentino F, Sill JC, Katusic ZS. Endothelial L-arginine pathway and relaxations to vasopressin in canine basilar artery. American Journal of Physiology. 1993;33:H413-18
51. Cosentino F, Rubattu S, Savoia C. et al. Endothelial Dysfunction and Stroke. Journal of Cardiovascular Pharmacology. 2001;38(2):S75-S78
52. Schiffrin EL, Park JB, Intengan HD. et al. Correction of arterial structure and endothelial dysfunction in human essential hypertension by the angiotensin receptor antagonist losartan. Circulation. 2000;101:1653-1659
53. Park JB, Charbonneau F, Schiffrin EL. Correlation of endothelial function in large and small arteries in human essential hypertension. Journal of Hypertenion. 2001;19:415-420
54. Moncada S, Vane JR. Prostacyclin (PGI2), the vascular wall and vasodilation. Mechanisms of Vasodilation, ed. by P.A. Vanhoutte and I. Leusen, pp 107-121. Karger, Basel. 1978
55. Panza JA, Quyyumi AA, Callahan TS. et al. Effect of antihypertensive treatment on endothelium-dependent vascular relaxation in patients with essential hypertension. Journal of the American College of Cardiology. 1993;21:1145-1151
56. Schiffrin EL, Park JB, Pu Q. Effect of crossing over hypertensive patients from a beta-blocker to an angiotensin receptor antagonist onresistance artery structure and on endothelial function. Journal of Hypertension. 2002;20:71-78
57. Taddei S, Virdis A, Ghiadoni L. et al. Vitamin C improves endothelium-dependent vasodilation by restoring nitric oxide activity in essential hypertension. Circulation. 1998;97:2222-2229
58. Taddei S, Virdis A, Mattei P. et al. Defective L-arginine-nitric oxide pathway in offspring of essential hypertensive patients. Circulation. 1996;94:1298-1303
59. Chen X, Touyz RM, Park JB. et al. Antioxidant effects of vitamins C and E are associated with altered activation of vascular NADPH oxidase and superoxide dismutase in stroke-prone SHR. Hypertension. 2001;38:606-611
60. Touyz RM, Chen X, Tabet F. et al. Expression of a functionally active gp91phoxcontaining neutrophil-type NAD(P)H oxidase in smooth muscle cells from human resistance arteries: regulation by angiotensin II. Circulation Research. 2002;90:1205-1213
61. Lockette W, Otsuka Y, Carretero O. The loss of endotheliumdependent vascular relaxation in hypertension. Hypertension. 1986;8:II61-II66
62. Yoshibayashi M, Nishioka M, Nakao K. et al. Plasma endothelin concentrations in patients with pulmonary hypertension associated with congenital heart defects. Evidence for increased production of endothelin in pulmonary circulation. Circulation. 1991;84:2280-2285
63. Channick RN, Sitbon O, Barst RJ. et al. Endothelin receptor antagonists in pulmonary arterial hypertension. Journal of American College of Cardiology. 2004;43:62S-67S
64. Landmesser U, Hornig B, Drexler H. Endothelial function: a critical determinant in atherosclerosis? Circulation. 2004;109:II27-II33
65. Landmesser U, Harrison DG. Oxidant stress as a marker for cardiovascular events: Ox marks the spot. Circulation. 2001;104:2638-2640
66. Landmesser U, Dikalov S, Price SR. et al. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. Journal of Clinical Investigation. 2003;111:1201-1209
67. White CR, Darley-Usmar V, Berrington WR. et al. Circulating plasma xanthine oxidase contributes to vascular dysfunction in hypercholesterolemic rabbits. Proceedings of the National Academy of Sciences. 1996;93:8745-8749
68. Maier W, Cosentino F, Lutolf RB. et al. Tetrahydrobiopterin improves endothelial function in patients with coronary artery disease. Journal of Cardiovascular Pharmacology. 2000;35:173-178
69. Pasceri V, Willerson JT, Yeh ET. Direct proinflammatory effect of C-reactive protein on human endothelial cells. Circulation. 2000;102:2165-2168
70. Eizawa Z, Yui Y, Inoue R. et al. Lysophosphatidylcholine inhibits endothelium-dependent hyperpolarization and N omega-nitro-l-arginine/indomethacinresistant endothelium-dependent relaxation in the porcine coronary artery. Circulation. 1995;92:3520-3526
71. Chan H, Lougheed M, Laher I. et al. Oxidized low-density lipoprotein inhibits endothelium-dependent vasodilation by an antioxidant-sensitive, lysophosphatidylcholine-independent mechanism. Journal of Cardiovascular Pharmacology. 2003;41:856-865
72. Galley HF, Webster NR. Physiology of the endothelium. British Journal of Anaesthesia. 2004;93:105-113
73. Cunningham ME, Huribal M, Bala RJ. et al. Endothelin-1 and endothelin-4 stimulate monocyte production of cytokines. Critical Care Medicine. 1997;25:958-964
74. Best PJ, Lerman A. Endothelin in cardiovascular disease: from atherosclerosis to heart failure. Journal of Cardiovascular Pharmacology. 2000;35:S61-S63
75. Halim A, Kanayama N, Maradny E, Maehara K. et al. Endothelin-1 evoked an increase and oscillations in cytosolic calcium concentration in adherent single human platelets and increased GMP-140 (P-selectin) in platelet suspension. Thrombosis Research. 1995;80:105-112
76. Behrendt Ganz P. Endothelial function. From vascular biology to clinical applications. American Journal of Cardiology. 2002;90:40L-48L
77. Daniele Versari, Elena Daghini, Agostino Virdis. et al. Endothelial Dysfunction as a Target for Prevention of Cardiovascular Disease. Diabetes Care. 2009;32(2):314-321
78. Mohanty P, Ghanim H, Hamouda W. et al. Both lipid and protein intakes stimulate increased generation of reactive oxygen species by polymorphonuclear leukocytes and mononuclear cells. American Journal of Clinical Nutrition. 2002;75:767-772
79. Mohanty P, Hamouda W, Garg R. et al. Glucose challenge stimulates reactive oxygen species (ROS) generation by leucocytes. Journal of Clinicla Endocrinology Metabolism. 2000;85:2970-2973
80. Vogel RA, Corretti MC, Plotnick GD. Effect of a single high-fat meal on endothelial function in healthy subjects. The American Journal of Cardiology. 1997;79:350-354
81. Tripathy D, Mohanty P, Dhindsa S. et al. Elevation of free fatty acids induces inflammation and impairs vascular reactivity in healthy subjects. Diabetes. 2003;52:2882- 2887
82. Calles-Escandon J, Cipolla M. Diabetes and endothelial dysfunction: a clinical perspective. Endocrinology Review. 2001;22:36-52
83. Baron AD, Brechtel G. Insulin differentially regulates systemic and skeletal muscle vascular resistance. American Journal of Physiology. 1993;265:E61-E67
84. Clerk LH, Vincent MA, Lindner JR. et al. The vasodilatory actions of insulin on resistance and terminal arterioles and their impact on muscle glucose uptake. Diabetes Metabolism Resesach and Reviews. 2004;20:3-12
85. Muniyappa R, Iantorno M, Quon M.J. An integrated view of insulin resistance and endothelial dysfunction. Endocrinology and Metabolism Clinics of North America. 2008;37:685-711
86. Aljada A, Dandona P. Effect of insulin on human aortic endothelial nitric oxide synthase. Metabolism. 2000;49:147-150
87. Mather K, Laakso M, Edelman S. et al. Evidence for physiological coupling of insulin-mediated glucose metabolism and limb blood flow. American Journal Physiology Endocrinology Metabolism. 2000;279:E1264-E1270
88. Taddei S, Virdis A, Mattei P. et al. Effect of insulin on acetylcholine-induced vasodilation in normotensive subjects and patients with essential hypertension. Circulation. 1995;92:2911-2918
89. Paradisi G, Steinberg HO, Hempfling A. et al. Polycystic ovary syndrome is associated with endothelial dysfunction. Circulation. 2001;103:1410-1415
90. Xu J, Zou MH. Molecular Insights and Therapeutic Targets for Diabetic Endothelial Dysfunction. Circulation. 2009;120:1266-1286
91. Zhu D. et al. Luteolin inhibits inflammatory response and improves insulin sensitivity in the endothelium. Biochimie. 2011;93:506-512
92. Li R. et al. Vascular resistance in prehypertensive rats: Role of PI3- kinase/Akt/eNOS signaling. European Journal of Pharmacology. 2010;628(1-3):140-147
93. Yang Z, Li JC. Stimulation of endothelin-1 gene expression by insulin via phosphoinositide-3 kinase-glycogen synthase kinase-3β signaling in endothelial cells. Life Sciences. 2008;82(9-10):512-518
94. Barrett EJ, Wang H, Upchurch CT. et al. Insulin regulates its own delivery to skeletal muscle by feed-forward actions on the vasculature. American Journal of Physiology Endocrinology Metabolism. 2011;301(2):E252-63
95. Barrett EJ, Eggleston EM, Inyard AC. et al. The vascular actions of insulin control its delivery to muscle and regulate the rate-limiting step in skeletal muscle insulin action. iabetologia. 2009;52(5):752-64
96. Genders AJ, Frison V, Abramson SR. et al. Endothelial cells actively concentrate insulin during its transendothelial transport. Microcirculation. 2013 doi: 10.1111/micc.12044
97. Kim JA, Montagnani M, Koh KK. et al. Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation. 2006;113:1888-1904
98. Jonathan H Pinkney, Coen DA Stehouwer, Simon W Coppack. et al. Endothelial Dysfunction: Cause of the Insulin Resistance Syndrome. Diabetes. 1997;46(2):9-13
99. Carmine Zoccali. The endothelium as a target in renal diseases. Journal of Nephrology. 2007;20(12):S39-S44
100. Keller G, Zimmer G, Mall G. et al. Nephron number in patients with primary hypertension. The New England Journal of Medicine. 2003;348:101-8
101. Jolanta Malyszko. Mechanism of endothelial dysfunction in chronic kidney disease. Clinical Chimica Acta. 2010;411:1412-1420
102. Bombeli T, Mueller M, Haeberli A. Anticoagulant properties of the vascular endothelium. The Journal of Thrombosis and Haemostasis. 1997;77:408-23
103. Perticone F, Sciacqua A, Maio R. et al. Asymmetric dimethylarginine, L-arginine, and endothelial dysfunction in essential hypertension. Journal of the American College of Cardiology. 2005;46:518-23
104. Raff U, Schwarz TK, Schmidt BM. et al. Renal resistive index—a valid tool to assess renal endothelial function in humans? Nephrology Dialysis Transplantation. 2010;25:1869-74
105. Faber DR, de Groot PG, Visseren FL. Role of adipose tissue in haemostasis, coagulation and fibrinolysis. Obesity Review. 2009;10:554-63
106. Malyszko J, Malyszko JS, Kozminski P. et al. Adipokines, linking adipocytes and vascular function in hemodialyzed patients, may also be possibly related to CD146, a novel adhesion molecule. Clinical and Applied Thrombosis/Hemostasis. 2008;14:338-45
107. Stam F, Van GC, Becker A, Dekker JM, Heine RJ, Bouter LM, Stehouwer CD. Endothelial dysfunction contributes to renal function-associated cardiovascular mortality in a population with mild renal insufficiency: the Hoorn study. J Am Soc Nephrol. 2006;17:537-45
108. Panichi V, Migliori M, De PS, Taccola D, Bianchi AM, Giovannini L, Norpoth M, Metelli MR, Cristofani R, Bertelli AA, Sbragia G, Tetta C, Palla R, Colombo R. C-reactive protein and interleukin-6 levels are related to renal function in predialytic chronic renal failure. Nephron. 2002;91:594-600
109. Landray MJ, Wheeler DC, Lip GY, Newman DJ, Blann AD, McGlynn FJ, Ball S, Townend JN, Baigent C. Inflammation, endothelial dysfunction, and platelet activation in patients with chronic kidney disease: the chronic renal impairment in Birmingham (CRIB) study. Am J Kidney Dis. 2004;43:244-53
110. Stam F, van Guldener C, Schalkwijk CG, Ter Wee PM, Donker AJ, Stehouwer CD. Impaired renal function is associated with markers of endothelial dysfunction and increased inflammatory activity. Nephrol Dial Transplant. 2003;18:892-8
111. Tonelli M, Sacks F, Pfeffer M, Jhangri GS, Curhan G. Biomarkers of inflammation and progression of chronic kidney disease. Kidney Int. 2005;68:237-45
112. Annuk M, Lind L, Linde T, Fellstrom B. Impaired endothelium-dependent vasodilatation in renal failure in humans. Nephrol Dial Transplant. 2001;16:302-6
113. Yilmaz MI, Saglam M, Caglar K, Cakir E, Sonmez A, Ozgurtas T, Aydin A, Eyileten T, Ozcan O, Acikel C, Tasar M, Genctoy G, Erbil K, Vural A, Zoccali C. The determinants of endothelial dysfunction in CKD: oxidative stress and asymmetric dimethylarginine. Am J Kidney Dis. 2006;47:42-50
114. Małyszko J, Małyszko JS, Myśliwiec M. Endothelial cell injury markers in chronic renal failure on conservative treatment and continuous ambulatory peritoneal dialysis (CAPD). Kidney Blood Pressure Research. 2004;27:71-7
115. Disanza A, Steffen A, Hertzog M. et al. Actin polymerization machinery: the finish line of signaling networks, the starting point of cellular movement. Cell Molecular Life Science. 2005;62:955-970
116. Small JV, Resch GP. The comings and goings of actin: coupling protrusion and retraction in cell motility. Current Opinion in Cell Biology. 2005;17:517-523
117. Small JV, Stradal T, Vignal E. et al. The lamellipodium: where motility begins. Trends in Cell Biology. 2002;12:112-120
118. Atanu Basu, Umesh C. Chaturvedi. Vascular endothelium: the battle¢eld of dengue viruses. FEMS Immunol Med Microbiol. 2008;53:287-299
119. Spiropoulou CF, Srikiatkhachorn A. The role of endothelial activation in dengue hemorrhagic fever and hantavirus pulmonary syndrome. Virulence. 2013Aug15;4(6):525-36
120. Higashi Y, Yoshizumi M. Exercise and endothelial function: Role of endothelium-derived nitric oxide and oxidative stress in healthy subjects and hypertensive patients. Pharmacology & Therapeutics. 2004;102:87- 96
121. Balakumar P. et al. Pharmacological Interventions to Prevent Vascular Endothelial Dysfunction:Future Directions. Journal of health science. 2008;54(1):1-16
Author contact
![]() Corresponding author: NPO-International Laboratory of Biochemistry,1-166, Uchide, Nakagawa-ku, Nagoya 454-0926, Japan; Phone: 81 5236 11601, Email: nishigakistarcat.ne.jp.
Corresponding author: NPO-International Laboratory of Biochemistry,1-166, Uchide, Nakagawa-ku, Nagoya 454-0926, Japan; Phone: 81 5236 11601, Email: nishigakistarcat.ne.jp.