Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(1):73-79. doi:10.7150/ijbs.5750 This issue Cite
Research Paper
Mismatch Repair Deficient Mice Show Susceptibility to Oxidative Stress-Induced Intestinal Carcinogenesis
Jingshu Piao1, Yoshimichi Nakatsu1, Mizuki Ohno1, Ken-ichi Taguchi2, Teruhisa Tsuzuki1 ![]()
1. Department of Medical Biophysics and Radiation Biology, Graduate School of Medical Sciences, Kyushu University,
2. Department of Cancer Pathology, Institute for Clinical Research, National Kyushu Cancer Center, Fukuoka, Japan
Received 2012-12-21; Accepted 2013-7-24; Published 2013-12-19
Abstract
We have previously established an experimental system for oxidative DNA damage-induced tumorigenesis in the small intestine of mice. To elucidate the roles of mismatch repair genes in the tumor suppression, we performed oxidative DNA damage-induced tumorigenesis experiments using Msh2-deficient mice. Oral administration of 0.2% Potassium Bromate, KBrO3, effectively induced epithelial tumors in the small intestines of Msh2-deficient mice. We observed a 22.5-fold increase in tumor formation in the small intestines of Msh2-deficient mice compared with the wild type mice. These results indicate that mismatch repair is involved in the suppression of oxidative stress-induced intestinal tumorigenesis in mice. A mutation analysis of the Ctnnb1 gene of the tumors revealed predominant occurrences of G:C to A:T transitions. The TUNEL analysis showed a decreased number of TUNEL-positive cells in the crypts of small intestines from the Msh2-deficient mice compared with the wild type mice after treatment of KBrO3. These results suggest that the mismatch repair system may simultaneously function in both avoiding mutagenesis and inducing cell death to suppress the tumorigenesis induced by oxidative stress in the small intestine of mice.
Keywords: HNPCC, oxidative DNA damage, Wnt signaling pathway, mutagenesis, cell death
Introduction
Reactive oxygen species (ROS) are generated by the normal cellular metabolism and also by exposure to environmental factors, such as radiation and chemicals. ROS constantly induce various lesions in the DNA of living organisms under physiological conditions, and the resulting DNA damage causes mutations and cell death, leading to aging-associated diseases, such as cancer and neurodegeneration (1). Among the various types of oxidative DNA damage, oxidized guanine, 8-oxo-7, 8-dihydroguanine (8-oxoG) is abundant and highly mutagenic because of its ambiguous base-pairing properties; it can be paired with adenine as well as with cytosine (2-6). Therefore, 8-oxoG in DNA causes G:C to T:A transversions after two rounds of DNA replication. In mammalian cells, the base excision repair (BER) pathway initiated by OGG1 or MUTYH plays a role in the suppression of 8-oxoG-related mutagenesis. OGG1, an 8-oxoG DNA glycosylase, excises the 8-oxoG paired with cytosine from DNA (7, 8). MUTYH is an adenine DNA glycosylase that excises the adenine incorporated opposite 8-oxoG from DNA (9-12). The synergistic actions of OGG1 and MUTYH suppress the mutagenesis caused by 8-oxoG in DNA.
In addition to DNA repair, a nucleotide pool sanitizing enzyme, MTH1, suppresses the mutagenesis induced by oxidative stress (13, 14). This enzyme can hydrolyze oxidized purine nucleotides, such as 8-oxo-dGTP, 2-OH-dATP and 8-OH-dATP, to prevent the incorporation of mutagenic nucleotides into DNA during replication (15, 16). In addition to 8-oxoG, a wide variety of oxidatively modified bases in DNA were removed by the BER pathway following initiation by various DNA glycosylases (17).
Besides BER, mismatch repair (MMR) is also involved in the repair of oxidative DNA damage. MMR is an evolutionarily conserved system that corrects replication errors such as mismatched bases and small insertions/deletions. The MSH2/MSH6 heterodimer (MutSα) and MSH2/MSH3 heterodimer (MutSβ) recognize mismatched bases and small insertions/deletions, respectively, and then recruit MutLα (MLH1 and PMS2 heterodimer) to initiate the MMR reaction (18). In addition to its role in correcting replication errors, MMR is known to involve in the induction of apoptosis in response to DNA lesions caused by alkylating agents (19-21). The human MMR genes are associated with hereditary non-polyposis colorectal cancer (HNPCC), which is a common cancer predisposition syndrome characterized by a dominant mode of transmission and high penetrance (22-25). deWeese et al reported that Msh2-deficient mouse embryonic stem (ES) cells showed the accumulation of oxidative DNA damage, such as 8-oxoG and thymine glycol, in their genomes, as well as tolerance to apoptosis caused by low dose gamma-ray irradiation (26). Based on the analysis of spontaneous mutational specificity of mice defective in the Mth1 and/or the Msh2 genes, we speculated that MMR might act to correct mispairs with the oxidized nucleotides (27). Furthermore, Colussi et al, and Russo et al reported that MMR could suppress the mutations caused by the incorporation of oxidized purine deoxynucleoside triphosphate (28, 29). These findings suggest the involvement of MMR in the suppression of oxidative stress-induced mutagenesis and tumorigenesis in mammals.
KBrO3 is an oxidizing agent that is known to induce 8-oxoG in the DNA of rats and mice, and is recognized as a renal carcinogen in rats (30-33). We previously established an experimental system for oxidative DNA damage-induced tumorigenesis in the intestinal tract of mice using this agent (34). The oral administration of KBrO3 for 16 weeks effectively induced epithelial tumors in the small intestines of Mutyh-deficient mice, indicating the significance of Mutyh in the suppression of tumorigenesis induced by oxidative stress (34).
In this study, we performed KBrO3-induced tumorigenesis experiments using Msh2-deficient mice to elucidate the roles of MMR in the suppression of oxidative stress-induced tumorigenesis. We found that MMR plays a significant role in the suppression of oxidative stress-induced intestinal tumorigenesis in mice.
Materials and Methods
Animals
The Msh2-deficient mice used in this study were generated as reported previously (27). The wild type, heterozygous and homozygous mutant mice used in this study were obtained by intercrossing the heterozygous mutant mice. All animals were maintained under specific pathogen free (SPF) conditions. All animal care and handling procedures were approved by the Institutional Animal Care and Use Committee of Kyushu University, and followed the Guideline for Proper Conduct of Animal Experiments, Science Council of Japan.
KBrO3 treatment
KBrO3 (Sigma-ALDRICH) was given to 6~8-week-old mice in their drinking water at a concentration of 0.2% for 16 weeks. The body weight and consumption of drinking water were measured every week. After 16 weeks, all the animals were sacrificed, and the intestines were fixed with 10% phosphate buffered-formalin, and then stored in 70% ethanol.
Histological analysis
The inspections for tumor formation in the intestinal mucosa were carefully performed under a dissecting microscope. The small intestinal tumors were carefully removed from intestines, embedded in paraffin and sectioned. The sections were stained with hematoxylin and eosin for the diagnosis of the tumors. The evaluation of the tumors was performed according to the Vienna classification (35).
Mutation analysis of the Ctnnb1 gene
The small intestinal tumors were carefully removed from the mucosa under a dissecting microscope. Genomic DNA was extracted using a DNeasy Tissue Kit (QIAGEN) according to the manufacturer's protocol. Eighty-nine small intestinal tumors obtained from five Msh2-deficient mice were analyzed for mutations in the Ctnnb1 (β-catenin) gene. Thirty to fifty nanograms of genomic DNA extracted from each small intestinal tumor was used as the template for PCR with rTaq DNA polymerase (TaKaRa). The entire coding sequence of the second exon of the Ctnnb1 gene was amplified using primers 5'-TCCTTGGCTGCCTTTCTAACAGTA-3' (upper) and 5'-GCATGCCCTCATCTAGCGTCT-3' (lower). Amplified DNA containing exon 2 of the Ctnnb1 gene was purified with a PCR purification kit (QIAGEN) according to the manufacturer's protocol. The purified DNA fragments were used as a template for direct sequencing with a BigDye Terminator v3.1 Cycle Sequencing kit (Applied Biosystems) and the sequences were determined with an ABI PRISM® 3100 Genetic Analyzer (Applied Biosystems).
TUNEL analysis
The intestines were removed from the wild type and mutant mice treated with KBrO3 for 16 weeks, and 3 μm sections were made after the samples were embedded in paraffin. We analyzed the cell death (apoptosis) in the crypts of the small intestine using a TUNEL kit (TaKaRa) as described in the manual supplied by the manufacturer. We counted the TUNEL-positive cells in more than 100 crypts from five mice of each genotype.
Results
Tumor formation induced by KBrO3 treatment in Msh2-deficient mice
In order to examine whether oxidative stress increases the intestinal tumorigenesis in mismatch repair-deficient mice, congenic wild type, heterozygous and homozygous Msh2-deficient (5, 6 and 7 animals for each genotype, respectively) mice were administered 0.2% KBrO3 in their drinking water for 16 weeks. At the same time, five mice of each genotype were kept under the same conditions except for the KBrO3-treatment. As previously observed, the KBrO3-treatment appeared to cause a slowdown in the increase of body weight at almost the same rate in all groups of animals during the period of KBrO3-treatment. We dissected the mice after the 16-week treatment with KBrO3, and inspected the intestines under a dissecting microscope. In the homozygous Msh2-deficient mice treated with KBrO3, the formation of small intestinal tumors was dramatically increased (Figure 1, Table 1). The mean number of tumors induced in the small intestines of the seven Msh2-deficient mice was 27.0, whereas it was 1.2 and 1.5 in the five wild type and six heterozygous mice, respectively. Tumor formation was also observed in the untreated homozygous Msh2-deficient mice, albeit at a much lower frequency (mean: 1.2 tumor/mouse, n=5) compared with the treated homozygous mice. As previously observed in the Mutyh-deficient mice (34), the KBrO3-induced tumors predominantly developed in the duodenum and in the upper region of the jejunum (Figure 1A). We found no other anomalies in the KBrO3-treatment mice.
Tumor formation in the intestine of Msh2-deficient mice
| Genotype | No treatment | KBrO3-treatment | ||
|---|---|---|---|---|
| No. of tumorsa | No. of tumorsa | Ratiob | ||
| Wild type | 0 | 1.20 ± 0.98 | 1.00 | |
| Heterozygote | 0 | 1.50 ± 1.26 | 1.25 | |
| Homozygote | 1.20 ± 0.75 | 27.00 ± 7.44 | 22.50 | |
a: The no. of tumors is the mean number of tumors per mouse, with the standard deviation. b: ratio to tumors in wild type mice
KBrO3-induced tumors in the small intestine of Msh2-deficient mice. A. The proximal regions of the small intestines of KBrO3-treated mice are shown; (+/+): wild type, (+/-): heterozygous Msh2-deficient, and (-/-): homozygous Msh2-deficient mice. Multiple polyp formations could be observed in the KBrO3-treated homozygous Msh2-deficient mice. B. A section of a KBrO3-induced tumor stained with hematoxylin and eosin (original magnification: objective 10X). The regions containing the neoplasia are encircled by a broken line.
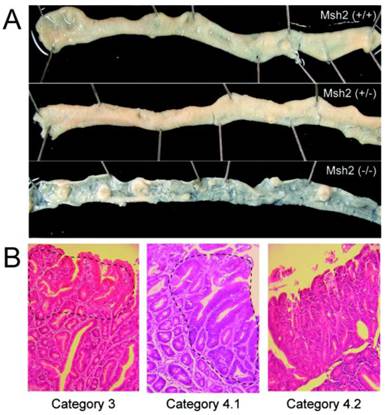
Pathological analysis of tumors induced by KBrO3 in the small intestines
We performed a pathological analysis of 25, three and two small intestinal tumors derived from five homozygous-deficient, three heterozygous Msh2-deficient mice and two wild type mice, respectively, according to the Vienna classification of gastrointestinal epithelial neoplasia (Table 2, Figure 1B). All tumors from homozygous Msh2-deficient mice were classified as category 4 (non-invasive high grade neoplasia), except for one case that was classified as category 3 (non-invasive low grade neoplasia). All tumors from wild type and heterozygous Msh2-deficient mice were also classified as category 4 (Table 2).
Classification of KBrO3-induced small intestinal tumors in mice
| Genotype | Category 3* | Category 4* | Total | ||
|---|---|---|---|---|---|
| 4.1 | 4.2 | 4.3 | |||
| Wild type | − | − | 2 | − | 2 |
| Heterozygote | − | − | 3 | − | 3 |
| Homozygote | 1 | 3 | 21 | − | 25 |
*Tumors were categorized according to the Vienna classification of gastrointestinal epithelial neoplasia
Mutation analysis of the Ctnnb1 gene
The β-catenin protein encoded by Ctnnb1 gene is a transcriptional activator functioning in the Wnt-signaling pathway (36). The phosphorylation of β-catenin by GSK3β in a complex with Axin and Apc is required for the ubiquitin-mediated degradation of β-catenin. Therefore, the presence of mutations affecting the phosphorylation of the protein lead to its stabilization and the accumulation of β-catenin in nuclei, inducing the expression of target genes such as c-myc and cyclin D1 without Wnt signaling. The mutations at four putative GSK3β-phosphorylation sites (S33, S37, T41, S45) and amino acids adjacent these sites have been detected in a wide variety of human cancers including HNPCC, as well as in chemically-induced tumors in model animals. Therefore, we analyzed the mutations in exon 2 of the Ctnnb1 gene encoding the GSK3β-phosphorylation sites of β-catenin. Among 89 tumors from five homozygous Msh2-deficient mice, 27 tumors (30.3%) showed a mutation in this region (Figure 2, Table 3). All the mutations were base substitutions and occurred at or in the vicinity of the codons for S33, S37 and T41. No mutations were observed at the codon for S45. Among them, G:C to A:T transitions predominantly occurred; 20 mutations (74.1%) were identified as G:C to A:T transitions, and the others were three A:T to G:C transitions (11.1%), two G:C to T:A transversions and two G:C to C:G transversions (7.4%, respectively). There is no clear hotspot for G:C to A:T transitions in this region, with there being seven at D32, five at S37, four at G34 and four at T41. However, besides one G:C to T:A transversion at S33, three other types of mutations were observed only at the codon for S37. The base substitutions observed at the codon for S37 were as follows; five G:C to A:T transitions, three A:T to G:C transitions, two G:C to C:G transversions and one G:C to T:A transversion.
Somatic mutations found in the Ctnnb1 gene of tumors. The amino acid sequence and the corresponding nucleotide sequence of GSK3β phosphorylation sites are shown at the top. The amino acids at phosphorylation sites are depicted in bold. The somatic mutations found in the KBrO3-induced intestinal tumors are shown below the nucleotide sequence of the Ctnnb1 gene; upper and lower panels show the nucleotide sequencing results from normal tissues and tumors, respectively.
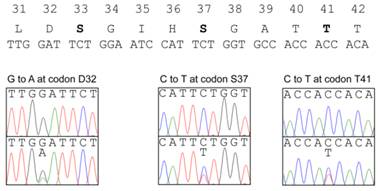
Mutations found in the Ctnnb1 gene
| Mouse ID | Sample ID | Nucleotide position | Wild type | Mutant | Mutation | Amino acid change |
|---|---|---|---|---|---|---|
| 22 | 1 | 94 | GAT | AAT | G:C→A:T | D32N |
| 38 | 1 | 109 | TCT | CCT | A:T→G:C | S37P |
| 4 | 109 | TCT | CCT | A:T→G:C | S37P | |
| 5 | 122 | ACC | ATC | G:C→A:T | T41I | |
| 7 | 101 | GGA | GAA | G:C→A:T | G34E | |
| 8 | 110 | TCT | TAT | G:C→T:A | S37Y | |
| 12 | 94 | GAT | AAT | G:C→A:T | D32N | |
| 13 | 100 | GGA | AGA | G:C→A:T | G34R | |
| 18 | 110 | TCT | TTT | G:C→A:T | S37F | |
| 20 | 110 | TCT | TTT | G:C→A:T | S37F | |
| 21 | 122 | ACC | ATC | G:C→A:T | T41I | |
| 22 | 94 | GAT | AAT | G:C→A:T | D32N | |
| 48 | 4 | 110 | TCT | TGT | G:C→C:G | S37C |
| 6 | 110 | TCT | TTT | G:C→A:T | S37F | |
| 10 | 94 | GAT | AAT | G:C→A:T | D32N | |
| 15 | 100 | GGA | AGA | G:C→A:T | G34R | |
| 22 | 109 | TCT | CCT | A:T→G:C | S37P | |
| 45 | 1 | 122 | ACC | ATC | G:C→A:T | T41I |
| 3 | 110 | TCT | TGT | G:C→C:G | S37C | |
| 9 | 110 | TCT | TTT | G:C→A:T | S37F | |
| 10 | 94 | GAT | AAT | G:C→A:T | D32N | |
| 50 | 5 | 122 | ACC | ATC | G:C→A:T | T41I |
| 8 | 100 | GGA | AGA | G:C→A:T | G34R | |
| 12 | 94 | GAT | AAT | G:C→A:T | D32N | |
| 14 | 94 | GAT | AAT | G:C→A:T | D32N | |
| 16 | 110 | TCT | TTT | G:C→A:T | S37F | |
| 24 | 98 | TCT | TAT | G:C→T:A | S33Y |
Analysis of cell death
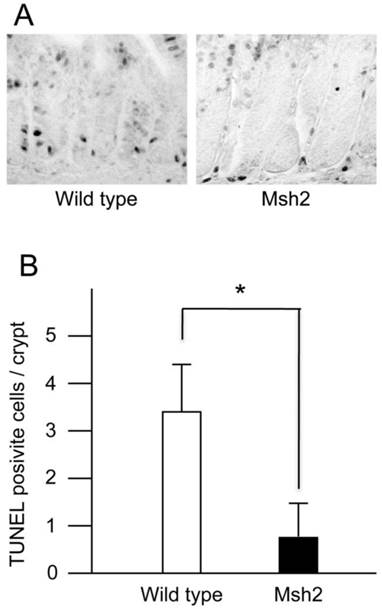
MMR is involved in the signaling for cell death induced by genotoxic chemicals, such as alkylating agents (19-21). A previous study showed that ES cells carrying disrupted Msh2 alleles displayed an increased survival following exposure to low-level ionizing radiation compared with wild type ES cells (26). The increased survival could be attributed to a failure of the cells to efficiently execute apoptosis in response to oxidative DNA damage induced by radiation exposure. These findings suggested that MMR is involved in the induction of apoptosis caused by oxidative DNA damage. It has been shown that intestinal cancer originates in the stem cells resided in the bottom of intestinal crypts (37). Thus, in the present study, we analyzed the cell death in the crypts of small intestines from wild type and Msh2-deficient mice treated with KBrO3 using a TUNEL method. A few TUNEL-positive cells were detected in the crypts of wild type mice, but not in those of Msh2-deficient mice (Figure 3A). We counted the TUNEL-positive cells in more than 100 crypts from each genotype of mice. The average numbers of crypts per mouse were as follows: wild type, 21.2 (min 14, max 31) and Msh2-deficient, 21.6 (min 17, max 30). We found that 3.36 ± 0.96 (mean ± SD) TUNEL-positive cells per crypt were present in wild type mice, while 0.80 ± 0.46 (mean ± SD) TUNEL-positive cell per crypt were present in Msh2-deficient mice (Figure 3B). This difference was statistically significant (p< 0.002; t-test).
TUNEL-positive crypt cells in the small intestines from KBrO3-treated mice. A. The sections stained with TUNEL. The crypts of small intestines from wild type (left) and Msh2-deficient (right) mice treated with KBrO3. B. The number of TUNEL-positive cells in the crypts. The mean numbers of TUNEL-positive cells with standard deviations are indicated by white (wild type mice) and black (homozygous Msh2-deficient mice) bars. * p<0.002 (Student's t-test).
Discussion
In the present study, we performed KBrO3-induced tumorigenesis experiments using Msh2-deficient mice to examine the involvement of mismatch repair (MMR) in the suppression of oxidative stress-induced tumorigenesis. The oral administration of KBrO3 at a dose of 0.2% in drinking water dramatically increased the formation of intestinal tumors in Msh2-deficient mice compared to untreated Msh2-deficient mice and treated wild type mice. Thus, we concluded that MMR plays a significant role preventing the intestinal tumorigenesis induced by oxidative stress in mice.
Several lines of evidence suggest that oxidative stress could be generated in the intestines of animals under physiological conditions. For example, it was reported that the incidence of G:C to T:A transversions increases significantly in the intestines of older mice compared with younger mice (38). Because G:C to T:A transversions are mainly caused by 8-oxoG, a major oxidative DNA damage, these observations indicate that the mutations caused by oxidative DNA damage would tend to accumulate in the intestines during the course of aging. Consistent with this notion, defects in MUTYH, the human DNA glycosylase suppressing 8-oxoG-induced mutagenesis, lead to a susceptibility to colorectal cancers with excess G:C to T:A transversions in humans (39). Furthermore, Mutyh-deficient mice also show susceptibility to spontaneous and KBrO3-induced intestinal adenoma/carcinoma (34). Therefore, based on these previous observations and our present results, it is likely that on a MMR-defective genetic background, oxidative stress generated in the intestine may enhance tumor development, thus leading to HNPCC in humans.
The mutation analyses of the tumor-related gene, Ctnnb1, revealed that more than 30% of the tumors that developed in KBrO3-treated Msh2-deficient mice had somatic mutations in the coding region for GSK3β phosphorylation sites. All the mutations detected were base substitutions; 20 G:C to A:T transitions (74.1%), three A:T to G:C transitions (11.1%), two G:C to T:A transversions and two G:C to C:G transversions (7.4%). These observations are consistent with the results obtained from mutation analyses of the CTNNB1 gene in cancers from HNPCC patients; 12 reported base substitutions were as follows: TCT to TTT, CCT, or TGT at codon S45; ACC to GCC at codon T41; TCT to TGT at codon S37; GGA to GAA at codon G34; GAC to GGC or TAC at codon D32 (40). The G:C to A:T transition at codon G34 and G:C to C:G transversion at codon S37 were commonly observed in both human and mouse tumors. The difference observed between human and mouse mutation spectra may due to the different nucleotide sequence context in this locus of these two species. The similarity of the mutation types and spectra suggests that the KBrO3-treatment of mice may mimic the oxidative stress in human to induce DNA damage in the intestine. We detected only two G:C to T:A transversions, thus suggesting that the DNA repair enzymes, including Ogg1 and Mutyh, may suppress 8-oxoG-related mutagenesis to some extent in KBrO3-treated Msh2-deficient mice. Besides the one G:C to T:A transversion at the codon for S33, three types of mutations other than the G:C to A:T transition were observed at only the codon for S37; three A:T to G:C transitions, two G:C to C:G transversions, and one G:C to T:A transversion. Because these types of mutations were suppressed by the overexpression of MTH1, which hydrolyzes oxidized purine nucleotide triphosphates, in MSH2-deficient human cells (28, 29), the nucleotide sequence context around the codon for S37 might be competent to enhance the incorporation of oxidatively-damaged purine nucleotide triphosphates by DNA polymerase.
MMR factors are well known to be involved in the induction of apoptosis caused by O6-methylguanine (O6-mG), a type of DNA damage produced by alkylating agents (19-21). O6-mG can pair with thymine during DNA replication, forming O6-mG:T mispairs. MutSα recognizes this mispair and forms a complex with MutLα and PCNA to induce apoptosis (20). The Msh2-deficiency also caused a failure to induce apoptosis effectively in response to oxidative DNA damage (26). Thus, in the present study, we analyzed the cell death of the crypts of small intestines from wild type and Msh2-deficient mice treated with KBrO3 using a TUNEL assay. A larger number of TUNEL-positive cells were observed in the crypts from wild type mice compared with Msh2-deficient mice, suggesting that the crypt cells with MMR-deficiencies showed an increased chance of surviving protracted exposure to KBrO3. The better survival of MMR-deficient cells with pre-mutagenic lesions in the genome induced by oxidative stresses may contribute to the increased cancer risk characteristic of the hereditary non-polyposis colorectal cancer syndrome.
In conclusion, we herein demonstrated that oxidative stress enhanced the tumor formation in the small intestines of Msh2-deficient mice, thereby providing experimental evidence of the association between oxidative stress and hereditary non-polyposis colorectal cancer caused by MMR-deficiency in humans. We propose that MMR suppresses spontaneous tumorigenesis in the intestines of mammals by simultaneously preventing the occurrence of mutations and the removal of precancerous cells containing pre-mutagenic oxidative lesions in the genome.
Acknowledgements
We thank Dr. Brian Quinn for comments on the manuscript. We also appreciate the technical support from the Research Support Center, Graduate School of Medical Sciences, Kyushu University. This work was supported by grants from Japan Society for the Promotion of Science, and the Ministry of Health, Labor and Welfare of Japan.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Ames BN, Shigenaga MK Hagen TM. Oxidants, antioxidants, and the degenerative diseases of aging. Proc Natl Acad Sci USA. 1993;90:7915-22
2. Gajewski E, Rao G, Nackerdien Z, Dizdaroglu M. Modification of DNA bases in mammalian chromatin by radiation-generated free radicals. Biochemistry. 1990;29:7876-82
3. Kasai H, Nishimura S. Hydroxylation of deoxyguanosine at the C-8 position by ascorbic acid and other reducing agents. Nucleic Acids Res. 1984;12:2137-45
4. Fraga CG, Shigenaga MK, Park JW, Degan P, Ames BN. Oxidative damage to DNA during aging: 8-hydroxy-2'-deoxyguanosine in rat organ DNA and urine. Proc Natl Acad Sci USA. 1990;87:4533-7
5. Shibutani S, Takeshita M, Grollman AP. Insertion of specific bases during DNA synthesis past the oxidation-damaged base 8-oxodG. Nature. 1991;349:431-4
6. Smith KC. Spontaneous mutagenesis: experimental, genetic and other factors. Mutat Res. 1992;277:139-162
7. Radicella JP, Dherin C, Desmaze C, Fox MS, Boiteux S. Cloning and characterization of hOGG1, a human homolog of the OGG1 gene of Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1997;94:8010-5
8. Rosenquist TA, Zharkov DO, Grollman AP. Cloning and characterization of a mammalian 8-oxoguanine DNA glycosylase. Proc Natl Acad Sci USA. 1997;94(14):7429-34
9. Ohtsubo T, Nishioka K, Imaiso Y. et al. Identification of human MutY homolog (hMYH) as a repair enzyme for 2-hydroxyadenine in DNA and detection of multiple forms of hMYH located in nuclei and mitochondria. Nucleic Acids Res. 2000;28:1355-64
10. Shinmura K, Yamaguchi S, Saitoh T. et al. Adenine excisional repair function of MYH protein on the adenine:8-hydroxyguanine base pair in double-stranded DNA. Nucleic Acids Res. 2000;28:4912-8
11. Slupska MM, Luther WM, Chiang JH, Yang H, Miller JH. Functional expression of hMYH, a human homolog of the Escherichia coli MutY protein. J Bacteriol. 1999;181:6210-3
12. Takao M, Zhang QM, Yonei S, Yasui A. Differential subcellular localization of human MutY homolog (hMYH) and the functional activity of adenine:8-oxoguanine DNA glycosylase. Nucleic Acids Res. 1999;27:3638-44
13. Mo JY, Maki H Sekiguchi M. Hydrolytic elimination of a mutagenic nucleotide, 8-oxodGTP, by human 18-kilodalton protein: sanitization of nucleotide pool. Proc Natl Acad Sci USA. 1992;89:11021-5
14. Sakumi K, Furuichi M, Tsuzuki T. et al. Cloning and expression of cDNA for a human enzyme that hydrolyzes 8-oxo-dGTP, a mutagenic substrate for DNA synthesis. J Biol Chem. 1993;268:23524-30
15. Fujikawa K, Kamiya H, Yakushiji H. et al. The oxidized forms of dATP are substrates for the human MutT homologue, the hMTH1 protein. J Biol Chem. 1999;274:18201-5
16. Fujikawa K, Kamiya H, Yakushiji H, Nakabeppu Y, Kasai H. Human MTH1 protein hydrolyzes the oxidized ribonucleotide, 2-hydroxy-ATP. Nucleic Acids Res. 2001;29:449-54
17. Bjelland S, Seeberg E. Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat Res. 2003;531:37-80
18. Iyer RR, Pluciennik A, Burdett V, Modrich PL. DNA mismatch repair: functions and mechanisms. Chem. Rev. 2006;106:302-23
19. Stojic L, Brun R, Jiricny J. Mismatch repair and DNA damage signalling. DNA Repair (Amst). 2004;3:1091-1101
20. Hidaka M, Takagi Y, Takano TY, Sekiguchi M. PCNA-MutSalpha-mediated binding of MutLalpha to replicative DNA with mismatched bases to induce apoptosis in human cells. Nucleic Acids Res. 2005;33:5703-12
21. Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol. 2006;7:335-46
22. Fishel R, Lescoe MK, Rao MR. et al. The human mutator gene homolog MSH2 and its association with hereditary nonpolyposis colon cancer. Cell. 1993;75:1027-38
23. Leach FS, Nicolaides NC, Papadopoulos N. et al. Mutations of a MutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75:1215-25
24. Bronner CE, Baker SM, Morrison PT. et al. Mutation in the DNA mismatch repair gene homologue hMLH1 is associated with hereditary non-polyposis colon cancer. Nature. 1994;368:258-61
25. Papadopoulos N, Nicolaides NC, Wei YF. et al. Mutation of a MutL homolog in hereditary colon cancer. Science. 1994;263:1625-9
26. DeWeese TL, Shipman JM, Larrier NA. et al. Mouse embryonic stem cells carrying one or two defective Msh2 alleles respond abnormally to oxidative stress inflicted by low-level radiation. Proc. Natl. Acad. Sci. USA. 1998;95:11915-20
27. Egashira A, Yamauchi K, Yoshiyama K. et al. Mutational specificity of mice defective in the MTH1 and/or the MSH2 genes. DNA Repair (Amst). 2002;1:881-93
28. Colussi C, Parlanti E, Degan P. et al. The mammalian mismatch repair pathway removes DNA 8-oxodGMP incorporated from the oxidized dNTP pool. Current Biol. 2002;12:912-8
29. Russo MT, Blasi MF, Chiera F. et al. The oxidized deoxynucleoside triphosphate pool is a significant contributor to genetic instability in mismatch repair-deficient cells. Mol Cell Biol. 2004;24:465-74
30. Kasai H, Nishimura S, Kurokawa Y, Hayashi Y. Oral administration of the renal carcinogen, potassium bromate, specifically produces 8-hydroxydeoxyguanosine in rat target organ DNA. Carcinogenesis. 1987;8:1959-1
31. DeAngelo AB, George MH, Kilburn SR, Moore TM, Wolf DC. Carcinogenicity of potassium bromate administered in the drinking water to male B6C3F1 mice and F344/N rats. Toxicol Pathol. 1998;26:587-94
32. Kurokawa Y, Maekawa A, Takahashi M, Hayashi Y. Toxicity and carcinogenicity of potassium bromate--a new renal carcinogen. Environ Health Perspect. 1990;87:309-35
33. Kawanishi S, Murata M. Mechanism of DNA damage induced by bromate differs from general types of oxidative stress. Toxicology. 2006;221:172-8
34. Sakamoto K, Tominaga Y, Yamauchi K. et al. MUTYH-null mice are susceptible to spontaneous and oxidative stress-induced intestinal tumorigenesis. Cancer Res. 2007;67:6599-604
35. Schlemper RJ, Riddell RH, Kato Y. et al. The Vienna classification of gastrointestinal epithelial neoplasia. Gut. 2000;47:251-5
36. Polakis P. Wnt signaling and cancer. Genes Dev. 2000;14:1837-51
37. Barker N, Ridgway RA, van Es JH. et al. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 2009;457:608-11
38. Dolle ME, Snyder WK, Gossen JA, Lohman PH, Vijg J. Distinct spectra of somatic mutations accumulated with age in mouse heart and small intestine. Proc Natl Acad Sci USA. 2000;97:8403-8
39. Al-Tassan N, Chmiel NH, Maynard J. et al. Inherited variants of MYH associated with somatic G:C-->T:A mutations in colorectal tumors. Nat Genet. 2002;30:227-32
40. Miyaki M, Iijima T, Kimura J. et al. Frequent mutation of beta-catenin and APC genes in primary colorectal tumors from patients with hereditary nonpolyposis colorectal cancer. Cancer Res. 1999;59:4506-9
Author contact
![]() Corresponding author: Teruhisa Tsuzuki, Department of Medical Biophysics and Radiation Biology, Faculty of Medical Sciences, Kyushu University, Fukuoka 812-8582, Japan. Phone: +81-92-642-6141: fax: +81-92-642-6145: E-mail: tsuzukikyushu-u.ac.jp
Corresponding author: Teruhisa Tsuzuki, Department of Medical Biophysics and Radiation Biology, Faculty of Medical Sciences, Kyushu University, Fukuoka 812-8582, Japan. Phone: +81-92-642-6141: fax: +81-92-642-6145: E-mail: tsuzukikyushu-u.ac.jp