Impact Factor ISSN: 1449-2288
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(1):90-102. doi:10.7150/ijbs.7738 This issue Cite
Research Paper
Knockdown of TRPM8 Suppresses Cancer Malignancy and Enhances Epirubicin-induced Apoptosis in Human Osteosarcoma Cells
Yongzhi Wang, Zhonghua Yang, Zhe Meng, Hong Cao, Guangbin Zhu, Tao Liu, Xinghuan Wang ![]()
Department of Urology, Zhongnan Hospital, Wuhan University, Wuhan, China, 430071
Received 2013-9-24; Accepted 2013-12-3; Published 2013-12-22
Abstract
As the function of transient receptor potential melastatin member 8 (TRPM8) in osteosarcoma is still unknown, we aim to investigate the possible effects and potential mechanisms of TRPM8 on cell proliferation, metastasis and chemosensitivity in osteosarcoma cells. We find that TRPM8 is aberrantly over-expressed in human osteosarcoma tissues and cell lines. Knockdown of TRPM8 by siRNA in osteosarcoma cells leads to the impaired regulation of intracellular Ca2+ concentration and then the Akt-GSK-3β pathway and the phosphorylation of p44/p42 and FAK are suppressed. Knockdown of TRPM8 not only negatively influences the cell proliferation and metastasis but also enhances epirubicin-induced cell apoptosis. Such results reveal that TRPM8 is worthy further investigation for its potential as a clinical biomarker and therapeutic target in osteosarcoma.
Keywords: TRPM8, osteosarcoma, epirubicin, apoptosis, MAPK
Introduction
Osteosarcoma is the most common type of primary malignant cancer originating from bone in children and young adolescents. By the mid-1980s, the application of neoadjuvant chemotherapy and surgery had improved the five year survival rate dramatically from 20% to approximately 70% [1]. However, despite the advances in multimodality treatments, the progress has been painfully slow and the overall survival of patients has reached a plateau in the past 20 years [2]. Local recurrence or distant metastasis and chemoresistance represent two important mechanisms for therapy failure. Therefore, developing more targeted treatment approaches is imperative as a plateau in efficacy has been reached with current treatments [3].
As we all know that doxorubicin has been widely used in the osteosarcoma treatment, however such use is limited by its side effects like cardiotoxicity and potential nephropathy [4]. Several randomized controlled trials (RCTs) suggest that epirubicin (EPI) can induce fewer severe side effects, and especially there is no significant difference in anti-tumor response rate and survival between epirubicin and doxorubicin [5]. So combination chemotherapy with epirubicin and other agents may be an active, reasonably well-tolerated regimen in some osteosarcoma patients.
The role of Ca2+ in global cancer-related cell signaling pathways is uncontested. Fluctuations in Ca2+ homeostasis may lead to an increase in cell proliferation [6, 7] and even may induce differentiation [8] and apoptosis [9]. According to a growing number of studies, cationic channels of the transient receptor potential (TRP) family represent key players in calcium homeostasis and cell physiopathology. Especially, further evidence indicates that the transient receptor potential melastatin member 8 (TRPM8), a member of the TRP family, plays an important role in human cancer.
TRPM8 is a Ca2+-permeable cation channel and also known as 'cold receptor', because it can be activated by cold temperature and menthol. Since the discovery of TRPM8 gene in 2001, it has been observed that the abnormal expression of TRPM8 is associated with the phenotype of cancers originating from breast, lung, colon, and prostate [10]. TRPM8 ion channel has been the target of various therapeutic applications ranging from cancer and urological disorder to cold hypersensitivity and pain. Especially in recent years, TRPM8 channel has emerged as a promising prognostic marker and putative therapeutic target in prostate cancer [11], and growing evidences further suggest that TRPM8 could be a novel biomarker and molecular target in other human cancers like breast cancer [12], pancreatic cancer [13], etc. Nevertheless, until now, there is only one study [14] has been performed to investigate the expression of TRPM8 in osteoblastic cells, but its roles were not addressed in his study. The function of TRPM8 in osteosarcoma is still unknown. Given this, this study was designed to investigate the possible effects and potential mechanisms of TRPM8 on proliferation, metastasis and chemosensitivity in human osteosarcoma cells.
Materials and Methods
Cell Lines and Culture
Human osteosarcoma cell lines MG-63, U2OS, SaOS2 and HOS were obtained from American Type Culture Collection (ATCC, Manassas, VA, USA) and human bone marrow mesenchymal stem cells (BMSCs) was obtained from ScienCell Research Laboratories (San Diego, CA). Cells were cultured in RPMI 1640 medium (Gibco BRL, Grand Island, NY, USA) supplemented with 100 IU mL-1 penicillin G sodium, 100µg mL-1 streptomycin sulphate and 10% foetal bovine serum (FBS) (Gibco BRL, Grand Island, NY, USA) in a humidified atmosphere consisting of 95% air and 5% CO2 at 37ºC.
Tissue Samples
Tissue samples were used with agreement of the patients treated by surgery during January 2010 to June 2012 in Zhongnan Hospital of Wuhan University, China. This study was approved by the local research ethics committee (Zhongnan Hospital of Wuhan University). The osteosarcoma specimens were collected from 10 patients who did not receive any preoperative chemotherapy or radiotherapy, with 10 osteochondroma specimens also collected as control. For the 10 osteosarcoma patients, 6 were male and 4 were female with average age of 29.1 ranging from 16-57 years; and for the 10 osteochondroma patients, 5 were male and 5 were female, with average age of 23.4 ranging from 15-28 years. The diagnosis of all samples has been reconfirmed by three experienced pathologists independently.
Immunohistochemistry
Immunohistochemical studies were performed by Streptavidin-peroxidase-biotin method. The sections were immersed with 3% H2O2 for 10min to block endogenous peroxidase. After antigen retrieval by microwave, new born calf serum was added as blocking agent, and10min later, rabbit polyclonal anti-TRPM8 antibody (1:100, Cat #: ACC-049, Alomone Labs, Jerusalem, Israel) was added to incubate overnight (4°C) and then anti-rabbit IgG (BOSTER, China) was added to incubate for 20min at room temperature. After that, streptavidin-biotin-peroxidase solution (Sigma) was used to incubate for 30min and 3, 3-diaminobenzidine (Gibco BRL, Grand Island, NY, USA) was added to chlorate for 15min. After hematoxylin staining, dehydration and hyalinization, the slides were covered. Positive staining would show brown.
Evaluation of Immunohistochemistry
The “immunohistochemical score” (IHS) was calculated based on previously published research [15]. Immunoreactivity was evaluated prior to the collection of patient identity and clinical information. The IHC classification of positivity was scored as follows: (1) ≤25% of cells staining positively; (2) 26%-50% of cells staining positively; (3) 51%-75% of cells staining positively; (4) 76%-100% of cells staining positively. The intensity of the immunoexpression was rated as negative (0), weak (1), moderate (2), or strong (3). A consensus was achieved by three of the authors in all cases. The final IHS was obtained by multiplying the score of extent and intensity. The IHS of all specimens was categorized into four groups: -, (0-2); +, (3-5); ++, (6-8); +++, (9-12). Scores in the range of 0-5 were designated as low expression, and scores in the range of 6-12 as high expression.
Transfection of siRNA
MG-63 and U2OS cells were seeded in 6-well plates at 40%-50% confluence and then were transfected with 10nM siTRPM8 and control siRNA (siCON) (synthesized by GenePharma, Shanghai, China) respectively by using 5µl Lipofectamine 2000 (Invitrogen) as recommended by the manufacturer's protocol. Untransfected cells were named Parental cells. The sense sequence of siTRPM8 was 5′-UCUCUGAGCGCACUAUUCA (dTdT)-3′[16], and the sequence of control siRNA was 5′-AAGGTGGTTGTTTTGTTCACT-3′. The expression of TRPM8 in MG-63 and U2OS cells transfected with siTRPM8 and siCON was evaluated by Ca2+ imaging and western blot.
Ca2+ Imaging
The cytosolic Ca2+ concentration ([Ca2+] c) was measured using the ratiometric dye Fura-3 (Molecular Probes, Leiden, The Netherlands). The temperature was maintained at 37ºC using a temperature controller (Cell Microcontrol System, Norfolk, VA, USA). For confocal Ca2+ imaging, the cells were loaded in the presence of 2.5 µmol L-1 Fluo-3AM dye in the culture medium at 37ºC for 2 h. During measurement, the cells were incubated in Hanks' balanced salt solution (HBSS) containing 150 mmol L-1 NaCl, 5.4 mmol L-1 KCl, 2 mmol L-1 CaCl2 , 1 mmol L-1 MgCl2, and 10 mmol L-1 Hepes (pH adjusted to 7.4 with 1 mol L-1 NaOH). The emission intensity was measured for 250s at 3-s intervals using excitation wavelengths of 488 nm at an emission wavelength of 525 nm. Menthol (Sigma) was added after 15s. The fluorescence intensity indicates the cytosolic Ca2+ concentration.
Immunofluorescence
The cells transfected with siTRPM8 and siCON were plated onto coverslips and processed for immunofluorescence analysis with antibody against Ki67 (1:100, Cat #: ab66155, rabbit polyclonal IgG, Abcam) and the secondary antibody is fluorescein isothiocyanate (FITC) Green goat anti-rabbit (Boster, Wuhan, China). The nuclei were counterstained with Hoechst 33258 (Beyotime, Shanghai, China) and the fluorescence was detected by Olympus inverted fluorescence microscope.
The Hoechst 33258 staining was also used to evaluate the cell apoptosis and whether SP600125 (10μM) (Beyotime Institute of Biotechnology, Shanghai, China), special inhibitor of JNK, can abolish the effect of EPI treatment. After the Parental, siCON and siTRPM8 cells were incubated with the indicated disposals, Hoechst 33258 (10μg/ml) was added. After incubation at room temperature for 15min, cells were washed by PBS for 3 times and finally observed under the Olympus inverted fluorescence microscope.
CCK8 Assay
Cell growth and viability were measured using cell proliferation and cytotoxicity reagent WST-8 (Roche Biochemicals, Mannheim, Germany). The non-transfected and transfected MG-63 and U2OS cells were plated in 96-well plates with 2×103/well for cell growth assay and 5×103/well for cell viability assay. For cell growth assay, the cells were incubated for the indicated time; while for the cell viability assay, the cells were treated with EPI at the indicated concentration for 48h. At the harvest time 10μl of CCK8 was added into each well and after one hour's incubation cellular viability was determined by measuring the absorbance of the converted dye at 450nm.
Cell Cycle and Cell Apoptosis Were Examined by Flow Cytometry
Approximately 1×106 cells for each sample were harvested and they were fixed with 70% ethanol at 4ºC overnight and then re-suspended in PBS containing 40μg mL-1 propidium iodide (PI), 0.1mg mL-1 RNase and 0.1% Triton X-100 in a dark room. After incubation at 37 ºC for 30min, the cells were analyzed through flow cytometry. The cell cycle stage was then determined and analyzed.
For cell apoptosis analysis, after receiving the indicated disposals, cells were harvested, stained with Annexin V-FITC apoptosis detection kit (Abcam, Cambridge, MA, USA) and then analyzed by flow cytometry.
Scratch migration Assay and Transwell Invasion Assay
In scratch assay, wound was created with a standard 200μL pipette tip [17], and the wounded monolayer was washed twice to remove the non-adherent cells. The wound closure was monitored using an inverted phase contrast microscope at the time the wound was created and 24h after incubation in 1% FBS medium. The distance between borderlines was measured at four different equidistant points in four independent fields of each sample to get a better estimate. The migration rate was expressed as a percentage of the control and was calculated as the proportion of the mean distance between the borderline caused by scratching and the borderline that remained cell-free after re-growing. Three independent series of experiments were performed.
In transwell assay, for each sample, 5×104 cells in 1% FBS medium were seeded into the matrigel-coated, porous upper chamber inserts (Bection Dickinson, San Jose, CA, USA) with 700μl completed medium in the lower chamber. After 24h incubation, cotton-tipped swabs were used to remove non-invasive cells in interior of the inserts. Next, inserts were incubated with 400μl 0.1% Crystal Violet for 10 min at room temperature. After washed in PBS for several times, inserts were placed under an inverted phase contrast microscope and eight fields of vision were randomly selected for observation.
Western blot assay
40μg lysates per sample was resolved in 10% SDS-PAGE with the indicated primary antibodies, as described earlier [18]. Antibodies against various proteins for western blot such as pAkt (1:1000, #4060), Akt (1:1000, #9272), pGSK-3β (1:1000, #9323), GSK-3β (1:1000, #12456), p-p44/p42 (1:1000, #4370P), p44/p42 (1:1000, #4695P), pJNK (1:1000, #4668P), JNK (1:1000, #9258P), pFAK (1:1000, #8556), FAK (1:1000, #3285) and MMP-2 (1:1000, #4022) were obtained from Cell Signaling Technologies (Danvers, MA). The other antibody against Cdk4 was purchased from Neomarkers (1:500, Cat. #MS-299-P0, Union city, CA, USA) and those against MKP-1 (1:1000, sc-1102), cyclinD1 (1:1000, sc-735), GAPDH (1:1000, sc-166574) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
Statistical Analysis
The SPSS version 11.5 for Windows (SPSS, Chicago, IL, USA) was used for the statistical analysis. The correlation of TRPM8 expression in osteosarcoma and osteochondroma specimens was assessed by chi-square test. Data were presented as means ± SD of the indicated number of experiments. Statistical analysis was performed through unpaired t-text, with P < 0.05 taken as statistically significant.
Results
The expression of TRPM8 in human osteosarcoma tissues and cell lines
The difference of TRPM8 expression between osteosarcoma and osteochondroma specimens was shown in Fig. 1A and the arrows indicate the positive staining. For the expression of TRPM8, 70% osteosarcoma cases were at high level (7/10) with 30% cases (3/10) at low level and the proportion values in osteochondroma cases, which were used as normal control, was about 10%(1/10) and 90%(9/10). The chi-square test demonstrated that the difference of TRPM8 expression between osteosarcoma and osteochondroma had statistical significance (p<0.05, Table 1). Besides, western blot was also performed to examine the protein expression of TRPM8 in human osteosarcoma cells, since the expression of TRPM8 in mRNA level has been investigated [14]. As for the normal control cell to MG-63 and U2OS cells, human osteoblast cell line hFOB1.19 is the best choice. However, hFOB1.19 cells should be cultured at 34°C, and different culture condition may have an influence on the protein expression. Based on this, we chose BMSCs as normal control. The protein expression of TRPM8 was at a high level in MG-63, U2OS, SaOS2 and HOS compared with BMSCs, among which, the expression was higher in U2OS and was approximately identical in the other three cell lines (Fig. 1B). Based on such results, MG-63 and U2OS cells were chosen as representatives in the following experiments.
The expression of TRPM8 in osteosarcoma and osteochondroma specimens
| Cases | High expression | Low expression | p | ||
|---|---|---|---|---|---|
| osteosarcoma | 10 | 7 (70.0%) | 3 (30.0%) | 0.0225* | |
| osteochondroma | 10 | 1 (10.0%) | 9 (90.0%) |
* The chi-square test demonstrated that the difference of TRPM8 expression between osteosarcoma and osteochondroma had statistical significance, p<0.05.
The expression of TRPM8 in osteosarcoma tissues and cell lines. A: TRPM8 is over-expressed in osteosarcoma tissues when compared with osteochondroma tissues used as normal control (magnification x400), arrows indicate the positive staining cells. Negative controls for immunostaining were performed in the absence of anti-TRPM8 antibody. The correlation of TRPM8 expression in osteosarcoma and osteochondroma specimens was assessed by chi-square test. B: The expression of TRPM8 in osteosarcoma cell lines was detected by western blot (a) and its abundance was expressed as normalized values over BMSCs (b). C: The knockdown efficiency of siRNA targeted on TRPM8 in MG-63 and U2OS cells. (a) Ca2+ imaging indicated that the response to menthol was clearly diminished in siTRPM8 cells and the knockdown of TRPM8 leads to impaired regulation of intracellular Ca2+ concentration. The fluorescence intensity in y-axis represents intracellular Ca2+ concentration. (b) The time course and dose dependent manner of TRPM8 siRNA in MG-63 and U2OS cells. (c) The western blot data in (b) was quantified and the results were expressed in histograms.
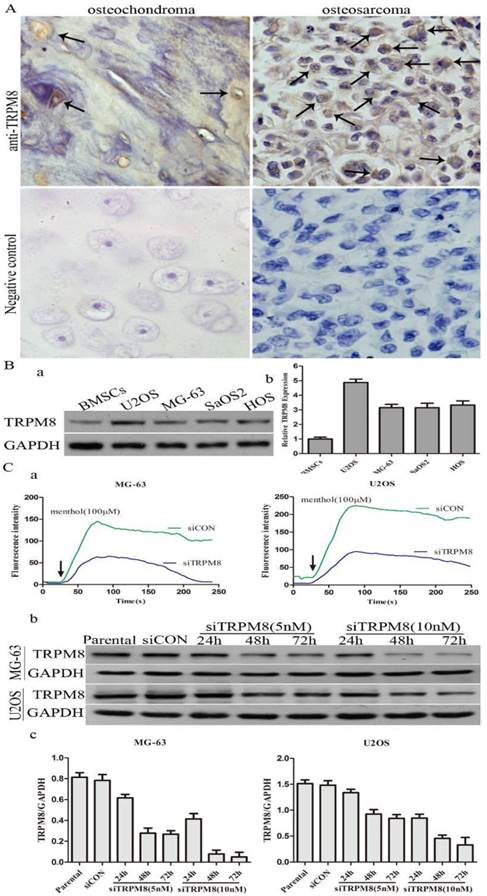
The efficiency of siTRPM8 was investigated by Ca2+ imaging and western blot. Compared with siCON cells, the elevation of intracellular Ca2+ levels induced by menthol was diminished by TRPM8 knockdown and the expression of TRPM8 was dramatically down-regulated in the cells transfected with siTRPM8 in a time and dose dependent manner (Fig. 1C).
Knockdown of TRPM8 inhibited cell proliferation
We first assessed the effect of TRPM8 knockdown on the proliferation of MG-63 and U2OS cells. According to the results of CCK8 assay, the siTRPM8 cells grew more slowly than the Parental and siCON cells and the difference was more pronounced after day 2. There was no statistical difference between Parental and siCON cells (Fig. 2A).
Knockdown of TRPM8 suppressed growth of MG-63 and U2OS cells. A: The effect of TRPM8 knockdown on MG-63 (a) and U2OS (b) cell proliferation was measured by CCK8 assay. Growth of siTRPM8 cells was significantly suppressed at day 2 compared with Parental and siCON cells. One-Way ANOVA was used for the data analysis. **P<0.01. OD in y-axis means A450nm and represents cell growth; bars, SD. B: Parental, siCON and siTRPM8 cells of MG-63 (a) and U2OS (b) were immunostained with anti-Ki67 antibody (green) and Hoechst33258 (blue), scale bars = 50 μm.
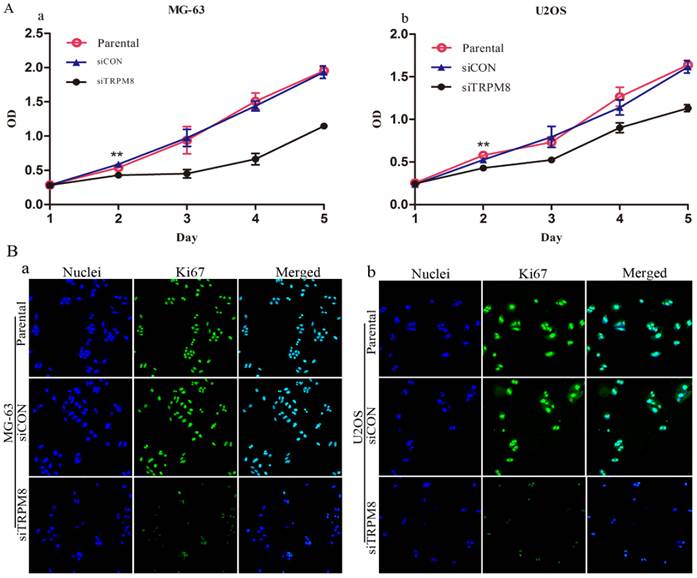
Immunofluorescence analysis targeting on the Ki67 also confirmed the proliferation inhibition effect. The Ki67 expression was more markedly reduced in siTRPM8 cells compared with the Parental and siCON cells (Fig. 2B).
Next, we examined whether the knockdown of TRPM8 has cell cycle arrest effect in MG-63 and U2OS cells. Different from the Parental and siCON cells, the siTRPM8 cells were blocked in G0/G1 phase (MG-63: siTRPM8 67.35% ± 2.14% vs. Parental 43.44% ±1.60%, siCON 42.73% ± 1.80%; U2OS: siTRPM8 64.18% ± 1.76% vs. Parental 45.67% ± 2.08%, siCON 46.43% ± 4.84%, P < 0.01, Fig. 3A). Moreover, the expressions of cyclinD1and Cdk4 as well as p-Akt and p-GSK-3β in siTRPM8 cells were markedly decreased. (Fig. 3B)
Knockdown of TRPM8 induced G0/G1 arrest and decreased the level of phospho-p44/p42. A: (a) Knockdown of TRPM8 induced G0/G1 arrest. 48h after the indicated transfection, cells were harvested and cell cycle was examined. (b) Compared with Parental and siCON cells, the accumulation of cells in G0/G1 phase was significantly increased in siTRPM8 cells, **P<0.01. B: (a) Knockdown of TRPM8 suppressed the Akt-GSK-3β pathway and decreased the expression of cyclinD1 and Cdk4. 48h after the indicated transfection, cells were harvested and western blot was performed. (b) The western blot results were quantified and statistic analysis was performed, *P<0.05. C: (a) Knockdown of TRPM8 decreased the level of phospho-p44/p42. (b) The western blot results were quantified and statistic analysis was performed. *P<0.05. All the data in this figure was analyzed by One-Way ANOVA.
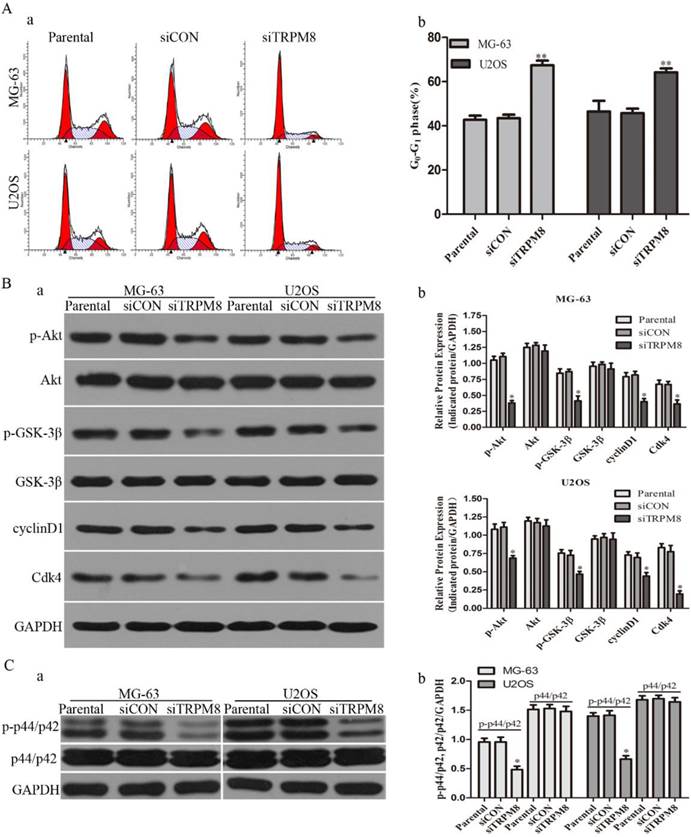
Apart from the cell cycle-related proteins, the level of phospho-p44/p42, which has been reported to be able to promote cell growth[19], was also significantly decreased in siTRPM8 cells compared with the Parental and siCON cells but the total p44/p42 was not affected (Fig. 3C).
Knockdown of TRPM8 inhibited cell migration and invasion
In scratch-wound assay, the migration of siTRPM8 cells was significantly suppressed after 24h incubation compared with Parental and siCON cells. The migration rate was expressed as a percentage with the value of Parental cells being 100% and there was no statistical difference between Parental cells and siCON cells (MG-63: siTRPM8 62.84% ± 6.59% vs. Parental 100%±5.13%, siCON 101.65%±4.12%; U2OS: siTRPM8 63.43%±8.92% vs. Parental 100%±5.50%, siCON 97.04%±2.24%, P < 0.01, Fig. 4A).
Knockdown of TRPM8 inhibited cell migration and invasion. A: Knockdown of TRPM8 inhibited cell migration in MG-63 (a) and U2OS (b) cells (magnification x200). (c) The migration rate was expressed as a percentage with the value of Parental cells being 100% and the migration was significantly suppressed in siTRPM8 cells, **P<0.01. B: Knockdown of TRPM8 inhibited cell invasion. (a) The invaded siTRPM8 cells were obviously less than the invaded Parental and siCON cells (magnification x200). (b) Values are presented as invaded cells/field, **P<0.01. C: (a) Knockdown of TRPM8 suppressed the expression of pFAK and MMP-2. (b) The western blot results were quantified and statistic analysis was performed, **P<0.01, *P<0.05. All the data in this figure was analyzed by One-Way ANOVA.
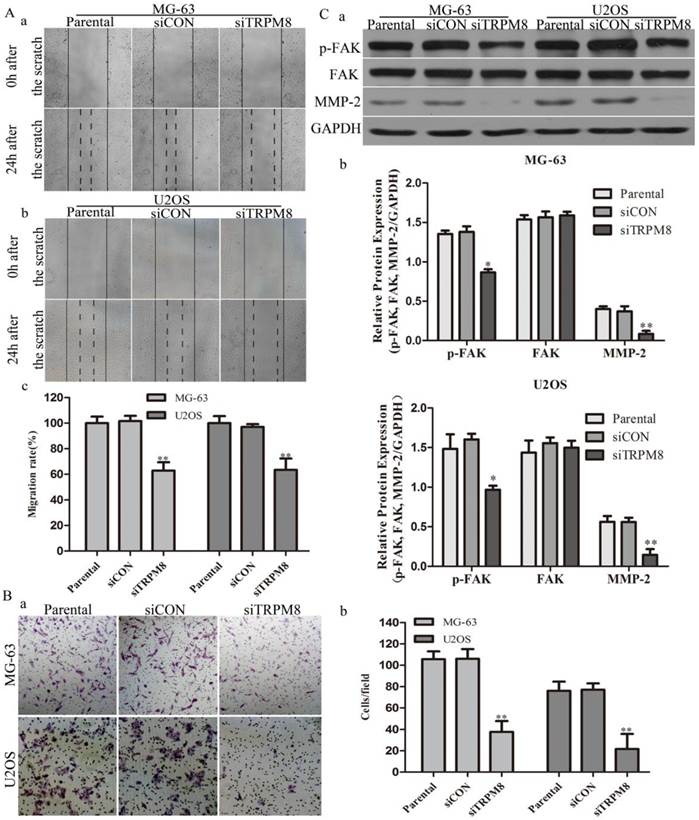
As for cell invasion assay, the number of the invaded siTRPM8 cells was significantly reduced compared with Parental and siCON cells. The cell invasion capability was represented as the number of the invaded cells per field (MG-63: siTRPM8 37.67±10.05 vs. Parental 105.67±7.35, siCON 106.0±9.10; U2OS: siTRPM8 21.67±14.1 vs. Parental 76.0±8.63, siCON 77.0±5.95, P < 0.01, Fig. 4B). Moreover, the expressions of p-FAK and MMP-2 were decreased in siTRPM8 cells, both of which play key roles in cell migration [20, 21], a function required for the invasion and metastasis of cancer cells (Fig. 4C).
In order to exclude the possibility that the inhibition of the invasion and metastasis of TRPM8 knockdown were caused by reduced cell viability and/or proliferation, CCK-8 assay and Annexin V-FITC assay were performed to examine the proliferation and apoptosis status of si-TRPM8 cells after incubation in 1% FBS medium for 24h. Compared with siCON cells, TRPM8 knockdown do not affect the proliferation and apoptosis of MG-63 and U2OS in 24h, indicating that the inhibitory effect of TRPM8 knockdown on the invasion and metastasis was not caused by reduced cell viability and/or proliferation (Supplementary Fig. 1).
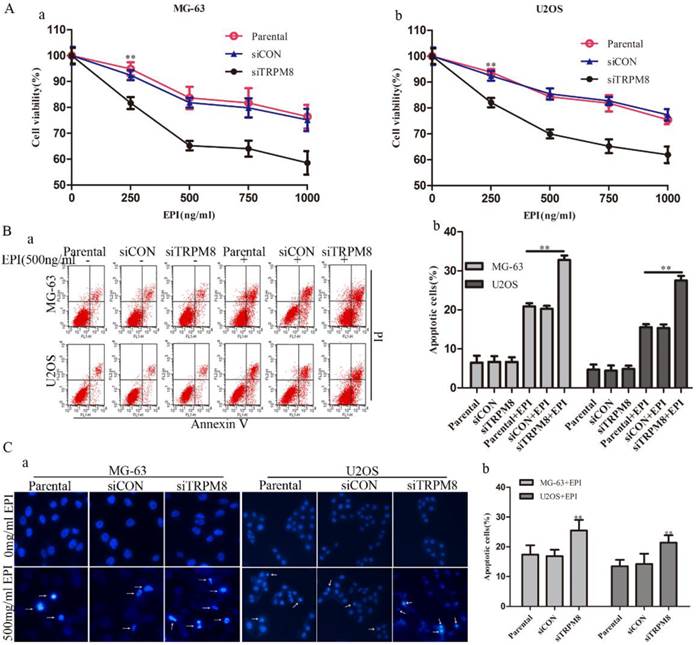
Knockdown of TRPM8 enhanced the epirubicin-induced apoptosis
Although the knockdown of TRPM8 failed to trigger cell apoptosis (Fig. 5B and 5C), it facilitated the cell apoptosis induced by EPI. This was confirmed by the CCK8, Annexin V-FITC and Hoechst 33258 staining assays. The CCK8 assay showed that, compared with the Parental and siCON cells, the viability of siTRPM8 cells was dramatically weakened in a dose-dependent manner after incubated with EPI at the indicated concentration for 48h (Fig. 5A). The concentration of EPI used in the following experiments was set at 500ng/ml according to the CCK8 assay. The Annexin V-FITC assay revealed that knockdown of TRPM8 facilitated EPI-induced cell apoptosis when compared with the Parental and siCON cells (MG-63: siTRPM8 32.83%±1.23% vs. Parental 20.90%±0.82%, siCON 20.27%±0.27%; U2OS: siTRPM8 27.55%±1.17% vs. Parental 15.54%±0.82%, siCON 15.36%±0.93%, P < 0.01, Fig. 5B). The Hoechst 33258 staining assay also demonstrated this result (Fig. 5C).
Knockdown of TRPM8 enhanced epirubicin-induced apoptosis. A: Knockdown of TRPM8 significantly reduced the viability of MG-63 (a) and U2OS (b) cells, **P<0.01. B: (a) Cell apoptosis was analyzed by Annexin V assay. (b) Knockdown of TRPM8 enhanced epirubicin-induced apoptosis, **P<0.01. C: (a) The Hoechst 33258 staining assay revealed that down regulation of TRPM8 facilitated cell apoptosis in MG-63 and U2OS cells. Arrows indicate apoptotic cells. (b) The quantitative data showed the percentage of apoptotic cells in EPI-treated MG-63 and U2OS cells, **P<0.01. All the data in this figure was analyzed by One-Way ANOVA.
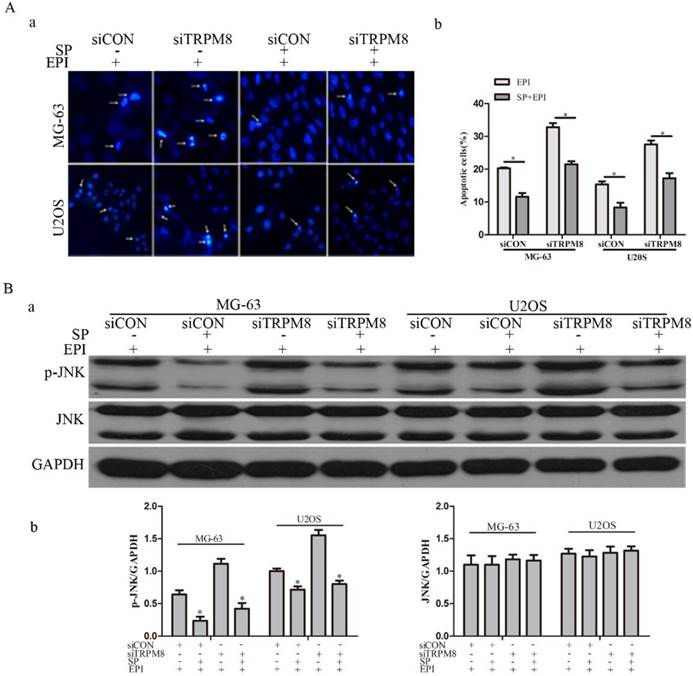
Studies suggest that the activation of JNK increases after EPI treatment and JNK depletion confers resistance to EPI-induced apoptosis [22]. To figure out the possible function of JNK in the EPI-induced apoptosis, its inhibitor SP600125 was applied. The efficiency of SP600125 was evaluated by western blot and the result revealed that the phosphorylation of JNK was obviously decreased (Fig. 6B). The Hoechst 33258 staining assay suggested that the EPI-induced apoptosis was partly abolished (Fig. 6A).
SP600125 (SP), the inhibitor of JNK, partly abolished the effect of EPI treatment. A: The Hochest 33258 assay (a) and its quantitative data (b) revealed that EPI-induced apoptosis was partly abolished by SP600125, *P<0.05. Arrows indicate apoptotic cells. B: (a) The efficiency of SP600125 was examined by western blot, and the phosphorylation of JNK induced by EPI was significantly suppressed by SP600125 in siCON and siTRPM8 cells. (b) The western blot results were quantified and the data was analyzed by t-text, *P<0.05.
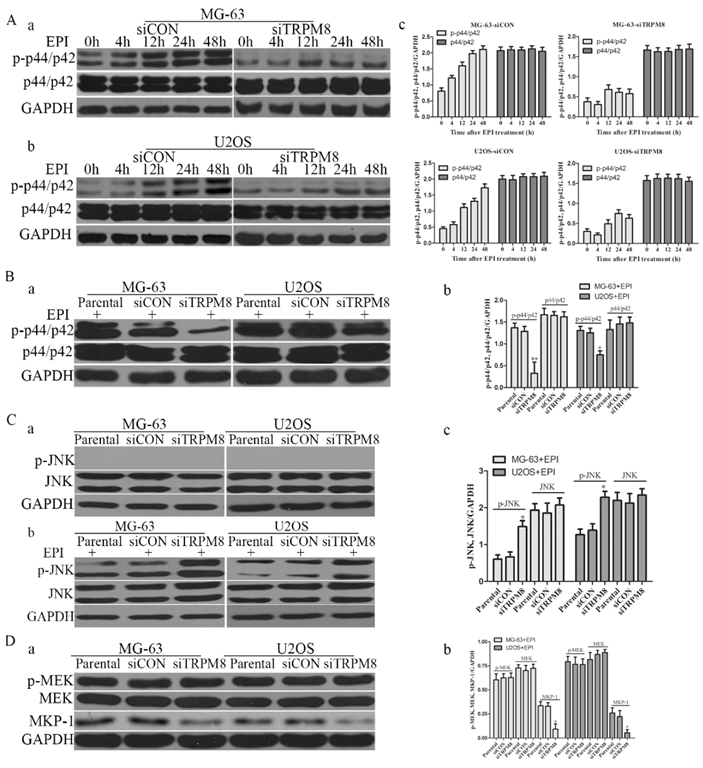
Knockdown of TRPM8 suppressed the phosphorylation of p44/p42 and facilitated epirubicin-induced JNK activation
Compared with the Parental and siCON cells, the level of phospho-p44/p42 was markedly reduced in siTRPM8 cells (Fig. 3C). However, phospho-JNK was detected neither in Parental and siCON cells nor in siTRPM8 cells (Fig. 7C a). EPI could activate p44/p42 in a time-dependent manner in siCON cells, but such manner disappeared when TRPM8 was knocked down (Fig. 7A). Especially, the level of p-p44/p42 in siTRPM8 cells was still lower than that in the Parental and siCON cells after EPI was applied (Fig. 7B). However, EPI could dramatically activate JNK especially in siTRPM8 cells (Fig. 7C b). Why the knockdown of TRPM8 can suppress the activation of p44/p42 and facilitate the activation of JNK induced by EPI? To figure out this question, we analyzed the MEK, an upstream factor of p44/p42, and the mitogen-activated protein kinase phosphatase-1(MKP-1), an endogenous negative regulator of MAPKs. The results revealed that knockdown of TRPM8 had no impact on phospho-MEK, but decreased the expression of MKP-1 (Fig. 7D).
The changes of p-p44/p42 and pJNK. A: After transfected with siCON and siTRPM8, MG-63 (a) and U2OS (b) cells were treated with 500ng/ml EPI for the indicated time and then western blot was performed to investigate the p-p44/p42 and p44/p42. (c) The results of the western blot were quantified and expressed in histograms. B: (a) After 48h EPI treatment, p-p44/p42 was analyzed in each sample by western blot. (b) The quantitative data of the western blot and statistic analysis was performed by One-Way ANOVA, **P<0.01, *P<0.05. C: Cells were prepared as indicated in material and methods, and p-JNK and JNK was analyzed by western blot. Knockdown of TRPM8 had no influence on the phosphorylation of JNK (a); however, after EPI treatment it enhanced the phosphorylation of JNK (b). (c) The results of western blot in (b) were quantified and One-Way ANOVA was applied for the statistic analysis, *P<0.05. D: (a) p-MEK, MEK and MKP-1 was investigated by western blot in Parental, siCON and siTRPM8 cells. (b) The results of western blot were quantified and One-Way ANOVA was applied for the statistic analysis, *P<0.05.
Discussion
TRPM8, a Ca2+-permeable cation channel, belongs to the melastatin subfamily of TRP channels and it is also known as the so-called 'cold' receptor protein for it can be activated by cold temperature and menthol. Researches on TRPM8 are growing rapidly, which mainly focus on thermosensation and cancer biomarker. For thermosensation, the role of TRPM8 in mediating the cold-evoked excitation has been fairly well established [23, 24]. Being a cancer biomarker, TRPM8 is associated with the phenotype of cancers originating from breast, lung, colon, prostate and bone as stated in this study. For prostate cancer, the expression of TRPM8 increases in early-stage [10] and then decreases as tumor progresses to androgen-independent stage [25]. Our former studies suggest that the overexpression of TRPM8 in PC-3 cells [26] or the activation of TRPM8 channel by menthol in DU145 cells [27] can reduce cell proliferation and migration. For breast cancer, TRPM8 is overexpressed and the expression level strongly correlates with proliferative parameters such as SBR grade, Ki67 proliferation index and tumor size [28]. For pancreatic adenocarcinoma, siRNA-mediated knockdown of TRPM8 does not induce apoptosis but induces replicative senescence, which suggests that targeted inhibition of TRPM8 may enhance tumor sensitivity to therapeutics [29]. In this study, we also found that TRPM8 was overexpressed in osteosarcoma and was required for cell proliferation and motility, especially, siRNA-mediated knockdown of TRPM8 enhanced EPI-induced apoptosis. More and more studies suggest that Ca2+ is a versatile intracellular messenger and it has an influence on the phosphorylation of Akt and p42/p44. The Ca2+ imaging assay revealed that knockdown of TRPM8 lead to the impaired regulation of intracellular Ca2+ concentration. So the aberrant intracellular Ca2+ levels may be responsible for the decrease of p-Akt and p-p42/p44.
The activated Akt can phosphorylate and inhibit GSK-3β function and leads to de-phosphorylation and stabilization of cyclin D1, which can form compound with Cdk4 to regulate cell cycle. Ca2+ could bind the PH domain of activated Akt to prevent or delay its inactivation by an Akt phosphatase, and then the inactivation of Akt suppressed its downstream GSK-3β and cyclinD1 and the cell cycle was arrested in the G0/G1 phase.
As for p44/p42, which has been reported to be able to promote cell growth, the phosphorylation suppression may be resulted from the aberrant intracellular Ca2+ levels rather than MEK and MKP-1. Because knockdown of TRPM8 had no influence on the activation of MEK, the upstream of p44/p42, and especially it decreased the expression of MKP-1 which can de-phosphorylate all three MAPKs. Studies suggest that elevated calcium concentration can either activate or, less frequently, inhibit the p44/p42-MAPK pathway [30, 31]. Seongho Ryu [32] et al found that aberrant Ca2+ levels decreased the activation of p44/p42 as well as FAK, because p44/p42 was able to phosphorylate FAK. And such results were also found in this study. So the aberrant Ca2+ levels may play an important role in the decrease of p-p44/p42 and p-FAK caused by TRPM8 knockdown and further inhibited cell proliferation and motility.
In addition to the above functions, knockdown of TRPM8 also enhanced epirubicin-induced apoptosis in osteosarcoma cells. This is associated with the MAPK pathway.
Many drugs used in cancer therapy activate not only the apoptosis but also the anti-apoptotic signal transduction pathways that promote survival, and possibly limit their own antitumor efficacy. One notable example is the nuclear factor κB pathway, whose activation results in enhanced transcription of Bcl-2 homologs such as Bcl-xL [33]. Another example is the p44/p42-MAPK pathway whose activation generally results in an increase of the threshold for cell death [34]. Anthracycline-based antitumor antibiotics have been reported to be able to activate p44/p42-MAPK in some cell systems, including primary rat ventricular myocytes [35, 36], neuroblastoma cells [37], rat hepatoma cells [38], human cervical carcinoma cells [39] and monoblasts [40]. And in this study, we found that EPI, one of the anthracycline-based antitumor antibiotics, can activate p44/p42-MAPK in siCON cells in a time-dependent manner, but such manner disappeared in siTRPM8 cells. Especially, the level of p-p44/p42 in siTRPM8 cells was still lower than that in the Parental and siCON cells after EPI treatment. George et al. found that the activation of p44/p42-MAPK, induced by anthracycline-based antitumor antibiotics, was anti-apoptotic [41]. Therefore the knockdown of TRPM8 may decrease the threshold for EPI-induced cell death.
Activation of JNK is very important, because studies suggest that the activation of JNK increases after EPI treatment and JNK depletion confers resistance to EPI-induced apoptosis [22]. Although the knockdown of TRPM8 failed to activate JNK, it facilitated the activation of JNK induced by EPI. Such facilitation may be resulted from the down-modulation of MKP-1, because it can de-phosphorylate JNK and p38 with a much higher affinity and de-phosphorylate p44/p42 with a much lower affinity [42]. MKP-1 is the prototypic member in the family of dual-specificity phosphatases that dephosphorylate tyrosine and threonine residues on target proteins. Growing evidences suggest that MKP-1 may play a role in chemotherapy resistance. Overexpression of MKP-1 is able to protect cancer cells from chemotherapy-mediated apoptosis by limiting JNK activity, such as cisplatin-induced apoptosis in human lung cancer cells [43] and doxorubicin-, mechlorethamine-, paclitaxel-induced apoptosis in breast cancer cells [44]. Studies have revealed that the repression of MKP-1 by anthracyclines or siRNA can increase phosphorylation of p44/p42 [41] and the induction of MKP-1 by proteasome inhibitor can decrease the phosphorylation of p44/p42 [45]. However, there are also studies indicating that MKP-1 expression is inversely correlated to JNK but not to p44/p42 enzymatic activity [46, 47], and our results agree with it. The knockdown of TRPM8 facilitated the EPI-induced activation of JNK and decreased the phosphorylation of p44/p42 and the expression of MKP-1, which suggests that the expression of MKP-1 is inversely correlated to the activation of JNK but not to p44/p42 in osteosarcoma cells.
When the activation of JNK was inhibited by its special inhibitor SP600125, the effect of EPI treatment was partly abolished. So the enhancement of EPI treatment caused by the knockdown of TRPM8 may be partly through JNK activation and perhaps the decrease of p-p44/p42 is also involved, because p44/p42 activation generally results in an increase of the threshold for cell death.
In conclusion, we are the first to report that TRPM8 is aberrantly overexpressed in osteosarcoma, and the knockdown of TRPM8 not only negatively influences the proliferation and malignant progression of osteosarcoma cells but also facilitates EPI-induced apoptosis. Therefore, TRPM8 is worthy further investigation for its potential as a clinical biomarker and therapeutic target in osteosarcoma. In spite of these findings, there are still deficiencies which deserve further investigation in this study, such as osteochondroma was used as normal control and the proofs of regulation between TRPM8 and these changed proteins induced by its downregulation as well as the in vivo mouse model experiments.
Abbreviations
TRPM8: transient receptor potential melastatin member 8; BMSCs: human bone marrow mesenchymal stem cells; EPI: epirubicin.
Supplementary Material
Supplementary Figure 1Acknowledgements
This study was supported by two grants of Natural Science Foundation of China (No.81172734 and No. 81202027)
Competing Interests
The authors have declared that no competing interest exists.
References
1. Longhi A, Errani C, De Paolis M. et al. Primary bone osteosarcoma in the pediatric age: state of the art. Cancer Treat Rev. 2006;32(6):423-36
2. Lee JA, Kim MS, Kim DH. et al. Relative tumor burden predicts metastasis-free survival in pediatric osteosarcoma. Pediatr Blood Cancer. 2008;50(2):195-200
3. Wachtel M, Schafer BW. Targets for cancer therapy in childhood sarcomas. Cancer Treat Rev. 2010;36(4):318-27
4. Janeway KA, Grier HE. Sequelae of osteosarcoma medical therapy: a review of rare acute toxicities and late effects. Lancet Oncol. 2010;11(7):670-8
5. van Dalen EC, Michiels EM, Caron HN. et al. Different anthracycline derivates for reducing cardiotoxicity in cancer patients. Cochrane Database Syst Rev. 2010(5):CD005006
6. Legrand G, Humez S, Slomianny C. et al. Ca2+ pools and cell growth. Evidence for sarcoendoplasmic Ca2+-ATPases 2B involvement in human prostate cancer cell growth control. J Biol Chem. 2001;276(50):47608-14
7. Thebault S, Flourakis M, Vanoverberghe K. et al. Differential role of transient receptor potential channels in Ca2+ entry and proliferation of prostate cancer epithelial cells. Cancer Res. 2006;66(4):2038-47
8. Vanoverberghe K, Vanden Abeele F, Mariot P. et al. Ca2+ homeostasis and apoptotic resistance of neuroendocrine-differentiated prostate cancer cells. Cell Death Differ. 2004;11(3):321-30
9. Skryma R, Mariot P, Bourhis XL. et al. Store depletion and store-operated Ca2+ current in human prostate cancer LNCaP cells: involvement in apoptosis. J Physiol. 2000;527(Pt 1):71-83
10. Tsavaler L, Shapero MH, Morkowski S. et al. Trp-p8, a novel prostate-specific gene, is up-regulated in prostate cancer and other malignancies and shares high homology with transient receptor potential calcium channel proteins. Cancer Res. 2001;61(9):3760-9
11. Zhang L. TRPM8 in prostate cancer cells: a potential diagnostic and prognostic marker with a secretory function? Endocrine Related Cancer. 2006;13(1):27-38
12. Ouadid-Ahidouch H, Dhennin-Duthille I, Gautier M. et al. [TRP calcium channel and breast cancer: expression, role and correlation with clinical parameters]. Bull Cancer. 2012;99(6):655-64
13. Yee NS, Zhou W, Lee M. Transient receptor potential channel TRPM8 is over-expressed and required for cellular proliferation in pancreatic adenocarcinoma. Cancer Letters. 2010;297(1):49-55
14. Abed E, Labelle D, Martineau C. et al. Expression of transient receptor potential (TRP) channels in human and murine osteoblast-like cells. Molecular Membrane Biology. 2009;26(3):146-58
15. Gun B, Bahadir B, Bektas S. et al. Clinicopathological significance of fascin and CD44v6 expression in endometrioid carcinoma. Diagnostic Pathology. 2012;7(1):80
16. Bidaux G, Flourakis M, Thebault S. et al. Prostate cell differentiation status determines transient receptor potential melastatin member 8 channel subcellular localization and function. Journal of Clinical Investigation. 2007;117(6):1647-57
17. Saadoun S, Papadopoulos MC, Hara-Chikuma M. et al. Impairment of angiogenesis and cell migration by targeted aquaporin-1 gene disruption. Nature. 2005;434(7034):786-92
18. Vanden Abeele F, Skryma R, Shuba Y. et al. Bcl-2-dependent modulation of Ca(2+) homeostasis and store-operated channels in prostate cancer cells. Cancer Cell. 2002;1(2):169-79
19. Wada T, Penninger JM. Mitogen-activated protein kinases in apoptosis regulation. Oncogene. 2004;23(16):2838-49
20. Cao J, Chiarelli C, Richman O. et al. Membrane type 1 matrix metalloproteinase induces epithelial-to-mesenchymal transition in prostate cancer. J Biol Chem. 2008;283(10):6232-40
21. Schlaepfer DD, Mitra SK, Ilic D. Control of motile and invasive cell phenotypes by focal adhesion kinase. Biochim Biophys Acta. 2004;1692(2-3):77-102
22. de Olano N, Koo CY, Monteiro LJ. et al. The p38 MAPK-MK2 axis regulates E2F1 and FOXM1 expression after epirubicin treatment. Mol Cancer Res. 2012;10(9):1189-202
23. McKemy DD, Neuhausser WM, Julius D. Identification of a cold receptor reveals a general role for TRP channels in thermosensation. Nature. 2002;416(6876):52-8
24. Peier AM, Moqrich A, Hergarden AC. et al. A TRP channel that senses cold stimuli and menthol. Cell. 2002;108(5):705-15
25. Henshall SM, Afar DE, Hiller J. et al. Survival analysis of genome-wide gene expression profiles of prostate cancers identifies new prognostic targets of disease relapse. Cancer Res. 2003;63(14):4196-203
26. Yang ZH, Wang XH, Wang HP. et al. Effects of TRPM8 on the proliferation and motility of prostate cancer PC-3 cells. Asian J Androl. 2009;11(2):157-65
27. Wang Y, Wang X, Yang Z. et al. Menthol Inhibits the Proliferation and Motility of Prostate Cancer DU145 Cells. Pathology & Oncology Research. 2012;18(4):903-10
28. Dhennin-Duthille I, Gautier M, Faouzi M. et al. High expression of transient receptor potential channels in human breast cancer epithelial cells and tissues: correlation with pathological parameters. Cell Physiol Biochem. 2011;28(5):813-22
29. Yee NS, Brown RD, Lee MS. et al. TRPM8 ion channel is aberrantly expressed and required for preventing replicative senescence in pancreatic adenocarcinoma: potential role of TRPM8 as a biomarker and target. Cancer Biol Ther. 2012;13(8):592-9
30. Agell N, Bachs O, Rocamora N. et al. Modulation of the Ras/Raf/MEK/ERK pathway by Ca(2+), and calmodulin. Cell Signal. 2002;14(8):649-54
31. Cullen PJ, Lockyer PJ. Integration of calcium and Ras signalling. Nat Rev Mol Cell Biol. 2002;3(5):339-48
32. Ryu S, McDonnell K, Choi H. et al. Suppression of miRNA-708 by Polycomb Group Promotes Metastases by Calcium-Induced Cell Migration. Cancer Cell. 2013;23(1):63-76
33. Orlowski RZ, Baldwin AS Jr. NF-kappaB as a therapeutic target in cancer. Trends Mol Med. 2002;8(8):385-9
34. Dent P, Grant S. Pharmacologic interruption of the mitogen-activated extracellular-regulated kinase/mitogen-activated protein kinase signal transduction pathway: potential role in promoting cytotoxic drug action. Clin Cancer Res. 2001;7(4):775-83
35. Adderley SR, Fitzgerald DJ. Oxidative damage of cardiomyocytes is limited by extracellular regulated kinases 1/2-mediated induction of cyclooxygenase-2. J Biol Chem. 1999;274(8):5038-46
36. Zhu W, Zou Y, Aikawa R. et al. MAPK superfamily plays an important role in daunomycin-induced apoptosis of cardiac myocytes. Circulation. 1999;100(20):2100-7
37. Guise S, Braguer D, Carles G. et al. Hyperphosphorylation of tau is mediated by ERK activation during anticancer drug-induced apoptosis in neuroblastoma cells. J Neurosci Res. 2001;63(3):257-67
38. Kim SG, Sung M, Kang KW. et al. DA-125, a novel anthracycline derivative showing high-affinity DNA binding and topoisomerase II inhibitory activities, exerts cytotoxicity via c-Jun N-terminal kinase pathway. Cancer Chemother Pharmacol. 2001;47(6):511-8
39. Yeh PY, Chuang SE, Yeh KH. et al. Increase of the resistance of human cervical carcinoma cells to cisplatin by inhibition of the MEK to ERK signaling pathway partly via enhancement of anticancer drug-induced NF kappa B activation. Biochem Pharmacol. 2002;63(8):1423-30
40. Mas VM, Hernandez H, Plo I. et al. Protein kinase Czeta mediated Raf-1/extracellular-regulated kinase activation by daunorubicin. Blood. 2003;101(4):1543-50
41. Small GW. Repression of Mitogen-Activated Protein Kinase (MAPK) Phosphatase-1 by Anthracyclines Contributes to Their Antiapoptotic Activation of p44/42-MAPK. Journal of Pharmacology and Experimental Therapeutics. 2003;307(3):861-69
42. Camps M, Nichols A, Arkinstall S. Dual specificity phosphatases: a gene family for control of MAP kinase function. FASEB J. 2000;14(1):6-16
43. Wang Z, Xu J, Zhou JY. et al. Mitogen-activated protein kinase phosphatase-1 is required for cisplatin resistance. Cancer Res. 2006;66(17):8870-7
44. Small GW, Shi YY, Higgins LS. et al. Mitogen-Activated Protein Kinase Phosphatase-1 Is a Mediator of Breast Cancer Chemoresistance. Cancer Research. 2007;67(9):4459-66
45. Orlowski RZ. Evidence That Inhibition of p44/42 Mitogen-activated Protein Kinase Signaling Is a Factor in Proteasome Inhibitor-mediated Apoptosis. Journal of Biological Chemistry. 2002;277(31):27864-71
46. Loda M, Capodieci P, Mishra R. et al. Expression of mitogen-activated protein kinase phosphatase-1 in the early phases of human epithelial carcinogenesis. Am J Pathol. 1996;149(5):1553-64
47. Magi-Galluzzi C, Mishra R, Fiorentino M. et al. Mitogen-activated protein kinase phosphatase 1 is overexpressed in prostate cancers and is inversely related to apoptosis. Lab Invest. 1997;76(1):37-51
Author contact
![]() Corresponding author: Xinghuan Wang. Address: Department of Urology, Zhongnan Hospital, Wuhan University, Wuhan, China, 430071. E-mail: urologistwxhcom; Tel: +86-15871805490; Fax: +86-27-6781-3090
Corresponding author: Xinghuan Wang. Address: Department of Urology, Zhongnan Hospital, Wuhan University, Wuhan, China, 430071. E-mail: urologistwxhcom; Tel: +86-15871805490; Fax: +86-27-6781-3090