Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(8):909-920. doi:10.7150/ijbs.9214 This issue Cite
Research Paper
Mouse Macrophage Galactose-type Lectin (mMGL) is Critical for Host Resistance against Trypanosoma cruzi Infection
Alicia Vázquez1, Juan de Dios Ruiz-Rosado1, Luis I. Terrazas1, Imelda Juárez1, Lorena Gomez-Garcia2, Elsa Calleja1, Griselda Camacho1, Ana Chávez1, Miriam Romero1, Tonathiu Rodriguez1, Bertha Espinoza3, Miriam Rodriguez-Sosa1 ![]()
1. Unidad de Biomedicina, Facultad de Estudios Superiores-Iztacala, Universidad Nacional Autónoma de México (UNAM), C. P. 54090, Estado de México, México.
2. Department of Immunology, Instituto Nacional de Cardiología “Ignacio Chávez,” México, D.F. 14080 México.
3. Department of Immunology, Instituto de Investigaciones Biomédicas, Universidad Nacional Autónoma de México. México, D.F. 04510 México.
Received 2014-3-25; Accepted 2014-7-5; Published 2014-8-23
Abstract
The C-type lectin receptor mMGL is expressed exclusively by myeloid antigen presenting cells (APC) such as dendritic cells (DC) and macrophages (Mφ), and it mediates binding to glycoproteins carrying terminal galactose and α- or β-N-acetylgalactosamine (Gal/GalNAc) residues. Trypanosoma cruzi (T. cruzi) expresses large amounts of mucin (TcMUC)-like glycoproteins. Here, we show by lectin-blot that galactose moieties are also expressed on the surface of T. cruzi. Male mMGL knockout (-/-) and wild-type (WT) C57BL/6 mice were infected intraperitoneally with 104 T. cruzi trypomastigotes (Queretaro strain). Following T. cruzi infection, mMGL-/- mice developed higher parasitemia and higher mortality rates compared with WT mice. Although hearts from T. cruzi-infected WT mice presented few amastigote nests, mMGL-/- mice displayed higher numbers of amastigote nests. Compared with WT, Mφ from mMGL-/- mice had low production of nitric oxide (NO), interleukin (IL)-12 and tumor necrosis factor (TNF)-α in response to soluble T. cruzi antigens (TcAg). Interestingly, upon in vitro T. cruzi infection, mMGL-/- Mφ expressed lower levels of MHC-II and TLR-4 and harbored higher numbers of parasites, even when mMGL-/- Mφ were previously primed with IFN-γ or LPS/IFN-γ. These data suggest that mMGL plays an important role during T. cruzi infection, is required for optimal Mφ activation, and may synergize with TLR-4-induced pathways to produce TNF-α, IL-1β and NO during the early phase of infection.
Keywords: mMGL, Trypanosoma cruzi, Proinflammatory cytokines, C-Type lectin receptor, Macrophages receptors.
Introduction
Chagas disease, also known as American trypanosomiasis, is a disease caused by the protozoan parasite Trypanosoma cruzi. This parasitic infection affects approximately 7-12 million people in Latin America, and 60-80 million more are at risk (1, 2). The natural transmission of the T. cruzi parasite to humans and other mammals depends on the triatomine insect belonging to the family (Reduviidae), known as the kissing bug (3). Inside the host, the parasite is internalized in the cells of the innate immune system. After multiplication by binary fission, the amastigotes are transformed into blood trypomastigotes, which are released into the bloodstream by cell lysis. This stage can infect a wide range of host cells or be taken up by the insect vector through its mouthparts, closing the life cycle (4).
Once an individual has acquired the infection, a progressive disease develops. The acute phase is characterized by a high number of trypomastigotes in the blood as well as, fever and hepatomegaly. The chronic phase presents fewer parasites in the blood. Many patients infected with T. cruzi remain asymptomatic. However, 10-20 years after the initial infection, 5-10% of people develop anatomical and functional abnormalities in the esophagus and colon, while approximately 30% develop myocarditis, leading to heart failure or sudden death (4).
The outcome of the immune response depends on the capacity of Antigen Presenting Cells (APCs) to sensing foreign molecular configurations, also known as pathogen-associated molecular patterns (PAMPs), through pattern recognition receptors (PRRs) expressed on the surface of macrophages (Mφ) or dendritic cells (DC). Activation of PRRs leads to intracellular signals that activate innate immunity and orchestrate the development of an acquired immune response, which is necessary for protection against re-infection (5, 6). Some of these highly specialized receptors include Toll-like receptors (TLRs) and C-type lectin receptors (CLRs) (6).
In phagocytic cells from both mouse and human, the molecule glycosylphosphatidylinositol (GPI) anchors to T. cruzi trypomastigotes, and is recognized as a potent activator of TLR2 (7-10). The glycoinositolphospholipids (GIPLs) and unmethylated CpG motifs on DNA that are present in all stages of the T. cruzi life cycle are recognized by TLR4 (11, 12) and TLR9 (13), respectively. For activation of TLR2 and TLR9, the nonsaturated fatty acid chains and periodate-sensitive components from the GPI anchor covalently linked to mucin-like glycoproteins are required to trigger the production of inflammatory cytokines by APCs (11, 14-17).
In contrast to TLRs, CLRs recognize and internalize specific carbohydrates by lectin-glycan interactions. It has been suggested that CLRs work as mediators of microbial recognition and initiators of immune responses (5). The most important molecules from the CLR family include macrophage galactose type C-lectin (MGL), specific for mannose or fucose dendritic cell-specific intercellular adhesion molecule-3-grabbing non-integrin (DC-SIGN), the mannose receptor (MR), DEC205, and Dectin-1 (18). Some of these receptors may function as adhesion, signaling or antigen-uptake receptors (19-21). Moreover, CLRs have been shown to contribute to the loading of endocytosed antigens on MHC class I or class II, thereby facilitating effective antigen-specific CD4+ and CD8+ T- cell responses (22, 23).
The CLRs are able to trigger distinct signaling pathways that modulate APC functions through the expression of specific molecules and cytokines. In most cases, CLRs promote antigen presentation and determine the polarization of T cells (24-26). However, most evidence about how CLRs shape the immune response and trigger signaling pathways has emerged using viral and bacterial pathogens, fungi or peptides (18). There is no clear evidence that parasites interact with CLRs and activate a specific signaling pathway.
The MGL molecule is a member of the CLR family. Human MGL (hMGL) is a type II C-Type lectin, which is selectively expressed in APCs such as immature dendritic cells (iDC) and Mφ and is overexpressed in subsets of DCs with tolerogenic functions and alternatively activated Mφ. MGL recognizes Gal/GalNAc residues of N- and O-glycans carried by glycoproteins and /or glycosphingolipids and it promotes endocytosis (27).
It is well-known that endogenous functions for MGLs include pattern recognition of tumor antigens, foreign glycoproteins derived from helminth parasites, and cleaning of apoptotic cell embryos (28). The mechanisms or pathways used for internalizing carbohydrates by MGL have not been fully described, but has been reported that phagocytic cells are able to endocytose antigens through MGL, which them are transported along the endosomal-lysosomal pathway and presented in MHC-II molecule (29). Mice have two homologous copies of hMGL, MGL1 and MGL2, while humans and rats have only one copy (30, 31). Mouse MGL (mMGL) is a transmembrane glycoprotein of 42KD that recognizes Gal/GalNAc and Lewis X and Lewis A structures (30, 32).
In this work, we investigated the role of mMGL in immune response to an acute T. cruzi infection using mMGL knockout mice (mMGL-/-). Our results demonstrate that mMGL plays an important role in T. cruzi infection and may be useful to prevent T. cruzi invasion and to induce inflammatory cytokines and NO production by macrophages.
Materials and Methods
Trypanosoma cruzi lysate antigen. Epimastigotes of T. cruzi that had been maintained at 28°C by sequential culture in a liver infusion tryptose medium (LIT) with 25 mg/l of hemin supplemented with 10% heat inactivated fetal bovine serum (FBS) and 100 U of penicillin/streptomycin were isolated, washed three times in PBS, and centrifuged at 20,000 g for 15 min (33). Protease inhibitors were added (0.1 to 2 µg/ml apotinin, 0.5 to 2 mM EDTA, 1 to 5 mM phenylmethyl fluoride, 1 μg/ml pepstatin, 50 μg/ml TLCK (a-p-tosyl-L-Lysine chloromethyl Ketone) (Sigma, St. Louis, MO), and parasites were sonicated six times for 10 s each at 50 W using a sonic Dimembrator 300 (Fisher). Parasite destruction was confirmed using a microscope. Parasite lysates were then centrifuged at 20, 000 g for 30 min to separate the soluble fraction, which was stored at -70oc until use. Total protein content was determined in the soluble fraction (34).
SDS-PAGE and Lectin-Blotting. Sodium dodecyl sulphate (SDS)-polyacrylamide gel electrophoresis (PAGE) and lectin blotting were performed by standard techniques. Briefly, antigen extracts in nonreducing sample buffer were boiled for 5 min at 95°C and separated on 12% polyacrylamide gels at a concentration of 40 μg/well. Separated proteins were transferred to a nitrocellulose membrane (Amersham, Piscataway, NJ, USA) using a Western blotting unit (Bio-Rad). The membrane was blocked overnight at 4°C with 2% (w/v) bovine serum albumin in PBS pH 7.2, washed thoroughly with PBS/Tween 0.1% and incubated with Artocarpus integrifolia lectin (Jacalin)-Peroxidase (Sigma-Aldrich), a lectin specific to Gal residues, for 3 h. After washing, bound peroxidase on the membrane was developed with 1: 1000 PBS/ H and diaminobenzidine at a concentration of 2 mg/ ml.
Mice. Eight to ten week-old male C57BL/6 mice were purchased from Harlan (México City, Mexico). The generation of mMGL-deficient mice from C57BL/6 mice by a disruption in mMGL exons 2 and 3 has been previously described (35). mMGL-/- mice on a C57BL/6 background were donated by Glycomics Consortium. All mice were genotyped by PCR. Briefly, total DNA was extracted from the tails of the mice (36), and DNA amplification was performed using 1 μg of DNA, oligo(dT) and specific primers for 35 cycles of 30 sec at 65oC. Oligos for the mMGL gene were (5'-ATGTCATGACTCAGGATC-3' and 5'-CTTGGTCCCAGATCCGTATC-3'), and for the neomicine gene (Neo), 5´-AGGATCTCCTGTCATCTCACCTTGCTCCTG-3' and 3'-AAGAACTCGTCAAGAAGGCGATAGAAGGCG-5´. A PCR fragment of 633 bp to mMGL and 492 bp to Neo were visualized to identify WT or mMGL-/- mice, respectively. Wild type C57BL/6 purchased from Harlan (México) were used as control mice. All animal studies were performed according to the guidelines for the Care and Use of Laboratory Animals, as adopted by the U.S. National Institutes of Health and the Mexican Regulation of Animal Care and maintenance (NOM-062-ZOO-1999, 2001).
Parasites and experimental infections. The Mexican T. cruzi TBAR/MX/0000/Queretaro strain belonging to DTU TcI was used in this work. Experimental infections of male mMGL-/- and WT mice were induced by intraperitoneal (i.p.) injection with 104 blood trypomastigotes that were obtained from previously infected mice, counted, and suspended in 100 μl of sterile phosphate-buffered saline (PBS). Parasitemia was determined every week by using hemocytometer counts of parasites in the blood diluted 1:10 in PBS with 3.8% sodium citrate.
Histopathology. Non-infected and T. cruzi infected hearts from mMGL-/- and WT mice were fixed overnight in formaldehyde and embedded in paraffin blocks, after which 5-μ-thick transverse sections were mounted on slides, stained with hematoxylin and eosin (H&E), and scored as previously described (37). The presence of inflammatory cells was scored as (0) - absent/none, (1) - focal or mild with ≤1 foci, (2) - moderate with ≥2 inflammatory foci, (3) - extensive with generalized coalescing of inflammatory foci or disseminated inflammation and (4) - severe with diffused inflammation, interstitial edema, and loss of tissue integrity. The foci of pseudocysts (Tc nests) were scored as (0) absent, (1) 0-1 foci, (2) 1-5 foci, and (3) 5 foci. Using an Olympus BX51 microscope (Olympus American, Melville, NY) equipped with a digital video camera, 4 mice per group with 10-slides per mouse were evaluated for each group.
Isolation of macrophages and activation. Peritoneal exudate cells (PECs) were obtained from the peritoneal cavity at 0, 21, 28 and 35 days post T. cruzi infection of mMGL-/- and WT mice under sterile conditions using 10 ml of ice-cold Hank´s balanced salt solution (Microlab, México). Following two washes with Hank's solution, red blood cells were lysed by resuspending the cells in Boyle's solution. After two washes, viable cells were counted by trypan blue exclusion (routinely over 95%) with a Neubauer hemocytometer. PECs were adjusted to 5x106 cells/ml in DMEM medium supplemented with 10% fetal calf serum (FCS), 100 U of penicillin/streptomycin, and 2 mM glutamine (all from Gibco-BRL, Grand Island, NY) and were cultured in 24-well plates (Costar, Cambridge, MA, USA). After 2 hours at 37oC and 5% CO2, non-adherent cells were removed by washing with warm supplemented DMEM medium. Adherent cells (Mφ) were removed from the plate by washing with 5 mM EDTA in warm PBS and were then readjusted to 1x106 cells/ml. Viability was checked again at this point (>90%), and samples were analyzed by FACS using the macrophage marker F4/80 (Mφ purity was estimated as >85%). One milliliter of Mφ was plated on 12-well plates (Costar), left untreated or stimulated with LPS (0.5 μg/ml; Escherichia coli 0111:B4 Sigma-Aldrich), or Poly:IC (25 μg/ml; Polyinosinic-polycytidylic acid potassium salt), or 25 μg/ml of total T. cruzi antigen (TcAg). Mφ were incubated for 48 hours at 37oC and 5% CO2. Supernatants were collected for quantification of cytokine production. Cytokine levels (IL-12, IFN-γ, TNF-α, IL-10, IL-4, and IL-13) were measured using the sandwich ELISA method according to the manufacturer's instructions (Preprotech, México).
Reverse transcriptase-PCR. Total RNA was extracted from Mφ obtained as described above from untreated or in vitro T. cruzi infected (21 days after infection) WT or mMGL-/- mice, using the TRIzol reagent (Sigma). cDNA was prepared using a first strand synthesis superscript II kit (Invitrogen) from 5 μg of total RNA. cDNA samples were standardized based on the content of the housekeeping gene Glyceraldehyde-3-Phosphate Dehydrogenase (GAPDH) cDNA. The primers for GAPDH were F-CTC ATG ACC ACA GTC CAT GC and R-CAC ATT GGG GGT AGG AAC AC (201 bp). The primers for Arginase-1 were F-CAG AAG AAT GGA AGA GTC AG and R-CAG ATA TGC AGG GAG TCA CC (250 bp). The primers for Ym1 were F-TCA CAG GTC TGG CAA TTC TTC TG and R-TTT GTC CTT AGG AGG GCT TCC TC (436 bp). The primers for TNF-α were F-GGC AGG TCT ACT TTG GAG TCA TTG C and R-ACA TTC GAG GCT CCA GTG AAT TCG (307 bp). The primers for inducible nitric oxide synthase were F-CTG GAG GAG CTC CTG CCT CAT G and R-GCA GCA TCC CCT CTG ATG GTG (449 bp). The primers for IL-1β were F-GAG TGT GGA TCC CAA GCA AT and R-CTC AGT GCA GGC TAT GAC CA (500 bp). The primers for TLR-4 were ACC TGG CTG GTT TAC ACG TC and R-CTG CCA GAG ACA TTG CAG AA (201 bp). The primers for TLR-3 F-CCC CCT TTG AAC TCC TCT TC and R-TTT CGG CTT CTT TTG ATG CT. The primers for TLR-2 F-AAG AGG AAG CCC AAG AAA GC and R-CGA TGG AAT CGA TGA TGT TG (199 bp). Polymerase chain reaction (PCR) was performed in a total volume of 50 μL in PCR buffer in the presence of 10 mM dNTPs, 15 pM each primer, and 1.5 U of kappa TaqDNA polymerase (kapabiosystems Boston M.A, USA) using a XP-cycler (Bioer, Switzerland). After 35 cycles of amplification, the PCR products were separated by electrophoresis on a 1.5% agarose gel and visualized by ethidium bromide staining.
In vitro T. cruzi infection and flow cytometry analysis of macrophages. One milliliter (1x106) of Mφ obtained as described above from WT or mMGL-/- mice was plated, and infected in vitro with epimastigores of T. cruzi (ratio 1: 10) for two hours. Following infection, parasites were washed off. Mφ were incubated at 4oC for 15 minutes in blocking buffer and 2% FCS, 10 μg/ml anti-CD16/32 in FACS buffer (PBS supplemented with 2mM EDTA and 0.5% BSA), followed by staining for 20 minutes on ice with the antibodies (Ab) of interest at the appropriate dilution as determined by titration. Abs included APC-conjugated anti-F4/80, FITC-conjugated MHC-II and PE-conjugated TLR-4 as well as appropriate isotype control Abs (all Abs from Biolegend, San Diego, CA, USA).
For infection analysis; 1x106 Mφ obtained from WT or mMGL-/- mice were plated, and infected in vitro with epimastigores of T. cruzi (ratio 1: 10) for two hours. For infection analysis, epimastigotes of T. cruzi were washed in PBS and resuspended at 1x107/ml in 5 mM carboxyfluorescein succinimidyl ester (CFSE) in serum-free DMEM for 15 min at 37oC. Following infection, parasites were washed off, and Mφ infected and not infected were incubated in DMEM medium supplemented with 10% FCS for 2, 6, 12 or 24 hours at 37oC and 5% CO2.
For infection analysis on activated Mφ, 1x106 thioglycollate-elicited Mφ were left untreated or treated for 24 h with IFN-γ or IFN-γ/LPS. Cells were washed and infected with CFSE labeled epimastigotes of T. cruzi for 2 h (10:1 parasite to Mφ ratio), after this the parasites were washed off.
All cells described in this section were washed 3x in FACS buffer and fixed in 0.8% paraformaldehyde before acquisition and analysis (BD FACStation and FlowJo software).
Statistical analysis. Comparisons between WT and mMGL-/- groups were made by using Student's unpaired t test. P values of ‹ 0.05 were considered significant. For survival assays, a log-rank test was used with the Graph Pad computer program (Graph Pad 6, San Diego, CA).
Results
Intact carbohydrates on total antigens of T. cruzi are rich in glycoproteins bearing galactose residues. Given that T. cruzi parasites are rich in carbohydrates (38), we developed a PAGE (Fig. 1A) and lectin-blot analysis (Fig. 1B) to determine whether the T. cruzi Queretaro strain contained glycoproteins bearing terminal Gal and GalNAc sugars, which are recognized by mMGL with high affinity. According to the lectin-blot analysis (Fig. 1B) the total soluble antigens of T. cruzi were highly recognized by Jacalin lectin, indicating the presence of Gal residues, similar to other reports with different T. cruzi strains (39).
Total T. cruzi antigen is rich in glycoproteins bearing galactose (Gal) and N-acetylgalactosamine (GALNAc). TcAg were separated by SDS-PAGE and transferred to NC sheets that were used to detect N-linked glycans with horseradish peroxidase (HRP)-conjugated Jacalin. T. cruzi antigen lectin blood, SDS-page A); Jacalin-Blot B). Antigens were from different infected mice. MW indicates the molecular weight markers in kilodaltons (kDa).

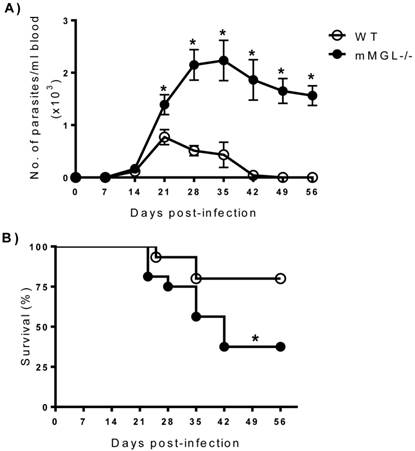
mMGL-/- mice develop high parasitemia levels and increased mortality to T. cruzi infection. To investigate the role of mMGL in immunity to T. cruzi, mMGL-/- and WT mice were i.p. infected with 104 metacyclic trypomastigotes of T. cruzi, and parasitemia was monitored weekly. T. cruzi-infected mMGL-/- mice developed significantly higher blood parasitemia levels than similarly infected WT mice, which contained nearly four-fold fewer parasites in their blood by days 21 to 35 post infection (pi) (Fig. 2A, P <0.05). Both groups developed blood parasitemia on day 14 pi, but on day 28 pi, mMGL-/- mice displayed significantly greater levels of parasitemia, which peaked at day 28 pi and was positively detected until day 42 pi (Fig. 2A). In contrast, the maximum peak of parasitemia in WT mice was observed on day 21 pi, and these mice controlled the infection by day 42. Furthermore, mMGL-/- mice succumbed to T. cruzi infection as early as day 23 pi, and only 37% of them survived to day 42, whereas 80% of the WT mice survived throughout the infection (Fig. 2B).
mMGL-/- mice succumb to acute T. cruzi infection. WT and mMGL-/- mice were infected with 104 T. cruzi parasites. Parasitemia A), data shown represent the mean ±SE of at least 21 mice per data point corresponding to three independent experiments (7 mice per experiment). Survival rate B), data shown represent the mean ±SE of 15 mice corresponding to three independent experiments (5 mice per experiment). Both experimental groups were monitored every week until seven weeks p.i. *P‹0.05 with respect to WT. Student's t test, and log rank test for parasite load and survival were used respectively (Graph Pad Prism 6).
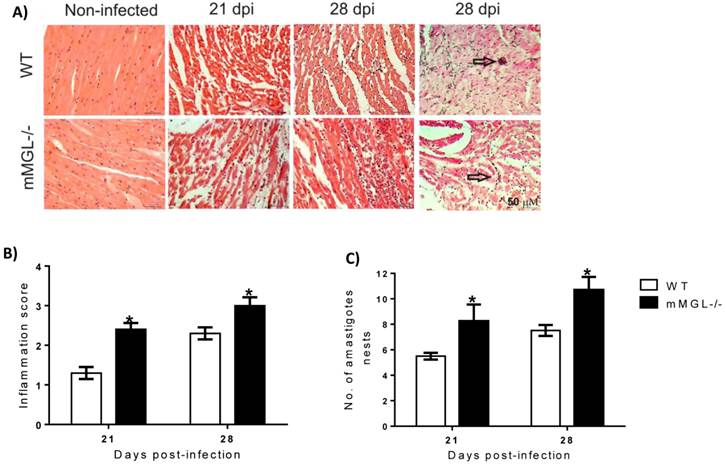
T. cruzi-infected mMGL-/- mice develop severe heart pathology. The heart is one of the main target organs affected by T. cruzi infection, therefore we evaluated whether the higher mortality of mMGL-/- mice was associated with heart damage. We found that on day 21 pi, the hearts of WT mice showed low tissue parasitism with moderate inflammatory mononuclear cell infiltration and no major histopathological signs of lesions (Fig. 3A at 21 and 28 days post infection -dpi and 3B). In contrast, heart histopathology sections from infected mMGL-/- mice showed an intense inflammatory reaction (Fig. 3A at 21 and 28 dpi and 3B), with a marked increase in amastigote nests (Fig. 3C) accompanied by severe heart injury due to large necrotic lesions. These results suggest that mMGL-/- mice succumb to the classical pathophysiology of the infection, which is typical of the severe acute phase of experimental Chagas' disease.
Hearts from mMGL-/- mice display higher T. cruzi parasitism and inflammation compared with WT mice. Histology of hearts post-T. cruzi infection A). Quantitative scoring of heart inflammation from H&E-stained tissue sections B). Number of amastigote nests in 25 histopathological fіelds C). Representative H&E images of heart tissue sections shown myocyte fibers cut in longitudinal (blue: nuclear, pink: muscle/cytoplasm/keratin) (arrows indicate parasite nests) (magnification: 40X, Olympus BX51 microscope). Data are expressed as the means ±SE of the means (n 4 to 5 animals per group). Mice were killed at 0, 21 and 28 days p.i. Arrows point to parasite nests. *P‹ 0.05 with respect to WT values obtained the same day. Student's t test was used (Graph Pad Prism 6).
mMGL-/- Mφ from T. cruzi infected-mice have impaired pro-inflammatory cytokine expression. Given that the inflammatory immune response is a well-defined mechanism to control early T. cruzi dissemination (16, 40), we decided investigate the pro-inflammatory profile of Mφ obtained from mMGL-/- and WT mice during acute T. cruzi infection. Peritoneal Mφ from uninfected and T. cruzi infected mice at 21 and 35 dpi were left untreated or treated for 48 hours with LPS, POLY:IC or TcAg, and supernatants were recovered for cytokine detection by ELISA. As expected, uninfected Mφ (day 0) from mMGL-/- or WT mice showed similar levels of NO, TNF-α, IL-12 and IL-10 in response to all stimuli (Fig. 4A-D). As the infection progressed, mMGL-/- Mφ left untreated exhibited lower levels of NO at 21 and 35 dpi (Fig. 4A) and lower levels of TNF-α, IL-12 and IL-10 at day 21 post-infection, compared with their similar WT Mφ (Figure 4A).
NO2-, TNF-α, and IL-12 production are reduced in response to LPS or TcAg in mMGL-/- Mφ compared with WT Mφ. Naïve or infected Mφ from WT or mMGL-/- mice were recovered after 21, 28 and 35 days post T. cruzi infection. Mφ were left untreated or treated for 48 hours with LPS (0.5 μg/ml), POLY: IC (25 μg/ml) or TcAg (25 μg/ml) as indicated. Supernatants were recovered and the levels of IL-12 A), TNF-α B), IFN-γ C) were measured by ELISA sandwich. Results are shown as the means of replicate samples +/- SEM and are representative of three experiments. *P˂0.05 by Student's t test (GraphPad Prism 6).
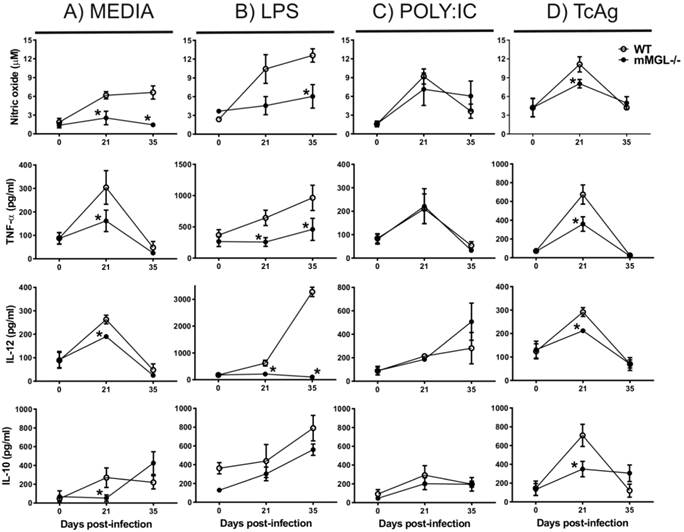
LPS is considered a Th1 activating signal that favors inflammatory cytokine production by Mφ (41). Here, we show that in response to LPS, mMGL-/- Mφ from 21 or 35 days after infection displayed impaired production of NO, TNF-α and IL-12 but not IL-10 compared with WT Mφ (Fig. 4B). In response to TcAg, mMGL-/- Mφ produced lower levels of NO, IL-12 and IL-10 only at 21 dpi (Fig. 4D), whereas no differences were observed with POLY:IC stimulation (Fig. 4C). These results indicate that mMGL-/- Mφ coming from T. cruzi infected animals may less activated, or may display an alternative activation Mφ (AAMφ) phenotype.
mMGL-/- Mφ from 21 days post-T. cruzi infection display a defect in their activation more than a phenotype switch. Signals encountered by developing Mφ during migration determine the development of highly divergent Mφ phenotypes with specific functional properties at the site of inflammation or infection (42). Classically activated (CA)Mφ, activated by Th1-type signals such as IFN-γ, produce high levels of nitric oxide (NO), enhancing antimicrobial and cytotoxic properties (43, 44). AAMφ are dependent on the products of activated Th2 cells, such as IL-4 and IL-13, and play important roles in allergy and the response to parasitic infection (45, 46). AAMφ express secretory lectin Ym-1 and Arginase-1 (Arg-1) over NO. Recently, mMGL has also been considered a marker for AAMφ elicited on Mφ and DCs during infection with Trypanosoma brucei or Taenia crassiceps (47, 48). In contrast, CAMφ produce high levels of NO from inducible nitric oxide synthase (iNOS), and they do not express Ym1 and Arg-1(41).
In order to determine whether the impaired inflammatory cytokine production in mMGL-/- Mφ was due to a phenotype switch from CAMφ to AAMφ, we analyzed the gene expression of Arg-1, Ym-1 and iNOS as Mφ phenotype markers, TNF-α and IL-1β as inflammatory markers, and TLR-2, TLR-3 and TLR-4 as activation markers. Peritoneal Mφ recovered from mMGL-/- and WT mice 21 days after infection and Mφ from non-infected mice were left untreated (media) or treated overnight with LPS, POLY:IC or TcAg for gene expression analysis by RT-PCR. We found that non-infected mMGL-/- Mφ showed high Arg-1, Ym-1 and TLR3 expression in response to LPS (Fig. 5 B, C and H). High Arg-1 and TLR3 expression was observed in response to TcAg (Fig. 5 B, H), but similar mRNA levels of TNF-α, iNOS, IL-1β, TLR-4 and TLR-2 in response to LPS compared with WT-Mφ (Fig. 5 D, E, F, G and I). No or low mRNA expression of Ym-1, IL-1β and TLR-4 was observed in response to TcAg compared with WT-Mφ (Fig. 5 C, F and G).
Mφ Phenotype from WT and mMGL-/- 21 days post-T. cruzi infection. Naïve or infected Mφ from WT or mMGL-/- mice were recovered 21 days after T. cruzi infection. Mφ were left untreated or treated for 24 hours with LPS (5 μg/ml), POLY: IC (25 μg/ml) or TcAg (25 μg/ml), as indicated. The cells were recovered for RNA expression analysis A) of Arginase1 B), Ym1 C), TNF-α D), iNOS E), IL-1β F), TLR-4 G), TLR-3 H) and TLR-2 I) by RT-PCR. Results are shown as the mean of triplicate samples (+/- S.E.M.) and are representative of three independent experiments. *P˂0.05 by Student's t test (GraphPad Prism 6).
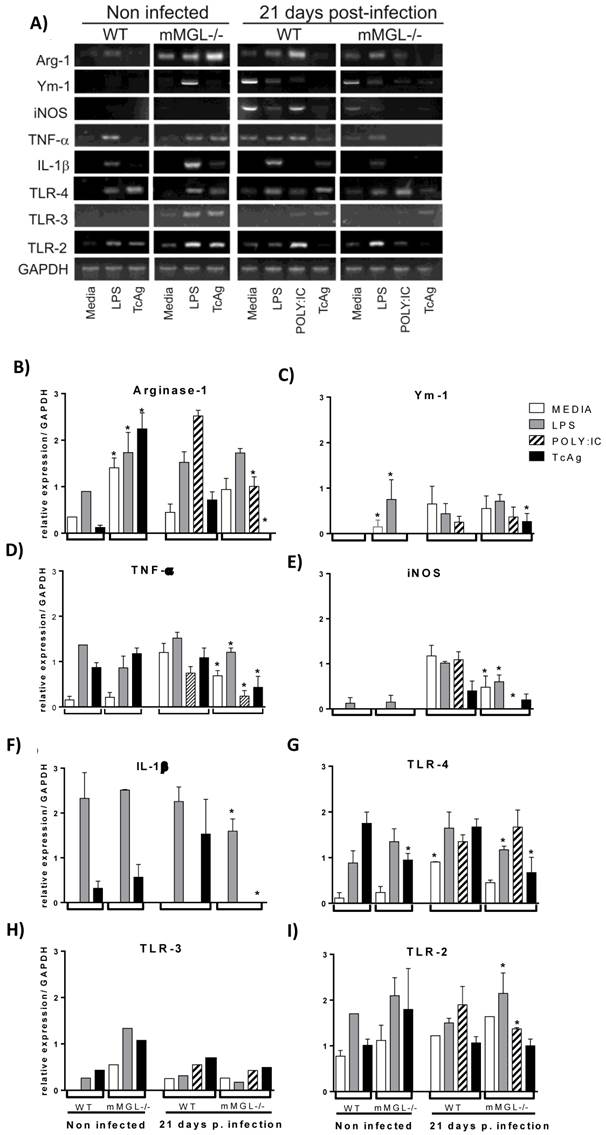
Untreated (media) mMGL-/- Mφ from 21 dpi showed similar expression of Arg-1, Ym-1, IL-1β, TLR3 and TLR2 (Fig. 5 B, C, F, H, and I -media) but low expression of iNOS, TNF-α and TLR-4 in these basal condition (Fig 5 D, E, G -media). In response to LPS, 21 day post-infection Mφ showed similar mRNA expression of Arg-1 and Ym-1 (Fig 5B, C) but low mRNA expression of iNOS, TNF-α, IL-1β and TLR-4 (Fig. 5 D, E, F, G). In response to POLY:IC, low mRNA expression of Arg-1, iNOS and TNF-α was observed (Fig. 5B, E, D, F and G -21 dpi). In response to TcAg, mMGL-/- Mφ showed high Ym-1 expression (Fig. 5C -21 dpi), similar levels of iNOS, TLR3 and TLR-2 (Fig. 5E, H and I -21 dpi), and no or low levels of Arg-1, TNF-α, IL-1β and TLR-4 compared with WT Mφ (Fig. 5B, D, F, and G. -21 dpi).
mMGL-/- Mφ display deficiencies in MHC-II and TLR-4 cell surface activation markers during in vitro infection with T. cruzi. In order to rule out the possibility that altered activation status could be present in mMGL-/- Mφ, we studied the surface expression of MHC class II (MHC-II), co-stimulatory molecules CD80 (B7.1), CD86 (B7.2) and TLR-4. Peritoneal naïve Mφ from WT or mMGL-/- mice were infected in vitro with trypomastigotes of T. cruzi at a ratio of 1:10 and marker expression was monitored in uninfected Mφ (0 hpi) and T. cruzi-infected Mφ at 2, 6, 12 and 24 hpi. Uninfected mMGL-/- Mφ expressed similar levels of MHC-II (Fig. 6A, 0 hpi), CD80 (data not shown), CD86 (data not-shown) and TLR-4 (Fig. 6B, 0 hpi).
Effect of T. cruzi infection on the cell surface activation markers of WT and mMGL-/- Mφ. Naïve Mφ from WT or mMGL-/- mice were infected in vitro with trypomastigotes of T. cruzi at 10:1parasite to Mφ ratio. The cells were recovered and double-stained for F4/80 and MHC-II A), and TLR-4 B). Flow cytometry graphs show dot plots of F4/80-gated Mφ. Results are representative of three experiments. *P˂0.05 by Student's t test (GraphPad Prism 6).
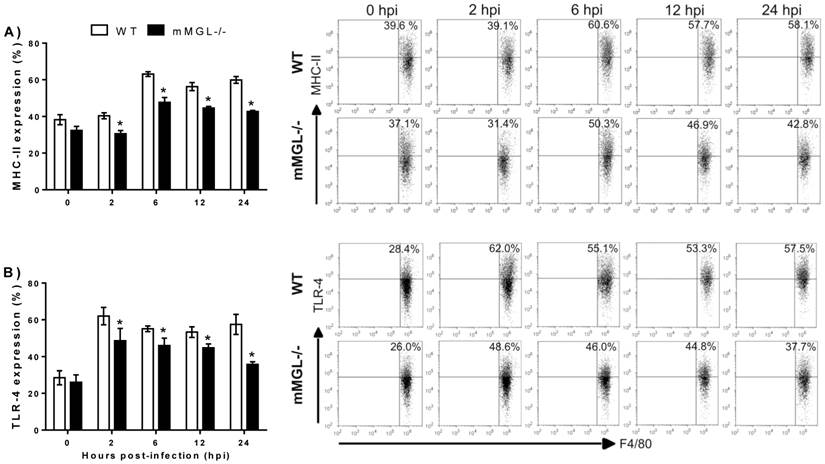
Upon T. cruzi infection, the percentage of all markers was increased in both mMGL-/- and WT-Mφ from two hours until 24 hpi. Similar expression levels of CD80 and CD86 were observed, but significantly less expression of MHC-II and TLR-4 was observed on mMGL-/- Mφ at 2, 6, 12 and 24 hpi compared with WT Mφ (Fig. 6 A and B, respectively).
Next, we asked whether on the altered activation of mMGL-/- Mφ could reflect their ability to control T. cruzi infection. To address this question, T. cruzi epimastigotes were labeled with CFSE and used to infect Mφ from healthy mMGL-/- and WT mice in vitro, then Mφ were recovered and analyzed by flow cytometry. This allowed the comparison of parasite uptake between mMGL-/- and WT Mφ. Interestingly mMGL-/- Mφ harbored significantly more parasites after 2, 6, 12 and 24 hours post-infection (hpi) (Fig. 7 A) than their WT counterparts, which suggested that the presence of mMGL is important in delaying the entry of parasites into the host cell.
Untreated, IFN-γ and LPS/IFN-γ primed mMGL-/- Mφ take up most Trypanosoma cruzi parasites. Peritoneal derived WT or mMGL-/- Mφ were untreated A) or treated 24 h with IFN-γ or LPS/IFN-γ Β), followed by infection with T. cruzi at 10:1 parasite to Mφ ratio. T. cruzi-CFSE-labelling that were not taken up were washed off 2 hours post infection, and parasite uptake determined by flow cytometry at 0, 2, 6 and 12 hours post infection A), or 0 and 2 hours post infection B). Flow cytometry graphs show dot plots of F4/80-gated Mφ. Results are representative of three experiments to A) and two experiments to B). *P˂0.05 by Student's t test (GraphPad Prism 6).
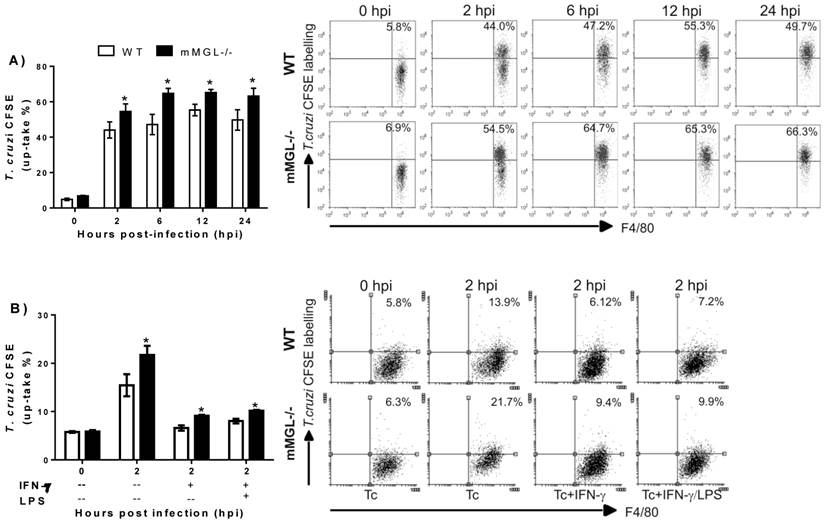
To investigate whether the Th1 activating signals, like IFN-γ or IFN-γ/LPS could alter the mMGL-/- Mφ activation. Thioglycollate-elicited Mφ mMGL-/- and WT were left untreated o treated 24 h with IFN-γ or LPS/IFN-γ, infected with CFSE labeled epimastigotes of T. cruzi and analyzed by cytometry. As expected, both stimulus IFN-γ and LPS/ IFN-γ act to reduce the parasite uptake on WT as mMGL-/- Mφ. However, this effect was significantly lesser on mMGL-/- Mφ, which showed higher number of parasites compared with WT Mφ (Fig. 7 B). This result demonstrates that mMGL is important to IFN-γ or LPS/IFN-γ stimulus works to activate efficiently Mφ.
Discussion
New evidence has recently emerged about the role of CLRs in modulating the activation and function of APC (18, 49, 50). Some of these studies suggest that MGL improves DC antigen presentation and triggers distinct signaling pathways that regulate APC functions through the expression of specific molecules and cytokines that modulate the innate and adaptive immune response (24, 51). This hypothesis has been driven by the knowledge that MGL works as a molecular target for Gal/GalNAc or Tn-carrying tumor-associated antigens on DCs (24, 25, 52) and is supported by subsequent data showing that mMGL recognizes Gal/GlalNAc residues carried by glycoproteins and/or glycosphingolipids present on the helminth Schistosoma mansoni inducing phagocytosis, endocytosis, enhancing parasite glycopeptide presentation and promoting the polarization to Th2 responses (27, 53). Despite evidence pointing to a role for MGL in the immune response to these parasites, the function of MGL in the host defense against protozoa or other helminth parasites rich in Gal/GalNAc residues has not been determined. Therefore, in this study, we evaluated the in vivo role of mMGL in T. cruzi infection using MGL-/- and WT mice.
First, we demonstrated by Jacalin-Peroxidase lectin blotting that a pathogenic T. cruzi strain (Queretaro) has Gal residues on its membrane antigens that can be recognized by mMGL receptors on APCs. This observation is in line with previous reports from other T. cruzi strains which demonstrated that this parasite displays highly glycosylated membrane proteins related to the invasion of host cells through recognition by CLRs (31, 54), mainly DEC205 and SIGN (18, 55). However, the role of CLRs in vivo has not been determined. Here, we used mMGL-/- mice to demonstrate for the first time that the CLR mMGL plays a critical role in the recognition of and resistance to a protozoan infection. We showed that mMGL-/- mice are highly susceptible to experimental T. cruzi infection, harboring prolonged and higher parasitemia levels, severe cardiac immunopathology and increased mortality. These findings indicate that mMGL plays a major role in the host defense against acute T. cruzi infection.
Mφ, specifically CAMφ, mediate microbial destruction and play an important role in controlling parasite replication during the acute phase of T. cruzi infection by enhancing their microbicidal activity by increasing NO and the proinflammatory cytokines IL-12, TNF-α and MIF (56, 57). We showed that the peritoneal Mφ population generated at 21 days after T. cruzi infection in mMGL-/- mice produced significantly less NO, TNF-α, IL-12 and IL-10. Moreover, this Mφ population did not respond as expected to ex vivo proinflammatory stimuli (LPS) or to TcAg, but they did respond efficiently to POLY:IC. The lack of proinflammatory cytokines and NO production in early infection in response to ex vivo stimuli with LPS or TcAg in mMGL-/- Mφ could be explained by the following: 1) The development of different Mφ phenotypes in mMGL-/- compared with WT infected T. cruzi mice. However, this possibility was discarded because we did not observe a phenotype switch in mMGL-/- Mφ. 2) Inadequate Mφ activation that could be dependent on a surface receptor because with POLY:IC stimuli, a ligand to intracellular TLR-3, the mMGL-/- Mφ produced similar levels of cytokines compared with WT Mφ. Thus, mMGL may be required for the efficient production of NO, TNF-α, IL-12 and IL-10 in innate immunity against T. cruzi infection.
These observations suggest there was no clear phenotype switch in mMGL-/- Mφ toward an AAM profile. However, our data may reflect a less mature activation state in mMGL-/- Mφ, independently of whether the Mφ come from T. cruzi infected mice, which suggests that mMGL-/- Mφ may require signaling through a pathogen recognition receptor such as mMGL before becoming responsive to LPS or TcAg.
The current model states that ligand binding to CLRs elicits signaling cascades that modulate immune responses. Some CLRs, such as Dectin-1 and CLEC9A/DNGR-1, clearly promote immunity through ITAM-like motifs within their cytoplasmic tails, leading to the production of several inflammatory cytokines (49). Strikingly, DC-SIGN and DCIR do not seem to act individually, as their signaling pathways require co-triggering of a TLR molecule for their effects to become apparent. Also, MGL induces IL-10 secretion after antibody crosslinking stimulation (58) and has been reported to trigger the phosphorylation of extracellular signal-regulated kinase 1, 2 (ERK1, 2) and nuclear factor-κB activation. However, this finding has not been tested with “natural ligands” but with strong activation such as direct antibody stimulation.
On the other hand, MGL engagement improved DC performance as antigen-presenting cells, promoting the up-regulation of maturation markers (HLAII-DR, CD83, CD86, CD40), enhancing motility, and increasing antigen-specific CD8+ activation (25). Recently, the capacity of MGL to modulate TLR-2 signaling has been reported (51). In accordance with this, we showed that upon in vitro T. cruzi infection, mMGL-/- Mφ displayed significantly less expression of MHC-II and TLR-4. This is consistent with previous studies showing that MCH-II and TLR-4 expression is essential to develop “activated” Mφ with the ability to kill T. cruzi parasites (10). Because TLR4-mediated responses to T. cruzi have been reported as one of the main pathways for inducing early cytokine production (12), the low expression levels of TLR-4 on mMGL-/- Mφ may explain the low proinflammatory cytokine production by these cells. It is possible that mMGL engagement may couple to TLR-4 signal transduction for increased TNF-α and IL-1β secretion by Mφ during T. cruzi infection. Consistent with this, mMGL-/- Mφ were infected more than WT Mφ. This defect on mMGL-/- Mφ was persistent even when mMGL-/- Mφ were previously primed with IFN-γ or LPS/IFN-γ. The ability of WT Mφ under IFN-γ or IFN-γ/LPS stimulation are able to control parasite number more effectively than mMGL-/- Mφ may be explained by signals that WT Mφ encounter with T. cruzi or IFN-γ/LPS causing the upregulation of TLR4. Interestingly, no changes were observed in CD80, CD86, TLR-3 and TLR-2 expression, which suggests that the role of mMGL in upregulating costimulatory molecules may depend on the type of triggering ligand and the time of exposure.
These results, together with the finding that mMGL-/- mice were more susceptible to T. cruzi infection, suggest that mMGL is required for optimal Mφ activation and may synergize with TLR-4-induced pathways to produce TNF-α, IL-1β and NO during the early phase of T. cruzi infection. These observations contribute to the understanding of the inflammatory properties of the mMGL molecule, pointing to its potential role as an important modulator of the immune response during T. cruzi infection, and perhaps in other parasitic diseases. Moreover, mMGL may act as a CLR that plays a critical role in determining the quality of the adaptive immune response to this parasite. A better characterization of the effects of mMGL on APCs involved in innate and adaptive immunity in response to parasitic diseases is therefore of great interest.
Acknowledgements
The authors would like to thank MVZ Leticia Flores for the excellent care of the animals and Thalia Pacheco and Pablo F. Moreno for assistance with the histology pictures. We also thank the awards program for academic staff (PASPA-UNAM-2013) for partially supporting the academic exchange with Ohio State University, USA for MRS, and CONACYT-México for supporting a PhD fellowship for the first author to obtain her degree in Biomedical Sciences, UNAM. This work was supported by grant 152224 from CONACYT, 2012-23 from PAPCA-FESI 2011-12, and IN212412 from UNAM-DGAPA-PAPIIT.
Conflict of Interests
The authors declare that there is no conflict of interests.
References
1. Carabarin-Lima A, Gonzalez-Vazquez MC, Rodriguez-Morales O, Baylon-Pacheco L, Rosales-Encina JL, Reyes-Lopez PA. et al. Chagas disease (American trypanosomiasis) in Mexico: an update. Acta Trop. 2013;127(2):126-35
2. Barrias ES, de Carvalho TM, De Souza W. Trypanosoma cruzi: Entry into Mammalian Host Cells and Parasitophorous Vacuole Formation. Front Immunol. 2013;4:186
3. Teixeira SM, Vieira LQ, Gazzinelli RT. Genomics, pathogenesis and control of infection with protozoan parasites. Trends Parasitol. 2002;18(2):52-4
4. Junqueira C, Caetano B, Bartholomeu DC, Melo MB, Ropert C, Rodrigues MM. et al. The endless race between Trypanosoma cruzi and host immunity: lessons for and beyond Chagas disease. Expert Rev Mol Med. 2010;12:e29
5. Davicino RC, Elicabe RJ, Di Genaro MS, Rabinovich GA. Coupling pathogen recognition to innate immunity through glycan-dependent mechanisms. Int Immunopharmacol.
6. Gazzinelli RT, Denkers EY. Protozoan encounters with Toll-like receptor signalling pathways: implications for host parasitism. Nat Rev Immunol. 2006;6(12):895-906
7. Camargo MM, Andrade AC, Almeida IC, Travassos LR, Gazzinelli RT. Glycoconjugates isolated from Trypanosoma cruzi but not from Leishmania species membranes trigger nitric oxide synthesis as well as microbicidal activity in IFN-gamma-primed macrophages. J Immunol. 1997;159(12):6131-9
8. Camargo MM, Almeida IC, Pereira ME, Ferguson MA, Travassos LR, Gazzinelli RT. Glycosylphosphatidylinositol-anchored mucin-like glycoproteins isolated from Trypanosoma cruzi trypomastigotes initiate the synthesis of proinflammatory cytokines by macrophages. J Immunol. 1997;158(12):5890-901
9. Talvani A, Ribeiro CS, Aliberti JC, Michailowsky V, Santos PV, Murta SM. et al. Kinetics of cytokine gene expression in experimental chagasic cardiomyopathy: tissue parasitism and endogenous IFN-gamma as important determinants of chemokine mRNA expression during infection with Trypanosoma cruzi. Microbes Infect. 2000;2(8):851-66
10. Campos MA, Almeida IC, Takeuchi O, Akira S, Valente EP, Procopio DO. et al. Activation of Toll-like receptor-2 by glycosylphosphatidylinositol anchors from a protozoan parasite. J Immunol. 2001;167(1):416-23
11. Oliveira AC, Peixoto JR, de Arruda LB, Campos MA, Gazzinelli RT, Golenbock DT. et al. Expression of functional TLR4 confers proinflammatory responsiveness to Trypanosoma cruzi glycoinositolphospholipids and higher resistance to infection with T. cruzi. J Immunol. 2004;173(9):5688-96
12. Medeiros MM, Peixoto JR, Oliveira AC, Cardilo-Reis L, Koatz VL, Van Kaer L. et al. Toll-like receptor 4 (TLR4)-dependent proinflammatory and immunomodulatory properties of the glycoinositolphospholipid (GIPL) from Trypanosoma cruzi. J Leukoc Biol. 2007;82(3):488-96
13. Shoda LK, Kegerreis KA, Suarez CE, Roditi I, Corral RS, Bertot GM. et al. DNA from protozoan parasites Babesia bovis, Trypanosoma cruzi, and T. brucei is mitogenic for B lymphocytes and stimulates macrophage expression of interleukin-12, tumor necrosis factor alpha, and nitric oxide. Infect Immun. 2001;69(4):2162-71
14. Campos MA, Gazzinelli RT. Trypanosoma cruzi and its components as exogenous mediators of inflammation recognized through Toll-like receptors. Mediators Inflamm. 2004;13(3):139-43
15. Bafica A, Santiago HC, Goldszmid R, Ropert C, Gazzinelli RT, Sher A. Cutting edge: TLR9 and TLR2 signaling together account for MyD88-dependent control of parasitemia in Trypanosoma cruzi infection. J Immunol. 2006;177(6):3515-9
16. Campos MA, Closel M, Valente EP, Cardoso JE, Akira S, Alvarez-Leite JI. et al. Impaired production of proinflammatory cytokines and host resistance to acute infection with Trypanosoma cruzi in mice lacking functional myeloid differentiation factor 88. J Immunol. 2004;172(3):1711-8
17. Ropert C, Gazzinelli RT. Regulatory role of Toll-like receptor 2 during infection with Trypanosoma cruzi. J Endotoxin Res. 2004;10(6):425-30
18. Vazquez-Mendoza A, Carrero JC, Rodriguez-Sosa M. Parasitic infections: a role for C-type lectins receptors. Biomed Res Int. 2013;2013:456352
19. Engering A, Geijtenbeek TB, van Vliet SJ, Wijers M, van Liempt E, Demaurex N. et al. The dendritic cell-specific adhesion receptor DC-SIGN internalizes antigen for presentation to T cells. J Immunol. 2002;168(5):2118-26
20. Brown GD, Herre J, Williams DL, Willment JA, Marshall AS, Gordon S. Dectin-1 mediates the biological effects of beta-glucans. J Exp Med. 2003;197(9):1119-24
21. Chakraborty P, Ghosh D, Basu MK. Modulation of macrophage mannose receptor affects the uptake of virulent and avirulent Leishmania donovani promastigotes. J Parasitol. 2001;87(5):1023-7
22. Svajger U, Anderluh M, Jeras M, Obermajer N. C-type lectin DC-SIGN: an adhesion, signalling and antigen-uptake molecule that guides dendritic cells in immunity. Cell Signal. 2010;22(10):1397-405
23. Lloyd DH, Viac J, Werling D, Reme CA, Gatto H. Role of sugars in surface microbe-host interactions and immune reaction modulation. Vet Dermatol. 2007;18(4):197-204
24. Geijtenbeek TB, Gringhuis SI. Signalling through C-type lectin receptors: shaping immune responses. Nat Rev Immunol. 2009;9(7):465-79
25. Napoletano C, Zizzari IG, Rughetti A, Rahimi H, Irimura T, Clausen H. et al. Targeting of macrophage galactose-type C-type lectin (MGL) induces DC signaling and activation. Eur J Immunol. 2012;42(4):936-45
26. Kingeter LM, Lin X. C-type lectin receptor-induced NF-kappaB activation in innate immune and inflammatory responses. Cell Mol Immunol. 2012;9(2):105-12
27. van Vliet SJ, van Liempt E, Saeland E, Aarnoudse CA, Appelmelk B, Irimura T. et al. Carbohydrate profiling reveals a distinctive role for the C-type lectin MGL in the recognition of helminth parasites and tumor antigens by dendritic cells. Int Immunol. 2005;17(5):661-9
28. Yuita H, Tsuiji M, Tajika Y, Matsumoto Y, Hirano K, Suzuki N. et al. Retardation of removal of radiation-induced apoptotic cells in developing neural tubes in macrophage galactose-type C-type lectin-1-deficient mouse embryos. Glycobiology. 2005;15(12):1368-75
29. van Vliet SJ, Aarnoudse CA, Broks-van den Berg VC, Boks M, Geijtenbeek TB, van Kooyk Y. MGL-mediated internalization and antigen presentation by dendritic cells: a role for tyrosine-5. Eur J Immunol. 2007;37(8):2075-81
30. Tsuiji M, Fujimori M, Ohashi Y, Higashi N, Onami TM, Hedrick SM. et al. Molecular cloning and characterization of a novel mouse macrophage C-type lectin, mMGL2, which has a distinct carbohydrate specificity from mMGL1. J Biol Chem. 2002;277(32):28892-901
31. Higashi N, Fujioka K, Denda-Nagai K, Hashimoto S, Nagai S, Sato T. et al. The macrophage C-type lectin specific for galactose/N-acetylgalactosamine is an endocytic receptor expressed on monocyte-derived immature dendritic cells. J Biol Chem. 2002;277(23):20686-93
32. Singh SK, Streng-Ouwehand I, Litjens M, Weelij DR, Garcia-Vallejo JJ, van Vliet SJ. et al. Characterization of murine MGL1 and MGL2 C-type lectins: distinct glycan specificities and tumor binding properties. Mol Immunol. 2009;46(6):1240-9
33. Lopez-Olmos V, Perez-Nasser N, Pinero D, Ortega E, Hernandez R, Espinoza B. Biological characterization and genetic diversity of Mexican isolates of Trypanosoma cruzi. Acta Trop. 1998;69(3):239-54
34. Markwell MA, Haas SM, Bieber LL, Tolbert NE. A modification of the Lowry procedure to simplify protein determination in membrane and lipoprotein samples. Anal Biochem. 1978;87(1):206-10
35. Onami TM, Lin MY, Page DM, Reynolds SA, Katayama CD, Marth JD. et al. Generation of mice deficient for macrophage galactose- and N-acetylgalactosamine-specific lectin: limited role in lymphoid and erythroid homeostasis and evidence for multiple lectins. Mol Cell Biol. 2002;22(14):5173-81
36. Laird PW, Zijderveld A, Linders K, Rudnicki MA, Jaenisch R, Berns A. Simplified mammalian DNA isolation procedure. Nucleic Acids Res. 1991;19(15):4293
37. Gupta S, Garg NJ. TcVac3 induced control of Trypanosoma cruzi infection and chronic myocarditis in mice. PLoS One. 2013;8(3):e59434
38. Ropert C, Ferreira LR, Campos MA, Procopio DO, Travassos LR, Ferguson MA. et al. Macrophage signaling by glycosylphosphatidylinositol-anchored mucin-like glycoproteins derived from Trypanosoma cruzi trypomastigotes. Microbes Infect. 2002;4(9):1015-25
39. Sastry MV, Banarjee P, Patanjali SR, Swamy MJ, Swarnalatha GV, Surolia A. Analysis of saccharide binding to Artocarpus integrifolia lectin reveals specific recognition of T-antigen (beta-D-Gal(1----3)D-GalNAc). J Biol Chem. 1986;261(25):11726-33
40. Kumar S, Tarleton RL. Antigen-specific Th1 but not Th2 cells provide protection from lethal Trypanosoma cruzi infection in mice. J Immunol. 2001;166(7):4596-603
41. Mylonas KJ, Nair MG, Prieto-Lafuente L, Paape D, Allen JE. Alternatively activated macrophages elicited by helminth infection can be reprogrammed to enable microbial killing. J Immunol. 2009;182(5):3084-94
42. Espinoza-Jimenez A, Peon AN, Terrazas LI. Alternatively activated macrophages in types 1 and 2 diabetes. Mediators Inflamm. 2012;2012:815953
43. Tomioka H, Tatano Y, Maw WW, Sano C, Kanehiro Y, Shimizu T. Characteristics of suppressor macrophages induced by mycobacterial and protozoal infections in relation to alternatively activated M2 macrophages. Clin Dev Immunol. 2012;2012:635451
44. Arnold CE, Whyte CS, Gordon P, Barker RN, Rees AJ, Wilson HM. A critical role for SOCS3 in promoting M1 macrophage activation and function in vitro and in vivo. Immunology. 2013
45. Van Dyken SJ, Locksley RM. Interleukin-4- and interleukin-13-mediated alternatively activated macrophages: roles in homeostasis and disease. Annu Rev Immunol. 2013;31:317-43
46. El Fiky A, Perreault R, McGinnis GJ, Rabin RL. Attenuated expression of interferon-beta and interferon-lambda1 by human alternatively activated macrophages. Hum Immunol. 2013
47. Terrazas CA, Alcantara-Hernandez M, Bonifaz L, Terrazas LI, Satoskar AR. Helminth-excreted/secreted products are recognized by multiple receptors on DCs to block the TLR response and bias Th2 polarization in a cRAF dependent pathway. FASEB J. 2013;27(11):4547-60
48. Raes G, Brys L, Dahal BK, Brandt J, Grooten J, Brombacher F. et al. Macrophage galactose-type C-type lectins as novel markers for alternatively activated macrophages elicited by parasitic infections and allergic airway inflammation. J Leukoc Biol. 2005;77(3):321-7
49. Sancho D, Reis e Sousa C. Signaling by myeloid C-type lectin receptors in immunity and homeostasis. Annu Rev Immunol. 2012;30:491-529
50. Lang R, Schoenen H, Desel C. Targeting Syk-Card9-activating C-type lectin receptors by vaccine adjuvants: findings, implications and open questions. Immunobiology. 2011;216(11):1184-91
51. van Vliet SJ, Bay S, Vuist IM, Kalay H, Garcia-Vallejo JJ, Leclerc C. et al. MGL signaling augments TLR2-mediated responses for enhanced IL-10 and TNF-alpha secretion. J Leukoc Biol. 2013;94(2):315-23
52. Napoletano C, Rughetti A, Agervig Tarp MP, Coleman J, Bennett EP, Picco G. et al. Tumor-associated Tn-MUC1 glycoform is internalized through the macrophage galactose-type C-type lectin and delivered to the HLA class I and II compartments in dendritic cells. Cancer Res. 2007;67(17):8358-67
53. van Liempt E, van Vliet SJ, Engering A, Garcia Vallejo JJ, Bank CM, Sanchez-Hernandez M. et al. Schistosoma mansoni soluble egg antigens are internalized by human dendritic cells through multiple C-type lectins and suppress TLR-induced dendritic cell activation. Mol Immunol. 2007;44(10):2605-15
54. van Vliet SJ, Saeland E, van Kooyk Y. Sweet preferences of MGL: carbohydrate specificity and function. Trends Immunol. 2008;29(2):83-90
55. Stafford JL, Neumann NF, Belosevic M. Macrophage-mediated innate host defense against protozoan parasites. Crit Rev Microbiol. 2002;28(3):187-248
56. Brener Z, Gazzinelli RT. Immunological control of Trypanosoma cruzi infection and pathogenesis of Chagas' disease. Int Arch Allergy Immunol. 1997;114(2):103-10
57. Reyes JL, Terrazas LI, Espinoza B, Cruz-Robles D, Soto V, Rivera-Montoya I. et al. Macrophage migration inhibitory factor contributes to host defense against acute Trypanosoma cruzi infection. Infect Immun. 2006;74(6):3170-9
58. Li D, Romain G, Flamar AL, Duluc D, Dullaers M, Li XH. et al. Targeting self- and foreign antigens to dendritic cells via DC-ASGPR generates IL-10-producing suppressive CD4+ T cells. J Exp Med. 2012;209(1):109-21
Author contact
![]() Corresponding author: Miriam Rodríguez-Sosa, Unidad de Biomedicina, FES-Iztacala, UNAM. Av. de los Barrios # 1, Los Reyes Iztacala, 54090 Tlalnepantla, Edo. de México. Mexico. Phone: (52-55) 5623-1333 ext. 39789. Fax: (52-55) 5623-1225. Email: rodriguezmiztacala.unam.mx.
Corresponding author: Miriam Rodríguez-Sosa, Unidad de Biomedicina, FES-Iztacala, UNAM. Av. de los Barrios # 1, Los Reyes Iztacala, 54090 Tlalnepantla, Edo. de México. Mexico. Phone: (52-55) 5623-1333 ext. 39789. Fax: (52-55) 5623-1225. Email: rodriguezmiztacala.unam.mx.