Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
The production of EPO and the...
EPO/EpoR system activity in...
EPO/EpoR in pancreatic beta cells
EPO/EpoR in skeletal muscle
EPO/EpoR in heart
EPO/EpoR and neural system
EPO and energy metabolism...
The crosstalk between EPO and...
The protective role of EPO in...
Conclusion
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2014; 10(8):921-939. doi:10.7150/ijbs.9518 This issue Cite
Review
Erythropoietin, a Novel Versatile Player Regulating Energy Metabolism beyond the Erythroid System
Li Wang1 ![]() , Lijun Di1
, Lijun Di1 ![]() , Constance Tom Noguchi2
, Constance Tom Noguchi2
1. Faculty of Health Sciences, University of Macau, SAR of People's Republic of China
2. Molecular Medicine Branch, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, Bethesda, MD 20892, U.S.A
Received 2014-4-27; Accepted 2014-6-4; Published 2014-8-23
Abstract
Erythropoietin (EPO), the required cytokine for promoting the proliferation and differentiation of erythroid cells to stimulate erythropoiesis, has been reported to act as a pleiotropic cytokine beyond hematopoietic system. The various activities of EPO are determined by the widespread distribution of its cell surface EPO receptor (EpoR) in multiple tissues including endothelial, neural, myoblasts, adipocytes and other cell types. EPO activity has been linked to angiogenesis, neuroprotection, cardioprotection, stress protection, anti-inflammation and especially the energy metabolism regulation that is recently revealed. The investigations of EPO activity in animals and the expression analysis of EpoR provide more insights on the potential of EPO in regulating energy metabolism and homeostasis. The findings of crosstalk between EPO and some important energy sensors and the regulation of EPO in the cellular respiration and mitochondrial function further provide molecular mechanisms for EPO activity in metabolic activity regulation. In this review, we will summarize the roles of EPO in energy metabolism regulation and the activity of EPO in tissues that are tightly associated with energy metabolism. We will also discuss the effects of EPO in regulating oxidative metabolism and mitochondrial function, the interactions between EPO and important energy regulation factors, and the protective role of EPO from stresses that are related to metabolism, providing a brief overview of previously less appreciated EPO biological function in energy metabolism and homeostasis.
Keywords: Erythropoietin, pleiotropic cytokine, erythroid system
Introduction
EPO, the important cytokine, is required and necessary for the growth, survival and differentiation of red blood progenitor cells. EPO exerts its physiological role by binding to its specific cell surface receptor (EpoR). The mice lacking EPO or EpoR are embryonic lethal in utero due to severe anemia, suggesting its primary role of EPO in the regulation of red cell production [1]. Human EPO was first purified in 1977 and cloned in 1985 [2-4], which lead to the production of recombinant EPO. For the past two decades, human recombinant EPO has been widely used clinically in the treatment of anemia and associated conditions including chronic kidney disease and chemotherapeutic cancer patients. However, the reason for EPO attracts more researchers' attention is not only its primary role in erythropoiesis but also a range of actions of EPO in nonhematopoietic system. The discovery of expression of EPO and EpoR in a number of nonhematopoietic tissues provides the strong support for the various biological function of EPO. EPO production has been detected in the brain, retina, reproductive tract, and skeletal muscle myoblasts. EpoR even can be detected in more widespread tissues including brain, endothelial cells, retina, muscle progenitor cells, lung, heart, liver, myocardium, adipocytes, macrophages and pancreas [5-7]. Importantly, EPO and EpoR are both hypoxia inducible, suggesting the potential protective role of EPO in dealing with stress. Accumulating evidences have demonstrated the neuroprotective activity during hypoxic conditions, antiapoptotic effect and increased angiogenesis properties of EPO. Consistently, EPO and EpoR knockout mice also show impaired angiogenesis and defective brain and heart development [8-10]. More recently, some new findings promote people to realize that EPO may act as a novel regulator of energy homeostasis. For example, the mice with EpoR restricted in erythroid system exhibited significant increase in body weight gain due to increased fat mass and a decrease in total activity and energy expenditure compared with Wild type (WT) mice [6]. These mice were also glucose intolerant and became insulin resistant with the development of obesity [6]. EPO treatment can protect against diabetes development in db/db mouse model (a model of obesity, diabetes, and dyslipidemia wherein leptin receptor activity is deficient) and streptozotocin-induced type 1 and type 2 diabetes mouse models via the direct antiapoptotic, anti-inflammatory, and angiogenic effects within the islets [7]. The loss of EpoR in adipocytes negatively regulates mitochondrial function of adipocytes and leads to obese mice with decreased energy expenditure [11]. In contrast, EPO administration in wild type mice protects the mice from high fat induced obesity and improves glucose intolerance and insulin resistance [6]. Moreover, the crosstalk between EPO/EpoR and the important energy sensors such as PGC-1α, Sirt1 and AMPK also provides evidence for EPO acting as an energy metabolism regulator [11-14]. In this review, we will discuss the important activity of EPO in nonhematopoietic system especially to highlight the tissue protective role and metabolic activity regulation function of EPO. We will also describe the novel activity of EPO in the metabolism related diseases including obesity, metabolism disorders and diabetes and the involvement of EPO in the energy sensing network.
The production of EPO and the expression of EpoR in different tissues
EPO protein and tissue specific production
EPO is a glycoprotein hormone and a single polypeptide of 166 amino acids folded into four α-helix with two dissulphide brides between cysteines 6 and 161 and between cysteines 29 and 33 [3, 4, 15]. EPO shares structural homology with growth hormone and other members of the hematopoietic cytokine superfamily. In erythroid progenitors, EPO binds to its cell surface receptor (EpoR) to activate the JAK2 and downstream signal transduction pathways including STAT5, PI3K and MAPK [16, 17]. EPO/EpoR is required for erythroid progenitor survival, proliferation and differentiation. Definitive erythropoiesis takes place in the fetal liver during development. Therefore, the major place of EPO production is fetal liver. In humans, EPO is also expressed in kidney begins from about 18 weeks before birth and then increases dramatically after 30 weeks of gestation. The EPO producing cells change from central veins surrounding hepatocytes to peritublar interstitial cells with neural characteristics [18-20]. After birth, the kidney becomes the principal EPO producing organ with the site of hematopoiesis switches to the bone marrow. However, EPO expression in liver is inducible and can be induced to 20%-50% of total body EPO mRNA by hypoxia in mice [21, 22]. In addition, in adult kidney, the EPO is also inducible. Hypoxia inducible factor (HIF) plays a primary regulatory role in hypoxia induced EPO expression in both liver and kidney although the regulation of EPO expression in the liver and kidney requires different flanking regulatory elements, which are located 3' for liver expression and 5' for kidney expression, respectively [23-25].
Besides the kidney and fetal liver, EPO production is also detected in the brain, female organs and skeletal muscle myoblasts. In the brain, EPO is produced by astrocytes and neurons [26-28]. EPO production in the brain is also hypoxia-inducible [27, 29]. In the human brain, EPO is observed in the fetal CNS and in the spinal fluid of normal preterm and term infants [30, 31]. EPO production in female reproductive organs can be stimulated by estrogen and contributes to the regulation of the cyclic changes in female reproductive organs [32-34]. Also, hypoxia can induce EPO production in female organs [35]. Reduced oxygen tension induced EPO is also detected in skeletal muscle myoblasts, which contributes to muscle maintenance and repair [36-38]. The expression of EPO in non-hematopoietic cells provides evidence for its pleiotropic biological function.
EpoR expression and regulation
The EPO response in hematopoietic cells relies not only on the EPO concentration but also on the extent of EpoR expression. EpoR is expressed in a cell-restricted fashion and expression depends on the stage of differentiation. EpoR expresses at a relative low level on early erythroid progenitor cells or BFU-E (erythroid burst-forming unit) stage [39]. The expression is then increased during erythroid differentiation by more than tenfold on the erythroid progenitor cells by the CFU-E stage, which requires EPO signaling for protection against apoptosis. During late erythropoiesis, EpoR expression is downregulated and EPO/EpoR signaling is no longer required for the erythroid cell survival [39]. EpoR expression is also regulated by erythroid specific transcription factors including GATA1, SCL/TAL1 and EKLF (KLF1) [40-42].
The expression of EpoR now has been demonstrated beyond hematopoietic cells, which provides the possibility for EPO function generally in proliferation, protective/survival activity in multiple tissue development, maintenance and or repair. The two well-documented roles of EPO beyond hematopoietic system are angiogenesis and neuroprotection that are related to the findings of EpoR expression in endothelial cells and brains (Figure 1). Expression of EpoR in nonhematopoietic tissues was first detected in endothelial cells [43, 44] and the expression can be induced by hypoxia when combined with EPO treatment [45, 46]. The expression of EpoR in endothelial cells mediates increased angiognenesis in vascular system (Figure 1), promotes survival of primary human endothelial cells and also importantly, contributes to cardioprotective effects of EPO in animal model [47-49]. In the developing mouse brain during mid gestation, EpoR was found to localize to the neural tube in the neuroepithelium that contains proliferating neuroprecursors [50, 51]. EpoR is expressed at a high level in the neural tube at E10.5, which is comparable to adult hematopoietic tissue in mice [51].With the neural progenitor cell differentiation to mature neurons, EpoR expression was down-regulated [51]. EpoR level persists at low levels through adulthood [51]. In adult brain from humans, nonhuman primates and rodents, EPO binding experiments provided the evidence of EpoR localization in the hippocampus, cortex and midbrain [52, 53]. In addition to neurons, EpoR is also expressed in astrocytes and microglia [54]. Harmful stress, such as inflammation and ischemia can up-regulate neuronal EpoR, suggesting that the EPO/EpoR signaling can provide preconditional protection against severe ischemia. Hypoxia also induced EpoR expression in neuronal cells and increases sensitivity to EPO (2007 review 7, 28). However, no evidence shows EpoR can be directly regulated by hypoxia inducible factor (HIF). Similar to the situation in erythroid cells, EpoR expression in neural NT2 cells can be transactivated by GATA-3, a transcription factor required for brain development and inducible by EPO (2007 review 7,32).
The pleiotropic activity of EPO in multiple tissues beyond hematopoietic tissues. EpoR expression was detected on erythroid cells, adipocytes, immune system cells such as macrophages, pancreatic beta cells, skeletal muscle myoblasts, neural cells, hypothalamus neurons and endothelial cells. The primary function of the EPO/EpoR system is to stimulate erythroid progenitor cell proliferation, survival and differentiation to provide adequate red blood cells. The well documented non hematopoietic effect of EPO is cytoprotection including cardioprotection and neuroprotection, which are also contributed by the EPO activity in endothelial cells such as angiogenesis. The newly revealed biological activity of EPO includes prevention from obesity and metabolic disorders and improvement of insulin resistance and glucose intolerance. These effects are contributed by EPO promoted energy metabolism in adipocytes, anti-inflammation in macrophages, antiapoptosis in pancreatic beta cells, and the central control of energy intake in hypothalamus neurons.
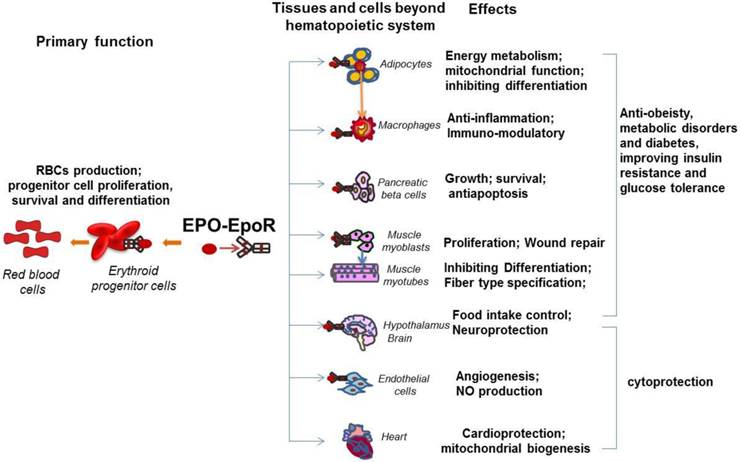
In addition to the important role of EPO in angiogenesis and neuroprotection, recently, the novel and important function of EPO has drawn researchers' attention, the effect of EPO in metabolism and energy homeostasis. This function is tightly related to the expression of EpoR in metabolism related tissues such as skeletal muscle, adipocytes, adipose tissue macrophage, POMC neurons, and pancreas (Figure 1). The widespread distribution of EPO and EpoR among a variety of organs beyond erythroid system suggests EPO is not only a required cytokine for erythropoiesis but also an important factor that is involved in many physiological activities and possibly, potential diseases therapeutic applications.
EPO/EpoR system activity in tissues with high energy demands
EPO/EpoR in adipose tissue
EpoR expression in adipocytes
Compared with other non-hematopoietic tissue, we found that EpoR is expressed at high level in white adipocyte tissue (WAT) (around 60% of hematopoietic tissue) (Figure 1). Also, the expression of EpoR in adipocytes is comparable to the expression in the stromal vascular cells in white adipose tissue [6]. In vitro, EpoR protein is also detected in cultured 3T3-L1 adipocytes and EpoR expression is increased during preadipocyte differentiation and significantly decreased in dexamethasone-induced insulin resistant 3T3-L1 adipocytes [55, 56]. Upregulated EpoR expression and activated PI3K/AKT and JAK/STAT5 pathways under EPO treatment were also observed in adipocytes [55]. And EpoR siRNA blocked EPO-induced activation of AKT and STAT5 [55]. In addition, the loss of EpoR in adipose tissue leads to the obese phenotype of mice with increased body weight and white fat accumulation [6, 11]. In contrast, EPO treatment in wild type mice, EPO over expression in skeletal muscle and mice expressing high transgenic levels of EPO demonstrated reduced body weight and adipose tissue mass [6, 57, 58]. These observations raise the possibility that endogenous EPO action in adipocytes may contribute importantly to protection against obesity and energy homeostasis.
EPO and energy homeostasis in adipose tissue
Since the demonstration of that EPO overexpression in mice reduced body weight gain and white adipose tissue accumulation, improved glucose tolerance and reduced insulin resistance compared with control mice [58], and importantly, mice with EpoR restricted to hematopoietic cells resulting in increased body weight and white fat accumulation [6, 11], all these EPO activities extend EPO activity to energy metabolism and energy homeostasis especially in adipocytes.
Fat specific deletion of EpoR (generated by mating EpoR-floxed mice and aP2-Cre mice on a C57Bl6 background as a model system for diet induced obesity) preferentially increased body weight and fat mass, and by 30 weeks, the gene knockout mice have 65% greater fat mass with reduced oxygen consumption and total respiratory exchange ratio (RER) on normal chow [11]. On a high fat diet, these mice also show a significantly greater increase in fat mass, increased glucose intolerance and insulin resistance. Wild type mice treated with EPO show increased hematocrit and a significant decrease in body weight, while mice with fat knockout of EpoR show the significant increase in hematocrit but no significant reduction in body mass. Serial experimental evidences in these mice also demonstrated that specific EPO activity in fat is strongly associated with the regulation of mitochondrial biogenesis, cellular oxygen consumption and fatty acid metabolism by EPO in adipocytes [11, 46]. Although the indirect effect of EPO on whole body cannot be entirely excluded, the study of fat specific knock out of EpoR demonstrates for the first time that the loss of EPO/EpoR signaling in adipose tissue is sufficient to develop metabolic syndrome phenotype including obesity, glucose intolerance and insulin resistance, and supports the idea that EPO activity in fat may contribute to energy homeostasis. These findings indicate that endogenous and exogenous EPO/EpoR in adipocytes plays an important role in the regulation of body weight, fat mass accumulation and glucose metabolism and insulin sensitivity. Another interesting and important finding is that EPO may contribute to the development of a brown fat-like gene program in white adipose tissue. EPO was found to promote brown fat-like characteristics including increased mitochondrial content and uncoupled respiration in white adipocytes, enhanced brown fat associated gene expression. Functionally active BAT is inversely correlated with BMI [59, 60], and a higher level of BAT may be protective against obesity and have stimulated interest concerning the therapeutic potential of augmenting brown fat to combat obesity and its associated metabolic disease. Thus, increasing these brown fat enriched characteristics and factors by EPO may be a plausible strategy for the treatment of obesity and its associated disease. C57BL/6 mouse is used as a suitable model for diet induced diabetes [61-63]. In contrast to the increased susceptibility to diet induced obesity in mice with EpoR fat specific knockout on the C57BL/6 background, mice with fat specific knockout of EpoR in mice on a mixed background were reported to behave metabolically similarly to control mice, although it was not demonstrated how control mice on this mixed background respond metabolically to EPO administration [64]. Nevertheless, the differences between these two studies underscore the complexities of genetic influences on obesity and EPO response.
EPO/EpoR in pancreatic beta cells
Emerging evidence has suggested that EPO contributes to glucose metabolism and insulin sensitivity that are closely associated with diabetes development. It is believed that insufficient functional pancreatic β-cell mass is one of pathogenic mechanisms of type 1 and 2 diabetes. Therefore, promoting β-cell growth and survival may be helpful for diabetes prevention and treatment. EPO, required for erythroid progenitor cell proliferation, survival and differentiation, has been demonstrated for the cytoprotective effects on nonerythroid cells. Although detailed expression regulation mechanism remains unknown, EpoR expression presence was demonstrated on pancreatic islets in human and rodents (Figure 1), which provides the possibility for pancreatic islet cell response to EPO [65]. The evidence that recombinant EPO treatment in culture or EPO overexpression in human islets protects islets from destruction and apoptosis confirmed the response of pancreatic islet cell to EPO [65, 66]. Further in vivo experiments demonstrated that EPO protects mice against streptozotocin (STZ) induced type 1 diabetes via reverse STZ-mediated β-cell destruction [67]. EPO also has protective effects against diabetes in db/db mouse model of type 2 diabetes [67]. Furthermore, the pancreatic islet specific knock out of EpoR demonstrated that EpoR is required for EPO mediated diabetes protection under STZ induced diabetes conditions but not under basal conditions, and this protection effect is mediated by EPO stimulated increase in β-cell mass and viability rather than the direct effects of EPO on β-cell function, suggesting that the pancreatic islet cell response to EPO resulted in promotion of β-cell growth and survival [7]. It is worth noting that JAK2, the immediate downstream kinase of EPO/EpoR signaling, is required for diabetes protection by EPO under induced diabetes conditions [7], indicating that pancreatic β-cells may use similar signaling pathway as that which is used in erythroid system to respond to EPO stimulation. Another report demonstrated that EPO may also protect pancreatic β-cells from apoptosis and increase survival of β-cells via PI3K-AKT pathway [68]. Interestingly, the pancreatic islet response to EPO was also demonstrated by promotion of gene expression associated with proliferation and angiogenesis such as c-myc, c-kit and vegf [7]. Collectively, pancreatic islet response to EPO suggests potential protection effect of EPO against diabetes via EPO mediated increase in β-cell mass including promoting proliferation, antiapoptosis and possibly increased angiogenesis. These observations strongly suggest that the nonhematopoietic effects of EPO may extend to encompass the regulation of fat and glucose metabolism, thereby implicating its signaling as a regulator of glucose tolerance and possibly insulin sensitivity.
EPO/EpoR in skeletal muscle
EpoR expression and regulation in myoblasts
We previously demonstrated that EPO stimulates myoblast proliferation and delays differentiation and contributes to myoblast survival and muscle repair from injury [36, 37, 69]. Consistently, the EpoR transgenic expression showed that EpoR expression follows the expression of basic-helix-loop-helix muscle transcription factors Myf5, a myoblast specific factor, but not MyoD and myogenin, required for muscle differentiation [36]. Importantly, only skeletal muscle myoblasts but not matured myotubes were found to show proliferative response to EPO stimulation (Figure 1). Further expression evidence demonstrated that EpoR is only expressed in muscle myoblasts and decreased with the skeletal muscle differentiation and no EPO response was detected in mature muscle fibers [12, 36]. EPO stimulation in myoblasts promoted EpoR expression and muscle specific transcription factors, such as Myf5 and MyoD [14, 36]. However, only Myf5 regulates EpoR expression in myoblasts. In addition, GATA factors including GATA3 and GATA4 and another basic-helix-loop-helix factor, TAL1 were found to be up-regulated by EPO stimulation in myoblasts and down-regulated with myoblast differentiation [14, 37]. The class III deacetylase, Sirt1, which is most homologous to yeast Sir2 and is a NAD+-dependent deacetylase, was observed to be increased by EPO stimulation in myoblast to regulate GATA4 and TAL1 expression, leading to inhibited muscle differentiation [14]. In addition, EpoR can be up-regulated by GATA3, GATA4 and TAL1 in myoblast, which explains partially the reason that these factors increase myoblast response to EPO and myoblast proliferation but inhibit myogenesis [14].
EPO function in muscle metabolism and muscle fiber type specification
Interestingly, ectopic EPO expression specifically in skeletal muscles resulted in reduced body weight and adipose tissue mass accompanied by improved fasting insulin levels and glucose tolerance in the high-fat fed mice [57]. The observation that increased expression of genes related to lipid metabolism and thermogenesis, also increased muscular fat oxidation in EPO overexpressed mice suggest the important role of EPO in muscle metabolism [57]. Although the absence of EpoR in mature muscle fiber indicates that the EPO effect in muscle fiber may be indirect and probably related to an increased oxygen delivery in this tissue, however, another study suggests EPO increases submaximal endurance performance to a greater extent than what can be explained by increased oxygen delivery, suggesting EPO may stimulate related metabolic activity in muscle via other unknown manners [70]. In human, EPO treatment enhances skeletal muscle mitochondrial oxidative phosphorylation capacity and electron transport capacity [71]. In addition, overexpressed EPO during muscle development may contribute to muscle type specification via EPO changed metabolic activity with the development. Skeletal muscles of vertebrates contain two types of myofibers, slow twitch (type I) and fast twitch (type II), that differ in function, mitochondrial density, and metabolic properties. Slow twitch (ST) myofibers contain a high concentration of mitochondria and high oxidative capacity. In contrast, fast-twitch myofibers such as type IIB fibers, show low mitochondrial density and low oxidative metabolism [72]. The percentage of ST fibers is reduced in obese and type 2 diabetic patients, and within each fiber type, obese and type 2 diabetic patients have lower oxidative enzyme activity and a corresponding greater lipid content and smaller mitochondria in skeletal muscle [73-75]. Defective insulin signaling has been suggested to be associated with mitochondrial dysfunction [76, 77]. Furthermore, mice engineered with increased type I muscle fibers exhibit resistance to obesity and improved metabolic profiles. It was observed that skeletal muscles from mice with high EPO production in vivo exhibit an increase in the proportion of slow twitch myofibers and increased mitochondrial activity [12]. In rats, erythropoietin treatment increases significantly the specific activities of oxidative enzyme and induces a shift of muscle phenotype from fast glycolytic to slow oxidative [78]. The important metabolism sensors, AMPK and PGC-1α, which also are demonstrated to promote the slow and oxidative fiber specification [79, 80], were activated by EPO in the muscle [12]. In comparison, skeletal muscle from wild type mice and mice with erythropoietin activity restricted to erythroid tissue has fewer slow twitch myofibers and reduced mitochondrial activity compared to the mice with overexpressed EPO [12]. These observations suggest that EPO contributes to skeletal muscle fiber programming and metabolism, which may link EPO activity to the potential of anti-obesity and metabolism disorder.
EPO/EpoR in heart
EPO and heart protection
Although early studies suggested that EPO/EpoR activity is required for heart development especially embryonic heart since EPO-/- and EpoR-/- mice show heart development defects including ventricular hypoplasia, epicardium detachment and abnormalities in the vascular network and these defects were speculated to be related to ineffective cell proliferation and expansion of myocardium [9], however, later studies using the mice with EpoR expression restricted to erythroid system found that these mice do not display severe defects including epicardium detachment and abnormalities in the vascular network [81], suggesting severe anemia might be pertinent to the blockage of heart development and EPO/EpoR system may do not relate directly to epicardium or capillary development in the embryonic heart. It is worth noting that EpoR expression was detected in the developing heart including in pericardium, epicardium and endocardium. The essential factor for cardioprotection, GATA4 has been reported to positively regulate EpoR expression [82], however, the regulatory mechanisms of EpoR expression in cardiomyocytes in health and disease are still largely unknown yet.
These observations indicate that endogenous EPO activity may still contribute to cardiac function although not crucial to heart development. Later studies using adult animal model demonstrated that EPO can directly protect the ischemic and infarcted heart independent on increased hematocrit by preventing myocyte apoptosis and attenuating postinfarct deterioration [83-85]. EPO also provides direct protection during ischemia-reperfusion injury that associates with preservation of ATP levels in the ischemic myocardium [83].
Another important contribution of EPO in the cardiac protection is that EPO stimulates endothelial nitric oxide (NO) synthase (eNOS) activity to promote NO production to provide cardioprotection. Especially during hypoxia, low oxygen tension increases endothelial cell capacity to produce NO in response to EPO by induction of both EpoR and eNOS [45, 86-88]. EPO promoted eNOS activation is associated with PI3K-AKT signaling and contributes to cardiac mitochondrial biogenesis by enhancing PGC-1α, the important energy metabolism sensor [13]. Ectopic expression of EPO in mice increased circulating and vascular tissue NO level that prevented cardiovascular diseases such as hypertension and thromboembolism [88]. The cardioprotection role of EPO is also mediated by suppressed inflammatory response. In rodents with myocardial ischemia-reperfusion injury and in the mice with chronic post-myocardial infarction associated heart failure, EPO pretreatment reduced inflammatory cell infiltration and fibrosis and the incidence of ventricular arrhythmia and decreased serum level of proinflammatory cytokines [86, 89, 90].
EPO/EpoR and neural system
EpoR expression in hypothalamus and the role of EPO in food intake
The hypothalamus is a neural organ responsible for regulating energy metabolism. Two populations of neurons in the arcuate nucleus of hypothalamus act in an opposing manner to control food intake, energy expenditure and glucose metabolism. Proopiomelanocortin (POMC)-expressing neurons sense the change in peripheral regulatory hormones, leptin and insulin, and respond by secreting α-melanocyte-stimulating hormone that stimulates melanocortin 3 and 4 receptors in target neurons to decrease food intake and increase energy expenditure [91, 92]. Activation of neuropeptide Y (NPY)/agouti-related protein (AGRP) neurons expressing orexigenic neuron peptides NPY and AGRP stimulates orexigens MCH and orexins expression in lateral hypothalamus target neurons to increase food intake and decrease energy expenditure [91-93]. An intriguing finding that EpoR expression is detected at a relative high level in hypothalamus supports the view that EPO may play a role in central regulation of energy homeostasis [6]. Furthermore, EPO treatment in WT-mice increased POMC mRNA by threefold and in vitro culture of hypothalamic neurons stimulated with EPO demonstrated that EPO can directly promote POMC expression, but not NPY, AGRP, pro-MCH and prepro-orexin [6]. Also the immunostaining of EpoR on POMC neurons in the hypothalamus provides further support for that endogenous EPO activity in regulation of food intake and energy metabolism homeostasis. In contrast, EpoR expression in the hypothalamus was absent in the mice with EpoR restricted in the erythroid system (Tg-mice) and significantly reduced in ob/ob-mice compared with control mice [6]. Consistently, the expression of POMC that regulates activity, food intake and energy homeostasis, decreased more than 50% in Tg-mice, suggesting that EPO regulates energy homeostasis at least through regulating its target, POMC expression, in the hypothalamus (Figure 1). Further experiments are required to investigate the detailed mechanism by which EPO regulates POMC expression and the role of EPO in the central regulation of energy homeostasis.
EPO and neuroprotection
Another important and well documented function of EPO is neuroprotective role (Figure 1). The protective function of EPO is mediated by blocking neuronal cell apoptosis, promoting cell survival and neurogenesis and regulating angiogenesis. EPO is neuroprotective against hypoxia, ischemic brain injury inflammation and glutamate toxicity [8, 94-98]. Neural cells from the mice with EpoR restricted in erythroid system (Tg-mice) proliferated more slowly and were less tolerant to hypoxia [99]. The Tg-mice also exhibited increased apoptosis in the developing brain, neural cells with increased sensitivity to hypoxia and decreased neural cell proliferation and increased sensitivity to glutamate toxicity in adult brain [50]. Furthermore, the mice with brain-specific deletion of EpoR showed defective neural progenitor cell migration to the peri-infarct cortex in response to ischemic stroke [50]. EPO administration in brain of rodents reduced injury and improved performance in the Morris water maze test [100, 101]. EPO was also neuroprotective against peripheral inflammatory pain in postnatal rats [102]. EPO neuroprotection may also result from the indirect effect of EPO on other cell type rather than neural cells such as endothelial cells. EPO may increases VEGF production and induced VEGFR2, resulting in enhanced angiogenesis [103]. EPO can also stimulate endothelial cells to increase eNOS activity to produce nitric oxide (NO), particularly at low pO(2) to provide neuroprotection function [104]. EPO is also protective in hypoxia-induced cell death in both embryonic and postnatal hippocampal neurons [8, 95].
EPO and energy metabolism regulation
EPO and cancer
Cancer-triggered systemic metabolic disease in the host, called cancer cachexia, leads to weight loss, muscular atrophy, adipose atrophy, and other systemic disorders. It is estimated that approximately 25% of all cancer patients eventually die of cachexia. The studies performed in animal cancer cachexia models suggested that in tumor-bearing rodents with reduced white adipose tissue, EPO administration partially preserved adipose tissue and increased lipoprotein lipase activity [105], suggesting that in animals with cachexia and markedly reduced fat stores, EPO increases fatty acid synthesis in adipocytes and provides a sparing effect and acts to normalize metabolic homeostasis under pathological state. However, some conflicting findings about the direct effect of EPO on cancer cells have been reported by various research groups, which restricted the clinical use of EPO in cancer patient therapy. For the past few decades, EPO has been used widely for treatment of cancer patients with anemia induced by chemotherapy. The EPO treatment in these clinical trials increased red blood cell level, hemoglobin concentration and decreased fatigue, leading to improved quality of life [106-108]. Controversially, EPO treatment for anemia in cancer patients has been challenged by some later studies. It was reported that EPO promotes tumor growth through regulating tumor angiogenesis such as in hepatocellular carcinomas, renal carcinoma and glioma [109-111]. A shorter survival and increased risk of mortality by EPO treatment were also demonstrated especially in solid cancers such as breast cancer and head and neck cancer [112-114] , where EPO treatment may facilitate tumor invasion and metastasis via modulating the tumor vasculature and increasing thrombin deposition [115, 116]. These conflicting clinical outcomes of EPO promote us to pay more attention to the EPO therapy in treatment of anemia in cancer patients.
The relationship of EPO and obesity/ diabetes
Diabetes, especially type 2 diabetes is a metabolic disorder related disease and is characterized by high blood glucose in the context of insulin resistance and relative insulin deficiency. Insulin is the principal hormone that regulates uptake and metabolism of glucose. Therefore, abnormal insulin secretion and improper insulin action are central and critical to the pathogenesis of diabetes. Numerous factors have been implicated in the insulin secretion and insulin signaling pathway regulation. Obesity and its associated metabolic syndrome including glucose intolerance and insulin resistance are also well-documented risk factors for type 2 diabetes. Obesity is tightly associated with energy metabolism including glucose and lipid metabolism. Understanding of the mechanism of the disrupt energy metabolism relating to obesity and abnormal metabolic processes can provide insight on the development of potential therapeutic anti-obesity strategies, which will also contribute to the protection against diabetes and metabolic syndrome.
Some early clinical trials have suggested the effects of EPO in metabolism since EPO was introduced into the clinic treatment of anemia associated with chronic renal failure and cancer in 1989. In addition, it was observed that EPO treatment improved glycemic control and insulin sensitivity in hemodialysis and diabetic patients [117-119]. Positive regulation of lipid metabolism was also accompanied together with EPO treatment in uremic patients with end-stage renal disease including improved plasma triglycerides and cholesterol levels [120]. Although some other studies suggested controversial observation that EPO treatment in hemodialysis patients and healthy human subjects did not improve lipid profile and glucose metabolism [121], significantly increased energy expenditure and augmented systemic and muscular oxygen consumption and delivery were observed [122, 123]. In addition, improvement in physical exercise capacity and muscle metabolic rate in predialytic uremic patients and hemodialysis patients has also been noted in some small clinical studies [124-127]. Improved metabolic parameters including fasting glucose level and insulin sensitivity were also observed in EPO treated diabetic and chronic renal disease patients [118, 120, 128, 129].
Since EPO and EpoR null mice die of severe anemia around embryonic day 13.5 [1, 130], the mice with endogenous EpoR expression restricted largely to hematopoietic and endothelial were made ( the normal erythropoiesis was restored by transgene EpoR driven by erythroid-specific GATA promoter into the EpoR knockout background mice to rescue mice [81]. These mice (Tg-mice) exhibit increased body weight from the first week after birth and become obese due to increased fat mas accumulation [6]. These obese mice also show insulin resistance, decreased energy expenditure and glucose intolerance. In addition, overexpression of EPO either in the muscle or human platelet-derived growth factor B-chain promoter driven human EPO transgenic mice (tg6) show improved glucose tolerance and insulin sensitivity, and lower body weight and fat mass [57, 58]. EPO treatment in WT mice and obese (ob/ob) mice (genetically deficient in leptin) improves overall metabolic characteristics, reduces blood glucose levels, improves glucose tolerance and attenuates weight gain and fat mass accumulation [6, 58]. EPO treatment in diabetes (db/db) mice provides protection for cardiomypathy and insulin producing pancreatic β-cells via antiapoptotic and angiogenic effects in the islets [7, 131, 132]. EPO was also protective against diabetic europathy in the streptozotocin induced diabetic rat model [67]. These findings of EPO activity in animals provide more insights on potential EPO regulation in energy metabolism and homeostasis.
EPO regulates oxygen consumption and oxidative metabolism
About 95% of the protein content of the red blood cells is hemoglobin that binds oxygen cooperatively and transports oxygen from the lungs to the tissues. Induction of EPO production in response to low oxygen or ischemic stress increases mature red blood cell production [133]. Increasing red cell number increases the oxygen carrying capacity of blood to enhance oxygen delivery to the tissues. Hence, the primary function of EPO is to regulate oxygen delivery via the production of red blood cells and is facilitated by the hypoxia induction of EPO gene transcription resulting in sensitivity of EPO production to the local oxygen environment [133]. Beyond erythroid tissue, EPO also facilitates oxygen delivery to brain, heart and other non-hematopoietic tissues via stimulation of nitric oxide production by vascular endothelium. Increased oxygen supply will contribute to energy metabolism. The disrupted EPO activity in Tg-mice resulted in decreased energy expenditure and reduced total oxygen consumption and respiratory quotient [6]. In contrast, EPO treatment of wild type mice increased total physical activity and oxygen consumption. The mice with EpoR knock out specifically in fat tissue also exhibited decreased physical activity, Oxygen consumption, and respiratory exchange ratios [11]. These observations suggest the important role of EPO in energy homeostasis. The regulation of EPO in energy homeostasis may be critical in the development of obesity in Tg-mice and in suppressed fat mass accumulation in EPO-treated wild type mice. Another direct evidence for EPO in energy homeostasis is that EPO stimulates cellular oxygen consumption rate (OCR) beyond whole animal level. EPO increased basal OCR in cultured adipocytes and in skeletal myoblasts [11, 12]. The EPO-stimulated increase in OCR became even more evident under stress. For example, in the presence of oligomycin, which uncouples phosphorylation from mitochondrial respiration by blocking mitochondrial complex V, and with treatment of carbonyl cyanide p-trifluoromethoxy phenylhydrazone (FCCP), which uncouples oxidative phosphorylation from ATP synthesis and is used to assess maximal oxidative phosphorylation capacity, the OCR increased even more with EPO treatment in adipocytes and myoblasts [11, 12].
It is known that hypoxia decreases OCR, however, EPO treatment overcomes the hypoxia adverse effects on OCR and increases OCR in cultured mice adipocytes under basal condition and in the presence of oligomycin and FCCP under hypoxia [11]. In cultured human adipocyte, the same observation was also found [11]. These findings highlight a previously unrecognized role for EPO/EpoR activity in directly increasing cellular mitochondrial respiration and oxidative metabolism capacity beyond its effect of increased erythropoiesis and oxygen transport capacity, leading to increased oxygen utilization capacity and energy oxidative metabolism efficiency, which will contribute to prevention against obesity and metabolism disorder. Consistent with increased oxygen consumption rate, cultured adipocytes with EPO stimulation showed increased energy metabolism such as fatty acid oxidation. In contrast, disrupted EPO activity in fat tissue resulted in decreased fatty acid metabolism of mice adipocytes [11]. The increased fatty acid metabolism by EPO may limit the storage of excess lipid to protect against obesity and diabetes.
EPO promotes mitochondrial biogenesis and mitochondrial function
Increased mitochondrial biogenesis and related gene expression correlate with a reduction of diet induced obesity [134]. Mitochondrial dysfunction in adipose tissue is linked to obesity and type 2 diabetes in humans, as indicated by reduced oxidative phosphorylation capacity and reduced fatty acid oxidation in several tissues, including adipocytes [135, 136]. Interestingly, EPO treatment enhances muscle mitochondrial capacity as indicated by an upregulation of oxidative phosphorylation (OXPHOS) and electron transport capacity (ETS) in human skeletal muscle [71]. In mice, EPO also stimulates mitochondrial biogenesis in heart as indicated by increased mitochondrial density, mitochondrial mass and mitochondrial biogenesis gene expression including NRF-1, PGC-1α, and mitochondrial transcription factor-A (Tfam) [13, 137]. Increased EPO activity in mice also promotes mitochondrial activity as indicated by increased Citrate synthase (CS) activity in skeletal muscle and myoblasts, which may be mediated by increased PGC-1α and AMPK activity, the two important energy metabolism regulators [12]. The increased mitochondrial content by EPO stimulation indicated as increased mitochondrial DNA was also demonstrated in primary myoblasts and cultured myoblasts [12]. In addition, EPO contributes to cytoprotection during ischemic vascular injury through direct modulation of mitochondrial membrane potential in endothelial cells [138]. In adipocytes, EPO also increases mitochondrial density reflected by increased mitochondrial staining and mitochondrial DNA [11]. EPO also promotes mitochondrial biogenesis related factor level at gene level and protein level and increased CS activity in cultured adipocytes and fat tissue isolated from EPO treated mice [11]. In contrast, the loss of EPO activity in fat tissue results in decreased mitochondrial activity and density [11]. Functionally, EPO's effect on the mitochondria was reflected by increased energy metabolism efficiency such as increased fatty acid oxidation. In adipocytes, the increased metabolic activity by EPO is mediated by Sirt1, a NAD+ -dependent type III deacetylase, and peroxisome proliferator-activated receptor a (PPARα), an important regulator in lipid metabolism [139]. In human, EPO administration also stimulates resting energy expenditure and fat oxidation [140]. All of these observations provide evidence for the function of EPO in regulating mitochondrial activity and energy homeostasis in the multiple tissues and highlight the new potential role of EPO in protection against metabolism related disease such as obesity, metabolism disorders and diabetes beyond its primary function in the erythroid system.
The crosstalk between EPO and energy sensors
EPO and PGC-1α
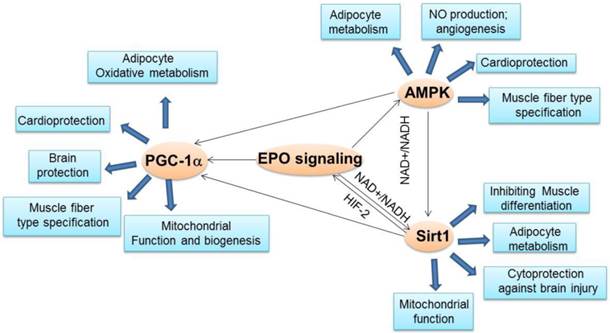
As we mentioned above, EPO activity is linked to some important energy sensors such as PGC -1α. The core function of PGC-1α is to stimulate mitochondrial biogenesis and oxidative metabolism to contribute to energy expenditure [141, 142]. PGC-1α is abundantly expressed in tissues with high energy demand, including the fat, heart, skeletal muscle, kidney and brain, and interacts with some other transcription factors to regulate the expression of a number of genes involved in diverse metabolic pathways such as fatty acid oxidation, glycolysis and gluconeogenesis. The change of the expression of these co-activators might strongly impact on whole-body energy homeostasis and on the pathogenesis of the metabolic syndrome [143, 144]. PGC-1α has also been demonstrated to be related to adipocyte differentiation, adaptive thermogenesis and muscle fiber development, which make the PGC-1α be involved in some diseases such as obesity, type 2 diabetes, and insulin-resistance [79, 145-148]. The function of EPO has been demonstrated to be linked to the PGC-1α activity in recent studies (Figure 2). EPO was found to increase PGC-1α expression in the cardiomyocytes to contribute to increased mitochondrial biogenesis and cardioprotection [13, 137]. The increased PGC-1α by EPO possibly requires eNOS function because eNOS knock out abolished EPO increased cardiac PGC-1α expression [13]. In skeletal muscle and myoblasts, EPO also stimulated PGC-1α expression via increasing AMPK activity, an upstream regulator of PGC-1α expression and activity [144, 149-151], which may contribute to the EPO effect in regulating skeletal muscle fiber programming and metabolism [12]. In addition, in adipocytes, EPO increased PGC-1α expression and regulated PGC-1α activity via activating Sirt1 deacetylation activity to deacetylate PGC-1α, leading to the increased PGC-1α activity [11, 151]. These effects of EPO in cultured adipocyte and white fat tissue is believed to contribute to maintaining energy homeostasis and developing a brown fat-like gene program in white fat tissue [11]. The neural PGC-1α was markedly decreased after acute ischemic stroke stimulation, but EPO can preserve PGC-1α expression and PGC-1α + neurons especially combined with cyclosporine treatment, which contribute to the protecting brain from ischemic damage [152]. The various mechanisms by which EPO regulates PGC-1α expression and activity in multiple tissues promote us to speculate that EPO activity is linked to energy sensing system to sense the energy change and maintain energy homeostasis via regulating PGC-1α. EPO is linked to energy sensing system is also confirmed by the observation that EPO regulates NAD+/NADH level and Sirt1 activity, a direct regulator of PGC-1α activity and a master metabolic regulator [153].
The crosstalk between EPO and important energy sensors. EPO regulated AMPK activity is involved in adipocyte energy metabolism, angiogenesis, cardioprotection and skeletal muscle fiber type specification. EPO production can be regulated by Sirt1 via HIF-2 under hypoxia condition. On the other hand, EPO modulates NAD+/NADH level and ratio to regulate Sirt1 activity, which contributes to energy metabolism in adipocytes, inhibition of skeletal muscle differentiation, and brain protection from injury and mitochondrial function. EPO regulated AMPK activity may regulate Sirt1 activity via modulating NAD+/NADH ratio. As the downstream target of Sirt1and AMPK, PGC-1α may also be directly regulated by EPO or via regulating Sirt1 and AMPK activity to promote adipocyte oxidative metabolism, cardioprotection, brain protection, mitochondrial biogenesis and function and muscle fiber type specification.
EPO and Sirt1
Sirt1 is one of the seven mammalian orthologs (sirtuins Sirt1-7) of the yeast protein silent information regulator 2 (Sirt2), a highly conserved NAD+-dependent protein deacetylases and/or ADP-ribosyltransferases [154-157]. Growing evidences in other model organisms have demonstrated that Sirt1 is also a crucial regulator of a variety of cellular processes, ranging from energy metabolism, and stress response, to tumorigenesis and aging. Sirt1 has a prominent role in metabolic tissues including liver, pancreatic islet, skeletal muscle, adipose tissues and brain, where it deacetylates a range of substrates, including P53, PGC-1α, LXR, Ucp2, NF-κB and FoxO1 proteins, which results in a pronounced effect on glucose homeostasis, insulin secretion, energy expenditure and homeostasis [151, 158-160]. Sirt1 regulates the activity of the nuclear receptor PPARγ and thus influences adipogenesis as well as fat storage in white adipose tissue [161]. Activating Sirt1 in mice contributes to protection against high-fat-induced obesity and metabolic disorders [162-164]. Sirt1 also promotes brown adipose tissue differentiation through repressing MyoD-mediated myogenic gene expression signature and stimulating PGC-1α-mediated mitochondrial gene expression [165]. Sirt1 is a vital regulator of pancreatic insulin secretion in response to the nutrient availability, which contributes to protection against obesity, glucose intolerance and insulin resistance [158, 166].
Sirt1 activity also appears to be an important player in the central regulation of nutrient sensing. Inhibition of Sirt1 activity in POMC neurons increases acetylation of FOXO1, resulting in increased POMC and decreased AgRP expressions, thereby decreasing food intake and body weight gain [167]. However, specific deletion of Sirt1 in POMC neurons in mice causes reduced energy expenditure, leading to hypersensitivity to diet-induced obesity [168]. Central administration of Sirt1 activator has shown promise in controlling of diet-induced obesity and diabetes [169].
In skeletal muscle, Sirt1-regulated activation of PGC-1α activates mitochondrial fatty-acid oxidation genes and upregulates GLUT4 to promote insulin sensitization [170]. Sirt1 may affect metabolic activity and energy homeostasis via its regulatory effect on mitochondrial function. The close relationship between Sirt1 and PGC-1α in multi tissues might provide some insights into the association between Sirt1 and mitochondrial function. Sirt1 can deacetylate PGC-1α at several lysine residues and thereby increase its ability to activate transcription of target genes involved in mitochondrial biogenesis. Indeed, feeding mice with resveratrol to activate Sirt1 upregulates the number of mitochondria in their muscle cells [163].
Interestingly, Sirt1 was demonstrated to transactivate EPO via activating HIF-2α in hypoxic hepatoma and human embryonic kidney cells [171]. On the other hand, EPO increases the expression and deacetylase activity of Sirt1 and its downstream target AKT and FoxO3a in the cerebral microvascular endothelial cells (ECs) to exert its cytoprotective ability against brain injury during oxygen-glucose deprivation [172]. The direct evidence for that EPO regulates Sirt1 activity is that EPO can modulate NAD+/NADH level, the regulator of Sirt1 activity [11, 14]. NAD+ or NADH levels are readout of cellular metabolic activity. NAD+ and NADH metabolism is also important in oxidative metabolism and energy homeostasis and has been linked to protection against dietary obesity and a therapeutic target for associated metabolic diseases [173-175]. Induced NAD+ level by pharmacological activation of NADH oxidation resolves diet induced obesity and related phenotypes in mice [176], in part analogous to the reduction in fat mass by EPO treatment in obese mice [6]. The accumulation of NADH within mitochondria further affects pyruvate metabolism, which contributes to metabolic acidosis. An increase in mitochondrial NADH can impair electron transport chain function. EPO treatment in a mouse model with severe sepsis can decrease NADH accumulation in skeletal muscle, which improves mitochondria oxidative phosphorylation and pyruvate metabolism [177]. Hypoxia can lead to an increase in intracellular NADH levels thereby decreasing the NAD+/NADH ratio by inhibiting NADH oxidation [178, 179]. Our study showed that EPO dramatically rescued the reduced NAD+/NADH ratio by hypoxia and decreased NADH level in adipocytes [11]. Modified NAD+ level and NAD+/NADH ratio by EPO were also observed in human and mice primary adipocytes in vitro and in white adipose tissue in vivo [11]. The increased NAD+ by EPO resulted in enhanced Sirt1 activity as shown by decreased PGC-1α acetylation in adipocytes, leading to the activation of PGC-1α [11]. Furthermore, Sirt1 knockdown attenuated the EPO-stimulated increase in brown fat associated and mitochondrial genes and in oxidative respiration and fatty acid oxidation, indicating Sirt1 mediates EPO activity in adipocyte metabolism [11]. EPO regulated Sirt1 activity is also illustrated by the effect of EPO in myoblasts where EPO increases NAD+ level and NAD+ /NADH ratio to regulate Sirt1 activity [14]. Sirt1 has been found to negatively regulate muscle differentiation by deacetylating MyoD and forming a complex with the acetyltransferase PCAF and MyoD in a NAD+-dependent manner [180]. EPO was found to upregulate Sirt1 expression and increase Sirt1 activity in myoblasts to regulate transcription factors GATA-4 and TAL1 and these factors together with Sirt1 coordinately regulate skeletal myoblast differentiation [14]. Given that the indispensable role of NAD+/NADH in cellular oxidation/reduction reactions and metabolic activity and the important role of Sirt1 in various cellular and pathophysiological processes, the regulation of EPO in NAD+/NADH and Sirt1 activity strongly suggest that EPO may exert its function in various processes such as sensing energy imbalance to change energy metabolic activity including lipid and glucose metabolism, which will contribute to protection from obesity, metabolism disorders and diabetes, regulating cell proliferation and differentiation and protecting from stresses (Figure 2). In addition, further experiments are required to clearly clarify the crosstalk among the EPO signaling, Sirt1, and PGC-1α factors.
EPO and AMPK
AMP-activated protein kinase (AMPK), a serine/threonine kinase, is an evolutionarily conserved energy metabolic sensor and an important regulator of energy homeostasis.
It has been demonstrated that AMPK agonists increased oxidative gene expression and protection against metabolic disease [181, 182]. AMPK activation induces fatty acid oxidation in liver and heart, inhibits hepatic lipogenesis and adipocytes differentiation, and stimulates glucose uptake and mitochondrial biogenesis to modulate energy balance in muscle [145, 149, 183]. AMPK can be activated to block body weight gain and increase fatty acid oxidation by inducing transcription regulators involved in energy homeostasis such as peroxisome proliferator-activated receptor alpha (PPAR-α) and peroxisome proliferator-activated receptor gamma (PPAR-γ) co-activator 1 (PGC-1α) [144, 149-151, 183, 184]. AMPK activator, metformin and thiazolidinediones have shown important therapeutic benefits in the treatment of type 2 diabetes and metabolic syndrome [150, 185, 186]. AMPK is also implicated in the appearance of brown features and increased mitochondrial activity in white adipose tissue via regulating or interacting with factors such as PRDM16, a master regulator of brown fat determination and uncoupled protein 1 (UCP1), a key regulator of brown fat thermogenesis [187-192]. In addition, AMPK also exerts its function in metabolic processes through interaction with Sirt1 [144, 160]. For example, Sirt1 directly deacetylates the AMPK protein kinase liver kinase B1 to increase the activity of AMPK to regulate hepatocyte lipid metabolism and improve systemic insulin sensitivity [193, 194]. Moreover, activation of AMPK by its synthetic activator, AICAR, increases cellular NAD+/NADH ratio, resulting in activation of Sirt1 and its downstream target including PGC-1α and FOXO1 to contribute to energy homeostasis through increasing fatty acid oxidation and mitochondrial biogenesis in skeletal muscle [160, 195]. These observations suggest that AMPK and Sirt1 may coherently regulate metabolic processes and energy homeostasis (Figure 2). Intriguingly, AMPK activity has been involved in EPO effect in various biological processes (Figure 2). EPO administration in endothelial cells and mice increased phosphorylation and activity of AMPK, leading to increased eNOS activity and NO production and, ultimately angiogenesis [196]. EPO also exerts its cardioprotective role in an AMPK-dependent manner [197]. EPO regulated skeletal muscle fiber programming and metabolism may also be contributed by increased AMPK activity by EPO [12]. Our newly published data also revealed that EPO facilitates energy expenditure by regulating AMPK via CaMKK in white adipocytes (unpublished data). EPO alters cellular NAD+/NADH levels through regulating AMPK activity in adipocytes, which may lead to increased Sirt1 activity in adipocytes as we demonstrated in our published paper [11]. EPO mediated activation of AMPK also contributes to energy expenditure and reduction of hypoxia induced oxidative stress of adipocytes (unpublished data). Further experiments are required to investigate the important regulation role of EPO on AMPK activity in multiple tissues.
Since AMPK, Sirt1 and PGC-1α activity are tightly associated in regulating metabolic processes and metabolism diseases resistance and EPO is also involved in the regulation of these three important metabolism regulators, EPO may be part of the AMPK-Sirt1-PGC-1α pathway where EPO activates this energy sensing network or EPO regulates each energy sensor activity, respectively (Figure 2). However, further studies are required before the definitive role of EPO in this pathway is established. It would be of interest to determine the mechanism by which EPO activates AMPK phosphorylation through EPO downstream signal pathway directly or indirectly.
The protective role of EPO in metabolism related stresses
EPO and hypoxia
The protective role of EPO from stresses has been well-documented in multiple tissues such as hypoxia, reactive oxygen species (ROS), ischemia/reperfusion injury and inflammation. Some of the stresses are also associated with diseases including obesity, diabetes and neurodegenerative disease. For example, white adipose tissue is hypoxia in obese condition and has reduced mitochondrial number and activity [198]. It is believed that mitochondria respiration and biogenesis are inhibited by hypoxia [199, 200]. This possibility is also supported by the reduced number of mitochondria in adipose tissue of obese people [198]. EPO improved mitochondrial function and number in obese white adipose tissue and EPO also modulated NAD+/NADH level and increased oxidative respiration under hypoxia in adipocytes [11]. These data suggested EPO can overcome the adverse effect induced by hypoxia in adipocytes, which contributes to EPO protection from obesity. The protective effect of EPO from hypoxia has also been revealed by many other studies. Under hypoxia conditions, the kidney will produce and secrete EPO to increase red blood cells. EPO also affects neuronal protection during hypoxic conditions. EPO provides protection to embryonic and postnatal hippocampal neurons from hypoxia-induced cell death [8, 95]. EPO can also prevent neuronal apoptosis in vitro in hypoxic neurons and in vivo in cerebral ischemia in rats [96].
EPO and reactive oxygen species (ROS)
Another stress related to disease is oxidative stress. Oxidative stress, defined as a pathological state characterized by increased reactive oxygen species (ROS) production or decreased ability to detoxify ROS, plays a causative role in tissue injury in many disease conditions, including cardiovascular disease, neurological disorder, cancers, and aging. Obesity can induce systemic oxidative stress and increased oxidative stress in accumulated fat and cause dysregulation of adipocytokines and development of metabolic syndrome [201]. Increased ROS in excessively accumulated fat also causes increased oxidative stress in blood and has deleterious effects on other organs including the liver, skeletal muscle and cardiovascular system. EPO was reported to reduce oxidative stress damage in heart disease to provide cardio protection [202, 203]. EPO also has antioxidant effect in thalassemic blood cells [204]. In end-stage renal failure patients undergoing continuous ambulatory peritoneal dialysis, EPO treatment significantly reduced ROS production in polymorphonuclear leukocytes, which may contribute to anti-inflammation effect [205]. It is also believed that increased oxidative stress in fat is important underlying cause of obesity-associated metabolic disorder [201]. Therefore, how to modulate the redox state and reduce ROS production in adipose tissue can be used as a potentially useful target to develop new therapies against obesity and its associated metabolism syndrome. In the diabetic condition, oxidative stress impairs glucose uptake in muscle and fat and decreases insulin secretion form pancreatic cells [206-209]. Both oxidative stress and hypoxia are known to occur in adipose tissue in response to diet induced obesity as well as in genetic models of obesity [210]. Insulin resistance has also been associated with the generation of ROS and hypoxia can induce ROS production in adipocytes [211]. Of noting, our unpublished data found that EPO inhibits ROS production and stimulates the expression of antioxidant enzyme in adipocytes. It is possible that the antioxidant stress effect of EPO mediates the protection effects of EPO against obesity and the improvement in insulin resistance in obese mice [6, 11]. It is now clear that the overproduction of ROS in diabetes is a direct consequence of hyperglycemia and that various types of cells including renal tubular cells can produce ROS under hyperglycemic conditions. Dang et.al revealed that EPO inhibited high glucose-induced ROS generation in renal tubular cell and protected the cells from apoptosis, suggesting EPO contributes to prevention of Diabetic kidney disease via suppressing ROS production [212].
EPO and its anti-inflammatory function
Recently, some studies have revealed another important role of EPO in anti-inflammatory effect. EPO treatment has been reported to stimulate anti-inflammatory signaling, which was suggested to contribute to its direct neuroprotective effect during cerebral ischemia [213]. The administration of EPO also inhibits the inflammatory response and delays the onset of an autoimmune disease of the CNS and multiple sclerosis [214-217]. In a rat model of renal injury and in liver ischemia-reperfusion injury, EPO was observed to show anti-inflammatory activity [218, 219], suggesting the potential for EPO therapy as an anti-inflammatory agent following injury. In cardiovascular system, EPO pretreatment can suppress the systemic inflammatory response in myocardial ischemia-reperfusion injury in rodents [86]. Moreover, in chemically induced colitis, EPO administration impaired the production of several macrophage-derived inflammatory mediators such as NO, TNF-α, and IL-6 both in vitro and in vivo and decreased the production of NF-κB-inducible immune mediators, thus limiting tissue damage and ameliorating disease severity [220].
Inflammation is also now realized to be tightly associated with metabolic stress. Typically, obesity is found to induce inflammation that is causally involved in the development of insulin resistance and glucose intolerance [221, 222]. In obese patients, a chronic inflammation occurs demonstrated by increased plasma levels of inflammatory cytokines [223], which will affect metabolic homeostasis over time. Inflammation is also observed in particular in diabetic elderly patients [224]. Inflammation in pancreatic islets can reduce insulin secretion and stimulate β cell apoptosis, which plays a critical role in the progression of diabetes [225]. Emerging studies have suggested that EPO can regulate inflammatory cytokines to regulate inflammation. For instance, EPO treatment decreased IL-1β and TNF-α to attenuate the inflammatory response in injured rat brain and EPO stimulated neuroblastoma cells became resistant to TNF-α induced cell death and apoptosis [226, 227]. EPO treatment in mice mitigated the high fat diet induced inflammatory TNF-α and IL-6 production, toll like receptor 4 expression and NF-κB and JNK in the liver [228]. EPO also inhibits the activation of NF-κB, one of the central transcription factors to initiate and perpetuate inflammation, to increase the sensitivity of chemotherapeutics to promote tumor cells to undergo apoptosis [229]. In addition, EPO inhibits the induction of proinflammatory genes including TNF-α, IL-6 and inducible nitric oxide synthase (iNOS/NOS2) in activated macrophages via the blockage of nuclear factor NF-κB p65 activation by EPO in the gut [220].
Experimental and clinical data have clearly established that adipose tissue itself is an important site of inflammation during obesity. In the adipose tissue of obese mice, it was observed increased production of TNF-α and upregulated genes of inflammatory factors and increased accumulation of immune cells, which play important role in the development of insulin resistance [230-233]. Given that the important role of EPO in the adipocytes in the protection from obesity and the improvement from glucose intolerance and insulin resistance [6, 11, 58], it is reasonable to speculate that EPO may exert its anti-inflammatory function in the adipose tissue to contribute to the protection from obesity and metabolic disorder including glucose intolerance and insulin resistance. In supporting our view, Teng et al. observed increased expression of proinflammatory adipokines, Ccl1 and TNF-α but reduced IL-10 in the adipocytes from the mice with EpoR knock out in non-hematopoietic tissues that showed obese phenotype, glucose intolerance and insulin resistance, suggesting loss of EPO activity in non-hematopoietic tissue contributes to increased inflammation giving rise to insulin resistance [48]. A newly published paper also reported that EPO signaling can act as a novel regulator of white adipose tissue inflammation during diet-induced obesity. The anti-inflammatory effects of EPO were found to be driven by EPO mediated Stat3 activation to increase “anti-inflammatory” M2-like macrophages and reduce ”pro-inflammatory” M1-like macrophages [234], suggesting that an anti-inflammatory effect of EPO in adipose tissue immune system may contribute to the protective role of EPO against obesity and associated metabolism disorders. Although the detection of EpoR in macrophages of atherosclerotic lesion and in bone marrow-derived macrophages provide further evidence for EPO effects in macrophages [235, 236], further experiments are still needed to reveal the EPO effect in anti-inflammation in adipose tissue and other tissues, which will highlight the therapeutic potential of EPO in many diseases via regulating inflammation beyond its primary function in hematopoietic system.
Conclusion
The primary physiological function of the EPO/EpoR system is to stimulate erythroid progenitor cell production to provide adequate red blood cells and oxygen delivery. However, emerging evidences revealed previously less realized roles of EPO/EpoR signaling beyond hematopoietic system. For example, EPO stimulated angiogenesis via endothelial cells in cardiovascular system contributes to cardioprotection to ischemic injury. EPO also provides neuroprotection during ischemic/hypoxic stress or disease via stress induced EPO production and EpoR expression in the brain to exert the antiapoptotic, oxygen delivery and anti-inflammatory effects. The protective function and the wound healing and repair role of EPO in skeletal muscle during ischemic/injury were also demonstrated. These pleiotropic activities of EPO in non-erythroid system strongly suggest therapeutic potential of EPO beyond its traditionally primary clinical application in hematopoietic system. More interestingly, recently published data confirm the tight link between EPO/EpoR signaling and energy metabolism and homeostasis. EPO administration in mice protects mice from diet induced obesity, promotes energy expenditure, reduces fat mass accumulation and improves glucose intolerance and insulin resistance. The loss of EPO activity in non-hematopoietic system in mice leads to glucose intolerant and insulin resistant with the development of obesity. EPO activity is also demonstrated to play a role in central regulation of appetite. The crosstalk between EPO and other important energy sensors including PGC-1α, Sirt1 and AMPK further provides a mechanism for EPO regulating energy homeostasis (Figure 2). Although these novel findings highlight the possible therapeutic potential of EPO in non-hematopoietic diseases, there is still a long way to go before EPO is really used in clinical trials for these diseases. Further studies are required to uncover the detailed mechanism by which EPO regulates central control of energy homeostasis and energy metabolism in multiple tissues.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wu H, Liu X, Jaenisch R, Lodish HF. Generation of committed erythroid BFU-E and CFU-E progenitors does not require erythropoietin or the erythropoietin receptor. Cell. 1995;83:59-67
2. Miyake T, Kung CK, Goldwasser E. Purification of human erythropoietin. The Journal of biological chemistry. 1977;252:5558-64
3. Lin FK, Suggs S, Lin CH, Browne JK, Smalling R, Egrie JC. et al. Cloning and expression of the human erythropoietin gene. Proceedings of the National Academy of Sciences of the United States of America. 1985;82:7580-4
4. Jacobs K, Shoemaker C, Rudersdorf R, Neill SD, Kaufman RJ, Mufson A. et al. Isolation and characterization of genomic and cDNA clones of human erythropoietin. Nature. 1985;313:806-10
5. Noguchi CT, Wang L, Rogers HM, Teng R, Jia Y. Survival and proliferative roles of erythropoietin beyond the erythroid lineage. Expert reviews in molecular medicine. 2008;10:e36. doi:10.1017/S1462399408000860
6. Teng R, Gavrilova O, Suzuki N, Chanturiya T, Schimel D, Hugendubler L. et al. Disrupted erythropoietin signalling promotes obesity and alters hypothalamus proopiomelanocortin production. Nature communications. 2011;2:520. doi:10.1038/ncomms1526
7. Choi D, Schroer SA, Lu SY, Wang L, Wu X, Liu Y. et al. Erythropoietin protects against diabetes through direct effects on pancreatic beta cells. The Journal of experimental medicine. 2010;207:2831-42 doi:10.1084/jem.20100665
8. Yu X, Shacka JJ, Eells JB, Suarez-Quian C, Przygodzki RM, Beleslin-Cokic B. et al. Erythropoietin receptor signalling is required for normal brain development. Development. 2002;129:505-16
9. Wu H, Lee SH, Gao J, Liu X, Iruela-Arispe ML. Inactivation of erythropoietin leads to defects in cardiac morphogenesis. Development. 1999;126:3597-605
10. Kertesz N, Wu J, Chen TH, Sucov HM, Wu H. The role of erythropoietin in regulating angiogenesis. Developmental biology. 2004;276:101-10 doi:10.1016/j.ydbio.2004.08.025
11. Wang L, Teng R, Di L, Rogers H, Wu H, Kopp JB. et al. PPARalpha and Sirt1 mediate erythropoietin action in increasing metabolic activity and browning of white adipocytes to protect against obesity and metabolic disorders. Diabetes. 2013;62:4122-31 doi:10.2337/db13-0518
12. Wang L, Jia Y, Rogers H, Suzuki N, Gassmann M, Wang Q. et al. Erythropoietin contributes to slow oxidative muscle fiber specification via PGC-1alpha and AMPK activation. The international journal of biochemistry & cell biology. 2013;45:1155-64 doi:10.1016/j.biocel.2013.03.007
13. Carraway MS, Suliman HB, Jones WS, Chen CW, Babiker A, Piantadosi CA. Erythropoietin activates mitochondrial biogenesis and couples red cell mass to mitochondrial mass in the heart. Circulation research. 2010;106:1722-30 doi:10.1161/CIRCRESAHA.109.214353
14. Wang L, Jia Y, Rogers H, Wu YP, Huang S, Noguchi CT. GATA-binding protein 4 (GATA-4) and T-cell acute leukemia 1 (TAL1) regulate myogenic differentiation and erythropoietin response via cross-talk with Sirtuin1 (Sirt1). The Journal of biological chemistry. 2012;287:30157-69 doi:10.1074/jbc.M112.376640
15. Lai PH, Everett R, Wang FF, Arakawa T, Goldwasser E. Structural characterization of human erythropoietin. The Journal of biological chemistry. 1986;261:3116-21
16. Quelle FW, Wang D, Nosaka T, Thierfelder WE, Stravopodis D, Weinstein Y. et al. Erythropoietin induces activation of Stat5 through association with specific tyrosines on the receptor that are not required for a mitogenic response. Molecular and cellular biology. 1996;16:1622-31
17. Zhao W, Kitidis C, Fleming MD, Lodish HF, Ghaffari S. Erythropoietin stimulates phosphorylation and activation of GATA-1 via the PI3-kinase/AKT signaling pathway. Blood. 2006;107:907-15 doi:10.1182/blood-2005-06-2516
18. Koury ST, Bondurant MC, Koury MJ. Localization of erythropoietin synthesizing cells in murine kidneys by in situ hybridization. Blood. 1988;71:524-7
19. Maxwell PH, Osmond MK, Pugh CW, Heryet A, Nicholls LG, Tan CC. et al. Identification of the renal erythropoietin-producing cells using transgenic mice. Kidney international. 1993;44:1149-62
20. Obara N, Suzuki N, Kim K, Nagasawa T, Imagawa S, Yamamoto M. Repression via the GATA box is essential for tissue-specific erythropoietin gene expression. Blood. 2008;111:5223-32 doi:10.1182/blood-2007-10-115857
21. Tan CC, Eckardt KU, Ratcliffe PJ. Organ distribution of erythropoietin messenger RNA in normal and uremic rats. Kidney international. 1991;40:69-76
22. Fandrey J, Bunn HF. In vivo and in vitro regulation of erythropoietin mRNA: measurement by competitive polymerase chain reaction. Blood. 1993;81:617-23
23. Semenza GL, Koury ST, Nejfelt MK, Gearhart JD, Antonarakis SE. Cell-type-specific and hypoxia-inducible expression of the human erythropoietin gene in transgenic mice. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:8725-9
24. Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE. Hypoxia-inducible nuclear factors bind to an enhancer element located 3' to the human erythropoietin gene. Proceedings of the National Academy of Sciences of the United States of America. 1991;88:5680-4
25. Kochling J, Curtin PT, Madan A. Regulation of human erythropoietin gene induction by upstream flanking sequences in transgenic mice. British journal of haematology. 1998;103:960-8
26. Masuda S, Okano M, Yamagishi K, Nagao M, Ueda M, Sasaki R. A novel site of erythropoietin production. Oxygen-dependent production in cultured rat astrocytes. The Journal of biological chemistry. 1994;269:19488-93
27. Marti HH, Gassmann M, Wenger RH, Kvietikova I, Morganti-Kossmann MC, Kossmann T. et al. Detection of erythropoietin in human liquor: intrinsic erythropoietin production in the brain. Kidney international. 1997;51:416-8
28. Bernaudin M, Bellail A, Marti HH, Yvon A, Vivien D, Duchatelle I. et al. Neurons and astrocytes express EPO mRNA: oxygen-sensing mechanisms that involve the redox-state of the brain. Glia. 2000;30:271-8
29. Chavez JC, Baranova O, Lin J, Pichiule P. The transcriptional activator hypoxia inducible factor 2 (HIF-2/EPAS-1) regulates the oxygen-dependent expression of erythropoietin in cortical astrocytes. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26:9471-81 doi:10.1523/JNEUROSCI.2838-06.2006
30. Juul SE, Anderson DK, Li Y, Christensen RD. Erythropoietin and erythropoietin receptor in the developing human central nervous system. Pediatric research. 1998;43:40-9 doi:10.1203/00006450-199804001-00243
31. Juul SE, Yachnis AT, Christensen RD. Tissue distribution of erythropoietin and erythropoietin receptor in the developing human fetus. Early human development. 1998;52:235-49
32. Chikuma M, Masuda S, Kobayashi T, Nagao M, Sasaki R. Tissue-specific regulation of erythropoietin production in the murine kidney, brain, and uterus. American journal of physiology Endocrinology and metabolism. 2000;279:E1242-8
33. Yasuda Y, Fujita Y, Musha T, Tanaka H, Shiokawa S, Nakamatsu K. et al. Expression of erythropoietin in human female reproductive organs. Italian journal of anatomy and embryology = Archivio italiano di anatomia ed embriologia. 2001;106:215-22
34. Yasuda Y, Masuda S, Chikuma M, Inoue K, Nagao M, Sasaki R. Estrogen-dependent production of erythropoietin in uterus and its implication in uterine angiogenesis. The Journal of biological chemistry. 1998;273:25381-7
35. Mukundan H, Resta TC, Kanagy NL. 17Beta-estradiol decreases hypoxic induction of erythropoietin gene expression. American journal of physiology Regulatory, integrative and comparative physiology. 2002;283:R496-504 doi:10.1152/ajpregu.00573.2001
36. Ogilvie M, Yu X, Nicolas-Metral V, Pulido SM, Liu C, Ruegg UT. et al. Erythropoietin stimulates proliferation and interferes with differentiation of myoblasts. The Journal of biological chemistry. 2000;275:39754-61 doi:10.1074/jbc.M004999200
37. Jia Y, Suzuki N, Yamamoto M, Gassmann M, Noguchi CT. Endogenous erythropoietin signaling facilitates skeletal muscle repair and recovery following pharmacologically induced damage. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2012;26:2847-58 doi:10.1096/fj.11-196618
38. Rundqvist H, Rullman E, Sundberg CJ, Fischer H, Eisleitner K, Stahlberg M. et al. Activation of the erythropoietin receptor in human skeletal muscle. European journal of endocrinology / European Federation of Endocrine Societies. 2009;161:427-34 doi:10.1530/EJE-09-0342
39. Broudy VC, Lin N, Brice M, Nakamoto B, Papayannopoulou T. Erythropoietin receptor characteristics on primary human erythroid cells. Blood. 1991;77:2583-90
40. Chiba T, Ikawa Y, Todokoro K. GATA-1 transactivates erythropoietin receptor gene, and erythropoietin receptor-mediated signals enhance GATA-1 gene expression. Nucleic acids research. 1991;19:3843-8
41. Fujiwara T, O'Geen H, Keles S, Blahnik K, Linnemann AK, Kang YA. et al. Discovering hematopoietic mechanisms through genome-wide analysis of GATA factor chromatin occupancy. Molecular cell. 2009;36:667-81 doi:10.1016/j.molcel.2009.11.001
42. Chin K, Oda N, Shen K, Noguchi CT. Regulation of transcription of the human erythropoietin receptor gene by proteins binding to GATA-1 and Sp1 motifs. Nucleic acids research. 1995;23:3041-9
43. Anagnostou A, Lee ES, Kessimian N, Levinson R, Steiner M. Erythropoietin has a mitogenic and positive chemotactic effect on endothelial cells. Proceedings of the National Academy of Sciences of the United States of America. 1990;87:5978-82
44. Anagnostou A, Liu Z, Steiner M, Chin K, Lee ES, Kessimian N. et al. Erythropoietin receptor mRNA expression in human endothelial cells. Proceedings of the National Academy of Sciences of the United States of America. 1994;91:3974-8
45. Beleslin-Cokic BB, Cokic VP, Yu X, Weksler BB, Schechter AN, Noguchi CT. Erythropoietin and hypoxia stimulate erythropoietin receptor and nitric oxide production by endothelial cells. Blood. 2004;104:2073-80 doi:10.1182/blood-2004-02-0744
46. Beleslin-Cokic BB, Cokic VP, Wang L, Piknova B, Teng R, Schechter AN. et al. Erythropoietin and hypoxia increase erythropoietin receptor and nitric oxide levels in lung microvascular endothelial cells. Cytokine. 2011;54:129-35 doi:10.1016/j.cyto.2011.01.015
47. Westenbrink BD, Lipsic E, van der Meer P, van der Harst P, Oeseburg H, Du Marchie Sarvaas GJ. et al. Erythropoietin improves cardiac function through endothelial progenitor cell and vascular endothelial growth factor mediated neovascularization. European heart journal. 2007;28:2018-27 doi:10.1093/eurheartj/ehm177
48. Teng R, Calvert JW, Sibmooh N, Piknova B, Suzuki N, Sun J. et al. Acute erythropoietin cardioprotection is mediated by endothelial response. Basic research in cardiology. 2011;106:343-54 doi:10.1007/s00395-011-0158-z
49. Zhande R, Karsan A. Erythropoietin promotes survival of primary human endothelial cells through PI3K-dependent, NF-kappaB-independent upregulation of Bcl-xL. American journal of physiology Heart and circulatory physiology. 2007;292:H2467-74 doi:10.1152/ajpheart.00649.2006
50. Tsai PT, Ohab JJ, Kertesz N, Groszer M, Matter C, Gao J. et al. A critical role of erythropoietin receptor in neurogenesis and post-stroke recovery. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26:1269-74 doi:10.1523/JNEUROSCI.4480-05.2006
51. Liu C, Shen K, Liu Z, Noguchi CT. Regulated human erythropoietin receptor expression in mouse brain. The Journal of biological chemistry. 1997;272:32395-400
52. Marti HH, Wenger RH, Rivas LA, Straumann U, Digicaylioglu M, Henn V. et al. Erythropoietin gene expression in human, monkey and murine brain. The European journal of neuroscience. 1996;8:666-76
53. Digicaylioglu M, Bichet S, Marti HH, Wenger RH, Rivas LA, Bauer C. et al. Localization of specific erythropoietin binding sites in defined areas of the mouse brain. Proceedings of the National Academy of Sciences of the United States of America. 1995;92:3717-20
54. Nagai A, Nakagawa E, Choi HB, Hatori K, Kobayashi S, Kim SU. Erythropoietin and erythropoietin receptors in human CNS neurons, astrocytes, microglia, and oligodendrocytes grown in culture. Journal of neuropathology and experimental neurology. 2001;60:386-92
55. Pan Y, Shu JL, Gu HF, Zhou DC, Liu XL, Qiao QY. et al. Erythropoietin improves insulin resistance via the regulation of its receptor-mediated signaling pathways in 3T3L1 adipocytes. Molecular and cellular endocrinology. 2013;367:116-23 doi:10.1016/j.mce.2012.12.027
56. Mikolas E, Cseh J, Pap M, Szijarto IA, Balogh A, Laczy B. et al. Effects of erythropoietin on glucose metabolism. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 2012;44:279-85 doi:10.1055/s-0032-1301901
57. Hojman P, Brolin C, Gissel H, Brandt C, Zerahn B, Pedersen BK. et al. Erythropoietin over-expression protects against diet-induced obesity in mice through increased fat oxidation in muscles. PloS one. 2009;4:e5894. doi:10.1371/journal.pone.0005894
58. Katz O, Stuible M, Golishevski N, Lifshitz L, Tremblay ML, Gassmann M. et al. Erythropoietin treatment leads to reduced blood glucose levels and body mass: insights from murine models. The Journal of endocrinology. 2010;205:87-95 doi:10.1677/JOE-09-0425
59. Nedergaard J, Bengtsson T, Cannon B. Unexpected evidence for active brown adipose tissue in adult humans. American journal of physiology Endocrinology and metabolism. 2007;293:E444-52 doi:10.1152/ajpendo.00691.2006
60. Cypess AM, Lehman S, Williams G, Tal I, Rodman D, Goldfine AB. et al. Identification and importance of brown adipose tissue in adult humans. The New England journal of medicine. 2009;360:1509-17 doi:10.1056/NEJMoa0810780
61. Surwit RS, Kuhn CM, Cochrane C, McCubbin JA, Feinglos MN. Diet-induced type II diabetes in C57BL/6J mice. Diabetes. 1988;37:1163-7
62. Surwit RS, Feinglos MN, Rodin J, Sutherland A, Petro AE, Opara EC. et al. Differential effects of fat and sucrose on the development of obesity and diabetes in C57BL/6J and A/J mice. Metabolism: clinical and experimental. 1995;44:645-51
63. Toye AA, Lippiat JD, Proks P, Shimomura K, Bentley L, Hugill A. et al. A genetic and physiological study of impaired glucose homeostasis control in C57BL/6J mice. Diabetologia. 2005;48:675-86 doi:10.1007/s00125-005-1680-z
64. Luk CT, Shi SY, Choi D, Cai EP, Schroer SA, Woo M. In vivo knockdown of adipocyte erythropoietin receptor does not alter glucose or energy homeostasis. Endocrinology. 2013;154:3652-9 doi:10.1210/en.2013-1113
65. Fenjves ES, Ochoa MS, Cabrera O, Mendez AJ, Kenyon NS, Inverardi L. et al. Human, nonhuman primate, and rat pancreatic islets express erythropoietin receptors. Transplantation. 2003;75:1356-60 doi:10.1097/01.TP.0000062862.88375.BD
66. Fenjves ES, Ochoa MS, Gay-Rabinstein C, Molano RD, Pileggi A, Mendez AJ. et al. Adenoviral gene transfer of erythropoietin confers cytoprotection to isolated pancreatic islets. Transplantation. 2004;77:13-8 doi:10.1097/01.TP.0000110422.27977.26
67. Bianchi R, Buyukakilli B, Brines M, Savino C, Cavaletti G, Oggioni N. et al. Erythropoietin both protects from and reverses experimental diabetic neuropathy. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:823-8 doi:10.1073/pnas.0307823100
68. Shuai H, Zhang J, Zhang J, Xie J, Zhang M, Yu Y. et al. Erythropoietin protects pancreatic beta-cell line NIT-1 cells against cytokine-induced apoptosis via phosphatidylinositol 3-kinase/Akt signaling. Endocrine research. 2011;36:25-34 doi:10.3109/07435800.2010.534753
69. Jia Y, Warin R, Yu X, Epstein R, Noguchi CT. Erythropoietin signaling promotes transplanted progenitor cell survival. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2009;23:3089-99 doi:10.1096/fj.09-130237
70. Thomsen JJ, Rentsch RL, Robach P, Calbet JA, Boushel R, Rasmussen P. et al. Prolonged administration of recombinant human erythropoietin increases submaximal performance more than maximal aerobic capacity. European journal of applied physiology. 2007;101:481-6 doi:10.1007/s00421-007-0522-8
71. Plenge U, Belhage B, Guadalupe-Grau A, Andersen PR, Lundby C, Dela F. et al. Erythropoietin treatment enhances muscle mitochondrial capacity in humans. Frontiers in physiology. 2012;3:50. doi:10.3389/fphys.2012.00050
72. Zierath JR, Hawley JA. Skeletal muscle fiber type: influence on contractile and metabolic properties. PLoS biology. 2004;2:e348. doi:10.1371/journal.pbio.0020348
73. Gaster M, Poulsen P, Handberg A, Schroder HD, Beck-Nielsen H. Direct evidence of fiber type-dependent GLUT-4 expression in human skeletal muscle. American journal of physiology Endocrinology and metabolism. 2000;278:E910-6
74. Gaster M, Staehr P, Beck-Nielsen H, Schroder HD, Handberg A. GLUT4 is reduced in slow muscle fibers of type 2 diabetic patients: is insulin resistance in type 2 diabetes a slow, type 1 fiber disease? Diabetes. 2001;50:1324-9
75. Szendroedi J, Phielix E, Roden M. The role of mitochondria in insulin resistance and type 2 diabetes mellitus. Nature reviews Endocrinology. 2012;8:92-103 doi:10.1038/nrendo.2011.138
76. Sleigh A, Raymond-Barker P, Thackray K, Porter D, Hatunic M, Vottero A. et al. Mitochondrial dysfunction in patients with primary congenital insulin resistance. The Journal of clinical investigation. 2011;121:2457-61 doi:10.1172/JCI46405
77. Hoeks J, van Herpen NA, Mensink M, Moonen-Kornips E, van Beurden D, Hesselink MK. et al. Prolonged fasting identifies skeletal muscle mitochondrial dysfunction as consequence rather than cause of human insulin resistance. Diabetes. 2010;59:2117-25 doi:10.2337/db10-0519
78. Cayla JL, Maire P, Duvallet A, Wahrmann JP. Erythropoietin induces a shift of muscle phenotype from fast glycolytic to slow oxidative. International journal of sports medicine. 2008;29:460-5 doi:10.1055/s-2007-965359
79. Lin J, Wu H, Tarr PT, Zhang CY, Wu Z, Boss O. et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797-801 doi:10.1038/nature00904
80. Ljubicic V, Miura P, Burt M, Boudreault L, Khogali S, Lunde JA. et al. Chronic AMPK activation evokes the slow, oxidative myogenic program and triggers beneficial adaptations in mdx mouse skeletal muscle. Human molecular genetics. 2011;20:3478-93 doi:10.1093/hmg/ddr265
81. Suzuki N, Ohneda O, Takahashi S, Higuchi M, Mukai HY, Nakahata T. et al. Erythroid-specific expression of the erythropoietin receptor rescued its null mutant mice from lethality. Blood. 2002;100:2279-88 doi:10.1182/blood-2002-01-0124
82. Salisch S, Klar M, Thurisch B, Bungert J, Dame C. Gata4 and Sp1 regulate expression of the erythropoietin receptor in cardiomyocytes. Journal of cellular and molecular medicine. 2011;15:1963-72 doi:10.1111/j.1582-4934.2010.01193.x
83. Wright GL, Hanlon P, Amin K, Steenbergen C, Murphy E, Arcasoy MO. Erythropoietin receptor expression in adult rat cardiomyocytes is associated with an acute cardioprotective effect for recombinant erythropoietin during ischemia-reperfusion injury. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2004;18:1031-3 doi:10.1096/fj.03-1289fje
84. Parsa CJ, Matsumoto A, Kim J, Riel RU, Pascal LS, Walton GB. et al. A novel protective effect of erythropoietin in the infarcted heart. The Journal of clinical investigation. 2003;112:999-1007 doi:10.1172/JCI18200
85. Calvillo L, Latini R, Kajstura J, Leri A, Anversa P, Ghezzi P. et al. Recombinant human erythropoietin protects the myocardium from ischemia-reperfusion injury and promotes beneficial remodeling. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:4802-6 doi:10.1073/pnas.0630444100
86. Rui T, Feng Q, Lei M, Peng T, Zhang J, Xu M. et al. Erythropoietin prevents the acute myocardial inflammatory response induced by ischemia/reperfusion via induction of AP-1. Cardiovascular research. 2005;65:719-27 doi:10.1016/j.cardiores.2004.11.019
87. Burger D, Lei M, Geoghegan-Morphet N, Lu X, Xenocostas A, Feng Q. Erythropoietin protects cardiomyocytes from apoptosis via up-regulation of endothelial nitric oxide synthase. Cardiovascular research. 2006;72:51-9 doi:10.1016/j.cardiores.2006.06.026
88. Ruschitzka FT, Wenger RH, Stallmach T, Quaschning T, de Wit C, Wagner K. et al. Nitric oxide prevents cardiovascular disease and determines survival in polyglobulic mice overexpressing erythropoietin. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:11609-13 doi:10.1073/pnas.97.21.11609
89. Shen Y, Wang Y, Li D, Wang C, Xu B, Dong G. et al. Recombinant human erythropoietin pretreatment attenuates heart ischemia-reperfusion injury in rats by suppressing the systemic inflammatory response. Transplantation proceedings. 2010;42:1595-7 doi:10.1016/j.transproceed.2009.11.050
90. Liu X, Shen J, Jin Y, Duan M, Xu J. Recombinant human erythropoietin (rhEPO) preconditioning on nuclear factor-kappa B (NF-kB) activation & proinflammatory cytokines induced by myocardial ischaemia-reperfusion. The Indian journal of medical research. 2006;124:343-54
91. Yamada M, Miyakawa T, Duttaroy A, Yamanaka A, Moriguchi T, Makita R. et al. Mice lacking the M3 muscarinic acetylcholine receptor are hypophagic and lean. Nature. 2001;410:207-12 doi:10.1038/35065604
92. Belgardt BF, Husch A, Rother E, Ernst MB, Wunderlich FT, Hampel B. et al. PDK1 deficiency in POMC-expressing cells reveals FOXO1-dependent and -independent pathways in control of energy homeostasis and stress response. Cell metabolism. 2008;7:291-301 doi:10.1016/j.cmet.2008.01.006
93. Mesaros A, Koralov SB, Rother E, Wunderlich FT, Ernst MB, Barsh GS. et al. Activation of Stat3 signaling in AgRP neurons promotes locomotor activity. Cell metabolism. 2008;7:236-48 doi:10.1016/j.cmet.2008.01.007
94. Morishita E, Masuda S, Nagao M, Yasuda Y, Sasaki R. Erythropoietin receptor is expressed in rat hippocampal and cerebral cortical neurons, and erythropoietin prevents in vitro glutamate-induced neuronal death. Neuroscience. 1997;76:105-16
95. Lewczuk P, Hasselblatt M, Kamrowski-Kruck H, Heyer A, Unzicker C, Siren AL. et al. Survival of hippocampal neurons in culture upon hypoxia: effect of erythropoietin. Neuroreport. 2000;11:3485-8
96. Siren AL, Fratelli M, Brines M, Goemans C, Casagrande S, Lewczuk P. et al. Erythropoietin prevents neuronal apoptosis after cerebral ischemia and metabolic stress. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:4044-9 doi:10.1073/pnas.051606598
97. Sola A, Rogido M, Lee BH, Genetta T, Wen TC. Erythropoietin after focal cerebral ischemia activates the Janus kinase-signal transducer and activator of transcription signaling pathway and improves brain injury in postnatal day 7 rats. Pediatric research. 2005;57:481-7 doi:10.1203/01.PDR.0000155760.88664.06
98. Kilic E, Kilic U, Soliz J, Bassetti CL, Gassmann M, Hermann DM. Brain-derived erythropoietin protects from focal cerebral ischemia by dual activation of ERK-1/-2 and Akt pathways. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2005;19:2026-8 doi:10.1096/fj.05-3941fje
99. Suzuki N, Ohneda O, Takahashi S, Higuchi M, Mukai HY, Nakahata T. et al. Erythroid-specific expression of the erythropoietin receptor rescued its null mutant mice from lethality. Blood. 2002;100:2279-88 doi:10.1182/blood-2002-01-0124
100. Sadamoto Y, Igase K, Sakanaka M, Sato K, Otsuka H, Sakaki S. et al. Erythropoietin prevents place navigation disability and cortical infarction in rats with permanent occlusion of the middle cerebral artery. Biochemical and biophysical research communications. 1998;253:26-32 doi:10.1006/bbrc.1998.9748
101. Bernaudin M, Nedelec AS, Divoux D, MacKenzie ET, Petit E, Schumann-Bard P. Normobaric hypoxia induces tolerance to focal permanent cerebral ischemia in association with an increased expression of hypoxia-inducible factor-1 and its target genes, erythropoietin and VEGF, in the adult mouse brain. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2002;22:393-403 doi:10.1097/00004647-200204000-00003
102. Mohamad O, Chen D, Zhang L, Hofmann C, Wei L, Yu SP. Erythropoietin reduces neuronal cell death and hyperalgesia induced by peripheral inflammatory pain in neonatal rats. Molecular pain. 2011;7:51. doi:10.1186/1744-8069-7-51
103. Wang L, Chopp M, Gregg SR, Zhang RL, Teng H, Jiang A. et al. Neural progenitor cells treated with EPO induce angiogenesis through the production of VEGF. Journal of cerebral blood flow and metabolism: official journal of the International Society of Cerebral Blood Flow and Metabolism. 2008;28:1361-8 doi:10.1038/jcbfm.2008.32
104. Chen ZY, Wang L, Asavaritkrai P, Noguchi CT. Up-regulation of erythropoietin receptor by nitric oxide mediates hypoxia preconditioning. Journal of neuroscience research. 2010;88:3180-8 doi:10.1002/jnr.22473
105. Penna F, Busquets S, Toledo M, Pin F, Massa D, Lopez-Soriano FJ. et al. Erythropoietin administration partially prevents adipose tissue loss in experimental cancer cachexia models. Journal of lipid research. 2013;54:3045-51 doi:10.1194/jlr.M038406
106. Littlewood TJ, Bajetta E, Nortier JW, Vercammen E, Rapoport B, Epoetin Alfa Study G. Effects of epoetin alfa on hematologic parameters and quality of life in cancer patients receiving nonplatinum chemotherapy: results of a randomized, double-blind, placebo-controlled trial. Journal of clinical oncology: official journal of the American Society of Clinical Oncology. 2001;19:2865-74
107. Vansteenkiste J, Pirker R, Massuti B, Barata F, Font A, Fiegl M. et al. Double-blind, placebo-controlled, randomized phase III trial of darbepoetin alfa in lung cancer patients receiving chemotherapy. Journal of the National Cancer Institute. 2002;94:1211-20
108. Hedenus M, Adriansson M, San Miguel J, Kramer MH, Schipperus MR, Juvonen E. et al. Efficacy and safety of darbepoetin alfa in anaemic patients with lymphoproliferative malignancies: a randomized, double-blind, placebo-controlled study. British journal of haematology. 2003;122:394-403
109. Ribatti D, Marzullo A, Gentile A, Longo V, Nico B, Vacca A. et al. Erythropoietin/erythropoietin-receptor system is involved in angiogenesis in human hepatocellular carcinoma. Histopathology. 2007;50:591-6 doi:10.1111/j.1365-2559.2007.02654.x
110. Wiesener MS, Munchenhagen P, Glaser M, Sobottka BA, Knaup KX, Jozefowski K. et al. Erythropoietin gene expression in renal carcinoma is considerably more frequent than paraneoplastic polycythemia. International journal of cancer Journal international du cancer. 2007;121:2434-42 doi:10.1002/ijc.22961
111. Nico B, Annese T, Guidolin D, Finato N, Crivellato E, Ribatti D. Epo is involved in angiogenesis in human glioma. Journal of neuro-oncology. 2011;102:51-8 doi:10.1007/s11060-010-0294-6
112. Henke M, Laszig R, Rube C, Schafer U, Haase KD, Schilcher B. et al. Erythropoietin to treat head and neck cancer patients with anaemia undergoing radiotherapy: randomised, double-blind, placebo-controlled trial. Lancet. 2003;362:1255-60 doi:10.1016/S0140-6736(03)14567-9
113. Leyland-Jones B, Investigators B, Study G. Breast cancer trial with erythropoietin terminated unexpectedly. The lancet oncology. 2003;4:459-60
114. Bohlius J, Schmidlin K, Brillant C, Schwarzer G, Trelle S, Seidenfeld J. et al. Recombinant human erythropoiesis-stimulating agents and mortality in patients with cancer: a meta-analysis of randomised trials. Lancet. 2009;373:1532-42 doi:10.1016/S0140-6736(09)60502-X
115. Hedley BD, Allan AL, Xenocostas A. The role of erythropoietin and erythropoiesis-stimulating agents in tumor progression. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17:6373-80 doi:10.1158/1078-0432.CCR-10-2577
116. Janmaat ML, Heerkens JL, de Bruin AM, Klous A, de Waard V, de Vries CJ. Erythropoietin accelerates smooth muscle cell-rich vascular lesion formation in mice through endothelial cell activation involving enhanced PDGF-BB release. Blood. 2010;115:1453-60 doi:10.1182/blood-2009-07-230870
117. Mak RH. Effect of recombinant human erythropoietin on insulin, amino acid, and lipid metabolism in uremia. The Journal of pediatrics. 1996;129:97-104
118. Allegra V, Mengozzi G, Martimbianco L, Vasile A. Early and late effects of erythropoietin on glucose metabolism in maintenance hemodialysis patients. American journal of nephrology. 1996;16:304-8
119. Bofill C, Joven J, Bages J, Vilella E, Sans T, Cavalle P. et al. Response to repeated phlebotomies in patients with non-insulin-dependent diabetes mellitus. Metabolism: clinical and experimental. 1994;43:614-20
120. Tuzcu A, Bahceci M, Yilmaz E, Bahceci S, Tuzcu S. The comparison of insulin sensitivity in non-diabetic hemodialysis patients treated with and without recombinant human erythropoietin. Hormone and metabolic research = Hormon- und Stoffwechselforschung = Hormones et metabolisme. 2004;36:716-20 doi:10.1055/s-2004-826021
121. Prata MM, Madeira C, Vicente O, Miguel MJ. Lipid profile in haemodialysis patients treated with recombinant human erythropoietin. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 1998;13:2345-7
122. Lundby C, Robach P, Boushel R, Thomsen JJ, Rasmussen P, Koskolou M. et al. Does recombinant human Epo increase exercise capacity by means other than augmenting oxygen transport? Journal of applied physiology. 2008;105:581-7 doi:10.1152/japplphysiol.90484.2008
123. Christensen B, Vendelbo MH, Krusenstjerna-Hafstrøm T, Madsen M, Pedersen SB, Jessen N. et al. Erythropoietin administration acutely stimulates resting energy expenditure in healthy young men. Journal of applied physiology (Bethesda, Md: 1985). 2012;112:1114-21 doi:10.1152/japplphysiol.01391.2011
124. McMahon LP, Johns JA, McKenzie A, Austin M, Fowler R, Dawborn JK. Haemodynamic changes and physical performance at comparative levels of haemoglobin after long-term treatment with recombinant erythropoietin. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 1992;7:1199-206
125. Clyne N, Jogestrand T. Effect of erythropoietin treatment on physical exercise capacity and on renal function in predialytic uremic patients. Nephron. 1992;60:390-6
126. Fagher B, Thysell H, Monti M. Effect of erythropoietin on muscle metabolic rate, as measured by direct microcalorimetry, and ATP in hemodialysis patients. Nephron. 1994;67:167-71
127. Fagher B, Thysell H, Monti M. Effect of erythropoietin on muscle metabolic rate, as measured by direct microcalorimetry, and ATP in hemodialysis patients. Nephron. 1994;67:167-71
128. Borissova AM, Djambazova A, Todorov K, Dakovska L, Tankova T, Kirilov G. Effect of erythropoietin on the metabolic state and peripheral insulin sensitivity in diabetic patients on haemodialysis. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 1993;8:93
129. Chagnac A, Weinstein T, Zevin D, Korzets A, Hirsh J, Gafter U. et al. Effects of erythropoietin on glucose tolerance in hemodialysis patients. Clinical nephrology. 1994;42:398-400
130. Lin CS, Lim SK, D'Agati V, Costantini F. Differential effects of an erythropoietin receptor gene disruption on primitive and definitive erythropoiesis. Genes & development. 1996;10:154-64
131. Menne J, Park JK, Shushakova N, Mengel M, Meier M, Fliser D. The continuous erythropoietin receptor activator affects different pathways of diabetic renal injury. Journal of the American Society of Nephrology: JASN. 2007;18:2046-53 doi:10.1681/ASN.2006070699
132. Shushakova N, Park JK, Menne J, Fliser D. Chronic erythropoietin treatment affects different molecular pathways of diabetic cardiomyopathy in mouse. European journal of clinical investigation. 2009;39:755-60 doi:10.1111/j.1365-2362.2009.02165.x
133. Bunn HF. Erythropoietin. Cold Spring Harbor perspectives in medicine. 2013;3:a011619. doi:10.1101/cshperspect.a011619
134. Bournat JC, Brown CW. Mitochondrial dysfunction in obesity. Current opinion in endocrinology, diabetes, and obesity. 2010;17:446-52 doi:10.1097/MED.0b013e32833c3026
135. Lowell BB, Shulman GI. Mitochondrial dysfunction and type 2 diabetes. Science. 2005;307:384-7 doi:10.1126/science.1104343
136. Petersen KF, Befroy D, Dufour S, Dziura J, Ariyan C, Rothman DL. et al. Mitochondrial dysfunction in the elderly: possible role in insulin resistance. Science. 2003;300:1140-2 doi:10.1126/science.1082889
137. Qin C, Zhou S, Xiao Y, Chen L. Erythropoietin enhances mitochondrial biogenesis in cardiomyocytes exposed to chronic hypoxia through Akt/eNOS signalling pathway. Cell biology international. 2014;38:335-42 doi:10.1002/cbin.10205
138. Chong ZZ, Kang JQ, Maiese K. Erythropoietin is a novel vascular protectant through activation of Akt1 and mitochondrial modulation of cysteine proteases. Circulation. 2002;106:2973-9
139. Li AC, Glass CK. PPAR- and LXR-dependent pathways controlling lipid metabolism and the development of atherosclerosis. Journal of lipid research. 2004;45:2161-73 doi:10.1194/jlr.R400010-JLR200
140. Christensen B, Vendelbo MH, Krusenstjerna-Hafstrom T, Madsen M, Pedersen SB, Jessen N. et al. Erythropoietin administration acutely stimulates resting energy expenditure in healthy young men. Journal of applied physiology. 2012;112:1114-21 doi:10.1152/japplphysiol.01391.2011
141. St-Pierre J, Lin J, Krauss S, Tarr PT, Yang R, Newgard CB. et al. Bioenergetic analysis of peroxisome proliferator-activated receptor gamma coactivators 1alpha and 1beta (PGC-1alpha and PGC-1beta) in muscle cells. The Journal of biological chemistry. 2003;278:26597-603 doi:10.1074/jbc.M301850200
142. Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. The Journal of clinical investigation. 2000;106:847-56 doi:10.1172/JCI10268
143. Wu Z, Puigserver P, Andersson U, Zhang C, Adelmant G, Mootha V. et al. Mechanisms controlling mitochondrial biogenesis and respiration through the thermogenic coactivator PGC-1. Cell. 1999;98:115-24 doi:10.1016/S0092-8674(00)80611-X
144. Canto C, Auwerx J. PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Current opinion in lipidology. 2009;20:98-105 doi:10.1097/MOL.0b013e328328d0a4
145. Chau MD, Gao J, Yang Q, Wu Z, Gromada J. Fibroblast growth factor 21 regulates energy metabolism by activating the AMPK-SIRT1-PGC-1alpha pathway. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:12553-8 doi:10.1073/pnas.1006962107
146. Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F. et al. Insulin-regulated hepatic gluconeogenesis through FOXO1-PGC-1alpha interaction. Nature. 2003;423:550-5 doi:10.1038/nature01667
147. Yoon JC, Puigserver P, Chen G, Donovan J, Wu Z, Rhee J. et al. Control of hepatic gluconeogenesis through the transcriptional coactivator PGC-1. Nature. 2001;413:131-8 doi:10.1038/35093050
148. Vega RB, Huss JM, Kelly DP. The coactivator PGC-1 cooperates with peroxisome proliferator-activated receptor alpha in transcriptional control of nuclear genes encoding mitochondrial fatty acid oxidation enzymes. Molecular and cellular biology. 2000;20:1868-76
149. Lage R, Dieguez C, Vidal-Puig A, Lopez M. AMPK: a metabolic gauge regulating whole-body energy homeostasis. Trends Mol Med. 2008;14:539-49
150. Hardie DG. AMP-activated/SNF1 protein kinases: conserved guardians of cellular energy. Nat Rev Mol Cell Biol. 2007;8:774-85
151. Rodgers JT, Lerin C, Haas W, Gygi SP, Spiegelman BM, Puigserver P. Nutrient control of glucose homeostasis through a complex of PGC-1alpha and SIRT1. Nature. 2005;434:113-8 doi:10.1038/nature03354
152. Yuen CM, Sun CK, Lin YC, Chang LT, Kao YH, Yen CH. et al. Combination of cyclosporine and erythropoietin improves brain infarct size and neurological function in rats after ischemic stroke. Journal of translational medicine. 2011;9:141. doi:10.1186/1479-5876-9-141
153. Menard R. Medicine: knockout malaria vaccine? Nature. 2005;433:113-4 doi:10.1038/433113a
154. Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature. 2000;403:795-800 doi:10.1038/35001622
155. Landry J, Sutton A, Tafrov ST, Heller RC, Stebbins J, Pillus L. et al. The silencing protein SIR2 and its homologs are NAD-dependent protein deacetylases. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:5807-11 doi:10.1073/pnas.110148297
156. Smith JS, Brachmann CB, Celic I, Kenna MA, Muhammad S, Starai VJ. et al. A phylogenetically conserved NAD+-dependent protein deacetylase activity in the Sir2 protein family. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:6658-63
157. Haigis MC, Mostoslavsky R, Haigis KM, Fahie K, Christodoulou DC, Murphy AJ. et al. SIRT4 inhibits glutamate dehydrogenase and opposes the effects of calorie restriction in pancreatic beta cells. Cell. 2006;126:941-54 doi:10.1016/j.cell.2006.06.057
158. Moynihan KA, Grimm AA, Plueger MM, Bernal-Mizrachi E, Ford E, Cras-Meneur C. et al. Increased dosage of mammalian Sir2 in pancreatic beta cells enhances glucose-stimulated insulin secretion in mice. Cell metabolism. 2005;2:105-17 doi:10.1016/j.cmet.2005.07.001
159. Kitamura YI, Kitamura T, Kruse JP, Raum JC, Stein R, Gu W. et al. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell metabolism. 2005;2:153-63 doi:10.1016/j.cmet.2005.08.004
160. Canto C, Gerhart-Hines Z, Feige JN, Lagouge M, Noriega L, Milne JC. et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458:1056-60 doi:10.1038/nature07813
161. Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R. et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature. 2004;429:771-6 doi:10.1038/nature02583
162. Baur JA, Pearson KJ, Price NL, Jamieson HA, Lerin C, Kalra A. et al. Resveratrol improves health and survival of mice on a high-calorie diet. Nature. 2006;444:337-42 doi:10.1038/nature05354
163. Lagouge M, Argmann C, Gerhart-Hines Z, Meziane H, Lerin C, Daussin F. et al. Resveratrol improves mitochondrial function and protects against metabolic disease by activating SIRT1 and PGC-1alpha. Cell. 2006;127:1109-22 doi:10.1016/j.cell.2006.11.013
164. Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ. et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature. 2007;450:712-6 doi:10.1038/nature06261
165. Timmons JA, Wennmalm K, Larsson O, Walden TB, Lassmann T, Petrovic N. et al. Myogenic gene expression signature establishes that brown and white adipocytes originate from distinct cell lineages. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:4401-6 doi:10.1073/pnas.0610615104
166. Bordone L, Motta MC, Picard F, Robinson A, Jhala US, Apfeld J. et al. Sirt1 regulates insulin secretion by repressing UCP2 in pancreatic beta cells. PLoS biology. 2006;4:e31. doi:10.1371/journal.pbio.0040031
167. Cakir I, Perello M, Lansari O, Messier NJ, Vaslet CA, Nillni EA. Hypothalamic Sirt1 regulates food intake in a rodent model system. PloS one. 2009;4:e8322. doi:10.1371/journal.pone.0008322
168. Ramadori G, Fujikawa T, Fukuda M, Anderson J, Morgan DA, Mostoslavsky R. et al. SIRT1 deacetylase in POMC neurons is required for homeostatic defenses against diet-induced obesity. Cell metabolism. 2010;12:78-87 doi:10.1016/j.cmet.2010.05.010
169. Ramadori G, Gautron L, Fujikawa T, Vianna CR, Elmquist JK, Coppari R. Central administration of resveratrol improves diet-induced diabetes. Endocrinology. 2009;150:5326-33 doi:10.1210/en.2009-0528
170. Gerhart-Hines Z, Rodgers JT, Bare O, Lerin C, Kim SH, Mostoslavsky R. et al. Metabolic control of muscle mitochondrial function and fatty acid oxidation through SIRT1/PGC-1alpha. The EMBO journal. 2007;26:1913-23 doi:10.1038/sj.emboj.7601633
171. Dioum EM, Chen R, Alexander MS, Zhang Q, Hogg RT, Gerard RD. et al. Regulation of hypoxia-inducible factor 2alpha signaling by the stress-responsive deacetylase sirtuin 1. Science. 2009;324:1289-93 doi:10.1126/science.1169956
172. Hou J, Wang S, Shang YC, Chong ZZ, Maiese K. Erythropoietin employs cell longevity pathways of SIRT1 to foster endothelial vascular integrity during oxidant stress. Current neurovascular research. 2011;8:220-35
173. Houtkooper RH, Canto C, Wanders RJ, Auwerx J. The secret life of NAD+: an old metabolite controlling new metabolic signaling pathways. Endocrine reviews. 2010;31:194-223 doi:10.1210/er.2009-0026
174. Houtkooper RH, Auwerx J. Exploring the therapeutic space around NAD+. The Journal of cell biology. 2012;199:205-9 doi:10.1083/jcb.201207019
175. Canto C, Houtkooper RH, Pirinen E, Youn DY, Oosterveer MH, Cen Y. et al. The NAD(+) precursor nicotinamide riboside enhances oxidative metabolism and protects against high-fat diet-induced obesity. Cell metabolism. 2012;15:838-47 doi:10.1016/j.cmet.2012.04.022
176. Hwang JH, Kim DW, Jo EJ, Kim YK, Jo YS, Park JH. et al. Pharmacological stimulation of NADH oxidation ameliorates obesity and related phenotypes in mice. Diabetes. 2009;58:965-74
177. Kao R, Xenocostas A, Rui T, Yu P, Huang W, Rose J. et al. Erythropoietin improves skeletal muscle microcirculation and tissue bioenergetics in a mouse sepsis model. Critical care. 2007;11:R58. doi:10.1186/cc5920
178. Di LJ, Fernandez AG, De Siervi A, Longo DL, Gardner K. Transcriptional regulation of BRCA1 expression by a metabolic switch. Nat Struct Mol Biol. 2010;17:1406-13
179. Nyengaard JR, Ido Y, Kilo C, Williamson JR. Interactions between hyperglycemia and hypoxia: implications for diabetic retinopathy. Diabetes. 2004;53:2931-8
180. Fulco M, Schiltz RL, Iezzi S, King MT, Zhao P, Kashiwaya Y. et al. Sir2 regulates skeletal muscle differentiation as a potential sensor of the redox state. Molecular cell. 2003;12:51-62
181. Narkar VA, Downes M, Yu RT, Embler E, Wang YX, Banayo E. et al. AMPK and PPARdelta agonists are exercise mimetics. Cell. 2008;134:405-15
182. Cool B, Zinker B, Chiou W, Kifle L, Cao N, Perham M. et al. Identification and characterization of a small molecule AMPK activator that treats key components of type 2 diabetes and the metabolic syndrome. Cell Metab. 2006;3:403-16
183. Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res. 2007;100:328-41
184. Giri S, Rattan R, Haq E, Khan M, Yasmin R, Won JS. et al. AICAR inhibits adipocyte differentiation in 3T3L1 and restores metabolic alterations in diet-induced obesity mice model. Nutr Metab (Lond). 2006;3:31
185. Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J. et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108:1167-74
186. Fryer LG, Parbu-Patel A, Carling D. The Anti-diabetic drugs rosiglitazone and metformin stimulate AMP-activated protein kinase through distinct signaling pathways. J Biol Chem. 2002;277:25226-32
187. Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S. et al. PRDM16 controls a brown fat/skeletal muscle switch. Nature. 2008;454:961-7
188. Bostrom P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC. et al. A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature. 2012;481:463-8
189. Sun L, Xie H, Mori MA, Alexander R, Yuan B, Hattangadi SM. et al. Mir193b-365 is essential for brown fat differentiation. Nat Cell Biol. 2011;13:958-65
190. Ahmadian M, Abbott MJ, Tang T, Hudak CS, Kim Y, Bruss M. et al. Desnutrin/ATGL is regulated by AMPK and is required for a brown adipose phenotype. Cell Metab. 2011;13:739-48
191. Kajimura S, Seale P, Tomaru T, Erdjument-Bromage H, Cooper MP, Ruas JL. et al. Regulation of the brown and white fat gene programs through a PRDM16/CtBP transcriptional complex. Genes Dev. 2008;22:1397-409
192. Seale P, Conroe HM, Estall J, Kajimura S, Frontini A, Ishibashi J. et al. Prdm16 determines the thermogenic program of subcutaneous white adipose tissue in mice. J Clin Invest. 2011;121:96-105
193. Lan F, Cacicedo JM, Ruderman N, Ido Y. SIRT1 modulation of the acetylation status, cytosolic localization, and activity of LKB1. Possible role in AMP-activated protein kinase activation. The Journal of biological chemistry. 2008;283:27628-35 doi:10.1074/jbc.M805711200
194. Hou X, Xu S, Maitland-Toolan KA, Sato K, Jiang B, Ido Y. et al. SIRT1 regulates hepatocyte lipid metabolism through activating AMP-activated protein kinase. The Journal of biological chemistry. 2008;283:20015-26 doi:10.1074/jbc.M802187200
195. Canto C, Jiang LQ, Deshmukh AS, Mataki C, Coste A, Lagouge M. et al. Interdependence of AMPK and SIRT1 for metabolic adaptation to fasting and exercise in skeletal muscle. Cell metabolism. 2010;11:213-9 doi:10.1016/j.cmet.2010.02.006
196. Su KH, Yu YB, Hou HH, Zhao JF, Kou YR, Cheng LC. et al. AMP-activated protein kinase mediates erythropoietin-induced activation of endothelial nitric oxide synthase. Journal of cellular physiology. 2012;227:3053-62 doi:10.1002/jcp.23052
197. Li XJ, Wang XW, Du YJ. Protective effects of erythropoietin on myocardial infarction in rats: the role of AMP-activated protein kinase signaling pathway. The American journal of the medical sciences. 2011;342:153-9 doi:10.1097/MAJ.0b013e318210041d
198. Keijer J, van Schothorst EM. Adipose tissue failure and mitochondria as a possible target for improvement by bioactive food components. Current opinion in lipidology. 2008;19:4-10 doi:10.1097/MOL.0b013e3282f39f95
199. Zhang H, Gao P, Fukuda R, Kumar G, Krishnamachary B, Zeller KI. et al. HIF-1 inhibits mitochondrial biogenesis and cellular respiration in VHL-deficient renal cell carcinoma by repression of C-MYC activity. Cancer Cell. 2007;11:407-20
200. Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177-85
201. Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y. et al. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752-61 doi:10.1172/JCI21625
202. Li Y, Takemura G, Okada H, Miyata S, Maruyama R, Li L. et al. Reduction of inflammatory cytokine expression and oxidative damage by erythropoietin in chronic heart failure. Cardiovasc Res. 2006;71:684-94 doi:10.1016/j.cardiores.2006.06.003
203. Kim KH, Oudit GY, Backx PH. Erythropoietin protects against doxorubicin-induced cardiomyopathy via a phosphatidylinositol 3-kinase-dependent pathway. The Journal of pharmacology and experimental therapeutics. 2008;324:160-9 doi:10.1124/jpet.107.125773
204. Amer J, Dana M, Fibach E. The antioxidant effect of erythropoietin on thalassemic blood cells. Anemia. 2010;2010:978710. doi:10.1155/2010/978710
205. Shurtz-Swirski R, Kristal B, Shasha SM, Shapiro G, Geron R, Sela S. Interaction between erythropoietin and peripheral polymorphonuclear leukocytes in continuous ambulatory dialysis patients. Nephron. 2002;91:759-61 doi:65044
206. Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813-20 doi:10.1038/414813a
207. Matsuoka T, Kajimoto Y, Watada H, Kaneto H, Kishimoto M, Umayahara Y. et al. Glycation-dependent, reactive oxygen species-mediated suppression of the insulin gene promoter activity in HIT cells. J Clin Invest. 1997;99:144-50 doi:10.1172/JCI119126
208. Maddux BA, See W, Lawrence JC Jr, Goldfine AL, Goldfine ID, Evans JL. Protection against oxidative stress-induced insulin resistance in rat L6 muscle cells by mircomolar concentrations of alpha-lipoic acid. Diabetes. 2001;50:404-10
209. Rudich A, Tirosh A, Potashnik R, Hemi R, Kanety H, Bashan N. Prolonged oxidative stress impairs insulin-induced GLUT4 translocation in 3T3-L1 adipocytes. Diabetes. 1998;47:1562-9
210. Ye J, Gao Z, Yin J, He Q. Hypoxia is a potential risk factor for chronic inflammation and adiponectin reduction in adipose tissue of ob/ob and dietary obese mice. Am J Physiol Endocrinol Metab. 2007;293:E1118-28
211. Regazzetti C, Peraldi P, Gremeaux T, Najem-Lendom R, Ben-Sahra I, Cormont M. et al. Hypoxia decreases insulin signaling pathways in adipocytes. Diabetes. 2009;58:95-103 doi:10.2337/db08-0457
212. Dang J, Jia R, Tu Y, Xiao S, Ding G. Erythropoietin prevents reactive oxygen species generation and renal tubular cell apoptosis at high glucose level. Biomedicine & pharmacotherapy = Biomedecine & pharmacotherapie. 2010;64:681-5 doi:10.1016/j.biopha.2010.06.011
213. Villa P, Bigini P, Mennini T, Agnello D, Laragione T, Cagnotto A. et al. Erythropoietin selectively attenuates cytokine production and inflammation in cerebral ischemia by targeting neuronal apoptosis. The Journal of experimental medicine. 2003;198:971-5 doi:10.1084/jem.20021067
214. Zhang J, Li Y, Cui Y, Chen J, Lu M, Elias SB. et al. Erythropoietin treatment improves neurological functional recovery in EAE mice. Brain research. 2005;1034:34-9 doi:10.1016/j.brainres.2004.11.036
215. Savino C, Pedotti R, Baggi F, Ubiali F, Gallo B, Nava S. et al. Delayed administration of erythropoietin and its non-erythropoietic derivatives ameliorates chronic murine autoimmune encephalomyelitis. Journal of neuroimmunology. 2006;172:27-37 doi:10.1016/j.jneuroim.2005.10.016
216. Agnello D, Bigini P, Villa P, Mennini T, Cerami A, Brines ML. et al. Erythropoietin exerts an anti-inflammatory effect on the CNS in a model of experimental autoimmune encephalomyelitis. Brain research. 2002;952:128-34
217. Yuan R, Maeda Y, Li W, Lu W, Cook S, Dowling P. Erythropoietin: a potent inducer of peripheral immuno/inflammatory modulation in autoimmune EAE. PloS one. 2008;3:e1924. doi:10.1371/journal.pone.0001924
218. Chang YK, Choi DE, Na KR, Lee SJ, Suh KS, Kim SY. et al. Erythropoietin attenuates renal injury in an experimental model of rat unilateral ureteral obstruction via anti-inflammatory and anti-apoptotic effects. The Journal of urology. 2009;181:1434-43 doi:10.1016/j.juro.2008.10.105
219. Sepodes B, Maio R, Pinto R, Sharples E, Oliveira P, McDonald M. et al. Recombinant human erythropoietin protects the liver from hepatic ischemia-reperfusion injury in the rat. Transplant international: official journal of the European Society for Organ Transplantation. 2006;19:919-26 doi:10.1111/j.1432-2277.2006.00366.x
220. Nairz M, Schroll A, Moschen AR, Sonnweber T, Theurl M, Theurl I. et al. Erythropoietin contrastingly affects bacterial infection and experimental colitis by inhibiting nuclear factor-kappaB-inducible immune pathways. Immunity. 2011;34:61-74 doi:10.1016/j.immuni.2011.01.002
221. Zeyda M, Stulnig TM. Obesity, inflammation, and insulin resistance--a mini-review. Gerontology. 2009;55:379-86 doi:10.1159/000212758
222. Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. The Journal of clinical investigation. 2011;121:2111-7 doi:10.1172/JCI57132
223. Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763-70 doi:10.1038/27376
224. Kalofoutis C, Piperi C, Zisaki A, Singh J, Harris F, Phoenix D. et al. Differences in expression of cardiovascular risk factors among type 2 diabetes mellitus patients of different age. Annals of the New York Academy of Sciences. 2006;1084:166-77 doi:10.1196/annals.1372.001
225. Ehses JA, Boni-Schnetzler M, Faulenbach M, Donath MY. Macrophages, cytokines and beta-cell death in Type 2 diabetes. Biochemical Society transactions. 2008;36:340-2 doi:10.1042/BST0360340
226. Pregi N, Wenker S, Vittori D, Leiros CP, Nesse A. TNF-alpha-induced apoptosis is prevented by erythropoietin treatment on SH-SY5Y cells. Experimental cell research. 2009;315:419-31 doi:10.1016/j.yexcr.2008.11.005
227. Chen G, Shi JX, Hang CH, Xie W, Liu J, Liu X. Inhibitory effect on cerebral inflammatory agents that accompany traumatic brain injury in a rat model: a potential neuroprotective mechanism of recombinant human erythropoietin (rhEPO). Neuroscience letters. 2007;425:177-82 doi:10.1016/j.neulet.2007.08.022
228. Meng R, Zhu D, Bi Y, Yang D, Wang Y. Erythropoietin inhibits gluconeogenesis and inflammation in the liver and improves glucose intolerance in high-fat diet-fed mice. PloS one. 2013;8:e53557. doi:10.1371/journal.pone.0053557
229. Carvalho G, Lefaucheur C, Cherbonnier C, Metivier D, Chapel A, Pallardy M. et al. Chemosensitization by erythropoietin through inhibition of the NF-kappaB rescue pathway. Oncogene. 2005;24:737-45 doi:10.1038/sj.onc.1208205
230. Hotamisligil GS, Shargill NS, Spiegelman BM. Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science. 1993;259:87-91
231. Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, Ferrante AW Jr. Obesity is associated with macrophage accumulation in adipose tissue. The Journal of clinical investigation. 2003;112:1796-808 doi:10.1172/JCI19246
232. Wu H, Ghosh S, Perrard XD, Feng L, Garcia GE, Perrard JL. et al. T-cell accumulation and regulated on activation, normal T cell expressed and secreted upregulation in adipose tissue in obesity. Circulation. 2007;115:1029-38 doi:10.1161/CIRCULATIONAHA.106.638379
233. Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ. et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. The Journal of clinical investigation. 2003;112:1821-30 doi:10.1172/JCI19451
234. Alnaeeli M, Raaka BM, Gavrilova O, Teng R, Chanturiya T, Noguchi CT. Erythropoietin signaling: A novel regulator of white adipose tissue inflammation during diet-induced obesity. Diabetes. 2014 doi:10.2337/db13-0883
235. De Luisi A, Binetti L, Ria R, Ruggieri S, Berardi S, Catacchio I. et al. Erythropoietin is involved in the angiogenic potential of bone marrow macrophages in multiple myeloma. Angiogenesis. 2013;16:963-73 doi:10.1007/s10456-013-9369-2
236. Lu KY, Ching LC, Su KH, Yu YB, Kou YR, Hsiao SH. et al. Erythropoietin suppresses the formation of macrophage foam cells: role of liver X receptor alpha. Circulation. 2010;121:1828-37 doi:10.1161/CIRCULATIONAHA.109.876839
Author contact
![]() Corresponding author: Li Wang at liwangmo and Lijun Di at lijundimo
Corresponding author: Li Wang at liwangmo and Lijun Di at lijundimo