Impact Factor ISSN: 1449-2288
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Conclusions
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(2):144-155. doi:10.7150/ijbs.10692 This issue Cite
Research Paper
Clinical Significance of a Point Mutation in DNA Polymerase Beta (POLB) Gene in Gastric Cancer
Xiaohui Tan1, 4, Hongyi Wang2, Guangbin Luo3, Shuyang Ren1, Wenmei Li1, Jiantao Cui1, Harindarpal S. Gill4, Sidney W. Fu4 ![]() , Youyong Lu1
, Youyong Lu1 ![]()
1. Laboratory of Molecular Oncology, Key Laboratory of Carcinogenesis and Translational Research (Ministry of Education);
2. Department of Sugary, Peking University School of Oncology, Beijing Cancer Hospital & Institute, Beijing, 100142, P.R. China.
3. Department of Genetics, Case Western Reserve University, Cleveland, OH, 44106, USA.
4. Department of Medicine, The George Washington University School of Medicine and Health Sciences, Washington, DC 20037, USA.
Received 2014-10-1; Accepted 2014-11-19; Published 2015-1-1
Abstract

Gastric cancer (GC) is a major cause of global cancer mortality. Genetic variations in DNA repair genes can modulate DNA repair capability and, consequently, have been associated with risk of developing cancer. We have previously identified a T to C point mutation at nucleotide 889 (T889C) in DNA polymerase beta (POLB) gene, a key enzyme involved in base excision repair in primary GCs. The purpose of this study was to evaluate the mutation and expression of POLB in a larger cohort and to identify possible prognostic roles of the POLB alterations in GC. Primary GC specimens and their matched normal adjacent tissues were collected at the time of surgery. DNA, RNA and protein samples were isolated from GC specimens and cell lines. Mutations were detected by PCR-RFLP/DHPLC and sequencing analysis. POLB gene expression was examined by RT-PCR, tissue microarray, Western blotting and immunofluorescence assays. The function of the mutation was evaluated by chemosensitivity, MTT, Transwell matrigel invasion and host cell reactivation assays. The T889C mutation was detected in 18 (10.17%) of 177 GC patients. And the T889C mutation was associated with POLB overexpression, lymph nodes metastases and poor tumor differentiation. In addition, patients with- the mutation had significantly shorter survival time than those without-, following postoperative chemotherapy. Furthermore, cell lines with T889C mutation in POLB gene were more resistant to the treatment of 5-fluorouracil, cisplatin and epirubicin than those with wild type POLB. Forced expression of POLB gene with T889C mutation resulted in enhanced cell proliferation, invasion and resistance to anticancer drugs, along with increased DNA repair capability. These results suggest that POLB gene with T889C mutation in surgically resected primary gastric tissues may be clinically useful for predicting responsiveness to chemotherapy in patients with GC. The POLB gene alteration may serve as a prognostic biomarker for GC.
Keywords: DNA polymerase beta (POLB), gastric cancer, DNA Repair, point mutation, chemotherapy.
Introduction
Gastric cancer (GC) is the second leading cause of cancer-related deaths worldwide [1]. It remains difficult to cure, primarily due to lack of reliable screening markers and advanced disease when diagnosed. Even patients who present the most favorable condition at diagnosis and subsequently undergo curative surgical resection often die of recurrent disease [2]. Genetic variations in DNA repair genes can modulate DNA repair capability and, consequently, have been associated with the risk of developing cancer [3]. DNA polymerase beta (POLB) is a DNA repair polymerase involved in base-excision repair (BER), and recombination and drug resistance [4-6]. Thirty percent of all tumors reported to date harbor mutations in the POLB gene [7]. Aberrant POLB expression resulted in an increased rate of spontaneous mutagenesis and a highly mutagenic tolerance phenotype [8, 9]. POLB gene mutation and overexpression have been reported in various cancers [10-14]. In our previous studies, we identified a novel homozygous T to C point mutation at nucleotide 889 (T889C) in POLB gene (GenBank Acc# M13140.1) from human primary GC. This mutation resulted in a substitution of the amino acid at 259 from Leucine to Serine in protein [15]. In the present work, we examined the T889C mutation and expression of POLB in a large cohort of GC patients with integral follow-up records, and analyzed the relationship between aberrant POLB expression and patients' clinicopathological features.
Materials and Methods
Cell lines and tissue samples
Eight human GC cell lines AGS, N-87, SUN1, SUN5, SNU16, RF1, RF48 and MKN45 were obtained from the American Type Culture Collection (ATCC). Cell lines BGC823, MGC803, SGC7901, PAMC82, and GES were established in China. Primary GC specimens and their matched normal adjacent tissues were collected and snap-frozen at the time of surgery. None of the patients had received any chemotherapy or radiotherapy before surgical resection. Clinical and histological features were obtained and evaluated (Table 1). Fifty peripheral blood samples were obtained from healthy volunteers. The study was conducted according to the Helsinki Declaration, approved by the Institutional Ethical Standards Committee of Peking University. Informed consent of all study subjects were obtained before the samples were collected.
Clinical and pathological parameters of the 177 patient samples used in mutation analysis
| Tissues parameters | Number |
|---|---|
| Tumor | 177 |
| Gender | |
| Male | 125 |
| Female | 52 |
| Age (years) | |
| Male | 28-86 |
| Female | 29-79 |
| Pathology | |
| Intestinal | 116 |
| Diffuse | 30 |
| Mixed | 20 |
| Adenosquamous | 11 |
| Tumour location | |
| GOJ * | 51 |
| Fundus | 12 |
| Greater curve-body | 18 |
| Lesser curve-body | 11 |
| Body non-specified | 20 |
| Antrum | 65 |
| Differentiation | |
| Well | 21 |
| Moderate to poor | 60 |
| Poor | 96 |
| Depth of invasion | |
| Mucosa | 11 |
| Submucosa | 10 |
| Muscularis propria | 31 |
| Serosa/full thickness | 125 |
| No. of involved Lymph nodes | |
| 0 | 60 |
| 1 | 5 |
| 1-6 | 19 |
| 7-15 | 30 |
| >15 | 63 |
* Gastro-esophageal junction
DNA isolation and PCR amplification
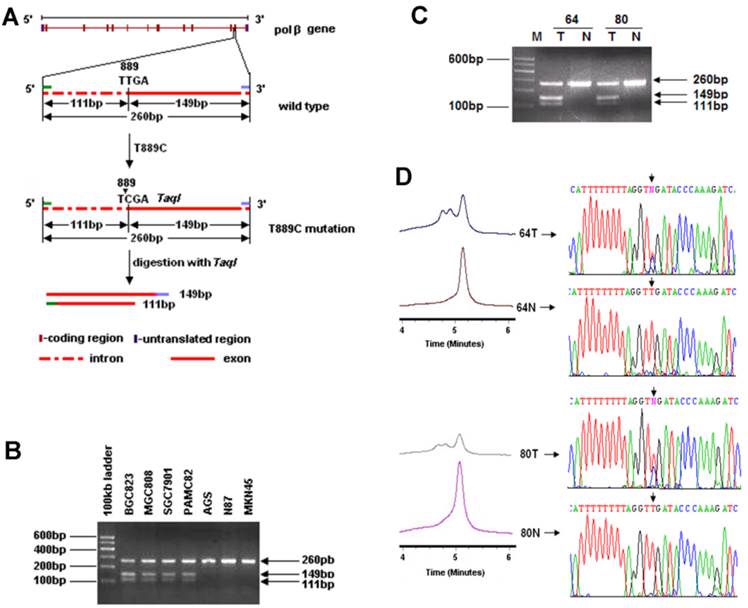
Genomic DNA was extracted from GC cell lines, primary GC tissues and their matched normal gastric tissues, and peripheral blood samples of healthy volunteers. Since T889C mutation results in a new Taq1 site from 888 to 891 (TTGA to TCGA) in POLB gene, which allows us to screen for the point mutation by Taq1 enzyme digestion (Figure 1A). Specifically, we used a PCR strategy that can distinguish the T889C mutation from the wild-type in POLB allele. The primers used were: POLB Forward: 5′-CCACTTAGGCTTATCTTTGGT-3′ and Reverse: 5′-GGA CAC TCA CCA GTG ACT CC-3′.
Identification of the T889C mutation in primary GCs with PCR-RFLP, DHPLC and DNA sequencing. A. Restriction analysis of the POLB gene fragment amplified by PCR. RFLP patterns obtained after digestion of PCR products with Taq1 endonucleases. Primers were designed to amplify the region spanning 3' region of intron 12 to 5' region of exon 13 in POLB gene and length of the PCR products were 260bp. T889C mutation formed of a new Taq1 site from nucleotide 888 to 891 (TTGA to TCGA) of POLB (NM_002690.1) cDNA and therefore, the amplified product could be digested with Taq1. The molecular weights of digested fragments were 149bp and 111bp. B. The digested fragments were separated by 2.0% agarose gel electrophoresis. 100-bp ladder was used as a size marker. PCR products (260bp) amplified from BGC823, MGC803, SGC7901 and PAMC82 cell lines (carrying the T889C mutation) could be digested by Taq1, the molecular weights of digested fragments were 149 and 111bp while those from AGS, N87 and MKN45 cell lines couldn't be digested by Taq1. C. Restriction patterns obtained by Taq1 digestion of amplified product from GC tissues. The digested fragments were separated by 2.0% agarose gel electrophoresis and 100bp ladder was used as a size marker. PCR products (260bp) with T889C mutation could be digested by Taq1, the molecular weights of digested fragments were 149 and 111bp while the PCR products with wild type POLB couldn't be digested by Taq1. D. Chromatograph of DHPLC and sequencing analysis.
Purification of PCR products and PCR-RFLP analysis
The PCR products of the expected sizes were purified with Geneclean Kit (Bio 101, Inc., La Jolla, Calif.). Ten μl of PCR products were digested with the restriction enzyme, TaqI (New England BioLabs, Beverly, Mass.).
PCR-DHPLC and DNA sequencing analysis
DHPLC (denaturing high-performance liquid chromatography) analysis was performed as described [16, 17]. PCR products showing aberrant PCR-RFLP banding patterns and exhibiting abnormal elution peak in DHPLC were subjected to sequencing analysis.
RT-PCR assays
Five μg of total RNA from each sample was used for cDNA synthesis, using the MMLV and random primers (Oligo dT). Two μg of reverse-transcription product was used as templates to amplify specific fragment of POLB gene. The expression of housekeeping gene glyceraldehydes-3-phosphate dehydrogenase (GAPDH) was used as an internal control. The average relative densities of bands were detected by Bandlead 3.0 program. The expression level was shown by the ratio of POLB to GAPDH in band density.
Tissue microarray construction and immunohistochemical (IHC) staining and evaluation
Tissue microarrays were constructed as described previously [18]. An affinity purified goat polyclonal antibody POLB (sc-5927, Santa Cruz) was used for IHC staining. For evaluation, the total fields were counted microscopically under high power (×200). The results of POLB expression were graded as: negative (-), <5% of cell stained; positive (+), >5% of cells stained.
Western blot analysis
Protein extraction and Western blot analysis with chemiluminescent detection were as described [19]. The following antibodies and dilution factors were used: affinity purified goat polyclonal antibody POLB (sc-5927, Santa Cruz, 1:800), rabbit anti-actin (H-196, Santa Cruz, 1:1000), donkey anti-goat IgG-HRP (sc-2020, Santa Cruz, 1:2000) and goat anti-rabbit IgG-HRP (sc-2030, Santa Cruz, 1:2000).
Association analysis between alterations of POLB gene and patients' clinicopathological features, and their responses to chemotherapy
The association between the point mutation and clinicopathological features such as gender, age, pathological diagnosis, differentiation, tumor location, depth of invasion, liver metastasis, and lymph metastases was analyzed by the paired chi-square test with SPSS 13.0. p < 0.05 was considered statistically significant.
To analyze the correlation between T889C mutation and patients' response to postoperative adjuvant chemotherapy, we compared progression-free time and overall survival time of the patients who received identical anticancer drug treatment, at the same pathological stage when initially diagnosed.
Regarding surgical operations, patients included for this study must have received complete surgical removal of the tumor, with negative pathologic margin, and with the caveat of the anatomical location of the tumor and the condition of the patients. The degree of surgical resection was assessed by a central review of operative reports, and pre- and post-operative imaging. When the results of scans and operative reports differed, the degree of surgical resection was assessed based on the results of computed tomography (CT) or magnetic resonance imaging (MRI). Patients with one of the following two conditions were excluded: 1. underwent unsuccessful surgical procedures (for instance, anastomotic leakage, fistula, abscess, hemorrhage); 2. had “medical” complications caused by non-malignant diseases or uncontrolled infections, which were detected by the following examinations: CT, MRI, height, weight, complete blood count, hemoglobin level, kidney/liver function, electrolyte levels, and calcium and magnesium concentrations, and a caloric intake per day by oral route (<1500 kcal were excluded).
Regarding postoperative adjuvant chemotherapy, patients eligible for the analysis should have a complete and detailed medical record and a report with gastroscopy, pathological, CT or MRI diagnosis, quality-of-life assessment, progression-free survival and overall survival time. And the information was obtained before chemotherapy and at specific times during and after drug treatment. Cases that were unclear whether abnormalities on postoperative scans represented residual tumor or postoperative artifacts, and cases with combination therapy of chemotherapy, immunotherapy and/or radiotherapy were all excluded.
Judgments of disease relapse or progression were based on clinical evidence of gastroscopy, pathological, CT or MRI diagnosis. Patients who had clinical evidence of disease progression, despite scans indicating otherwise, were considered disease progression. The site and date of first relapse, and the cause and date of death were recorded. The sites of relapse were classified as follows: the relapse was coded as: loco-regional if tumor was detected within the operation fields (including gastric bed, remnant stomach, and surgical anastomosis); peritoneal if tumor was detected in the peritoneal cavity; and distant if the metastases were diagnosed outside the peritoneal cavity or liver metastasis.
Real time qRT-PCR analysis
First-strand cDNA was synthesized using the Bio-Rad RT kit. Primer sequences are available upon request. Real-time qRT-PCR assays were carried out on a Bio-Rad iCycler iQ system (Bio-Rad, Hercules, CA, USA) using SYBR Green reagent as described [19].
Construction of eukaryotic expression vector containing wild type and T889C mutants of POLB gene and transient transfection
The entire coding region of POLB cDNA containing wild type or T889C mutant, and site-directed mutagenesis were amplified by RT-PCR, and cloned respectively in a pcDNA3.1 vector before transfecting into AGS cells. 5 X105 cells were plated in 10 cm2 culture dishes, 25 pg of plasmid DNA/dish were transfected using the calcium phosphate precipitation method.
Immunofluorescence assays
Cells were seeded at 2 × 104 cells per well on glass coverslips in six-well plates and fixed in 2% paraformaldehyde as described previously [19]. Confocal images were obtained using a LSM 510 Confocal microscope (Carl Zeiss). The number of nuclei containing at least one localized area of immunofluorescence was determined by examination of the confocal images. Antibodies for immunofluorescence were as follows: goat polyclonal antibody POLB (sc-5927, Santa Cruz) at final concentration of 1:800, Alexa Flour 568 goat anti-mouse IgG (Invitrogen), 1:500 and Alexa Flour 568 goat anti-mouse IgG (Invitrogen), 1:500.
POLB knockdown assays
The siRNA targeting POLB was purchased from Qiagen (SI02663605: 5′-TACGAGTTCATCCATCAATTT-3′). siRNAs against GAPDH (SI02653266 and SI03650325, Qiagen) were used as positive and negative controls, respectively. Cells were transfected with 40 pmol of siRNA using the HiPerFect lipid transfection reagent (Qiagen), and harvested at 24h, 48h and 72 h post-transfection. POLB expression was then analyzed at both mRNA and protein level by qRT-PCR and Western blotting, respectively.
Chemosensitivity and MTT assays
To determine the sensitivity of GC cell lines to chemotherapy, BGC823, SGC7901, AGS and N87 cells were seeded in a 96-well plate as described [20]. 5-FU, cisplatin and epirubicin were obtained from the pharmacy at Beijing Cancer Hospital (Beijing, China). Test drug concentrations were 5 to 400 µg/mL for 5-FU, 0.5 to 8 µg/mL for cisplatin and 0.025 to 1.6 µg/mL for epirubicin. The cells were treated by either single or a variety of combinations for analysis. Cells were seeded in a 96-well plate at the density of 3× 104 per well, and were incubated in medium containing 5% serum. The attached cells in the plates were washed once with PBS, and then replaced with fresh medium containing various concentrations of drugs to treat for 24h and 48 h. To further confirm the effect of the T889C on chemotherapy, the wild type, T889C mutant or empty vector transfected AGS cells were subject to MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assays at 24h, 48h and 72h. At each time point, 10 µl of MTT reagent (MILLIPORE, Billerica, MA, USA) was added to each well. After 4 h of incubation, cells were lysed by addition of 200 μl DMSO. Absorbance was measured at 570 nm wavelength. Experiments were repeated three times, and the percentage of growth was the average of the experiments.
Transwell matrigel invasion assays
The cell line AGS transfected with wild-type and mutant POLB was harvested using a cell-dissociation solution at 90% confluence. The cells (1 × 105) were suspended in 100 μl of serum-free medium before adding to the upper chamber of the Transwell insert. The lower chamber was filled with 500 μl of the medium with 0.1% bovine serum albumin. After 24 h of incubation at 37°C, the cells on the upper surface of the filter were removed using a cotton swab. The cells that penetrated to the lower surface of the filter were stained with hematoxylin and counted under an Olympus IX70 microscope (Olympus) in 13 randomized fields at ×400 magnification. The assay was performed four times as described earlier [20, 21].
Plasmid treatment with UV/anticancer drugs and host cell reactivation (HCR) assays
DNA repair capability of cells with wild-type and T889C mutant POLB were assessed using post-UV HCR as described previously [19] [20]. pCMVLuc reporter gene plasmid (a kind gift from Dr. Kenneth H. Kraemer, National Cancer Institute ) was dissolved in 10 mm Tris-HCl, 1 mm EDTA, pH 8 (TE buffer) to a final concentration of 100 μg/ml and poured in a Petri dish to form 1D 2 mm thick layer. The Petri dish was placed on ice and irradiated by 1000 J/m2 of UV light. For the drug treatment, 1 μl aliquots of a stock solution of 1 μg / μl cisplatin, 10 μg / μl 5-FU and 0.01 μg / μl cisplatin (Sigma-Aldrich, St. Louis, MO) in TE were added to 10 μg plasmid DNA before dissolving in 200 μl TE buffer, and then the samples were incubated at 37°C for 6 h. At the end of the incubation, 1 M NaCl was added to a final concentration of 0.2 M NaCl. The plasmid DNA was precipitated with 2 volumes of 100% ethanol, and then washed with 70% ethanol. The pellets were air-dried and dissolved in TE buffer. DNA repair capability of cells was assessed using HCR with the CsCl-purified pCMVLuc plasmid treated by UV or drugs. Four microliters (200 ng) of damaged or undamaged pCMVLuc were co-transfected with 0.4 µl (200 ng) of wild-type, and T889C mutant POLB effector plasmids into cells using Lipofectamine 2000 (Invitrogen). To estimate the DNA repair capacity after POLB knockdown, we co-transfected pCMVLuc with 40 pmol of siRNA, relative luciferase activities were determined by a percentage of activities obtained with treated versus untreated control plasmids.
Statistical analysis
Means ± SD were calculated, and statistical analysis was performed using one-way ANOVA, followed by the nonparametric Kruskal-Wallis test; p < 0.05 was considered statistically significant.
Protein-structure modelling
A three-dimensional model of the L259S mutation was created by comparative protein modelling methods [22]. The crystal structure of human POLB (PDBid: 4JWM) at 2.0-Å resolution [23] was used as a template as justified by a 98.5% identity in amino-acid sequence alignment. Using the model-building program O [24, 25], residue E256 was replaced with aspartate to restore the active site back to that of the wild type, and residue L259 was replaced with serine to model the natural occurring mutation in cancer patients. The peptide backbones were not adjusted. Side chains were adjusted based on likelihood of nearby hydrogen-bonds partners within a distance of ~3.5 Å. Protein residue numbering corresponds to the coding sequence within genbank accession M13140.1.
Results
T889C point mutation screening in a new cohort of GC patients
In order to investigate the incidence of T889C mutation in GC, we established a new cohort of 177 GC patients (Table 1). To facilitate large-scale T889C point mutation screening, a strategy based on PCR-RFLP was designed to characterize the mutation (Figure 1A). The feasibility of this strategy was firstly tested in 13 GC cell lines. PCR products from cell lines with T889C mutant POLB, BGC823, MGC803, SGC7901 and PAMC82 [15] were able to be cut by Taq1, but not for those with wild type POLB, AGS, N87 and MKN45. Thus, the PCR-RFLP indeed provided a reliable and simple approach for detecting the T889C mutation (Figure 1B).
We examined 177 primary GCs. The T889C point mutation was detected in 18 (10.17%) of them (Figure 1C), but not in any of the matched normal gastric tissues or the 50 normal volunteer controls (Figure 1D). These data indicate that T889C mutation may represent an important genetic risk factor for human GCs.
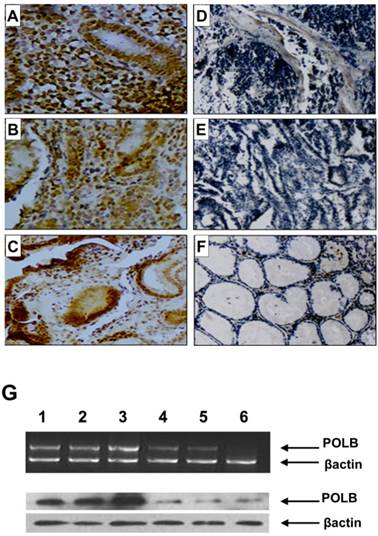
Since point mutations could alter gene function by either reducing (loss-of-function mutations) or increasing (gain-of-function mutations) its expression level, we examined the relationship between T889C mutation and expression of POLB in GC tissues. We constructed tissue microarrays consisting of samples from 70 GC patients, including 18 with- and 52 without- T889C mutation, and 20 normal adjacent tissues (5 from patients with- and 15 without- T889C mutation). The positive hybridization signals of POLB protein were observed in 47 (67.1%) of the 70 GCs, of which, 16 (88.9%) of 18 with T889C mutation, and 31 (59.6 %) of 52 without. There was a significant positive association between T889C point mutation and positive signals of POLB protein (p = 0.023, Figure 2A-F and Supplementary Material: Table S1). The concordance between T889C mutation and overexpression of POLB was confirmed by RT-PCR and Western blot analysis, respectively (Figure 2G & 4C).
POLB expression on tissue array by immunohistochemistry, RT-PCR and Western blot analysis in GC patients. A, B and C. With T889C mutation; D and E. Without T889C mutation; F. Normal tissue. G. RT-PCR (top) and Western blot (bottom) analysis of POLB gene in GC tissues with-T889C mutation (lanes 1-3) and without- (lanes 5-6).
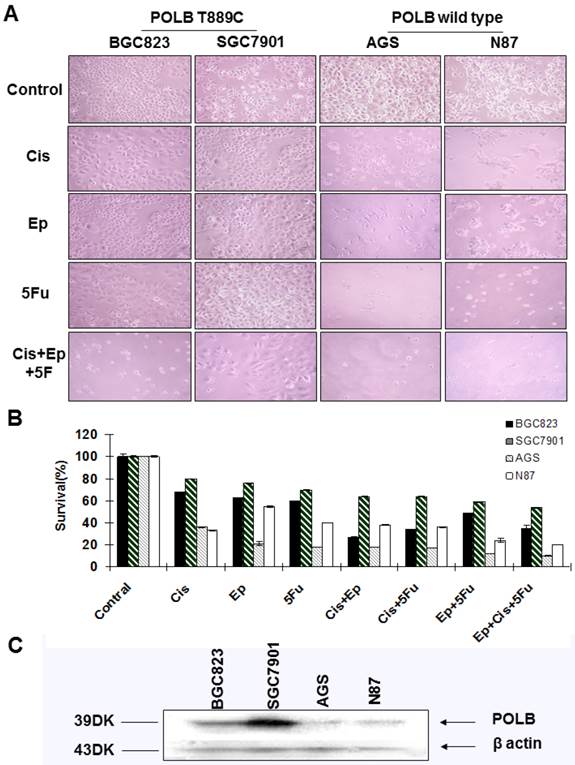
Correlation between T889C mutation and cytotoxicity of 5-fluorouracil, cisplatin and epirubicin. A. Phenotypic comparison of BGC823, SGC7901 and AGS cell lines before and after treatment. B. Sensitivity of cell lines to cisplatin, epirubicin, 5-fluorouracil and combination treatment with three drugs. Concentration of 5-fluorouracil, cisplatin, epirubicin, and was 5ug/ml, 0.5ug/ml and 0.1ug/ml respectively. The concentration of combination treatment of three drugs is cisplatin 0.5ug/ml, epirubicin 0.1ug/ml, and 5-fluorouracil 5ug/ml. Survival is expressed as the relative plating efficiency of treated cells to the control. The values shown in the figures are mean readings from six wells in each experiment and representative of at least three independent experiments. C. Protein expression of POLB in cell lines BGC823, SGC7901, AGS and N87.
Association of clinicopathological features with T889C mutation and POLB overexpression
We analyzed the relationship between T889C mutation and gender, age, tumour location, depth of invasion, differentiation and metastasis in 177 primary GCs. Lymph node metastases was found in all 18 GC patients carrying T889C mutation (100%), whereas in 99 of the rest of the 159 patients (62.26%) . There were significant differences between the patients with- and without- T889C mutation in regard to the propensity of lymph nodes metastasis (p = 0.002, Supplementary Material: Table S2). GC harboring T889C mutation was more likely to metastasize to lymph nodes. Furthermore, mutation occurred in 14 (14.58%) of 96 poorly differentiated primary GCs and 4 (4.94%) of 81 moderately/well differentiated tumors (p = 0.034, Supplementary Material: Table S2). Thus the T889C mutation is more prevalent in poorly differentiated primary GCs compared with those of moderately/well differentiated tumors. However, we did not find any significant correlation between T889C mutation and gender, age, tumour location or depth of invasion (p > 0.05). These findings suggest that T889C mutation is strongly associated with metastasis and differentiation in human GC.
We next analyzed the relationship between POLB expression and clinicopathological features. By analyzing the 70 samples on our tissue array, we found that POLB positivity was not associated with lymph nodes metastasis, poorly differentiation, age, gender, tumour location and depth of invasion (p > 0.05), despite that T889C mutation was significantly associated with poor differentiation (p = 0.009) and lymph nodes metastasis (p = 0.018). Our data suggested that POLB expression alone is not associated with clinicopathological features.
T889C mutation is associated with the response to postoperative chemotherapy
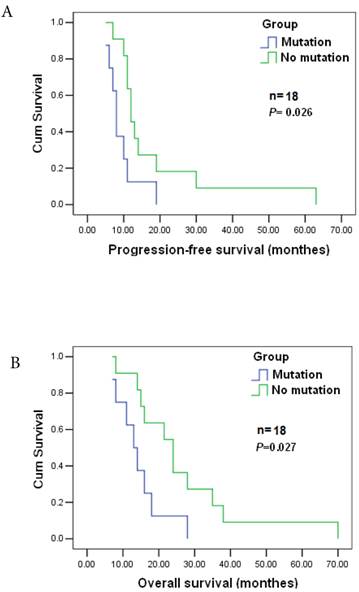
POLB plays a significant role in chemotherapeutic agent resistance [26]. Among the 177 cases, 143 patients received postoperative adjuvant chemotherapy. Nineteen GC patients were analyzed to reveal the relationship between the mutation and the response to postoperative chemotherapy as they were diagnosed at the same clinicopathological stage) before surgery, and treated with the same anticancer drugs/doses. They were all at stage T4N2M0, and treated with cisplatin (60 mg / m2 i.v.) and epirubicin (50 mg / m2 i.v.), every 3 weeks for four cycles together with a continuous 12 week infusion of 5-FU (200 mg / m2 /day). The estimated median progression-free and overall survival times were 11±1.405 and 16±4.265 months, respectively. Significant differences in progression-free and survival time were found between patients with T889C mutation (8) and those without the mutation (11) (p=0.026), and significant difference also existed in overall survival (16±4.265 vs. 24±8.34, p=0.027) between the two groups (Figure 3A&B). Patients with T889C mutation had significantly shorter survival than those without the mutation following chemotherapy. Our data indicates that T889C mutation is associated with poor response to postoperative chemotherapy.
Correlation between T889C mutation and survival. Progression-free survival (top) and overall survival (bottom) for all 19 patients enrolled. The statistical significance of the difference was p = 0.027 and p = 0.026. Patients with T889C mutation had significantly shorter survival than did patients without the mutation after postoperative chemotherapy.
To further examine the relationship between alternative POLB and poor response to anticancer drugs, we assessed the sensitivity of cell lines BGC823, SGC7901, AGS and N87 to the single-agent and combinations of 5-FU, cisplatin and epirubicin by MTT assays. We found that the cell lines with T889C mutation (BGC823, SGC7901) were more resistant to 5-FU, cisplatin and epirubicin than those with wild type POLB (Figure 4A&B). These results support the clinical observation that T889C mutation is associated with poor response to postoperative chemotherapy.
Overexpression of POLB gene stimulates proliferation and promotes invasion and migration in human GC cell lines
Having demonstrated the relationship between T889C mutation, and lymph node metastasis and differentiation in clinical samples, we further examined the role of T889C mutation in metastasis, differentiation and resistance to anticancer drugs in GC cell lines. Relative POLB mRNA and protein levels in transfected AGS cell lines with wild type or T889C were significantly increased (p<0.01) than that of the vector control lines, and the receptor genes expressing both the wild-type and mutant were localized to the nucleus (Figure 5A). We compared the number of cells expressing wild type or mutated POLB at 24, 48 and 72 h after transfection. The number of mutant POLB-transfected cells increased more significantly than that of the wild-type (Figure 5B). Conversely, knockdown of POLB by siRNA resulted in a decreased cell growth in AGS cells. Our data suggested that cells transfected with T889C mutated POLB increased cell proliferation. Next, we examined the invasive and spontaneous metastatic potential by matrigel assays. The invasion of the mutant POLB-transfected cells was significantly increased compared with the wild type POLB-transfected cells. In addition, knockdown of POLB by siRNA reduced cell growth in AGS cells (Figure 5C). These results suggest that GC cells with T889C mutation are more proliferative and invasive.
The effect of POLB overexpression. AGS cells were transfected with empty pcDNA3.1, wild type POLB, T889C mutant and POLB siRNA. A. Expression of transfected POLB genes in human GC cell lines. Left panel, representative results of the QRT-PCR analysis of POLB mRNA levels in AGS cell lines. Each reaction was run twice in triplicates. POLB expression was normalized by GAPDH. Right panel, representative results of the Immunofluorescence for POLB protein expression. POLB protein of both the wild-type and mutant localized to the nucleus. B. The effect of POLB wild type, mutant and siRNA in AGS cell proliferation. MTT assays indicate cell proliferation rate was significantly higher in T889C overexpressors (left). The experiments were done three times in triplicates. Right panel shows phenotypic comparison of AGS cell lines before and after transfection of POLB wild type and mutant and siRNA. C. Matrigel analysis. Three fields of unit area on each membrane were counted for cell numbers, and the experiments were repeated twice. Data analysis was based on the average of parallel repeats. T889C mutation overexpressing cell lines exhibited very significantly higher invasiveness than wild type and empty vector control cell lines. D. Resistance assay. The cells were treated with or without 5-fluorouracil, cisplatin, and epirubicin for 24 h. The values are represented as a mean from three separate experiments; bars, ±SD. E. HCR assay showing increased post-UV DNA repair capability in T889C overexpressors. *p < 0.05.
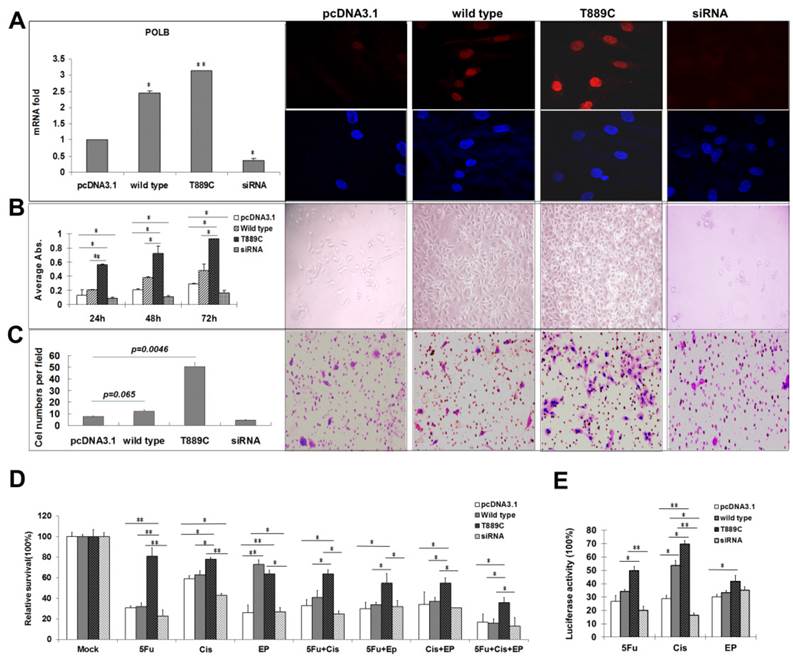
Expression of POLB with T889C mutation leads to resistance to chemotherapeutic agents
In order to further elucidate relationship between T889C mutation and resistance to chemotherapy, we transfected the AGS cell line with either wild type or T889C mutated POLB, and treated them with various concentrations of anticancer drugs. We found that cells transfected with T889C mutant were significantly more resistant to cisplatin and 5-FU compared to those transfected with empty vectors (Figure 5D). Inhibition of POLB expression by siRNA restored chemosensitivity in AGS cells. Our data further corroborates that T889C mutation correlated with poor response to postoperative chemotherapy.
Overexpression of POLB with T889C mutation significantly increased post-drugs host cell reactivation activity in GC cell line
POLB participates in regulation of DNA repair during carcinogen exposure [8]. We then asked whether overexpression of POLB increases the resistance of the GC cell lines to anticancer drugs via DNA repair pathway. We measured the luciferase activity by co-transfecting wild type or T889C mutant POLB, along with pCMU-Luc vector [19], which was pre-treated by anticancer drugs respectively, into the AGS cell line. We found that AGS exhibited significantly increased luciferase activity when the pCMU-Luc vector was treated with cisplatin and 5-FU. There was no difference in luciferase activity when the pCMU-Luc vector was treated with epirubicin (Figure 5E). Our data indicate that the T889C mutation enhances the DNA repair capability in GC cell line.
When co-transfected AGS with POLB siRNA and pCMU-Luc vector pre-treated with anticancer drugs, DNA repair capability was decreased when pre-treated with cisplatin and 5-FU. However, there were no significant DNA repair capability changes when co-transfected with POLB siRNA and pCMU-Luc vector pre-treated with epirubicin. These data demonstrate that the T889C mutation exerts a distinct effect on DNA repair toward anticancer drugs, such as cisplatin and 5-FU, but not epirubicin in GC cell line. Therefore, T889C mutation may be a gain-of-function mutation affecting chemotherapy response via DNA repair pathway.
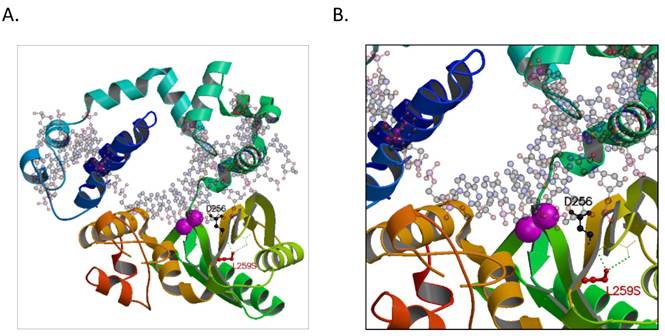
A hypothetical model of the human POLB with T889C mutation
To elucidate the possible mechanism of the POLB T889C mutation associated functional changes, we modeled its protein crystallographic structure. The T889C mutation in the nucleotide sequence of genbank accession M13140.1 would result in an encoded serine at residue position 259 that would be in hydrogen-bonding proximity to carboxyl groups of residues 232 and 233 on the backbone chain in an adjacent β-strand (Figure 6). These interactions would distort the β-strand where the serine resides resulting in a positional shift of the catalytic residue, D256. In wild type, D256 is stabilized by one of the two bound metal ions (Mg2+) and is thought to deprotonate the O3' group of the primer or newly forming DNA strand during synthesis [23]. A change in the active site could either result in a lower activation barrier for catalysis or simply loosen the constraints in the active site for the DNA strand to pass with greater flexibility and speed. There may be additional precedence for a more efficient active site as other hydrophobic substituted residues, now capable of electrostatic interactions, are also present around the active site, such as L228R, I260Y, and possibly L287K when comparing genbank accession number M13140.1 with AAA60133.1 used in our model (PDBid:4JWM).
A hypothetical model of the human POLB enzyme is shown in complex with DNA, and containing the L259S mutation that is found commonly in gastric cancer patients. The two known metal ions bound in the active site are shown as magenta spheres. A. The entire enzyme is shown revealing the relative location of the serine in proximity to the active site. B. A close-up view of additional hydrogen-bond networks is shown in a β-sheet that would lead to a predicted small distortion in the active site. The figure was made with the program, MOLSCRIPT [38].
Discussion
This study presents a comprehensive picture of the T889C mutation in POLB gene and its expression in relation with GC patients' clinicopathological features. We deciphered the functional relationship between mutant and wild type POLB gene, in association with DNA repair pathway. The T889C mutation was associated with POLB overexpression, clinicopathological features, such as lymph node metastasis and differentiation, and response to chemotherapy. Our clinical and experimental data indicate an oncogenic property in mutant POLB gene in comparison to the wild type POLB gene in GC. Previous study suggested that POLB exhibits dichotomous functions depending on its expression, either as an oncogene or as a tumor suppressor [27-30]. Our data indicate that T889C mutation in POLB is a relatively frequent phenomenon and this may be an underlying mechanism for gastric cell mutagenesis via enhanced DNA repair pathway. There are two possible reasons where the L259S substitution may arise naturally. First, it could allow for higher expression levels. We found a significant positive association between T889C mutation and overexpression of POLB gene. However, the exact mechanism by which T889C mutation leads to POLB gene overexpression is not clear. We hypothesize that epigenetic factors may be involved, which would be our direction for future investigation. Second, beyond epigenetics, a serine substitution could conceivably increase the specific activity of the polymerase in crisis. Detailed X-ray crystallographic studies are needed to determine the effects of the substitutions in the active site.
DNA repair and the fidelity of DNA relegation have been found to be correlated with metastatic potential [31-33]. We discovered that T889C mutation, but not POLB overexpression alone, was significantly correlated to increased lymph nodes metastasis. Our data suggests that abnormally enhanced DNA repair capability increases the risk of tumor metastasis for GC.
Previous studies suggested that POLB may play a fundamental role in cellular differentiation [34, 35]. Our results further demonstrated that poorly-differentiated tumors carried higher frequency of T889C mutation than moderately/well differentiated tumors. We hypothesize that the poor differentiation associated with T889C mutation may be due to the disruption of DNA repair synthesis or DNA translation synthesis during differentiation, or by facilitating accumulation of mutations in differentiation-related genes.
POLB has been found to efficiently bypass the cisplatin adduct [36]. Studies showed that abnormal POLB expression associated with the cytotoxic effects of DNA damaging chemotherapeutic agents [37]. In this study, we showed that T889C mutation of POLB gene was correlated closely with a poor response to 5-FU, cisplatin and epirubicin in GC patients. It is suggested that T8889C mutation might facilitate cell survival by enhancing DNA repair capability, to bypass various DNA damages by commonly used chemotherapeutic drugs.
Conclusions
The T889C mutation in POLB gene is a prevalent event among GC patients. The alteration of POLB gene mutation and expression is associated with the pathological features including differentiation, lymph nodes metastasis, and poor prognosis via DNA repair pathway. Thus, the POLB gene with T889C mutation in surgically resected primary gastric tissues may be clinically useful for predicting responsiveness to chemotherapy in patients with GC. The POLB gene alteration may serve as a prognostic biomarker for GC.
Abbreviations
GC, Gastric cancer; POLB, DNA polymerase beta; PCR-RFLP, PCR-restriction fragment length polymorphism.
Supplementary Material
Tables S1 -S2.
Acknowledgements
We would like to thank Dr. Kenneth H. Kraemer (NCI/NIH) for providing us with the pCMVLuc reporter gene plasmid and thank the individual patients for participating in this study. This work was supported by grants from the National Key Basic Research Program Project of China (Grant# 2004CB518708), the National Natural Science Foundation of China (NSFC) (Grant# 39625016), the National Bio-Tech 863 Program (Grant# 2012-AA02A203), the NIDDK/NIH (K01DK082646 to HSG) and the Elaine H. Snyder Cancer Research Award (to SWF).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Shi Y, Li L, Hu Z, Li S, Wang S, Liu J. et al. A genome-wide association study identifies two new cervical cancer susceptibility loci at 4q12 and 17q12. Nat Genet. 2011;45:918-22 doi:10.1038/ng.2687
2. Liu X, Cai H, Shi Y, Wang Y. Prognostic factors in patients with node-negative gastric cancer: a single center experience from China. J Gastrointest Surg. 2012;16:1123-7 doi:10.1007/s11605-012-1881-y
3. Wilson DM 3rd, Bohr VA. The mechanics of base excision repair, and its relationship to aging and disease. DNA Repair (Amst). 2007;6:544-59 doi:10.1016/j.dnarep.2006.10.017
4. Barakat K, Gajewski M, Tuszynski JA. DNA repair inhibitors: the next major step to improve cancer therapy. Curr Top Med Chem. 2012;12:1376-90 doi:CTMC-12-12-1376 [pii]
5. Masaoka A, Horton JK, Beard WA, Wilson SH. DNA polymerase beta and PARP activities in base excision repair in living cells. DNA Repair (Amst). 2009;8:1290-9 doi:10.1016/j.dnarep.2009.08.004
6. Zmudzka BZ, Fornace A, Collins J, Wilson SH. Characterization of DNA polymerase beta mRNA: cell-cycle and growth response in cultured human cells. Nucleic Acids Res. 1988;16:9587-96
7. Dalal S, Chikova A, Jaeger J, Sweasy JB. The Leu22Pro tumor-associated variant of DNA polymerase beta is dRP lyase deficient. Nucleic Acids Res. 2008;36:411-22 doi:10.1093/nar/gkm1053
8. Canitrot Y, Hoffmann JS, Calsou P, Hayakawa H, Salles B, Cazaux C. Nucleotide excision repair DNA synthesis by excess DNA polymerase beta: a potential source of genetic instability in cancer cells. FASEB J. 2000;14:1765-74
9. Srivastava DK, Husain I, Arteaga CL, Wilson SH. DNA polymerase beta expression differences in selected human tumors and cell lines. Carcinogenesis. 1999;20:1049-54
10. Bhattacharyya N, Chen HC, Comhair S, Erzurum SC, Banerjee S. Variant forms of DNA polymerase beta in primary lung carcinomas. DNA Cell Biol. 1999;18:549-54 doi:10.1089/104454999315097
11. Dobashi Y, Shuin T, Tsuruga H, Uemura H, Torigoe S, Kubota Y. DNA polymerase beta gene mutation in human prostate cancer. Cancer Res. 1994;54:2827-9
12. Miyamoto H, Miyagi Y, Ishikawa T, Ichikawa Y, Hosaka M, Kubota Y. DNA polymerase beta gene mutation in human breast cancer. Int J Cancer. 1999;83:708-9 doi:10.1002/(SICI)1097-0215(19991126)83:5<708::AID-IJC24>3.0.CO;2-C [pii]
13. Wang L, Patel U, Ghosh L, Banerjee S. DNA polymerase beta mutations in human colorectal cancer. Cancer Res. 1992;52:4824-7
14. Zhao GQ, Wang T, Zhao Q, Yang HY, Tan XH, Dong ZM. Mutation of DNA polymerase beta in esophageal carcinoma of different regions. World J Gastroenterol. 2005;11:4618-22
15. Tan XH, Zhao M, Pan KF, Dong Y, Dong B, Feng GJ. et al. Frequent mutation related with overexpression of DNA polymerase beta in primary tumors and precancerous lesions of human stomach. Cancer Lett. 2005;220:101-14 doi:10.1016/j.canlet.2004.07.049
16. Betsalel OT, van de Kamp JM, Martinez-Munoz C, Rosenberg EH, de Brouwer AP, Pouwels PJ. et al. Detection of low-level somatic and germline mosaicism by denaturing high-performance liquid chromatography in a EURO-MRX family with SLC6A8 deficiency. Neurogenetics. 2008;9:183-90 doi:10.1007/s10048-008-0125-5
17. Ponti G, Losi L, Martorana D, Priola M, Boni E, Pollio A. et al. Clinico-pathological and biomolecular findings in Italian patients with multiple cutaneous neurofibromas. Hered Cancer Clin Pract. 2011;9:6. doi:10.1186/1897-4287-9-6
18. Tang Z, Zhao M, Ji J, Yang G, Hu F, He J. et al. Overexpression of gastrin and c-met protein involved in human gastric carcinomas and intestinal metaplasia. Oncol Rep. 2004;11:333-9
19. Tan X, Anzick SL, Khan SG, Ueda T, Stone G, Digiovanna JJ. et al. Chimeric negative regulation of p14ARF and TBX1 by a t(9;22) translocation associated with melanoma, deafness, and DNA repair deficiency. Hum Mutat. 2013;34:1250-9 doi:10.1002/humu.22354
20. Tan X, Peng J, Fu Y, An S, Rezaei K, Tabbara S. et al. miR-638 mediated regulation of BRCA1 affects DNA repair and sensitivity to UV and cisplatin in triple negative breast cancer. Breast cancer research: BCR. 2014;16:435. doi:10.1186/s13058-014-0435-5
21. Fu Y, Lian Y, Kim KS, Zhang L, Hindle AK, Brody F. et al. BP1 Homeoprotein Enhances Metastatic Potential in ER-negative Breast Cancer. Journal of Cancer. 2010;1:54-62
22. Marti-Renom MA, Stuart AC, Fiser A, Sanchez R, Melo F, Sali A. Comparative protein structure modeling of genes and genomes. Annu Rev Biophys Biomol Struct. 2000;29:291-325 doi:10.1146/annurev.biophys.29.1.291
23. Batra VK, Perera L, Lin P, Shock DD, Beard WA, Pedersen LC. et al. Amino acid substitution in the active site of DNA polymerase beta explains the energy barrier of the nucleotidyl transfer reaction. J Am Chem Soc. 2013;135:8078-88 doi:10.1021/ja403842j
24. Kleywegt GJ, Jones TA. Model building and refinement practice. Methods Enzymol. 1997;277:208-30
25. Kleywegt GJ, Jones TA. Model-building and refinement practice. Methods In Enzymology. 1999;277:173
26. Barakat K, Tuszynski J. Relaxed complex scheme suggests novel inhibitors for the lyase activity of DNA polymerase beta. J Mol Graph Model. 2011;29:702-16 doi:10.1016/j.jmgm.2010.12.003
27. Poltoratsky V, Prasad R, Horton JK, Wilson SH. Down-regulation of DNA polymerase beta accompanies somatic hypermutation in human BL2 cell lines. DNA Repair (Amst). 2007;6:244-53 doi:10.1016/j.dnarep.2006.10.003
28. Singhal RK, Prasad R, Wilson SH. DNA polymerase beta conducts the gap-filling step in uracil-initiated base excision repair in a bovine testis nuclear extract. J Biol Chem. 1995;270:949-57
29. Starcevic D, Dalal S, Sweasy JB. Is there a link between DNA polymerase beta and cancer? Cell Cycle. 2004;3:998-1001 doi:1062 [pii]
30. Sweasy JB, Lang T, DiMaio D. Is base excision repair a tumor suppressor mechanism? Cell Cycle. 2006;5:250-9 doi:2414 [pii]
31. Kasakura Y, Mochizuki F, Wakabayashi K, Kochi M, Fujii M, Takayama T. An evaluation of the effectiveness of extended lymph node dissection in patients with gastric cancer: a retrospective study of 1403 cases at a single institution. J Surg Res. 2002;103:252-9 doi:10.1006/jsre.2002.6368
32. Usmani BA, Lunec J, Sherbet GV. DNA repair and repair fidelity in metastatic variants of the B16 murine melanoma. J Cell Biochem. 1993;51:336-44 doi:10.1002/jcb.240510313
33. Wei Q, Cheng L, Xie K, Bucana CD, Dong Z. Direct correlation between DNA repair capacity and metastatic potential of K-1735 murine melanoma cells. J Invest Dermatol. 1997;108:3-6 doi:S0022202X97819276 [pii]
34. Shadan FF, Villarreal LP. Potential role of DNA polymerase beta in gene therapy against cancer: a case for colorectal cancer. Med Hypotheses. 1996;47:1-9 doi:S0306-9877(96)90033-X [pii]
35. Sugo N, Niimi N, Aratani Y, Takiguchi-Hayashi K, Koyama H. p53 Deficiency rescues neuronal apoptosis but not differentiation in DNA polymerase beta-deficient mice. Mol Cell Biol. 2004;24:9470-7 doi:10.1128/MCB.24.21.9470-9477.2004
36. Hoffmann JS, Pillaire MJ, Maga G, Podust V, Hubscher U, Villani G. DNA polymerase beta bypasses in vitro a single d(GpG)-cisplatin adduct placed on codon 13 of the HRAS gene. Proc Natl Acad Sci U S A. 1995;92:5356-60
37. Horton JK, Watson M, Stefanick DF, Shaughnessy DT, Taylor JA, Wilson SH. XRCC1 and DNA polymerase beta in cellular protection against cytotoxic DNA single-strand breaks. Cell Res. 2008;18:48-63 doi:10.1038/cr.2008.7
38. Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. Journal of Applied Crystallography. 1991;24:5
Author contact
![]() Corresponding author: Sidney W. Fu (sfuedu), or Youyong Lu (youyonglu66com)
Corresponding author: Sidney W. Fu (sfuedu), or Youyong Lu (youyonglu66com)