International Journal of Biological Sciences 10
Impact Factor
ISSN: 1449-2288
Impact Factor
ISSN: 1449-2288
- Issue 6; 2026
- Issue 5; 2026
- Issue 4; 2026
- Issue 3; 2026
- Issue 2; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Top
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References
Introduction
Materials and Methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(5):546-558. doi:10.7150/ijbs.11084 This issue Cite
Research Paper
Evidence for Fungal Infection in Cerebrospinal Fluid and Brain Tissue from Patients with Amyotrophic Lateral Sclerosis
Ruth Alonso1, Diana Pisa1, Ana Isabel Marina1, Esperanza Morato1, Alberto Rábano2, Izaskun Rodal2, Luis Carrasco1 ![]()
1. Centro de Biología Molecular “Severo Ochoa”. c/Nicolás Cabrera, 1. Universidad Autónoma de Madrid. Cantoblanco. 28049 Madrid. Spain.
2. Department of Neuropathology and Tissue Bank, Unidad de Investigación Proyecto Alzheimer, Fundación CIEN, Instituto de Salud Carlos III, Madrid. Spain.
Received 2014-11-17; Accepted 2015-2-10; Published 2015-4-2
Citation:
Alonso R, Pisa D, Marina AI, Morato E, Rábano A, Rodal I, Carrasco L. Evidence for Fungal Infection in Cerebrospinal Fluid and Brain Tissue from Patients with Amyotrophic Lateral Sclerosis. Int J Biol Sci 2015; 11(5):546-558. doi:10.7150/ijbs.11084. https://www.ijbs.com/v11p0546.htm
Other stylesAbstract

Among neurogenerative diseases, amyotrophic lateral sclerosis (ALS) is a fatal illness characterized by a progressive motor neuron dysfunction in the motor cortex, brainstem and spinal cord. ALS is the most common form of motor neuron disease; yet, to date, the exact etiology of ALS remains unknown. In the present work, we have explored the possibility of fungal infection in cerebrospinal fluid (CSF) and in brain tissue from ALS patients. Fungal antigens, as well as DNA from several fungi, were detected in CSF from ALS patients. Additionally, examination of brain sections from the frontal cortex of ALS patients revealed the existence of immunopositive fungal antigens comprising punctate bodies in the cytoplasm of some neurons. Fungal DNA was also detected in brain tissue using PCR analysis, uncovering the presence of several fungal species. Finally, proteomic analyses of brain tissue demonstrated the occurrence of several fungal peptides. Collectively, our observations provide compelling evidence of fungal infection in the ALS patients analyzed, suggesting that this infection may play a part in the etiology of the disease or may constitute a risk factor for these patients.
Keywords: Amyotrophic lateral sclerosis, fungal infection, fungal PCR, brain histological analysis, proteomic analysis.
Introduction
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease that usually leads to death within 1-5 years period of diagnosis [1] ALS is a neuromuscular disease in which motor neurons located in the brain or spinal cord degenerate, giving rise to muscular weakness and atrophy. This loss of motor neurons leads to fatal paralysis and death and is the most frequent disease of motor neurons [1]. The vast majority of ALS cases are sporadic (about 95%), whereas only a few are familial. In familial ALS, a number of mutations in several genes have been described, including Cu/Zn-superoxide dismutase (SOD1) [2, 3]. Relationships between ALS and other neurodegenerative diseases have been noted, particularly with frontotemporal dementia (FTD) [4]. As occurs with most neurodegenerative diseases, the etiology of ALS remains unknown despite intensive research. The existence of aberrant protein aggregates inside neurons and glial cells constitutes an important hallmark for the classification of neurodegenerative diseases. In ALS, several proteins have been identified forming aggregates inside the cells in most patients. This is exemplified by ubiquitinated inclusions, as well as detection of transactive response (TAR)-DNA binding protein 43 (TDP-43) and the fused in sarcoma protein (FUS) in cytoplasmic granules [5-7]. TDP-43 exhibits a predominant nuclear localization, shuttling between the nucleus and cytoplasm, and is widely expressed in most cell types [8]. TDP-43 can interact with both DNA and RNA, and also with some members of the heterogeneous nuclear ribonucleoprotein family, participating in the process of transcription and splicing [9, 10]. In this way, TDP-43 modulates a number of cellular functions including apoptosis [11]. FUS protein is also expressed in a wide array of cells, and shuttles between nucleus and cytoplasm with the capacity to interact with nucleic acids. FUS is involved in a number of cellular processes, including transcription and RNA processing, cell proliferation and DNA repair [12]. Together with TDP-43 and FUS, a number of related proteins are under investigation as potential participants in ALS pathogenesis, including hnRNPA1, TAF15 and EWSR1 [6, 13-15].
The presence of cytoplasmic aggregates constitutes the basis for classification of different subtypes of both ALS and FTD, and it has been suggested that different protein aggregates may govern pathological mechanisms [4]. However, it is still unclear what practical value this classification provides for diagnosis and for future potential therapies. The central concept of ALS research is that mutations in some genes may lead to the synthesis of aberrant proteins that are misfolded, giving rise to cytoplasmic aggregates that are pathological for the correct functioning of motor neurons [16]. Accordingly, a number of mutations have been identified in TARDBP, the gene encoding TDP-43, and in the FUS gene [16]. These mutations only account for a small percentage of ALS cases; consequently, the number of candidate ALS genes is steadily increasing [16]. An intriguing question in the field is why mutations in different genes manifest after many years in the patient´s lifetime and also why these protein inclusions are specifically pathological for motor neurons and are not present in other cell types.
Similarities exist between ALS and Alzheimer's disease (AD), with regards to the presence of hyperphosphorylated tau protein in some neural cells and the formation of amyloid peptide [17, 18]. Recently, we have reported that fungal infection may play a part in the development of AD [19-21]. Fungal proteins and polysaccharides are present in the blood of AD patients and, more significantly, fungal proteins and DNA can be detected in brain samples from AD patients. Further, direct visualization of intracellular fungal infection in neurons from the frontal cortex of AD patients could be demonstrated with fungal-specific antibodies. Interestingly, chitin-like polysaccharides have been detected in AD brains and chitinase is elevated in cerebrospinal fluid (CSF) from AD patients [22-24]. To our knowledge, the possibility that a microbial infection could be the cause of ALS has thus far not been considered, although some correlations has been found between sporadic ALS and enterovirus infections [25]. More recently, elevated chitinase levels have been detected in CSF from ALS patients [26, 27]. In the present work, we have evaluated the existence of fungal proteins and DNA in CSF and brain samples from ALS patients. Our findings provide evidence for disseminated fungal infection in the central nervous system (CNS) in ALS patients.
Materials and Methods
Description of patients
The samples analyzed in this study correspond to cerebrospinal fluid (CSF) and tissue sections obtained from brain donors diagnosed with ALS. Details about the age and gender of each patient are listed in Supplementary Tables SI and SII. All samples were supplied by Banco de Tejidos, Fundación CIEN (Centro de Investigación de Enfermedades Neurologicas). The Ethics Committee of Universidad Autónoma de Madrid approved the study. The transfer of samples was performed according to national regulations concerning research on human biological samples. For all cases, written informed consent is available. All ethico-legal documents of the brain bank, including written informed consent, have been approved by an ethics committee external to the bank. The data obtained in this study was analyzed by an observer blinded to the diagnosis.
Preparation of antifungal antibodies
Rabbit antisera against Candida famata, C. albicans, C. parapsilosis, C. glabrata, and Penicillium notatum were obtained by inoculation of 1 or 2 mg of dried fungi (after autoclaving and lyophilization) in 0.5 ml PBS. Rabbit antiserum against peptide B tubulin-KLH (keyhole limpet hemocyanin) was purchased from PolyPeptide Group (Strasbourg, France). Each inoculum had been previously mixed with an equal volume of Freund's adjuvant. Rabbits were inoculated up to three times every three weeks and the antibody titer and specificity of the sera were tested by immunohistochemistry and immunoblotting. For antiserum against recombinant enolase of C. famata, pMAL-enolase was cloned using 5´Eno and 3´Eno primers (CGCCGCGGATCCATGGCCGTCACTAAGTTATT and CGCCGCGTCGACTTATAATTGAGAAGCAGCGT, respectively) and expressed in E. coli BL21 for purification via Ni-NTA Superflow (Promega) chromatography. Purified protein was then inoculated in rats, as described with rabbits.
Slot-blot assay of fungal proteins
To estimate the presence of fungal protein antigens, CFS was diluted and filtered through nitrocellulose membranes. A 200 µl volume of different CFS dilutions (1:10) in TBS was added to each well. Samples were blotted onto a 0.45-microns nitrocellulose membrane (Bio-Rad) previously hydrated in TBS for 10 minutes, using the Bio-Dot SF apparatus (Bio-Rad). After blotting, the membrane was processed and developed as described [28, 29]. The primary antibodies, rabbit polyclonal antibodies raised against C. famata, C. albicans, C. parapsilosis, C. glabrata, P. notatum and peptide of beta tubulin KLH, were used at a 1:500 dilution. A donkey anti-rabbit IgG horseradish peroxidase-conjugated secondary antibody (Amersham Biosciences) was used at a 1:5000 dilution. Rat polyclonal antibody raised against C. famata enolase of was used at a 1:200 dilution. A rabbit anti-rat IgG horseradish peroxidase-conjugated secondary antibody (Sigma) was used at a 1:5000 dilution. Densitometric analysis of the film was carried out using a GS-800 Calibrated Densitometer (Bio-Rad). The median of three different experiments and the cut-off of these values are indicated in the Results section. In previous studies, we established that values below 10 in this assay should be considered negative, between 10 and 20 as uncertain, and values above 20 as positive. In the latter case, values between 20 and 50 are considered low, between 50 and 80 as moderate and above 80 as high.
DNA Extraction from CSF
To extract DNA from CSF samples we followed this procedure: 20 µl of proteinase K (>600mAU/ml) was added to 150 µl of CSF and 200 µl buffer AL (QIAmp Kit, Qiagen) and mixed for 15 s. 200 µl ethanol was added to each sample after incubation at 56ºC for 10 min, followed by mixing by pulse-vortexing for 15 s. The mixture was then applied to the QIAamp Mini spin column and centrifuged at 8,000 rpm for 1 min. Afterwords, 500 µl buffer AW was added and samples were centrifuged at 8,000 rpm for 1 min. Next, 500 µl buffer AW2 was applied, followed by centrifugation at 14,000 rpm for 3 min. Finally, each sample was suspended in 40 µl distilled water. DNA from extracts was quantified with a NanoDrop® ND-1000 UV-Vis Spectrophotometer. As negative controls we employed three samples of tri-distilled filtered water.
DNA Extraction from tissue
Brain samples were used to extract DNA using the QIAmp (Qiagen) genomic DNA isolation kit as follows: 20 µl proteinase K (>600mAU/ml) and 180 µl of buffer ATL were added to 25 mg of brain tissue, followed by pulse-vortexing for 15 s. Then, it was incubated at 56ºC for 1-3 h with agitation. A 200 µl volume of buffer AL was added to each sample followed by vortexing for 15 s and incubation at 70ºC for 10 min. 200 µl ethanol was added to each sample with vortexing for 15 s. The mixture was applied to the QIAamp Mini spin column and centrifuged at 8,000 rpm for 1 min. After, 500 µl buffer AW was applied to the column, which was centrifuged at 8,000 rpm for 1 min. 500 µl buffer AW2 was applied and the column was centrifuged at 14,000 rpm for 3 min. Finally, each sample was collected in 40 µl distilled water and quantification of DNA was carried out with a NanoDrop® ND-1000 UV-Vis Spectrophotometer and. negative controls three samples of tri-distilled filtered water were employed.
Design of oligonucleotides
ITS oligonucleotides. The gene sequences of 5.8S and 18S rRNA, as well as internal transcribed spacer 1 (ITS 1) region from several organisms (C. albicans, C. famata, C. parapsilosis, C. glabrata, Rhodotorula mucilaginosa, Pichia guilliermondii, Cryptococcus neoformans and Homo sapiens) were accessed via the GenBank database and were aligned using the Clustal W sequence analysis package. Multiple potential primer-binding sites for this panfungal primer pair were chosen by comparing regions of Candida homologous with regions of the fungal group within the fungal kingdom with the most divergent DNA sequences and regions of Candida incongruous with the human DNA sequence. The primer selection was optimized for melting temperature equivalence, lack of duplex, hairpin, or primer-dimer formation, and internal stability by using OLIGO software (Amplify).
Nested PCR
A number of measures were used to avoid PCR assay contamination. Thus, we used separate rooms and glassware supplies for PCR set-up and products, aliquoted reagents, positive-displacement pipettes, aerosol-resistant tips, and multiple negative controls. DNA samples obtained from CSF or brain tissue were analyzed by nested PCR using the ITS-1 primers. The first PCR was carried out with 4 μl of DNA incubated at 95°C for 10 min and amplified with 30 cycles of 45 s at 94°C, 1 min at 57.3°C, and 45 s at 72°C. The oligonucleotides used were forward 14485´ GTTCTGGGCCGCACGGG 3′1465 and reverse 106R5′ GGCAAAGATTCGATGATT 3´88R The second PCR was carried out using one of two primer sets; in one instance, 2A ITS-1 primers were employed, whereas in the second instance 2B ITS-1 primers ( ITS1-ITS2, panfungal primers) were used. The second PCR was carried out with 0.5 μl of the product obtained in the first PCR, and was amplified with 2A ITS-1 primers with 30 cycles of 45 s at 94°C, 1 min at 52°C, and 45 s at 72°C. The oligonucleotides used were forward 17815′ GCGTCTA GACCTGCGGAAGGATCA 3´1798 and reverse 20R5′ GCGAAGCTT GATCCGTTGTTGAAA 3´5R. These primers contained extra nucleotides to provide a cloning site for the amplified PCR product. As an alternative second PCR, 2B ITS1-ITS2 primers were used. The second PCR was carried out with 0.5 μl of the product obtained in the first PCR and was amplified with 35 cycles of 45 s at 94°C, 1 min at 55°C, and 45 s at 72°C. The oligonucleotides used were forward 17715´ TCCGTAGGTGAACCTGCGG 3´1790 and reverse 50R5´ GCTGCGTTCTTCATCGATGC 3´30R which are the commonly used ITS1 and ITS2 primers developed by [30]. Amplified products were analyzed by agarose gel electrophoresis and stained with ethidium bromide. We wish to indicate that these PCR assays can amplify the vast majority of fungal DNA, however some fungal species may not be detected because of the primers or the conditions employed. Moreover, different combinations of these primers may provide a better coverage of other species.
Mass Spectrometry Analysis
Initially, proteins from brain samples were separated by polyacrylamide gel electrophoresis (PAGE) under reducing conditions according to Laemmli, with a 12.5% separating gel and a 5% stacking gel. Protein staining was carried out with GelCode Blue Stain Reagent (Thermo Scientific). Separation of proteins and gel excision were carried out essentially as previously described [19]. Twelve stained sections (about 0.5-0.8 cm) were excised from the gel, and proteins were in-gel digested with trypsin, after reduction and alkylation. This procedure was performed after drying, gel bands were destained in acetonitrile:water (ACN:H2O, 1:1) and were reduced and alkylated. Thus, disulfide bonds from cysteinyl residues were reduced with 10 mM DTT for 1 h at 60ºC, and then thiol groups were alkylated with 55 mM iodoacetamide for 1 h at room temperature in the dark. The proteins were then digested in situ with sequencing grade trypsin (Promega, Madison, WI). The gel pieces were shrunk by dehydration in acetonitrile, which was then removed. The gel pieces were then dried in a vacuum centrifuge. Afterwards, the dried gel pieces were re-swollen in 50 mM ammonium bicarbonate pH 8.8 with 12.5 ng/μl trypsin for 1 h in an ice-bath. The digestion buffer was removed and gels were covered again with 50 mM NH4HCO3 and incubated at 37°C for 12 h. Digestion was stopped by the addition of 1% TFA. Whole supernatants were dried down and then desalted on OMIX C18 Pipette tips (Agilent) for the mass spectrometry analysis. The peptide extracts were combined into 3 pools of discontinuous bands, to reduce the analysis by LC-MS to 4 runs per sample. The desalted protein digest was dried, resuspended in 10 μl of 0.1% formic acid and analyzed by RP-LC-MS/MS in an Easy-nLC II system coupled to a LTQ-Orbitrap-Velos-Pro hybrid mass spectrometer (Thermo Scientific). Concentration of peptides (on-line) was carried out by reverse phase chromatography using a 0.1 mm × 20 mm C18 RP precolumn (Thermo Scientific), and then separated using a 0.075mm x 150 mm C18 RP column (Thermo Scientific) operating at 0.3 μl/min. Elution of peptides was performed using two different gradients: a 120 or 180-min gradient from 5 to 40% solvent B (Solvent A: 0.1% formic acid in water, solvent B: 0.1% formic acid, 80% acetonitrile in water). ESI ionization was done using a Nano-bore emitter Stainless Steel ID 30 μm (Proxeon) interface. The Orbitrap resolution was set at 30,000. Peptides were detected in survey scans from 400 to 1600 amu (1 μscan), followed by twenty data dependent MS/MS scans (Top 20), using an isolation width of 2 u (in mass-to-charge ratio units), normalized collision energy of 35%, and dynamic exclusion applied during 30 second periods.
Mass spectrometry data processing
Processing of the MS data was carried out as previously described [19]. Briefly, peptide identification from raw data was carried out using the SEQUEST algorithm (Proteome Discoverer 1.3, Thermo Scientific). Database searching was performed against uniprot-homo fasta and uniprot-fungi fasta.
Histological analysis
Brain sections (5 μm) were fixed in 10% buffered formalin for 24 h and then embedded in paraffin following standard protocols. For immunohistochemical analysis, paraffin was removed and the tissues were rehydrated and boiled for 2 min in citrate buffer. After, samples were incubated 10 min with ammonium chloride. Subsequently, tissue sections were incubated 10 min with PBS/Triton X-100 0.1% and further incubated for 20 min with PBS/BSA 2%. Sections were then incubated overnight at 4oC with mouse monoclonal antibody raised against human α-tubulin at a 1:50 dilution, or rabbit polyclonal antibody raised against proteins obtained from C. glabrata at a 1:500 dilution. After this incubation, the sections were washed with PBS and incubated for 1 hour at 37ºC with donkey anti-mouse IgG secondary antibody conjugated to Alexa 555 (Invitrogen) for α-tubulin and donkey anti-rabbit IgG secondary antibody conjugated to Alexa 555 or Alexa 488 (Invitrogen) at a 1:500 dilution for anti-C. glabrata. Afterwards, tissue sections were stained with DAPI (Merck). Then, samples were treated with Autofluorescence Eliminator Reagent (Merck). Finally, sections were observed using a LSM710 confocal laser scanning microscope combined with the upright microscope stand AxioImager.M2 (Zeiss). The spectral system employed was Quasar + 2 PMTs. Zeiss ZEN 2010 Program.
Results
Search of fungal infection in CSF from ALS patients. Presence of fungal antigens
A highly sensitive slot-blot assay to analyze the presence of fungi in blood serum or CSF has been developed using specific antibodies against fungal antigens [20, 28, 31]. Samples are blotted onto a nitrocellulose membrane and then incubated with the corresponding primary antibody at the indicated dilution. Subsequently, the membrane is incubated with a peroxidase-conjugated secondary antibody and the film obtained is scanned. We have previously established the cut-off values to determine negative or positive results [31]. We first tested a number of antibodies raised against total fungal cells or purified enolase from C. famata, or a β-tubulin peptide specific for fungi. CSF samples from five ALS patients and from three controls (see Supplementary Table SI) were tested. Figure 1 shows representative results obtained with anti-C. albicans and enolase antibodies. A positive control using yeast proteins and a negative control containing TBS were used. Certainly, CSF samples from ALS patients rendered a clear positive result. Densitometric quantification of this assay employed a panel of different antibodies is presented in Table I indicating the positive reactivity for fungal antigens in all five patients. As expected, values obtained in control CSF samples for a range of fungi were rather low. In contrast, slot-blot values from CSF of ALS patients were generally considerably higher. For example, CSF from patient 1 exhibited high values with several antibodies employed, including anti-C. famata, C. albicans, C. glabrata, enolase and β-tubulin. Further, high values were obtained in most cases with the anti-C. albicans, enolase and β-tubulin antibodies. Not all the antibodies exhibited a similar behavior, since in some instances the differences found with a given antibody were more pronounced between ALS and control CSFs. This may indicate that the fungal antigens present in ALS CSF immunoreact much better than other anti-fungal antibodies. For example, this is the case of values obtained with anti-C. albicans antibodies that rendered a median of 131.2 in ALS and 11.3 in controls, with a P=0.18 and the odds ratio of 8.0. Considering all the values of Table I, the results obtained are: P=0.01 and odds ratio=4.8. Overall, these results indicate that fungal antigens are present in CSF from ALS patients.
Figure 1
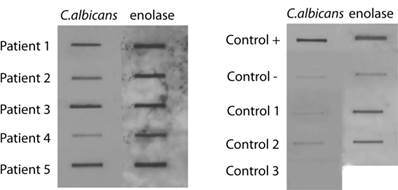
Analysis of C. albicans and enolase antigens in CSF by slot-blot. 20 μl CSF samples were diluted with 180 μl TBS and were blotted onto a nitrocellulose membrane, which was incubated with the rabbit antiserum against C. albicans or enolase or recombinant MBP-enolase (primary antibody) as indicated and afterwards incubated with a rabbit anti rat IgG (secondary antibody). Positive control: control + 200 ng yeast protein or purified MBP-enolase. Negative control: control - corresponds to TBS alone.
Table I
Fungal antigens present in CSF from ALS patients immunoreacting with antibodies against different yeast species.
| Patients | C.famata | C.albicans | C.glabrata | C.parapsilosis | Penicillium | Enolase | β tubulin peptide |
|---|---|---|---|---|---|---|---|
| Patient 1 | 88±2.7 | 164±6.6 | 118±4.7 | 42±2.9 | 56±3.9 | 183±5.5 | 280±8.4 |
| Patient 2 | 17±1.4 | 65±4.1 | 58±4.0 | 32±2.2 | 15±1.2 | 150±4.5 | 52±3.6 |
| Patient 3 | 2±0.2 | 180±7.2 | 14±1.1 | 5±0.4 | 129±5.2 | 130±3.9 | 144±5.8 |
| Patient 4 | 39±3.1 | 56±3.8 | 1±0.1 | 48±3.4 | 17±1.4 | 60±4.2 | 153±6.1 |
| Patient 5 | 58±4.1 | 191±7.6 | 7±0.6 | 13±1.0 | 20±1.6 | 78±3.6 | 55±3.8 |
| Mean value | 40.8 | 131.2 | 39.6 | 28 | 47.4 | 120.2 | 136.8 |
| Control 1 | 26±2.1 | 13±0.9 | 15±1.2 | 55±4.4 | 4±0.3 | 79±3.9 | 10±0.7 |
| Control 2 | 9±0.7 | 20±1.5 | 88±2.8 | 30±2.4 | 59±4.1 | 79±3.9 | 81±3.2 |
| Control 3 | 1±0.1 | 1±0.2 | 5±0.4 | 3±0.2 | 1±0.1 | ND | 1±0.2 |
| Mean value | 12.0 | 11.3 | 33.3 | 29.3 | 21.3 | 79 | 30.6 |
The median of the densitometric values of three experiments and the standard deviation are indicated.
ND : not done
PCR assay of DNA isolated from CSF
An additional sensitive method to assess fungal infection is PCR [32, 33]. This technique has the added advantage that, after amplification of variable internal transcribed spacer 1 (ITS1) present between the rRNA genes (see scheme Figure 2), followed by agarose electrophoresis, sequencing of the amplified products reveals the fungal species present. However, it must be considered that the bulk of the DNA obtained is from human origin and only a very small amount would be fungal. Due to the limited volume of CSF, DNA could not be obtained from one of the patient samples; nonetheless, DNA was obtained from four CSF samples and the ITS1 sequences were amplified by nested PCR. After the first PCR, the second amplification was carried out with two independent primer pairs, as detailed in materials and methods (see scheme, Figure 2). The first primer pair, 2A ITS1, has been previously employed successfully by us [19, 31]. The second primer pair, 2B ITS1, have been described as panfungal primers [34], although they do not completely hybridize with Malassezia spp. The use of different primers may serve to a better coverage of several fungal species. Moreover, future studies in this regard may benefit from the use of a wider range of primers to perform PCR studies. Notably, several products were amplified using 2A ITS1 primers in DNA extracted from CSF of ALS patients (Figure 2). In contrast, no products were obtained in control CSF samples or in controls for DNA extraction or PCR amplification without DNA, indicating that these amplified products were not due to contamination during DNA extraction or PCR amplification. DNA sequencing of the amplified products revealed the fungal species listed in Figure 2. Of interest, 2A ITS1 primers amplifed Malassezia spp. (Figure 2). More importantly, C. albicans was also detected in two patients (2 and 3). The use of the panfungal primers 2B ITS1 also revealed the presence of C. albicans DNA in these two patients. Additionally, Rhodotorula mucilaginosa was detected in patients 1 and 4 using the 2B ITS1 primer pair, but Malassezia spp. was not amplified. The possibility that DNA from other fungal species was present in lower amounts is not excluded by these results. Moreover, it is possible that fungal cells from some species, which are present in brain tissue, are not released to the CSF. Taken together, these results strongly suggest that DNA from several fungi is present in CSF from ALS patients.
Figure 2
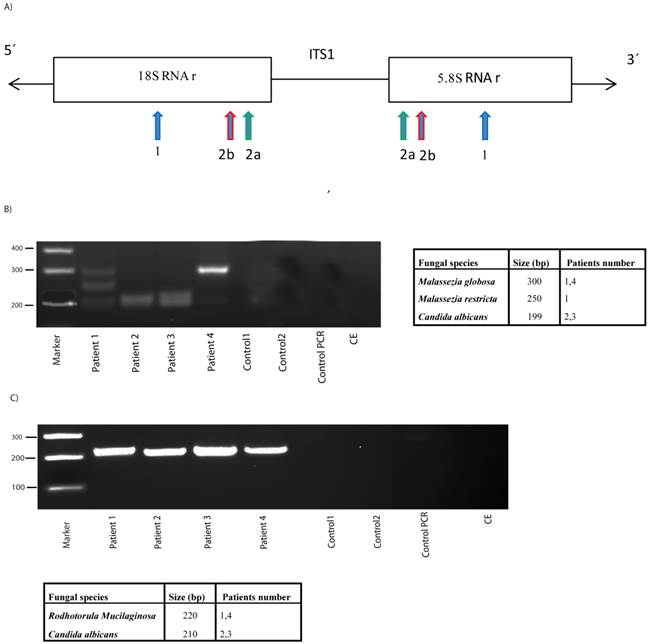
PCR analysis of DNA obtained from CSF. A) Schematic representation of fungal rRNA genes and the ITS1 sequence. Location of the primers employed for the PCR: primers 1 employed in the first PCR; primers 2A employed in the second PCR; primers 2B employed in the second PCR and previously described as panfungal primers. B) PCR was carried out as described from DNA samples obtained from CSF of ALS patients or controls. The primers employed were primers 1 for the first round PCR and primers 2A for the second round. After PCR, the samples were separated on agarose gels and stained with ethidium bromide. DNA size markers are shown on the left. Fungal species detected after sequencing each product is shown on the right. C) The samples obtained after the first PCR (primers 1) were amplified using primers 2B. PCR products were separated on agarose gels, extracted and sequenced. Fungal species detected are depicted on the right. Control PCR: PCR without DNA. CE: Control of DNA extraction without CFS DNA. 1: external primers. 2A: internal primers. 2B: panfungal primers.
Visualization of fungal infection in brain by immunohistochemistry
Given the above results in CSF, we next examined sections from the frontal cortex by immunohistochemistry using polyclonal rabbit antibodies raised against C. glabrata. Two individual fields are depicted for each sample (Figure 3). No immunofluorescence was found in the frontal cortex from three control individuals (Supplementary Figure S1). In contrast, several neurons were immunopositive for C. glabrata and stained intracellular punctate bodies in the cytoplasm in ALS brain sections (Figure 3). These bodies resemble the intracellular morphology of other fungi, as reported previously [35, 36]. This fungal material was not observed in all neurons, because some of them were devoid of it. A wider film of one control and one ALS patient is shown in Figure 4. In this case, double immunofluorescence with anti-tubulin and anti-C. glabrata antibodies was carried out. The subcellular distribution of tubulin was similar to that reported for neurons in some neurodegenerative diseases [37, 38]. The control sample was devoid of fungal bodies whereas they were clearly observed in the ALS patient. Apart from the intracellular punctate fungal material some extracellular bodies were also apparent. The morphological appearance is similar to our recently described findings in neurons from patients diagnosed with AD and these fungal bodies were known as intramycosomes [21] or perhaps we can name them more adequately, endomycosomes. Both, ALS and AD share parallels, but it is possible that the fungal species that affect both types of patients are different, and the genetic background of these patients also differs. The most important conclusion from these results is that fungal bodies can be observed in neurons from brains of ALS patients. Obviously, these studies do not indicate the exact fungal species present in these samples.
Fungal DNA in brain samples
To analyze the potential fungal species present in brain samples from ALS patients, DNA was extracted from different CNS regions as indicated in Figure 5, and nested PCR was carried out as described using primers 1 in the first round and primers 2A in the second round. Interestingly, several DNA products were amplified in the three ALS patients analyzed, whereas no amplification was found in control sample (Figure 3). DNA sequencing of the isolated products resulted in the identification of the species, depicted in the bottom of Figure 5. Notably, several species could be identified from one patient and these species varied according to the CNS region analyzed. Further, different fungal species were detected in patients, although some were present in all three ALS patients. Accordingly, Malassezia globosa was present in most of the samples tested, whereas Cryptococcus neoformans was only detected in one sample. In our opinion, the most striking finding was that C. albicans appeared in all three patients. This yeast was also present in the CSF of several ALS patients (Figure 2) and it is acknowledged that C. albicans can be highly pathogenic; this also applies to the presence of C. neoformans in one patient. Possibly, the existence of different fungal species may account for the differences in the severity and evolution of disease observed between ALS patients. Results from the second PCR using panfungal primers (2B), revealed that, as before, different fungal species could be detected. Trychoderma viridae was evident in all samples, whereas Cryptococcus magnus appeared in the four samples of patient 6. Collectively, these findings show that fungal infection of the CNS in ALS patients can be detected by PCR analysis, and different fungal species can also be determined. However, we do not know if the species identified by PCR are exactly the ones observed by immunofluorescence.
Proteomic analyses of brain samples
Determination of the proteome of CSF or CNS from patients with neurodegenerative disease may serve to identify biomarkers for diagnosis, prognosis and evolution of each particular disease [39-42]. To our knowledge, no attempts have been made to identify fungal proteins in samples from ALS patients. Therefore, frontal cortex proteins from three ALS patients were initially separated by polyacrylamide gel electrophoresis and stained with Coomassie blue (Supplementary Figure S2). After gel staining, each lane was cut in 12 slices and digested with trypsin. Peptide extracts from three non-consecutive slices were pooled and analyzed by mass spectrometry. Consequently, four samples were analyzed by MS from each patient. Each sample was analyzed twice; the first analysis was carried out with a 120 min gradient and the second analysis with 180 min gradient, in order to obtain more peptides by increasing the gradient time. Each run was analyzed with the Proteome Discoverer (PD) 1.3 software, allowing the search for human and fungal proteins (Uniprot). In this manner, we could compare the peptides that are common to both human and fungi, to better discriminate which peptides were specific for fungi. Additionally, the peptides obtained from the 180 min gradient were analyzed with the Peaks 6 software, using the tool called PEAKS PTM which identifies post-translational modifications and variations due to potential mutations compiled in the Unimod library,as well as custom PTMs [43]. Each peptide identified as fungal by PD 1.3 was subjected to further scrutiny by: 1) confirmation of the identification quality by visual inspection of the spectra; 2) search of homology to homo sapiens peptides by BLAST (ExPASY BLAST) to assess the differences between the corresponding human and fungal peptides; 3) peaks 6 study of the sequences assigned for fungi and human.
Figure 3
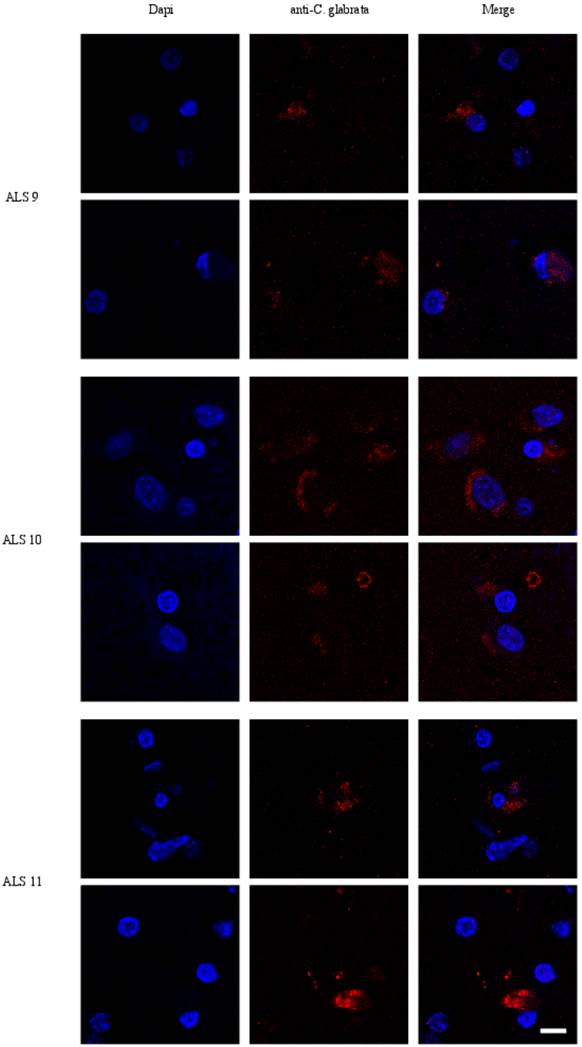
Immunohistochemistry analysis of brain sections from the frontal cortex of ALS patients. Brain sections (frontal cortex) from ALS patients 9, 10 and 11 were observed with a confocal laser scanning microscope. Sections were obtained from fixed tissue and immunohistochemistry analyses were carried out. Sections were incubated with anti-C. glabrata antibodies (1:500 dilution), followed by incubation with secondary antibody donkey anti-rabbit IgG conjugated to Alexa 555 (1:500). Subsequently, sections were incubated with DAPI (1 μg/ml). The different panels in the figure are indicated. Scale bar: 10 μm.
Figure 4
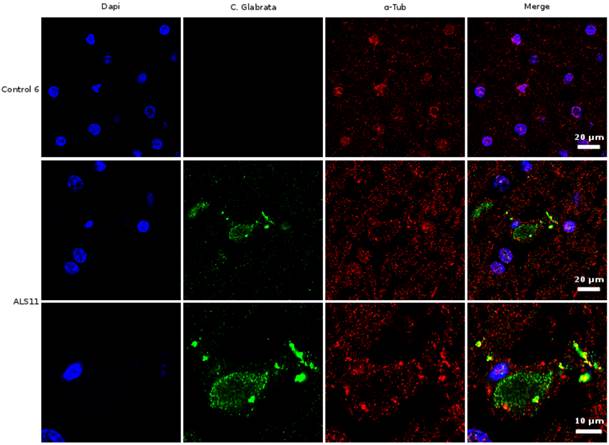
Immunohistochemistry analysis of brain sections from the frontal cortex of an ALS patient and a control. Brain sections (frontal cortex) from ALS patient 11 and control 6 were observed with a confocal laser scanning microscope. Sections were obtained from fixed tissue and immunohistochemistry analyses were carried out. Double immunofluorescence assay using anti-tubulin and anti-C. glabrata antibodies was carried out as detailed in Materials and Methods. DAPI appears in blue and anti-C. glabrata is shown in green. Human tubulin appears in red. The different panels in the figure are indicated.
Figure 5
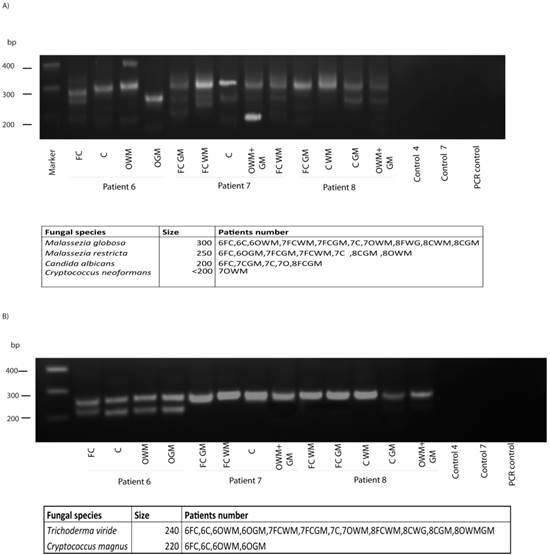
PCR analysis of different brain regions from three ALS patients. PCR was carried out as described. A) PCR analysis of different brain regions and controls using primers 1 and 2a. Table shows fungal species detected. B) PCR analysis of different brain regions and controls using primers 1 and 2b. Table shows fungal species detected. Control PCR: PCR without DNA. CE: Control of DNA extraction without CFS DNA. 1: external primers. 2A: internal primers. 2B: panfungal primers. FC: Frontal cortex. C: Cerebellum. O: Occipital cortex. WM: White matter. GM: Grey matter.
The human proteins common to the three patients that were not present in a control recently published by our laboratory are listed in Supplementary Table SIII. These proteins can be grouped according to their cellular function (Figure 6). These proteins may constitute the basis for future ALS biomarkers.
Regarding the identification of fungal proteins, Table II summarizes the fungal peptides obtained in the four fractions A-D from the three ALS patients. Although many more peptides may be present, we only indicate in Table II the peptides that belong to fungi beyond any doubt. We are confident that the peptides corresponding to α- and β-tubulin are of fungal origin. The identification of tubulin by proteomic analyses is consistent with the fact that tubulin is an abundant cellular protein. Fungal peptides corresponding to actin have similarities to their human counterparts and could not therefore be unequivocally identified. Nevertheless, the important finding from proteomic analysis is that fungal tubulin is present in brain samples from ALS patients, further supporting the notion that fungal infection exists in the CNS of ALS patients.
Figure 6
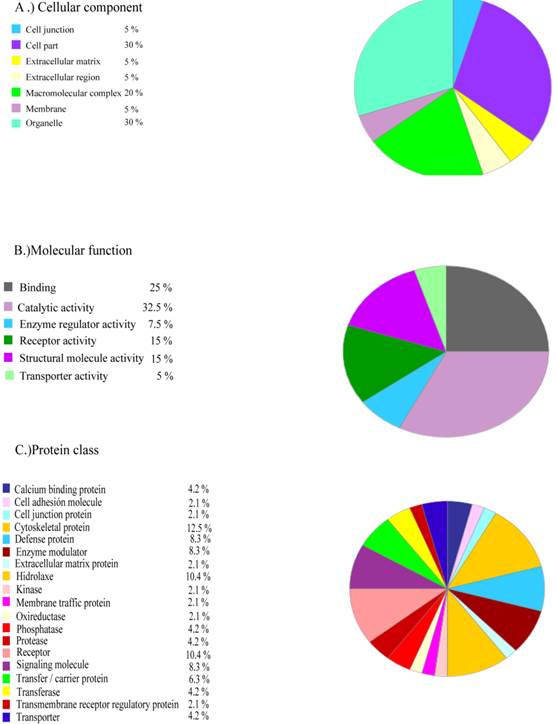
Functional grouping of human proteins specifically present in the frontal cortex from ALS patients. Human proteins which are common in the three ALS patients that are not present in control brain grouped by: a) cellular component; b) molecular function; c) protein class.
Table II
Proteomic analysis of fungal peptides present in brain samples from ALS patients.
| Patients | Total peptides fungi identified with PD 1.3 | Peptides fungi not present in human database | Peptides fungi confirmed with peaks PTM and manual inspection | Certain fungal peptides |
|---|---|---|---|---|
| Patient 6 | ||||
| Fraction A | 159 | 27 | 0 | |
| Fraction B | 188 | 50 | 2 | AVLDLEPGTMDNVR β-tubulin AVCMLSNTTAIAEAWSR. α-tubulin |
| Fraction C | 192 | 37 | 0 | |
| Fraction D | 103 | 15 | 0 | |
| Patient 7 | ||||
| Fraction A | 212 | 48 | 0 | |
| Fraction B | 210 | 49 | 1 | AVCMLSNTTAIAEAWSR. α-tubulin |
| Fraction C | 213 | 33 | 0 | |
| Fraction D | 170 | 27 | 0 | |
| Patient 8 | ||||
| Fraction A | 266 | 68 | 0 | |
| Fraction B | 207 | 48 | 1 | CVSMLSNTTAIAEAWSR. α-tubulin |
| Fraction C | 210 | 34 | 0 | |
| Fraction D | 187 | 31 | 0 |
PD1.3: Proteome Discoverer 1.3 software
Discussion
The involvement of microbial infections in neurodegenerative diseases has been deliberated by several laboratories [44-47]. However, these studies have focused on viral or bacterial infections and less attention has been given to the possibility that fungal infections may be responsible for some of these diseases [48]. The notion that fungal infection may be involved in the etiology of multiple sclerosis (MS) and AD has been considered in several reports from our laboratory [19-21, 28, 31, 49]. This concept is consistent with a number of clinical symptoms and observations reported for these diseases. Similar to neurodegenerative disease, fungal infections are usually chronic and progressive if untreated. Neuroinflammation is also repeatedly observed in the majority of neurodegenerative diseases, including ALS, and the continuous presence of fungal pathogens in the CNS can provoke not only chronic inflammation, but also the sustained production of microbial products. Both inflammation and microbial products, or even intracellular fungal cells (endomycosomes), could lead to neuronal degeneration and death. Additionally, fungal infection could also account for the generation of granules of misfolded or aberrantly-processed proteins. Granular type may depend on a number of variables: 1) the subset of neural cells affected; 2) the individual fungal species infecting the CNS and the route of this infection; 3) the genetic background of the patient and, 4) the state of the immune system and the lifestyle of each individual. Differences in any of these parameters could give rise to different pathologies. Conversely, if some neurodegenerative diseases are caused by fungal infections, it is also logical that they will share several similarities [50-52]. Our previous reports have provided evidence for fungal infections in MS and AD patients. Strikingly, chitin-like polysaccharides have been found in brains from MS and AD patients [22, 23] and most probably as a consequence there are elevated levels of chitinase in AD CSF [24]. However, these researchers did not suggest the existence of fungal infection. The present work provides the first description of fungal proteins and DNA both in CSF and CNS from ALS patients. Furthermore, immunofluorescence analysis clearly demonstrated fungal infection within some neurons, whereas other neighboring cells were devoid of this material. These observations provide direct evidence for the concept that ALS patients suffer fungal infections in some neurons of the CNS and are in accord with proteomic and PCR results identifying several fungal species. The recent finding that CSF from ALS patients contains elevated levels of chitinase [26, 27] do also support the idea of fungal infection in these patients.
ALS is a heterogeneous disease in which the evolution and clinical symptoms vary widely between patients; consequently, the disease can progress rapidly in some patients, who die within one year from diagnosis, while other patients can survive several years after the onset of clinical symptoms. The presence of different fungal species in each patient may account for this behavior. A number of similarities have been observed between different neurodegenerative diseases, particularly between MS, AD and Parkinson's disease, as well as between ALS and FTD [4, 53-55]. Notably, some likenesses have also been reported between AD and ALS, including tangles of hyperphosphorylated tau in some neural cells from ALS patients with or without cognitive impairment, although in the latter there is a more widely distributed tau pathology [18, 56]. The existence of fungal infections both in AD and ALS patients also provides an explanation for the similarities reported between these diseases. It is well established that individual genetic background can determine the likelihood of fungal infections [57-59]. Also, the presence of particular mutations in different genes in ALS patients may dictate their predisposition for ALS. In familial ALS cases, the most represented gene mutations are in the SOD1 gene [3]. SOD, an antioxidant defense gene, is involved in the innate immune response to a number of pathogens including fungi [60, 61]. Therefore, it is perhaps not surprising that SOD1 mutations that may affect the immune response in ALS patients [62, 63] might confer a greater sensitivity to fungal infection.
An additional symptom shared by AD and ALS is the occurrence of neuroinflammation and infiltration of T-lymphocytes in some areas of the CNS or spinal cord [52, 64-68]. Also, modifications in the number of different subsets of circulating T-lymphocytes have been noted in both disorders [69-71], suggesting that there is a systemic alteration of the immune system in patients suffering from these diseases. Supporting the concept that neuroinflammation plays a part in ALS pathology, the number of regulatory T-lymphocytes correlates inversely with disease progression [72]. Neuroinflammation and T cell infiltration may be consistent with microbial infection [73]. In this regard, our present findings reveal fungal infection both in CSF and brain samples from patients with ALS. Strikingly, the immunofluorescence results are also consistent with the concept that some neural cells contain intracellular fungal material or endomycosomes. This low burden of infection, together with the fact that endomycosomes are very small (about 1 μm in diameter), provides an explanation for the hitherto elusive detection of this type of infection in ALS. If disseminated mycoses are responsible for ALS, this might explain why the disease appears suddenly in patients. Also, differences in the severity and evolution of clinical symptoms might be rationalized by the different fungal species, or even combinations of these species. Another possibility should be that immune tolerance to fungal infection is lost in these patients and inflammation provokes injury and clinical symptoms [74]. Our present observations not only open a new field for the investigation of ALS, but if they are correct, ALS patients would immediately benefit from antifungal therapies. In this sense, clinical trials should definitively clarify the potential participation of fungal infection in the pathology of ALS.
Supplementary Material
Table SI, SII, SIII, and Fig.S1-S2.
Acknowledgements
The financial support of Fundación ONCE (Organización Nacional de Ciegos Españoles) is acknowledged. We acknowledge an institutional grant to Centro de Biología Molecular “Severo Ochoa” from the Fundación Ramón Areces.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mitchell JD, Wokke JH, Borasio GD. Recombinant human insulin-like growth factor I (rhIGF-I) for amyotrophic lateral sclerosis/motor neuron disease. The Cochrane database of systematic reviews. 2007:CD002064
2. Leblond CS, Kaneb HM, Dion PA, Rouleau GA. Dissection of genetic factors associated with amyotrophic lateral sclerosis. Experimental neurology. 2014
3. Tan W, Pasinelli P, Trotti D. Role of mitochondria in mutant SOD1 linked amyotrophic lateral sclerosis. Biochimica et biophysica acta. 2014;1842:1295-301
4. Mackenzie IR, Rademakers R, Neumann M. TDP-43 and FUS in amyotrophic lateral sclerosis and frontotemporal dementia. Lancet neurology. 2010;9:995-1007
5. Kwiatkowski TJ Jr, Bosco DA, Leclerc AL, Tamrazian E, Vanderburg CR, Russ C. et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science (New York, NY. 2009;323:1205-8
6. Neumann M, Bentmann E, Dormann D, Jawaid A, DeJesus-Hernandez M, Ansorge O. et al. FET proteins TAF15 and EWS are selective markers that distinguish FTLD with FUS pathology from amyotrophic lateral sclerosis with FUS mutations. Brain. 2011;134:2595-609
7. Vance C, Rogelj B, Hortobagyi T, De Vos KJ, Nishimura AL, Sreedharan J. et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science (New York, NY. 2009;323:1208-11
8. Wang HY, Wang IF, Bose J, Shen CK. Structural diversity and functional implications of the eukaryotic TDP gene family. Genomics. 2004;83:130-9
9. Buratti E, Brindisi A, Giombi M, Tisminetzky S, Ayala YM, Baralle FE. TDP-43 binds heterogeneous nuclear ribonucleoprotein A/B through its C-terminal tail: an important region for the inhibition of cystic fibrosis transmembrane conductance regulator exon 9 splicing. The Journal of biological chemistry. 2005;280:37572-84
10. Buratti E, Dork T, Zuccato E, Pagani F, Romano M, Baralle FE. Nuclear factor TDP-43 and SR proteins promote in vitro and in vivo CFTR exon 9 skipping. The EMBO journal. 2001;20:1774-84
11. Ghavami S, Shojaei S, Yeganeh B, Ande SR, Jangamreddy JR, Mehrpour M. et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Progress in neurobiology. 2014;112:24-49
12. Lagier-Tourenne C, Polymenidou M, Cleveland DW. TDP-43 and FUS/TLS: emerging roles in RNA processing and neurodegeneration. Human molecular genetics. 2010;19:R46-64
13. Le Ber I, Van Bortel I, Nicolas G, Bouya-Ahmed K, Camuzat A, Wallon D. et al. hnRNPA2B1 and hnRNPA1 mutations are rare in patients with "multisystem proteinopathy" and frontotemporal lobar degeneration phenotypes. Neurobiology of aging. 2014;35:934 e5-6
14. Thomsen C, Grundevik P, Elias P, Stahlberg A, Aman P. A conserved N-terminal motif is required for complex formation between FUS, EWSR1, TAF15 and their oncogenic fusion proteins. Faseb J. 2013;27:4965-74
15. Ugras SE, Shorter J. RNA-Binding Proteins in Amyotrophic Lateral Sclerosis and Neurodegeneration. Neurology research international. 2012;2012:432780
16. Iguchi Y, Katsuno M, Ikenaka K, Ishigaki S, Sobue G. Amyotrophic lateral sclerosis: an update on recent genetic insights. Journal of neurology. 2013;260:2917-27
17. Muresan V, Villegas C, Ladescu Muresan Z. Functional interaction between amyloid-beta precursor protein and peripherin neurofilaments: a shared pathway leading to Alzheimer's disease and amyotrophic lateral sclerosis? Neuro-degenerative diseases. 2013;13:122-5
18. Yang W, Strong MJ. Widespread neuronal and glial hyperphosphorylated tau deposition in ALS with cognitive impairment. Amyotroph Lateral Scler. 2012;13:178-93
19. Alonso R, Pisa D, Marina AI, Morato E, Rabano A, Carrasco L. Fungal infection in patients with Alzheimer's disease. J Alzheimers Dis. 2014;41:301-11
20. Alonso R, Pisa D, Rabano A, Carrasco L. Alzheimer's disease and disseminated mycoses. Eur J Clin Microbiol Infect Dis. 2014;33:1125-32
21. Pisa D, Alonso R, Juarranz A, Rabano A, Carrasco L. Direct Visualization of Fungal Infection in Brains from Patients with Alzheimer's Disease. J Alzheimers Dis. 2014
22. Castellani RJ, Siedlak SL, Fortino AE, Perry G, Ghetti B, Smith MA. Chitin-like polysaccharides in Alzheimer's disease brains. Current Alzheimer research. 2005;2:419-23
23. Sotgiu S, Musumeci S, Marconi S, Gini B, Bonetti B. Different content of chitin-like polysaccharides in multiple sclerosis and Alzheimer's disease brains. Journal of neuroimmunology. 2008;197:70-3
24. Watabe-Rudolph M, Song Z, Lausser L, Schnack C, Begus-Nahrmann Y, Scheithauer MO. et al. Chitinase enzyme activity in CSF is a powerful biomarker of Alzheimer disease. Neurology. 2012;78:569-77
25. Cermelli C, Vinceti M, Beretti F, Pietrini V, Nacci G, Pietrosemoli P. et al. Risk of sporadic amyotrophic lateral sclerosis associated with seropositivity for herpesviruses and echovirus-7. European journal of epidemiology. 2003;18:123-7
26. Pagliardini V, Pagliardini S, Corrado L, Lucenti A, Panigati L, Bersano E. et al. Chitotriosidase and lysosomal enzymes as potential biomarkers of disease progression in amyotrophic lateral sclerosis: A survey clinic-based study. J Neurol Sci. 2015;348:245-50
27. Varghese AM, Sharma A, Mishra P, Vijayalakshmi K, Harsha HC, Sathyaprabha TN. et al. Chitotriosidase - a putative biomarker for sporadic amyotrophic lateral sclerosis. Clinical proteomics. 2013;10:19
28. Pisa D, Alonso R, Carrasco L. Fungal infection in a patient with multiple sclerosis. Eur J Clin Microbiol Infect Dis. 2011;30:1173-80
29. Pisa D, Ramos M, Garcia P, Escoto R, Barraquer R, Molina S. et al. Fungal infection in patients with serpiginous choroiditis or acute zonal occult outer retinopathy. Journal of clinical microbiology. 2008;46:130-5
30. White TJ, Bruns T, Lee S, Taylor J. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. PCR Protocols: a Guide to Methods and Applications, Orlando Florida. 1990:315-322
31. Pisa D, Alonso R, Jimenez-Jimenez FJ, Carrasco L. Fungal infection in cerebrospinal fluid from some patients with multiple sclerosis. Eur J Clin Microbiol Infect Dis. 2013;32:795-801
32. Chen SC, Halliday CL, Meyer W. A review of nucleic acid-based diagnostic tests for systemic mycoses with an emphasis on polymerase chain reaction-based assays. Medical mycology. 2002;40:333-57
33. Nguyen MH, Wissel MC, Shields RK, Salomoni MA, Hao B, Press EG. et al. Performance of Candida real-time polymerase chain reaction, beta-D-glucan assay, and blood cultures in the diagnosis of invasive candidiasis. Clin Infect Dis. 2012;54:1240-8
34. Lau A, Chen S, Sorrell T, Carter D, Malik R, Martin P. et al. Development and clinical application of a panfungal PCR assay to detect and identify fungal DNA in tissue specimens. Journal of clinical microbiology. 2007;45:380-5
35. Pacheco M, Pisa D, Garcia-Gomez P, Carrasco L, Juarranz A. Attachment and entry of Candida famata in monocytes and epithelial cells. Microscopy research and technique. 2007;70:975-86
36. Rai MN, Borah S, Bairwa G, Balusu S, Gorityala N, Kaur R. Establishment of an in vitro system to study intracellular behavior of Candida glabrata in human THP-1 macrophages. J Vis Exp. 2013:e50625
37. D'Angelo MA, Raices M, Panowski SH, Hetzer MW. Age-dependent deterioration of nuclear pore complexes causes a loss of nuclear integrity in postmitotic cells. Cell. 2009;136:284-95
38. Woulfe JM. Abnormalities of the nucleus and nuclear inclusions in neurodegenerative disease: a work in progress. Neuropathology and applied neurobiology. 2007;33:2-42
39. Brettschneider J, Lehmensiek V, Mogel H, Pfeifle M, Dorst J, Hendrich C. et al. Proteome analysis reveals candidate markers of disease progression in amyotrophic lateral sclerosis (ALS). Neuroscience letters. 2010;468:23-7
40. Pal R, Alves G, Larsen JP, Moller SG. New insight into neurodegeneration: the role of proteomics. Molecular neurobiology. 2014;49:1181-99
41. Pasinetti GM, Ungar LH, Lange DJ, Yemul S, Deng H, Yuan X. et al. Identification of potential CSF biomarkers in ALS. Neurology. 2006;66:1218-22
42. Teunissen CE, Koel-Simmelink MJ, Pham TV, Knol JC, Khalil M, Trentini A. et al. Identification of biomarkers for diagnosis and progression of MS by MALDI-TOF mass spectrometry. Multiple sclerosis (Houndmills, Basingstoke, England). 2011;17:838-50
43. Han X, He L, Xin L, Shan B, Ma B. PeaksPTM: Mass spectrometry-based identification of peptides with unspecified modifications. Journal of proteome research. 2011;10:2930-6
44. De Chiara G, Marcocci ME, Sgarbanti R, Civitelli L, Ripoli C, Piacentini R. et al. Infectious agents and neurodegeneration. Molecular neurobiology. 2012;46:614-38
45. Holmes C, Cotterell D. Role of infection in the pathogenesis of Alzheimer's disease: implications for treatment. CNS drugs. 2009;23:993-1002
46. Reis HJ, Mukhamedyarov MA, Rizvanov AA, Palotas A. A new story about an old guy: is Alzheimer's disease infectious? Neuro-degenerative diseases. 2010;7:272-8
47. Tselis A. Evidence for viral etiology of multiple sclerosis. Seminars in neurology. 2011;31:307-16
48. Starakis I, Panos G, Koutras A, Mazokopakis EE. Pathogens and chronic or long-term neurologic disorders. Cardiovascular & hematological disorders drug targets. 2011;11:40-52
49. Benito-Leon J, Pisa D, Alonso R, Calleja P, Diaz-Sanchez M, Carrasco L. Association between multiple sclerosis and Candida species: evidence from a case-control study. Eur J Clin Microbiol Infect Dis. 2010;29:1139-45
50. Amor S, Puentes F, Baker D, van der Valk P. Inflammation in neurodegenerative diseases. Immunology. 2010;129:154-69
51. Behari M, Shrivastava M. Role of platelets in neurodegenerative diseases: a universal pathophysiology. The International journal of neuroscience. 2013;123:287-99
52. He F, Balling R. The role of regulatory T cells in neurodegenerative diseases. Wiley interdisciplinary reviews. 2013;5:153-80
53. Dhib-Jalbut S, Arnold DL, Cleveland DW, Fisher M, Friedlander RM, Mouradian MM. et al. Neurodegeneration and neuroprotection in multiple sclerosis and other neurodegenerative diseases. Journal of neuroimmunology. 2006;176:198-215
54. Hoglund K, Salter H. Molecular biomarkers of neurodegeneration. Expert review of molecular diagnostics. 2013;13:845-61
55. Ramanan VK, Saykin AJ. Pathways to neurodegeneration: mechanistic insights from GWAS in Alzheimer's disease, Parkinson's disease, and related disorders. American journal of neurodegenerative disease. 2013;2:145-75
56. Revett TJ, Baker GB, Jhamandas J, Kar S. Glutamate system, amyloid ss peptides and tau protein: functional interrelationships and relevance to Alzheimer disease pathology. J Psychiatry Neurosci. 2013;38:6-23
57. Cunha C, Aversa F, Bistoni G, Casagrande A, Rodrigues F, Romani L. et al. Immunogenetic profiling to predict risk of invasive fungal diseases: where are we now? Immunological investigations. 2011;40:723-34
58. Plantinga TS, Johnson MD, Scott WK, Joosten LA, van der Meer JW, Perfect JR. et al. Human genetic susceptibility to Candida infections. Medical mycology. 2012;50:785-94
59. Smeekens SP, van de Veerdonk FL, Kullberg BJ, Netea MG. Genetic susceptibility to Candida infections. EMBO molecular medicine. 2013;5:805-13
60. Ding C, Festa RA, Sun TS, Wang ZY. Iron and copper as virulence modulators in human fungal pathogens. Molecular microbiology. 2014;93:10-23
61. Leal SM Jr, Vareechon C, Cowden S, Cobb BA, Latge JP, Momany M. et al. Fungal antioxidant pathways promote survival against neutrophils during infection. The Journal of clinical investigation. 2012;122:2482-98
62. Bendotti C, Marino M, Cheroni C, Fontana E, Crippa V, Poletti A. et al. Dysfunction of constitutive and inducible ubiquitin-proteasome system in amyotrophic lateral sclerosis: implication for protein aggregation and immune response. Progress in neurobiology. 2012;97:101-26
63. Moisse K, Strong MJ. Innate immunity in amyotrophic lateral sclerosis. Biochimica et biophysica acta. 2006;1762:1083-93
64. Engelhardt JI, Tajti J, Appel SH. Lymphocytic infiltrates in the spinal cord in amyotrophic lateral sclerosis. Archives of neurology. 1993;50:30-6
65. Grammas P. Neurovascular dysfunction, inflammation and endothelial activation: implications for the pathogenesis of Alzheimer's disease. Journal of neuroinflammation. 2011;8:26
66. Graves MC, Fiala M, Dinglasan LA, Liu NQ, Sayre J, Chiappelli F. et al. Inflammation in amyotrophic lateral sclerosis spinal cord and brain is mediated by activated macrophages, mast cells and T cells. Amyotroph Lateral Scler Other Motor Neuron Disord. 2004;5:213-9
67. Kawamata T, Akiyama H, Yamada T, McGeer PL. Immunologic reactions in amyotrophic lateral sclerosis brain and spinal cord tissue. The American journal of pathology. 1992;140:691-707
68. Mantovani S, Garbelli S, Pasini A, Alimonti D, Perotti C, Melazzini M. et al. Immune system alterations in sporadic amyotrophic lateral sclerosis patients suggest an ongoing neuroinflammatory process. Journal of neuroimmunology. 2009;210:73-9
69. Larbi A, Pawelec G, Witkowski JM, Schipper HM, Derhovanessian E, Goldeck D. et al. Dramatic shifts in circulating CD4 but not CD8 T cell subsets in mild Alzheimer's disease. J Alzheimers Dis. 2009;17:91-103
70. Rentzos M, Evangelopoulos E, Sereti E, Zouvelou V, Marmara S, Alexakis T. et al. Alterations of T cell subsets in ALS: a systemic immune activation? Acta neurologica Scandinavica. 2011;125:260-4
71. Saresella M, Calabrese E, Marventano I, Piancone F, Gatti A, Calvo MG. et al. PD1 negative and PD1 positive CD4+ T regulatory cells in mild cognitive impairment and Alzheimer's disease. J Alzheimers Dis. 2010;21:927-38
72. Henkel JS, Beers DR, Wen S, Rivera AL, Toennis KM, Appel JE. et al. Regulatory T-lymphocytes mediate amyotrophic lateral sclerosis progression and survival. EMBO molecular medicine. 2013;5:64-79
73. Fung A, Vizcaychipi M, Lloyd D, Wan Y, Ma D. Central nervous system inflammation in disease related conditions: mechanistic prospects. Brain research. 2012;1446:144-55
74. Romani L. Immunity to fungal infections. Nature reviews. 2004;4:1-23
Author contact
![]() Corresponding author: lcarrascocsic.es, Telephone number: +34 1 497 84 50
Corresponding author: lcarrascocsic.es, Telephone number: +34 1 497 84 50
Citation styles
APA
Alonso, R., Pisa, D., Marina, A.I., Morato, E., Rábano, A., Rodal, I., Carrasco, L. (2015). Evidence for Fungal Infection in Cerebrospinal Fluid and Brain Tissue from Patients with Amyotrophic Lateral Sclerosis. International Journal of Biological Sciences, 11(5), 546-558. https://doi.org/10.7150/ijbs.11084.
ACS
Alonso, R.; Pisa, D.; Marina, A.I.; Morato, E.; Rábano, A.; Rodal, I.; Carrasco, L. Evidence for Fungal Infection in Cerebrospinal Fluid and Brain Tissue from Patients with Amyotrophic Lateral Sclerosis. Int. J. Biol. Sci. 2015, 11 (5), 546-558. DOI: 10.7150/ijbs.11084.
NLM
Alonso R, Pisa D, Marina AI, Morato E, Rábano A, Rodal I, Carrasco L. Evidence for Fungal Infection in Cerebrospinal Fluid and Brain Tissue from Patients with Amyotrophic Lateral Sclerosis. Int J Biol Sci 2015; 11(5):546-558. doi:10.7150/ijbs.11084. https://www.ijbs.com/v11p0546.htm
CSE
Alonso R, Pisa D, Marina AI, Morato E, Rábano A, Rodal I, Carrasco L. 2015. Evidence for Fungal Infection in Cerebrospinal Fluid and Brain Tissue from Patients with Amyotrophic Lateral Sclerosis. Int J Biol Sci. 11(5):546-558.
This is an open access article distributed under the terms of the Creative Commons Attribution (CC BY-NC) License. See http://ivyspring.com/terms for full terms and conditions.