Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Multiple Forms and Genomic...
Mediators of Cytosine DNA...
DNA Methylation and Cancer...
Final Remark
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(5):604-617. doi:10.7150/ijbs.11218 This issue Cite
Review
DNA Methylation, Its Mediators and Genome Integrity
Huan Meng1,2 ![]() , Ying Cao2, Jinzhong Qin2, Xiaoyu Song1, Qing Zhang2, Yun Shi2, Liu Cao1
, Ying Cao2, Jinzhong Qin2, Xiaoyu Song1, Qing Zhang2, Yun Shi2, Liu Cao1 ![]()
1. Key Laboratory of Medical Cell Biology, Ministry of Education, China Medical University, Shenyang 110001, China;
2. MOE Key Laboratory of Model Animal for Disease Study, Model Animal Research Center, Nanjing Biomedical Research Institute, Nanjing University, China.
Received 2014-12-2; Accepted 2015-3-2; Published 2015-4-8
Abstract

DNA methylation regulates many cellular processes, including embryonic development, transcription, chromatin structure, X-chromosome inactivation, genomic imprinting and chromosome stability. DNA methyltransferases establish and maintain the presence of 5-methylcytosine (5mC), and ten-eleven translocation cytosine dioxygenases (TETs) oxidise 5mC to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC), which can be removed by base excision repair (BER) proteins. Multiple forms of DNA methylation are recognised by methyl-CpG binding proteins (MeCPs), which play vital roles in chromatin-based transcriptional regulation, DNA repair and replication. Accordingly, defects in DNA methylation and its mediators may cause silencing of tumour suppressor genes and misregulation of multiple cell cycles, DNA repair and chromosome stability genes, and hence contribute to genome instability in various human diseases, including cancer. Thus, understanding functional genetic mutations and aberrant expression of these DNA methylation mediators is critical to deciphering the crosstalk between concurrent genetic and epigenetic alterations in specific cancer types and to the development of new therapeutic strategies.
Keywords: DNA methylation, DNA methyltransferases, methyl-CpG binding proteins, DNA glycosylases, BRCA1, genome instability.
Introduction
The presence of 5-methylcytosine (5mC) was first demonstrated in tubercle bacillus DNA in 1925 [1] and in calf thymus DNA two decades later [2]. Although biological functions of this cytosine modification remained uncharacterised for decades, in 1975, two studies demonstrated important roles of 5mC as an epigenetic modification that influences gene expression [3, 4] and highlighted the significance of the 'fifth nucleotide' in eukaryotic biology [5]. DNA methylation is now widely recognised as a typical epigenetic mark because it satisfies the stringent criterion of an epigenetic system that is mitotically and meiotically heritable as redefined from Waddington [6] by Riggs in 1996 [7]. In prokaryotes, methylation at both adenine (A) and cytosine (C) residues contributes to host restriction systems and protects the cell from foreign genetic materials such as bacterial and viral genomes [8]. In contrast, DNA methylation in multicellular eukaryotes occurs predominantly but not exclusively at cytosine residues within CpG dinucleotides [9]. In vertebrates, DNA methylation is a major form of epigenetic modification and is regulated during development to control tissue and differentiation states [10]. Moreover, DNA methylation patterns are altered in cancers and in embryos produced by somatic cell nuclear transfer [11, 12]. These changes contribute significantly to the molecular pathology of these disease states [12-14].
In this review, we introduce various forms of DNA methylation in terms of distributions and transcriptional mechanisms in mammals. We summarise recent advances in the understanding of mediators of DNA methylation and demethylation, including DNA methyltransferases (DNMTs), methyl-CpG binding proteins (MeCPs), ten-eleven translocation cytosine dioxygenases (TETs) and base excision repair (BER) DNA glycosylases. Aberrant transcription of cell cycle, DNA repair and chromosome stability genes are associated with promoter hypermethylation of corresponding transcriptional start sites (TSSs) with CpG islands (CGIs) in human cancers and were recently linked with germ-line and somatic mutations in their gene bodies without CGIs. Moreover, a major role of DNA methylation in modelling chromatin structure, which generally regulates gene expression states and thus the accessibility of DNA for damage, is discussed. We emphasise the current understanding of genetic and epigenomic alterations involving DNA methylation mediators in human cancers and discuss their potential influence on carcinogenesis by providing selective growth advantages for tumour transformation and aggression.
Multiple Forms and Genomic Distribution of Cytosine DNA Methylation
Multiple forms of DNA methylation have been identified in mammals, including 5mC, the recently discovered 5-hydroxymethylcytosine (5hmC) and the ensuing oxidation products 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) [15-17]. The major epigenetic modification 5mC and its hydroxylated derivative 5hmC are relatively stable and abundant in mammalian genomes [18] (Fig. 1A). In contrast, 5fC and 5caC are extremely rare and can be transiently removed by thymine DNA glycosylase (TDG) and are therefore speculated as active DNA demethylation intermediates [17, 19, 20]. DNMTs produce 5mC by covalently adding a methyl group to the 5-position of the cytosine ring, predominantly occurring at CpG dinucleotides in somatic cells [21, 22]. Mounting associations of 5mC with gene silencing indicate important roles in normal mammalian genomic imprinting, X-chromosome inactivation, repetitive element suppression and lineage-specific gene expression regulation [13, 23, 24]. However, non-CpG methylation also occurs with high frequency in mouse and human embryonic stem (ES) cells and induced pluripotent stem cells [21, 22]. The 5hmC intermediate was recently discovered as a second modification in vertebrate DNA and is formed by addition of a hydroxy group to 5mC by TETs [16, 25] (Fig. 1A). These enzymes are enriched in Purkinje neurons and ES cells [15, 16], and because 5hmC is more stable than its oxidation products 5fC and 5caC, the hydroxymethyl group is likely to have biological properties and may be an epigenetic mark [26, 27]. Because 5mC and 5hmC are only distinguishable in experiments using 5hmC specific antibodies [28, 29], recent developments have been aimed at resolving 5hmC sites in the genome [30, 31]. Nonetheless, it is accepted that 5mC is the most prominent modification in vertebrate DNA in the majority of mammalian tissues [32].
Major forms and distribution of DNA methylation. (A) The three major forms of cytosine bases in mammalian DNA. The 5-position of cytosine is covalently methylated by DNA cytosine methyltransferases (DNMTs) with the presence of co-factor S-adenosyl methionine (SAM). The resulting 5-methylcytosine (5mC) is mostly found on CpG dinucleotides in somatic cells. 5-hydroxymethylcytosine (5hmC) is formed by methylation and subsequent hydroxylation and is mediated by the ten-eleven translocation cytosine dioxygenases (TETs). (B) Distribution of CpG dinucleotides in mammalian genomes. In vertebrate genomes, CpG dinucleotides are generally highly methylated, whereas CpG islands (CGIs) that are associated with gene promoters have exceptional global unmethylated patterns. Exceptions include CGIs on inactive X-chromosomes in female cells, where CGIs are hypermethylated. In addition to canonical CGIs located at annotated transcription start sites (TSSs), orphan CGIs of unknown function are found within gene bodies (intragenic) and between annotated genes (intergenic). Unmethylated CGIs at 5' ends of multiple genes are positively correlated with transcriptional activity (active, left), whereas a small number of genes are hypermethylated at their promoter CGIs and are repressed in specific cell types (inactive, right). Gene bodies are often methylated with higher DNA methylation at exons than introns, and 5hmC is present at expressed gene bodies and are the proposed 5mC oxidation products of TET enzymes (labelled white squares at body of gene). White circles, nonmethylated CpGs; black circles, methylated CpGs; white squares, hydroxylmethylated CpGs; red boxes, active and transcribed exons; black boxes, inactive and silenced exons; transcriptional states of these genes are represented by the red arrow (active) and the black cross (inactive).
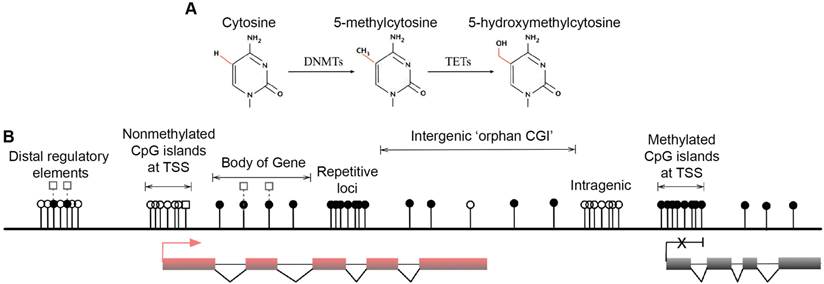
Genome-wide studies demonstrated direct regulation of a number of developmental genes by DNA methylation [33, 34]. Most (70%-80%) mammalian CpG sites are methylated, and highly methylated sequences are found in satellite DNAs, repetitive elements, gene bodies and non-repetitive intergenic DNA [35]. However, CpG islands (CGIs), the CpG-rich regions that are located in more than half of the promoters of mammalian genes (approximately 60% in human genes) have exceptional global unmethylated patterns [36-38]. Unmethylated CGIs in the vicinity of promoter regions are normally associated with tissue-specific expression of corresponding genes in early embryos and ensuing somatic cells. However, these CGIs become largely de novo methylated during X-chromosome inactivation, resulting in legitimate gene silencing on the inactivated chromosome, which is required for dosage compensation [39]. Moreover, although only 100-200 annotated CGIs were thought to be present in somatic cells [33], a recent study identified approximately 23,000 and 25,500 non-annotated 'orphan' CGIs in mouse and human genomes, respectively [14] (Fig. 1B. Top). A large number of these under-represented CGIs were found in the proximity of annotated TSSs of constitutively expressed genes and at intergenic regions and gene bodies [14]. These orphan CGIs may act as promoters but are often methylated during development [14], suggesting limited transcriptional activity. Associations of DNA methylation at these regulatory sequences and transcriptional repression in somatic cells are well established, and the potential long-term impact on the stability of gene expression profiles is widely accepted [40]. In general, DNA methylation of GC-rich gene promoter regions is associated with gene repression, whereas transcribed genes usually correlate with low DNA methylation levels around TSSs and high levels in the gene body [21, 41, 42] (Fig. 1B. Lower left and right). In pathophysiological states, both global DNA hypomethylation at repetitive and satellite regions of the genome and site-specific hypermethylation of CGIs at promoters of tumour suppressor genes are associated with whole genome instability, a hallmark of human cancers [43-47].
Mediators of Cytosine DNA Methylation
DNA Methyltransferases, the Canonical 'Writers'
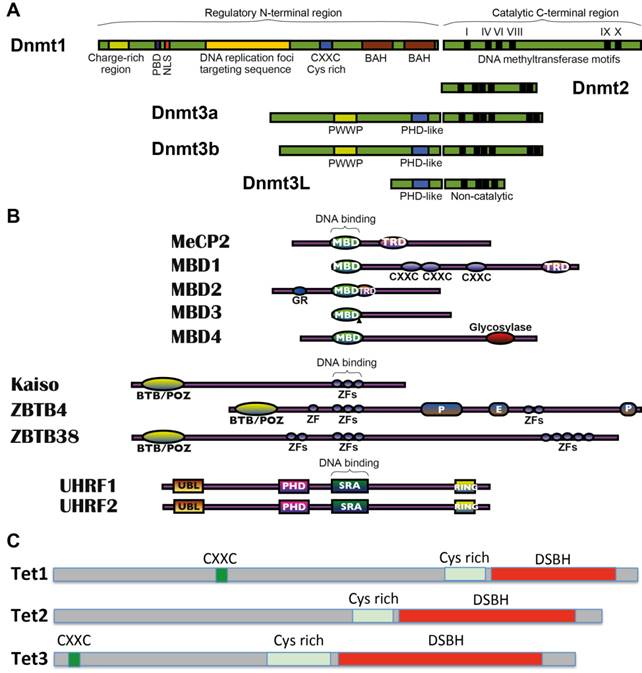
The methyltransferase enzymes DNMT1, DNMT3A and DNMT3B harmonise in the establishment and maintenance of DNA methylation patterns in mammals (Fig. 2A and Table 1). DNMT3A and DNMT3B are de novo methyltransferases that target cytosines of previously unmethylated CpG dinucleotides. These enzymes have an equal preference for hemimethylated and unmethylated DNA, which are essential for their roles in de novo methylation of the genome during development and for newly integrated retroviral sequences [48, 49]. Following the first wave of genome-wide demethylation in the preimplantation embryo, Dnmt3a and Dnmt3b are highly expressed at implantation and re-establish a bimodal methylation pattern that effects more than 80% of the genome [48], whereas most CGIs are protected by unknown mechanisms and therefore remain unmodified [41]. Genetic and functional analyses indicate that Dnmt3a and Dnmt3b have non-overlapping functions during development with different phenotypes and lethality stages [48], suggesting that each enzyme has regional specificity that reflects their respective N-terminal domains. Accordingly, Dnmt3a is necessary for maternal imprinting at differentially methylated regions, and Dnmt3b is required for methylation of pericentromeric repeats and CGIs on inactive X-chromosomes [50]. Established DNA methylation patterns are stably preserved over cell divisions by DNA methyltransferase-1 (DNMT1), which is known as a maintenance enzyme that guards existing methylated sites through its preference for hemimethylated DNA [51]. Dnmt1 is particularly present at high concentrations in dividing cells [51], localising perpetually to replication foci [52]. Dnmt1 operates with its methylation co-factor UHRF1 (Np95) in protein complexes that constitute an enzymatic platform, providing a maintenance methyltransferase function for CpG methylation [53-55]. In addition to its methyltransferase activity, DNMT1 has a proliferating cell nuclear antigen-interacting domain, replication-targeting region, cysteine-rich Zn2+-binding domain, nuclear localisation signal and polybromo-1 like protein domain [56, 57]. It also contains an N-terminal region that is associated with various chromatin-associated proteins, including de novo methyltransferases, histone modifying enzymes and MeCPs. Among DNMTs, DNMT2 shows weak methyltransferase activity in vitro, and its depletion has little impact on global CpG methylation levels and no discernible effects on developmental phenotypes [51]. Moreover, although DNMT3L (DNMT3-like) is catalytically inactive, it is highly expressed in germ and ES cells and acts as an obligatory cofactor for de novo methyltransferase in ES cells [58]. Dnmt3L stimulates the methyltransferase activity of Dnmt3a or Dnmt3b through physical interaction [59-62]. Crystallographic analyses of Dnmt3a and Dnmt3L indicate that these interactions may be mediated by a heterotetrameric complex formation [63], which may prevent Dnmt3a oligomerisation and heterochromatic localisation [64]. A recent study showed that DNMT3L is a positive regulator of DNA methylation at gene bodies of housekeeping genes and a negative regulator of DNA methylation at promoters of bivalent genes in mouse ES cells, suggesting a dual role in ES cell differentiation [65].
Mediators of DNA methylation machinery. (A) Domain structures of mammalian DNA methyltransferases (DNMTs). Functional domains in the N-terminal regions of DNMTs are shown and the conserved motifs in the C-terminal region are labelled. In the N-terminal region, the sub-domains include a proliferating cell nuclear antigen binding site (PBD), nuclear localisation signal region (NLS), plant homeo domain (PHD) like domain and PWWP domain (highly conserved proline-tryptophan-tryptophan-proline motif that is involved in protein-protein interactions) and bromo-adjacent homology domains (BAH). N- and C-terminal domains are linked by Gly-Lys dipeptides. Highly conserved C-terminal methyltransferase motifs are shown as thick black lines (indicated as I-X). (B) Domain structures of methyl-CpG binding proteins (MeCPs). Three families of characterised mammalian MeCPs include (1) the methyl-CpG binding domain proteins (MBDs) MBD1, MBD2, MBD3, MBD4 and MeCP2. (2) the structurally unrelated methyl-CpG binding zinc-finger proteins of the Kaiso family KAISO/ZBTB33, ZBTB4 and ZBTB38 and (3) the methyl-CpG binding SRA domain proteins of the UHRF family UHRF1 and its homologue UHRF2. Labelled sub-domains include MBD, methyl-CpG binding domain; TRD, trans-repressor domain; GR, E, P, amino acid repeats; BTB/POZ, broad complex, tramtrack, and bric à brac domains; ZF, zinc finger motifs; UBL, ubiquitin-like motif; PHD, Plant homeodomain and SRA, SET and Ring-associated domain. DNA binding regions are indicated. (C) Domain structures of ten-eleven translocation methylcytosine dioxygenases (TETs). Schematic representation of conserved domains of mouse Tet proteins is shown, including a double-stranded-helix (DSBH) fold (all Tets), cysteine-rich (Cys-rich) domain (all Tets) and CXXC zinc fingers (Tet1 and Tet3).
Functions and specificities of human DNA methyltransferases and methyl-CpG binding proteins
| Gene symbol | Gene name | Function | DNA Specificity | Human Tumours [67, 68] | Mouse knock-out phenotype (MGI database) |
|---|---|---|---|---|---|
| Human DNA methyltransferases | |||||
| DNMT1 | DNA methyltransferase 1 | Maintenance DNA methyltransferase | Hemimethylated DNA | Gene body Mutations in colorectal cancers [69]. Overexpression in leukaemia [70], gliomas [71], non-small cell lung [72], pancreatic [73], gastric [74], hepatocellular [75] and breast cancers [76]. | Stunted and delayed development and embryonic lethal by E9.5 [77]. Lack of appropriate genomic imprinting [78]. |
| DNMT2 | DNA methyltransferase 2 | low DNA methyltransferase activity; RNA (tRNA) methyltransferase activity | Cytosine 38 of transfer RNAAsp | Reduced expression in hepatocellular [79], colorectal and stomach cancers [80]. | No phenotype observed [81]. Direct data from MGI reported a decreased proportion of natural killer cells in the peripheral blood. |
| DNMT3A | DNA methyltransferase 3A | De novo DNA methyltransferase | Equal preference for unmethylated and hemimethylated DNA | Mutations in AML [82, 83]. Overexpression in gastric [84], hepatocellular [75], pancreatic [85] and colon cancers [86]. | Normal development at birth but become runted and die around four weeks of age [48]. |
| DNMT3B | DNA methyltransferase 3B | De novo DNA methyltransferase | Equal preference for unmethylated and hemimethylated DNA | Mutations in ICF syndrome [87], SNP in lung cancer [88]. Overexpression in leukaemia [70], glioblastoma [71], gastric [84], hepatocellular [75], colon [86, 89], prostate [90] and breast cancers [91] | Embryonic lethal before E9.5 with growth retardation and rostral neural tube defects. Slight under-methylation of endogenous viral DNA, substantial demethylation of minor satellite DNA [48]. |
| DNMT3L | DNA methyltransferase 3L | Cofactor; required for de novo methyltransferase activity in ES cells | / | Potential biomarker for cervical cancer [92] and embryonal carcinoma [93]. | Lack appropriate methylation of the maternal allele and cause azoospermia in homozygous males [94]; heterozygous progeny of homozygous females die by midgestation [95]. |
| Human methyl-CpG binding proteins | |||||
| MeCP2 | Methyl CpG binding protein 2 | Methyl-CpG binding; transcriptional repression | Symmetric 5meCpG; A/T-rich sequence adjacent to 5meCpG for efficient DNA binding shown by in vitro assays | / | Female mice homozygous or male mice hemizygous for a null allele and heterozygous mice exhibit neural, Rett syndrome-like symptoms [96, 97]. |
| MBD1 | Methyl-CpG binding domain protein 1 | Methyl-CpG binding; transcriptional repression | 5meCpG within TCGCA and TGCGCA sequence context | Mutations in lung and breast cancers [67]. | Defects in adult hippocampal neurogenesis and function, impaired spatial learning, reduced neuronal differentiation and increased genomic instability [98]. |
| MBD2 | Methyl-CpG binding domain protein 2 | Methyl-CpG binding; transcriptional repression and activation | 5meCpG in a single orientation | Mutations in lung and breast cancers [67]. | Viable and fertile; defective maternal nurturing behaviour, decreased tumourigenesis [99]. |
| MBD3 | Methyl-CpG binding domain protein 3 | Component of Mi-2/NuRD complex; transcriptional repression | / | Decreased expression in gastric carcinogenesis [100]. | Embryonic lethal because of failure in differentiation of pluripotent cells [99, 101]. |
| MBD4 | Methyl-CpG binding domain protein 4 | Methyl-CpG binding; BER DNA glycosylase; apoptosis; transcriptional repression | Symmetric 5meCpG | Mutations in colon, endometrial and pancreatic cancers [102, 103]. | Viable and fertile. Increased rate of C to T mutation at CpG dinucleotides [104, 105]. |
| KAISO/ ZBTB33 | KAISO/ zinc finger and BTB domain containing 33 | Methyl-CpG binding; transcriptional repression; Wnt signalling suppression | two 5meCpG motifs in close proximity preferably in tandem | Indicator of aggressive prostate cancers, associate with high grade and triple-negative invasive breast cancer, poor prognosis in non-small-cell lung cancer. | viable, fertile and overtly normal with no detectable changes; reduced tumourigenesis [106]. |
| ZBTB4 | Zinc finger and BTB domain containing 4 | Methyl-CpG binding; transcriptional repression | Unmethylated consensus sequence CC/TGCCATC; strong binding specificity to single methylated CpG in a surrounding nucleotide-specific manner | Possible prognostic marker and potential therapeutic target for breast cancer survival [107]. | / |
| ZBTB38 | Zinc finger and BTB domain containing 38 | Methyl-CpG binding; transcriptional repression | Bind to a single methylated CpG | / | / |
| UHRF1 | Ubiquitin-like with PHD and ring finger domains 1 | Cofactor for the DNA methylation maintenance; transcriptional regulation; E3 ubiquitin ligase activity for histone H3 | hemimethylated DNA | Overexpression in a variety of human cancers including those of the breast, liver, lung, bladder, which often correlate with a poor outcome [108-110]. | Embryonic lethal in gestation showing growth retardation and various malformations because of essential defects of global and local DNA methylation [111]. |
| UHRF2 | Ubiquitin-like with PHD and ring finger domains 2 | Ubiquitin E3 ligase; SUMO E3 ligase; specific recogniser of 5hmC | hemimethylated DNA | Possible predictor of survival and potential therapeutic target in colon cancer [112]. | / |
Methyl-CpG Binding Proteins, the Invited 'Interpreters'
The 'written' methylation marks at CpG dinucleotides can be specifically recognised by various MeCPs, which may in principle 'read' the established methylated DNA sequences and recruit histone-modifying complexes to regulate higher order chromatin structure, stabilise patterns of gene expression and maintain genome integrity [66]. Among the three major characterised families of mammalian MeCPs (Fig. 2B and Table 1), methyl-CpG binding domain (MBD) proteins, including MeCP2, MBD1, MBD2 and MBD4, but not MBD3, specifically recognise 5-methyl-CpG (5meCpG) dinucleotides via novel MBD domain. The second KAISO family comprises three structurally unrelated zinc-finger proteins KAISO/ZBTB33, ZBTB4 and ZBTB38, and KAISO proteins have been shown to bind to methylated DNA through zinc-finger motifs. The third family includes two 5meCpG-binding ubiquitin-like proteins UHRF1 and UHRF2, which recognise methylated DNA via RING finger-associated (SRA) domains.
Although all MeCPs share binding specificity for symmetrical 5meCpG dinucleotides, some have additional binding preferences. For example, the MBD domain of MBD1 recognises 5meCpG dinucleotides more efficiently within TCGCA and TGCGCA sequences [113], whereas MBD2 in chickens (96% homology to human MBD2) binds to 5meCpG dinucleotides in a single orientation, suggesting that additional sequences outside of the 5meCpG dinucleotides are necessary for MBD2 specificity [114]. In addition, efficient DNA binding of MeCP2 requires an A/T-rich sequence adjacent to 5meCpG dinucleotides, and in vitro assays showed the involvement of few amino acids upstream of the MBD domain of MeCP2 [115]. In the second MeCP family, the zinc finger domain of KAISO proteins was shown to have a binding preference for two 5meCpG motifs in close proximity, preferably in tandem [116-118]. The third UHRF family recognises 5meCpG using a distinct base-flipping mechanism [119, 120], which was demonstrated for the SRA domain of UHRF1 that recognises and binds hemi-methylated DNA and acts in conjunction with DNMT1 to maintain DNA methylation [120, 121]. UHRF2 has an SRA domain with 75% homology to UHRF1 and was shown to have a similar binding preference to hemimethylated DNA in vitro [122]. Moreover, a recent study showed the presence of a weak but specific affinity for hemi- and fully-hydroxymethylated DNA [123].
DNA methylation per se alters transcriptional binding sites to prevent transcriptional activation and the binding of transcriptional factors such as E2F or CREB [124, 125]. In addition, MeCPs can act as 'interpreters' that specifically recognise 5meCpG marks and subsequently recruit various chromatin modifiers to establish a repressive chromatin environment [126-129]. Almost all MeCPs have been demonstrated to associate with transcriptional repressors, implying an additional layer of regulation between DNA methylation and transcription. As an example, MeCP2 associates with co-repressor complexes such as Sin3A and NCoR via its trans-repressor domain (TRD) and induces strong global transcriptional repression [127, 130]. Moreover, MBD3 is an essential subunit of the Mi-2/NuRD chromatin remodelling complex, which was shown to function as a transcriptional repressor complex both in vivo and in cell culture assays [66].
Cytosine Dioxygenase TETs and Its 5mC Product 5hmC
The TET enzyme family comprises three cytosine dioxygenases, including TET1 and its two dioxygenase paralogues TET2 and TET3 (Fig. 2C and Table 1). These proteins are Fe(II)/α-ketoglutarate (αKG)-dependent dioxygenases of the AlkB family, which share a double-stranded β-helix (DSBH) catalytic domain. The DSBH domain of two AlkB family J-binding proteins JBP1 and JBP2 oxidise the 5-methyl group on thymine (T) to 5-hydroxymethyluracil (5hmU) [131]. JBP protein homology and similarities of 5mC and T at the 5-position of the cytosine ring suggested that TET proteins convert 5mC to 5hmC [25]. Accordingly, TET1 was shown to generate 5hmC from 5mC by oxidation of the methyl group in a Fe(II)/αKG-dependent manner [16], and all TET proteins were subsequently shown to catalyse stepwise conversions of 5mC to 5hmC, 5fC and 5caC in vitro and in vivo [19, 132, 133]. C-terminal catalytic domains of TET proteins contain an indispensable cysteine-rich region adjacent to their DSBH domain [134]. In addition, TET1 and TET3 carry a cysteine-X-X-cysteine (CXXC) domain at the N-terminus that strongly binds to unmethylated DNA [135].
Among the three 5mC oxidation forms, 5hmC is the most stable, but its presence significantly varies among tissues [136]. Recent studies suggested that 5hmC is predominantly enriched in the vicinity of transcription factor binding sites, including distal-regulatory elements and gene bodies of highly expressed genes, and is less abundant at gene promoter regions [30, 137] (Fig. 1B). In principle, 5hmC distribution at these gene regulatory loci may be associated with stable regulation of gene expression across the whole genome. As noted above, 5mC modification, particularly at promoter regions, is associated with gene repression. Accordingly, substitution of 5mC residues with 5hmC may inhibit recruitment of the classic 5mC interpreter MeCPs and undermine subsequent transcriptional repression activities. In support of this idea, in vitro studies showed that MeCP2 has a markedly reduced binding affinity for 5hmC in contrast to its strong binding affinity for 5mC [138-140]. The less bound MeCP2 may release the associated histone modifying enzymes that produce overall histone deacetylation or specific lysine methylation [126-128], deregulating the repressive transcription environment. Another study demonstrated an equal binding affinity of MeCP2 for both 5mC and 5hmC [141], perhaps owing to the experimental differences in terms of selected DNA probes and/or truncated protein. Thus, further investigations are required to test the possibility that conversion of 5mC to 5hmC inhibits potentially the binding to MeCPs. The relative abundance of 5hmC in brain and ES cells suggested that it has additional DNA demethylation-independent functions that are 'read' via specific 5hmC interpreters [142]. Several 5hmC candidate 'readers' have recently been identified, including UHRF2, UHRF1 and MeCP2 [139, 141-143], although the last two have been shown to bind to 5mC-containing DNA with equal or greater affinity in vitro [139]. A recent crystallographic study with structural and biochemical analyses confirmed that UHRF2 specifically recognises 5hmC with approximately 1.5 and 3.2 times affinity in hemi- and fully-hydroxymethylated DNA, respectively, than in hemimethylated controls [123]. Moreover, the conformation of a phenylalanine within the SRA domain of UHRF2 was shown to optimise the preferential binding pocket for 5hmC [123]. This study also used electrophoretic mobility shift assays to examine the binding affinity of SRA domain and a longer version of UHRF2 to DNA probes containing 15 CpGs with unmodified or modified cytosines and showed that UHRF2 preferentially binds 5hmC over 5mC modified probes via its SRA domain. Because numerous proteins containing zinc-fingers or SRA domains are encoded in mammalian genomes, novel classes of 5hmC-binding proteins with distinct binding specificities may be identified in future studies [123, 142]. As for 5fC and 5caC, two recent studies used proteomics approaches to identify proteins that show a strong binding preference for 5hmC and further oxidation products [142, 144]. Consistent with the hypothesis that separate functions exist for these unmodified or modified cytosines as epigenetic marks, both studies reported a large number of proteins that can be specifically recruited by 5fC and 5caC probes in vitro, including transcription regulators and DNA glycosylases, supporting that 5hmC and further oxidation products 5fC and 5caC may function separately.
BER Glycosylases Act as Mediators of 5mC Enzymatic Removal
Dynamic changes of 5mC patterns in developmental and pathophysiological states suggested that active removal of 5mC occurs in an enzyme-dependent manner [145]. Current lines of investigation favour mechanisms that involve conversion of 5mC to the deamination products T and 5hmU [146, 147] and oxidation products 5fC and 5caC [19, 133, 147], which can be subsequently excised and repaired by DNA glycosylases following activation of BER pathways [145]. In principle, DNA glycosylases, such as TDG and MBD4, initiate BER repair by cleavage of the glycosidic bond between the 5mC base and deoxyribose [148]. This activity uses a base-flipping mechanism and generates abasic apurinic/apyrimidinic (AP) sites, which can be efficiently recognised and processed by AP endonucleases, DNA polymerases and DNA ligases. Subsequently, deoxyribose is removed and replaced by a nonmethylated cytosine, which restores the original DNA sequence. At least four bifunctional DNA glycosylases have been identified in plants, including ROS1, DEMETER (DME), and the DME-like proteins 2 and 3, which remove 5mC from both CpG and non-CpG regions [145]. In 1995, vertebrate DNA glycosylases were shown to actively reverse DNA methylation, and weak 5mC glycosylase activity was observed in chicken embryo nuclear extracts. A later study demonstrated the involvement of the chicken homologue of human TDG [145]. Whereas precise global and gene-specific mechanisms of DNA demethylation have not been demonstrated in mammals, recent evidence strongly suggested the mammalian DNA glycosylases TDG and MBD4 activate the BER repair pathway to remove intermediate residues that are generated in proposed pathways of oxidation, deamination or both [35, 145, 149]. Nonetheless, direct removal of 5mC by these mammalian DNA glycosylases is not exclusive, and further investigations of putative 5mC glycosylase co-factors and/or post-translational modifications are required to define these mechanisms [145]. The glycosylases SMUG1, UNG2, NEIL1 and NTHL1 have also been implicated in the processing of mismatched hmU:G and T and in the processing of intermediate substrates such as 5-carboxyU (Smug1) and 5-formyl-U (Nthl1) [148, 150] (Table 2).
Human DNA glycosylases and their known substrates and functions
| Gene symbol | Gene name | Expression | DNA Substrates | Mouse Knock-out phenotype |
|---|---|---|---|---|
| TDG | Thymine DNA glycosylase | Nucleus | T:G [151] 5hmU:G [152] dsDNA | Embryonic lethal between E10.5-11.5, abnormal DNA methylation and impaired heart, vascular and limb development [146, 153]. |
| MBD4 | Methyl-CpG binding domain protein 4 | Nucleus | TpG [151] 5hmU [154] 5fU [154] dsDNA | Viable and fertile. Increased rate of C to T mutation at CpG dinucleotides [104, 105]. |
| SMUG1 | Single-strand-specific monofunctional uracil-DNA glycosylase 1 | Nucleus | 5hmU [154, 155] 5fU [154] 5caU [156] ss and dsDNA | Ablation of base-excision repair in hmU excision and reduced cellular sensitivity to 5-hydroxymethyluridine toxicity [157]. |
| UNG | Uracil DNA glycosylase | Nucleus (UNG2) Mitochondria (UNG1) | 5hmU [132] ss and dsDNA | Increased post-ischemic brain injury [158]. Elevated level of uracil into DNA of dividing cells; mutations at C/G pairs are shifted towards transitions in hypermutation of immunoglobulin genes; reduced class-switch recombination [159]. |
| NEIL1 | Endonuclease VIII-like glycosylase 1 | Nucleus, cytoplasm Mitochondria | 5hmU [132] ss and dsDNA | Severe obesity, dyslipidemia and fatty liver disease; tend to develop hyperinsulinemia; elevated mtDNA damage and deletions; sporadic symptoms of decreased subcutaneous fat, skin ulcers, joint inflammation, infertility and tumours; obesity in male heterozygotes [160]. |
| NTHL1 | Endonuclease III-like 1 | Nucleus and Mitochondria | T [151] 5fU [154] 5hmU [132] dsDNA | Viable and fertile; slower hepatic repair of thymine glycol DNA lesions under X-ray irradiation [161]. |
There are other five human DNA glycosylases OGG1, MYH, MPG, NEIL2 and NEIL3 not included in this table as they have not been reported to excise DNA substrates that are involved in model pathways of active DNA demethylation to date.
Functional Interaction of DNA Methylation Mediators in DNA Damage Response and Regulation of DNA Methylation Activity
Aberrant expression of oncogenes and/or silencing of tumour suppressors require additional factors to synergistically break DNA damage response (DDR) barriers for tumour initiation and progression [162]. Functional interaction of DNA methylation mediators may have important roles in these processes. For example, DNMT1 is a positive transcriptional target of BRCA1, and its decreased expression was shown in Brca1∆11/∆11;p53+/- mouse mammary glands and in several human clinical samples [163]. These data support an association between the direct role of DNMT1 methyltransferase activity and global DNA hypomethylation, genomic imprinting loss and an open chromatin configuration [163]. In addition to this direct impact via DNMT1 methyltransferase activity, DNMT1 is associated with the p53 apoptosis pathway via interactions with MBD4 and its protein partner MLH1 in Xenopus embryos and mammalian cells [164-166]. DNMT1 forms a trimeric complex with UHRF1 and the deubiquitinating enzyme USP7 on chromatin during cell proliferation [167, 168]. The methyl-CpG binding glycosylase MBD4 interacts with and recruits USP7 to heterochromatic foci, where it physically associates with UHRF1 and DNMT1, indicating that MBD4 regulates DNMT1 activity [169]. These data indicated a prospective functional link between interactions of DNA methylation mediators and BRCA1-associated genome instability via the p53 DDR pathway, alluding to possible epigenetic roles in transformation and aggression of BRCA1-deficient cancers.
Activation of the DDR and apoptosis, G2/M arrest and enhanced radiosensitivity (ATM-p53 apoptosis) were also demonstrated in cancer cells with depleted UHRF1 [170-172]. In these experiments, UHRF1 and USP7 participate in M phase-specific signalling during cell proliferation, which regulates UHRF1 stability according to the deubiquitinase activity of USP7 and the counteracting site-specific phosphorylation of UHRF1 within its SRA domain [168]. Previous studies showed that the SRA domain of UHRF1 is responsible for binding to methylated CpG [111], and its preferential affinity is dependent on the presence of hemimethylated DNA. Accordingly, UHRF1 and DNMT1 complexes co-localise to replicating heterochromatin and play essential in vivo roles in maintaining global and local DNA methylation. Genetic studies showed that Np95 (UHRF1) depletion resultes in a lethal phenotype during early gestation, which resembles multiple defects observed in Dnmt1-/- embryos, with growth retardation and excess apoptosis [78, 111]. Further experiments are required to examine the roles of these functional protein complexes in the propagation and preservation of epigenetic signatures and in cellular surveillance systems that respond to intrinsic and extrinsic DDR signals. Because defects or imbalances of epigenetic systems are known to undermine cell viability [40, 173], consequent epigenome instability and impaired DDR in BRCA1-deficient cells with genetic instability may contribute to disease susceptibility, loss of heterozygosity and/or increased phenotypic variation in certain subsets of human breast cancers
DNA Methylation and Cancer Genome Instability
DNA Methylation and Chromatin Structure
A more general role of DNA methylation in genome stability may achieve through chromatin structure modelling, which is the major effect of methyl groups [174]. Although the precise mechanisms by which DNA methylation affects chromatin structure remain elusive, it is accepted that sequence-independent methyl moieties have a direct role in generating a closed chromatin structure [175, 176]. DNA methylation may shape chromatin and gene expression states through an intrinsic effect on nucleosome structure and/or by regulating other factors that displace nucleosomes [177-179]. Additionally, MeCPs specifically recognise DNA methylation and these factors may recruit histone modifiers or chromatin remodellers to shape local chromatin structure. DNA methylation inhibits the binding of chromatin protein CTCF [180]. Moreover, recent studies showed that a group of unmethylated CpG binding proteins that carry CXXC domains, including CFP1, interpret recruitment signals of hypomethylated CGIs and act as transcriptional activators [181, 182]. In principle, an open chromatin structure that determines active gene expression states may increase DNA accessibility to damage and potentially destabilise enzymatic transactions. Therefore, the major role of DNA methylation in shaping chromatin structure places its association with genome integrity in perspective. It has been estimated that hydrolytic deamination of cytosine in single-stranded regions of DNA occurs at least 100-fold more rapidly than in double-stranded DNA [183, 184], indicating that chromatin structure configuration at replication forks or transcription bubbles may determine local DNA vulnerability to damage factors. Moreover, recent studies have discovered and established the significance of many destabilising enzymatic transactions at replication forks following replication stress [185-187]. Some epigenetic factors including demethylase complexes have been implicated at these open chromatic sites [186, 187], but their functional roles in coordinating DNA replication and transcription for genome stability have not been resolved. A better understanding of the significance of DNA methylation machinery and chromatin structure in maintaining genome integrity will facilitate future investigations to target DNA methylation and its mediators for novel drugs and chemotherapeutic combinations.
Promoter Hypermethylation of Tumour Suppressors
It is widely accepted that promoter hypermethylation of key tumour suppressor genes is a driving phenomenon in tumourigenesis [188] and that several DNA repair and cell cycle regulatory genes are common targets of promoter methylation in cancers and are extensively associated with genomic instability [67]. For example, a single-nucleotide variant in the promoter of the mismatch repair gene MLH1 reduces its activity in transfection reporter assays [189]. Moreover, the comparatively lower activity of the MLH1 allele was correlated with hypermethylation of its promoter, and transcriptional silencing was demonstrated in somatic cells of some colorectal cancer patients [189]. Epigenetic inactivation of MLH1 causes the microsatellite instability phenotype in association with colorectal, endometrial and other cancers, resulting in downstream genetic mutations that contribute to genome-wide instability [67]. Therefore, MLH1 silencing by promoter hypermethylation may be an early carcinogenic event and represents an epigenetic character of colorectal cancer patients with the CpG island methylator phenotype [190]. This epigenetic mechanism was demonstrated in the human colon cancer cell line HCT116, in which epigenetic silencing of MLH1 by promoter hypermethylation in one allele and genetic mutation in the other results in complete inactivation of MLH1 [191]. Although enhanced methylation (3%) occurs during in vitro passaging of culture cells [192], de novo MLH1 promoter methylation in cell lines is representative of that in primary tumour cells [193]. Similarly, CDKN2A, which encodes the cyclin-dependent kinase inhibitor p16, bears genetic mutations in one allele in HCT116 cells, and promoter hypermethylation of the other allele leads to complete inactivation of p16 [191] and misregulation of the cell cycle [67]. Moreover, MeCPs bind to methylated promoters of MLH1, p16(INK4a) and master cell cycle regulator and tumour suppressor BRCA1 and recruit repressive complexes that specifically repress target genes [194, 195]. Accordingly, promoter hypermethylation and silencing of MLH1 and CDKN2A and the key tumour suppressors BRCA1, p53 and RB1 represent a paradigmatic association between DNA methylation and cancer genome instability.
Gene Body Methylation
Gene body methylation, which occurs mostly in CpG-poor gene exons, is responsible for frequent C to T transition mutations in germ-line and somatic cells of many cancers [193]. Unlike DNA promoter hypermethylation, gene body methylation is not associated with transcriptional repression, but causes gene activation [196], as confirmed in a human active X-chromosome model [197]. Methylation of gene body and orphan CGI promoters benefits transcriptional elongation by suppressing alternative promoter activation [14]. However, CpG methylation blocks elongation in the lower organism Neurospora crassa, suggesting that the positive correlation between transcription elongation and CGI methylation in gene bodies is limited in higher organisms such as mammals [193]. In contrast, gene body methylation outside CGIs is considered a major mechanism for silencing repetitive DNA elements such as transposons [198]. Recently, control of alternative splicing by gene body methylation was proposed and is supported by whole-genome studies that show greater methylation in exons than introns and exon-intron boundaries with distinguishing methylation levels [41, 199]. Taken with whole exome sequencing studies that identified numerous gene body mutations of genomic and epigenomic significance, these studies suggested that gene body methylation plays more prominent roles than previously thought [67] Additionally, the presence of 5hmC in gene bodies was consistently associated with gene expression in a number of studies [200-203], further supporting the hypothesis that 5hmC functions as a separate epigenetic mark.
5mC as Mutagen and Cancer-Causing Mutation
5mC is prone to spontaneous deamination and is a prominent source of germ-line and somatic mutations [204, 205]. Specifically, mutation of 5mC to T occurs with 10-50-fold greater frequency than other transitions [206], and mutation rates at CpG sites are estimated to be at least 10-18-fold more frequent than at non-CpG dinucleotides [207-211]. Moreover, DNA bases in CpG dinucleotides are vulnerable to chemical reactions, and endogenous and exogenous stress may favour spontaneous deamination via direct or indirect influence. The extracyclic amino group at C4 position of cytosine is subject to hydrolysis that is sensitive to salts, ATP and pH under physiological conditions [212]. Additionally hydrolysis efficiency can also be changed by exposure to exogenous stimuli such as xenobiotics or reagents generating reactive oxygen species [213]. Hydrolytic deamination of cytosine and 5mC results in pairing of uracil (U) and T with guanine (G), leading to the mismatches G:U and G:T, respectively. A failure to repair these mismatches prior to DNA replication causes cell acquiescence and mutation from C:G to T:A. Mutations at methylated CpG sites reportedly contribute one-third of all pathophysiological mutations and include familial mutations and single nucleotide polymorphisms (SNPs) in somatic cells [67]. A number of mutation hotspots are frequently found in DNA methylation mediators such as DNMT3A in acute myeloid leukaemia (AML) [82], DNMT1 in colorectal cancer [69] and DNMT3B in immunodeficiency-centromeric instability-facial anomaly (ICF) and chromosome instability syndromes [87]. Moreover, C to T transitions at methylated CpG sites increase numbers of natural p53 point mutations by as much as 50% in colorectal cancers and significantly increase the incidence of predominant p53 mutations in breast and ovarian cancers [205, 214]. In addition, promoter methylation of BRCA1 and hotspot mutations in the BRCA1 gene body (due to methylation of exons and subsequent deamination of 5mC) are frequent in breast and ovarian cancers [215], indicating another association between DNA methylation, the p53 DDR pathway and BRCA1-associated breast cancers.
Final Remark
Genome integrity is an absolute requirement of intact systems in higher organisms. Various forms of DNA methylation and respective mediator proteins may act as a multi-reacting processor and reflector of other epigenetic events such as histone modification and chromatin remodelling. These DNA modifications respond to extra-nuclear signals via sensor proteins and control gene expression and chromatin changes accordingly, providing feedback mechanisms that recruit effectors of transcription and DNA repair and replication and that play vital roles in both development and disease. Functional characterisation of the associated proteins continues to be an area of high interest, and accumulating in vivo data contribute an increasingly precise understanding of DNA methylation modes. These insights linking the genetic instability and epigenetic perturbations such as aberrant DNA methylation machinery may ultimately form the basis for novel therapeutic strategies and targets for the treatment of inherited, acquired and malignant diseases.
Abbreviations
5mC: 5-methylcytosine; DNMTs: DNA methyltransferases; MeCPs: methyl-CpG binding proteins; BER: base excision repair; TETs: ten-eleven translocation cytosine dioxygenases; TSSs: transcriptional start sites; CGIs: CpG islands; 5hmC: 5-hydroxymethylcytosine; 5fC: 5-formylcytosine; 5caC: 5-carboxylcytosine; TDG: thymine DNA glycosylase; ES cell: embryonic stem cells; DNMT1: DNA methyltransferase-1; MBD proteins: methyl-CpG binding domain proteins; 5meCpG: 5-methyl-CpG; SRA domain: SET and RING finger-associated domain; PHD domain: plant homeo domain; TRD domain: trans-repressor domain; DSBH: double-stranded β-helix; 5hmU: 5-hydroxymethyluracil; CXXC: cysteine-X-X-cysteine; αKG: α-ketoglutarate; AP: abasic apurinic/apyrimidinic; DME: DEMETER; SNP: single nucleotide polymorphism; AML: acute myeloid leukaemia; ICF: immunodeficiency-centromeric instability-facial anomaly; DDR: DNA damage response; BRCA1: breast cancer1, early onset.
Acknowledgements
This project was supported by grants from the National Natural Science Foundation of P.R.China (No. 81130042, 3117132) and the Ministry of Education Innovation Team Development Plan (No. IRT13101).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Johnson TB, Coghill RD. The Discovery of 5-methyl-cytosine in Tuberculinic Acid, The Nucleic Acid of The Tubercle Bacillus. J Am Chem Soc. 1925;47:2838-44 doi:10.1021/ja01688a030
2. Hotchkiss RD. The quantitative separation of purines, pyrimidines, and nucleosides by paper chromatography. J Biol Chem. 1948;175:315-32
3. Riggs AD. X inactivation, differentiation, and DNA methylation. Cytogenet Cell Genet. 1975;14:9-25
4. Holliday R, Pugh JE. DNA modification mechanisms and gene activity during development. Science. 1975;187:226-32
5. Doerfler W. De novo methylation, long-term promoter silencing, methylation patterns in the human genome, and consequences of foreign DNA insertion. Curr Top Microbiol Immunol. 2006;301:125-75
6. Waddington CH. The epigenotype. Endeavour. 1942:18-20
7. Russo E, Martienssen R, Riggs AD. Epigenetic Mechanisms of Gene Regulation. Cold Spring Harbor Lab Press, Plainview, NY. 1996
8. Bickle TA, Kruger DH. Biology of DNA restriction. Microbiol Rev. 1993;57:434-50
9. Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9:2395-402
10. Baranzini SE, Mudge J, van Velkinburgh JC, Khankhanian P, Khrebtukova I, Miller NA. et al. Genome, epigenome and RNA sequences of monozygotic twins discordant for multiple sclerosis. Nature. 2010;464:1351-6 doi:10.1038/nature08990
11. Sproul D, Nestor C, Culley J, Dickson JH, Dixon JM, Harrison DJ. et al. Transcriptionally repressed genes become aberrantly methylated and distinguish tumors of different lineages in breast cancer. Proc Natl Acad Sci U S A. 2011;108:4364-9 doi:10.1073/pnas.1013224108
12. Beaujean N, Taylor J, Gardner J, Wilmut I, Meehan R, Young L. Effect of limited DNA methylation reprogramming in the normal sheep embryo on somatic cell nuclear transfer. Biol Reprod. 2004;71:185-93 doi:10.1095/biolreprod.103.026559
13. Illingworth RS, Bird AP. CpG islands--'a rough guide'. FEBS Lett. 2009;583:1713-20 doi:10.1016/j.febslet.2009.04.012
14. Illingworth RS, Gruenewald-Schneider U, Webb S, Kerr AR, James KD, Turner DJ. et al. Orphan CpG islands identify numerous conserved promoters in the mammalian genome. PLoS genetics. 2010 6. doi:10.1371/journal.pgen.1001134
15. Kriaucionis S, Heintz N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science. 2009;324:929-30 doi:10.1126/science.1169786
16. Tahiliani M, Koh KP, Shen Y, Pastor WA, Bandukwala H, Brudno Y. et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science. 2009;324:930-5 doi:10.1126/science.1170116
17. Maiti A, Drohat AC. Thymine DNA glycosylase can rapidly excise 5-formylcytosine and 5-carboxylcytosine: potential implications for active demethylation of CpG sites. J Biol Chem. 2011;286:35334-8 doi:10.1074/jbc.C111.284620
18. Wu SC, Zhang Y. Active DNA demethylation: many roads lead to Rome. Nat Rev Mol Cell Biol. 2010;11:607-20 doi:10.1038/nrm2950
19. He YF, Li BZ, Li Z, Liu P, Wang Y, Tang Q. et al. Tet-mediated formation of 5-carboxylcytosine and its excision by TDG in mammalian DNA. Science. 2011;333:1303-7 doi:10.1126/science.1210944
20. Meng H, Chen G, Gao HM, Song X, Shi Y, Cao L. The Emerging Nexus of Active DNA Demethylation and Mitochondrial Oxidative Metabolism in Post-Mitotic Neurons. Int J Mol Sci. 2014;15:22604-25 doi:10.3390/ijms151222604
21. Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J. et al. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature. 2009;462:315-22 doi:10.1038/nature08514
22. Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, Jaenisch R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci U S A. 2000;97:5237-42
23. Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet. 2002;3:662-73 doi:10.1038/nrg887
24. Bernstein BE, Meissner A, Lander ES. The mammalian epigenome. Cell. 2007;128:669-81 doi:10.1016/j.cell.2007.01.033
25. Iyer LM, Tahiliani M, Rao A, Aravind L. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell cycle. 2009;8:1698-710
26. Inoue A, Shen L, Dai Q, He C, Zhang Y. Generation and replication-dependent dilution of 5fC and 5caC during mouse preimplantation development. Cell Res. 2011;21:1670-6 doi:10.1038/cr.2011.189
27. Song CX, Szulwach KE, Dai Q, Fu Y, Mao SQ, Lin L. et al. Genome-wide profiling of 5-formylcytosine reveals its roles in epigenetic priming. Cell. 2013;153:678-91 doi:10.1016/j.cell.2013.04.001
28. Nestor C, Ruzov A, Meehan R, Dunican D. Enzymatic approaches and bisulfite sequencing cannot distinguish between 5-methylcytosine and 5-hydroxymethylcytosine in DNA. Biotechniques. 2010;48:317-9 doi:10.2144/000113403
29. Huang Y, Pastor WA, Shen Y, Tahiliani M, Liu DR, Rao A. The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS One. 2010;5:e8888. doi:10.1371/journal.pone.0008888
30. Song CX, Szulwach KE, Fu Y, Dai Q, Yi C, Li X. et al. Selective chemical labeling reveals the genome-wide distribution of 5-hydroxymethylcytosine. Nat Biotechnol. 2011;29:68-72 doi:10.1038/nbt.1732
31. Nestor CE, Ottaviano R, Reddington J, Sproul D, Reinhardt D, Dunican D. et al. Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res. 2012;22:467-77 doi:10.1101/gr.126417.111
32. Wu H, Zhang Y. Mechanisms and functions of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev. 2011;25:2436-52 doi:10.1101/gad.179184.111
33. Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A. et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature. 2008;454:766-70 doi:10.1038/nature07107
34. Lister R, Mukamel EA, Nery JR, Urich M, Puddifoot CA, Johnson ND. et al. Global epigenomic reconfiguration during mammalian brain development. Science. 2013;341:1237905. doi:10.1126/science.1237905
35. Li E, Zhang Y. DNA methylation in mammals. Cold Spring Harb Perspect Biol. 2014;6:a019133. doi:10.1101/cshperspect.a019133
36. Bird A, Taggart M, Frommer M, Miller OJ, Macleod D. A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell. 1985;40:91-9
37. Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci U S A. 2002;99:3740-5 doi:10.1073/pnas.052410099
38. Bird AP. CpG-rich islands and the function of DNA methylation. Nature. 1986;321:209-13 doi:10.1038/321209a0
39. Wolf SF, Dintzis S, Toniolo D, Persico G, Lunnen KD, Axelman J. et al. Complete concordance between glucose-6-phosphate dehydrogenase activity and hypomethylation of 3' CpG clusters: implications for X chromosome dosage compensation. Nucleic Acids Res. 1984;12:9333-48
40. Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27-36 doi:10.1093/carcin/bgp220
41. Laurent L, Wong E, Li G, Huynh T, Tsirigos A, Ong CT. et al. Dynamic changes in the human methylome during differentiation. Genome Res. 2010;20:320-31 doi:10.1101/gr.101907.109
42. Zhang Y, Rohde C, Tierling S, Jurkowski TP, Bock C, Santacruz D. et al. DNA methylation analysis of chromosome 21 gene promoters at single base pair and single allele resolution. PLoS genetics. 2009;5:e1000438. doi:10.1371/journal.pgen.1000438
43. Berman BP, Weisenberger DJ, Aman JF, Hinoue T, Ramjan Z, Liu Y. et al. Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains. Nat Genet. 2012;44:40-6 doi:10.1038/ng.969
44. Hansen KD, Timp W, Bravo HC, Sabunciyan S, Langmead B, McDonald OG. et al. Increased methylation variation in epigenetic domains across cancer types. Nat Genet. 2011;43:768-75 doi:10.1038/ng.865
45. Suter CM, Martin DI, Ward RL. Hypomethylation of L1 retrotransposons in colorectal cancer and adjacent normal tissue. Int J Colorectal Dis. 2004;19:95-101 doi:10.1007/s00384-003-0539-3
46. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57-70
47. Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability--an evolving hallmark of cancer. Nat Rev Mol Cell Biol. 2010;11:220-8 doi:10.1038/nrm2858
48. Okano M, Bell DW, Haber DA, Li E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99:247-57
49. Okano M, Xie S, Li E. Cloning and characterization of a family of novel mammalian DNA (cytosine-5) methyltransferases. Nat Genet. 1998;19:219-20 doi:10.1038/890
50. Kim JK, Samaranayake M, Pradhan S. Epigenetic mechanisms in mammals. Cellular and molecular life sciences: CMLS. 2009;66:596-612 doi:10.1007/s00018-008-8432-4
51. Goll MG, Bestor TH. Eukaryotic cytosine methyltransferases. Annu Rev Biochem. 2005;74:481-514 doi:10.1146/annurev.biochem.74.010904.153721
52. Leonhardt H, Page AW, Weier HU, Bestor TH. A targeting sequence directs DNA methyltransferase to sites of DNA replication in mammalian nuclei. Cell. 1992;71:865-73
53. Bostick M, Kim JK, Esteve PO, Clark A, Pradhan S, Jacobsen SE. UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science. 2007;317:1760-4 doi:10.1126/science.1147939
54. Sharif J, Koseki H. Recruitment of Dnmt1 roles of the SRA protein Np95 (Uhrf1) and other factors. Prog Mol Biol Transl Sci. 2011;101:289-310 doi:10.1016/B978-0-12-387685-0.00008-1
55. Achour M, Jacq X, Ronde P, Alhosin M, Charlot C, Chataigneau T. et al. The interaction of the SRA domain of ICBP90 with a novel domain of DNMT1 is involved in the regulation of VEGF gene expression. Oncogene. 2008;27:2187-97 doi:10.1038/sj.onc.1210855
56. Smallwood A, Esteve PO, Pradhan S, Carey M. Functional cooperation between HP1 and DNMT1 mediates gene silencing. Genes Dev. 2007;21:1169-78 doi:10.1101/gad.1536807
57. Esteve PO, Chin HG, Smallwood A, Feehery GR, Gangisetty O, Karpf AR. et al. Direct interaction between DNMT1 and G9a coordinates DNA and histone methylation during replication. Genes Dev. 2006;20:3089-103 doi:10.1101/gad.1463706
58. Ooi SK, Wolf D, Hartung O, Agarwal S, Daley GQ, Goff SP. et al. Dynamic instability of genomic methylation patterns in pluripotent stem cells. Epigenetics & chromatin. 2010;3:17. doi:10.1186/1756-8935-3-17
59. Chedin F, Lieber MR, Hsieh CL. The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. Proc Natl Acad Sci U S A. 2002;99:16916-21 doi:10.1073/pnas.262443999
60. Chen ZX, Mann JR, Hsieh CL, Riggs AD, Chedin F. Physical and functional interactions between the human DNMT3L protein and members of the de novo methyltransferase family. J Cell Biochem. 2005;95:902-17 doi:10.1002/jcb.20447
61. Gowher H, Liebert K, Hermann A, Xu G, Jeltsch A. Mechanism of stimulation of catalytic activity of Dnmt3A and Dnmt3B DNA-(cytosine-C5)-methyltransferases by Dnmt3L. J Biol Chem. 2005;280:13341-8 doi:10.1074/jbc.M413412200
62. Kareta MS, Botello ZM, Ennis JJ, Chou C, Chedin F. Reconstitution and mechanism of the stimulation of de novo methylation by human DNMT3L. J Biol Chem. 2006;281:25893-902 doi:10.1074/jbc.M603140200
63. Jia D, Jurkowska RZ, Zhang X, Jeltsch A, Cheng X. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature. 2007;449:248-51 doi:10.1038/nature06146
64. Jurkowska RZ, Rajavelu A, Anspach N, Urbanke C, Jankevicius G, Ragozin S. et al. Oligomerization and binding of the Dnmt3a DNA methyltransferase to parallel DNA molecules: heterochromatic localization and role of Dnmt3L. J Biol Chem. 2011;286:24200-7 doi:10.1074/jbc.M111.254987
65. Neri F, Krepelova A, Incarnato D, Maldotti M, Parlato C, Galvagni F. et al. Dnmt3L antagonizes DNA methylation at bivalent promoters and favors DNA methylation at gene bodies in ESCs. Cell. 2013;155:121-34 doi:10.1016/j.cell.2013.08.056
66. Klose RJ, Bird AP. Genomic DNA methylation: the mark and its mediators. Trends Biochem Sci. 2006;31:89-97 doi:10.1016/j.tibs.2005.12.008
67. You JS, Jones PA. Cancer genetics and epigenetics: two sides of the same coin? Cancer Cell. 2012;22:9-20 doi:10.1016/j.ccr.2012.06.008
68. Subramaniam D, Thombre R, Dhar A, Anant S. DNA methyltransferases: a novel target for prevention and therapy. Front Oncol. 2014;4:80. doi:10.3389/fonc.2014.00080
69. Kanai Y, Ushijima S, Nakanishi Y, Sakamoto M, Hirohashi S. Mutation of the DNA methyltransferase (DNMT) 1 gene in human colorectal cancers. Cancer Lett. 2003;192:75-82
70. Mizuno S, Chijiwa T, Okamura T, Akashi K, Fukumaki Y, Niho Y. et al. Expression of DNA methyltransferases DNMT1, 3A, and 3B in normal hematopoiesis and in acute and chronic myelogenous leukemia. Blood. 2001;97:1172-9
71. Rajendran G, Shanmuganandam K, Bendre A, Muzumdar D, Goel A, Shiras A. Epigenetic regulation of DNA methyltransferases: DNMT1 and DNMT3B in gliomas. J Neurooncol. 2011;104:483-94 doi:10.1007/s11060-010-0520-2
72. Xing J, Stewart DJ, Gu J, Lu C, Spitz MR, Wu X. Expression of methylation-related genes is associated with overall survival in patients with non-small cell lung cancer. Br J Cancer. 2008;98:1716-22 doi:10.1038/sj.bjc.6604343
73. Li A, Omura N, Hong SM, Goggins M. Pancreatic cancer DNMT1 expression and sensitivity to DNMT1 inhibitors. Cancer Biol Ther. 2010;9:321-9 doi:10.4161/cbt.9.4.10750
74. Etoh T, Kanai Y, Ushijima S, Nakagawa T, Nakanishi Y, Sasako M. et al. Increased DNA methyltransferase 1 (DNMT1) protein expression correlates significantly with poorer tumor differentiation and frequent DNA hypermethylation of multiple CpG islands in gastric cancers. Am J Pathol. 2004;164:689-99 doi:10.1016/S0002-9440(10)63156-2
75. Nagai M, Nakamura A, Makino R, Mitamura K. Expression of DNA (5-cytosin)-methyltransferases (DNMTs) in hepatocellular carcinomas. Hepatol Res. 2003;26:186-91
76. Mirza S, Sharma G, Parshad R, Gupta SD, Pandya P, Ralhan R. Expression of DNA methyltransferases in breast cancer patients and to analyze the effect of natural compounds on DNA methyltransferases and associated proteins. J Breast Cancer. 2013;16:23-31 doi:10.4048/jbc.2013.16.1.23
77. Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69:915-26
78. Howell CY, Bestor TH, Ding F, Latham KE, Mertineit C, Trasler JM. et al. Genomic imprinting disrupted by a maternal effect mutation in the Dnmt1 gene. Cell. 2001;104:829-38
79. Saito Y, Kanai Y, Sakamoto M, Saito H, Ishii H, Hirohashi S. Expression of mRNA for DNA methyltransferases and methyl-CpG-binding proteins and DNA methylation status on CpG islands and pericentromeric satellite regions during human hepatocarcinogenesis. Hepatology. 2001;33:561-8 doi:10.1053/jhep.2001.22507
80. Kanai Y, Ushijima S, Kondo Y, Nakanishi Y, Hirohashi S. DNA methyltransferase expression and DNA methylation of CPG islands and peri-centromeric satellite regions in human colorectal and stomach cancers. Int J Cancer. 2001;91:205-12
81. Goll MG, Kirpekar F, Maggert KA, Yoder JA, Hsieh CL, Zhang X. et al. Methylation of tRNAAsp by the DNA methyltransferase homolog Dnmt2. Science. 2006;311:395-8 doi:10.1126/science.1120976
82. Ley TJ, Ding L, Walter MJ, McLellan MD, Lamprecht T, Larson DE. et al. DNMT3A mutations in acute myeloid leukemia. N Engl J Med. 2010;363:2424-33 doi:10.1056/NEJMoa1005143
83. Yan XJ, Xu J, Gu ZH, Pan CM, Lu G, Shen Y. et al. Exome sequencing identifies somatic mutations of DNA methyltransferase gene DNMT3A in acute monocytic leukemia. Nat Genet. 2011;43:309-15 doi:10.1038/ng.788
84. Yang J, Wei X, Wu Q, Xu Z, Gu D, Jin Y. et al. Clinical significance of the expression of DNA methyltransferase proteins in gastric cancer. Mol Med Rep. 2011;4:1139-43 doi:10.3892/mmr.2011.578
85. He S, Wang F, Yang L, Guo C, Wan R, Ke A. et al. Expression of DNMT1 and DNMT3a are regulated by GLI1 in human pancreatic cancer. PLoS One. 2011;6:e27684. doi:10.1371/journal.pone.0027684
86. Robertson KD, Uzvolgyi E, Liang G, Talmadge C, Sumegi J, Gonzales FA. et al. The human DNA methyltransferases (DNMTs) 1, 3a and 3b: coordinate mRNA expression in normal tissues and overexpression in tumors. Nucleic Acids Res. 1999;27:2291-8
87. Wijmenga C, Hansen RS, Gimelli G, Bjorck EJ, Davies EG, Valentine D. et al. Genetic variation in ICF syndrome: evidence for genetic heterogeneity. Hum Mutat. 2000;16:509-17
88. Shen H, Wang L, Spitz MR, Hong WK, Mao L, Wei Q. A novel polymorphism in human cytosine DNA-methyltransferase-3B promoter is associated with an increased risk of lung cancer. Cancer Res. 2002;62:4992-5
89. Ibrahim AE, Arends MJ, Silva AL, Wyllie AH, Greger L, Ito Y. et al. Sequential DNA methylation changes are associated with DNMT3B overexpression in colorectal neoplastic progression. Gut. 2011;60:499-508 doi:10.1136/gut.2010.223602
90. Kobayashi Y, Absher DM, Gulzar ZG, Young SR, McKenney JK, Peehl DM. et al. DNA methylation profiling reveals novel biomarkers and important roles for DNA methyltransferases in prostate cancer. Genome Res. 2011;21:1017-27 doi:10.1101/gr.119487.110
91. Girault I, Tozlu S, Lidereau R, Bieche I. Expression analysis of DNA methyltransferases 1, 3A, and 3B in sporadic breast carcinomas. Clin Cancer Res. 2003;9:4415-22
92. Gokul G, Gautami B, Malathi S, Sowjanya AP, Poli UR, Jain M. et al. DNA methylation profile at the DNMT3L promoter: a potential biomarker for cervical cancer. Epigenetics. 2007;2:80-5
93. Minami K, Chano T, Kawakami T, Ushida H, Kushima R, Okabe H. et al. DNMT3L is a novel marker and is essential for the growth of human embryonal carcinoma. Clin Cancer Res. 2010;16:2751-9 doi:10.1158/1078-0432.CCR-09-3338
94. Webster KE, O'Bryan MK, Fletcher S, Crewther PE, Aapola U, Craig J. et al. Meiotic and epigenetic defects in Dnmt3L-knockout mouse spermatogenesis. Proc Natl Acad Sci U S A. 2005;102:4068-73 doi:10.1073/pnas.0500702102
95. Bourc'his D, Xu GL, Lin CS, Bollman B, Bestor TH. Dnmt3L and the establishment of maternal genomic imprints. Science. 2001;294:2536-9 doi:10.1126/science.1065848
96. Guy J, Hendrich B, Holmes M, Martin JE, Bird A. A mouse Mecp2-null mutation causes neurological symptoms that mimic Rett syndrome. Nat Genet. 2001;27:322-6 doi:10.1038/85899
97. Chen RZ, Akbarian S, Tudor M, Jaenisch R. Deficiency of methyl-CpG binding protein-2 in CNS neurons results in a Rett-like phenotype in mice. Nat Genet. 2001;27:327-31 doi:10.1038/85906
98. Zhao X, Ueba T, Christie BR, Barkho B, McConnell MJ, Nakashima K. et al. Mice lacking methyl-CpG binding protein 1 have deficits in adult neurogenesis and hippocampal function. Proc Natl Acad Sci U S A. 2003;100:6777-82 doi:10.1073/pnas.1131928100
99. Hendrich B, Guy J, Ramsahoye B, Wilson VA, Bird A. Closely related proteins MBD2 and MBD3 play distinctive but interacting roles in mouse development. Genes Dev. 2001;15:710-23 doi:10.1101/gad.194101
100. Pontes TB, Chen ES, Gigek CO, Calcagno DQ, Wisnieski F, Leal MF. et al. Reduced mRNA expression levels of MBD2 and MBD3 in gastric carcinogenesis. Tumour Biol. 2014;35:3447-53 doi:10.1007/s13277-013-1455-y
101. Kaji K, Caballero IM, MacLeod R, Nichols J, Wilson VA, Hendrich B. The NuRD component Mbd3 is required for pluripotency of embryonic stem cells. Nat Cell Biol. 2006;8:285-92 doi:10.1038/ncb1372
102. Riccio A, Aaltonen LA, Godwin AK, Loukola A, Percesepe A, Salovaara R. et al. The DNA repair gene MBD4 (MED1) is mutated in human carcinomas with microsatellite instability. Nat Genet. 1999;23:266-8 doi:10.1038/15443
103. Bader SA, Walker M, Harrison DJ. A human cancer-associated truncation of MBD4 causes dominant negative impairment of DNA repair in colon cancer cells. Br J Cancer. 2007;96:660-6 doi:10.1038/sj.bjc.6603592
104. Millar CB, Guy J, Sansom OJ, Selfridge J, MacDougall E, Hendrich B. et al. Enhanced CpG mutability and tumorigenesis in MBD4-deficient mice. Science. 2002;297:403-5 doi:10.1126/science.1073354
105. Wong E, Yang K, Kuraguchi M, Werling U, Avdievich E, Fan K. et al. Mbd4 inactivation increases Cright-arrowT transition mutations and promotes gastrointestinal tumor formation. Proc Natl Acad Sci U S A. 2002;99:14937-42 doi:10.1073/pnas.232579299
106. Prokhortchouk A, Sansom O, Selfridge J, Caballero IM, Salozhin S, Aithozhina D. et al. Kaiso-deficient mice show resistance to intestinal cancer. Mol Cell Biol. 2006;26:199-208 doi:10.1128/MCB.26.1.199-208.2006
107. Kim K, Chadalapaka G, Lee SO, Yamada D, Sastre-Garau X, Defossez PA. et al. Identification of oncogenic microRNA-17-92/ZBTB4/specificity protein axis in breast cancer. Oncogene. 2012;31:1034-44 doi:10.1038/onc.2011.296
108. Mudbhary R, Hoshida Y, Chernyavskaya Y, Jacob V, Villanueva A, Fiel MI. et al. UHRF1 overexpression drives DNA hypomethylation and hepatocellular carcinoma. Cancer Cell. 2014;25:196-209 doi:10.1016/j.ccr.2014.01.003
109. Unoki M, Kelly JD, Neal DE, Ponder BA, Nakamura Y, Hamamoto R. UHRF1 is a novel molecular marker for diagnosis and the prognosis of bladder cancer. Br J Cancer. 2009;101:98-105 doi:10.1038/sj.bjc.6605123
110. Unoki M, Daigo Y, Koinuma J, Tsuchiya E, Hamamoto R, Nakamura Y. UHRF1 is a novel diagnostic marker of lung cancer. Br J Cancer. 2010;103:217-22 doi:10.1038/sj.bjc.6605717
111. Sharif J, Muto M, Takebayashi S, Suetake I, Iwamatsu A, Endo TA. et al. The SRA protein Np95 mediates epigenetic inheritance by recruiting Dnmt1 to methylated DNA. Nature. 2007;450:908-12 doi:10.1038/nature06397
112. Lu S, Yan D, Wu Z, Jiang T, Chen J, Yuan L. et al. Ubiquitin-like with PHD and ring finger domains 2 is a predictor of survival and a potential therapeutic target in colon cancer. Oncol Rep. 2014;31:1802-10 doi:10.3892/or.2014.3035
113. Clouaire T, de Las Heras JI, Merusi C, Stancheva I. Recruitment of MBD1 to target genes requires sequence-specific interaction of the MBD domain with methylated DNA. Nucleic Acids Res. 2010;38:4620-34 doi:10.1093/nar/gkq228
114. Scarsdale JN, Webb HD, Ginder GD, Williams DC Jr. Solution structure and dynamic analysis of chicken MBD2 methyl binding domain bound to a target-methylated DNA sequence. Nucleic Acids Res. 2011;39:6741-52 doi:10.1093/nar/gkr262
115. Klose RJ, Sarraf SA, Schmiedeberg L, McDermott SM, Stancheva I, Bird AP. DNA binding selectivity of MeCP2 due to a requirement for A/T sequences adjacent to methyl-CpG. Mol Cell. 2005;19:667-78 doi:10.1016/j.molcel.2005.07.021
116. Filion GJ, Zhenilo S, Salozhin S, Yamada D, Prokhortchouk E, Defossez PA. A family of human zinc finger proteins that bind methylated DNA and repress transcription. Mol Cell Biol. 2006;26:169-81 doi:10.1128/MCB.26.1.169-181.2006
117. Prokhortchouk A, Hendrich B, Jorgensen H, Ruzov A, Wilm M, Georgiev G. et al. The p120 catenin partner Kaiso is a DNA methylation-dependent transcriptional repressor. Genes Dev. 2001;15:1613-8 doi:10.1101/gad.198501
118. Ruzov A, Dunican DS, Prokhortchouk A, Pennings S, Stancheva I, Prokhortchouk E. et al. Kaiso is a genome-wide repressor of transcription that is essential for amphibian development. Development. 2004;131:6185-94 doi:10.1242/dev.01549
119. Hashimoto H, Horton JR, Zhang X, Bostick M, Jacobsen SE, Cheng X. The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature. 2008;455:826-9 doi:10.1038/nature07280
120. Arita K, Ariyoshi M, Tochio H, Nakamura Y, Shirakawa M. Recognition of hemi-methylated DNA by the SRA protein UHRF1 by a base-flipping mechanism. Nature. 2008;455:818-21 doi:10.1038/nature07249
121. Avvakumov GV, Walker JR, Xue S, Li Y, Duan S, Bronner C. et al. Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature. 2008;455:822-5 doi:10.1038/nature07273
122. Pichler G, Wolf P, Schmidt CS, Meilinger D, Schneider K, Frauer C. et al. Cooperative DNA and histone binding by Uhrf2 links the two major repressive epigenetic pathways. J Cell Biochem. 2011;112:2585-93 doi:10.1002/jcb.23185
123. Zhou T, Xiong J, Wang M, Yang N, Wong J, Zhu B. et al. Structural Basis for Hydroxymethylcytosine Recognition by the SRA Domain of UHRF2. Mol Cell. 2014 doi:10.1016/j.molcel.2014.04.003
124. Campanero MR, Armstrong MI, Flemington EK. CpG methylation as a mechanism for the regulation of E2F activity. Proc Natl Acad Sci U S A. 2000;97:6481-6 doi:10.1073/pnas.100340697
125. Iguchi-Ariga SM, Schaffner W. CpG methylation of the cAMP-responsive enhancer/promoter sequence TGACGTCA abolishes specific factor binding as well as transcriptional activation. Genes Dev. 1989;3:612-9
126. Fuks F, Hurd PJ, Wolf D, Nan X, Bird AP, Kouzarides T. The methyl-CpG-binding protein MeCP2 links DNA methylation to histone methylation. J Biol Chem. 2003;278:4035-40 doi:10.1074/jbc.M210256200
127. Jones PL, Veenstra GJ, Wade PA, Vermaak D, Kass SU, Landsberger N. et al. Methylated DNA and MeCP2 recruit histone deacetylase to repress transcription. Nat Genet. 1998;19:187-91 doi:10.1038/561
128. Nan X, Ng HH, Johnson CA, Laherty CD, Turner BM, Eisenman RN. et al. Transcriptional repression by the methyl-CpG-binding protein MeCP2 involves a histone deacetylase complex. Nature. 1998;393:386-9 doi:10.1038/30764
129. Wade PA, Gegonne A, Jones PL, Ballestar E, Aubry F, Wolffe AP. Mi-2 complex couples DNA methylation to chromatin remodelling and histone deacetylation. Nat Genet. 1999;23:62-6 doi:10.1038/12664
130. Kokura K, Kaul SC, Wadhwa R, Nomura T, Khan MM, Shinagawa T. et al. The Ski protein family is required for MeCP2-mediated transcriptional repression. J Biol Chem. 2001;276:34115-21 doi:10.1074/jbc.M105747200
131. Borst P, Sabatini R. Base J: discovery, biosynthesis, and possible functions. Annu Rev Microbiol. 2008;62:235-51 doi:10.1146/annurev.micro.62.081307.162750
132. Pfaffeneder T, Hackner B, Truss M, Munzel M, Muller M, Deiml CA. et al. The discovery of 5-formylcytosine in embryonic stem cell DNA. Angew Chem Int Ed Engl. 2011;50:7008-12 doi:10.1002/anie.201103899
133. Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA. et al. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine. Science. 2011;333:1300-3 doi:10.1126/science.1210597
134. McDonough MA, Loenarz C, Chowdhury R, Clifton IJ, Schofield CJ. Structural studies on human 2-oxoglutarate dependent oxygenases. Curr Opin Struct Biol. 2010;20:659-72 doi:10.1016/j.sbi.2010.08.006
135. Xu Y, Xu C, Kato A, Tempel W, Abreu JG, Bian C. et al. Tet3 CXXC domain and dioxygenase activity cooperatively regulate key genes for Xenopus eye and neural development. Cell. 2012;151:1200-13 doi:10.1016/j.cell.2012.11.014
136. Munzel M, Lercher L, Muller M, Carell T. Chemical discrimination between dC and 5MedC via their hydroxylamine adducts. Nucleic Acids Res. 2010;38:e192. doi:10.1093/nar/gkq724
137. Yu M, Hon GC, Szulwach KE, Song CX, Zhang L, Kim A. et al. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell. 2012;149:1368-80 doi:10.1016/j.cell.2012.04.027
138. Valinluck V, Tsai HH, Rogstad DK, Burdzy A, Bird A, Sowers LC. Oxidative damage to methyl-CpG sequences inhibits the binding of the methyl-CpG binding domain (MBD) of methyl-CpG binding protein 2 (MeCP2). Nucleic Acids Res. 2004;32:4100-8 doi:10.1093/nar/gkh739
139. Hashimoto H, Liu Y, Upadhyay AK, Chang Y, Howerton SB, Vertino PM. et al. Recognition and potential mechanisms for replication and erasure of cytosine hydroxymethylation. Nucleic Acids Res. 2012;40:4841-9 doi:10.1093/nar/gks155
140. Khrapunov S, Warren C, Cheng H, Berko ER, Greally JM, Brenowitz M. Unusual characteristics of the DNA binding domain of epigenetic regulatory protein MeCP2 determine its binding specificity. Biochemistry. 2014;53:3379-91 doi:10.1021/bi500424z
141. Mellen M, Ayata P, Dewell S, Kriaucionis S, Heintz N. MeCP2 binds to 5hmC enriched within active genes and accessible chromatin in the nervous system. Cell. 2012;151:1417-30 doi:10.1016/j.cell.2012.11.022
142. Spruijt CG, Gnerlich F, Smits AH, Pfaffeneder T, Jansen PW, Bauer C. et al. Dynamic readers for 5-(hydroxy)methylcytosine and its oxidized derivatives. Cell. 2013;152:1146-59 doi:10.1016/j.cell.2013.02.004
143. Frauer C, Hoffmann T, Bultmann S, Casa V, Cardoso MC, Antes I. et al. Recognition of 5-hydroxymethylcytosine by the Uhrf1 SRA domain. PLoS One. 2011;6:e21306. doi:10.1371/journal.pone.0021306
144. Iurlaro M, Ficz G, Oxley D, Raiber EA, Bachman M, Booth MJ. et al. A screen for hydroxymethylcytosine and formylcytosine binding proteins suggests functions in transcription and chromatin regulation. Genome Biol. 2013;14:R119. doi:10.1186/gb-2013-14-10-r119
145. Zhu JK. Active DNA demethylation mediated by DNA glycosylases. Annu Rev Genet. 2009;43:143-66 doi:10.1146/annurev-genet-102108-134205
146. Cortellino S, Xu J, Sannai M, Moore R, Caretti E, Cigliano A. et al. Thymine DNA glycosylase is essential for active DNA demethylation by linked deamination-base excision repair. Cell. 2011;146:67-79 doi:10.1016/j.cell.2011.06.020
147. Pfaffeneder T, Spada F, Wagner M, Brandmayr C, Laube SK, Eisen D. et al. Tet oxidizes thymine to 5-hydroxymethyluracil in mouse embryonic stem cell DNA. Nat Chem Biol. 2014;10:574-81 doi:10.1038/nchembio.1532
148. Svilar D, Goellner EM, Almeida KH, Sobol RW. Base excision repair and lesion-dependent subpathways for repair of oxidative DNA damage. Antioxid Redox Signal. 2011;14:2491-507 doi:10.1089/ars.2010.3466
149. Shen L, Song CX, He C, Zhang Y. Mechanism and function of oxidative reversal of DNA and RNA methylation. Annu Rev Biochem. 2014;83:585-614 doi:10.1146/annurev-biochem-060713-035513
150. Jacobs AL, Schar P. DNA glycosylases: in DNA repair and beyond. Chromosoma. 2012;121:1-20 doi:10.1007/s00412-011-0347-4
151. Trivedi RN, Almeida KH, Fornsaglio JL, Schamus S, Sobol RW. The role of base excision repair in the sensitivity and resistance to temozolomide-mediated cell death. Cancer Res. 2005;65:6394-400 doi:10.1158/0008-5472.CAN-05-0715
152. Hardeland U, Bentele M, Jiricny J, Schar P. The versatile thymine DNA-glycosylase: a comparative characterization of the human, Drosophila and fission yeast orthologs. Nucleic Acids Res. 2003;31:2261-71
153. Cortazar D, Kunz C, Selfridge J, Lettieri T, Saito Y, MacDougall E. et al. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature. 2011;470:419-23 doi:10.1038/nature09672
154. Bjelland S, Seeberg E. Mutagenicity, toxicity and repair of DNA base damage induced by oxidation. Mutat Res. 2003;531:37-80
155. Wibley JE, Waters TR, Haushalter K, Verdine GL, Pearl LH. Structure and specificity of the vertebrate anti-mutator uracil-DNA glycosylase SMUG1. Mol Cell. 2003;11:1647-59
156. Darwanto A, Theruvathu JA, Sowers JL, Rogstad DK, Pascal T, Goddard W 3rd. et al. Mechanisms of base selection by human single-stranded selective monofunctional uracil-DNA glycosylase. J Biol Chem. 2009;284:15835-46 doi:10.1074/jbc.M807846200
157. Kemmerich K, Dingler FA, Rada C, Neuberger MS. Germline ablation of SMUG1 DNA glycosylase causes loss of 5-hydroxymethyluracil- and UNG-backup uracil-excision activities and increases cancer predisposition of Ung-/-Msh2-/- mice. Nucleic Acids Res. 2012;40:6016-25 doi:10.1093/nar/gks259
158. Endres M, Biniszkiewicz D, Sobol RW, Harms C, Ahmadi M, Lipski A. et al. Increased postischemic brain injury in mice deficient in uracil-DNA glycosylase. J Clin Invest. 2004;113:1711-21 doi:10.1172/JCI20926
159. Nilsen H, Rosewell I, Robins P, Skjelbred CF, Andersen S, Slupphaug G. et al. Uracil-DNA glycosylase (UNG)-deficient mice reveal a primary role of the enzyme during DNA replication. Mol Cell. 2000;5:1059-65
160. Vartanian V, Lowell B, Minko IG, Wood TG, Ceci JD, George S. et al. The metabolic syndrome resulting from a knockout of the NEIL1 DNA glycosylase. Proc Natl Acad Sci U S A. 2006;103:1864-9 doi:10.1073/pnas.0507444103
161. Takao M, Kanno S, Shiromoto T, Hasegawa R, Ide H, Ikeda S. et al. Novel nuclear and mitochondrial glycosylases revealed by disruption of the mouse Nth1 gene encoding an endonuclease III homolog for repair of thymine glycols. EMBO J. 2002;21:3486-93 doi:10.1093/emboj/cdf350
162. Deng CX. BRCA1: cell cycle checkpoint, genetic instability, DNA damage response and cancer evolution. Nucleic Acids Res. 2006;34:1416-26 doi:10.1093/nar/gkl010
163. Shukla V, Coumoul X, Lahusen T, Wang RH, Xu X, Vassilopoulos A. et al. BRCA1 affects global DNA methylation through regulation of DNMT1. Cell Res. 2010;20:1201-15 doi:10.1038/cr.2010.128
164. Ruzov A, Shorning B, Mortusewicz O, Dunican DS, Leonhardt H, Meehan RR. MBD4 and MLH1 are required for apoptotic induction in xDNMT1-depleted embryos. Development. 2009;136:2277-86 doi:10.1242/dev.032227
165. Laget S, Miotto B, Chin HG, Esteve PO, Roberts RJ, Pradhan S. et al. MBD4 cooperates with DNMT1 to mediate methyl-DNA repression and protects mammalian cells from oxidative stress. Epigenetics. 2014;9:546-56 doi:10.4161/epi.27695
166. Meng HX, Hackett JA, Nestor C, Dunican DS, Madej M, Reddington JP. et al. Apoptosis and DNA Methylation. Cancers (Basel). 2011;3:1798-820 doi:10.3390/cancers3021798
167. Felle M, Joppien S, Nemeth A, Diermeier S, Thalhammer V, Dobner T. et al. The USP7/Dnmt1 complex stimulates the DNA methylation activity of Dnmt1 and regulates the stability of UHRF1. Nucleic Acids Res. 2011;39:8355-65 doi:10.1093/nar/gkr528
168. Ma H, Chen H, Guo X, Wang Z, Sowa ME, Zheng L. et al. M phase phosphorylation of the epigenetic regulator UHRF1 regulates its physical association with the deubiquitylase USP7 and stability. Proc Natl Acad Sci U S A. 2012;109:4828-33 doi:10.1073/pnas.1116349109
169. Meng H, Harrison DJ, Meehan RR. MBD4 Interacts with and Recruits USP7 to heterochromatic foci. J Cell Biochem. 2014 doi:10.1002/jcb.25001
170. Tien AL, Senbanerjee S, Kulkarni A, Mudbhary R, Goudreau B, Ganesan S. et al. UHRF1 depletion causes a G2/M arrest, activation of DNA damage response and apoptosis. Biochem J. 2011;435:175-85 doi:10.1042/BJ20100840
171. Mistry H, Tamblyn L, Butt H, Sisgoreo D, Gracias A, Larin M. et al. UHRF1 is a genome caretaker that facilitates the DNA damage response to gamma-irradiation. Genome Integr. 2010;1:7. doi:10.1186/2041-9414-1-7
172. Yang C, Wang Y, Zhang F, Sun G, Li C, Jing S. et al. Inhibiting UHRF1 expression enhances radiosensitivity in human esophageal squamous cell carcinoma. Mol Biol Rep. 2013;40:5225-35 doi:10.1007/s11033-013-2559-6
173. Whitelaw NC, Chong S, Morgan DK, Nestor C, Bruxner TJ, Ashe A. et al. Reduced levels of two modifiers of epigenetic gene silencing, Dnmt3a and Trim28, cause increased phenotypic noise. Genome Biol. 2010;11:R111. doi:10.1186/gb-2010-11-11-r111
174. Cedar H, Bergman Y. Programming of DNA methylation patterns. Annu Rev Biochem. 2012;81:97-117 doi:10.1146/annurev-biochem-052610-091920
175. Buschhausen G, Wittig B, Graessmann M, Graessmann A. Chromatin structure is required to block transcription of the methylated herpes simplex virus thymidine kinase gene. Proc Natl Acad Sci U S A. 1987;84:1177-81
176. Keshet I, Lieman-Hurwitz J, Cedar H. DNA methylation affects the formation of active chromatin. Cell. 1986;44:535-43
177. Razin A, Cedar H. Distribution of 5-methylcytosine in chromatin. Proc Natl Acad Sci U S A. 1977;74:2725-8
178. Solage A, Cedar H. Organization of 5-methylcytosine in chromosomal DNA. Biochemistry. 1978;17:2934-8
179. Chodavarapu RK, Feng S, Bernatavichute YV, Chen PY, Stroud H, Yu Y. et al. Relationship between nucleosome positioning and DNA methylation. Nature. 2010;466:388-92 doi:10.1038/nature09147
180. Bell AC, Felsenfeld G. Methylation of a CTCF-dependent boundary controls imprinted expression of the Igf2 gene. Nature. 2000;405:482-5 doi:10.1038/35013100
181. Jin SG, Kadam S, Pfeifer GP. Examination of the specificity of DNA methylation profiling techniques towards 5-methylcytosine and 5-hydroxymethylcytosine. Nucleic Acids Res. 2010;38:e125. doi:10.1093/nar/gkq223
182. Thomson JP, Skene PJ, Selfridge J, Clouaire T, Guy J, Webb S. et al. CpG islands influence chromatin structure via the CpG-binding protein Cfp1. Nature. 2010;464:1082-6 doi:10.1038/nature08924
183. Barnes DE, Lindahl T. Repair and genetic consequences of endogenous DNA base damage in mammalian cells. Annu Rev Genet. 2004;38:445-76 doi:10.1146/annurev.genet.38.072902.092448
184. Jimeno S, Rondon AG, Luna R, Aguilera A. The yeast THO complex and mRNA export factors link RNA metabolism with transcription and genome instability. EMBO J. 2002;21:3526-35 doi:10.1093/emboj/cdf335
185. Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol. 2010;11:208-19 doi:10.1038/nrm2852
186. Knott SR, Viggiani CJ, Aparicio OM. To promote and protect: coordinating DNA replication and transcription for genome stability. Epigenetics. 2009;4:362-5
187. Aguilera A, Garcia-Muse T. R loops: from transcription byproducts to threats to genome stability. Mol Cell. 2012;46:115-24 doi:10.1016/j.molcel.2012.04.009
188. Baylin SB, Jones PA. A decade of exploring the cancer epigenome - biological and translational implications. Nat Rev Cancer. 2011;11:726-34 doi:10.1038/nrc3130
189. Hitchins MP, Rapkins RW, Kwok CT, Srivastava S, Wong JJ, Khachigian LM. et al. Dominantly inherited constitutional epigenetic silencing of MLH1 in a cancer-affected family is linked to a single nucleotide variant within the 5'UTR. Cancer Cell. 2011;20:200-13 doi:10.1016/j.ccr.2011.07.003
190. Hinoue T, Weisenberger DJ, Lange CP, Shen H, Byun HM, Van Den Berg D. et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2012;22:271-82 doi:10.1101/gr.117523.110
191. Baylin SB, Ohm JE. Epigenetic gene silencing in cancer - a mechanism for early oncogenic pathway addiction? Nat Rev Cancer. 2006;6:107-16 doi:10.1038/nrc1799
192. Markl ID, Cheng J, Liang G, Shibata D, Laird PW, Jones PA. Global and gene-specific epigenetic patterns in human bladder cancer genomes are relatively stable in vivo and in vitro over time. Cancer Res. 2001;61:5875-84
193. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484-92 doi:10.1038/nrg3230
194. Fraga MF, Ballestar E, Montoya G, Taysavang P, Wade PA, Esteller M. The affinity of different MBD proteins for a specific methylated locus depends on their intrinsic binding properties. Nucleic Acids Res. 2003;31:1765-74
195. Ballestar E, Wolffe AP. Methyl-CpG-binding proteins. Targeting specific gene repression. European journal of biochemistry / FEBS. 2001;268:1-6
196. Wolf SF, Jolly DJ, Lunnen KD, Friedmann T, Migeon BR. Methylation of the hypoxanthine phosphoribosyltransferase locus on the human X chromosome: implications for X-chromosome inactivation. Proc Natl Acad Sci U S A. 1984;81:2806-10
197. Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science. 2007;315:1141-3 doi:10.1126/science.1136352
198. Yoder JA, Walsh CP, Bestor TH. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997;13:335-40
199. Shukla S, Kavak E, Gregory M, Imashimizu M, Shutinoski B, Kashlev M. et al. CTCF-promoted RNA polymerase II pausing links DNA methylation to splicing. Nature. 2011;479:74-9 doi:10.1038/nature10442
200. Ficz G, Branco MR, Seisenberger S, Santos F, Krueger F, Hore TA. et al. Dynamic regulation of 5-hydroxymethylcytosine in mouse ES cells and during differentiation. Nature. 2011;473:398-402 doi:10.1038/nature10008
201. Xu Y, Wu F, Tan L, Kong L, Xiong L, Deng J. et al. Genome-wide regulation of 5hmC, 5mC, and gene expression by Tet1 hydroxylase in mouse embryonic stem cells. Mol Cell. 2011;42:451-64 doi:10.1016/j.molcel.2011.04.005
202. Wu H, D'Alessio AC, Ito S, Wang Z, Cui K, Zhao K. et al. Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. 2011;25:679-84 doi:10.1101/gad.2036011
203. Pastor WA, Pape UJ, Huang Y, Henderson HR, Lister R, Ko M. et al. Genome-wide mapping of 5-hydroxymethylcytosine in embryonic stem cells. Nature. 2011;473:394-7 doi:10.1038/nature10102
204. Cooper DN, Youssoufian H. The CpG dinucleotide and human genetic disease. Hum Genet. 1988;78:151-5
205. Rideout WM 3rd, Coetzee GA, Olumi AF, Jones PA. 5-Methylcytosine as an endogenous mutagen in the human LDL receptor and p53 genes. Science. 1990;249:1288-90
206. Duncan BK, Miller JH. Mutagenic deamination of cytosine residues in DNA. Nature. 1980;287:560-1
207. Campbell CD, Chong JX, Malig M, Ko A, Dumont BL, Han L. et al. Estimating the human mutation rate using autozygosity in a founder population. Nat Genet. 2012;44:1277-81 doi:10.1038/ng.2418
208. Campbell CD, Eichler EE. Properties and rates of germline mutations in humans. Trends Genet. 2013;29:575-84 doi:10.1016/j.tig.2013.04.005
209. Kondrashov AS. Direct estimates of human per nucleotide mutation rates at 20 loci causing Mendelian diseases. Hum Mutat. 2003;21:12-27 doi:10.1002/humu.10147
210. Kong A, Frigge ML, Masson G, Besenbacher S, Sulem P, Magnusson G. et al. Rate of de novo mutations and the importance of father's age to disease risk. Nature. 2012;488:471-5 doi:10.1038/nature11396
211. Lynch M. Rate, molecular spectrum, and consequences of human mutation. Proc Natl Acad Sci U S A. 2010;107:961-8 doi:10.1073/pnas.0912629107
212. Sousa MM, Krokan HE, Slupphaug G. DNA-uracil and human pathology. Mol Aspects Med. 2007;28:276-306 doi:10.1016/j.mam.2007.04.006
213. De Bont R, van Larebeke N. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis. 2004;19:169-85
214. Kleihues P, Schauble B, zur Hausen A, Esteve J, Ohgaki H. Tumors associated with p53 germline mutations: a synopsis of 91 families. Am J Pathol. 1997;150:1-13
215. Caputo S, Benboudjema L, Sinilnikova O, Rouleau E, Beroud C, Lidereau R. et al. Description and analysis of genetic variants in French hereditary breast and ovarian cancer families recorded in the UMD-BRCA1/BRCA2 databases. Nucleic Acids Res. 2012;40:D992-1002 doi:10.1093/nar/gkr1160
Author contact
![]() Corresponding authors: E-mail: menghuancmu.edu.cn (H.M.) or caoliucmu.edu.cn (L.C.); Tel.: +86-24-232-56666; Fax: +86-24-232-64417
Corresponding authors: E-mail: menghuancmu.edu.cn (H.M.) or caoliucmu.edu.cn (L.C.); Tel.: +86-24-232-56666; Fax: +86-24-232-64417