Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(7):794-802. doi:10.7150/ijbs.12248 This issue Cite
Research Paper
Smooth Muscle Hgs Deficiency Leads to Impaired Esophageal Motility
Jicheng Chen1,2,*, Ning Hou2,*, Chong Zhang1,2, Yan Teng2, Xuan Cheng2, Zhenhua Li2, Jie Ren3, Jian Zeng2, Rui Li2, Wei Wang3, Xiao Yang1,2, ![]() , Yu Lan2,
, Yu Lan2, ![]()
1. Model Organism Division, E-institutes of Shanghai Universities, Shanghai Jiaotong University, Shanghai 200025, China
2. State Key Laboratory of Proteomics, Collaborative Innovation Center for Cardiovascular Disorders, Genetic Laboratory of Development and Disease, Institute of Biotechnology, Beijing 100071, China
3. Department of Cardiovascular Electrophysiology Research, College of Basic Medicine, Capital Medical University, Beijing 100069, China
*These authors contributed equally to this work.
Received 2015-3-26; Accepted 2015-4-13; Published 2015-5-22
Abstract

As a master component of endosomal sorting complex required for transport proteins, hepatocyte growth factor-regulated tyrosine kinase substrate (Hgs) participates multiple cellular behaviors. However, the physiological role of Hgs in smooth muscle cells (SMCs) is by far unknown. Here we explored the in vivo function of Hgs in SMCs by using a conditional gene knockout strategy. Hgs deficiency in SMCs uniquely led to a progressive dilatation of esophagus with a remarkable thinning muscle layer. Of note, the mutant esophagus showed a decreased contractile responsiveness to potassium chloride and acetylcholine stimulation. Furthermore, an increase in the inhibitory neurites along with an intense infiltration of T lymphocytes in the mucosa and muscle layer were observed. Consistently, Hgs deficiency in SMCs resulted in a disturbed expression of a set of genes involved in neurotrophin and inflammation, suggesting that defective SMC might be a novel source for excessive production of cytokines and chemokines which may trigger the neuronal dysplasia and ultimately contribute to the compromised esophageal motility. The data suggest potential implications in the pathogenesis of related diseases such as gastroesophageal reflux disease.
Keywords: hepatocyte growth factor-regulated tyrosine kinase substrate, smooth muscle cell, esophageal motility, inhibitory neurite, inflammation
Introduction
Transport of food through esophagus to stomach is carried out by ordered esophageal peristalsis. Esophagus is composed of striated and smooth muscle cells (SMCs) [1]. As a major functional component in esophageal motility, SMCs play critical executor role in esophageal peristalsis, especially in the relaxation and contraction of lower esophageal sphincter (LES). Smooth muscle abnormality is often observed in many gastrointestinal tract dysmotility diseases. The myogenic weakness is thought to be a critical cause of hypotensive peristalsis and hypotensive LES in scleroderma [2]. In addition, significantly decreased numbers of SMCs are observed in LES muscle samples taken from patients with gastroesophageal reflux disease (GERD) [3]. Up-to-now, few literature has investigated the specific role of SMCs in gastrointestinal tract motility. Cdo knockout mice exhibit megaesophagus with mispatterned musculature and abnormal motility due to the disturbed transdifferentiation of smooth muscle to skeletal muscle [4]. However, the exact function of SMCs in the regulation of gastrointestinal tract motility needs to be further elucidated.
The contraction of smooth muscle in esophagus is controlled by myenteric plexus neurons which consist of inhibitory vasoactive intestinal polypeptide (VIP) and neuronal nitric oxide synthase (nNOS) and excitatory (acetylcholine releasing) neurons [5]. In previous reports, deficiency of Lsc/p115 or Sprouty2 both leads to hypercontraction of esophagus, but the former is caused by neuronal hypoplasia and the latter is caused by neuronal hyperplasia [6,7]. Mice with nNOS gene disruption have achalasia-like LES dysfunction [8]. These reports suggest the imbalance of inhibitory and the excitatory nerves as an important cause in esophageal motility disorders, but the mechanisms regulating the balance of myenteric plexus neurons are still largely unknown.
Pro-inflammatory factors are thought to be critical mediators in esophageal motility disorders. In GERD patient specimens, the expression level of several inflammation mediators is increased in mucosa and muscle layers [9-11]. The inflammatory factors affect esophageal motility through multiple ways. Except for inducing chemotaxis of leukocyte, they also directly impair the SMCs contractility [12,13]. In addition, the inflammation factors also mediate the interaction between SMC and myenteric plexus neurons in esophagus by inducing glial cell line-derived neurotrophic factor (GDNF) and nerve growth factor (NGF) expression in SMCs [14]. GDNF and NGF are important neurotrophic factors for the enteric nervous system [15-17]. Whether immune reaction could be directly triggered by SMCs is largely unknown.
Hepatocyte growth factor-regulated tyrosine kinase substrate (Hgs) participates protein sorting and recycling and affects multiple cellular functions through regulating various signaling pathways [18]. Physiologically, Hgs is required in the peripheral development and function of B lymphocytes [19]. It also regulates antigen-presentation activity of dendritic cells through exosome secretion [20]. In addition, Hgs deficiency in the neurons leads to increased apoptosis and a loss of hippocampal CA3 pyramidal neurons proteinaceous aggregates [21]. It has been reported that Hgs sustains tumor growth and metastasis by regulating degradation of E-cadherin [22]. GEF-1, a rat homolog of Hgs, could induce myogenesis in C3H10T1/2 mouse fibroblasts [23]. Nevertheless, the role of Hgs in SMCs and esophageal motility has not been explored.
In this study, using Cre-LoxP system, we generated a mutant mouse model in which Hgs was specifically knocked out in SMCs. Hgs deficiency leads to severely attenuated esophageal motility, which may result from a multifactorial mechanism between SMCs, myenteric plexus neurons and inflammatory mediators. Our results have important implications in the pathogenesis of esophageal hypomotility diseases.
Materials and methods
Mice
To construct Hgs-floxed allele, a 8.5 kb genomic fragment, comprising the LoxP-floxed exon 5, was cloned into the Pfrt1 vector containing a FRT-floxed neomycin resistance cassette and a TK cassette. The neomycin cassette was inserted 1128 bp upstream of exon 5, and the other LoxP was inserted 588 bp downstream of exon 5. NotI-linearized targeting vector was electroporated into TC1 mouse embryonic stem (ES) cells [24]. The chimeric mice were generated by microinjection of targeted ES cells into C57BL/6J blastocysts. α-SMA-Cre transgenic mice have been reported previously [25]. ROSA-EYFP reporter mice were purchased from The Jackson Laboratory. All experiments were carried out in accordance with the Guide for the Care and Use of Laboratory Animals published by the United States National Institutes of Health after securing the approval of the Committee of Animal Care of the Beijing Institute of Biotechnology.
Histology and immunohistology
Mice esophagus were harvested by section, rinsed with ice-cold phosphate-buffered saline (PBS), fixed in 4% paraformaldehyde overnight, paraffin-embedded, sectioned, and stained with hematoxylin and eosin (H&E) and masson trichrome following standard protocols. Immunohistochemistry was performed on 5 μm paraffin sections using standard procedures. Primary antibodies were used as follows: anti-α-SMA (Sigma-Aldrich), anti-GFP (Cell Signaling), anti-PGP9.5 (Abgent), anti-E-cadherin (Cell Signaling), anti-SM22α (Abcam), anti-Ki-67 (Abcam), anti-CD3 (Dako), anti-CD117 (eBioscience), For double immunohistochemistry staining, Klear Mouse Ap GBI-permanent Red Kit (GBI Labs) and HighDef blue IHC chromogen (Enzo) double staining were used according to the instructions of the manufacturer. Apoptosis was monitored using a terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling (TUNEL) Apoptosis Detection Kit (Millipore) according to the instructions of the manufacturer.
Quantitative real-time PCR
RNA was extracted from esophagus of control and α-SMA-Cre;Hgsfl/fl mice or primary esophageal SMCs isolated from control and α-SMA-Cre;Hgsfl/fl mice by Trizol (Invitrogen) according to the manufacturer's instructions. Reactions were run with the 7500 Real-Time PCR System (Applied Biosystems). The primers used for the quantitative polymerase chain reactions are listed in Supplementary Table 1. PCR reaction in triplicate using SYBR Green PCR Master Mix (Toyobo), relative expression levels were calculated using the ΔΔCt method, with normalization to the level of Hprt1 mRNA in the same samples.
Whole-mount immunostaining
Whole-mount immunostaining was done as described [6]. Briefly, anesthetized 1-month-old animals were sacrificed, and dissected esophagus were fixed by soaking in cold acetone at -20°C for 30 minutes. After washing in PBS for 15 minutes and then in PBS containing 0.3% Triton X-100 (PBST) for 30 minutes, the muscularis was exfoliated from the mucosa with sharp tweezers under a dissecting microscope as described. After blocking with 1% skim milk in PBST overnight at 4°C, specimens were incubated with anti-PGP9.5 (1:1000, Cell signaling) in the blocking buffer for 48 hours at 4°C. After 4 washes in PBST for 30 minutes each, the tissues were incubated with DyLight 594-conjugated goat anti-rabbit IgG (1:200 dilution) for 3 hours at room temperature. Finally, after washing in PBST overnight at 4°C for 30 min, the specimens were mounted under cover slips with PermaFluor.
Cell culture and treatment
Primary esophageal SMCs were isolated from the esophagus of control and α-SMA-Cre;Hgsfl/fl by collagenase II digestion. In briefly, the mice were anesthetized, the esophagus were dissected and the mucosa were removed. Then the strips were cut into pieces and digested with 1.4 mg/ml collagenase solution at 37°C for 1 hour. After digestion, the cells were collected, suspended with 20% DMEM and plated in 12-well plate. The cells used in the experiments were from passage 3 to 5.
Measurement of contraction force of the esophageal strips
The mice were sacrificed with ether and the whole esophageal strips were isolated from mice. Mucosa and submucosa were removed under microscope and circular and longitudinal muscle layer were left. The LES strips were cut for about 3mm near stomach to determine the circular muscle contraction responsive to acetylcholine (Ach, Sigma-Aldrich) and KCL stimulating. The rest of esophageal body strips were used to measure the responsive of longitudinal muscle. Each strip was mounted vertically in a quartz organ bath. One end of each strip was connected to a fixed hook, and the other end was connected to a strain gauge (TB-612-T; Nihon Koden, Japan). Recording of the force development in response to 10 μM ACh and 60 mM KCL and data analysis was carried out as described [6].
Western blot
The proteins were collected from the esophagus of control or α-SMA-Cre;Hgsfl/fl mice using Ripa lysis. To determine the phosphorylation status of MLC20, 1-month-old control and α-SMA-Cre;Hgsfl/fl mice whole esophageal strips were isolated and placed in 10 μM ACh Kreb's buffer at 37°C for 10 minutes before protein collection. 20 micrograms of proteins were electrophoresed on 10% SDS-PAGE and transferred onto polyvinylidene difluoride membranes. Immunoblotting was performed according to the instructions of the manufacturer using the following antibodies: anti-Hgs (Enzo), anti-MLC20 (Abcam), anti-p-MLC20 (Abcam), anti-Calponin, anti-SM α-actin, anti-SM22a, anti-PGP9.5, and anti-GAPDH (ZSBIO).
Statistical Analysis
The data are presented as mean values ± SD. The Student t-test was used for comparisons between 2 groups. P < 0.05 was considered statistically significant.
Results
Deletion of Hgs in SMCs resulted in progressive esophageal dilatation with reduced thickness of the muscle layer
We generated a Hgs-floxed mouse line with LoxP sites flanking exon 5 of the mouse Hgs locus (Supplementary Fig. 1). To delete Hgs gene specifically in SMCs, we used the α-SMA-Cre transgenic mice we developed previously [25]. We firstly determined the localization of the α-SMA-Cre-mediated ablation in the esophagus using the ROSA-EYFP reporter mice. Double immunostaining showed the co-localization of EYFP and SMC marker α-SMA in the muscularis mucosa and partially in muscle layer, while the EYFP expression was not detected in the PGP9.5-positive neurons (Fig. 1A). Western blot showed the significant decrease of Hgs in the esophagus of α-SMA-Cre;Hgsfl/fl mice (Fig. 1B), confirming the specific knockout of Hgs in esophageal SMCs.
Deletion of Hgs in SMCs led to progressive esophageal dilatation with reduced thickness of the muscle layer. (A) Double immunohistochemistry staining of the esophagus from 1-month-old α-SMA-Cre;ROSA-EYFP (Cre;ROSA-EYFP) mice. Purple arrows indicate α-SMA and EYFP double positive signals. Red arrow indicates EYFP single positive staining. Blue arrow indicates neuron cell marker PGP9.5 single positive staining. mc, mucous; mm, muscularis mucosa; ml, muscle layer. (B) Western blot analysis of Hgs expression in the esophagus from 1-month-old α-SMA-Cre;Hgsfl/+ (Cre;fl/+) control and α-SMA-Cre;Hgsfl/fl (Cre;fl/fl) mutant mice. (C) Representative gross view of the esophagus from 1- (upper) and 3-month-old (lower) mice. (D) H&E staining of the esophagus from 1- (upper) and 3-month-old (lower) mice. A dilation of esophagus was observed in 3-month-old mutant mice. (E) SM22α and α-SMA immunohistochemistry staining of the esophagus from 1-month-old mice. (F) Western blot analysis of the smooth muscle markers, SM22α, α-SMA and CNN1, in esophagus from 3-month-old mice. Scale bars: 200 μm.
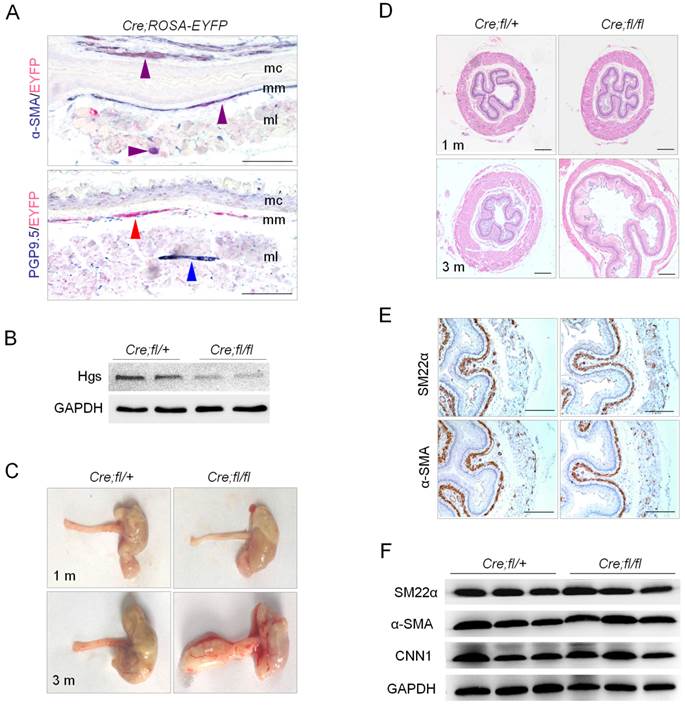
The esophagus of α-SMA-Cre;Hgsfl/fl mutant mice was normal in appearance within 1 month after birth compared to control mice (Fig. 1C). A progressive dilation of esophagus was observed in the mutants as early as 3 months with 100% penetrance (Fig. 1C). The mutant mice showed weight loss, and died within 5 months after birth. By using H&E staining, we did not found any significant structural abnormalities both in esophageal body and in LES of mutant mice at 1 month of age. At 3 months of age, the mutant esophagus showed a dilated lumen and a remarkably reduced thickness of the muscularis (Fig. 1D). Masson-Goldner trichrome staining confirmed the reduced thickness of muscularis layer with obviously increased collagenous fibrosis in the mutant esophagus (Supplementary Fig. 2). The reduced muscularis in mutant mice throughout esophageal body and LES was also shown by Masson-Goldner trichrome staining on longitudinal sections (Supplementary Fig. 2). Interestingly, the proliferation and apoptosis, revealed by Ki-67 immunostaining and TUNEL staining, respectively, both showed apparently increased in the muscularis layer of the mutant esophagus as compared to that of the littermate controls (Supplementary Fig. 3). The results suggested a disturbed balance of cell cycle and survival status in the muscularis layer, which might partially explain the esophageal thinning and dilation at later stage in the mutants. The structure of esophageal mucosa, evidenced by E-cadherin immunostaining, and that of submucosa appeared normal in the mutants (Supplementary Fig. 4). We did not find obvious morphological abnormality in the intestinal tract of the mutant mice at 3 months (Supplementary Fig. 5).
We next determined whether Hgs deficiency affects SMC differentiation. Immunohistochemical staining showed the comparable localization and expression of smooth muscle markers SM22a and α-SMA in the esophagus of 1-month-old mutant and control mice (Fig. 1E). Western blot confirmed comparable expression levels of SM22a, α-SMA and Calponin-1 between mutant mice and littermate controls at the age of 3 months (Fig. 1F). The data indicated that the differentiation of SMCs was normal in the absence of Hgs.
Hgs mutant mice showed reduced esophageal motility
SMC is a critical functional component in esophageal motility, and circular and longitudinal muscle layer is the master motor in esophageal peristalsis. We then investigated whether esophageal motility function was changed in Hgs deficient mice. Contractility measurement of isolated esophageal muscle strips was performed in vitro at 1 month of age, when no obvious histological abnormality was observed. The nerve-independent and nerve-dependent contraction, induced by KCL and Ach, respectively, was measured. The esophageal body but not LES strips from the mutant mice manifested decreased contractile response to KCL, as compared to those from the wild-type mice (Fig. 2, A and B). Notably, both esophageal body and LES strips from the mutant mice showed significantly reduced contractile response to ACh, with former exhibiting a dramatic 70% reduction (Fig. 2, A and B).
Hgs deficiency in SMCs resulted in reduced esophageal motility. (A) Graphs showing the representative measurement records of the contractility of the isolated esophageal strips. Esophageal body (EB) and LES were isolated from 1-month-old α-SMA-Cre;Hgsfl/+ (Cre;fl/+) and α-SMA-Cre;Hgsfl/fl (Cre;fl/fl) mice. The response of strips to 60 mM KCL or 10 μM ACh were recorded. (B) Quantification results of the esophageal contractility measurement. n = 6. Data are means ± SD. **P < 0.001; *P < 0.05. (C) Western blot analysis showing decreased p-MLC20 expression in the esophagus from 1-month-old mice upon ACh stimulation.
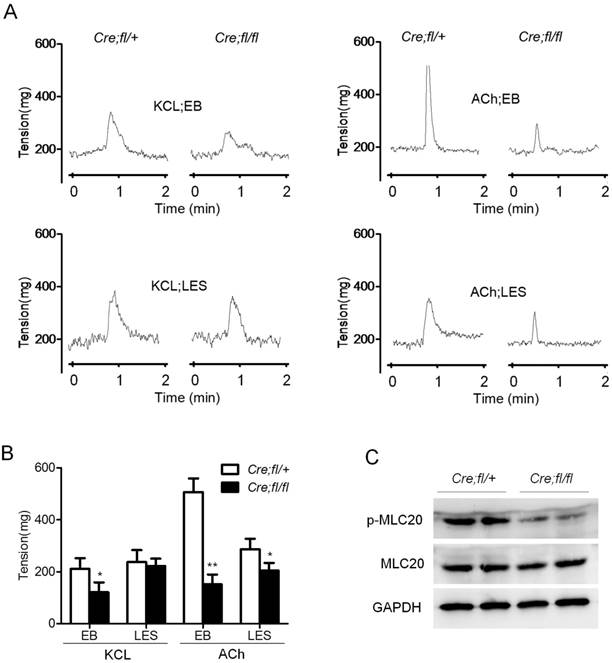
As a critical component in SMCs contraction, MLC20 phosphorylation is always used to reflect the status of contractility. So we determined the protein levels of p-MLC20 after ACh stimulation for 10 minutes in wild-type and mutant esophagus. Comparable total MLC20 but an obviously decreased p-MLC20 level was observed in the mutant esophageal after ACh stimulation (Fig. 2C). This result was consistent with the markedly decreased contractility of isolated muscle strips from mutant mice. Together, these data indicated that Hgs deficiency in SMCs led to the decreased contraction of esophagus, which happened as early as 1 month of age.
Increased inhibitory neurons in the esophagus of Hgs mutant mice
The contraction of esophageal SMCs is controlled by myenteric plexus neurons. The remarkably decreased nerve-dependent contraction of the mutant esophageal strip suggested a potential defect in the neurons. So we explored whether myenteric plexus neurons were influenced in the mutant mice. We firstly performed whole-mount staining and observed an obviously increased density of PGP9.5-positive neurons in the mutant esophagus (Fig. 3A). The data were further confirmed by western blot analysis of esophageal proteins (Fig. 3B). Quantitative analysis showed an about 2-fold increase in the expression of PGP9.5 in the esophagus of mutant mice (Fig. 3C). Real-time PCR analysis showed the increased expression of acetylcholinesterase (ACHE) and vasoactive intestinal polypeptide (VIP), markers of inhibitory neurons (Fig. 3D). Whereas the inhibitory neuron markers nNOS and the stimulatory neuron marker CHAT showed no change as compared to the controls (Fig. 3D). All these data revealed that loss of Hgs in SMCs led to an unexpected increase in the inhibitory neurons in esophagus.
Increased inhibitory neurons in the esophagus of Hgs mutant mice. (A) Whole-mount immunostaining of PGP9.5 in esophagus from 1-month-old α-SMA-Cre;Hgsfl/ (Cre;fl/+) control and α-SMA-Cre;Hgsfl/fl (Cre;fl/fl) mutant mice. (B) Western blot analysis of PGP9.5 in the esophagus from 1-month-old mice. (C) Quantification analysis of the western blot data showing increased expression of PGP9.5 in mutant esophagus. n = 3. Data are means ± SD. (D) Real-time PCR analysis of the gene expression in the esophagus from 1-month-old mice (n = 3). (E) Real-time PCR analysis of the gene expression in the primary mouse esophageal SMCs (n = 3). Data are means ± SD. **P < 0.001; *P < 0.05. Scale bars: 200 μm.
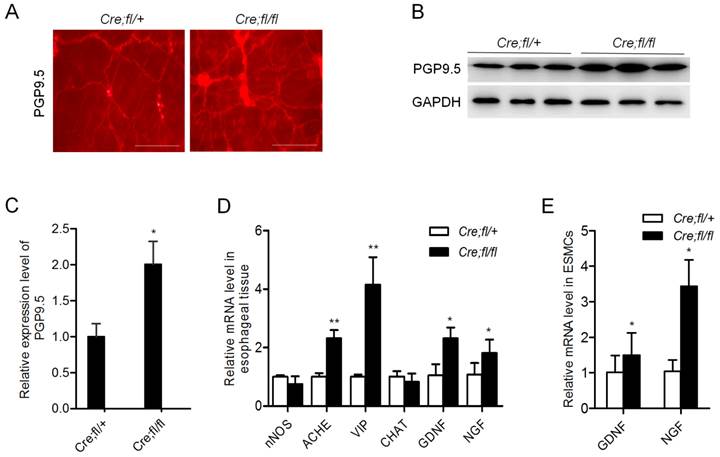
Of note, neurotrophic factors GDNF and NGF were also evidently increased in the mutant esophagus (Fig. 3D). Consistently, the increased expression of GDNF and NGF was also demonstrated in the esophageal SMCs from mutant mice (Fig. 3E). This result suggested a SMC derivation of the excessive secretion of neurotrophic factors, which might contribute to the increased neurons in the mutant esophagus.
Increased cytokine/chemokine expression and T lymphocyte infiltration in the esophagus of Hgs mutant mice
Pro-inflammation cytokines and chemokines have been shown to play roles in esophageal motility disorders by regulating neuron growth and extension [26,27]. We next checked whether the mRNA expression of pro-inflammation cytokines and chemokines was altered in the esophagus of 1-month-old mutant mice. We found the expression of cytokines IL-1β and TNF-α was increased 1.9-fold and 4.0-fold, respectively, as compared to the controls (Fig. 4A). The T cell chemokines Cxcl9, Cxcl10 and Cxcl16 all showed an obvious increase in the mutant esophagus (Fig. 4A). To find out the initial immune events and largely exclude the influence of infiltrated inflammation cells in esophagus, we isolated primary mouse esophageal SMCs with at least 95% purity, and further verified the up-regulation of the cytokines and chemokines in the Hgs-deficient SMCs (Fig. 4B).
Increased inflammatory factors expression and T lymphocyte infiltration in the esophagus of Hgs mutant mice. (A) Real-time PCR analysis of the cytokines and chemokines in the esophagus from 1-month-old α-SMA-Cre;Hgsfl/+ (Cre;fl/+) control and α-SMA-Cre;Hgsfl/fl (Cre;fl/fl) mutant mice. n = 5. (B) Real-time PCR analysis of the gene expression in the primary mouse esophageal SMCs. n = 3. (C) Immunohistochemical staining of mast cell marker (CD117) and T-cell marker (CD3) in esophagus from 3-month-old mice. (D) Immunohistochemical staining of CD3 on the longitudinal sections of esophagus from 3-month-old mice. Data are means ± SD. **P < 0.001; *P < 0.05. Scale bars: 200 μm.
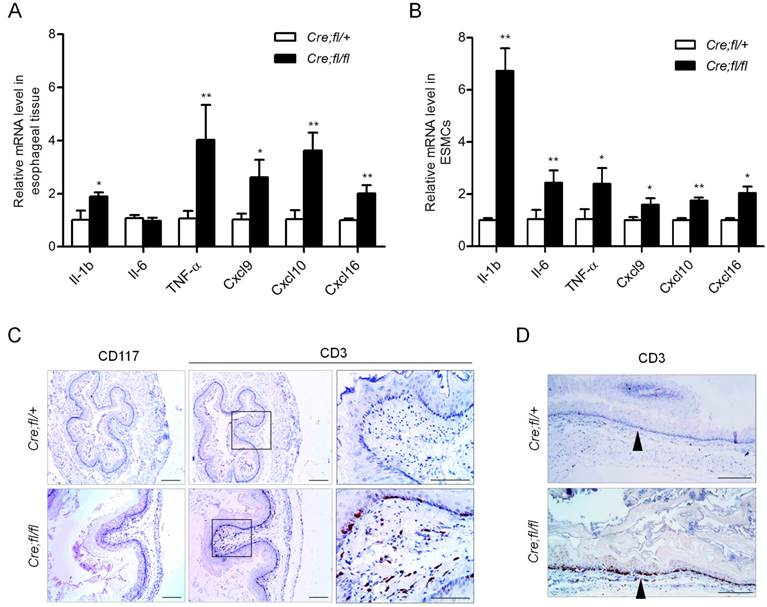
The up-regulated chemokines in the Hgs-deficient SMCs might induce chemotaxis of leukocytes. Therefore, we explored whether inflammatory cell infiltration occurred in the Hgs mutant esophagus. In the esophagus of 6-week-old mutant mice, CD3-positive T lymphocytes scattered in the muscularis layer but not the mucosal layer (Supplementary Fig. 6). At the age of 3 months, the mutant mice showed intense CD3-positive T lymphocytes localizing along the edges of mucosa (Fig. 4C). The infiltration pattern of T cells throughout the whole esophagus was also confirmed by the immunohistochemistry on the longitudinal sections (Fig. 4D). Such infiltration pattern was largely consistent with that in most esophageal motility disorders.
Discussion
In this study, for the first time we demonstrated a critical role of Hgs in SMCs in maintaining esophageal motility. Our data further suggested that the increase of the inhibitory neurons, presumably resulting from the aberrant neurotrophic factors and cytokines secretion from the Hgs-deficient SMCs, was likely contributing to the hypocontraction of the mutant esophagus.
Specific knockout of Hgs in SMCs uniquely led to a progressive dilatation of esophagus. Clinically, esophagus dilatation is a common performance of the diseases with esophageal dysmotility [6,7,28]. The significantly decreased esophageal motility in the Hgs mutant mice happened as early as 1 month of age, when no obvious structural or differentiation abnormality was detected in the muscularis. The compromised esophageal contraction force of esophageal body and LES in the mutant mice highly resembled that in GERD and systemic sclerosis patients [29]. This provides us a good genetic mouse model to explore the detailed mechanisms underlying the esophageal motility hypotensive.
Myenteric plexus neurons disturbance is acknowledged as an important cause of the esophageal motility diseases, such as achalasia, GERD and systemic sclerosis. So we explored whether myenteric plexus neurons were disturbed in our mouse model, although the signaling pathway was intact within the neurons. Surprisingly, significantly increased inhibitory neurons in the esophageal muscle layer were observed as early as 1 month of age, when the structure and SMC differentiation of the mutant esophagus appeared quite normal. In previous reports, the inhibitory neurons are critical for the contraction of the esophageal muscularis [8]. So the data at least partially explain the decreased motility in Hgs mutants. Our results further suggest Hgs-deficient SMCs as an important derivation of the excessive production of neurotrophic factors, which are pivotal for maintaining neuron survive and neurite growth. Whether and how the increased neurotrophic factors selectively induce the growth of the inhibitory neurons in the Hgs mutants needs further investigations.
Up-regulated pro-inflammatory factors in esophagus is common in esophageal motility disorders and is thought to be a critical cause of the disturbance of myenteric plexus neurons [30,31]. But all experimental esophagitis models focus on esophageal epithelium cell-mediated inflammation reaction. In our study, we revealed Hgs-deficient SMCs as a previously unknown derivation of the immune reaction within esophagus. We demonstrated that Hgs deficiency in SMCs led to a significantly increased expression of pro-inflammation cytokines and chemokines, which might trigger the inflammatory reaction within the mutant esophagus. We proposed that the excessive production of the cytokines and chemokines from mutant SMCs might contribute to the impaired esophageal motility either by executing direct damage on the contraction ability of SMCs, or through inducing the expression of NGF and GDNF to affect the density of myenteric plexus.
In summary, the esophageal motility disorder we observed in SMC-specific Hgs knockout mice may result from a multifactorial mechanism through interaction between SMCs and myenteric plexus neurons. Our findings suggest potential implications in the pathogenesis of the esophageal motility disorders such as GERD and systemic sclerosis esophagus.
Supplementary Material
Supplementary Table 1, Supplementary Figures 1-6.
Abbreviations
Hgs, hepatocyte growth factor-regulated tyrosine kinase substrate; SMCs, smooth muscle cells; LES, lower esophageal sphincter; GERD, gastroesophageal reflux disease; VIP, vasoactive intestinal polypeptide; nNOS, neuronal nitric oxide synthase; GDNF, glial cell line-derived neurotrophic factor; NGF, nerve growth factor; ACh, acetylcholine.
Acknowledgements
We thank Xizhi Guo for ROSA-EYFP reporter mice. We also thank Jiangkun Jia for technical assistance on the analysis of contraction force measurement.
Financial support
This study was supported by the Chinese National Key Program on Basic Research (2012CB945103 and 2011CB964803), and the National Natural Science Foundation of China (91439128, 31430057, 81370596, and 31171410).
Competing interests
The authors declare no competing financial interests.
References
1. Goyal RK, Chaudhury A. Physiology of normal esophageal motility. J Clin Gastroenterol. 2008;42:610-619
2. Altorjay A, Juhasz A, Kellner V, Sohar G, Fekete M. et al. Metabolic changes in the lower esophageal sphincter influencing the result of anti-reflux surgical interventions in chronic gastroesophageal reflux disease. World J Gastroenterol. 2005;11:1623-1628
3. Tian XP, Zhang X. Gastrointestinal complications of systemic sclerosis. World J Gastroenterol. 2013;19:7062-7068
4. Romer AI, Singh J, Rattan S, Krauss RS. Smooth muscle fascicular reorientation is required for esophageal morphogenesis and dependent on Cdo. J Cell Biol. 2013;201:309-323
5. Neuhuber WL, Raab M, Berthoud HR, Worl J. Innervation of the mammalian esophagus. Adv Anat Embryol Cell Biol. 2006;185:1-73
6. Taketomi T, Yoshiga D, Taniguchi K, Kobayashi T, Nonami A. et al. Loss of mammalian Sprouty2 leads to enteric neuronal hyperplasia and esophageal achalasia. Nat Neurosci. 2005;8:855-857
7. Zizer E, Beilke S, Bauerle T, Schilling K, Mohnle U. et al. Loss of Lsc/p115 protein leads to neuronal hypoplasia in the esophagus and an achalasia-like phenotype in mice. Gastroenterology. 2010;139:1344-1354
8. Sivarao DV, Mashimo HL, Thatte HS, Goyal RK. Lower esophageal sphincter is achalasic in nNOS(-/-) and hypotensive in W/W(v) mutant mice. Gastroenterology. 2001;121:34-42
9. Fitzgerald RC, Onwuegbusi BA, Bajaj-Elliott M, Saeed IT, Burnham WR. et al. Diversity in the oesophageal phenotypic response to gastro-oesophageal reflux: immunological determinants. Gut. 2002;50:451-459
10. Isomoto H, Wang A, Mizuta Y, Akazawa Y, Ohba K. et al. Elevated levels of chemokines in esophageal mucosa of patients with reflux esophagitis. Am J Gastroenterol. 2003;98:551-556
11. Isomoto H, Saenko VA, Kanazawa Y, Nishi Y, Ohtsuru A. et al. Enhanced expression of interleukin-8 and activation of nuclear factor kappa-B in endoscopy-negative gastroesophageal reflux disease. Am J Gastroenterol. 2004; 99:589-597. patients with reflux esophagitis. Am J Gastroenterol. 2003;98:551-556
12. Rieder F, Cheng L, Harnett KM, Chak A, Cooper GS. et al. Gastroesophageal reflux disease-associated esophagitis induces endogenous cytokine production leading to motor abnormalities. Gastroenterology. 2007;132:154-165
13. Tugtepe H, Tugay M, Bozkurt S, Yildiz F, Utkan T. et al. Esophageal smooth muscle reactivity is impaired in chronic reflux esophagitis by both receptor- and nonreceptor-mediated mechanisms. J Pediatr Surg. 2007;42:641-646
14. Gougeon PY, Lourenssen S, Han TY, Nair DG, Ropeleski MJ. et al. The pro-inflammatory cytokines IL-1beta and TNFalpha are neurotrophic for enteric neurons. J Neurosci. 20. 2013;33:3339-3351
15. Anitha M, Gondha C, Sutliff R, Parsadanian A, Mwangi S. et al. GDNF rescues hyperglycemia-induced diabetic enteric neuropathy through activation of the PI3K/Akt pathway. J Clin Invest. 2006;116:344-356
16. Rodrigues DM, Li AY, Nair DG, Blennerhassett MG. Glial cell line-derived neurotrophic factor is a key neurotrophin in the postnatal enteric nervous system. Neurogastroenterol Motil. 2011;23:e44-56
17. Wang H, Hughes I, Planer W, Parsadanian A, Grider JR. et al. The timing and location of glial cell line-derived neurotrophic factor expression determine enteric nervous system structure and function. J Neurosci. 2010;30:1523-1538
18. Raiborg C, Bache KG, Gillooly DJ, Madshus IH, Stang E. et al. Hrs sorts ubiquitinated proteins into clathrin-coated microdomains of early endosomes. Nat Cell Biol. 2002;4:394-398
19. Nagata T, Murata K, Murata R, Sun SL, Saito Y. et al. Hepatocyte growth factor regulated tyrosine kinase substrate in the peripheral development and function of B-cells. Biochem Biophys Res Commun. 2014;443:351-356
20. Tamai K, Tanaka N, Nakano T, Kakazu E, Kondo Y. et al. Exosome secretion of dendritic cells is regulated by Hrs, an ESCRT-0 protein. Biochem Biophys Res Commun. 2010;399:384-390
21. Tamai K, Toyoshima M, Tanaka N, Yamamoto N, Owada Y. et al. Loss of hrs in the central nervous system causes accumulation of ubiquitinated proteins and neurodegeneration. Am J Pathol. 2008;173:1806-1817
22. Toyoshima M, Tanaka N, Aoki J, Tanaka Y, Murata K. et al. Inhibition of tumor growth and metastasis by depletion of vesicular sorting protein Hrs: its regulatory role on E-cadherin and beta-catenin. Cancer Res. 2007;67:5162-5171
23. Ogura K, Niino YS, Tai T. Galactosylceramide expression factor-1 induces myogenesis in MDCK and C3H10T1/2 cells. Arch Biochem Biophys. 2004;426:279-285
24. Qi X, Yang G, Yang L, Lan Y, Weng T. et al. Essential role of Smad4 in maintaining cardiomyocyte proliferation during murine embryonic heart development. Dev Biol. 2007;311:136-146
25. Weinstein M, Yang X, Li C, Xu X, Gotay J. et al. Failure of egg cylinder elongation and mesoderm induction in mouse embryos lacking the tumor suppressor smad2. Proc Natl Acad Sci U S A. 1998;95:9378-9383
26. Carlson NG, Wieggel WA, Chen J, Bacchi A, Rogers SW. et al. Inflammatory cytokines IL-1 alpha, IL-1 beta, IL-6, and TNF-alpha impart neuroprotection to an excitotoxin through distinct pathways. J Immunol. 1999;163:3963-3968
27. Diem R, Hobom M, Grotsch P, Kramer B, Bahr M. Interleukin-1 beta protects neurons via the interleukin-1 (IL-1) receptor-mediated Akt pathway and by IL-1 receptor-independent decrease of transmembrane currents in vivo. Mol Cell Neurosci. 2003;22:487-500
28. Holmes TW Jr. Chalasia, peptic esophagitis, and hiatal hernia. A common syndrome inpatientswith central nervous system disease. Chest. 1971;60:441-445
29. Spechler SJ, Castell DO. Classification of oesophageal motility abnormalities. Gut. 2001;49:145-151
30. Altomare A, Ma J, Guarino MP, Cheng L, Rieder F. et al. Platelet-activating factor and distinct chemokines are elevated in mucosal biopsies of erosive compared with non-erosive reflux disease patients and controls. Neurogastroenterol Motil. 2012;24:943-e463
31. Rieder F, Nonevski I, Ma J, Ouyang Z, West G. et al. T-helper 2 cytokines, transforming growth factor beta1, and eosinophil products induce fibrogenesis and alter muscle motility in patients with eosinophilic esophagitis. Gastroenterology. 2014;146:1266-1277
Author contact
![]() Corresponding authors: Xiao Yang, Ph.D., Tel/Fax: 86-10-63895937; E-mail: yangxac.cn. Yu Lan, Ph.D., Tel: 86-10-66948884; E-mail: rainyblue_1999com
Corresponding authors: Xiao Yang, Ph.D., Tel/Fax: 86-10-63895937; E-mail: yangxac.cn. Yu Lan, Ph.D., Tel: 86-10-66948884; E-mail: rainyblue_1999com