Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(7):833-844. doi:10.7150/ijbs.10564 This issue Cite
Research Paper
ROS/Autophagy/Nrf2 Pathway Mediated Low-Dose Radiation Induced Radio-Resistance in Human Lung Adenocarcinoma A549 Cell
Ni Chen1,2, Lijun Wu1,2, Hang Yuan2, Jun Wang2, ![]()
1. School of Nuclear Science and Technology, University of Science and Technology of China, Hefei 230027, PR China;
2. Key Laboratory of Ion Beam Bioengineering, Hefei Institute of Physical Science, Chinese Academy of Sciences and Anhui Province, No. 350 of Shushanhu Road, Hefei 230031, PR China
Received 2014-9-16; Accepted 2015-5-5; Published 2015-5-30
Abstract

Low-dose ionizing radiation (LDIR) can induce radio-resistance to following high dose radiation in various mammalian cells. The protective role of LDIR has been thought to be associated with the overall outcomes of cancer radiotherapy. NF-E2 related factor 2 (Nrf2) is a transcription factor that plays pivotal roles in maintaining cellular oxidative equilibrium. Since oxidative stress has been indicated to be a mediator of LDIR induced radio-resistance, the role of Nrf2 in this process was investigated in this research. Our results showed that in human lung adenocarcinoma A549 cell, 5cGy alpha particle induced radio-resistance to following 75cGy alpha particle radiation. The expression level of Nrf2 and its target Heme Oxygenase-1(HO-1) increased after 5cGy radiation. Both the shRNA of Nrf2 and the chemical inhibitor of HO-1 suppressed the induced radio-resistance, indicating the involvement of Nrf2 antioxidant pathway in this process. Further, we found 5cGy radiation stimulated autophagy process in A549. Inhibition of the autophagy process resulted in suppression of the radio-resistance and the induced expression of Nrf2 and HO-1. ROS scavenger N-acetyl-L-cysteine (NAC) blocked the autophagy process induced by 5cGy alpha particle, the upregulation of Nrf2 and HO-1, as well as the induced radio-resistance. In conclusion, ROS elevation caused by LDIR promoted Autophagy/Nrf2-HO-1 and conferred radio-resistance in A549.
Keywords: Low dose ionizing radiation, radio-resistance, Nrf2, Autophagy, ROS
Introduction
More than 50% of cancer patients receive radiotherapy at some time during the course of their disease. Recent technical developments in diagnostic imaging such as CT scanning have significantly increased the precision of dose delivery to the target tumor, making radiotherapy more efficient in cancer treatment [1]. However, they also garner raised interests and importance in the biological effects of low-dose ionizing radiation (LDIR) [2]. Especially, more and more studies have shown that exposure of cells to LDIR induces an elevated resistance when subsequently exposed to a much higher dose of radiation, which may be linked to increased recurrence and treatment failure in many patients after radiotherapy [3-6]. Even though this phenomenon has been generally accepted in mammalian cells and animals [3, 4, 7, 8], the underlying mechanisms remain to be explored. LDIR induced radio-resistance has been shown to be associated with increased cellular functions such as stress responses and DNA repair capabilities [3]. For example, during the process of LDIR induced radio-resistance, STAT1 was found to be profoundly up-regulated [9], the activation of poly (ADP-ribose) polymerase (PARP) which is well known to participate in DNA repair was required [10], and ATM/ERK/NF-κB pathway was involved [11].
It has been well established that generation of reactive oxygen species (ROS) is a key event involved in ionizing radiation (IR) induced biological effects [12]. ROS is important in regulating both cell survival and cell death. Excessively high levels of ROS can lead to significant DNA damage and various cellular responses, including cell cycle arrest, senescence and apoptosis. However, supra-basal levels of ROS generated in response to LDIR may bring about untoward or even beneficial cellular responses [13]. There is evidence that the generation of ROS confers radio-resistance to directly irradiated cells and the bystander cells [3]. Antioxidant responses participate in LDIR induced protection in cells [14]. For example, Eldridge et al. reported that manganese superoxide dismutase (MnSOD), an important mitochondrial antioxidant in mammalian cells, played a key role in the LDIR-induced radio-resistance [15]. NF-E2 related factor 2 (Nrf2) is a member of the cap 'n collar family of basic leucine zipper transcription factors and can be induced by elevated oxidative stress. By transcriptional regulation of a variety of detoxification enzymes such as heme oxyegenase-1 (HO-1), NAD(P)H: quinone oxidoreductase 1 (NQO1), Nrf2 protects cells against oxidative stress caused damages [16-20]. Nrf2 is aberrantly overexpressed in several types of cancer cell lines or tumor tissues and antioxidant pathways regulated by Nrf2 are thought to play pivotal roles in maintaining favorable redox balance therefore promote cell survival, evade apoptosis and tolerate therapeutic intervention [21, 22]. Accumulated clues show that Nrf2 can be stimulated by IR [23-26]. And pre-activation of Nrf2 by chemical inducer such as sulforaphane and naphthoquinone results in amelioration of IR induced DNA double strand breaks [27, 28]. However, whether Nrf2 mediated antioxidative response participates in LDIR induced radio-resistance has not been elucidated. To this point, we performed research and our results were presented in the present article.
Materials and methods
Materials and chemicals
Human lung carcinoma A549 was kept by this laboratory. Dulbecco's Modified Eagle Medium (DMEM), superoxide indicator Hydroethidine and Amplex® Red Hydrogen Peroxide/Peroxidase Assay Kit were purchased from Life Technology (Shanghai Office, China). Fetal bovine serum (FBS) was purchased from Hyclone (Beijing, China). Earle's Balanced Salts solution was from Sigma (MS, USA). shRNA plasmids and transfection reagent MegaTran1.0 were purchased from OriGene (Shanghai China). Primary antibodies were Nrf2 (Cat. No. sc-722, Santa Cruz, TX, USA), Heme Oxygenase 1 (HO-1, Cat. No. 10701-1, ProteinTech, IL, USA), LC3B (Cat. No. L7543, Sigma, MS, USA), Beta Actin (Cat. No. M20010, Abmart, Shanghai, China), Alpha Tubulin (Cat. No. 2871-1, Epitomics, CA, USA), phospho-ERK 1/2 (Cat. No.4370, Cell Signal Technology, MA, USA), Histone H3 (Cat. No. H9289, Sigma, MS, USA). Both HRP linked goat anti-rabbit and HRP linked goat anti-mouse secondary antibodies were bought from Jackson ImmunoResearch Laboratories (PA, USA). MEK inhibitor U0126 was from Promega (WI, USA). HO-1 chemical inhibitor protoporphyrin IX zinc (II) (ZnPP), autophagy inhibitor 3-Methyladenine (3-MA), E-64d, pepstatin, dansylcadaverine, N-Acetyl-L-cysteine (NAC) and protein phosphotase inhibitor cocktail were from Sigma (MS, USA). Hemoglobin was purchased from Worthington Biochemical Corporation (NJ, USA). Etoposide was from J&K Chemicals (Shanghai, China). Protease inhibitor cocktail was purchased from Roche (Mannheim, Germany). Chemiluminescence substrate Superstar ECL for western blotting was from Boster (Wuhan, China). Protein amount was determined with bicinchoninic acid (BCA) kit which was purchased from Sangon (Shanghai, China). Other chemicals were reagent grade.
Cell culture and radiation
Complete medium for A549 culturing was DMEM with 10% FBS. Cells were incubated at 37℃ in a humidified 5% CO2 incubator. Radiation was performed on an alpha particle emitter which delivers 3.5 MeV α particles derived from 241Am at a dose rate of 1cGy per second. Cells were seeded into specially designed circular dish with a 3.0μm thick mylar film bottom on which the cells attached. Culture medium was refreshed every other day and the cells were irradiated by α particles when reaching 70%~80% confluence. The alpha particle emitter used in this research is assembled by this institute. The dose rate was calculated by putting a CR-39 on the mylar bottomed dish (the same as that used for cell culturing) and counting particle numbers after different exposure time. Then the received particle number was converted to dose and plotted against exposure time to determine dose rate. For radiation of cells, all the region of the dish was exposed for a total time calculated based on desired dose and the dose rate. The actual dose received by cultured cells was mentioned in Results section.
Clonogenic assay
Cells were seeded into mylar-bottomed dish. After alpha particle radiation, cells were cultured for 6 hours. Then the cells were washed carefully with PBS and trypsinized to produce single-cell suspension. 300 cells from each treatment group were plated into 6-cm cell culture dish respectively and incubated for 10 days. After washed with PBS, a mixture of 6% glutaraldehyde and 0.5% crystal violet was added into the dish. Four hours later, the glutaraldehyde crystal violet mixture was removed and the dish was washed with de-ionized water. After dried at room temperature, colonies in each dish were counted to calculate the plating efficiency and colony formation rate.
Protein extraction and western blot analysis
Cells were washed with PBS and then harvested using ice-cold RIPA lysis buffer containing protease inhibitor and protein phosphotase inhibitor. Protein concentration of total cell lysate was measured using BCA reagent and determined on microplate reader at 562nm. 60μg proteins were resolved by 10% or 12% Tris-Glycine SDS-PAGE according to the protocol described by Laemmli [29] and transferred to a PVDF membrane. After blocked in PBST with 1% BSA (bovine serum albumin), PVDF membrane was incubated with primary antibody at 4℃ overnight with gentle agitation. Then the membrane was washed with PBST 3 times at room temperature for 5 minutes and incubated with corresponding HRP-linked secondary antibody for 2 hours at room temperature. After washed with PBST 3 times at room temperature for 5 minutes, the membrane was incubated with chemiluminescence substrate for 3 minutes at 37℃. Protein band intensity was visualized and captured in FCE (Protein Simple) image analyzer.
One-step qRT-PCR
To determine the mRNA levels of Nrf2 in Scr-shRNA A549 and Nrf2-shRNA A549 cells, one-step qRT-PCR analysis was performed. Total RNA was prepared with RNAiso reagent purchased from TaKaRa (Dalian, China). One-step qRT-PCR was done with TaKaRa One Step SYBR® PrimeScriptTM PLUS RT-PCR Kit on StepOne real time PCR machine by ΔΔCt method. mRNA level of actin was used as endogenous control. The primers used for determine the mRNA level of Nrf2 were P1: ATGGATTTGATTGACATACTTT and P2: ACTGAGCCTGATTAGTAGCAAT. The primers used for determine the mRNA level of Actin were P3: CCTGGCACCCAGCACAAT and P4: GGGCCGGACTCGTCATAC. Reaction parameters were step 1: 42℃ for 5minutes, step 2: 95℃ for 10 seconds, step 3: 95℃ for 10 seconds, step 4: 50℃ for 30 seconds, step 5: 72℃ for 30 seconds. Step 3 to step 5 were repeated for 35 cycles. The level of Nrf2 mRNA was analyzed by StepOne Software version2.1.
Measurement of intracellular ROS
For measurement of superoxide, final concentration of 2μM hydroethidine was added into cell culture medium 30 minutes prior to 5cGy alpha particles radiation. 15 minutes after radiation, the cells were gently washed with warm PBS and the fluorescence was captured under fluorescence microscope. The averaged fluorescence intensity per cell for each experimental group was calculated with NIH Image software. For detection of H2O2 release, the Amplex® Red Hydrogen Peroxide/Peroxidase Assay Kit was used. At different time points post 5cGy radiation or sham radiation, the content of H2O2 in the growth medium (phenol red-free) from each group was measured with the kit on Molecular Device fluorescent plate reader according to the product manual.
shRNA plasmids transfection
shRNA plasmid targeted to specific human gene or scrambled shRNA plasmid were transfected into A549 by MegaTran 1.0 Transfection Reagent according to the manufacturer's recommendation respectively. Complete medium with 1μg/ml puromycin was used as the selective medium. Transfectants were isolated and maintained in complete medium containing 0.2μg/ml puromycin for further experiments. Knockdown of specific gene was confirmed by determining the expression level of the target protein with western blotting and the level of the corresponding mRNA by qRT-PCR.
Dansylcadaverine (MDC) staining of autophagic vacuoles
6 hours after exposure to 5cGy α particles, cells were washed with PBS. Then, the cells were incubated in PBS with 50μM MDC in incubator for 10 minutes. After that, cells were washed 3 times with PBS and immediately observed under fluorescence microscope with ultraviolet as excitation source.
Statistical analysis
Statistical data were represented as mean ± standard deviation from at the least three independent experiments performed in triplicate. Statistical significance of differences between two groups was determined by one way ANOVA with SPSS software version 20. P value <0.05 between groups was considered to be of statistical significance.
Results
Low dose α particle inducing radio-resistance in A549 cell line
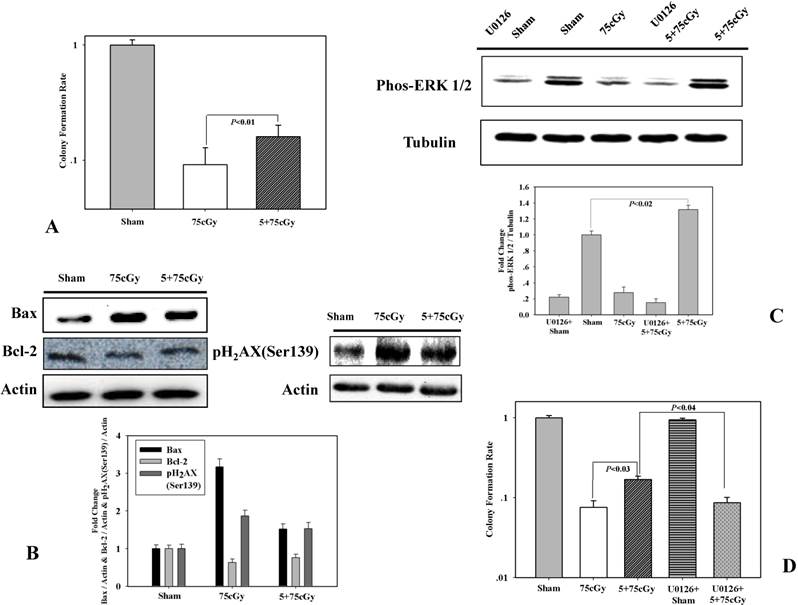
Firstly, we detected the induction of radio-resistance in human lung carcinoma A549 cell. Cells were irradiated with 5cGy α particle. 6 hours later, the irradiated cells were exposed to 75cGy α particle. The control group was sham irradiated and 6 hours later, irradiated with 75cGy α particle. By colony formation assay, it was observed that the surviving fraction after a single dose of 75cGy α particle treatment was 9% compared to that of the non-irradiated group (Fig 1A). Pretreatment with 5cGy α particle 6 hours before 75cGy radiation ameliorated the influence of 75cGy α particle on A549. As shown in figure 1A, the surviving fraction of 5+75cGy group was 16%. Levels of pro-apoptosis protein Bax and anti-apoptosis protein Bcl-2 were detected by western blotting (Fig 1B). Compared to sham irradiated group, single dose 75cGy α particle significantly increased the level of Bax. While for 5+75cGy α particle treatment, the level of Bax was higher than that in sham irradiated group, but lower than that in 75cGy α particle treatment group. An opposite tendency was observed for Bcl-2 level compared to that of Bax. pH2AX(Ser139) is a marker of DNA double strand breaks (DSBs). As shown in figure 1B, 5cGy pretreatment resulted in amelioration of DSBs formation after a further 75cGyα particle radiation compared to single 75cGy radiation. These confirmed that 5cGy radiation conferred A549 resistance to a higher dose radiation. Extracellular signal regulated protein kinases (ERK) was reported to play important role in controlling cell death induced by ionizing radiation. In this work, it was found that the level of phosphorylated ERK 1/2 in 5+75cGy α particle treatment group was significantly higher than that in the 75cGy α particle treatment group (Fig 1C). U0126 is a chemical inhibitor of MEK 1/2, the upstream regulator of ERK 1/2. Final concentration of 5μM U0126 was added into the culture medium 30 minutes before 5cGy α particle radiation. Under this concentration, U0126 diminished the activation of ERK 1/2 remarkably (Fig 1C). Colony formation assay showed that inhibition of ERK 1/2 resulted in the diminishment of radio-resistance induced by 5cGy α particle, indicating the role of activation of ERK 1/2 in the process (Fig 1D).
(A) LDIR induced radio-resistance in A549 detected by clonogenic assay. Cells were either pre-irradiated with 5cGy α particle or 0Gy α particle. 6 hours later, these cells were irradiated with 75cGy α particle. 6 hours after the 75cGy radiation, clonogenic assay was used to determine the survival fraction. Sham represented non-irradiated group. (B) Detection of apoptosis related proteins and pH2AX(Ser139) by WB. Radiation treatment steps were same as above mentioned. 3 hours after the 75cGy radiation, cell samples were collected for WB analysis of pH2AX(Ser139). 24 hours after the 75cGy radiation, cell samples were collected for WB analysis of Bax and Bcl-2. (C) Activation of ERK 1/2 in LDIR induced radio-resistance. Detection of phosphor-ERK 1/2 by WB was performed 24 hours after the 75cGy radiation. To evaluate the effect of ERK 1/2 inhibitor, final concentration of 5μM U0126 was added into the culture medium 30 minutes before 5cGy radiation. (D) Confirmation of ERK 1/2 activation in LDIR induced radio-resistance by clonogenic assay. Final concentration of 5μM U0126 was added into the culture medium 30 minutes before 5cGy radiation. 6 hours after 5cGy radiation, cells were irradiated with 75cGy. And after 6 hours incubation, clonogenic assay was performed to determine the survival fraction.
Nrf2 antioxidant pathway was involved in low dose α particle induced radio-resistance
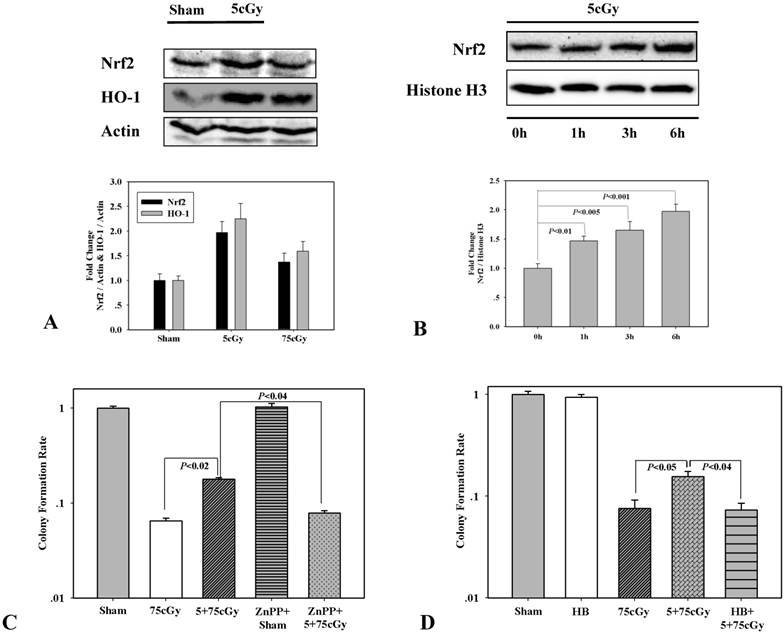
To make it clear whether Nrf2 mediated antioxidant response participated in low dose α particle induced radio-resistance, the expression of Nrf2 was detected 6 hours after 5cGy α particle radiation. As shown in figure 2A, the levels of Nrf2 and its target HO-1 were obviously stimulated by both 5cGy and 75cGy α particle in A549. However, the induction levels of both Nrf2 and HO-1 were higher in 5cGy irradiated cells those in 75cGy irradiated cells. Nuclear accumulation of Nrf2 after exposure to 5cGy was evaluated by western blotting detection. It can be seen from figure 2B, within 6 hours after 5cGy radiation, the presence of Nrf2 in nuclear fraction increased, indicating the induction of translocation of Nrf2 into nucleus where it stimulated the transcription of its target genes. Then the effect of HO-1 inhibitor on LDIR induced radio-resistance was investigated. 5μM ZnPP, a chemical inhibitor of HO-1, was added into culture medium 30 minutes prior to 5cGy α particle radiation and removed from culture medium just before 75cGy α particle radiation. Cell survival result showed that administration of ZnPP suppressed the induced radio-resistance (Fig 2C). Since HO-1 catalyzes the degradation of heme to produce carbon monoxide (CO), we further tested whether CO was involved in 5cGy α particle induced radio-resistance. 10ug/ml CO scavenger hemoglobin was added into culture medium 1 hour before 5cGy radiation. Cell survival result showed that this treatment resulted in the diminishment of the induced radio-resistance by 5cGy radiation, thus indicating the role of HO-1 mediated production of CO in this process.
(A) A549 cell were either irradiated with 5cGy or 0cGy. 6 hours later, cells were collected for WB detection of the expression level of Nrf2 and HO-1. (B) A549 cell were irradiated with 5cGy. Then proteins in nuclear fraction were extracted at time points including right after radiation, 1, 3 and 6 hours post exposure. The levels of Nrf2 in nuclear fractions were analyzed by WB. (C) Effects of HO-1 inhibition on LDIR induced radio-resistance. 5μM HO-1 inhibitor ZnPP was added into culture medium 30 minutes prior to 5cGy or 0cGy α particle radiation and removed from culture medium just before 75cGy α particle radiation. 6 hours after 75cGy radiation, clonogenic assay was used to determine survival fraction. (D) 10ug/ml hemoglobin was added into culture medium 1 hour before 5cGy radiation. 6 hours after 5cGy radiation, cells were irradiated with 75cGy. And after 6 hours incubation, clonogenic assay was performed to determine the survival fraction.
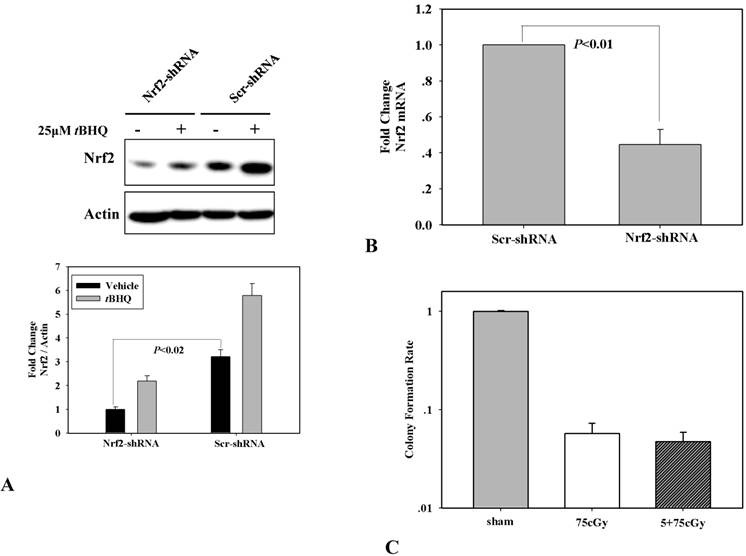
Through shRNA mediated RNA interference, Nrf2 knock-down A549 was constructed, which was testified by the expression of Nrf2 and its induction by 25μM chemical inducer tBHQ in scramble shRNA and Nrf2 shRNA transfected A549 respectively (Fig 3A). By one-step qRT-PCR analysis, the mRNA level of Nrf2 in Nrf2-shRNA A549 cells was decreased to about 44% compared to that in Scr-shRNA A549 cells (Fig 3B). In the Nrf2 knock-down A549, the induced radio-resistance by 5cGy α particle was suppressed (Fig 3C). Combining these results, it was concluded that low dose α particle stimulated Nrf2 was involved in inducing radio-resistance to high dose radiation.
(A) Determination of Nrf2 knockdown in A549 transfected with Nrf2 shRNA or its control counterpart which was transfected with scramble shRNA. Two cell lines were treated or mocked treated with Nrf2 activator tBHQ for 16 hours, and then the expression level of Nrf2 was detected by WB. (B) Suppression of 5cGy radio-resistance in Nrf2 knockdown A549 which was determined by clonogenic assay.
Autophagy conferring A549 LDIR induced radio-resistance
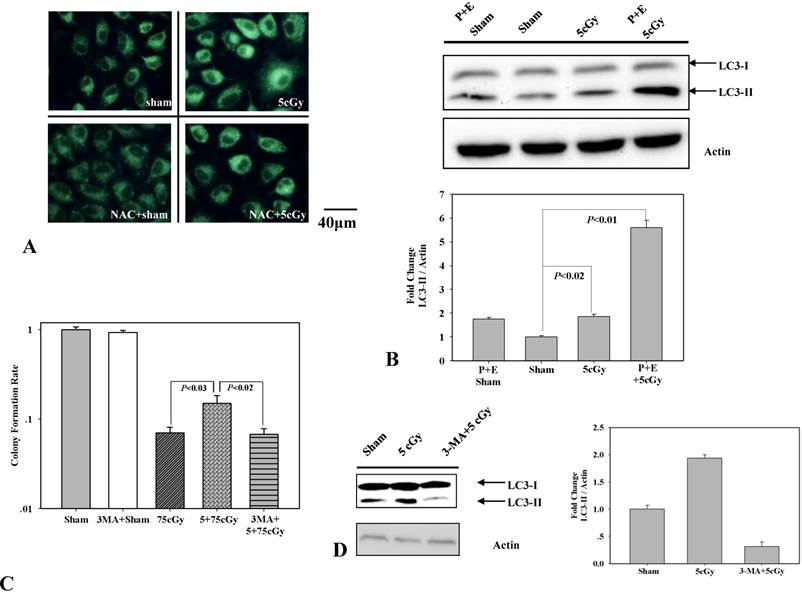
To test the role of autophagy in 5cGy α particle induced radio-resistance in A549, we firstly determined whether 5cGy α particle could induce autophagy process. 6 hours after radiation, A549 cells were stained with MDC, an autofluorescent dye which accumulates in autophagic vacuole. Compared to non-irradiated cells (Fig 4A upper left), cells irradiated with 5cGy α particle emitted enhanced fluorescence (Fig 4A upper right). The level of LC3-II, a biomarker of autophagy, was also detected by western blotting (Fig 4B). Compared to that in sham irradiated group, accumulation of LC3-II in 5cGy α particle treated group increased. Combined administration of lysosome inhibitors pepstantin and E-64D further enhanced the accumulation of LC3-II. These results confirmed that 5cGy α particle stimulated the autophagy process in A549. Then, the effect of stimulated autophagy process in LDIR induced radio-resistance was analyzed. Final concentration of 5mM autophagy inhibitor 3-MA was added into culture medium 1 hour before 5cGy α particle radiation and removed from culture medium before 75cGy α particle radiation. Inhibition of 5cGy α particle induced autophagy by 3-MA was evidenced by the decrease of LC3-II accumulation (Fig 4D). Cell survival result showed that inhibition of autophagy suppressed the radio-resistance induced by 5cGy α particle (Fig 4C), indicating the participation of autophagy in LDIR induced radio-resistance.
(A) MDC staining of autophagic vacuoles in A549. Upper left: 6 hours after A549 cell were sham irradiated; Upper right: 6 hours after A549 cell were irradiated with 5cGy; Lower left: 6 hours after A549 cell were sham irradiated under the existence of 1mM NAC; Lower right: 6 hours after A549 cell were irradiated with 5cGy under the existence of 1mM NAC. (B) Detection of the formation of LC3-Ⅱ. A549 cells were irradiated with 5cGy or 0cGy. 6 hours later, cells were collected for WB analysis. P represented pepstatin. E represented E-64d. Combining administration of pepstatin and E-64d suppressed autophagy mediated degradation of LC3-Ⅱ, which confirmed the activation of autophagy process. (C) Determination of the effect of autophagy inhibitor 3-MA on LDIR induced radio-resistance by clonogenic assay. 5mM 3-MA was added into culture medium 1 hours before 5cGy or 0cGy radiation. 6 hours later, cells were irradiated with 75cGy. And 6 hours after 75cGy radiation, cell samples were subjected to clonogenic assay. (D) Confirmation of the inhibition of 5cGy induced autophagy by 3-MA. Samples were collected 6 hours after 5cGy radiation. (E) Effects of 3-MA on the activation of ERK1/2. Radiation steps were the same as that described in (C) and protein samples were collected 24 hours after 75cGy radiation.
Low dose α particle activated Nrf2 was regulated by autophagy
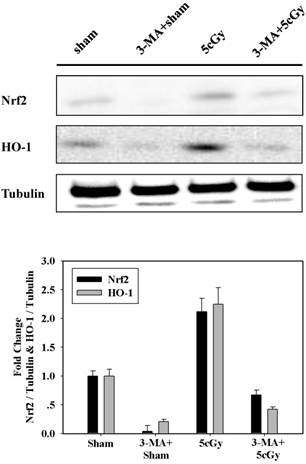
Based on above results, we assumed that autophagy participated in LDIR induced radio-resistance process by controlling the activation of Nrf2. To address this, the effect of 3-MA on 5cGy α particle stimulated Nrf2 expression was examined and the result was displayed in figure 5. It was obvious that 5cGy α particle elevated the level of Nrf2 and HO-1. Administration of 5mM 3-MA blocked the stimulation of both Nrf2 and HO-1 not only in 5cGy α particle treated cells, but also in sham irradiated cells. Together with the observation that 5cGy α particle stimulated autophagy process, these results indicated that autophagy mediated Nrf2 upregulation was involved in LDIR induced radio-resistance.
Inhibition of 5cGy induced autophagy by 3-MA suppressed the upregulation of Nrf2 and HO-1. A549 cells were irradiated with 5cGy or 0cGy under the existence or absence of 5mM 3-MA. 6 hours later, samples were collected for WB detection of the expression level of Nrf2 and HO-1.
LDIR induced ROS initiating the autophagy/Nrf2 signaling
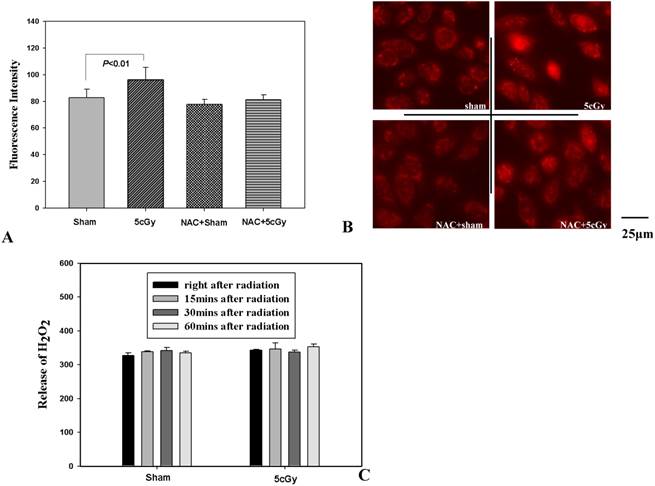
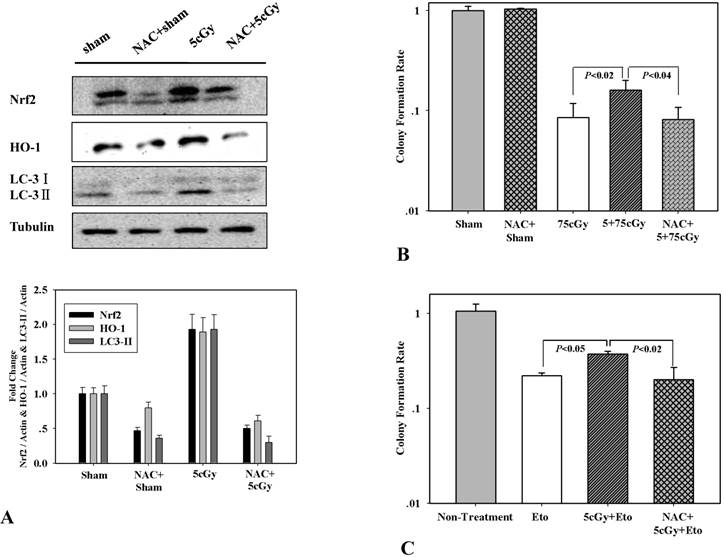
ROS is a well-known factor that mediating ionizing radiation induced biological effects. The role of LDIR induced generation of ROS in the activation of autophagy and Nrf2 in A549 was then investigated. Firstly, production of superoxide and release of H2O2 after LDIR was measured with hydroethidine and Amplex® Red respectively. As shown in figure 6A and 6B, the superoxide in 5cGy treated cells increased by 16.5% compared to that in non-irradiated cells 15 minutes after radiation. By using final concentration of 1mM N-Acetyl-L-cysteine (NAC), a kind of ROS scavenger, the increased superoxide level in 5cGy treated cells was suppressed. However, as to the released H2O2, no significant effect was observed by 5cGy treatment within 60 minutes after radiation (Fig 6C). Under the existence of 1mM NAC, the autophagy process stimulated by 5cGy α particle was inhibited as evidenced by the accumulation of LC-3II (Fig 6A). MDC staining of autophagic vacuole testified the role of NAC in inhibiting 5cGy α particle induced autophagy (Fig 4A lower left and lower right). These results indicated that the ROS level induced by low dose α particle promoted autophagy process. Furthermore, activation of Nrf2 and its target HO-1 was blocked by NAC concomitantly (Fig 7A), confirming that the autophagy regulated activation of Nrf2 antioxidant response was initiated by LDIR induced ROS level. Cell survival result revealed that NAC treatment abolished the induced radio-resistance to 75cGy α particle irradiation in A549 by 5cGy α particle (Fig 7B). We then tested whether 5cGy radiation conferred A549 resistance to etoposide, a chemical agent which led to DNA double strand breaks. As shown in figure 7C, a survival fraction of 22% was observed in A549 after 2μg/ml etoposide treatment. However, if A549 was radiated with 5cGy and 6 hours later, challenged with 2μg/ml etoposide, the survival fraction increased to 38%. And NAC treatment before 5cGy radiation resulted in the diminishment of the resistance to etoposide induced by the LDIR. This further confirmed the protective role of LDIR induced ROS.
(A) Effects of NAC on generation of intracellular superoxide after 5cGy radiation. A final concentration of 2μM hydroethidine was added into cell culture medium 30 minutes prior to radiation under the existence or absence of 1mM NAC. 15 minutes later, intracellular superoxide level was detected under fluorescence microscope. The histogram showed the statistic data of averaged fluorescence intensity per cell. (B) Representative fluorescence microscope images of superoxide detection. Upper left: sham irradiated; Upper right: 5cGy treatment; Lower left: sham irradiated under the existence of 1mM NAC; Lower right: 5cGy treatment under the existence of 1mM NAC. (C) Release of H2O2 measured with Amplex® Red Hydrogen Peroxide/Peroxidase Assay Kit. At different time points post radiation or sham radiation with 5cGy, the content of H2O2 in growth medium (phenol red-free) from each group was measured and normalized to cell numbers. The histogram represented the fluorescence intensity of 100μL culture medium from 0.4 million cells.
(A) WB detection of Nrf2, HO-1 and LC3Ⅱ expression level in A549 cells irradiated with 5cGy or 0cGy under the existence or absence of 1mM NAC. (B) Effect of NAC treatment on LDIR induced radio-resistance. 1mM NAC was added into culture medium 1 hour before 5cGy or 0cGy radiation. 6 hours later, cells were irradiated with 75cGy. And 6 hours after 75cGy radiation, cells were subjected to clonogenic assay. (C) Effect of NAC treatment on LDIR induced resistance to etoposide. 1mM NAC was added into culture medium 1 hour before 5cGy or 0cGy radiation. 6 hours later, 2μg/ml etoposide was added into culture medium. Clonogenic assay was then performed to determine the survival fraction.
Discussion
LDIR induced resistance to high dose radiation is a well recognized phenomenon which in some circumstances was called low dose radiation induced adaptive response. Upon LDIR treatment, cellular protective systems will be activated and thus confer cells resistance or adaptability to further high dose challenging radiation. This kind of phenomenon was not only observed with ionizing radiation, but also with many types of stress [30]. Radiotherapy has been widely applied for the treatment of tumors, independently or in combination with anti-tumor drugs. With the findings that LDIR resulted in resistance of cells to following high dose radiation, it was suggested that CT scanning might be one of the reasons responsible for the recurrence and failure of cancer after radiotherapy. Therefore, investigation on the mechanisms underlying LDIR induced radio-resistance is of importance for the optimization of cancer radiotherapy.
LDIR induced radio-resistance was observed in human lung carcinoma A549 cell line with α particle radiation by clonogenic assay and apoptosis assay in the current research. ERK 1/2 is one of the highly conserved mitogen-activated protein kinases (MAPKs) family which respond to diverse extracellular stimuli, including radiation. It has been suggested that ERK 1/2 plays an important role in determining the cellular outcomes between a balance of survival and death after ionizing radiation [31]. Low levels of DNA damage induced by radiation led to pro-survival signaling regulated by the activation of ERK 1/2 and its upstream regulator ATM. However, excessive level of DNA damage induced by radiation resulted in the ATM-independent dephosphorylation of ERK 1/2, thus shut down the pro-survival signaling. In this work, the pro-survival function of ERK 1/2 activation was observed in LDIR induced radio-resistance process, as evidenced by the obvious activation of ERK 1/2 in the 5cGy plus 75cGy treated cells compared to the non-irradiated or single 75cGy treated cells. Pretreatment with U0126 disturbed the 5cGy induced radio-resistance, further confirming the pro-survival role of ERK 1/2. Accumulating evidence demonstrated that the MEK-ERK pathway also contributes to the proper execution of cellular DNA damage response. Radiation induced ERK 1/2 activation has been linked to increased expression of the DNA repair proteins ERCC1, XRCC1 and XPC [32]. Besides, there is evidence that DNA damage induced ERK 1/2 activation contributes to the activation of ATM and ATR [33]. The role of ERK 1/2 in regulating DNA damage repair might also be responsible for LDIR induced radio-resistance.
Nrf2 mediated antioxidant response is considered to be the core machinery for maintaining cellular redox balance. Under basal conditions, the Nrf2 inhibitor KEAP1 binds and sequesters Nrf2 in cytoplasm where Nrf2 will be degraded through ubiquitin-mediated process. Upon oxidative or electrophilic stress, Nrf2 dissociates from KEAP1 and transfers into the nucleus where it interacts with other transcription factors to form transcription complex, binds to and activates ARE (Antioxidant Response Element)-containing genes [20]. Nrf2 has been reported to be associated with chemo-resistance of cancer cell lines. For instance, Wang et al. found that knockdown of endogenous Nrf2 by siRNA or by over-expressing KEAP1 sensitized A549 to cisplatin, doxorubicin or etoposide. Meanwhile, stable over-expression of Nrf2 led to an enhancement of resistance to these chemotherapeutic drugs in breast carcinoma cell line MDA-MB-231, and similar results were observed with neuroblastoma cell line SH-SY5Y [34]. Ionizing radiation was a stimulus for Nrf2 activation. McDonald et al. reported that doses from 2 to 8Gy gamma ray radiation activated ARE-dependent transcription in breast cancer cells in a dose-dependent manner. They also confirmed that Nrf2 deficient mouse embryonic fibroblast cells and C57BL/6 mice showed enhanced radio-sensitivity [25]. Mathew et al. reported that repeated administration of sulforaphane significantly ameliorated 2 or 4Gy gamma radiation induced cell death in mouse embryonic fibroblasts, and the radio-protective role of sulforaphane depended on the status of Nrf2 [27]. Therefore, based on the result that 5cGy α particle induced resistance to high dose radiation, we detected the induction of Nrf2 by LDIR in A549. The result showed an obvious elevation of Nrf2 6 hours after 5cGy radiation, which was consistent with the findings of Lee et al. [24] and Tsukimoto et al. that LDIR activated Nrf2-antioxidant pathway [26]. To further test whether the induced Nrf2 conferred resistance to high dose radiation, A549 stably expressing Nrf2 shRNA and scrambled shRNA were established. Loss of LDIR induced radio-resistance in A549 harboring Nrf2 shRNA validated that Nrf2 played a key role in the process. Nrf2 mediated protective role induced by LDIR was also indicated in diabetes mouse model. Xing et al. suggested that total dose of 7.5cGy X-ray radiation upregulated Nrf2 expression and function in the tested mouse model, and attenuated diabetes-induced renal inflammation, oxidative damage, remodeling, and dysfunction [35]. HO-1, which is also designated as Hsp32, is highly inducible in response to oxidative stress. Its expression is mainly upregulated by Nrf2 and it plays a pivotal role in maintaining cellular redox homeostasis. Vile et al. found that pretreatment of human skin fibroblast with ultraviolet A resulted in protective role against a following ultraviolet A radiation. HO-1 was upregulated in this process, and antisense oligonucleotide of HO-1 blocked both the induced expression of HO-1 and the protective role of ultraviolet A pretreatment [36]. In our work, similar results for HO-1 were observed. LDIR increased the expression of HO-1, and chemical inhibitor of HO-1 ZnPP treatment abolished LDIR induced radio-resistance. HO-1 is the rate-limiting enzyme in the degradation of heme into biliverdin, CO and free iron. It has been reported that CO exposure could facilitate homologous recombination repair of DNA damage through an ATM/ATR dependent pathway [37]. In this work, treatment with CO scavenger hemoglobin before 5cGy radiation led to diminishment of LDIR induced radio-resistance, showing that HO-1 contributed to the process partially through its activity to producing CO.
Autophagy, a cellular cytoplasm and organelle turnover process to maintain homeostasis, has evoked a number of correlation with radio-sensitivity and radio-resistance [38]. However, it is not known whether autophagy process participated in LDIR induced radio-resistance yet. Our results showed that LDIR promoted autophagy process. And if autophagy was blocked, the induction of radio-resistance by LDIR was inhibited, indicating the role of autophagy in this process. Since the role of Nrf2 in LDIR induced radio-resistance was established, we further investigated whether LDIR induced autophagy was responsible for Nrf2 upregulation. By inhibiting autophagy process, we proved that LDIR induced autophagy was a stimulus of Nrf2. Concomitant change of the expression level of HO-1 with Nrf2 was observed, confirming the role of Nrf2/HO-1 antioxidant pathway in LDIR induced radio-resistance. It was reported that autophagy deficiency resulted in a competitive inhibition of Nrf2-KEAP1 binding which was due to the sequestration of KEAP1 by the selective autophagy substrate p62, therefore leading to the stabilization of Nrf2 and its downstream pathways [39]. Taguchi et al. found another regulatory pathway for the stabilization of Nrf2 that was autophagy led to the degradation of KEAP1 [40]. Denicola et al. reported that transcriptional upregulation of Nrf2 by oncogene-dependent signaling contributes to the increase of Nrf2 protein level [41]. Chen et al. showed that a direct interaction between p21 and Nrf2 upregulated the Nrf2 mediated antioxidant pathways [42]. It is well known that autophagy regulation was influenced by networks of tumor suppressors and oncogenes [43]. Since it was reported that in A549 cell line, there was a somatic mutation in KEAP1 gene which resulted in its function deficiency and constitutive stabilization of Nrf2 [34], KEAP1 level might not be directly related to the level of Nrf2. Thus, complicated regulations between autophagy and Nrf2 may exist in A549 after LDIR treatment, which deserve further investigation.
It has been well accepted that over-generation of free radical species is pivotal for radiation induced cellular damages. Besides, there were studies showed that reactive oxygen species and nitric oxide radicals were important for radiation induced protective roles [13, 44]. Activities of detoxification enzymes such as MnSOD, Catalase, Gpx and GST had been reported to increase in LDIR induced radio-resistance [14]. The protective role of LDIR induced ROS was also observed in our work, as shown by the fact that ROS scavenger NAC treatment abolished 5cGy radiation induced radio-resistance. However, it is still largely unclear how the moderately induced ROS by LDIR leads to the protection against high dose radiation. We clarified that LDIR induced autophagy and Nrf2/HO-1 expression in A549 was responsible for its resistance to following high dose radiation. Therefore, whether ROS elevation after LDIR treatment promoted autophagy was investigated. NAC treatment blocked the 5cGy radiation induced formation of LC3Ⅱ, indicating the ROS induction was responsible for LDIR stimulated autophagy process. Studies have confirmed that ROS is an important regulator of autophagy under various conditions [45]. For example, deprivation of nerve growth factor led to accumulation of mitochondrial ROS, which further resulted in autophagy and cell death [46]. ROS is essential for starvation induced autophagy by oxidative inactivation of Atg4 and facilitating autophagosome formation [47]. In this work, similar to that observed with starvation, LDIR induced ROS and autophagy was shown to be protective not only to high-dose radiation, but also to etoposide treatment, a chemotherapy agent for cancers such as lung cancer, Kaposi's sarcoma, Ewing's sarcoma and et al. Consistent with the previous findings that autophagy upregulated Nrf2 and HO-1, NAC treatment suppressed of elevated expression of the two proteins, further confirmed ROS promoted autophagy and its downstream events.
In conclusion, this study indicated the following findings: (1) LDIR conferred radio-resistance in human lung adenocarcinoma A549 cell through activating the Nrf2/HO-1 antioxidant pathway; (2) Moderate elevation of ROS caused by LDIR promoted autophagy process, which resulted in the activation of Nrf2/HO-1. Our results implicated that ROS/autophagy might be an intervention target for improving radiotherapy outcomes. Further studies are needed for better understanding of the underlying mechanisms of ROS/autophagy/Nrf2 pathway in LDIR induced radio-resistance.
Acknowledgements
This work was supported by grants from the National Natural Science Foundation of China (31370842, 81273004, 31200644) and ITER program from the Ministry of Science and Technology of China (2014GB112006)
Conflict of interest
The authors have declared that no competing interest exists.
References
1. Fuentes-Orrego JM, Sahani DV. Low-dose CT in clinical diagnostics. Expert Opin Med Diagn. 2013;7:501-510
2. Martin LM, Marples B, Lynch TH, Hollywood D, Marignol L. Exposure to low dose ionising radiation: molecular and clinical consequences. Cancer Lett. 2013;338:209-218
3. Tapio S, Jacob V. Radioadaptive response revisited. Radiat Environ Biophys. 2007;46:1-12
4. Tang FR, Loke WK. Molecular mechanisms of low dose ionizing radiation induced hormesis, adaptive responses, radioresistance, bystander effects, and genomic instability. Int J Radiat Biol. 2015;91:13-27
5. Ahmed KM, Li JJ. NF-kappa B-mediated adaptive resistance to ionizing radiation. Free Radic Biol Med. 2008;44:1-13
6. Blankenbecler R. Low-dose pretreatment for radiation therapy. Dose Response. 2010;8:534-542
7. Lall R, Ganapathy S, Yang M, Xiao S, Xu T, Su H, Shadfan M, Asara JM, Ha CS, Ben-Sahra I, Manning BD, Little JB, Yuan ZM. Low-dose radiation exposure induces a HIF-1-mediated adaptive and protective metabolic response. Cell Death Differ. 2014;21:836-844
8. Seong J, Kim SH, Pyo HR, Chung EJ, Suh CO. Low-dose radiation-induced adaptive response in the murine system. Ann N Y Acad Sci. 2002;973:255-257
9. Khodarev NN, Beckett M, Labay E, Darga T, Roizman B, Weichselbaum RR. STAT1 is overexpressed in tumors selected for radioresistance and confers protection from radiation in transduced sensitive cells. Proc Natl Acad Sci USA. 2004;101:1714-1719
10. Zhang H, Zhao W, Wang Y, Li N, Wu Z, Liu Y, Chen J, Cai Y. Induction of cytogenetic adaptive response in spermatogonia and spermatocytes by pre-exposure of mouse testis to low-dose (12)C(6+) ions. Mutat Res. 2008;653:109-112
11. Ahmed KM, Nantajit D, Fan M, Murley JS, Grdina DJ, Li JJ. Coactivation of ATM/ERK/NF-kappaB in the low-dose radiation-induced radioadaptive response in human skin keratinocytes. Free Radic Biol Med. 2009;46:1543-1550
12. Caputo F, Vegliante R, Ghibelli L. Redox modulation of the DNA damage response. Biochem Pharmacol. 2012;84:1292-1306
13. Lehnert BE, Iyer R. Exposure to low-level chemicals and ionizing radiation: reactive oxygen species and cellular pathways. Hum Exp Toxicol. 2002;21:65-69
14. Miura Y. Oxidative stress, radiation-adaptive responses, and aging. J Radiat Res. 2004;45:357-372
15. Eldridge A, Fan M, Woloschak G, Grdina DJ, Chromy BA, Li JJ. Manganese superoxide dismutase interacts with a large scale of cellular and mitochondrial proteins in low-dose radiation-induced adaptive radioprotection. Free Radic Biol Med. 2012;53:1838-1847
16. Jaiswal AK. Regulation of genes encoding NAD(P)H:quinone oxidoreductases. Free Radic Biol Med. 2000;29:254-262
17. Kobayashi M, Yamamoto M. Molecular mechanisms activating the Nrf2-Keap1 pathway of antioxidant gene regulation. Antioxid Redox Signal. 2005;7:385-394
18. Srisook K, Kim C, Cha YN. Molecular mechanisms involved in enhancing HO-1 expression: de-repression by heme and activation by Nrf2, the "one-two" punch. Antioxid Redox Signal. 2005;7:1674-1687
19. Ma Q. Role of Nrf2 in oxidative stress and toxicity. Annu Rev Pharmacol Toxicol. 2013;53:401-426
20. Sporn MB, Liby KT. NRF2 and cancer: the good, the bad and the importance of context. Nat Rev Cancer. 2012;12:564-571
21. Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27:2179-2191
22. Shelton P, Jaiswal AK. The transcription factor NF-E2-related factor 2 (Nrf2): a protooncogene? FASEB J. 2013;27:414-423
23. Kim JH, Thimmulappa RK, Kumar V, Cui W, Kumar S, Kombairaju P, Zhang H, Margolick J, Matsui W, Macvittie T, Malhotra SV, Biswal S. NRF2-mediated Notch pathway activation enhances hematopoietic reconstitution following myelosuppressive radiation. J Clin Invest. 2014;124:730-741
24. Lee EK, Kim JA, Park SJ, Kim JK, Heo K, Yang KM, Son TG. Low-dose radiation activates Nrf1/2 through reactive species and the Ca(2+)/ERK1/2 signaling pathway in human skin fibroblast cells. BMB Rep. 2013;46:258-263
25. McDonald JT, Kim K, Norris AJ, Vlashi E, Phillips TM, Lagadec C, Della Donna L, Ratikan J, Szelag H, Hlatky L, McBride WH. Ionizing radiation activates the Nrf2 antioxidant response. Cancer Res. 2010;70:8886-8895
26. Tsukimoto M, Tamaishi N, Homma T, Kojima S. Low-dose gamma-ray irradiation induces translocation of Nrf2 into nuclear in mouse macrophage RAW264.7 cells. J Radiat Res. 2010;51:349-353
27. Mathew ST, Bergström P, Hammarsten O. Repeated Nrf2 stimulation using sulforaphane protects fibroblasts from ionizing radiation. Toxicol Appl Pharmacol. 2014;276:188-194
28. Khan NM, Sandur SK, Checker R, Sharma D, Poduval TB, Sainis KB. Pro-oxidants ameliorate radiation-induced apoptosis through activation of the calcium-ERK1/2-Nrf2 pathway. Free Radic Biol Med. 2011;51:115-128
29. Laemmli UK. Cleavage of Structural Proteins during the Assembly of the Head of Bacteriophage T4. Nature. 1970;227:680-685
30. Subhash Peter MC. Understanding the adaptive response in vertebrates: the phenomenon of ease and ease response during post-stress acclimation. Gen Comp Endocrinol. 2013;181:59-64
31. Dent P, Yacoub A, Fisher PB, Hagan MP, Grant S. MAPK pathways in radiation responses. Oncogene. 2003;22:5885-5896
32. Yacoub A, Park JS, Qiao L, Dent P, Hagan MP. MAPK dependence of DNA damage repair: ionizing radiation and the induction of expression of the DNA repair genes XRCC1 and ERCC1 in DU145 human prostate carcinoma cells in a MEK1/2 dependent fashion. Int J Radiat Biol. 2001;77:1067-1078
33. Wei F, Yan J, Tang D. Extracellular signal-regulated kinases modulate DNA damage response - a contributing factor to using MEK inhibitors in cancer therapy. Curr Med Chem. 2011;18:5476-5482
34. Wang XJ, Sun Z, Villeneuve NF, Zhang S, Zhao F, Li Y, Chen W, Yi X, Zheng W, Wondrak GT, Wong PK, Zhang DD. Nrf2 enhances resistance of cancer cells to chemotherapeutic drugs, the dark side of Nrf2. Carcinogenesis. 2008;29:1235-1243
35. Xing X, Zhang C, Shao M, Tong Q, Zhang G, Li C, Cheng J, Jin S, Ma J, Wang G, Li X, Cai L. Low-dose radiation activates Akt and Nrf2 in the kidney of diabetic mice: a potential mechanism to prevent diabetic nephropathy. Oxid Med Cell Longev. 2012 doi: 10.1155/2012/291087
36. Vile GF, Basu-Modak S, Waltner C, Tyrrell RM. Heme oxygenase 1 mediates an adaptive response to oxidative stress in human skin fibroblasts. Proc Natl Acad Sci USA. 1994;91:2607-2610
37. Otterbein LE, Hedblom A, Harris C, Csizmadia E, Gallo D, Wegiel B. Heme oxygenase-1 and carbon monoxide modulate DNA repair through ataxia-telangiectasia mutated (ATM) protein. Proc Natl Acad Sci USA. 2011;108:14491-14496
38. Palumbo S, Comincini S. Autophagy and ionizing radiation in tumors: the "survive or not survive" dilemma. J Cell Physiol. 2013;228:1-8
39. Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou YS, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol. 2010;12:213-223
40. Taguchi K, Fujikawa N, Komatsu M, Ishii T, Unno M, Akaike T, Motohashi H, Yamamoto M. Keap1 degradation by autophagy for the maintenance of redox homeostasis. Proc Natl Acad Sci USA. 2012;109:13561-13566
41. DeNicola GM, Karreth FA, Humpton TJ, Gopinathan A, Wei C, Frese K, Mangal D, Yu KH, Yeo CJ, Calhoun ES, Scrimieri F, Winter JM, Hruban RH, Iacobuzio-Donahue C, Kern SE, Blair IA, Tuveson DA. Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 2011;475:106-109
42. Chen W, Sun Z, Wang XJ, Jiang T, Huang Z, Fang D, Zhang DD. Direct interaction between Nrf2 and p21(Cip1/WAF1) upregulates the Nrf2-mediated antioxidant response. Mol Cell. 2009;34:663-673
43. Lorin S, Hamaï A, Mehrpour M, Codogno P. Autophagy regulation and its role in cancer. Semin Cancer Biol. 2013;23:361-379
44. Matsumoto H, Takahashi A, Ohnishi T. Nitric oxide radicals choreograph a radioadaptive response. Cancer Res. 2007;67:8574-8579
45. Azad MB, Chen Y, Gibson SB. Regulation of autophagy by reactive oxygen species (ROS): implications for cancer progression and treatment. Antioxid Redox Signal. 2009;11:777-790
46. Kirkland RA, Adibhatla RM, Hatcher JF, Franklin JL. Loss of cardiolipin and mitochondria during programmed neuronal death: evidence of a role for lipid peroxidation and autophagy. Neuroscience. 2002;115:587-602
47. Scherz-Shouval R, Shvets E, Fass E, Shorer H, Gil L, Elazar Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007;26:1749-1760
Author contact
![]() Corresponding author: wangjun0457ac.cn (Jun Wang), Fax: 86-551-65595670
Corresponding author: wangjun0457ac.cn (Jun Wang), Fax: 86-551-65595670