Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(9):992-1005. doi:10.7150/ijbs.10918 This issue Cite
Research Paper
MDC1 Enhances Estrogen Receptor-mediated Transactivation and Contributes to Breast Cancer Suppression
Renlong Zou1,*, Xinping Zhong2,*, Chunyu Wang1, Hongmiao Sun1, Shengli Wang1, Lin Lin1, Shiying Sun1, Changci Tong1, Hao Luo1, Peng Gao1, Yanshu Li1, Tingting Zhou1, Da Li3, Liu Cao1, Yue Zhao1 ![]()
1. Department of Cell Biology, Key laboratory of Cell Biology, Ministry of Public Health, and Key laboratory of Medical Cell Biology, Ministry of Education, China Medical University, Shenyang, Liaoning 110122, China
2. Department of General Surgery, the First Affiliated Hospital, China Medical University, Shenyang, Liaoning 110001, China
3. Department of Obstetrics and Gynecology, Shengjing Hospital of China Medical University, Shenyang, Liaoning 110003, China
* These authors contributed equally to this work.
Received 2014-10-27; Accepted 2015-5-28; Published 2015-7-3
Abstract

Estrogen receptor α (ERα) is a key transcriptional factor in the proliferation and differentiation in mammary epithelia and has been determined to be an important predictor of breast cancer prognosis and therapeutic target. Meanwhile, diverse transcriptional co-regulators of ERα play crucial and complicated roles in breast cancer progression. Mediator of DNA damage checkpoint 1 (MDC1) has been identified as a critical upstream mediator in the cellular response to DNA damage, however, some non-DNA damage responsive functions of MDC1 haven't been fully defined. In this study, we have identified MDC1 as a co-activator of ERα in breast cancer cells and demonstrated that MDC1 associates with ERα. MDC1 was also recruited to estrogen response element (ERE) of ERα target gene. Knockdown of MDC1 reduced the transcription of the endogenous ERα target genes, including p21. MDC1 depletion led to the promotion of breast cancer progression, and the expression of MDC1 is lower in breast cancer. Taken together, these results suggested that MDC1 was involved in the enhancement of ERα-mediated transactivation in breast cancer cells. This positive regulation by MDC1 might contribute to the suppression of breast cancer progression by acting as a barrier of positive to negative ERα function transformation.
Keywords: estrogen receptor α, MDC1, co-activator, breast cancer, tumor suppression
Introduction
Estrogen receptor α (ERα) is a ligand-dependent transcription factor that regulates a variety of genes involved in a variety of biological processes, including reproduction, development, and breast cancer progression [1, 2]. As a member of the nuclear receptor family, in the presence of ligand, ERα can regulate gene transcription by binding directly or indirectly to the estrogen response elements (ERE) in the enhancer and/or promoter of target genes [3, 4]. A series of ERα target genes are particularly relevant in breast cancer, such as cyclinD1[5], E2F1[6], pS2 (TFF1), p21[7, 8], c-Myc [9], and so on. The liganded ERα then recruits diverse co-regulators, thereby forming a transcriptional complex and regulating transcription of target genes [10]. ERα co-regulators (co-activators or co-repressors) play pivotal roles in breast cancer progression. A number of ERα co-activators including p160 family members (SRC-1, TIF2 or GRIP-1, and AIB1) [11-13], the histone acetylases CBP/ p300, the p300/CBP-associated factor (pCAF) [14, 15]; the co-repressors including N-CoR, SMRT [16], DACH1 [17], CARM1 [18], and NFAT3 [19] have been identified. These co-regulators affect the transcription of ERα-induced target genes transcription through a variety of different mechanisms and promote or inhibit breast cancer growth, invasion, metastasis and resistance to tamoxifen therapy.
About 70% of breast cancer patients are ERα-positive upon initial diagnosis. In the majority of those cancers, ERα status is of dominant importance for the selection of appropriate hormonal therapy and as a predictive marker [20-22]. Although higher ERα levels lead to higher hormone sensitivity and might predispose to malignant transformation, they also confer a higher success rate to antiestrogen treatment. In addition, a majority of ERα-positive breast tumors become hormone-resistant through various mechanisms and patients suffer recurrence and metastasis within 5 years. It has been proved that loss of ERα expression promotes progression and invasive growth of breast cancer through the aberrant regulation of MTA3, snail and E-cadherin [23]. Twist contributes to hormone resistance in breast cancer by downregulating ERα expression [24]. Furthermore, MTA1 or LMO4 identified as a co-repressor of ERα negatively regulates the expression of endogenous ERα target genes and contributes to invasive breast cancer progression [25, 26], suggesting that the loss of ERα activity may contribute to the generation of hormone resistance and invasion in breast tumors. Thus, positively modulating ERα activity may donate to suppression of breast cancer.
MDC1 is a critical component of the DNA damage response (DDR) machinery that participates in DNA damage checkpoint and protects genome integrity [27]. MDC1 is also known as nuclear factor with BRCT domains protein 1 (NFBDl). It contains several distinct domains, mainly including the forkhead-associated (FHA) domain in the N terminus, two BRCA1 carboxyl-terminal (BRCT) domains in the C terminus. When the DNA damage occurs, the activated form of ATM phosphorylates histone variant H2AX (γH2AX), then MDC1 interacts directly with γH2AX through its BRCT domains and recruits MRN (MTE11, RAD50, and NBS1) complex onto the sites of DNA damage to facilitate the efficient DNA repair [28, 29]. It has been shown that MDC1-deficient mice displayed chromosome instability, DNA repair defects, increase of tumor incidence, and male infertility [30]. MDC1 plays an important role in the decision of cell survival and death after DNA damage through the regulation of p53 [31]. Previous evidence also indicates that aberrant reduction or lack of MDC1 or 53BP1 in lung and breast cancer by immunohistochemiscal analysis supports that candidacy of both proteins for tumor suppressors [32]. It is imperative to better understand the mechanisms and biological function of MDC1 in breast cancer.
In the present study, we have identified MDC1 as a potential co-activator of ERα-induced transactivation. We have also demonstrated that MDC1 associates with ERα, and MDC1 is recruited to the ERE region of ERα target gene (pS2). It is found that MDC1 positively regulates the expression of partial endogenous ERα target genes in breast cancer cells. Functionally, down-expression of MDC1 is able to promote breast cancer progression. The potential indications of these regulatory interactions and the biological function for MDC1 in regulating ERα signaling in breast cancer cells are presented.
Materials and Methods
Plasmids and Antibodies
Human MDC1 cDNA coding sequence was amplified by PCR using KIAA0170 plasmid from the HUGE database (generous gift from Dr. T. Nagase) as a template. The human full length wild-type MDC1 cDNA (MDC1wt) or a series of truncated mutants (MDC1 N1, MDC1 N2, MDC1 N3, MDC1 C) were cloned in a pcDNA3.1-Flag vector containing a sequence encoding a Flag epitope upstream of the cloning site, to generate Flag-MDC1, Flag-MDC1 N1~N3, Flag-MDC1 C. The identities of constructs were verified by sequencing. The expression plasmid for human ERα (pSG5-ERα) and pGL-ERE-AdML reporter plasmid carrying three consensus estrogen response elements (3×ERE) were from Dr. Shigeaki Kato [33, 34].
The antibodies used in this study were: anti-MDC1 (Bethyl laboratories), anti-ERα (F10, Santa Cruz Biotechnology), anti-Flag (M2 or rabbit, Sigma), anti-c-Myc (Santa Cruz Biotechnology), anti-pS2 (Shanghai Sangon Biotech), anti-p21 (thermo), anti-GAPDH (Shanghai Kangchen).
Cell Culture, siRNA transfection, and Lentiviral Production
Human Embryonic Kidney cell lines HEK293 and Breast cancer cell lines MCF-7 were routinely cultured in Dulbercco's modified Eagle's medium (DMEM; GIBCO) supplemented with 10% fetal bovine serum (FBS; GIBCO). All the cell lines were maintained at 37ºC in the presence of 5% CO2.
siRNA against ERα (siERα) and a control siRNA (Ambion) were transfected in MCF-7 cells using LipofectamineTM 2000 (Invitrogen) following the manufacture's instruction. siERα sequence: 5'-UCAUCGCAUUCCUUGCAAAdTdT-3' [35]. For lentiviral production and infection, control shRNA (shCtrl) lentivirus and shRNA against MDC1 (shMDC1) lentivirus targeting the same sequence as siMDC1 as the previous paper [36] were supplied by Shanghai GeneChem Company. shMDC1 sequence: 5'-GUCUCCCAGAAGACAGUGATT-3'. The MCF-7 cell lines infected with lentivirus carrying shMDC1 or shCtrl were performed according to the manufacturer's instructions.
Luciferase Assay
Cells were co-transfected with ERα, Flag-MDC1 or a series of truncated mutants expression plasmids, and pGL-ERE-AdML reporter plasmid by using LipofectamineTM 2000 (Invitrogen). pRL-CMV vector was used as the internal control. 4 h later, the cells were rinsed and incubated in DMEM supplemented with 5% coal treated fetal bovine serum in the absence or presence of 17β-estradiol (E2) (10-8M), and ethanol was used as control. After an additional 21 h, cells were assayed using the Dual-Luciferase Reporter Assay System (Promega) as previously described [37].
RNA Isolation and Quantitative Real-Time PCR Assays
Total RNA was isolated using Trizol reagent (Invitrogen) and Reverse Trancription was performed using PrimeScript™ RT Master Mix (Perfect Real Time) (TAKARA) according to the manuscription's instructions. cDNAs were quantified by quantitative real-time PCR using SYBR®Premix Ex Taq™ II (TAKARA) on a Mx3000P instrument (Agilent StrataGene). Primers used to detect mRNA expression were as follows: MDC1, 5'-AGAGGAGGAGACAGAGCAATC-3' and 5'-CACAGAGCAGTCAGGCATTC-3'; ERα, 5'-CTAACTTGCTCTTGGAC AGGAA-3' and 5'-CAGGACTCGGTGGATATGGT-3'; c-Myc, 5'-GCTGCTT AGACGCTGGA TT-3' and 5'-TGCTGCTGCTGCTGGTA-3'; efp, 5'-ATCGC TCTGTTCACCACTCTT-3' and 5'-ATGCCACACGGACTGAGAC-3'; GREB1, 5'-ACGCAGAACCTCCTCAACTC-3' and 5'-GTCTCCACTCCACAT CCTCAAT-3'; p21, 5'-GGACAGCAGAGGAAGACCAT-3' and 5'-G AGACTAAGGCAGAAGATGTAGAG-3'; and GAPDH, 5'-GCACCGTCAAG GCTGAGAAC-3' and 5'-TGGTGAAGACGCCAGTGGA-3'. Gene expression levels were calculated relative to GAPDH using Stratagene Mx3000P software.
Immunoprecipitation and Western blotting
For immunoprecipitation (IP) experiments, the Flag-MDC1 and ERα expression plasmids were transiently transfected into HEK293 cells using liperfectamineTM 2000 (Invitrogen) and whole cell extracts were prepared 48 h after transfection, and equal protein amounts were immunoprecipitated with anti-MDC1, anti-ERα or anti-Flag M2 resin (Sigma). The immunoprecipitated protein complexes were washed three times with IP buffer containing 0.5M KCl and twice with IP buffer containing 100mM KCl. The crude extracts and immune complexes were analyzed by western blotting using anti-Flag, anti- ERα, or anti-MDC1[38].
GST Pull-down Assay
The GST alone and GST fusion proteins, including GST- ERα 29-180aa and GST- ERα 282-595aa [37], were expressed in BL21 bacteria and bound to GST-Sepharose beads according to the manufacturer's instructions (GE Healthcare). The expression plasmid for Flag-MDC1 was used for synthesis Flag-MDC1 protein in Vitro with transcription and translation in the TNT system (Promega). Equal amounts of GST alone, or GST-fusion proteins coupled with GST-Sepharose beads were incubated with in vitro-translated Flag-MDC1 protein at 4°C overnight. The precipitated proteins were washed thoroughly 4 times with binding buffer (20 mM Tris, pH 7.5, 50 mM NaCl, 10% glycerol, 1% Nonidet P-40). The bound proteins were detected by western blot and stained by Coomassie Brilliant Blue.
Immunofluorescence
Immunofluorescence analysis was performed as described [38], cells grown on the 12-well chamber slides were fixed in 4% paraformaldehyde (PFA) for 20 min, permeabilized in 0.2%TritonX-100 for 20 min and blocked in 1% donkey serum albumin, incubated with primary antibodies and secondary antibody conjugated to FITC or Cy5 (Jackson Immunoresearch Laboratories Inc). Cells were stained with DAPI (Roche) to visualize the nuclei.
ChIP and ChIP re-ChIP
ChIP was performed as previously described [38, 39]. The cells were grown in estrogen-free media for 48h and treated with 10-7M E2 or vehicle for 4 h. Cells were cross-linked with 1% formaldehyde at room temperature for 10 min, resuspended in lysis buffer with 1× protease inhibitor cocktail (Roche Molecular Biochemicals) and sonicated three times for 10 s each at the maximum setting (Handy Sonic, Model UR-20P) followed by centrifugation for 10 min. Supernatants were collected and diluted in buffer (1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris-HCl, pH 8.1) followed by immunoclearing with 2 μg sheared salmon sperm DNA-20 μl preimmune serum and protein A-Sepharose (45 μl of 50% slurry in 10 mM Tris-HCl, pH 8.1, 1 mM EDTA) for 2 hr at 4oC. Immunoprecipitations were performed overnight at 4oC with specific antibodies. Protein A-Sepharose beads were added and then washed sequentially with low-salt buffer, high-salt buffer, LiCl buffer, and TE buffer. The protein-DNA complexes were eluted and the crosslinking was reversed at 65 oC for 6 h. DNA fragments were purified with a DNA purification kit (DIAquick, Qiagen) and analyzed by regular PCR. Primer sequences for pS2 gene were forward: 5'-GGCCATCTCTCACTA TGAATCACTTCTGC-3' and 5'-GGCAGGCTCTGTTTGCTTAAAGAGCG-3' [40]. Primer sequences for c-Myc were forward: 5'-AGGCGCGCGTAGTTAATTCAT-3' and 5'-CGCCCTCTGCTTTGGGA-3'[41]. The precipitated DNA was analyzed by regular PCR. The relative enrichment was shown using agarose gel electrophoresis.
ChIP re-ChIP experiments were performed as described previously [38, 42]. Complexes were eluted from the first ChIP by incubation with 10mM DTT at 37oC for 30 min and diluted 1:50 in buffer as previously described followed by re-ChIP with the antibodies as indicated.
Cell Proliferation and Colony Formation Assay
Cells were plated at a density of 7×104 cells per well in 12-well plates. Cells were trypsinized and counted using a hemocytometer stained with trypan blue each day. For colony formation assay, 3×104 cells were maintained in medium 10%CSS supplemented with 10-7M E2 for 7 days. Cell cultures were then fixed and stained with Coomassie blue dye.
FACS Analysis
The cells were grown in six-well plates for 24 h with 10-7M E2 or ethanol vehicle. Then, the cells were dissociated with trypsin, resuspended in PBS, and fixed in ice-cold 70% ethanol. Next, the cells were incubated in propidium iodide/RNAse solution (5mg propidium iodide, 3.7 mg EDTA, 0.1ml Triton X-100, and 0.2ml of 10mg/ml RNAse dissolved in 100 ml PBS) at 37°C for 1 h. The cell-cycle analysis was performed by a FACS flow cytometer.
Transwell Assays
Migration and invasion assays were performed using Boyden chambers as described [25]. Cells were plated on the upper well of a Boyden chamber at a concentration of 104 cells per well in 100 μl serum-free DMEM, the lower compartments were filled with 600 μl DMEM containing 10% serum. After incubating at 37°C for 20 h, non-invaded cells were removed from the upper surface of the filter with a cotton swab, and the invaded cells on the lower surface of the filter were fixed with 95% ethanol, stained with Coomassie blue dye, and photographed. Cell numbers were manually counted in five random fields (×40) per filter.
Xenograft Tumor Growth
MCF-7 cells infected with lentivirus carrying shCtrl or shMDC1 were collected in 100 μl sterile PBS and matrigel (Sigma) mixture and inoculated subcutaneously into the 5-week-old female BALB/C-null mice at 5×106 cells per injection site. E2 pellets (0.72mg per pellet, 60 days release, Innovative Research of America) were subcutaneously implanted 1 day before the MCF-7 cell injection (N=8). Tumors was measured weekly using a vernier caliper and the volume was calculated according to the formula π/6×length×width2. 8 weeks after inoculation, mice were killed in keeping with the policy of the humane treatment of tumor-bearing animals. All procedures involved in animal experiments were approved by the Animal Ethics Committee of China Medical University.
Immunohistochemical Analysis
Formalin-fixed paraffin-embedded sections of tissue specimens were prepared from Shengjing hospital affiliated to China Medical University. Multicentre ethical approval for data collection and tissue use was granted by the Human Research Ethics Committee of the above hospital. Tissue sections were deparaffinized, rehydrated, removed endogenous peroxidase, boiled for antigen retrieval, followed by incubation with anti-MDC1 antibody and streptavidin-perosidase-conjugated second antibodies (Fuzhou Maixin Biotech. Co., Ltd.). The signals were visualized with diaminobenzidine and the nuclei were counterstained with hematoxylin as previously described [43]. Immunohistochemistry scores (IHC scores) derive from a semiquantitative assessment of both staining intensity (scale 0-3) and the percentage of positive cancer cells (scale 0%-100%). These two numbers were multiplied to generate an IHC score with a range of 0 to 3. Slides were viewed and scored using standard light microscopy.
Statistical Analysis
Statistical analyses of this study were performed using the SPSS (17.0) statistical software program. For immunohistochemisty, Mann-Whitney U test was used to determinate the significant difference between hyperplasia and breast cancer. For other results, two-sided Student's t-test was used to determinate the significant difference. *P<0.05, **P<0.01.
Results
Expression of MDC1 in Clinical Breast Cancer Samples
It is proved that MDC1-deficient mice exhibited the increased spontaneous tumor incidence, indicating that MDC1 may be a potential cancer suppressor. In order to determine the role of MDC1 in breast cancer, we first detected MDC1 protein expression levels in 28 paired breast cancer tissues and their matched adjacent noncancerous breast tissues by western blotting. As shown in Figure 1A and B, the expression levels of MDC1 in breast cancer tissues were significantly lower than those in the matched adjacent tissues (p<0.01). We further examined the expression of MDC1 in clinical breast biopsies including 70 cases of breast cancer in different clinical stages and 30 cases of benign breast hyperplasia. Compared with benign breast hyperplasia, immunohistochemistry (IHC) results using a well characterized MDC1 antibody showed lower expression intensity in breast cancer samples (p<0.01). Moreover, the decreased of MDC1 protein levels in breast cancer tissues concomitant with the increase in clinical stages (Figure 1E and F). Specificity of antibody against MDC1 was confirmed by IHC (Figure 1D) and western blotting experiments (Figure 5B).
MDC1 is low-expressed in breast cancer tissues. (A) Expression of MDC1 and ERα proteins in breast tumor tissues (T) and adjacent noncancerous tissues (N). GAPDH was used as a loading control. (B) MDC1 expression levels in breast tumor tissues (T) and adjacent noncancerous tissues (N) were quantified by densitometry. ** P <0.01. (C) MDC1 expression levels in ERα positive (ERα+) as well as ERα negative (ERα-) breast tumor tissues compared with adjacent noncancerous tissues (p=0.022). (D) Immunohistochemical (IHC) staining of benign breast hyperplasia with MDC1 antibody (a) and IgG (b). (E) MDC1 IHC staining images in clinical breast specimen of hyperplasia and breast cancer in different clinical stages. (F) Quantitative analysis of MDC1 IHC intensities in benign breast hyperplasia and breast cancer specimens of clinical stage I-III. * P<0.05, ** P<0.01.
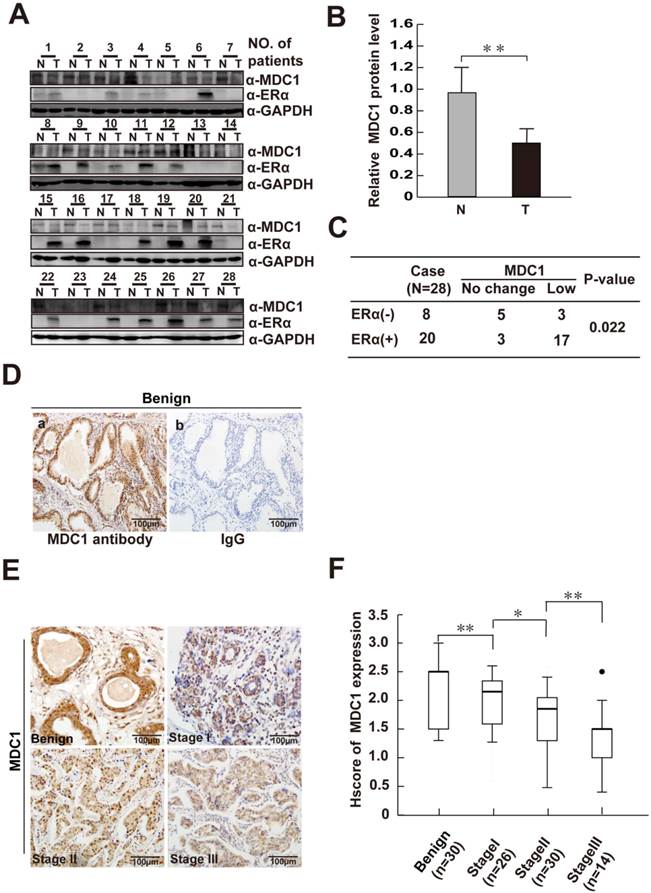
ERα and its co-regulators play crucial and complicated roles in breast cancer progression. We further ask whether or not MDC1 is correlated with ERα in breast cancer samples. Western blotting was performed with the antibody against ERα using the same lysates from the clinical tissues for detecting MDC1 expression as indicated in Figure 1A. We further analyzed the MDC1 expression in ERα positive (ERα+, 20 cases) and negative breast cancer samples (ERα-, 8 cases) as shown in Figure 1C. The low expression rate of MDC1 in ERα+ breast cancer tissues (17/20) is obviously higher than that in ERα- breast cancer tissues (3/8). Taken together, these data showed that the expression of MDC1 is lower in breast cancer, and the diminished of MDC1 protein levels in breast cancer tissues correlative with the increase in clinical stages. Moreover, MDC1 is under expressed in ERα+ breast cancer patients. These results suggest that MDC1 might contribute to the suppression of breast cancer.
MDC1 physically Associates with ERα in Human Cells
Given that there is a lower expression of MDC1 in breast cancer in different clinical stages, and ERα is an important predictor of breast cancer prognosis and therapeutic target. We next investigated whether MDC1 associates with ERα. Co-immunoprecipitation (Co-IP) experiments were performed (Figure 2A and B). HEK293 cells were transiently transfected with Flag-tagged MDC1 (Flag-MDC1) and ERα expression plasmids. The antibody against ERα or Flag was separately used for Co-IP and precipitated proteins were detected by western blotting as indicated (Figure 2A). To confirm the association between the endogenous MDC1 and ERα, Co-IP experiments were performed in MCF-7 cells using the antibody against ERα or MDC1 as indicated (Figure 2B). The above results indicated that exogenously expressed or the endogenous MDC1 interacts with ERα in E2-independent and -dependent manner, and the association between MDC1 and ERα with the treatment of E2 is stronger than that without E2 treatment.
MDC1 interacts with ERα. (A) MDC1 interacts with ERα in HEK293 cells. Expression plasmids encoding Flag-MDC1 and ERα were cotransfected in HEK293 cells. Equal amounts of cell lysates were subjected to immunoprecipitation experiments and western blot with anti-Flag or anti-ERα antibodies as indicated. (B) Endogenous MDC1 associates with ERα in MCF-7 cells. The cell lysates were immunoprecipitated with the antibodies against ERα or MDC1 as indicated. Precipitates were detected by western blot using the indicated antibodies. A 10% fraction of the cell lysates was loaded as input. IgG was used as a negative control. (C) Flag-MDC1 directly interacts with GST-ERα 282-595aa fragment. GST Pull-down experiments using synthesized Flag-MDC1 with in vitro transcript and translate Kit were incubated with different GST-ERα fusion proteins as indicated. Bound proteins were analyzed by Flag antibody and equal loading of GST-ERα fusion proteins was assessed by coomassie brilliant blue staining. Flag-MDC1 bound to GST-ERα 282-595aa, but not to GST-ERα 29-180aa, demonstrating that the C-terminus of ERα is required for MDC1 interaction. * is placed to indicate GST or the special GST-ERα deletion mutant proteins.
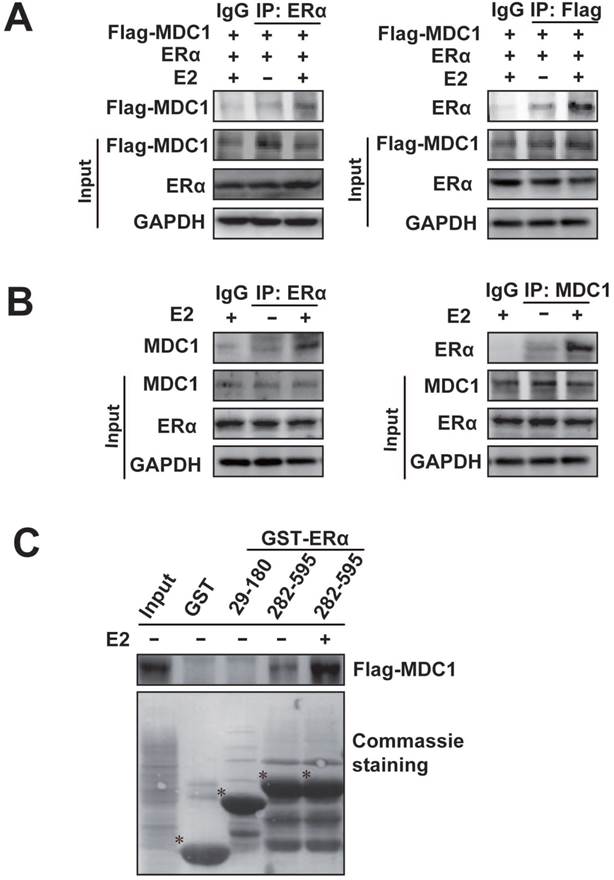
We further asked which domains of ERα interact with MDC1. GST pull-down experiments were performed using two GST fused fragments of ERα. The results demonstrated that MDC1 directly interacted with ERα 282-595aa fragment covering the ligand binding domain, not ERα 29-180aa fragment covering AF-1 in the absence or presence of E2. In agreement with the results from IP experiments, the interaction between MDC1 and ERα 282-595aa fragment in the presence of E2 was stronger than that in the absence of E2.
Subcellular Distribution of MDC1 and ERα in Cells
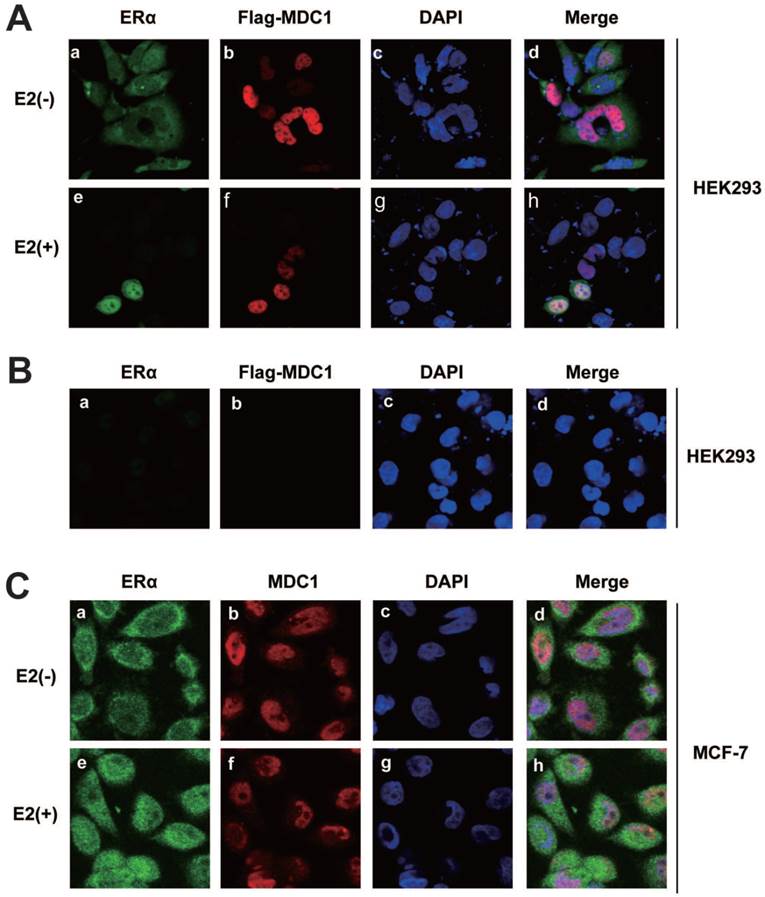
To study the subcellular localization of MDC1 and ERα, HEK293 cells were co-transfected with ERα and Flag-MDC1 expression plasmids. By immunofluorescence experiments, we observed that MDC1 was distributed in the nucleus (Figure 3A b and f). ERα was mainly distributed in cytoplasm in the absence of E2, whereas most of ERα was located in the nucleus and co-located with MDC1 in the presence of E2 (Figure 3A a, e, and h). HEK293 cell itself immunofluorescence staining was used as a negative control for the subcellular localization of Flag-MDC1 and ERα (Figure 3B). We further examine the endogenous subcellular localization of MDC1 and ERα in MCF-7 cells, and in agreement with the above results, we observed that the endogenous MDC1 co-localizes with ERα in the nucleus in the presence of E2 (Figure 3C). Taken together, these data demonstrated that MDC1 and ERα were distributed in the nucleus with the treatment of E2.
Subcellular localization of MDC1 and ERα. (A) HEK293 cells were cotransfected with Flag-MDC1 and ERα and then cultured in E2-free medium for 4 hours. Cells were then treated with or without E2 (10-7M). 24 hours later, cells were fixed and stained with the indicated antibodies, and then detected by the 2nd antibodies, including anti-rabbit FITC (a and e in green) and anti-mouse Cy5 (b and f in red). DAPI was used to visualize the nucleus (c and g in blue). Merged images were shown as indicated (d and h). Original magnification, ×60. (B) HEK293 cells were cultured in E2-free medium for 24 hours and then fixed and stained as (A). (C) Subcellular localizations of the endogenous MDC1 and ERα in breast cancer cells. MCF-7 cells were cultured in E2-free medium for 4 hours and then treated with or without E2 for 24 hours. Cells were then fixed and stained with the indicated antibodies, and then detected as (A).
MDC1 Enhances ERα-induced Transactivation
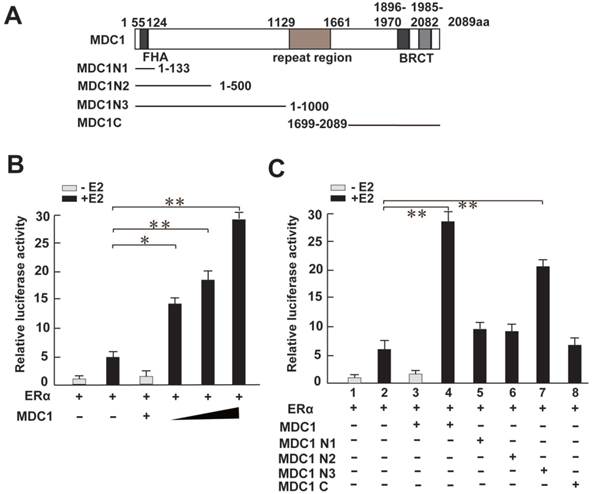
Having shown MDC1 associates with ERα in breast cancer cells, we next asked whether MDC1 functionally regulates ERα-mediated transactivation in breast cancer cells. To this end, we turned to perform a luciferase reporter assay. We generated MDC1 full length (MDC1) and a series of MDC1 truncated mutant expression plasmids as indicated (Figure 4A). HEK293 cells were co-transfected MDC1 or its truncated mutants, ERα expression plasmids, and pGL-ERE-AdML reporter plasmid followed by luciferase assay. The results demonstrated that MDC1 enhanced ERα-induced transactivation in the presence of E2, and MDC1 leaded to up to 5-fold increase in ERα function in a dose-dependent manner (Figure 4B). Compared with the positive regulation function of MDC1 on ERα-mediated transcriptional activity, MDC1 N1 (1-133aa), MDC1 N2 (1-500aa), or MDC1 C (1699-2089aa) had no visible effect on ERα-induced transactivation, while MDC1 N3 (1-1000aa) significantly enhanced ERα function (Figure 4C). These results suggested that MDC1 enhanced ERα-induced transactivation in a dose-dependent manner, moreover, MDC1 500-1000aa fragment was most, if not all, required for co-activator function of MDC1 on ERα action.
MDC1 enhances the ERα-induced transactivation. (A) Diagram representation of MDC1 and its truncated mutants, including MDC1 N1, N2, N3, and C fragments as indicated. (B and C) HEK293 cells were cotransfected with ERα, pGL-ERE-AdML reporter plasmid, and increased amounts of Flag-MDC1 or several MDC1 truncated mutants in the presence or absence of E2 (10-8M). Cells were then lysated and assayed for luciferase activity. MDC1 enhances ERα-mediated transactivation in a dose-dependent manner, and MDC1 N3, but not N1, N2, and C fragments increases ERα-induced transcriptional activity. In (B)-(C), the error bars represent mean ± SD (N≥4). * P<0.05, ** P<0.01.
To further confirm whether MDC1 up-regulates the endogenous ERα target genes in MCF-7 cells, we analyzed the estrogen-induced expression of ERα target genes, including c-Myc [3, 9], efp, GREB1, pS2, and p21 [7, 8] after shRNA-mediated silencing of MDC1 gene expression by using lentivirus containing MDC1 shRNA (shMDC1) [36]. The knockdown efficacy of lentivirus containing shMDC1 infected MCF-7 cells was analyzed by quantitative real-time PCR (qRT-PCR) and western blot (Figure 5A and B). As shown in Figure 5C, qRT-PCR results demonstrated that the induction of pS2 and p21 by E2 was significantly reduced by knockdown of MDC1 in MCF-7 cells, whereas mRNA expression of ERα or other ERα target genes such as c-Myc, efp, GREB1 had no obvious alteration. Moreover, western blotting results showed that MDC1 depletion obviously reduced p21 or pS2, but not the expression of ERα or c-Myc protein in MCF-7 cells (Figure 5D). These results suggested that knockdown of MDC1 reduced partial ERα target genes, including p21, which is required for tumor suppression in the transcription level.
MDC1 depletion decreases the expression levels of partial ERα target genes and MDC1 is recruited to the cis-regulatory element of ERα target gene. (A and B) Verification of down-regulation of MDC1 expression. MCF-7 cells were infected with shRNA against MDC1 (shMDC1) lentivirus or a control shRNA lentivirus. Expression levels of MDC1 were analyzed by qRT-PCR (A) and western blotting (B). (C) Induction of partial ERα target genes mRNA expression is expressed as the ratio of ERα target genes mRNA levels normalized to GAPDH levels in the absence or presence of E2 (10-7M). (D) The expression levels of ERα target genes (p21, pS2, and c-Myc) were analyzed by western blot in the whole cell lysates with knockdown of MDC1 by shMDC1 with or without E2. The blot was reprobed with anti-GAPDH as a loading control. (E-F) MCF-7 cells carrying shMDC1 or shCtrl were incubated with or without E2. ChIP assay was performed with the indicated antibodies. The precipitated chromatin was amplified by PCR using primers flanking the cis-regulatory element of pS2 or c-Myc. (G) MDC1 and ERα are predominantly recruited to the ERE of pS2 in the presence of E2. ChIP re-ChIP experiments were performed with the anti-MDC1 and anti-ERα antibodies as indicated. (H and I) Confirmation of knockdown of ERα expression. MCF-7 cells were transfected with siRNA against ERα (siERα). Expression levels of ERα were analyzed by qRT-PCR (H) and western blotting (I). (J) ERα depletion reduces the recruitment of MDC1 to the ERE of pS2. MCF-7 cells transfected with siERα were incubated with or without E2, ChIP assay was performed as E. All experiments were repeated at least 3 times. Results represent mean ± SD of three independent experiments. * P<0.05, ** P<0.01.
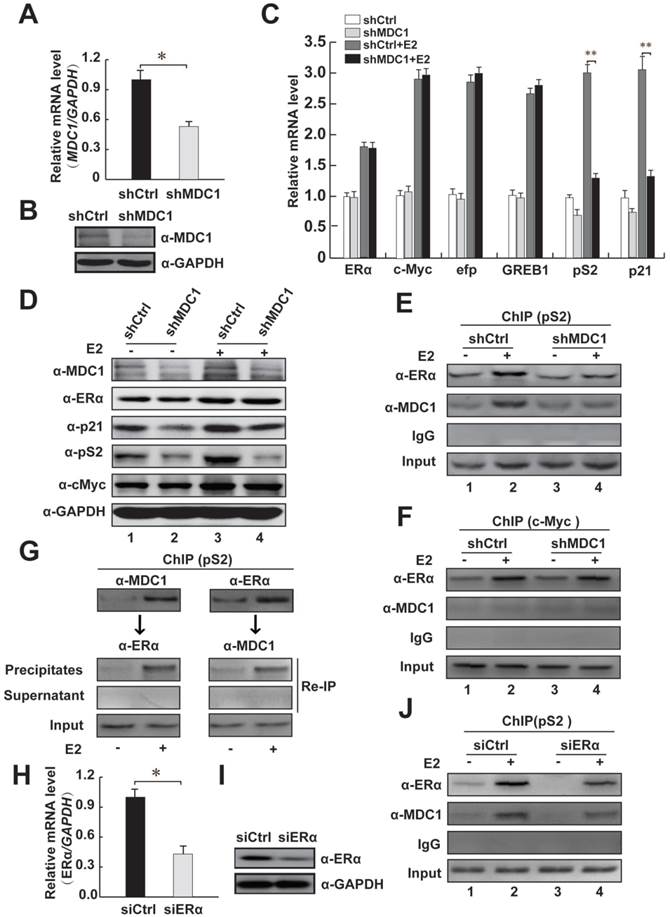
MDC1 is selectively Recruited to Estrogen Response Elements of ERα Target Genes
We thus ask whether MDC1 is recruited to estrogen response elements (ERE) of its target genes, ChIP assays were performed in MCF-7 cells. Our data showed that MDC1 or ERα was recruited to the ERE region of ERα target gene pS2 (Figure 5E). Meanwhile, our ChIP assay data also demonstrated that MDC1 was not recruited to ERα target gene c-Myc without expression alterations in MDC1 knockdown cells (Figure 5F). The recruitment of MDC1 to the promoter of pS2 was remarkably diminished by MDC1 knockdown. Interestingly, we observed that the recruitment of ERα was reduced by down-expression of MDC1, suggesting that MDC1 might be required for the binding of ERα to the ERE of pS2 (Figure 5E). We further perform ChIP assay to examine whether ERα knockdown would influence MDC1 recruitment to the ERE of pS2 in MCF-7 cells. The results indicated that ERα depletion reduced MDC1 recruitment to ERE of pS2 (Figure 5J). The efficiency of knockdown of ERα expression by siERα was confirmed by qRT-PCR and western blotting (Figure 5H and I). Moreover, to assess whether MDC1 and ERα are recruited together to the ERE of pS2, ChIP re-ChIP experiments were performed using the antibodies as indicated (Figure 5G). Together with the results that MDC1 was identified as a coactivator of ERα, MDC1 was able to be recruited to the ERE of pS2 together with ERα and enhanced the transcription level of ERα target gene p21, which plays important roles in tumor suppression, these results implied that MDC1 may suppress breast cancer progression, if not all, at least partially through influencing of the status of ERα target genes.
MDC1 Inhibits Breast Cancer Cell Progression in Vitro
To further investigate the potential biological function of MDC1 in breast cancer progression, we analyzed the influence of MDC1 expression on cell growth of MCF-7 cell line. We compared the proliferative capability between shMDC1-MCF-7 cells and shCtrl-MCF-7 cells. Colony formation assay demonstrated that knockdown of MDC1 increased colony numbers in MCF-7 cells (Figure 6A). The growth rate assay also showed that knockdown of MDC1 resulted in a significant increase in MCF-7 cell numbers (Figure 6B). These results suggest that MDC1 inhibits cell growth in MCF-7 cells, and the effect of MDC1 on suppression of breast cancer cell growth is stronger in the presence of E2 than that in the absence of E2.
Knockdown of MDC1 promotes MCF-7 cell progression. (A-B) Knockdown of MDC1 promotes the proliferation of MCF-7 cells. MCF-7 cells infected with lentiviruses harboring shRNA against MDC1 (shMDC1) were grown with or without treatment of E2 (10-7M) and subjected to colony formation (A) and cell account assays (B). A representative colony formation assay is shown in A. Each bar represents the mean ± SD for triplicate experiments. (C-D) Knockdown of MDC1 promotes S-phase entry in MCF-7 cells. MCF-7 cells carrying shMDC1 were grown in the presence or absence of E2 and subjected to flow cytometry assays. The average percentages of triplicate experiments were shown in histogram in D. (E) MDC1 depletion promotes cell migration of MCF-7 cells. MCF-7 cells with stably knockdown of MDC1 were plated into the upper chamber of the filters with or without treatment of E2, and detected after 24 hours. The migrated cells were quantified with Coomassie blue staining. (F) MDC1 depletion promotes cell invation of MCF-7 cells. A transwell invasion assay was similar performed as described in E, the chambers were pre-coated with matrigel (30µl/well) and invasive cells were detected after 36 hours. Original magnification, ×40. Each bar represents the mean ± SD for triplicate experiments. * P<0.05.
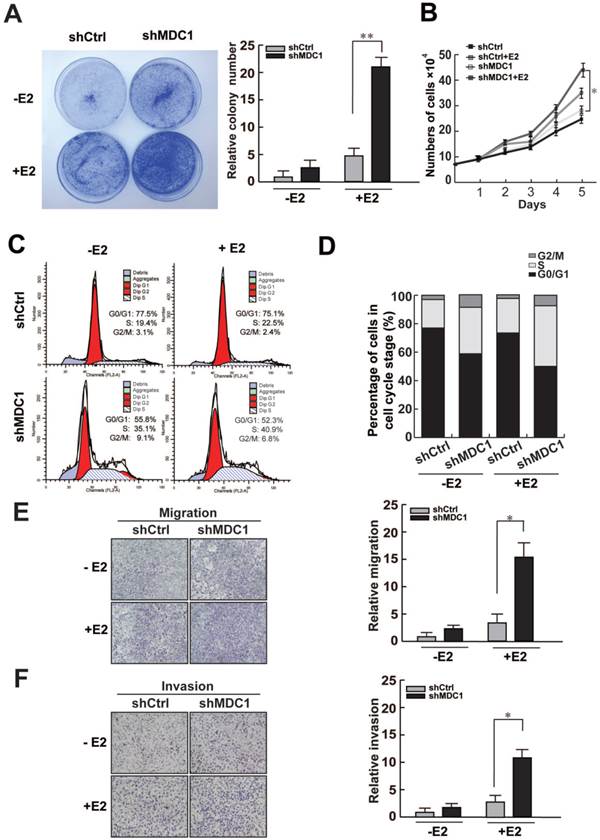
Since MDC1 can enhance expression of p21, which is a negative regulator of cell cycle, we assess whether MDC1 would influence the cell cycle progression. Flow cytometry was performed with or without the treatment of E2. The results demonstrated that knockdown of MDC1 significantly increased S-phase entry, and slightly increased the proportion of G2/M phase cells in MCF-7 cells (Figure 6C and D). Moreover, transwell experiments were also be performed to examine the function of MDC1 on the migration and invasion of breast cancer cells. The results showed that knockdown of MDC1 exhibited significantly stronger ability of migration and invasion in MCF-7 cells in the presence of E2 (Figure 6E and F). Our data demonstrated that MDC1 participates in the suppression of proliferation, invasion and migration of breast cancer cells.
MDC1 Suppresses Breast Cancer Cell Growth in Vivo
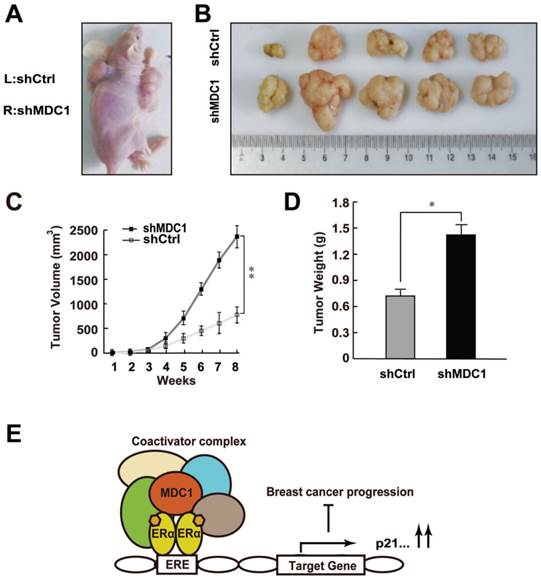
Since MDC1 was able to inhibit the growth of breast cancer cells in vitro, we want to further examine the role of MDC1 in estrogen-stimulated cell proliferation in vivo. MCF-7 cells infected with lentivirus carrying shMDC1 or shCtrl were individually injected into the right (shMDC1) or left (shCtrl) flank of the 5-week-old female BALB/C-null mice (n = 8) at 5×106cells per injection site (Figure 7A). E2 pellets were subcutaneously implanted as described in Materials and Methods. Subsequently, we measured the tumor volume every week after hypodermic injection. As shown in Figure 7B, the size of tumor grown from shMDC1-MCF-7 cells was mostly much bigger than that from shCtrl-MCF-7 cells. Furthermore, the tumor volume of shMDC1-MCF-7 cells exhibited markedly greater rate of grown than that of shCtrl-MCF-7 cells (Figure 7C). After 8 weeks, tumors were harvested and weighed. The tumor weight of shMDC1-MCF-7 cells was also higher than that of shCtrl-MCF-7 cells (Figure 7D). These results indicated that MDC1 plays an important role in suppression of estrogen-stimulated growth of MCF-7 tumors.
Knockdown of MDC1 promotes breast cancer cell growth in null mice. (A) The effect of MDC1 depletion on the growth of transplanted tumor in null mice. 5-week-old female BALB/C-null mice were subcutaneously injected with 5×106 MCF-7 cells infected with lentiviruses harboring shCtrl or shMDC1. (B) Representative photographs of tumors in each group isolated from nude mice after 8 weeks injection. Tumor growth was monitored with the treatment of E2 pellets. (C) Tumors was measured weekly using a vernier caliper and the volume was calculated according to the formula π/6×length×width2. MDC1 depletion causes an increase in tumor volume. Each point represents the mean ± SD for different animal measurements (n=8). (D) Tumor was excised and weighed after 8 weeks. Average tumor weight of two groups was shown. Results represent the mean ± SD. P values were determined by student's t test. * P<0.05, ** P<0.01. (E) Schematic representation of MDC1 co-activator functions on ERα-induced transactivation and its role in suppression of breast cancer.
Discussion
Recent studies have demonstrated that loss of MDC1 expression in lung and breast cancer and the increase of tumor incidence in MDC1 knockout mice [32, 44]. However, not much is known regarding the mode of MDC1 functioning, MDC1 interacting molecular partner, and the effect of MDC1 upon ERα-induced transactivation. Our data showed that overexpression of MDC1 led to a dramatic increase of ERα target gene transcription as measured by ERE-luciferase function assay in a dose-dependent manner, and this raises the possibility that MDC1 could act as a positive regulator of ERα pathway. Indeed, we observed that down-regulation of MDC1 decreased the expression of the endogenous estrogen-responsive genes. In addition, ChIP assay and ChIP re-ChIP experiments showed that MDC1 together with ERα was recruited to the ERE of ERα target gene. This would open a new avenue of study recognizing other interacting partners of ERα-co-activator complex.
Estrogens have long been known to have mitogenic function in breast cancer cell lines and in breast tumor. The modulation of ERα-induced transactivation has been a topic of interest for large number of researchers, due to its important roles in the development of effective therapeutic target to treat breast cancer [2, 45]. Approximately 30% of breast carcinomas lack ERα expression. Presumably, these breast cancers become estrogen independent through genetic alterations that bypass the requirement for ERα-dependent stimulation of cell proliferation. In this paradigm, loss of ERα expression or down-regulation of ERα function should provide selective growth advantage in the tumor microenvironment by increasing the probability of invasion and metastasis. It has been proved that loss of ERα expression promotes tumor progression through modulation of snail and E-cadherin expression [23]. Previous work has documented that MTA1 and LMO4 acting as co-repressors of ERα mainly participate in the invasiveness of breast cancer [25, 26]. In agreement with the important observation as above in the contrary direction, our results showed that expression of MDC1 was lower in the breast cancer, and down-expression of MDC1 promoted the breast cancer cell growth in mice. The effect of MDC1 upon ERα signaling described in this work demonstrates that co-activator of ERα may contribute to the suppression of breast cancer. It suggests the significant implications for the co-regulator of ERα in breast cancer therapy.
In the present study, we have identified MDC1 functions as an interacting partner of ERα. MDC1 effectively co-activated ERα-mediated transactivation functions, and down-regulation of MDC1 expression resulted in a significant inhibition of ERα actions. On the other hand, our recent studies have demonstrated that MDC1 also associates with AR and co-activates AR-mediated transactivation via increasing the recruitment of histone acetyltransferase GCN5 [46]. Thus, in addition to the role of MDC1 in DNA damage checkpoint, our findings provide the new function of MDC1 on modulation of steroid hormone receptor-mediated transcriptional activity. Our data further indicates that MDC1 directly interacts with ERα 282-595aa fragment covering ERα AF-2. As for the functional domain of MDC1 in regulating ERα action, the results in this study demonstrated that MDC1 500-1000aa fragment is most, if not all, required for co-activator function of MDC1 on ERα function. While, our recent published data showed that MDC1 directly interacts with AR 1-532aa fragment covering AR AF-1, and the functional domains of MDC1 for co-activating AR action are located in MDC1 500-1000aa or fragment C-terminal domain containing 1699-2089aa [46]. Furthermore, in agreement with the previous studies [32], our current results also demonstrated that MDC1 is lower expressed in breast cancer or prostate cancer [46] tissues from the clinical patients. However, very little is known regarding the mechanistic role played by it at the molecular level as a tumor suppressor. Our data raise the possibility that MDC1 up-regulation might suppress the process of breast cancer/prostate cancer by enhancing ERα or AR-mediated transactivation functions, finally, leading to decreased invasion and migration of breast or prostate cancer.
Abbreviations
ERα: estrogen receptor α; ERE: estrogen response element; MDC1: Mediator of DNA damage checkpoint 1; NFBD1: nuclear factor with BRCT domains protein 1; DDR: DNA damage response; FHA: forkhead-associated domain; BRCT: BRCA1 carboxyl-terminal domains; γH2AX: histone variant H2AX; E2: 17β-estradiol
Acknowledgements
We appreciate Dr. Yunlong Huo, Dr. Tao Wen and Ms Hongyan Zhang for helpful technique support.
This work was supported by 973 Program Grant 2013CB945201 from the Ministry of Science and Technology of China and the grants from the National Natural Science Foundation of China (30871390, 31171259, 31271364 for Yue Zhao; 31401115 for Chunyu Wang). This work was also supported by the Ministry of Education fund innovation team (IRT 13101) and Ministry of Education Science and technology research projects (No 213008A).
Conflict of interest
We confirm that none of the authors have a financial interest related to this work and there are no conflicts of interest associated with this publication.
References
1. McKenna NJ, O'Malley BW. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell. 2002;108:465-74
2. Jordan VC. Selective estrogen receptor modulation: concept and consequences in cancer. Cancer cell. 2004;5:207-13
3. Shang Y, Brown M. Molecular determinants for the tissue specificity of SERMs. Science. 2002;295:2465-8
4. Liang J, Shang Y. Estrogen and cancer. Annual review of physiology. 2013;75:225-40
5. Prall OWJ, Rogan EM, Musgrove EA, Watts CW, Sutherland RL. c-Myc or cyclin D1 mimics estrogen effects on cyclin E-Cdk2 activation and cell cycle reentry. Molecular and cellular biology. 1998;18:4499-508
6. Frietze S, Lupien M, Silver PA, Brown M. CARM1 regulates estrogen-stimulated breast cancer growth through up-regulation of E2F1. Cancer research. 2008;68:301-6
7. Margueron R, Licznar A, Lazennec G, Vignon F, Cavailles V. Oestrogen receptor alpha increases p21(WAF1/CIP1) gene expression and the antiproliferative activity of histone deacetylase inhibitors in human breast cancer cells. The Journal of endocrinology. 2003;179:41-53
8. Mandal S, Davie JR. Estrogen regulated expression of the p21 Waf1/Cip1 gene in estrogen receptor positive human breast cancer cells. Journal of cellular physiology. 2010;224:28-32
9. Dubik D, Dembinski TC, Shiu RP. Stimulation of c-myc oncogene expression associated with estrogen-induced proliferation of human breast cancer cells. Cancer research. 1987;47:6517-21
10. Green KA, Carroll JS. Oestrogen-receptor-mediated transcription and the influence of co-factors and chromatin state. Nature Reviews Cancer. 2007;7:713-22
11. Ofiate SA, Tsai SY, Tsai M-J, O'Malley BW. Sequence and characterization of a coactivator for the steroid hormone receptor superfamily. Science. 1995;270:1354-7
12. Ding XF, Anderson CM. et al. Nuclear receptor-binding sites of coactivators glucocorticoid receptor interacting protein 1 (GRIP1) and steroid receptor coactivator 1 (SRC-1) multiple motifs with different binding specificities. Molecular Endocrinology. 1998;12:302-13
13. Anzick SL, Kononen J, Walker RL, Azorsa DO, Tanner MM, Guan X-Y. et al. AIB1, a Steroid Receptor Coactivator Amplified in Breast and Ovarian Cancer. Science. 1997;277:965-8
14. Chakravarti D, LaMorte V. et al. Role of CBPP300 in nuclear receptor signalling. Nature. 1996;383:99-103
15. Blanco JCG, Minucci S. et al. The histone acetylase PCAF is a nuclear receptor coactivator. Genes&development. 1998;12:1638-51
16. Lavinsky RM, Jepsen K, Heinzel T. et al. Diverse signaling pathways modulate nuclear receptor recruitment of N-CoR and SMRT complexes. Proceedings of the National Academy of Sciences. 1998;95:2920-5
17. Popov VM, Zhou J, Shirley LA, Quong J, Yeow WS, Wright JA. et al. The cell fate determination factor DACH1 is expressed in estrogen receptor-alpha-positive breast cancer and represses estrogen receptor-alpha signaling. Cancer research. 2009;69:5752-60
18. Al-Dhaheri M, Wu J, Skliris GP. et al. CARM1 is an important determinant of ERalpha-dependent breast cancer cell differentiation and proliferation in breast cancer cells. Cancer research. 2011;71:2118-28
19. Zhang H, Xie X, Zhu X. et al. Stimulatory cross-talk between NFAT3 and estrogen receptor in breast cancer cells. The Journal of biological chemistry. 2005;280:43188-97
20. DeSantis C, Ma J, Bryan L, Jemal A. Breast cancer statistics, 2013. CA Cancer J Clin. 2014:52-62
21. Manavathi B, Dey O, Gajulapalli VN, Bhatia RS, Bugide S, Kumar R. Derailed estrogen signaling and breast cancer: an authentic couple. Endocr Rev. 2013;34:1-32
22. Le Romancer M, Poulard C, Cohen P, Sentis S, Renoir JM, Corbo L. Cracking the estrogen receptor's posttranslational code in breast tumors. Endocr Rev. 2011;32:597-622
23. Fujita N, Jaye DL, Kajita M, Geigerman C, Moreno CS, Wade PA. MTA3, a Mi-2/NuRD complex subunit, regulates an invasive growth pathway in breast cancer. Cell. 2003;113:207-19
24. Vesuna F, Lisok A, Kimble B, Domek J, Kato Y, van der Groep P. et al. Twist contributes to hormone resistance in breast cancer by downregulating estrogen receptor-alpha. Oncogene. 2012;31:3223-34
25. Mazumdar A, Wang RA, Mishra SK, Adam L, Bagheri-Yarmand R, Mandal M. et al. Transcriptional repression of oestrogen receptor by metastasis-associated protein 1 corepressor. Nature cell biology. 2001;3:30-7
26. Singh RR, Barnes CJ, Talukder AH, Fuqua SA, Kumar R. Negative regulation of estrogen receptor alpha transactivation functions by LIM domain only 4 protein. Cancer research. 2005;65:10594-601
27. Stewart GS, Wang B, Bignell CR, Taylor AMR, Elledge SJ. MDC1 is a mediator of the mammalian DNA damage checkpoint. Nature. 2003;421:961-6
28. Goldberg M, Stucki M, Falck J. et al. MDC1 is required for the intra-S-phase DNA damage checkpoint. Nature. 2003;421:952-6
29. Lou Z, Minter-Dykhouse K, Wu X, Chen J. MDC1 is coupled to activated CHK2 in mammalian DNA damage response pathways. Nature. 2003;421:957-61
30. Lou Z, Minter-Dykhouse K, Franco S, Gostissa M, Rivera MA, Celeste A. et al. MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Molecular cell. 2006;21:187-200
31. Nakanishi M, Ozaki T, Yamamoto H, Hanamoto T, Kikuchi H, Furuya K. et al. NFBD1/MDC1 associates with p53 and regulates its function at the crossroad between cell survival and death in response to DNA damage. The Journal of biological chemistry. 2007;282:22993-3004
32. Bartkova J, Horejsi Z, Sehested M, Nesland JM, Rajpert-De Meyts E, Skakkebaek NE. et al. DNA damage response mediators MDC1 and 53BP1: constitutive activation and aberrant loss in breast and lung cancer, but not in testicular germ cell tumours. Oncogene. 2007;26:7414-22
33. Yanagisawa J, Kitagawa H, Yanagida M, Wada O, Ogawa S, Nakagomi M. et al. Nuclear receptor function requires a TFTC-type histone acetyl transferase complex. Molecular cell. 2002;9:553-62
34. Tateishi Y, Kawabe Y, Chiba T, Murata S, Ichikawa K, Murayama A. et al. Ligand-dependent switching of ubiquitin-proteasome pathways for estrogen receptor. The EMBO journal. 2004;23:4813-23
35. Chang EC, Charn TH, Park SH, Helferich WG, Komm B, Katzenellenbogen JA. et al. Estrogen Receptors alpha and beta as determinants of gene expression: influence of ligand, dose, and chromatin binding. Mol Endocrinol. 2008;22:1032-43
36. Chapman JR, Jackson SP. Phospho-dependent interactions between NBS1 and MDC1 mediate chromatin retention of the MRN complex at sites of DNA damage. EMBO reports. 2008;9:795-801
37. Zhao Y, Goto K, Saitoh M, Yanase T, Nomura M, Okabe T. et al. Activation function-1 domain of androgen receptor contributes to the interaction between subnuclear splicing factor compartment and nuclear receptor compartment. Identification of the p102 U5 small nuclear ribonucleoprotein particle-binding protein as a coactivator for the receptor. The Journal of biological chemistry. 2002;277:30031-9
38. Zhao Y, Takeyama K, Sawatsubashi S, Ito S, Suzuki E, Yamagata K. et al. Corepressive action of CBP on androgen receptor transactivation in pericentric heterochromatin in a Drosophila experimental model system. Molecular and cellular biology. 2009;29:1017-34
39. Zhao Y, Lang G, Ito S, Bonnet J, Metzger E, Sawatsubashi S. et al. A TFTC/STAGA module mediates histone H2A and H2B deubiquitination, coactivates nuclear receptors, and counteracts heterochromatin silencing. Molecular cell. 2008;29:92-101
40. Mitra P, Pereira LA, Drabsch Y, Ramsay RG, Gonda TJ. Estrogen receptor-alpha recruits P-TEFb to overcome transcriptional pausing in intron 1 of the MYB gene. Nucleic acids research. 2012;40:5988-6000
41. Yang X, Liu D, Murray TJ, Mitchell GC, Hesterman EV, Karchner SI. et al. The aryl hydrocarbon receptor constitutively represses c-myc transcription in human mammary tumor cells. Oncogene. 2005;24:7869-81
42. Shang Y, Hu X, DiRenzo J, Lazar MA, Brown M. Cofactor dynamics and sufficiency in estrogen receptor-regulated transcription. Cell. 2000;103:843-52
43. Wang C, Li Y, Zhang H, Liu F, Cheng Z, Wang D. et al. Oncogenic PAK4 regulates Smad2/3 axis involving gastric tumorigenesis. Oncogene. 2014;33:3473-84
44. Minter-Dykhouse K, Ward I, Huen MS, Chen J, Lou Z. Distinct versus overlapping functions of MDC1 and 53BP1 in DNA damage response and tumorigenesis. The Journal of cell biology. 2008;181:727-35
45. Zhou W, Slingerland JM. Links between oestrogen receptor activation and proteolysis: relevance to hormone-regulated cancer therapy. Nature reviews Cancer. 2014;14:26-38
46. Wang C, Sun H, Zou R, Zhou T, Wang S, Sun S. et al. MDC1 functionally identified as an androgen receptor co-activator participates in suppression of prostate cancer. Nucleic acids research. 2015;43:4893-908
Author contact
![]() Corresponding author: Yue Zhao. Department of Cell Biology, Key laboratory of Cell Biology, Ministry of Public Health, and Key laboratory of Medical Cell Biology, Ministry of Education, China Medical University, Liaoning, Shenyang, 110122, PR China. E-mail: zhaoyuecmu.edu.cn
Corresponding author: Yue Zhao. Department of Cell Biology, Key laboratory of Cell Biology, Ministry of Public Health, and Key laboratory of Medical Cell Biology, Ministry of Education, China Medical University, Liaoning, Shenyang, 110122, PR China. E-mail: zhaoyuecmu.edu.cn