Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Warburg effect: aberrant...
3. PDC / PDKs regulates the...
4. PDKs as cancer biomarkers
5. PDKs inhibitors with emphasis...
6. Conclusion and perspective
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(12):1390-1400. doi:10.7150/ijbs.13325 This issue Cite
Review
Targeting Tumor Metabolism for Cancer Treatment: Is Pyruvate Dehydrogenase Kinases (PDKs) a Viable Anticancer Target?
Wen Zhang, Shao-Lin Zhang, Xiaohui Hu, Kin Yip Tam ![]()
Drug Development Core, Faculty of Health Sciences, University of Macau, Macau, China
Received 2015-7-23; Accepted 2015-9-7; Published 2015-11-1
Abstract

Cancer remains a lethal threat to global lives. Development of novel anticancer therapeutics is still a challenge to scientists in the field of biomedicine. In cancer cells, the metabolic features are significantly different from those of normal ones, which are hallmarks of several malignancies. Recent studies brought atypical cellular metabolism, such as aerobic glycolysis or the Warburg effect, into the scientific limelight. Targeting these altered metabolic pathways in cancer cells presents a promising therapeutic strategy. Pyruvate dehydrogenase kinases (PDKs), key enzymes in the pathway of glucose metabolism, could inactivate the pyruvate dehydrogenase complex (PDC) by phosphorylating it and preserving the substrates pyruvate, lactate and alanine for gluconeogenesis. Overexpression of PDKs could block the oxidative decarboxylation of pyruvate to satisfy high oxygen demand in cancer cells, while inhibition of PDKs could upregulate the activity of PDC and rectify the balance between the demand and supply of oxygen, which could lead to cancer cell death. Thus, inhibitors targeting PDKs represent a promising strategy for cancer treatment by acting on glycolytic tumors while showing minimal side effects on the oxidative healthy organs. This review considers the role of PDKs as regulator of PDC that catalyzes the oxidative decarboxylation of pyruvate in mitochondrion. It is concluded that PDKs are solid therapeutic targets. Inhibition of PDKs could be an attractive therapeutic approach for the development of anti-cancer drugs.
Keywords: aerobic glycolysis, pyruvate dehydrogenase complex, pyruvate dehydrogenase kinases, tumor metabolism.
1. Introduction
According to World Health Organization (WHO), cancer figures among the leading cause of death worldwide, accounting for 8.2 million deaths in 2012. It is estimated that the annual cancer cases will rise to 14 million within the next two decades [1]. With the serious threats of cancer posed to human health, development of promising anticancer agents to overcome cancer threatens is extremely urgent. Surgery, chemotherapy and radiation therapy are regarded as the conventional approaches to treat cancer. However, not all cancer patients are suitable for surgical resection, while chemotherapy using cytotoxic agents and radiation therapy usually associate with considerable side effects and drug resistance, which could be serious on some situations.
Kinases deregulation is considered as a principal mechanism of cancer cell proliferation and survival [2]. Accordingly, kinases emerged as one of the most highly investigated drug targets [3]. Great efforts have been devoted to investigate molecular mechanism by which aberrant kinases are inhibited selectively in cancers. The popularity of disordered kinases as drug targets is attributed to several reasons. Firstly, phosphotransfer cascade wired the signal transduction pathway, indicating that suppression of kinase could induce a physiological reaction. Secondly, molecular inhibitors with high selectivity and desirable pharmaceutical activity could be developed although high level of conservation exist in the adenosine triphosphate (ATP) binding pocket [4,5]. Thirdly, inhibition of kinase could usually be tolerated in nonneoplastic cells, which presents a therapeutic window to kill cancer cells selectively. Dasatinib is a good example, which is approved for treating imatinib-resistant CML [6,7].
The phenomena, mechanisms and consequences of atypical cellular metabolism in cancer patients have been brought into limelight these years [8,9]. The metabolic profiles of cancer cells divert significantly from those of nonneoplastic cells. Since all tumors rely on these metabolic alterations for growth, metastasis and survival, the atypical pathways might be potential targets of antineoplastic drugs [10]. Carbohydrate metabolism is the major pathway in the cell to provide ATP and building blocks for other macromolecules biosynthesis. The glucose metabolism in cancerous cells is remarkably different from that in their normal counterparts [11]. Proliferating cancer cells prefer to obtain ATP via aerobic glycolysis rather than oxidative phosphorylation even in the presence of ample oxygen [12]. During fermentation, most pyruvate is reduced to lactate in cytoplasm instead of being conveyed into the mitochondrion [13], a phenomenon known as Warburg effect.
Pyruvate dehydrogenase complex (PDC) and pyruvate dehydrogenase kinase PDKs are key mitochondrial enzymes in the metabolic pathway of glucose and their interaction could regulate the proportion between aerobic respiration and the Warburg effect [14]. Accumulating evidence suggests that PDKs are closely related to the initiation of the Warburg effect. Inhibition of PDKs increases oxidative phosphorylation (OXPHOS) by activating PDC [15], which might be an attractive therapeutic strategy to reverse the Warburg effect and then limit cancerous cell proliferation. Thus, investigating the function of PDKs is crucial in understanding their effects in regulating metabolic pathways and how PDKs are affected by other signaling molecules and transcription factors. In this article, we will discuss whether targeting PDKs would be a viable strategy for halting tumor development. We will direct our attention to this notion and discuss the previous attempts in the development of PDKs inhibitors. Finally, the limitations and challenges in existing PDKs inhibitors as well as their perspectives will be discussed.
2. Warburg effect: aberrant metabolic pathways in cancer cells
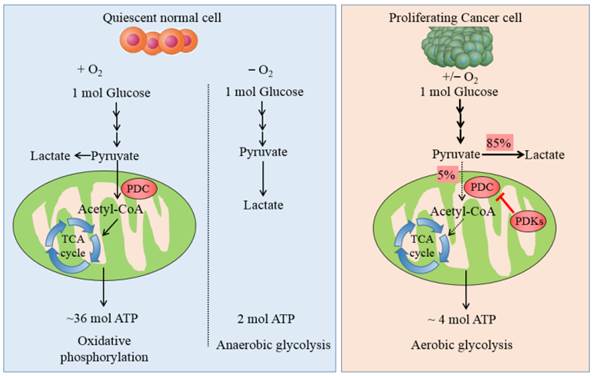
Carbohydrate/glucose metabolism is the most important pathway to provide ATP in human body. After glucose is transported into cells, it is metabolized by several steps to pyruvate by glycolysis. Majority of cancer cells transform most pyruvate into lactate in cytoplasm even in the presence of adequate oxygen rather than oxidized via the tricarboxylic acid (TCA) cycle. This phenomenon was named after Otto Warburg or referred to as aerobic glycolysis, which is the most prominent metabolic difference between normal cells and tumor ones. Non-neoplastic cells depend predominantly on ATP/energy produced by pyruvate oxidation in the mitochondria, 36 ATPs is generated per molecule glucose oxidized completely, whereas proliferating cancer cells predominately rely on aerobic glycolysis in the cytoplasm, with up to 4 ATPs produced from each glucose molecule (see Fig. 1). It is still an enigma why cancer cells divert to less efficient pathway even in the aerobic microenvironment.
The metabolic pathway of glucose in normal cells and tumor ones. The schematic illustration shows the metabolic pathway of glucose in normal cells and cancer ones. It manifests the differences between OXPHOS, anaerobic glycolysis and aerobic glycolysis. In quiescent normal cells, with the existence of sufficient oxygen, glucose is metabolized to pyruvate by which is transformed to acetyl-CoA by PDC for entering into the mitochondrial respiration. In the anaerobic microenvironment, pyruvate is reduced to lactate in cytosol. In proliferating cancer cells, enforced expression of PDKs phosphorylate and inactivate PDC, thereby shunting the pyruvate away from the OXPHOS by retarding its conversion to acetyl-CoA. 85% pyruvate in malignant cells are fermented into lactate and only 5% pyruvate enter into TCA cycle. The dashed arrows reflect the decreased mitochondrial respiration.
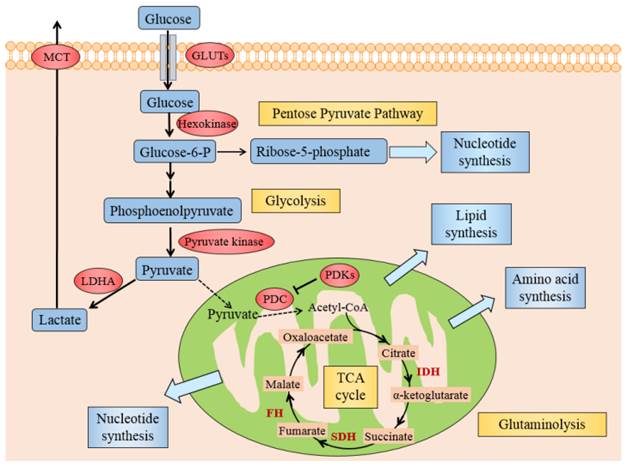
Several mechanisms might elucidate why cancer cells mostly utilize glycolysis for ATP under aerobic conditions. Generally, these could originate from either deregulated signals that enhance glycolysis or hinder the TCA cycle by the mitochondria, causing lactate fermentation the principal source of energy. Activated oncogenes and hypoxia-inducible factor (HIF) induce the forced expression of glycolytic enzymes, like glucose transporters (GLUTs), hexokinase, pyruvate kinase, lactate dehydrogenase A (LDHA) and monocarboxylate transporter (MCT), promote glycolysis in cancer cells (see Fig. 2) [16]. The dysfunctional TCA cycle might be suppressed by mutations in mitochondrial DNA [17], a loss of function of tumor suppressor genes [18] and also a deficiency of respiratory substrates, which is the foremost one [19].
Illustration of Warburg effect in cancer cells. The figure manifests the prominent aspects of Warburg effect in cancer cell, including glycolysis, lactate fermentation, TCA cycle, pentose pyruvate pathway, glutamine metabolism, and use of TCA cycle intermediates to synthesize lipids, amino acid and nucleotides. Pivotal metabolic pathways are shown in yellow boxes and the enzymes controlling key steps in glycolysis are labeled in red. Abbreviations: GLUTs: glucose transporters; MCT: monocarboxylate transporter; PDC: pyruvate dehydrogenase complex; PDKs: pyruvate dehydrogenase kinases; LDHA: lactate dehydrogenase A; HIF1: hypoxia inducible factor 1, IDH: isocitrate dehydrogenase; SDH: succinate dehydrogenase, FH: fumarate hydratase.
Pyruvate is the end product of glycolysis and then catalyzed to acetyl-CoA by PDC. The latter process is irreversible and therefore represents a critical regulatory point in cellular energy metabolism. PDKs, a primary mitochondrial enzyme, is able to inhibit PDC by phosphorylating it. Kim et al [20] and Papandreou et al [21] found that forced PDK1 expression inactivate PDC which limits entry of glycolytic carbon into the mitochondrial respiration and facilitates lactate generation from pyruvate. Acetyl-CoA is a key substrate for the TCA cycle and the ensuing OXPHOS, such that its deficiency may lead to decreased mitochondrial respiration (Fig. 2). As the TCA cycle is coupled to electron transport chain (Complex I - IV) and oxygen is the terminal electron acceptor in OXPHOS, the resulting attenuation of oxidative respiration is critical to prevent the production of reactive oxygen species (ROS) resulting from effective electron transport. In all, PDKs, locates at the central site of crossroads of glycolysis and OXPHOS, which plays a key role in cancer metabolism.
3. PDC / PDKs regulates the critical switch between glycolysis and OXPHOS
3.1 Pyruvate dehydrogenase complex
PDC locates in the mitochondrial intermembrane, which provides a link between OXPHOS and glycolysis by catalyzing the rate-limiting step in the oxidative decarboxylation of pyruvate to acetyl-CoA. PDC is a 9.5 M Da eukaryotic complex which consists of six enzymatic subunits divided into mainly two parts: one is catalytic enzymes including pyruvate dehydrogenases (PDH, E1), dihydrolipoamide transacetylase (E2), dihydrolipoamide dehydrogenase (E3) and an additional structural component, E3 binding protein (E3BP) [22]; the other is dedicated regulatory components containing PDKs and pyruvate dehydrogenase phosphatases (PDPs). The catalytic constituents function sequentially, catalyzing the oxidative decarboxylation of pyruvate with the formation of acetyl CoA, CO2 and NADH (H+). E1 pyruvate dehydrogenases component is a heterotetramer of 2α and 2β subunits (α2β2) and it catalyzes the first step of pyruvate decarboxylation. The gene coded E1α is situated on the X chromosome and all proteins constituting PDC are encoded by nuclear genes [15].
PDC plays a crucial part in the pathway of glucose metabolism and ATP production. Suppression of PDC reduces the aerobic oxidation of pyruvate and promotes its transformation to lactate in cytoplasm. Decreased flux of pyruvate via PDC also diminishes the amount of acetyl-CoA entering into the TCA cycle. Accordingly, defects in both PDC and OXPHOS activities result in lactic acidosis and the initiation of Warburg effect. The activity of PDC is controlled to a large extent by reversible phosphorylation of E1α subunit on residue sites serine 293 (Site 1), 300 (Site 2), 232 (Site 3) which are mediated by PDKs and PDP [23]. Phosphorylation of E1α by PDKs inactivates PDC, while PDP activate/reactivate catalytic activity by dephosphorylating the complex [24].
3.2 PDK isoforms
Four isomeric forms of PDKs, namely PDK 1, 2, 3 and 4 [25], express in a tissue specific manner in mammalian [26]. PDK2 is universally expressed in almost all tissues, particularly in the liver and kidney. PDK1 is dominantly found in heart and pancreatic islet. Most of PDK3 exist in testis and manifests minimum sensitivity to inhibitor molecules. PDK4 is present in small amounts in skeletal muscle, heart and liver [27].
PDKs take part in the regulation of PDC in which E1α1 is the first element. The serine residues at three sites on E1α1 are phosphorylated by PDKs at different rates and with different specificity. Site 1 is preferentially phosphorylated and sites 2 and 3 are sequentially phosphorylated, leading to slower reaction rate of the complex. E1α1 with single site mutation can all lead to E1α1 inactivation, suggesting that each site could be phosphorylated separately. Phosphorylation at site 1 almost inhibits PDC completely, however, phosphorylation at sites 2 and 3 attribute only a little to the deactivation. Thus, phosphorylation of site 1 controls E1α1 inactivation [28]. The specificities of PDKs to the three serine residues in E1 are distinct [29], accompanying with different kinetic parameters of phosphorylation. PDK1 is the only isoform which is able to phosphorylate all the three sites, whereas PDKs 2-4 could phosphorylate only sites 1 and 2 at different rates in vitro [23,30]. It is insufficient for PDKs to suppress PDC by phosphorylating only one of any E1α sites.
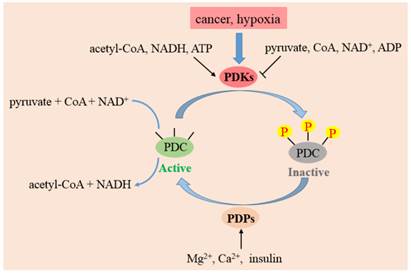
The activities, regulatory properties and phosphorylation rates of PDK1-4 are different from each other. These lead to a dynamic and tissue-specific control of mitochondrial function. As shown in Fig. 3, substances in the surroundings could regulate the activity of PDKs. NADH, acetyl-CoA and ATP in mitochondrion augment the activities of PDKs, while ADP, CoA-SH, NAD+ and pyruvate inactivate them [31]. This system controls the acute regulation of PDKs activity during short-term food intake-deprivation cycles. The expression of PDKs varies in different pathological conditions. Starvation, diabetes and hyperthyroidism could promote the expression of PDK2 and PDK4. The expressions of PDK1 and PDK3 are stimulated in cancer cells or hypoxia microenvironment, while insulin increases the expression of PDK4.
The function of PDKs and PDPs in the regulation of PDC. The model indicates that PDC catalyzes the decarboxylation of pyruvate to acetyl-CoA. PDC activity is mainly regulated by reversible phosphorylation of three serine sites on E1 subunit. Four PDKs isoforms phosphorylate up to three residues on E1 and inactivate the complex. PDP 1 and 2 could reactivate PDC by dephosphorylating serine 293, 300 and 232. The activity of PDKs are increased by a rise in the ratio of acetyl-CoA/CoA, NADH/NAD+ and ATP/ADP in mitochondria. PDPs are activated by magnesium ions, calcium ions and insulin.
3.3 PDKs governs the activity of PDC and controls the pyruvate flux
Glucose metabolism is at the heart of energy source in most mammal cells. Glucose is metabolized to pyruvate, a critical checkpoint leading two distinct destinies: one is transformed to acetyl CoA by PDC and further oxidized via TCA cycle in the mitochondrion [32], the other is converted to lactate by LDHA in the cytoplasm [8].
PDC is a pivotal regulator of mitochondrial function, since it catalyzes the pyruvate decarboxylation and controls the amount of carbons derived from carbohydrate oxidized via TCA cycle [33,34]. Therefore, the appropriate function of metabolic pathways in mitochondrion could be attained by the sustained real-time regulation of PDC activity. PDKs controls the activity of PDC: stimulation in PDKs could suppress PDC, accordingly restrain pyruvate from entering into mitochondrion, increase pyruvate phosphorylation and aggravate the Warburg effect in cancer cells [28]. To sum up, PDC and PDK are major gate-keeping enzymes which could dominate the critical switch between TCA cycle and glycolysis. Consequently, inhibiting PDKs might be a promising approach to reverse the Warburg effect in cancer cells [21].
4. PDKs as cancer biomarkers
4.1 Aberrant expression of PDKs in various cancers
It has been detected that PDKs were remarkably overexpressed in multiple human tumor samples (listed in Table 1). Higher PDKs activity, lower PDC activity and a dependence on glycolytic pathways correlate with increased tumorigenicity [35]. As stated in section 3, PDC is a predominant determinant of pyruvate flux entering into the mitochondrion. The upregulated expression of PDKs partly inactivates PDC in cancer cells and pushes more pyruvate reduced to lactate rather than oxidized by mitochondrial respiration. This metabolic alteration of decreased OXPHOS and increased lactate fermentation is closely associated with a poor prognosis in various types of cancers.
Association of PDKs overexpression with patient mortality a
| Cancer type | PDK isoforms | Prognosis | Citation |
|---|---|---|---|
| Breast cancer | PDK2 | Poor | [36] |
| Colon cancer | PDK3 and PDKs | Poor | [37,38] |
| Gastric cancer | PDK1 | Poor | [39] |
| Glioblastoma | PDKs | Poor | [35] |
| Head and neck, HNSCC | PDK1 and PDK2 | Poor | [15,40,41] |
| Hepatocellular carcinoma | PDKs | Poor | [42] |
| Lung, NSCLS | PDK2 and PDKs | ND | [36,43] |
| Melanoma | PDK1 and PDKs | Poor | [14,44] |
| Myeloma | PDK1 | Poor | [45] |
| Renal cell carcinoma, RCC | PDK1 | NC | [46] |
a HNSCC, head and neck squamous cell carcinoma; NSCLS, non-small cell lung carcinoma; ND, not determined; NC, no correlation.
Both McFate and Wigfield groups indicated that the enhanced expression of PDK1 controlled malignant phenotype and was associated with poor prognosis in HNSCC [15,40]. Hur et al found that PDK1 might serve as a biomarker of poor prognosis in gastric cancer [39]. Kaplon et al manifested that PDK1 was not only required for tumor initiation, but also for tumor maintenance and progression, suggesting that it may be beneficial to target PDK1 for therapeutic intervention of melanoma [14]. Fujiwara et al reported that the high expression of PDK1 in multiple myeloma generated high LDHA concentration which was an indicator of poor prognosis. This finding suggested that chemicals inhibiting PDK1 could be useful for cancer therapy [45]. Recently, Baumunk et al indicated that up-regulation of PDK1 might be an early event in renal cell carcinoma development, but less relevant for the progression toward an aggressive phenotype [46]. Sun et al suggested that the overexpression of PDK2 contributed to HIF1 accumulation and mitochondrial mutations, both of which leaded to the HNSCC development and deterioration [41]. Lu et al showed that the expression of PDK3 which contributed to drug resistance positively correlated with severity of colon cancer and was negatively relevant to disease-free survival [37]. Some other literatures also showed that the expression of PDKs in human cancers associated with tumor histological grades positively and with disease-free survival negatively [37,40]. Taken together, these findings suggest that PDKs is potentially an independent prognostic indicator of patient outcome and an attractive therapeutic approach for future studies.
4.2 Forced expression of PDKs in cancer cells
As reported by literatures, quite a number of established factors, such as HIF (hypoxia-inducible factor), MYC and Estrogen-related receptors (ERRs), exert an effect on the overexpression of PDKs, which further influence cellular metabolism, especially the Warburg effect.
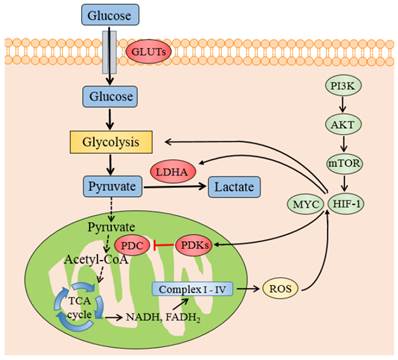
HIF-1 is a noteworthy determinant of the metabolic conversion from mitochondrial-based respiration to cytoplasmic-based fermentation in cancer cells. Its transcription is enhanced by the PI3K/AKT/mTOR pathway, which is one of the most general alterations found in tumor. HIF-1 could be activated in not only hypoxic but also pseudo-hypoxia microenvironment. Once activated, HIF-1 induces the expression of GLUTs [47] hexokinase [48] and pyruvate kinase [49] to promote the pumping of pyruvate. HIF-1 also actively suppresses OXPHOS by trans-activating the genes encoding PDK1 [20,21] and 3 [50,51] directly which phosphorylate and inactivate PDC, thereby shunting pyruvate away from the TCA cycle by retarding its conversion to acetyl-CoA. Meanwhile HIF1 induces LDHA to switch pyruvate from OXPHOS to lactate formation, consequently strengthens the glycolytic phenomenon in cancerous cells.
The oncogenic transcription factor MYC, which is often highly activated in a diverse set of malignancies, could affect the cell metabolism besides its role in regulating cell growth and proliferation [9]. MYC collaborates with HIF1 in augmenting the transcription of genes encoding several glucose transporters and glycolytic enzymes, such as PDK1 and LDHA [53], to confer metabolic advantages to tumorigenic milieu. MYC was testified to regulate PDK1 coding genes potentially through chromatin immunoprecipitation (ChIP) method in a various tissues and cell lines [54]. When MYC is mimicked to be overexpressed in human P493-6 Burkitt's lymphoma model, MYC was identified to cooperate with HIF1 to induce the expression of PDK1, which inactivates PDC and diminishes the mitochondrial OXPHOS [55].
The transcription factors and genes that regulate PDKs regulation. Schematic representation shows the metabolic switch from mitochondrial respiration to lactate production. PDKs retard the oxidative decarboxylation of pyruvate to acetyl-CoA by inhibiting PDC. HIF-1 and MYC promotes glycolysis and increases the expression of PDKs and LDHA. The transcription of HIF-1 is enhanced by the PI3K/AKT/mTOR pathway and ROS accumulation by electron transport chain Abbreviations: PI3K, phosphatidylinositol 3-kinase; mTOR, mammalian target of rapamycin; PDC: pyruvate dehydrogenase complex; PDKs, pyruvate dehydrogenase kinases; LDHA, lactate dehydrogenase A; HIF1, hypoxia inducible factor 1.
It is also reported that ERRs strengthen the contributions of HIF1 and MYC to the Warburg effect [56,57]. ERRs suppress oxidative metabolism by upregulating PDK4 [58], and hence control the fuel switch between glucose and fatty acids, which is related to cancer progression [59,60]. Another transcription factor, OCT1 (also known as POU2F1) might collaborate with p53 in adjusting the balance between OXPHOS and glycolysis [61]. The expression of OCT1 is augmented in several kinds of human tumor. The experiments on knockout mice in vivo and cancer cells in vitro indicate that OCT1 regulates genes of PDK4 to accelerate glucose metabolism and decrease oxidative respiration [62]. Some other transcription factors like Forehead Box O (FOXOs) [63], peroxisome proliferator-activated receptors (PPARs) [64] and PPAR coactivator PGC1 [65] could also stimulate the transcription of PDK4 in skeletal or muscle cells, especially during starvation or fasting.
Other than the oncogenes which induce PDKs expression as stated above, some other tumor suppressors could also down regulate PDKs levels to inhibit the glycolytic pathway. The transcription factor and tumor suppressor gene p53, which acts by regulating genes included in both of glucose oxidation and glycolytic phenotype, is found to be mutated in most human cancers [66]. p53 could suppress the expression of PDK2 to augment the entrance of pyruvate into mitochondrion for accelerating oxidative respiratory [67].
To sum up, HIF-dependent transcriptional program upregulates PDKs in cancer cells and further attenuates PDC activity, which decreases TCA-mediated aerobic respiration and efficiently accelerates lactate fermentation. However, HIF-1 actively regulates transcription of so many target genes involved in cell proliferation, angiogenesis and glucose metabolism [68] that down tuned HIF1 might cause undesired side effects in pathophysiological situations [69]. From that perspective, it seems more reasonable to develop inhibitors targeting PDKs.
5. PDKs inhibitors with emphasis as anticancer agents
Efforts are underway to develop novel inhibitors targeting PDKs to fuel the mitochondrial respiration. As PDKs show low expression in normal tissues, adverse effects caused by inhibition should be minimal, so they are favorable targets for cancer treatment. A number of agents targeting PDKs have been reported, although their efficacy remains to be confirmed clinically. So far, DCA was the only agent that enter into Phase II trial in treating cancer patients [70].
5.1 Dicholoroacetate (DCA)
DCA (Fig. 5) has been identified as a PDC activator, which acts by inhibiting the action of the PDKs [71,72]. The crystal structures of PDK1-DCA (PDB code: 2Q8H) and PDK2-DCA complex (PDB code: 2BU8) [30,73] have been reported, and showed that DCA occupied the pyruvate binding site in the N-terminal regulatory (R) domain of the enzyme [30,73]. DCA has attracted attention as a potentially simple and economical molecule to target glycolytic tumors while producing limited side effects in the oxidative healthy organs [74]. The interest in this drug by the cancer patients, scientific community and the media was kindled in 2007, when the Michelakis's group reported that DCA was uniquely toxic to human cancer cell lines and inhibited the growth of human non-small cell lung cancer (A549) lung tumor xenografts in rats [75]. The results by other researchers were largely in accordance with Michelakis's; DCA therapy slowed down the formation of cancer, reduced the rate of cell proliferation, decreased tumor volume and accelerated cancerous cell apoptosis [76-78]. In a clinical research, the tumor tissue from patients with glioblastoma was showed to downregulate HIF1 expression, reactivate mitochondrial function and remodel cellular metabolism after the treatment of DCA [79].
Chemical structures of some PDKs inhibitors. Structures of reported pyruvate dehydrogenase kinase (PDK) inhibitors
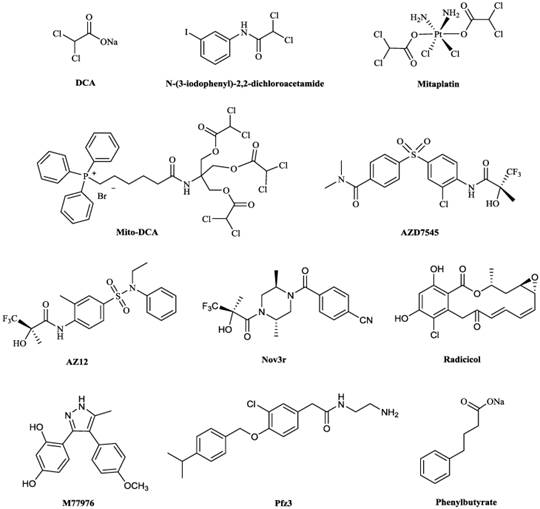
The binding affinity (Kd value) of DCA to PDK2 was 226 μM while it inhibited PDKs isoforms with Ki values ranging from 200-8000 μM [80]. DCA inhibited A549 cells proliferation at an IC50 of greater than 1000 μM [81]. Therefore, despite DCA shows promises, its applications to anticancer treatment are limited by its non-specific, low-potency and requirement of high doses (25-100 mg/kg body weight [81]) to exhibit therapeutic effects [82], which always leads to peripheral neurological toxicity [83]. The toxic effects caused by DCA include neuropathic effects, cataract formation, and testicular degeneration [84-87], which are due to the production of its principle metabolite, oxalic acid [88]. Halides in the α-position of a carbonyl group could also cause toxic effects [89,90]. Therefore, development of PDKs inhibitors with much more anticancer potency is imperative.
5.2 DCA derivatives and some other PDKs inhibitors
Researchers are investigating PDKs inhibitors based on the structure of DCA in order to minimize adverse effects and some progress have been achieved. A series of DCA derivatives were developed by Yang et al, among them, N-(3-iodophenyl)-2,2-dichloroacetamide (Fig. 5) showed an IC50 of 4.76 μM in A549 cells and LD50 of about 1.12g/kg on mice in toxicity experiment, which indicated this compound had improved anticancer potency and less adverse effects comparing with DCA [81]. Mitaplatin, c,t,c-[Pt(NH3)2(O2CHCl2)2Cl2] (Fig. 5) was synthesized based on the structure of DCA and cisplatin to treat solid tumors. The effect of mitaplatin was obviously enhanced with an IC50 of 14.0 μM against A549 cells, promoting cancer cell apoptosis but with little influence on normal cells [91]. Recently, Pathak et al. designed and constructed Mito-DCA (Fig. 5), the anticancer potency and mitochondrial targeting specificity of which was three orders of magnitude better than that of DCA [92].
Some PDKs inhibitors binding to three other pockets have also been developed. AZD7545, AZ12 and Nov3r (Fig. 5), all of which are dihydrolipoamide mimetics, bind at the lipoamide-binding pocket and were developed for treating Type 2 Diabetes Mellitus (T2DM) [93]. They have been shown to inhibit the activity of PDK2 by impeding PDKs binding to the inner lipoyl-bearing domains (L2) of E2 [30]. The IC50 values for inhibition of PDK2 in an in vitro kinase assay by AZD7545 and Nov3r were 87 nM [94] and 16.5 nM [95] respectively. AZD7545 bound to PDK2 strongly with Kd value of 24 nM [34]. Radicicol and M77976 (Fig. 5), both of which possess a dihydroxyphenyl ring, were developed to treat obesity and diabetes by binding to ATP-binding pocket. Radicicol was found to inhibit PDK4 with IC50 of 1079 μM [96] and bind to PDK2 with Kd of 18.6 μM [5]. M77976 was much more potent than radicicol with IC50 of 648 μM inhibiting PDK4 [96]. Pfz3 (Fig. 5) was developed as an analog of CoA by Pfizer and inhibited the PDK2 with an EC50 of 2 μM by binding to the allosteric domain [73]. Phenylbutyrate (Fig. 5) was another PDK2 inhibitor by binding to this pocket with a Ki of 330 μM [97].
With the small and simple structure of DCA, developing derivatives based on DCA to target the pyruvate binding pocket possesses potential and attracts much attention in pharmaceutical industry [98]. On the other hand, inhibitors (AZD7545, AZ12 and Nov3r) targeting lipoamide-binding domain in PDK2 with robust affinity have been developed to treat diabetes or obesity. It may be possible to tune the selectivity of these compounds to target the same pocket in other PDKs isoforms for cancer therapy. As far as we are aware, no successful attempts have been reported in the public domain.
6. Conclusion and perspective
Investigators have attempted to inhibit PDK enzymes in an order to reverse the metabolic disturbance in cancer cells. Cancer cells depend on aerobic glycolysis for energy supply. Altered metabolism is indispensable for the conversion from normally differentiating cells to cancer ones; in the meantime, aberrant metabolism is considered to be involved in most tumors, such as solid tumors, gliomas and leukemia. PDKs, a gate-keeping mitochondrial enzyme, plays as a checkpoint in regulating metabolic pathways. The initiation of tumor is associated with augmented PDKs activity, decreased PDC activity and a prevalent phenotype of the Warburg effect. Inhibiting PDKs could upregulate PDC activity, increase the conversion of pyruvate into acetyl-CoA, accelerate the rate of the TCA cycle and raise the proportion of oxidative phosphorylation in cancerous cells. Hence, the metabolic targeting of PDKs in cancer cells appears to be a fruitful effort for developing anticancer agents. Since expression of PDKs is elevated in a variety of malignancies, investigating novel drugs that target PDKs may be possible to treat various cancer types and may provide a new kind of antineoplastic agents.
Despite PDKs are rational targets for cancer treatment, several problems are to be confronted in developing PDKs inhibitors. They are summarized as follows:
a) The development of PDK inhibitors has been undertaken by various research groups as stated above. Unfortunately, none of these attempts other than DCA led to clinical evaluation. The crystal structures of PDK protein have been reported [73]. By exploitation of all the known drug binding sites in the target proteins, it would be possible to rationally design potent PDK inhibitors [99]. However, it remains to be seen if it is feasible to develop PDKs inhibitors with anticancer effects that are much more potent than DCA.
b) Given the disparate effects of PDK1, 2, 3 and 4 on cancer and diabetes, it is critical to identify whether potential candidates inhibit all PDKs isoforms equally. Under certain conditions, selective inhibition of one isoform would be of particular advantage. For instance, targeting PDK 2 and 4 might be especially beneficial for Type 2 Diabetes Mellitus (T2DM) or obesity [100,101], whereas inhibiting some other isoforms may be beneficial to tumor therapy.
c) PDKs inhibition could be useful only up to a certain point. As described above, the overexpression of PDKs decreases the activity of PDC, thus blocking TCA cycle and oxidative respiration. It is noteworthy that care must be exercised to ensure the glycolysis pathway is not completely switched off, as this is likely to impact on proliferating cells (such as lymphocytes) that rely on aerobic glycolysis [102]. Thus, inhibition of PDKs proteins might result in a narrower therapeutic window.
d) Inhibition of PDKs alone may not be sufficient to cause extensive apoptosis in cancer cells, so adding combination of inhibitors directed against other aberrant metabolic processes may be more effective. For instance, LDHA inhibitor could prevent the conversion of pyruvate to lactate in cytoplasm. Meanwhile inhibition of PDKs is able to convey more pyruvate into mitochondrion. Thus, the combined use of LDHA and PDK inhibitors could redirect pyruvate from glycolysis into the TCA cycle and then impair cancer cell proliferation more efficiently.
e) Although Warburg effect is the most significant phenotype, it is not a ubiquitous characteristic of all human tumors. According to literatures, cancer cells are able to use other energy sources to maintain proliferation, such as glutamine, amino acids and fatty acids [103]. Accordingly, for the sake of cutting down the fuel source of cancerous cells, some compensating pathways such as pentose phosphate pathway are also required to be inhibited besides suppressing the aerobic glycolysis [104]. Hopefully, with a thorough knowledge about the metabolic alterations and regulatory mechanisms, it will be possible to develop more efficient oncogenic agents with better selectivity to relieve patients from cancer suffering.
f) Unwanted toxicity and detrimental side effects are big challenges in developing drugs that target cancer cell metabolism. Since all cells depend on the mitochondrial metabolism or cytoplasmic glycolysis to synthesize ATP, agents that target altered metabolic pathways should be selective with the aim to minimize any adverse effects in normal tissues. Meanwhile, as kinase signaling cascades contains a complicated series of interconnected pathways, researches are required to exploit sophisticated modeling to investigate the cancer cell response to therapeutic treatments [2]. A better and deeper understanding of the molecular mechanisms that underlie why cancer cells are more dependent on some specific pathways may provide new approaches to target altered metabolic sites more selectively.
Acknowledgements
We thank the financial support from the Science and Technology Development Fund, Macao S.A.R (FDCT) (project reference no. 086/2014/A2) and the University of Macau (grant no. SRG2013-00055-FHS).
Competing Interests
The authors have declared that no competing interest exists.
References
1. American Cancer Society. Global Cancer Facts & Figures (2nd edn). American Cancer Society. 2011 Atlanta
2. Zhang J, Yang PL, Gray NS. Targeting cancer with small molecule kinase inhibitors. Nat Rev Cancer. 2009;9:28-39
3. Malik SM, Maher VE, Bijwaard KE, Becker RL, Zhang L, Tang SW, Song P, Liu Q, Marathe A, Gehrke B, Helms W, Hanner D, Justice R, Pazdur R. U.S. food and drug administration approval: Crizotinib for treatment of advanced or metastatic non-small cell lung cancer that is anaplastic lymphoma kinase positive. Clin Cancer Res. 2014;20:2029-34
4. Davies SP, Helen R, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95-105
5. Tso SC, Qi X, Gui WJ, Wu CY, Chuang JL, Wernstedt-Asterholm I, Morlock LK, Owens KR, Scherer PE, Williams NS, Tambar UK, Wynn RM, Chuang DT. Structure-guided development of specific pyruvate dehydrogenase kinase inhibitors targeting the ATP-binding pocket. J Biol Chem. 2014;289:4432-43
6. Lombardo LJ, Lee FY, Chen P, Norris D, Barrish JC, Behnia K, Castaneda S, Cornelius LA, Das J, Doweyko AM, Fairchild C, Hunt JT, Inigo I, Johnston K, Kamath A, Kan D, Klei H, Marathe P, Pang S, Peterson R, Pitt S, Schieven GL, Schmidt RJ, Tokarski J, Wen ML, Wityak J, Borzilleri RM. Discovery of N-(2-chloro-6-methylphenyl)-2-(6-(4-(2-hydroxyethyl)- piperazin-1-yl)-2-methylpyrimidin-4-ylamino)thiazole-5-carboxamide (BMS-354825), a dual Src/Abl kinase inhibitor with potent antitumor activity in preclinical assays. J Med Chem. 2004;47:6658-61
7. Shah NP, Tran C, Lee FY, Chen P, Norris D, Sawyers CL. Overriding imatinib resistance with a novel ABL kinase inhibitor. Science. 2004;305:399-401
8. Cairns RA, Harris IS, Mak TW. Regulation of cancer cell metabolism. Nat Rev Cancer. 2011;11:85-95
9. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646-74
10. Zhao Y, Butler EB, Tan M. Targeting cellular metabolism to improve cancer therapeutics. Cell Death Dis. 2013;4:e532
11. Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029-33
12. Warburg O, Wind F, Negelein E. The metabolism of tumors in the body. J Gen Physiol. 1927;8:519-30
13. Warburg O. On the origin of cancer cells. Science. 1956;123:309-14
14. Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, van der Burg SH, Verdegaal EM, Cascante M, Shlomi T, Gottlieb E, Peeper DS. A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature. 2013;498:109-12
15. McFate T, Mohyeldin A, Lu H, Thakar J, Henriques J, Halim ND, Wu H, Schell MJ, Tsang TM, Teahan O, Zhou S, Califano JA, Jeoung NH, Harris RA, Verma A. Pyruvate dehydrogenase complex activity controls metabolic and malignant phenotype in cancer cells. J Biol Chem. 2008;283:22700-8
16. Levine AJ, Puzio-Kuter AM. The control of the metabolic switch in cancers by oncogenes and tumor suppressor genes. Science. 2010;30:1340-4
17. Ju YS, Alexandrov LB, Gerstung M. et al. Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. Elife. 2014;3:1-28
18. Zhang LJ, Xiong Y, Nilubol N, He M, Bommareddi S, Zhu X, Jia L, Xiao Z, Park JW, Xu X, Patel D, Willingham MC, Cheng SY, Kebebew E. Testosterone regulates thyroid cancer progression by modifying tumor suppressor-genes and tumor immunity. Carcinogenesis. 2015;36:420-8
19. Simon MC. Coming up for air: HIF-1 and mitochondrial oxygen consumption. Cell Metab. 2006;3:150-1
20. Kim JW, Tchernyshyov I, Semenza GL, Dang C V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006;3:177-85
21. Papandreou I, Cairns R A, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab. 2006;3:187-97
22. Yu X, Hiromasa Y, Tsen H, Stoops JK, Roche TE, Zhou ZH. Structures of the human pyruvate dehydrogenase complex cores: a highly conserved catalytic center with flexible N-terminal domains. Structure. 2008;16:104-14
23. Kolobova E, Tuganova A, Boulatnikov I, Popov KM. Regulation of pyruvate dehydrogenase activity through phosphorylation at multiple sites. Biochem J. 2001;358:69-77
24. Fan J, Shan C, Kang HB, Elf S, Xie J, Tucker M, Gu TL, Aguiar M, Lonning S, Chen H, Mohammadi M, Britton LM, Garcia BA, Alečković M, Kang Y, Kaluz S, Devi N, Van Meir EG, Hitosugi T, Seo JH, Lonial S, Gaddh M, Arellano M, Khoury HJ, Khuri FR, Boggon TJ, Kang S, Chen J. Tyr phosphorylation of PDP1 toggles recruitment between ACAT1 and SIRT3 to regulate the pyruvate dehydrogenase complex. Mol Cell. 2014;53:534-48
25. Patel MS, Korotchkina LG. Regulation of the pyruvate dehydrogenase complex. Biochem Soc Trans. 2006;34:217-22
26. Devedjiev Y, Steussy CN, Vassylyev DG. Crystal structure of an asymmetric complex of pyruvate dehydrogenase kinase 3 with lipoyl domain 2 and its biological implications. J Mol Biol. 2007;370:407-16
27. Rardin MJ, Wiley SE, Naviaux RK, Murphy AN, Dixon JE. Monitoring phosphorylation of the pyruvate dehydrogenase complex. Anal Biochem. 2009;389:157-64
28. Holness MJ, Sugden MC. Regulation of pyruvate dehydrogenase complex activity by reversible phosphorylation. Biochem Soc Trans. 2003;31:1143-51
29. Rardin MJ, Wiley SE, Naviaux RK, Murphy AN, Dixon JE. Monitoring phosphorylation of the pyruvate dehydrogenase complex. Anal Biochem. 2009;389:157-64
30. Kato M, Li J, Chuang JL, Chuang DT. Distinct structural mechanisms for inhibition of pyruvate dehydrogenase kinase isoforms by AZD7545, Dichloroacetate, and Radicicol. Structure. 2007;15:992-1004
31. Houten SM, Chegary M, Te Brinke H, Wijnen WJ, Glatz JF, Luiken JJ, Wijburg FA, Wanders RJ. Pyruvate dehydrogenase kinase 4 expression is synergistically induced by AMP-activated protein kinase and fatty acids. Cell Mol Life Sci. 2009;66:1283-94
32. Roche TE, Baker JC, Yan X, Hiromasa Y, Gong X, Peng T, Dong J, Turkan A, Kasten SA. Distinct regulatory properties of pyruvate dehydrogenase kinase and phosphatase isoforms. Prog Nucleic Acid Res Mol Biol. 2001;70:33-75
33. Kroemer G, Pouyssegur J. Tumor cell metabolism: cancer's achilles' heel. Cancer Cell. 2008;13:472-82
34. Li J, Kato M, Chuang DT. Pivotal role of the C-terminal DW-motif in mediating inhibition of pyruvate dehydrogenase kinase 2 by dichloroacetate. J Biol Chem. 2009;284:34458-67
35. Jha MK, Suk K. Pyruvate dehydrogenase kinase as a potential therapeutic target for malignant gliomas. Brain Tumor Res Treat. 2013;1:57-63
36. Sutendra G, Dromparis P, Kinnaird A, Stenson TH, Haromy A, Parker JM, McMurtry MS, Michelakis ED. Mitochondrial activation by inhibition of PDKII suppresses HIF1a signaling and angiogenesis in cancer. Oncogene. 2012;32:1638-50
37. Lu CW, Lin SC, Chien CW, Lin SC, Lee CT, Lin BW, Lee JC, Tsai SJ. Overexpression of pyruvate dehydrogenase kinase 3 increases drug resistance and early recurrence in colon cancer. Am J Pathol. 2011;179:1405-14
38. Blouin JM, Penot G, Collinet M, Nacfer M, Forest C, Laurent-Puig P, Coumoul X, Barouki R, Benelli C, Bortoli S. Butyrate elicits a metabolic switch in human colon cancer cells by targeting the pyruvate dehydrogenase complex. Int J Cancer. 2011;128:2591-601
39. Hur H, Xuan Y, Kim YB, Lee G, Shim W, Yun J, Ham IH, Han SU. Expression of pyruvate dehydrogenase kinase-1 in gastric cancer as a potential therapeutic target. Int J Oncol. 2013;42:44-54
40. Wigfield SM, Winter SC, Giatromanolaki A, Taylor J, Koukourakis ML, Harris AL. PDK-1 regulates lactate production in hypoxia and is associated with poor prognosis in head and neck squamous cancer. Br J Cancer. 2008;98:1975-84
41. Sun W, Zhou S, Chang SS, McFate T, Verma A, Califano J A. Mitochondrial mutations contribute to HIFIα accumulation via increased reactive oxygen species and up-regulated pyruvate dehydrogenease kinase 2 in head and neck squamous cell carcinoma. Clin Cancer Res. 2009;15:476-84
42. Shen YC, Ou DL, Hsu C, Lin KL, Chang CY, Lin CY, Liu SH, Cheng AL. Activating oxidative phosphorylation by a pyruvate dehydrogenase kinase inhibitor overcomes sorafenib resistance of hepatocellular carcinoma. Br J Cancer. 2013;108:72-81
43. Koukourakis MI, Giatromanolaki A, Sivridis E, Gatter KC, Harris AL. Pyruvate dehydrogenase and pyruvate dehydrogenase kinase expression in non small cell lung cancer and tumor-associated stroma. Neoplasia. 2005;7:1-6
44. Abildgaard C, Dahl C, Basse AL, Ma T, Guldberg P. Bioenergetic modulation with dichloroacetate reduces the growth of melanoma cells and potentiates their response to BRAFV600E inhibition. J Transl Med. 2014;12:247-258
45. Fujiwara S, Kawano Y, Yuki H, Okuno Y, Nosaka K, Mitsuya H, Hata H. PDK1 inhibition is a novel therapeutic target in multiple myeloma. Br J Cancer. 2013;108:170-8
46. Daniel B, Uta R, Jannis H, Hans K, Jan E, Hannes C, Kurt M, Martin S, Steffen W. Expression parameters of the metabolic pathway genes pyruvate dehydrogenase kinase-1 (PDK-1) and DJ-1/PARK7 in renal cell carcinoma (RCC). World J Urol. 2013;31:1191-6
47. Starska K, Forma E, Jóźwiak P, Bryś M, Lewy-Trenda I, Brzezińska-Błaszczyk E, Krześlak A. Gene and protein expression of glucose transporter 1 and glucose transporter 3 in human laryngeal cancer-the relationship with regulatory hypoxia-inducible factor-1α expression, tumor invasiveness, and patient prognosis. Tumor Biol. 2015;36:2309-21
48. Mergenthaler P, Kahl A, Kamitz A, van Laak V, Stohlmann K, Thomsen S, Klawitter H, Przesdzing I, Neeb L, Freyer D, Priller J, Collins TJ, Megow D, Dirnagl U, Andrews DW, Meisel A. Mitochondrial hexokinase II (HKII) and phosphoprotein enriched in astrocytes (PEA15) form a molecular switch governing cellular fate depending on the metabolic state. Proc Natl Acad Sci USA. 2012;109:1518-23
49. Palsson-McDermott EM, Curtis AM, Goel G, Lauterbach MA, Sheedy FJ, Gleeson LE, van den Bosch MW, Quinn SR, Domingo-Fernandez R, Johnston DG, Jiang JK, Israelsen WJ, Keane J, Thomas C, Clish C, Vander Heiden M, Xavier RJ, O'Neill LA. Pyruvate kinase M2 regulates hif-1α activity and IL-1β induction and is a critical determinant of the Warburg effect in LPS-activated macrophages. Cell Metab. 2015;21:65-80
50. Denko NC, Fontana LA, Hudson KM, Sutphin PD, Raychaudhuri S, Altman R, Giaccia AJ. Investigating hypoxic tumor physiology through gene expression patterns. Oncogene. 2003;22:5907-14
51. Lu CW, Lin SC, Chen KF, Lai YY, Tsai SJ. Induction of pyruvate dehydrogenase kinase-3 by hypoxia-inducible factor-1 promotes metabolic switch and drug resistance. J Biol Chem. 2008;283:28106-14
52. Dang CV. MYC on the path to cancer. Cell. 2012;149:22-35
53. Dang CV. The interplay between MYC and HIF in the Warburg effect. Ernst Schering Found Symp Proc. 2008;4:35-53
54. Li Z, Van Calcar S, Qu C, Cavenee WK, Zhang MQ, Ren B. A global transcriptional regulatory role for c-Myc in Burkitt's lymphoma cells. Proc Natl Acad Sci U S A. 2003;100:8164-9
55. Kim JW, Gao P, Liu YC, Semenza GL, Dang CV. Hypoxia-inducible factor 1 and dysregulated c-Myc cooperatively induce vascular endothelial growth factor and metabolic switches hexokinase 2 and pyruvate dehydrogenase kinase 1. Mol Cell Biol. 2007;27:7381-93
56. Ao A, Wang H, Kamarajugadda S, Lu J. Involvement of estrogen-related receptors in transcriptional response to hypoxia and growth of solid tumors. Proc Natl Acad Sci U S A. 2008;105:7821-6
57. Cai Q, Lin T, Kamarajugadda S, Lu J. Regulation of glycolysis and the Warburg effect by estrogen-related receptors. Oncogene. 2012;32:2079-86
58. Zhang Y, Ma K, Sadana P, Chowdhury F, Gaillard S, Wang F, McDonnell DP, Unterman TG, Elam MB, Park EA. Estrogen-related receptors stimulate pyruvate dehydrogenase kinase isoform 4 gene expression. J Biol Chem. 2006;281:39897-906
59. Deblois G, Giguère V. Oestrogen-related receptors in breast cancer: control of cellular metabolism and beyond. Nat Rev Cancer. 2013;13(1):27-36
60. Chang CY, McDonnell DP. Molecular pathways: the metabolic regulator estrogen-related receptor α as a therapeutic target in cancer. Clin Cancer Res. 2012;18:6089-95
61. Almeida R, Almeida J, Shoshkes M, Mendes N, Mesquita P, Silva E, Van Seuningen I, Reis CA, Santos-Silva F, David L. OCT-1 is over-expressed in intestinal metaplasia and intestinal gastric carcinomas and binds to, but does not transactivate, CDX2 in gastric cells. J Pathol. 2005;207:396-401
62. Arvind Shakya, Robert Cooksey, James E. Cox, Victoria Wang, Donald A. McClain DT. Oct1 loss of function induces a coordinate metabolic shift that opposes tumorigenicity. Nat Cell Biol. 2009;11:320-7
63. Furuyama T, Kitayama K, Yamashita H, Mori N. Forkhead transcription factor FOXO1 (FKHR)-dependent induction of PDK4 gene expression in skeletal muscle during energy deprivation. Biochem J. 2003;375:365-71
64. Wu P, Peters JM, Harris RA. Adaptive increase in pyruvate dehydrogenase kinase 4 during starvation is mediated by peroxisome proliferator-activated receptor alpha. Biochem Biophys Res Commun. 2001;287:391-6
65. Abbot EL, McCormack JG, Reynet C, Hassall DG, Buchan KW, Yeaman SJ. Diverging regulation of pyruvate dehydrogenase kinase isoform gene expression in cultured human muscle cells. FEBS J. 2005;272:3004-14
66. Berkers CR, Maddocks OD, Cheung EC, Mor I, Vousden KH. Metabolic regulation by p53 family members. Cell Metab. 2013;18:617-33
67. Contractor T, Harris CR. p53 negatively regulates transcription of the pyruvate dehydrogenase kinase Pdk2. Cancer Res. 2012;72:560-7
68. Semenza GL. The hypoxic tumor microenvironment: a driving force for breast cancer progression. Biochim Biophys Acta-Mol Cell Res. 2015 dio: 10.1016/j.bbamcr.2015.05.036
69. Dromparis P, Sutendra G, Paulin R, Proctor S, Michelakis ED, McMurtry MS. Pioglitazone inhibits HIF-1α-dependent angiogenesis in rats by paracrine and direct effects on endothelial cells. J Mol Med. 2014;92:497-507
70. Dunbar EM, Coats BS, Shroads AL, Langaee T, Lew A, Forder JR, Shuster JJ, Wagner DA, Stacpoole PW. Phase 1 trial of dichloroacetate (DCA) in adults with recurrent malignant brain tumors. Invest New Drugs. 2014;32:452-64
71. Kluza J, Corazao-Rozas P, Touil Y, Jendoubi M, Maire C, Guerreschi P, Jonneaux A, Ballot C, Balayssac S, Valable S, Corroyer-Dulmont A, Bernaudin M, Malet-Martino M, de Lassalle EM, Maboudou P, Formstecher P, Polakowska R, Mortier L, Marchetti P. Inactivation of the HIF-1α/PDK3 signaling axis drives melanoma toward mitochondrial oxidative metabolism and potentiates the therapeutic activity of pro-oxidants. Cancer Res. 2012;72:5035-47
72. Velpula KK, Bhasin A, Asuthkar S, Tsung AJ. Combined targeting of PDK1 and EGFR triggers regression of glioblastoma by reversing the Warburg effect. Cancer Res. 2013;73:7277-89
73. Knoechel TR, Tucker AD, Robinson CM, Phillips C, Taylor W, Bungay PJ, Kasten SA, Roche TE, Brown DG. Regulatory roles of the N-terminal domain based on crystal structures of human pyruvate dehydrogenase kinase 2 containing physiological and synthetic ligands. Biochemistry. 2006;45:402-15
74. Kankotia S, Stacpoole PW. Dichloroacetate and cancer: new home for an orphan drug? Biochim Biophys Acta. 2014;1846:617-29
75. Bonnet S, Archer SL, Allalunis-Turner J, Haromy A, Beaulieu C, Thompson R, Lee CT, Lopaschuk GD, Puttagunta L, Bonnet S, Harry G, Hashimoto K, Porter CJ, Andrade MA, Thebaud B, Michelakis ED. A mitochondria-K+ channel axis is suppressed in cancer and its normalization promotes apoptosis and inhibits cancer growth. Cancer Cell. 2007;11:37-51
76. Shahrzad S, Lacombe K, Adamcic U, Minhas K, Coomber BL. Sodium dichloroacetate (DCA) reduces apoptosis in colorectal tumor hypoxia. Cancr Lett. 2010;297:75-83
77. Lefort N, Brown A, Lloyd V, Ouellette R, Touaibia M, Culf AS, Cuperlovic-Culf M. 1H NMR metabolomics analysis of the effect of dichloroacetate and allopurinol on breast cancers. J Pharm Biomed Anal. 2014;93:77-85
78. Kumar A, Kant S, Singh SM. Antitumor and chemosensitizing action of dichloroacetate implicates modulation of tumor microenvironment: a role of reorganized glucose metabolism, cell survival regulation and macrophage differentiation. Toxicol Appl Pharmacol. 2013;273:196-208
79. Michelakis ED, Sutendra G, Dromparis P, Webster L, Haromy A, Niven E, Maguire C, Gammer TL, Mackey JR, Fulton D, Abdulkarim B, McMurtry MS, Petruk KC. Metabolic modulation of glioblastoma with dichloroacetate. Sci Transl Med. 2010;2:31ra34
80. Bowker-Kinley MM, Davis WI, Wu P, Harris RA, Popov KM. Evidence for existence of tissue-specific regulation of the mammalian pyruvate dehydrogenase complex. Biochem J. 1998;329:191-6
81. Yang Y, Shang P, Cheng C, Wang D, Yang P, Zhang F, Li T, Lu A, Zhao Y. Novel N-phenyl dichloroacetamide derivatives as anticancer reagents: design, synthesis and biological evaluation. Eur J Med Chem. 2010;45:4300-6
82. Stacpoole PW, Kurtz TL, Han Z, Langaee T. Role of dichloroacetate in the treatment of genetic mitochondrial diseases. Adv Drug Deliv Rev. 2008;60:1478-87
83. Jiang DK, Sun J, Cao G, Liu Y, Lin D, Gao YZ, Ren WH, Long XD, Zhang H, Ma XP, Wang Z, Jiang W, Chen TY, Gao Y, Sun LD, Long JR, Huang HX, Wang D, Yu H, Zhang P, Tang LS, Peng B, Cai H, Liu TT, Zhou P, Liu F, Lin X, Tao S, Wan B, Sai-Yin HX, Qin LX, Yin J, Liu L, Wu C, Pei Y, Zhou YF, Zhai Y, Lu PX, Tan A, Zuo XB, Fan J, Chang J, Gu X, Wang NJ, Li Y, Liu YK, Zhai K, Zhang H, Hu Z, Liu J, Yi Q, Xiang Y, Shi R, Ding Q, Zheng W, Shu XO, Mo Z, Shugart YY, Zhang XJ, Zhou G, Shen H, Zheng SL, Xu J, Yu L. Genetic variants in STAT4 and HLA-DQ genes confer risk of hepatitis B virus-related hepatocellular carcinoma. Nat Genet. 2013;45:72-5
84. Stacpoole PW, Moore GW, Kornhauser DM. Toxicity of chronic dichloroacetate. N Engl J Med. 1979;300:372-37
85. Yount EA, Felten SY, O'Connor BL, Peterson RG, Powell RS, Yum MN, Harris RA. Comparison of the metabolic and toxic effects of 2-chloropropionate and dichloroacetate. J Pharmacol Exp Ther. 1982;222:501-8
86. Katz R, Tai CN, Diener RM, McConnell RF, Semonick DE. Dichloroacetate, sodium: 3-month oral toxicity studies in rats and dogs. Toxicol Appl Pharmacol. 1987;57:273-87
87. Toth GP, Kelty KC, George EL, Read EJ, Smith MK. Adverse male reproductive effects following subchronic exposure of rats to sodium dichloroacetate. Fundam Appl Toxicol. 1992;19:57-63
88. Aicher TD, Anderson RC, Gao J, Shetty SS, Coppola GM, Stanton JL, Knorr DC, Sperbeck DM, Brand LJ, Vinluan CC, Kaplan EL, Dragland CJ, Tomaselli HC, Islam A, Lozito RJ, Liu X, Maniara WM, Fillers WS, DelGrande D, Walter RE, Mann WR. Secondary amides of (R)-3,3,3-trifluoro-2-hydroxy-2-methylpropionic acid as inhibitors of pyruvate dehydrogenase kinase. J Med Chem. 2000;43:236-49
89. Halpert JR, Balfour C, Miller NE, Kaminsky LS. Dichloromethyl compounds as mechanism-based inactivators of rat liver cytochromes P-450 in vitro. Mol Pharmacol. 1986;30:19-24
90. Bookstaff RC, Moore RW, Ingall GB, Peterson RE. Androgenic deficiency in male rats treated with perfluorodecanoic acid. Toxicol Appl Pharmacol. 1990;104:322-33
91. Dhar S, Lippard SJ. Mitaplatin, a potent fusion of cisplatin and the orphan drug dichloroacetate. Proc Natl Acad Sci USA. 2009;106:22199-204
92. Pathak RK, Marrache S, Harn DA, Dhar S. Mito-DCA: A mitochondria targeted molecular scaffold for efficacious delivery of metabolic modulator dichloroacetate. ACS Chem Biol. 2014;9:1178-87
93. Mayers RM, Butlin RJ, Kilgour E, Leighton B, Martin D, Myatt J, Orme JP, Holloway BR. AZD7545, a novel inhibitor of pyruvate dehydrogenase kinase 2 (PDHK2), activates pyruvate dehydrogenase in vivo and improves blood glucose control in obese (fa/fa) Zucker rats. Biochem Soc Trans. 2003;31:1165-7
94. Morrell JA, Orme J, Butlin RJ, Roche TE, Mayers RM, Kilgour E. AZD7545 is a selective inhibitor of pyruvate dehydrogenase kinase 2. Biochem Soc Trans. 2003;31:1168-70
95. Aicher TD, Anderson RC, Bebernitz GR, Coppola GM, Jewell CF, Knorr DC, Liu C, Sperbeck DM, Brand LJ, Strohschein RJ, Gao J, Vinluan CC, Shetty SS, Dragland C, Kaplan EL, DelGrande D, Islam A, Liu X, Lozito RJ, Maniara WM, Walter RE, Mann WR. (R)-3,3,3-Trifluoro-2-hydroxy-2-methylpropionamides are orally active inhibitors of pyruvate dehydrogenase kinase. J Med Chem. 1999;42:2741-6
96. Kukimoto-Niino M, Tokmakov A, Terada T, Ohbayashi N, Fujimoto T, Gomi S, Shiromizu I, Kawamoto M, Matsusue T, Shirouzu M, Yokoyama S. Inhibitor-bound structures of human pyruvate dehydrogenase kinase 4. Acta Crystallogr Sect D Biol Crystallogr. 2011;67:763-73
97. Ferriero R, Manco G, Lamantea E, Nusco E, Ferrante MI, Sordino P, Stacpoole PW, Lee B, Zeviani M, Brunetti-Pierri N. Phenylbutyrate therapy for pyruvate dehydrogenase complex deficiency and lactic acidosis. Sci Transl Med. 2013;5:175ra31
98. Saha S, Ghosh M, Dutta SK. A potent tumoricidal co-drug 'Bet-CA'--an ester derivative of betulinic acid and dichloroacetate selectively and synergistically kills cancer cells. Sci Rep. 2015;5:7762
99. Zhang S-L, Hu X, Zhang W, Yao H, Tam KY. Development of pyruvate dehydrogenase kinase inhibitors in medicinal chemistry with particular emphasis as anticancer agents. Drug Discov Today. 2015 doi: org/10.1016/j.drudis.2015.03.012
100. McAinch AJ, Cornall LM, Watts R, Hryciw DH, O'Brien PE, Cameron-Smith D. Increased pyruvate dehydrogenase kinase expression in cultured myotubes from obese and diabetic individuals. Eur J Nutr. 2014 doi: 10.1007/s00394-014-0780-2
101. Holness MJ, Zariwala G, Walker CG, Sugden MC. Adipocyte pyruvate dehydrogenase kinase 4 expression is associated with augmented PPARγ upregulation in early-life programming of later obesity. FEBS Open Bio. 2012;2:32-6
102. Lunt SY, Vander Heiden MG. Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol. 2011;27:441-64
103. Pan T, Gao L, Wu G, Shen G, Xie S, Wen H, Yang J, Zhou Y, Tu Z, Qian W. Elevated expression of glutaminase confers glucose utilization via glutaminolysis in prostate cancer. Biochem Biophys Res Commun. 2015;456:452-8
104. Zhang J, Fan J, Venneti S, Cross JR, Takagi T, Bhinder B, Djaballah H, Kanai M, Cheng EH, Judkins AR, Pawel B, Baggs J, Cherry S, Rabinowitz JD, Thompson CB. Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol Cell. 2014;56:205-18
Author contact
![]() Corresponding author: Tam, K.Y. (kintammo). Tel.: +853-88224988; fax: +853-88222314
Corresponding author: Tam, K.Y. (kintammo). Tel.: +853-88224988; fax: +853-88222314