Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Advance in genomic research...
Non-coding RNAs can be...
Other ribonomic advances also...
Advances in proteomic research...
Ectopic expression of cDNA skips...
Then, how should we redefine...
Concluding remarks
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(12):1413-1423. doi:10.7150/ijbs.13436 This issue Cite
Review
To Know How a Gene Works, We Need to Redefine It First but then, More Importantly, to Let the Cell Itself Decide How to Transcribe and Process Its RNAs
Yuping Jia1 ![]() , Lichan Chen2, Yukui Ma1, Jian Zhang3
, Lichan Chen2, Yukui Ma1, Jian Zhang3 ![]() , Ningzhi Xu4
, Ningzhi Xu4 ![]() , Dezhong Joshua Liao2
, Dezhong Joshua Liao2 ![]()
1. Shandong Academy of Pharmaceutical Sciences, Ji'nan, Shandong, 250101, P.R. China
2. Hormel Institute, University of Minnesota, Austin, MN 55912, USA
3. Center for Translational Medicine, Pharmacology and Biomedical Sciences Building, Guangxi Medical University, 22 Shuangyong Road, Nanning, Guangxi 530021, P.R. China.
4. Laboratory of Cell and Molecular Biology, Cancer Institute, Chinese Academy of Medical Science, Beijing 100021, P.R. China
Received 2015-8-3; Accepted 2015-10-12; Published 2015-11-19
Abstract

Recent genomic and ribonomic research reveals that our genome produces a stupendous amount of non-coding RNAs (ncRNAs), including antisense RNAs, and that many genes contain other gene(s) in their introns. Since ncRNAs either regulate the transcription, translation or stability of mRNAs or directly exert cellular functions, they should be regarded as the fourth category of RNAs, after ribosomal, messenger and transfer RNAs. These and other research advances challenge the current concept of gene and raise a question as to how we should redefine gene. We can either consider each tiny part of the classically-defined gene, such as each mRNA variant, as a “gene”, or, alternatively and oppositely, regard a whole genomic locus as a “gene” that may contain intron-embedded genes and produce different types of RNAs and proteins. Each of the two ways to redefine gene not only has its strengths and weaknesses but also has its particular concern on the methodology for the determination of the gene's function: Ectopic expression of complementary DNA (cDNA) in cells has in the past decades provided us with great deal of detail about the functions of individual mRNA variants, and will make the data less conflicting with each other if just a small part of a classically-defined gene is considered as a “gene”. On the other hand, genomic DNA (gDNA) will better help us in understanding the collective function of a genomic locus. In our opinion, we need to be more cautious in the use of cDNA and in the explanation of data resulting from cDNA, and, instead, should make delivery of gDNA into cells routine in determination of genes' functions, although this demands some technology renovation.
Keywords: Gene definition, non-coding RNA, Complementary DNA, Gene function, Genomic locus
Introduction
In the history of biology, it was long thought that each individual gene served a single phenotypic trait, which could be rephrased more mechanistically as “one gene for one function”. However, we now know that most, if not all, mammalian genes have multiple functions. One major mechanism for a gene to differentiate one function from another is for itself to be present in a different form. As described previously [1-3], this “different form” could be a polymorphism or mutation, a different RNA transcript initiated from or terminated at a different genomic site, a different cis- or trans-spliced product of a transcript, a different protein isoform due to usage of a different start codon or stop codon, or a different product of post-translational process such as proteolysis, phosphorylation or glycosylation. In this regard, the traditional concept of “one gene for one function” still, generally speaking, holds true if “one gene” is considered as one form of the gene. In other words, it may be the definition of gene, but not this concept, that needs to be amended instead. If a small part of a currently-defined gene, which is actually a genomic locus, is redefined as a “gene”, many advances in genomic, ribonomic and proteomic research that cannot be accommodated by the current gene definition become easily understandable, and a lot of data on the functions of most mammalian genes that are conflicting with each other will no longer be controversial. For example, it is understandable that two splice-derived mRNA variants would have different functions because they are two different “genes”. However, if we consider part of a classically-defined gene, such as an mRNA variant, as a “gene” and continue to mainly use complementary DNA (cDNA) to characterize its function, as we have done for decades, we will not be able to learn the collective function of the gene as a genomic locus and in turn the function of the whole mammalian genome. In this essay, we discuss these fundamental issues raised by the recent genomic, ribonomic and proteomic findings and by the omnipresent controversial data on genes' functions.
Advance in genomic research requires a new definition of gene
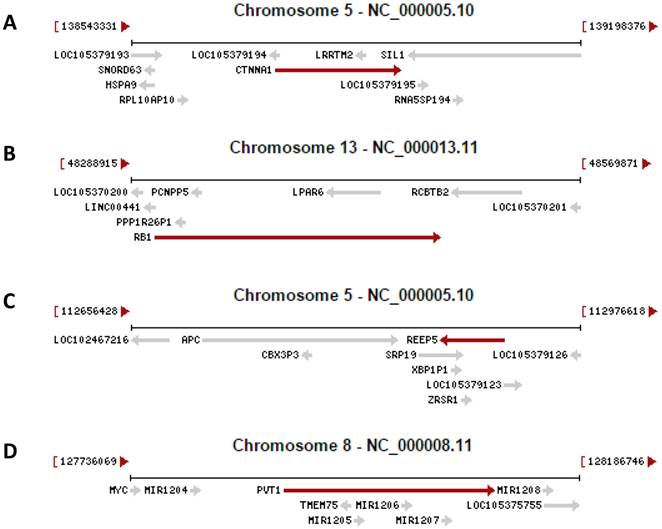
Recent research on the human genome has two revelations. One is that only about 1.5% of the human genomic sequence belongs to the exome, which is the sum of exons for encoding proteins, while the remaining 98.5% of the genome belongs to introns or intergenic sequence [4-7]. However, without a strong rationale, currently only those genomic loci that encode a peptide of 100 or more amino acids (AAs) are considered as genes [8-12], although even much shorter peptides, as short as 11 AAs [13-16], are known to be functional as well [17-22]. If we consider that all genomic loci are genes as long as they encode a peptide, no matter how short it is, the number of genes will be greatly increased [14;23], in turn increasing the protein-coding regions and reciprocally decreasing the intergenic regions. Actually, we anticipate that many currently unannotated genes as shown in figure 1 may later be confirmed to be authentic. Another new finding is that in the human genome there are many more genes than we had previously realized that contain one or more other genes in their introns, either on the Watson strand or the Crick strand of the DNA double helix, with some examples illustrated in figure 1. On many occasions these intron-embedded or nested genes are pseudogenes. Since virtually the whole genome is transcribed [4-7], virtually all pseudogenes are expressed. Actually, increasing evidence suggests that most pseudogenes may be functional, either via their protein products [24;25] or via their non-coding RNA (ncRNA) products as detailed below [26-28]. There is another situation wherein the nested gene is not located in an intron but is on the opposite strand of the DNA double helix, with exemplar cases indicated by the arrows pointing to the opposite direction in figure 1. A well-characterized example is that the DNA strand opposite to the one coding for N-myc gene, i.e. its Crick strand, also encodes a gene that is thus termed N-Cym according the nomenclature of antisense [29]. The existence of gene(s) within another gene challenges the current definition of gene, as in this case the gene is actually a genomic locus that harbors two or more genes. We herein refer those genes that contain other gene(s) to as “parental genes”. A parental gene and the other gene(s) it contains, especially when they are encoded on the same strand of the DNA double helix and thus have the same orientation, are likely to be expressed concomitantly. Exploration of the biological consequence of simultaneous expression of the genes in the same genomic locus is still largely lacking, but would be intriguing. Moreover, whether the hypothetical 3'-to-5' polymerization of DNA or RNA [30;31] widely exists in some organisms awaits exploration, as it, if confirmed to widely exist, also is incongruous with the current concept of gene.
Examples copied from the NCBI database in which a gene (indicated by a red arrow) contains other gene(s) (indicated by grey arrow). In the cases that two arrows point to the opposite directions, the genes are encoded by the opposite strands of the DNA double helix. A: The CTNNA1 (β-catenin) gene has the LRRTM2 gene and an unannotated gene (LOC105379193) embedded in its introns. B: The RB1 gene has the LPAR6 gene and several pseudogenes embedded in its introns. C: The REEP5 gene has the SRP19 and ERSP1 genes and the XBP1P1 pseudogene embedded in its introns. D: The genomic region for the oncogenic PVT1 ncRNA harbors the TMEM75 gene and several microRNAs as well.
Non-coding RNAs can be considered the fourth category of RNA
RNAs are traditionally categorized to 1) ribosomal RNAs (rRNAs), 2) messenger RNAs (mRNAs), and 3) transfer RNAs (tRNAs) that are synthesized by RNA polymerases I, II and III, respectively. Exceptions exist but are traditionally considered rare. For instance, the 5S rRNA is synthesized by RNA polymerase III instead, and the human mitochondrial 12S rRNA encodes a 16-amino-acid peptide called MOTS-c [32] whereas the 16S rRNA encodes another short peptide called humanin [33;34]. However, recent advance in ribonomic research reveals that besides these three classical categories, a huge number of RNA polymerase II products do not encode proteins and thus are not mRNAs. Based on the length of their sequences, these non-coding RNAs as RNA polymerase II products are divided into the long and short groups. Short ncRNAs are mainly regulatory, i.e. regulating transcription, translation, or degradation of mRNAs, such as microRNAs (miRNAs) and small-interference RNAs (siRNAs). Long ncRNAs can be further dichotomized into one group that are regulatory, similar to the short ncRNAs, and another group that directly exert cellular functions, such as the PVT-1 RNA that is oncogenic [35] and the Xist-and-Tsix pair of RNAs that regulate the inactivation of the X chromosome [36-38]. Most short and long ncRNAs are processed from intergenic transcripts or are byproducts of cis-splicing that processes pre-mRNAs to mRNAs [39], although one may argue that circular RNAs (circRNAs), which are also long ncRNAs and are recently gaining momentum in ribonomics, are products, but not byproducts, of cis-splicing [40-42]. Moreover, as we have discussed before [43;44], it is now a well-accepted notion that mammalian genomes are pervasively transcribed from both strands of the DNA double helix and thus produce a huge amount of antisense RNAs, as best exemplified by the aforementioned Xist and Tsix, each of which is the antisense of the other. Many of these antisenses are long and non-coding but have regulatory functions [45].
One caveat that needs to be given is that there is actually no clear demarcation between regulatory and functional ncRNAs, not only because “regulation” itself is a function as well but also because some long ncRNAs, such as some antisense RNAs and circRNAs, are not only regulatory but also functional. Another caveat is that there are other types of ncRNAs that are not described above, such as PIWI-interacting RNAs (piRNAs) [46;47], extracellular RNAs (exRNAs) [48;49], and small nuclear RNAs (snRNAs) that include small nucleolar RNAs (snoRNAs) [50;51]. These and the abovementioned ncRNAs are overlapping in the mechanisms for their production and their functions, have been extensively reviewed in the literature, and thus will not be described herein in detail in order to avoid digression. We propose that all ncRNAs should be considered as the fourth group of RNAs, although some of them, such as some snRNAs, may initially be transcribed by RNA polymerase III, but not II.
Other ribonomic advances also suggest a need of a new gene definition
A gene should be studied at the transcriptional, post-transcriptional (e.g. splicing), translational, post-translational, and functional steps. Each of these steps has steps of its own as well. The study of transcription analyzes the regulatory element of the gene, which is mainly localized at the upstream, i.e. 5', side of the transcription initiation site, although for many genes the 5'-untranslational region (UTR), introns, 3'-UTR and even the 3'-intergenic region are also involved [52;53]. It is common that transcription of a gene can be initiated from or terminated at an alternative site, as seen in the RSK4, CDK4 and Smarca2 genes [54-56]. Such alternative initiation or termination of transcription, when combined with alternative splicing, can generate not only different mRNA variants of the same gene but also different genes' mRNAs, as best exemplified by the p15, p16 and p19 tumor suppressors that are generated in this way from the human INK4A locus [57;58]. Sometimes, the alternative initiation site resides in an upstream gene while the alternative termination site resides in a downstream gene, as described and illustrated before [44;59]. These situations are irreconcilable with the current concept of gene and make it debatable whether the resulting RNA is a chimeric product of the two neighboring genes or a product of an unannotated gene [44].
Over 95% of the human genes contain exons and introns [60], with a gene having about 9 exons and 8 introns on average [61]. During cis-splicing in human cells, on average about 91% of the transcript is severed to be introns, with the remaining 9% of the transcript as exons encoding proteins [62], which collectively corresponds to about 1.5% of the human genomic sequence as aforementioned [4-7]. Although many more genes will likely be identified or annotated in the future, as discussed above, the non-coding sequence will still remain the overwhelming majority. This does not mean that almost the whole genome is junk, but, instead, it indicates that its vast majority is assigned to be regulatory, i.e. of control. Reiterated, one of the reasons for only such a short portion of the genome being used to encode proteins is because the cells need to create a deluge of regulatory RNAs. In some way, the RNA world resembles a departmental community in the biomedical academy in the aspect of the relationship between the ncRNAs and the coding mRNAs: ncRNAs as the regulators or controllers act as professors wearing neat suits and ties to give instructions, while their lab members (graduate students, postdocs and technicians), who are the academic counterpart of the blue-collar working class wearing gloves and lab coats (although lab coats are usually white), act like mRNA-derived proteins to produce data as told, as depicted in figure 2.
Illustration of the relationship between ncRNAs and mRNAs with an academic biomedical department as an analogy. Our genome assigns only 1.5% of its sequence to mRNAs that encode proteins, which conduct cellular functions and thus resemble the cellular counterpart of the blue-collar working class. The remaining 98.5% of the genome is non-coding but is also transcribed to RNAs as regulators of cellular functions, mainly via control of mRNAs, thus resembling the white-collar class. In a biomedical department, most scientific achievements, with their various rewards, are credited to professors, with the tiny leftover credited with acknowledgements (such as diplomas) to those graduate students, postdocs or technicians who are the academic counterpart of the blue-collar working class employed and told by the professors to produce the actual data in the labs or animal rooms. Therefore, those who provide the direction are considered more important than those who provide the labor. Today the main focus of the biomedical fraternity is still on proteins as before, but it is probably time to shift more attention to the governing 98.5%.

RNA transcripts from nearly 95% of the human genes undergo alternative cis-splicing to produce multiple mRNA variants [60]. In disease situations that have mutations, such as in cancers, many more genes' transcripts undergo alternative cis-splicing, because at least 14% of the mutations affect or occur at splice sites [63;64], and some cancer-specific cis-splices are recurrent [65]. The human gene that produces the largest number of cis-spliced products may be titin, as alternative cis-splicing of its 363 exons in the skeleton muscle results in over one million RNA variants by estimation [66]. The Drosophila gene Dscam also has 95 exons that undergo alternative cis-splicing to produce over 38,000 mRNA variants [67]. We therefore surmise that in animal cells most pre-mRNAs can be cis-spliced to many more mature mRNAs than we can imagine or can find in the literature or in the database of the US National Center for Biotechnology Information (NCBI). This is very possible considering that an exon could be as short as only three nucleotides, such as an exon in some mRNA variants of the mouse Ncam gene [68] shown in the NCBI database. A lot of genes have a huge number of expression sequence tags (ESTs) showing many unannotated mRNA variants (http://www.ncbi.nlm.nih.gov/ieb/research/acembly/), which could provide unofficial support for this conjecture.
For some genes, trans-splicing may be involved, which can produce mRNAs with duplicated exons [69;70], as seen in some estrogen receptor alpha variants [71-73]. It has been estimated that about 10% of the genes in the human, fly and worm contain tandem duplication of exons [74], although we opine that the actual figure may be smaller. Trans-splicing can also engender chimeric RNA [44], which may be bicistronic and may contain a transcript from the opposite strand of the DNA double helix or even a transcript from another chromosome. For instance, in mouse testis the Msh4 gene is expressed to seven mRNA variants that involve transcripts from four different chromosomes, and one of the mRNAs is bicistronic while another contains antisense sequence [75]. Obviously, the current concept of gene cannot accommodate any of the above-described situations.
Advances in proteomic research suggest a need of new gene definition as well
The human genome encompasses only slightly over 20,000 genes by the current estimation [76;77]. This number seems to be too small to explain the many complex biological functions and the very diverse social activities of the human being. For this discrepancy, besides the aforementioned reason that there are many genes awaiting identification or annotation, a major reason may be that many genomic loci are not considered as genes because their transcripts do not contain a long-enough open reading frame (ORF). However, this may be a problem of the translation algorithm that is formulated based on the current concept of gene. Indeed, today's algorithm cannot translate some human mitochondrial RNAs [78] and cannot explain why some RNAs with multiple stop codons, such as the Mig-7 mRNA [79-81], can still be expressed to a protein. The current algorithm cannot translate the mRNAs containing the CAG and GGGGCC repeats either [82-84].
About half of the human mRNAs contain upstream ORFs (uORFs) in the region regarded by the current algorithm as the 5'-URT (Fig 3), and in most cases one mRNA contains several uORFs [85]. Many uORFs may be translated to peptides [85-87], such as the one in the yeast SPO24 mRNA [88;89] and the one in the dendritically localized Shank1 mRNA [90], but the peptides may be too short to be noticed [91]. Besides regulation of the mRNA stability [85], these uORFs determine which start codon should be used [86], including non-AUG ones [92], such as CUG and even the questionable AUA [93-95]. Some uORFs may lead to translation of an N-terminally extended or truncated protein isoform as well [10;91].
Illustration of multiple ORFs in a given mRNA. Top panel: In the wt human CDK4 mRNA (copied from the NCBI database as a DNA sequence), as an example, all ATGs and CTGs as the most possible start codons are highlighted in red color while the three canonical stop codons (TAA, TAG and TGA) are shaded with yellow color. The ATG and TGA of the annotated CDK4 ORF are italicized and boldfaced with green color, while all in-frame downstream ATGs that may initiate N-terminally truncated CDK4 protein isoforms are highlighted in green color. Some (but not all, to avoid overwhelming the picture) ORFs that are initiated from out-of-frame ATGs and thus encode non-CDK4 peptides or proteins are underlined, with red underlining indicating the ORFs outside the CDK4 coding region, green underlining indicating an ORF overlapping with the CDK4 C-terminus, and black underlining indicating the ORFs within the CDK4 coding region. Some of these non-CDK4 AltORFs also contain some shorter out-of-frame AltORFs, which are displayed in yellow letters. Bottom panel: Although the current translation algorithm assigns only one ORF (long red bar, referred herein to as “annotated” ORF) to one mRNA (long black arrow), the mRNA also has two uORFs (short green bar) at the 5'UTR and an out-of-frame AltORF at the 3'UTR (long green bar). Moreover, there are many other short AltORFs (blue bar) that are not in frame with one another or with the annotated one. Some of these AltORFs may overlap with the nearby ones and contain some even shorter AltORFs (short yellow bar).
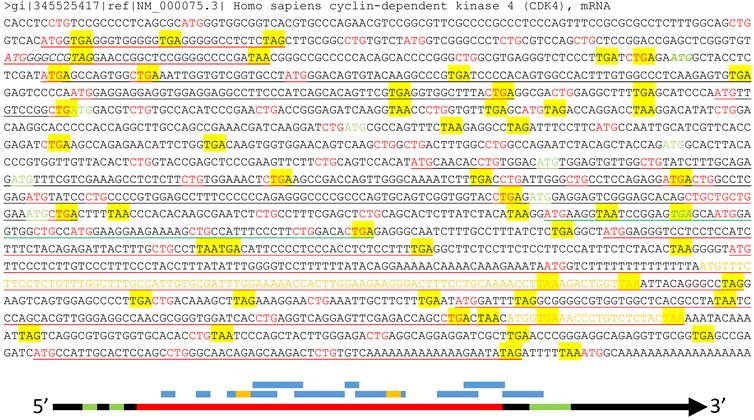
Some mRNAs have been known to contain alternative ORFs (AltORFs) that are irrelevant to the annotated or wild type (wt) ORF, as illustrated in figure 3, although the current algorithm allows only one authentic ORF for one mRNA and considers these AltORFs untranslatable. For example, the XL-exon of the XLαs/Gαs gene in the human and rat encodes a protein completely different from the XLαs/Gαs protein [96]. In the wt human Ataxin-1 mRNA, an out-of-frame ORF overlaps with the wt one [97]. There are a few other known cases of AltORFs in the literature, including the PRNP and the T cell epitopes [98;99]. We surmise that AltORFs may pervasively exist in human mRNAs to dramatically enlarge the protein repertoire (Fig 3), and this conjecture is supported by some bioinformatic data [100]. Unfortunately, like those peptides translated from short uORFs [91], many proteins or peptides translated from AltORFs may be too short to be catchable as well [10].
Even within the annotated ORF of a given mRNA, on most occasions there are many in-frame start codons that are downstream of the canonical ATG and may initiate translation via different mechanisms, such as the use of an IRES (internal ribosome entry site), to produce N-terminally truncated protein isoforms, as we discussed before [54-56;101] and as shown in figure 3 for the human CDK4. Some short protein isoforms of c-Myc, P53 and RB1 are good examples [102-104]. In addition, translation initiation may occur via the so-called +1 or -1 frame-shift mechanisms [105-107], which convert some ncRNAs to mRNAs and may even lead to production of a completely different protein. Most of these alternative initiations of translation occur more often in stressed situations such as in cancer and other diseases [85;108-110].
Termination of translation is as sophisticated as the above-described initiation and elongation, in part because in some situations the three canonical stop codons (UAA, UAG and UGA) may be read through or may instead encode glutamine, tyrosine, pyrrolysine, leucine, cysteine, tryptophan or selenocysteine [111;112]. The NCBI has already listed tryptophan, selenocysteine and pyrrolysine as three new AAs in proteins [112], but few translation algorithms have included them. AGG and AGA encode not only arginine but also glycine and serine and can, at least in human mitochondria, serve as stop codons as well [113;114]. Moreover, AAG and AAA encode not only lysine but also asparagine, whereas CUG encodes not only leucine but also serine, besides as a start codon [112]. Translation termination can be different in different subcellular locations as well. For instance, in human cells the humanin mRNA is translated to a 24-AA peptide in the cytoplasm but to a 21-AA peptide instead in the mitochondria [115;116]. It is worth noting that a drug called G418 is often used to assist selection of cell clones in cell culture but many peers do not realize that G418 is a potent stop codon suppressor that helps the cells in selecting another stop codon [117-120]. Just like the initiation and elongation of translation, stop codon read-through and alternative codon usage occur more often in stressed situations as well [121].
In most proteomic studies, including ours [1], there is always a large portion, often varying between 10-50% [122], of the peptides that cannot be matched to the references and thus are unknown. Although the reasons behind this phenomenon are multiple, including technical ones and gene mutations or polymorphisms, the above-described unannotated genes, imperfect translation algorithms, and alternative codon usages may also be countable for many of these unmatchable peptides. A new definition of gene is required as the basis for constructing a better algorithm to solve these problems of protein translation. An imperative task is to determine whether the omnipresent uORFs and AltORFs are really translated to functional proteins or peptides. For example, our conjecture awaits determination as to whether the wt CDK4 mRNA is translated to not only the wt CDK4 protein but also, in different situations, different N-terminally truncated CDK4 isoforms and many other proteins encoded by those uORFs and AltORFs underlined in figure 3.
Ectopic expression of cDNA skips RNA process, thus often misleading
Ever since the start of the widespread use of the techniques of reverse transcription and polymerase chain reactions three decades ago, genes have been identified from mRNA, and rarely from genomic DNA (gDNA) as was done previously. Nowadays, after a gene is identified, its function is determined using two opposite strategies, i.e. to increase and to decrease its expression in vitro or in vivo with the original level as the reference. Decreased levels are achieved by knocking out the gene (often just interrupting its ORF) or knocking down its mRNA levels by ectopically expressing miRNAs or siRNAs, whereas increased levels are reached by ectopically expressing a cDNA (Fig 4). Almost without exception, one cDNA of the gene is inserted into a vector and then delivered into cells in culture or in an animal by such as transfection, infection or a transgenic-technique. Inside the cells, the cDNA is transcribed to RNA again. In most cases, the cDNA contains only one ORF and thus the RNA is translated to only one protein isoform, although sometimes the cDNA construct is bicistronic with the other cistron encoding a tracer peptide. If the gene in question is known to express multiple mRNA variants, each of the corresponding cDNAs is separately constructed into a vector and then delivered to the target cells for study of its function.
Illustration of how a gene functions by producing different RNAs. Left panel: Flowchart of the routine in studying a gene's function, with emphasis on the ectopic expression approach. Sequencing RT products leads to identification of a gene's mRNA in a cDNA form. Aligning its sequence with gDNA will localize it to a chromosome, which allows us to knock-in or knockout the gene. Continuing to sequence more cDNAs will identify other mRNA variants, which allows us not only to knock down the expression of one, some or all of the variants using such as siRNA but also to ectopically express the mRNAs using cDNAs. For ectopic expression, each cDNA will be cloned into a vector and introduced to cells in culture or in an animal, and the resulting data are used to evaluate the function of this cDNA. Right panel: A gene, which may be expressed in two different cell types (A and B), has two alternative initiation sites and two alternative termination sites for transcription, permitting it to produce four different transcripts. One, some or all four transcripts may have a long 5'-UTR that may harbor multiple uORFs and/or an even-longer 3'-UTR that may contain AltORFs. In one cell type, e.g. normal cells, splicing of one transcript retains all five exons, thus annotated as the wt mRNA, or alternative splicing produces three mRNA variants. In another cell type, e.g. in cancer or another organ or at another developmental stage, the transcripts are spliced to a partly different spectrum of mRNA variants. Some of the mRNAs encode AltORFs as well, resulting in a total of six AltORFs in the two cell types. Moreover, the intron 2 encodes another gene, and its transcripts may be spliced to a wt mRNA with 3 exons (I1, I2 and I3) or, alternatively, to two other mRNA variants in the two cell types. The intron sequences may be processed to different ncRNAs, although only miRNAs and siRNAs are shown for simplicity. More complexly, part of the Crick strand of the DNA may be transcribed to some antisense RNAs as well. Therefore, the global picture about the function of this gene or genomic locus is a collective (but not simply additive) effect of the six mRNA variants and six AltORFs of the parental gene, the three mRNA variants of the nested gene, and all the ncRNAs (miRNAs, siRNAs, piRNAs, snRNAs, exRNAs, circRNAs, and antisense RNAs) in these two cell types. If the parental or the nested gene encodes a transcription factor or a membrane receptor, different heterodimers may be formed among the protein isoforms of the same gene to exert functions as well.
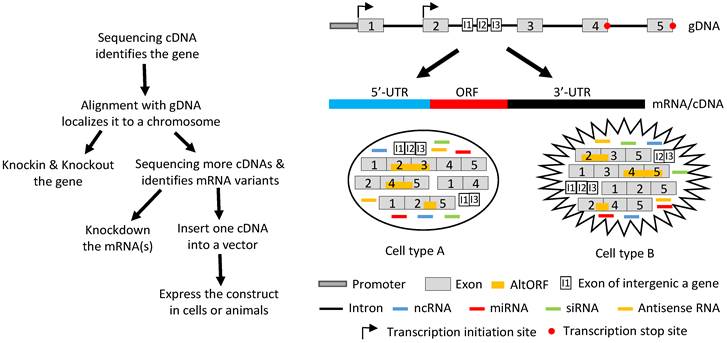
The above described procedure for study of genes' functions actually deprives the cell of its right to regulate the RNA production and various RNA processes, especially splicing. In other words, the cell may have its preferred mRNA variant(s) and ratio(s) among different variants to be expressed to better exert the function of the gene in the particular situation, but we do not allow the cell to decide. Instead, we force the cell to express one variant at a time and later piece together all the shreds of result from single cDNAs as the “global” picture of the gene's function. This current routine has so many flaws that makes it close to malpractice on, probably, most occasions: First, ectopic expression of a cDNA elides the alternative initiation or termination of transcription, thus depriving the cell of its right to produce different transcripts to cope with the particular situation. Second, in the case where there exists intron-embedded or nested gene(s), the function of the to-be-studied gene, or more correctly the genomic locus that contains two or more genes, should be partly contributed by the nested gene(s). Ectopic expression of an intron-less cDNA deletes this contribution. Third, intron sequences, once nipped during splicing, may be processed to regulatory RNAs, especially miRNAs and siRNAs, to elicit functions, but these functions are slipped because cDNAs lack introns. It seems to be possible for us to add back the regulatory RNAs such siRNAs to correct this weakness. However, we have shown that during cis-splicing of the mouse p53 pre-mRNA, different introns are spliced in different orders to produce different variants with retention of different introns [123]. Therefore, we have no way of knowing in any situation which regulatory RNAs from which introns should be produced, and thus should be added back to the system when we ectopically express a cDNA. Fourth, each individual mRNA (cDNA) may have different functions when it is present alone and when it is accompanied by its sibling mRNAs and/or intron-derived regulatory RNAs. This possibility is greatly increased when the gene encodes a transcription factor or a membrane receptor that often functions by forming heterodimers among different isoforms. A cDNA-derived single protein isoform cannot form such heterodimers and may heterodimerize with the protein isoforms of the endogenous origin to muddle things up. Fifth, in general we still know much less about translation, relative to transcription [9]. The existence of multiple uORFs in the 5'-UTR and the existence of the IRESs within the ORF can greatly affect the selection of the start codon, resulting in an N-terminally extended or truncated protein isoform, whereas a +1/-1 frame-shift mechanism may make ncRNA coding and may even produce a completely different protein. These three mechanisms are under the sway of the length of 5'-UTR. Moreover, the 3'-UTR also greatly influences the decision on whether the translation elongation needs to 1) read through the canonical stop codon, 2) use the stop codon to encode an AA, or 3) utilize a downstream stop codon that can be a canonical one or an alternative one, as described above. All these options are often trimmed away when the cDNA is cloned into the vector, not only because usually very short 5'- and 3'-URTs are retained in the construct but also because the artificial promoter in the vector, usually from a cytomegalovirus (CMV), ignores different translation algorithms. In addition, trimming away the 5'- and 3'-UTRs trims away many AltORFs as well. Therefore, when a cDNA is expressed from a vector, it is very likely to function differently from its corresponding mRNA that has a full length of the 5'- and 3'-UTRs. Sixth, it is likely that a gene's function is synergistically or antagonistically different from the simple addition of all single mRNAs. If trans-splicing is also involved, chimeric RNAs and bicistronic mRNAs may be engendered. Probably on many occasions, all the proteins translated from different annotated ORFs, different uORFs and different AltORFs interact in a complex manner to determine the gene's function (Fig 4).
The six scenarios described above, plus many others unmentioned, raise the complexity to a much higher order and make it impossible for us to produce a genuine picture of the gene's functions by piecing together the results from cDNAs that are expressed piecemeal. Addition of the data from the opposite approach, i.e. down-regulating the expression (Fig 4), will greatly help but will still not be able to correct the above described flaws of cDNA use. Besides, knockdown techniques have their own defects and weaknesses as well, such as the off-target problems of the routine RNA-silencing methods [124-127]. Moreover, in many gene-knockout animals, only the ORF for the target (usually the wt) protein is interrupted whereas the RNAs or part of the RNAs may still be expressed. These defects are great concerns in today's ribonomic research but will not be detailed herein to avoid distraction. To use an analogy, the achievements of each research group are made via collaborations among all teammates (technicians, graduate students, postdocs and the principal investigator) in a highly synergistic manner. Therefore, it is largely wrong to divide and distribute the achievements to different teammates at the proportions that we think are reasonable, and it is even more wrong if we attribute all achievements only to the principal investigator who somewhat is equivalent to the wt mRNA.
Then, how should we redefine gene and determine its function?
Today's routine practice of the expression of cDNAs by piecemeal in target cells is actually a good and efficient strategy for determining the function of individual mRNA variants and corresponding protein isoforms, i.e. individual forms, of a “gene”. It is undeniable that we have learned a great many details about the functions of our genome by using cDNA, and much of the knowledge so obtained has been verified using a variety of means such as clinical observations and manipulations in patients or in experimental animals. If we consider part of a classically-defined gene, such as an RNA variant (coding or non-coding) or a phosphorylated status of a protein, as an individual “gene”, our data on the function of “gene” would be much less conflicting with each other. For instance, it will become understandable that a truncated protein isoform translated from a cis-spliced mRNA variant differs in function from the wt protein as they are products of two different “genes”. However, considering a small section of the classically-defined gene as a “gene” may make it more difficult to understand the collective function of a genomic locus as a whole, because, as discussed above, each genomic locus functions via complex synergies and antagonisms among different types of RNAs and among different proteins or protein isoforms produced from the locus. These complex collaborative and antagonistic interactions among various gene elements are also the main reasons for the omnipresent controversy of the data on the functions of most genes documented in the literature. Actually, because of these omnipresent pros and cons, the whole biomedical fraternity has become used to, and enjoys, saying “on one hand…, but on the other hand…” Even worse, although we know that the information controversy may be largely due to the use of cDNA and that data resulting from cDNA use somewhat contort the picture of the functions of the currently-defined gene in question, we have no way of knowing the extent of the distortion. We opine that ectopic expression of single cDNAs by piecemeal will not lead us to an undistorted picture of the functions of genes defined as individual genomic loci, and probably not even close, in many cases.
We favor the gene definition given by the current Wikipedia: “a broad, modern working definition of a gene is any discrete region of heritable, genomic sequence which affects an organism's traits by being expressed as a functional product or by regulating expression.” For simplicity, a gene is herein referred to as a genomic locus, with its activities depicted in an oversimplified manner in figure 4, which shows that it functions in a much higher scale of complexity than what cDNAs can tell us.
Concluding remarks
It is now a post-genomic era, which requires a new gene definition to accommodate the recent advances in genomic, ribonomic and proteomic research. In the past decades, we have learned a great detail from cDNA about the functions of individual mRNA variants and protein isoforms. However, in most cases, our knowledge about the function of each genomic locus as a whole gene is a distorted and unfaithful picture with plentiful controversial information. The image distortion and the data controversy may mainly be because alternatives occur at all levels, including alternative initiation and termination of transcription and translation, alternative codon usage during translation elongation, alternative ORFs within a given mRNA, etc. In most cases, piecemeal ectopic expression of cDNA cannot mimic these alternatives, and piecing together the resulting data cannot lead us to the collective function of a gene as a genomic locus. To obtain an undistorted picture of a gene's function, we should take much greater caution when using a cDNA and when interpreting the data resulting from use of a cDNA. Instead, we should ectopically express gDNA, so that the cells can decide how to transcribe and then to process (mainly splice) the transcript(s) in order to better cope with the particular situation. The gDNA may be constructed under the control of a physiological or a viral (such as CMV) promoter, so as to address different aspects of transcription initiation. Delivery of a gDNA into cells is still difficult because gDNAs usually are of giant size, but it is doable with the available technology [128;129]. For instance, clones of bacterial [130-132], mouse and human artificial chromosomes [133-136] are available for this purpose. Actually, delivery of gDNA into cells in culture and even in an animal has already been used to correct genetic disorders in the lab [137]. It is probably time for us to put more efforts into renovating gDNA delivery technology and to make such delivery into cells routine in our exploration of genes' functions in both physiological and pathological situations.
Acknowledgements
We want to give special thanks to Fred Bogott, M.D. and Ph.D., at the Medical Center, Austin of Minnesota, for his valuable comments on, and excellent English editing of, this manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zhang J, Lou X, Shen H, Zellmer L, Sun Y, Liu S. et al. Isoforms of wild type proteins often appear as low molecular weight bands on SDS-PAGE. Biotechnol J. 2014;9(8):1044-1054
2. Yuan C, Xu N, Liao J. Switch of FANCL, a key FA-BRCA component, between tumor suppressor and promoter by alternative splicing. Cell Cycle. 2012;11(18):3355-3356
3. Lou X, Zhang J, Liu S, Xu N, Liao DJ. The other side of the coin: The tumor-suppressive aspect of oncogenes and the oncogenic aspect of tumor-suppressive genes, such as those along the CCND-CDK4/6-RB axis. Cell Cycle. 2014;13(11):1677-1693
4. Gingeras TR. Implications of chimaeric non-co-linear transcripts. Nature. 2009;461(7261):206-211
5. Pennisi E. Genomics. ENCODE project writes eulogy for junk DNA. Science. 2012;337(6099):1159 1161-doi: 10.1126/science
6. Skipper M, Dhand R, Campbell P. Presenting ENCODE. Nature. 2012;489(7414):45-doi 10.1038/489045a
7. Venter JC, Adams MD, Myers EW, Li PW, Mural RJ, Sutton GG. et al. The sequence of the human genome. Science. 2001;291(5507):1304-1351
8. Kageyama Y, Kondo T, Hashimoto Y. Coding vs non-coding: Translatability of short ORFs found in putative non-coding transcripts. Biochimie. 2011;93(11):1981-1986
9. Pauli A, Valen E, Schier AF. Identifying (non-)coding RNAs and small peptides: challenges and opportunities. Bioessays. 2015;37(1):103-112
10. Landry CR, Zhong X, Nielly-Thibault L, Roucou X. Found in translation: functions and evolution of a recently discovered alternative proteome. Curr Opin Struct Biol. 2015;32:74-80
11. Bazzini AA, Johnstone TG, Christiano R, Mackowiak SD, Obermayer B, Fleming ES. et al. Identification of small ORFs in vertebrates using ribosome footprinting and evolutionary conservation. EMBO J. 2014;33(9):981-993
12. Cheng H, Chan WS, Li Z, Wang D, Liu S, Zhou Y. Small open reading frames: current prediction techniques and future prospect. Curr Protein Pept Sci. 2011;12(6):503-507
13. Ladoukakis E, Pereira V, Magny EG, Eyre-Walker A, Couso JP. Hundreds of putatively functional small open reading frames in Drosophila. Genome Biol. 2011;12(11):R118-doi 10.1186
14. Chu Q, Ma J, Saghatelian A. Identification and characterization of sORF-encoded polypeptides. Crit Rev Biochem Mol Biol. 2015;50(2):134-141
15. Kondo T, Hashimoto Y, Kato K, Inagaki S, Hayashi S, Kageyama Y. Small peptide regulators of actin-based cell morphogenesis encoded by a polycistronic mRNA. Nat Cell Biol. 2007;9(6):660-665
16. Kondo T, Plaza S, Zanet J, Benrabah E, Valenti P, Hashimoto Y. et al. Small peptides switch the transcriptional activity of Shavenbaby during Drosophila embryogenesis. Science. 2010;329(5989):336-339
17. Magny EG, Pueyo JI, Pearl FM, Cespedes MA, Niven JE, Bishop SA. et al. Conserved regulation of cardiac calcium uptake by peptides encoded in small open reading frames. Science. 2013;341(6150):1116-1120
18. Andrews SJ, Rothnagel JA. Emerging evidence for functional peptides encoded by short open reading frames. Nat Rev Genet. 2014;15(3):193-204
19. Pauli A, Norris ML, Valen E, Chew GL, Gagnon JA, Zimmerman S. et al. Toddler: an embryonic signal that promotes cell movement via Apelin receptors. Science. 2014;343(6172):1248636-doi 10.1126/science
20. Anderson DM, Anderson KM, Chang CL, Makarewich CA, Nelson BR, McAnally JR. et al. A micropeptide encoded by a putative long noncoding RNA regulates muscle performance. Cell. 2015;160(4):595-606
21. Zanet J, Benrabah E, Li T, Pelissier-Monier A, Chanut-Delalande H, Ronsin B. et al. Pri sORF peptides induce selective proteasome-mediated protein processing. Science. 2015;349(6254):1356-1358
22. Hashimoto Y, Kondo T, Kageyama Y. Lilliputians get into the limelight: novel class of small peptide genes in morphogenesis. Dev Growth Differ. 2008;50(Suppl 1):S269-S276
23. Ma J, Ward CC, Jungreis I, Slavoff SA, Schwaid AG, Neveu J. et al. Discovery of human sORF-encoded polypeptides (SEPs) in cell lines and tissue. J Proteome Res. 2014;13(3):1757-1765
24. Kim MS, Pinto SM, Getnet D, Nirujogi RS, Manda SS, Chaerkady R. et al. A draft map of the human proteome. Nature. 2014;509(7502):575-581
25. Wilhelm M, Schlegl J, Hahne H, Moghaddas GA, Lieberenz M, Savitski MM. et al. Mass-spectrometry-based draft of the human proteome. Nature. 2014;509(7502):582-587
26. Grander D, Johnsson P. Pseudogene-Expressed RNAs: Emerging Roles in Gene Regulation and Disease. Curr Top Microbiol Immunol. 2015 [Epub ahead of print]:-DOI 10.1007/82_2015_442
27. Milligan MJ, Lipovich L. Pseudogene-derived lncRNAs: emerging regulators of gene expression. Front Genet. 2014;5:476-doi 10.3389/fgene
28. Goodhead I, Darby AC. Taking the pseudo out of pseudogenes. Curr Opin Microbiol. 2015;23:102-109
29. Armstrong BC, Krystal GW. Isolation and characterization of complementary DNA for N-cym, a gene encoded by the DNA strand opposite to N-myc. Cell Growth Differ. 1992;3(6):385-390
30. Seligmann H. Overlapping genes coded in the 3'-to-5'-direction in mitochondrial genes and 3'-to-5' polymerization of non-complementary RNA by an 'invertase'. J Theor Biol. 2012;315:38-52
31. Seligmann H. Systematic asymmetric nucleotide exchanges produce human mitochondrial RNAs cryptically encoding for overlapping protein coding genes. J Theor Biol. 2013;324:1-20
32. Lee C, Zeng J, Drew BG, Sallam T, Martin-Montalvo A, Wan J. et al. The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell Metab. 2015;21(3):443-454
33. Gong Z, Tas E, Muzumdar R. Humanin and age-related diseases: a new link? Front Endocrinol (Lausanne). 2014;5:210-doi 10.3389/fendo
34. Cohen P. New role for the mitochondrial peptide humanin: protective agent against chemotherapy-induced side effects. J Natl Cancer Inst. 2014;106(3):dju006-doi 10.1093/jnci/dju006
35. Takahashi Y, Sawada G, Kurashige J, Uchi R, Matsumura T, Ueo H. et al. Amplification of PVT-1 is involved in poor prognosis via apoptosis inhibition in colorectal cancers. Br J Cancer. 2014;110(1):164-171
36. Shibata S, Wutz A. Transcript versus transcription? Epigenetics. 2008;3(5):246-249
37. Thorvaldsen JL, Verona RI, Bartolomei MS. X-tra! X-tra! News from the mouse X chromosome. Dev Biol. 2006;298(2):344-353
38. Brown CJ, Chow JC. Beyond sense: the role of antisense RNA in controlling Xist expression. Semin Cell Dev Biol. 2003;14(6):341-347
39. Audas TE, Lee S. Stressing out over long noncoding RNA. Biochim Biophys Acta. 2015 pii: S1874-9399(15)00133-9.-doi: 10.1016/j.bbagrm
40. Jeck WR, Sharpless NE. Detecting and characterizing circular RNAs. Nat Biotechnol. 2014;32(5):453-461
41. Rybak-Wolf A, Stottmeister C, Glazar P, Jens M, Pino N, Giusti S. et al. Circular RNAs in the Mammalian Brain Are Highly Abundant, Conserved, and Dynamically Expressed. Mol Cell. 2015;58(5):870-885
42. Ebbesen KK, Kjems J, Hansen TB. Circular RNAs: Identification, Biogenesis and Function. Biochim Biophys Acta. 2015 -pii: S1874-9399(15)00145-5. doi: 10.1016/j.bbagrm
43. Yuan C, Liu Y, Yang M, Liao DJ. New methods as alternative or corrective measures for the pitfalls and artifacts of reverse transcription and polymerase chain reactions (RT-PCR) in cloning chimeric or antisense-accompanied RNA. RNA Biol. 2013;10(6):958-967
44. Peng Z, Yuan C, Zellmer L, Liu S, Xu N, Liao DJ. Hypothesis: Artifacts, Including Spurious Chimeric RNAs with a Short Homologous Sequence, Caused by Consecutive Reverse Transcriptions and Endogenous Random Primers. J Cancer. 2015;6(6):555-567
45. Villegas VE, Zaphiropoulos PG. Neighboring gene regulation by antisense long non-coding RNAs. Int J Mol Sci. 2015;16(2):3251-3266
46. Moyano M, Stefani G. piRNA involvement in genome stability and human cancer. J Hematol Oncol. 2015;8:38-doi 10.1186/s13045-015-0133-5
47. Sato K, Siomi MC. Functional and structural insights into the piRNA factor Maelstrom. FEBS Lett. 2015;589(14):1688-1693
48. Janas T, Janas MM, Sapon K, Janas T. Mechanisms of RNA loading into exosomes. FEBS Lett. 2015;589(13):1391-1398
49. Redzic JS, Balaj L, van der Vos KE, Breakefield XO. Extracellular RNA mediates and marks cancer progression. Semin Cancer Biol. 2014;28:14-23
50. Stepanov GA, Filippova JA, Komissarov AB, Kuligina EV, Richter VA, Semenov DV. Regulatory Role of Small Nucleolar RNAs in Human Diseases. Biomed Res Int. 2015;2015:206849-doi 10.1155/2015/206849
51. Dupuis-Sandoval F, Poirier M, Scott MS. The emerging landscape of small nucleolar RNAs in cell biology. Wiley Interdiscip Rev RNA. 2015;6(4):381-397
52. Gallegos JE, Rose AB. The enduring mystery of intron-mediated enhancement. Plant Sci. 2015;237:8-15
53. Gallegos-Garcia V, Pan SJ, Juarez-Cepeda J, Ramirez-Zavaleta CY, Martin-del-Campo MB, Martinez-Jimenez V. et al. A novel downstream regulatory element cooperates with the silencing machinery to repress EPA1 expression in Candida glabrata. Genetics. 2012;190(4):1285-1297
54. Sun Y, Cao S, Yang M, Wu S, Wang Z, Lin X. et al. Basic anatomy and tumor biology of the RPS6KA6 gene that encodes the p90 ribosomal S6 kinase-4. Oncogene. 2013;32(14):1794-1810
55. Sun Y, Lou X, Yang M, Yuan C, Ma L, Xie BK. et al. Cyclin-dependent kinase 4 may be expressed as multiple proteins and have functions that are independent of binding to CCND and RB and occur at the S and G 2/M phases of the cell cycle. Cell Cycle. 2013;12(22):3512-3525
56. Yang M, Sun Y, Ma L, Wang C, Wu JM, Bi A. et al. Complex alternative splicing of the smarca2 gene suggests the importance of smarca2-B variants. J Cancer. 2011;2:386-400
57. Tian X, Azpurua J, Ke Z, Augereau A, Zhang ZD, Vijg J. et al. INK4 locus of the tumor-resistant rodent, the naked mole rat, expresses a functional p15/p16 hybrid isoform. Proc Natl Acad Sci U S A. 2015;112(4):1053-1058
58. Sherr CJ. Divorcing ARF and p53: an unsettled case. Nat Rev Cancer. 2006;6(9):663-673
59. Yang W, Wu JM, Bi AD, Ou-Yang YC, Shen HH, Chirn GW. et al. Possible Formation of Mitochondrial-RNA Containing Chimeric or Trimeric RNA Implies a Post-Transcriptional and Post-Splicing Mechanism for RNA Fusion. PLoS One. 2013;8(10):e77016-doi 10.1371/journal.pone.0077016
60. Kornblihtt AR, Schor IE, Allo M, Dujardin G, Petrillo E, Munoz MJ. Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat Rev Mol Cell Biol. 2013;14(3):153-165
61. Sakharkar MK, Chow VT, Kangueane P. Distributions of exons and introns in the human genome. In Silico Biol. 2004;4(4):387-393
62. Stamm S, Ben-Ari S, Rafalska I, Tang Y, Zhang Z, Toiber D. et al. Function of alternative splicing. Gene. 2005;344:1-20
63. Kim E, Goren A, Ast G. Insights into the connection between cancer and alternative splicing. Trends Genet. 2008;24(1):7-10
64. Kim E, Goren A, Ast G. Alternative splicing and disease. RNA Biol. 2008;5(1):17-19
65. Sebestyen E, Zawisza M, Eyras E. Detection of recurrent alternative splicing switches in tumor samples reveals novel signatures of cancer. Nucleic Acids Res. 2015;43(3):1345-1356
66. Guo W, Bharmal SJ, Esbona K, Greaser ML. Titin diversity--alternative splicing gone wild. J Biomed Biotechnol. 2010;2010:753675-doi 10.1155/2010/753675
67. Park JW, Graveley BR. Complex alternative splicing. Adv Exp Med Biol. 2007;623:50-63
68. Santoni MJ, Barthels D, Vopper G, Boned A, Goridis C, Wille W. Differential exon usage involving an unusual splicing mechanism generates at least eight types of NCAM cDNA in mouse brain. EMBO J. 1989;8(2):385-392
69. Dixon RJ, Eperon IC, Samani NJ. Complementary intron sequence motifs associated with human exon repetition: a role for intragenic, inter-transcript interactions in gene expression. Bioinformatics. 2007;23(2):150-155
70. Rigatti R, Jia JH, Samani NJ, Eperon IC. Exon repetition: a major pathway for processing mRNA of some genes is allele-specific. Nucleic Acids Res. 2004;32(2):441-446
71. Flouriot G, Brand H, Seraphin B, Gannon F. Natural trans-spliced mRNAs are generated from the human estrogen receptor-alpha (hER alpha) gene. J Biol Chem. 2002;277(29):26244-26251
72. Pink JJ, Wu SQ, Wolf DM, Bilimoria MM, Jordan VC. A novel 80 kDa human estrogen receptor containing a duplication of exons 6 and 7. Nucleic Acids Res. 1996;24(5):962-969
73. Pink JJ, Fritsch M, Bilimoria MM, Assikis VJ, Jordan VC. Cloning and characterization of a 77-kDa oestrogen receptor isolated from a human breast cancer cell line. Br J Cancer. 1997;75(1):17-27
74. Letunic I, Copley RR, Bork P. Common exon duplication in animals and its role in alternative splicing. Hum Mol Genet. 2002;11(13):1561-1567
75. Hirano M, Noda T. Genomic organization of the mouse Msh4 gene producing bicistronic, chimeric and antisense mRNA. Gene. 2004;342(1):165-177
76. Bamshad MJ, Ng SB, Bigham AW, Tabor HK, Emond MJ, Nickerson DA. et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12(11):745-755
77. Belizario JE. The humankind genome: from genetic diversity to the origin of human diseases. Genome. 2013;56(12):705-716
78. Faure E, Delaye L, Tribolo S, Levasseur A, Seligmann H, Barthelemy RM. Probable presence of an ubiquitous cryptic mitochondrial gene on the antisense strand of the cytochrome oxidase I gene. Biol Direct. 2011;6:56-doi 10.1186/1745-6150-6-56
79. Ho MY, Liang SM, Hung SW, Liang CM. MIG-7 controls COX-2/PGE2-mediated lung cancer metastasis. Cancer Res. 2013;73(1):439-449
80. Petty AP, Dick CL, Lindsey JS. Translation of an atypical human cDNA requires fidelity of apurine-pyrimidine repeat region and recoding. Gene. 2008;414(1-2):49-59
81. Petty AP, Wright SE, Rewers-Felkins KA, Yenderrozos MA, Vorderstrasse BA, Lindsey JS. Targeting migration inducting gene-7 inhibits carcinoma cell invasion, early primary tumor growth, and stimulates monocyte oncolytic activity. Mol Cancer Ther. 2009;8(8):2412-2423
82. La Spada AR, Taylor JP. Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat Rev Genet. 2010;11(4):247-258
83. Mori K, Weng SM, Arzberger T, May S, Rentzsch K, Kremmer E. et al. The C9orf72 GGGGCC repeat is translated into aggregating dipeptide-repeat proteins in FTLD/ALS. Science. 2013;339(6125):1335-1338
84. Pearson CE. Repeat associated non-ATG translation initiation: one DNA, two transcripts, seven reading frames, potentially nine toxic entities!. PLoS Genet. 2011;7(3):e1002018-doi 10.1371/journal.pgen.1002018
85. Barbosa C, Peixeiro I, Romao L. Gene expression regulation by upstream open reading frames and human disease. PLoS Genet. 2013;9(8):e1003529-doi 10.1371/journal.pgen
86. Oyama M, Kozuka-Hata H, Suzuki Y, Semba K, Yamamoto T, Sugano S. Diversity of translation start sites may define increased complexity of the human short ORFeome. Mol Cell Proteomics. 2007;6(6):1000-1006
87. Oyama M, Itagaki C, Hata H, Suzuki Y, Izumi T, Natsume T. et al. Analysis of small human proteins reveals the translation of upstream open reading frames of mRNAs. Genome Res. 2004;14(10B):2048-2052
88. Hurtado S, Kim Guisbert KS, Sontheimer EJ. SPO24 is a transcriptionally dynamic, small ORF-encoding locus required for efficient sporulation in Saccharomyces cerevisiae. PLoS One. 2014;9(8):e105058-doi 10.1371/journal.pone
89. Brar GA, Yassour M, Friedman N, Regev A, Ingolia NT, Weissman JS. High-resolution view of the yeast meiotic program revealed by ribosome profiling. Science. 2012;335(6068):552-557
90. Studtmann K, Olschlager-Schutt J, Buck F, Richter D, Sala C, Bockmann J. et al. A non-canonical initiation site is required for efficient translation of the dendritically localized Shank1 mRNA. PLoS One. 2014;9(2):e88518-doi 10.1371/journal.pone
91. Somers J, Poyry T, Willis AE. A perspective on mammalian upstream open reading frame function. Int J Biochem Cell Biol. 2013;45(8):1690-1700
92. Ivanov IP, Firth AE, Michel AM, Atkins JF, Baranov PV. Identification of evolutionarily conserved non-AUG-initiated N-terminal extensions in human coding sequences. Nucleic Acids Res. 2011;39(10):4220-4234
93. Raschke S, Elsen M, Gassenhuber H, Sommerfeld M, Schwahn U, Brockmann B. et al. Evidence against a beneficial effect of irisin in humans. PLoS One. 2013;8(9):e73680-doi 10.1371/journal.pone
94. Albrecht E, Norheim F, Thiede B, Holen T, Ohashi T, Schering L. et al. Irisin - a myth rather than an exercise-inducible myokine. Sci Rep. 2015;5:8889-doi 10.1038/srep08889
95. Erickson HP. Irisin and FNDC5 in retrospect: An exercise hormone or a transmembrane receptor? Adipocyte. 2013;2(4):289-293
96. Klemke M, Kehlenbach RH, Huttner WB. Two overlapping reading frames in a single exon encode interacting proteins--a novel way of gene usage. EMBO J. 2001;20(14):3849-3860
97. Bergeron D, Lapointe C, Bissonnette C, Tremblay G, Motard J, Roucou X. An out-of-frame overlapping reading frame in the ataxin-1 coding sequence encodes a novel ataxin-1 interacting protein. J Biol Chem. 2013;288(30):21824-21835
98. Vanderperre B, Staskevicius AB, Tremblay G, McCoy M, O'Neill MA, Cashman NR. et al. An overlapping reading frame in the PRNP gene encodes a novel polypeptide distinct from the prion protein. FASEB J. 2011;25(7):2373-2386
99. Ho O, Green WR. Alternative translational products and cryptic T cell epitopes: expecting the unexpected. J Immunol. 2006;177(12):8283-8289
100. Vanderperre B, Lucier JF, Bissonnette C, Motard J, Tremblay G, Vanderperre S. et al. Direct detection of alternative open reading frames translation products in human significantly expands the proteome. PLoS One. 2013;8(8):e70698. doi: 10.1371/journal.pone.0070698
101. Bollig-Fischer A, Thakur A, Sun Y, Wu J-S, Liao DJ. The predominant proteins that react to the MC-20 estrogen receptor alpha antibody differ in molecular weight between the mammary gland and uterus in the mouse and rat. Int J Biomed Sci. 2012;8(1):51-63
102. Liao DJ, Dickson RB. c-Myc in breast cancer. Endocr Relat Cancer. 2000;7(3):143-164
103. Weingarten-Gabbay S, Khan D, Liberman N, Yoffe Y, Bialik S, Das S. et al. The translation initiation factor DAP5 promotes IRES-driven translation of p53 mRNA. Oncogene. 2014;33(5):611-618
104. Xu HJ, Xu K, Zhou Y, Li J, Benedict WF, Hu SX. Enhanced tumor cell growth suppression by an N-terminal truncated retinoblastoma protein. Proc Natl Acad Sci U S A. 1994;91(21):9837-9841
105. Atkins JF, Bjork GR. A gripping tale of ribosomal frameshifting: extragenic suppressors of frameshift mutations spotlight P-site realignment. Microbiol Mol Biol Rev. 2009;73(1):178-210
106. Xie P. Dynamics of +1 ribosomal frameshifting. Math Biosci. 2014;249:44-51
107. Temperley R, Richter R, Dennerlein S, Lightowlers RN, Chrzanowska-Lightowlers ZM. Hungry codons promote frameshifting in human mitochondrial ribosomes. Science. 2010;327(5963):301-doi 10.1126/science
108. Wethmar K. The regulatory potential of upstream open reading frames in eukaryotic gene expression. Wiley Interdiscip Rev RNA. 2014;5(6):765-778
109. von Arnim AG, Jia Q, Vaughn JN. Regulation of plant translation by upstream open reading frames. Plant Sci. 2014;214:1-12
110. Leprivier G, Rotblat B, Khan D, Jan E, Sorensen PH. Stress-mediated translational control in cancer cells. Biochim Biophys Acta. 2015;1849(7):845-860
111. Jungreis I, Lin MF, Spokony R, Chan CS, Negre N, Victorsen A. et al. Evidence of abundant stop codon readthrough in Drosophila and other metazoa. Genome Res. 2011;21(12):2096-2113
112. Lobanov AV, Turanov AA, Hatfield DL, Gladyshev VN. Dual functions of codons in the genetic code. Crit Rev Biochem Mol Biol. 2010;45(4):257-265
113. Richter R, Pajak A, Dennerlein S, Rozanska A, Lightowlers RN, Chrzanowska-Lightowlers ZM. Translation termination in human mitochondrial ribosomes. Biochem Soc Trans. 2010;38(6):1523-1526
114. Lightowlers RN, Chrzanowska-Lightowlers ZM. Terminating human mitochondrial protein synthesis: a shift in our thinking. RNA Biol. 2010;7(3):282-286
115. Nishimoto I, Matsuoka M, niikura T. Unravelling the role of Humanin. Trends Mol Med. 2004;10(3):102-105
116. Guo B, Zhai D, Cabezas E, Welsh K, Nouraini S, Satterthwait AC. et al. Humanin peptide suppresses apoptosis by interfering with Bax activation. Nature. 2003;423(6938):456-461
117. Keeling KM, Xue X, Gunn G, Bedwell DM. Therapeutics based on stop codon readthrough. Annu Rev Genomics Hum Genet. 2014;15:371-394
118. Floquet C, Deforges J, Rousset JP, Bidou L. Rescue of non-sense mutated p53 tumor suppressor gene by aminoglycosides. Nucleic Acids Res. 2011;39(8):3350-3362
119. Heier CR, DiDonato CJ. Translational readthrough by the aminoglycoside geneticin (G418) modulates SMN stability in vitro and improves motor function in SMA mice in vivo. Hum Mol Genet. 2009;18(7):1310-1322
120. Nudelman I, Glikin D, Smolkin B, Hainrichson M, Belakhov V, Baasov T. Repairing faulty genes by aminoglycosides: development of new derivatives of geneticin (G418) with enhanced suppression of diseases-causing nonsense mutations. Bioorg Med Chem. 2010;18(11):3735-3746
121. Endres L, Dedon PC, Begley TJ. Codon-biased translation can be regulated by wobble-base tRNA modification systems during cellular stress responses. RNA Biol. 2015;12(6):603-614
122. Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4(3):207-214
123. Yang M, Wu J, Wu SH, Bi AD, Liao DJ. Splicing of mouse p53 pre-mRNA does not always follow the "first come, first served" principle and may be influenced by cisplatin treatment and serum starvation. Mol Biol Rep. 2012;39(9):9247-9256
124. Wade M. High-Throughput Silencing Using the CRISPR-Cas9 System: A Review of the Benefits and Challenges. J Biomol Screen. 2015 -pii: 1087057115587916
125. Fellmann C, Lowe SW. Stable RNA interference rules for silencing. Nat Cell Biol. 2014;16(1):10-18
126. Ishida M, Selaru FM. miRNA-Based Therapeutic Strategies. Curr Anesthesiol Rep. 2013;1(1):63-70
127. Nana-Sinkam SP, Croce CM. Clinical applications for microRNAs in cancer. Clin Pharmacol Ther. 2013;93(1):98-104
128. Hibbitt OC, Wade-Martins R. Delivery of large genomic DNA inserts >100 kb using HSV-1 amplicons. Curr Gene Ther. 2006;6(3):325-336
129. Laner A, Goussard S, Ramalho AS, Schwarz T, Amaral MD, Courvalin P. et al. Bacterial transfer of large functional genomic DNA into human cells. Gene Ther. 2005;12(21):1559-1572
130. White RE, Wade-Martins R, Hart SL, Frampton J, Huey B, Desai-Mehta A. et al. Functional delivery of large genomic DNA to human cells with a peptide-lipid vector. J Gene Med. 2003;5(10):883-892
131. Kao BR, McColl B, Vadolas J. Generation of BAC reporter cell lines for drug discovery. Methods Mol Biol. 2015;1227:323-343
132. Holmes S, Lyman S, Hsu JK, Cheng J. Making BAC transgene constructs with lambda-red recombineering system for transgenic animals or cell lines. Methods Mol Biol. 2015;1227:71-98
133. Kazuki Y, Oshimura M. Human artificial chromosomes for gene delivery and the development of animal models. Mol Ther. 2011;19(9):1591-1601
134. Oshimura M, Uno N, Kazuki Y, Katoh M, Inoue T. A pathway from chromosome transfer to engineering resulting in human and mouse artificial chromosomes for a variety of applications to bio-medical challenges. Chromosome Res. 2015;23(1):111-133
135. Kouprina N, Tomilin AN, Masumoto H, Earnshaw WC, Larionov V. Human artificial chromosome-based gene delivery vectors for biomedicine and biotechnology. Expert Opin Drug Deliv. 2014;11(4):517-535
136. Kouprina N, Earnshaw WC, Masumoto H, Larionov V. A new generation of human artificial chromosomes for functional genomics and gene therapy. Cell Mol Life Sci. 2013;70(7):1135-1148
137. Appledorn DM, Seregin S, Amalfitano A. Adenovirus vectors for renal-targeted gene delivery. Contrib Nephrol. 2008;159:47-62
Author contact
![]() Corresponding authors: Yuping Jia, Shangdong Academy of Pharmaceutical Sciences, Ji'nan City, Shangdong 250101, P.R. China. Email: jiayupingygscom. Jian Zhang, Center for Translational Medicine Pharmacology and Biomedical Sciences Building, Guangxi Medical University, 22 Shuangyong Road, Nanning, Guangxi 530021, P.R. China. Email: jzhangqicom. Ningzhi Xu, Laboratory of Cell and Molecular Biology, Cancer Institute, Chinese Academy of Medical Science, Beijing 100021, P.R. China. Email: xuningzhiac.cn. D. Joshua Liao, Hormel Institute, University of Minnesota, Austin, MN 55912, USA. Email: djliaoumn.edu
Corresponding authors: Yuping Jia, Shangdong Academy of Pharmaceutical Sciences, Ji'nan City, Shangdong 250101, P.R. China. Email: jiayupingygscom. Jian Zhang, Center for Translational Medicine Pharmacology and Biomedical Sciences Building, Guangxi Medical University, 22 Shuangyong Road, Nanning, Guangxi 530021, P.R. China. Email: jzhangqicom. Ningzhi Xu, Laboratory of Cell and Molecular Biology, Cancer Institute, Chinese Academy of Medical Science, Beijing 100021, P.R. China. Email: xuningzhiac.cn. D. Joshua Liao, Hormel Institute, University of Minnesota, Austin, MN 55912, USA. Email: djliaoumn.edu