Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(12):1424-1435. doi:10.7150/ijbs.12930 This issue Cite
Research Paper
Inhibition of mTOR Pathway by Rapamycin Reduces Brain Damage in Rats Subjected to Transient Forebrain Ischemia
Xiao Yang*,1,2, Changhun Hei*,2,3, Ping Liu2,4, Yaozu Song1, Taylor Thomas2, Sylvie Tshimanga2, Feng Wang1, Jianguo Niu1, Tao Sun1, P. Andy Li2 ![]()
1. Neuroscience Center, General Hospital of Ningxia Medical University, Key Laboratory of Craniocerebral Diseases of Ningxia Hui Autonomous Region, Yinchuan 75004, China
2. Department of Pharmaceutical Sciences, Biomanufacturing Research Institute Biotechnology Enterprise (BRITE), North Carolina Central University, 1801 Fayetteville Street, Durham, NC 27707, USA
3. Department of Human Anatomy, Histology and Embryology, Ningxia Medical University, Yinchuan 75004, China
4. Department of Endocrinology, General Hospital of Ningxia Medical University, Yinchuan 750004, China
* These authors contributed equally to this work.
Received 2015-6-11; Accepted 2015-10-21; Published 2015-11-24
Abstract

The aims of this study are to clarify the role of mTOR in mediating cerebral ischemic brain damage and the effects of rapamycin on ischemic outcomes. Ten minutes of forebrain ischemia was induced in rats, and their brains were sampled after 3 h, 16 h, and 7 days reperfusion for histology, immunohistochemistry and biochemical analysis. Our data demonstrated that cerebral ischemia resulted in both apoptotic and necrotic neuronal death; cerebral ischemia and reperfusion led to significant increases of mRNA and protein levels of p-mTOR and its downstream p-P70S6K and p-S6; elevation of LC3-II, and release of cytochrome c into the cytoplasm in both the cortex and hippocampus. Inhibition of mTOR by rapamycin markedly reduced ischemia-induced damage; suppressed p-Akt, p-mTOR, p-P70S6K and p-S6 protein levels; decreased LC3-II and Beclin-1; and prevented cytochrome c release in the two structures. All together, these data provide evidence that cerebral ischemia activates mTOR and autophagy pathways. Inhibition of mTOR deactivates the mTOR pathway, suppresses autophagy, prevents cytochrome c release and reduces ischemic brain damage.
Keywords: Autophagy, Cerebral ischemia, Cytochrome c, mTOR, Rapamycin, Reperfusion damage
Introduction
Stroke continues to present a significant public health challenge for society. Approximately 80% of all strokes are ischemic [1]. Although various neuroprotective agents have been shown to be efficacious in reducing ischemic brain damage in animal stroke models, none of these drugs has demonstrated efficacy in clinical stroke patients. Recanalization of the occluded blood vessel using thrombolytic drugs or mechanical device remains the major therapeutic approach for cerebral ischemic stroke [2]. However, reperfusion itself may exacerbate brain injury, resulting in an extension of damage after ischemia [3-6]. It has been reported that cardiopulmonary resuscitation for victims of cardiac arrest within and outside of the hospital can successfully restore spontaneous circulation in approximately 70,000 patients a year in the United States. Unfortunately, approximately 60% of these patients subsequently die in the hospital as a result of extensive brain damage introduced by reperfusion [7].
The mammalian target of rapamycin (mTOR) pathway is an essential cellular signaling pathway involved in a number of important physiological functions, including cell growth, proliferation, metabolism, protein synthesis, and autophagy [8]. There is accumulating evidence that the mTOR pathway may be involved in the pathophysiology of certain neurological diseases, such as the tuberous sclerosis complex, genetic and acquired epilepsy, brain tumors as well as neurodegenerative diseases [9-18], in most of which the mTOR pathway is excessively activated. Rapamycin is a macrolide antibiotic possessing potent immunosuppressive and anti-proliferative properties [19]. Rapamycin binds to mTOR and prevents the phosphorylation of its downstream substrates p70 ribosomal protein S6 kinase (P70S6K), eukaryotic translation initiation factor 4E-binding protein 1 (4E-BP1) and other proteins involved in transcription, translation, and cell cycle control and further affecting cell survival or death [20]. Rapamycin associates with its intracellular receptor, FK 506-binding protein of 12 kDa (FKBP12), and the resulting complex interacts with the FKBP12-rapamycin binding (FRB) domain located in the C-terminus of mTOR [21]. To date, FK506 binding protein 12 is the only known target of rapamycin [22].
Given the potential role of mTOR in a number of neurologic disorders, enthusiasm is now focusing upon the mTOR pathway as a potential therapeutic target for ischemic cerebrovascular diseases. However, the results obtained so far are controversial. On the one hand, it has been reported that cerebral ischemia and oxygen glucose deprivation (OGD) suppress the mTOR pathway and that upregulating mTOR offers neuroprotection [23-28]. On the other hand, there is a growing body of evidence suggesting that rapamycin promotes neuronal viability and reduces neurological damage in several ischemic injury models through inhibiting mTOR pathway [29-33]. The objectives of this study were to clarify the role of mTOR in ischemic brain damage and the effects of rapamycin on ischemic outcome. To that end, rats were subjected to 10 min of forebrain ischemia, activation of the mTOR pathway was examined by phosporylation of mTOR, P70S6K and S6, cell death was assessed using routine histology and TUNEL staining, and autophagy was evaluated by microtubule-associated protein 1 light chain 3 (LC3) II and the Bcl2-interacting protein Beclin-1. The results demonstrated that cerebral ischemia activated the mTOR pathway, increased autophagy markers, and led to the release of cytochrome c into the cytoplasm. Treatment with rapamycin significantly reduced neuronal death, and its protective effects were associated with the suppression of mTOR signaling and the inhibition of autophagy and cytochrome c release.
Materials and methods
Animals
Forty-eight male Wistar rats (6-8 weeks, 240±10 g body weight), purchased from Charles River Laboratories, were used in this study. The animal procedures were approved by the Institutional Animal Care and Usage Committee (IACUC) at North Carolina Central University and follows the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All animal procedures were conducted under anesthesia. Animals were divided into vehicle- and rapamycin-treated groups (n=24 in each group), each consisting of a sham-operated control, ischemia plus 3 h, 16 h and 7 days of reperfusion (n=6, in each subgroup).
Rapamycin injection
Rapamycin (LC Laboratories, USA) stock solution was prepared by dissolving 100 mg in 5 ml of 100% ethanol (20 mg/ml) and stored at -20°C. Working solution is made by diluting the stock solution in 5% Tween 80 and 5% polyethyleneglycol 400 to a final concentration of 4% ethanol immediately before intraperitoneal (i.p) injection. Rapamycin (6 mg/kg/d) was injected 7 days prior to the induction of ischemia and continued daily until the animals were sacrificed.
Operative Procedures
The animals were fasted overnight prior to surgery but had free access to water. Anesthesia was induced by inhalation of 3.0% isoflurane in a mixture of N2O and O2 (70:30) and maintained at 1.0-2.0% during surgery. A midline incision was made in the ventral cervical area to expose the common carotid arteries. A central catheter was inserted into the right jugular vein for the withdrawal of blood. A tail artery was cannulated for blood pressure monitoring. A rectal thermometer was inserted to monitor body temperature. Head temperature was monitored by placing another needle thermometer subcutaneously on the temporal muscle. Both rectal and head temperatures were maintained close to 37 ± 0.5°C by lamp heating combined with an Automated Thermal Control Unit (DC Temperature Controller, FHS 40-90-8D, Bowdoin, ME) during the whole experimental process. Blood pressure was monitored using a pressure monitor (BP-1, World Precision Instruments).
Induction of Ischemia
Transient forebrain cerebral ischemia (10 min) was induced using a two-vessel occlusion (2-VO) method plus systemic hypotension, with which both the common carotid arteries were occluded with vascular clamps for 10 min and blood was withdrawn through the central catheter inserted into the jugular vein to maintain the blood pressure at 40 to 50 mm Hg during the ischemic period. Ischemia was terminated by reinfusion of the shed blood and removal of the carotid clamps. Animals that experienced the same surgical procedure but without occlusion of the common carotid arteries and withdrawal of blood were used as sham-operated controls. The rats were euthanatized at 3 h, 16 h and 7 days after reperfusion, and their brains were removed. In order to reduce the animal numbers, one hemisphere from each animal was used for morphological studies and the other for biochemical analyses.
Histology and Immunohistochemistry
Brains were placed into a pre-cooled rat brain matrix, cut into 3 coronal blocks of 4 mm each and fixed with 4% paraformaldehyde for 24 h. The blocks were dehydrated in ascending concentration of ethanol, embedded in paraffin, and sectioned at 5 μm thickness. For routine histology, the sections were stained with celestine blue and acid fuchsin. Neuronal death was examined under a light microscope at 400× magnification. Bright red-stained acidophilic neurons with shrunken, triangular dense purple nuclei were considered as dead neurons. The numbers of dead cells were counted in 5 microscopic fields in the parietal cortex and throughout the hippocampal CA1 subregion. The results are presented as percentage of dead neurons,
For immunohistochemistry, the sections were de-paraffinized and rehydrated. The sections were immersed in 0.1 M citric acid and steamed in a high-pressure cooker for 10 min for antigen retrieval. Endogenous non-specific biding sites were blocked using 3% hydrogen peroxide at 37°C for 60 min. The sections were incubated with a monoclonal anti-phospho-S6 primary antibody (dilution 1:200) at 4°C overnight. After sequential incubation with peroxidase conjugated donkey anti-rabbit IgG. 3,3'-Diaminobenzidine (DAB) was then used as a chromogen to view the reaction using a light microscope. The sections were counterstained by hematoxylin. All incubations were performed in a humidified chamber. Negative controls were run in parallel using adjacent sections incubated without primary antibody.
TUNEL Staining
The sections were de-paraffinized, rehydrated, and fixed with 4% formaldehyde for 15 min. The sections were permeabilized using 100 μl of a 20 μg/ml Proteinase K solution (10 min at room temperature). The sections were then stained using a TUNEL kit (DeadEndTM Fluorometric TUNEL System, G3250, USA). In brief, 100 μl Equilibration Buffer was added onto the sections at room temperature for 10 min, followed by application of 50 μl TdT reaction mix to the tissue. After a 60-min incubation period at 37°C in a humidified chamber, the reaction was stopped using 2X SSC. The slide was washed and mounted with Vectashield® mounting medium containing propidium iodide (PI). The slides were examined under a Nikon Eclipse E600 FN fluorescent confocal microscope. The number of positively stained cells was counted in 5 fields in the parietal cortex and throughout the CA1 region at Bregma -3.8 mm level. The results are presented as the percentage of TUNEL positive neurons in all dead neurons.
Real-time quantitative PCR
TRIzol kit (Invitrogen, Carlsbad, California, USA) was used to extract the total RNA from the cortical sample. The total RNA concentration was measured using a Nano Drop Spectrophotometer (ND-1000, Thermo Scientific, USA) at 260 nm absorbance. Two micrograms total RNA treated with M-MLV reverse transcriptase were used for synthesizing the first-strand of cDNA according to the manufacturer's recommendations (Promega, USA). The resulting cDNA was then subjected to real-time quantitative PCR for evaluation of the relative mRNA levels of mTOR and GAPDH (as an internal control). Gene-specific amplification was performed using an ABI 7900HT real-time PCR system (Life Technologies, Carlsbad, California, USA) with a 15-μl PCR mix containing 0.5 μl of cDNA, 7.5 μl of 2X SYBR Green master mix (Invitrogen, Carlsbad, California, USA), and 200 nM of the appropriate oligonucleotide primers. After 40 cycles of amplifications, the Ct (threshold cycle) value of each sample was calculated from the threshold cycles using the instrument's software (SDS 2.3), and the relative expression of the mTOR mRNA was normalized to the GAPDH value. Relative expression level was calculated by ΔCt = (the Ct values of mTOR in experimental group - the Ct values of GAPDH in experimental group) - (the Ct values of mTOR in sham control group-the Ct values of GAPDH in sham control group).
Western blotting
The cortical and hippocampal tissues were separated on ice-cold plate, frozen in liquid nitrogen and stored in a -80ºC freezer till use. The tissues were individually homogenized using a suspension buffer containing 15 mM Tris base/HCL (pH 7.7), 0.25 M sucrose, 15 mM NaCl, 1.5 mM MgCl2, 2.5 mM EDTA, 0.25 mM Na3VO4, 25 mM NaF, 1 mM EGTA, 2 mM NaPPi, and with addition of a protease inhibitor cocktail (Complete Tablets, Roche), 1 mM DTT, and 0.5 mM PMSF. Cytosolic and nuclear fractions were separated through a series of centrifugations. Protein concentrations were measured using a Microplate BCA Protein Assay kit (Thermo Scientific). Twenty-five micrograms of protein from each sample were loaded to 4%-12% NuPAGE gels (Invitrogen, Carlsbad, CA, USA) for electrophoresis. The proteins were transferred to PVDF membranes (Millipore), and probed with the primary antibodies listed in Table 1. Both target protein bands and internal loading control protein bands were viewed on LI-COR Biosciences Odyssey Infrared Fluorescent scanner (Lincoln, NE, USA), band intensities were calculated using LI-COR software, and results were presented as a ratio of the target protein band fluorescent intensity over the loading control protein band intensity.
Primary antibodies used in this study.
| Antibodies | Source | Dilutions | Manufacturer | Application |
|---|---|---|---|---|
| p-mTOR (Ser 2448) | Rabbit | 1:200 | Cell Signaling | WB |
| p-P70S6K (T421/S424) | Rabbit | 1:800 | Cell Signaling | WB |
| p-S6 (Ser235+236) | Rabbit | 1:200 | Thermo | WB,IHC-P |
| p-AKT (Ser473) | Rabbit | 1:800 | Cell Signaling | WB |
| Beclin-1 | Rabbit | 1:800 | Cell Signaling | WB |
| LC3A/B | Rabbit | 1:800 | Abcam | WB |
| Cyt c | Rabbit | 1:1000 | Cell Signaling | WB |
| β-actin | Mouse | 1:1000 | Cell Signaling | WB |
IHC-P, Immunohistochemistry for paraffin section; WB, Western blot.
Statistical analysis
Data are represented as mean±s.d. Parametric pathological data and semi- quantitative data were analyzed with variance (ANOVA) with Bonferroni post hoc comparison. Student's t test was employed when comparisons were made between vehicle- and rapamycin-treated groups at an identical reperfusion endpoint. P < 0.05 was considered statistically significant. All statistical analyses were performed using SPSS statistical software package version 13.0.
Results
Rapamycin Significantly Ameliorated Ischemia-Induced Neuronal Death
In the cortical area, there were only few scattered dead neurons detected in sham-operated animals (sham). Number of dead neurons increased after 3 and 16 h of reperfusion following a 10 min forebrain ischemic episode. As a result, the percentage of dead cells increased from 8% to 12% at 3 and 16 h of reperfusion, respectively. The number of dead neurons increased drastically to 30% after 7 days of reperfusion. Rapamycin treatment significantly ameliorated neuronal death after 7 days but not at 3 or 16 hrs of reperfusion. Thus, the percentages of dead neurons in the cortex were 7.8% and 9.3% at 3 and 16 h of reperfusion, respectively, while this value was lowered to 9% at 7 days (p<0.01 versus vehicle group). A set of representative microphotographs is presented in Fig. 1 A, and a summarized bar graph is presented in Fig. 1B.
Brain damage in the cortical and hippocampal CA1 areas at various reperfusion stages following 10 min forebrain ischemia and reperfusion (I/R) injury. A,C. Representative microphotographs showing histological outcomes in the cortex (A) and hippocampal CA1 (C) area; B,D. summarized percentage of death in each group in the cortex (B) and CA1 (D). Celestine blue and acid fuchsin staining. Arrows indicate dead neurons. Bar=50 μm. Data are presented as means ± s.d. *p<0.05, ** p< 0.01 vs. sham in the same treatment group, ## p<0.01 between rapamycin- (RAPA) and vehicle-treated groups at an identical reperfusion stage.
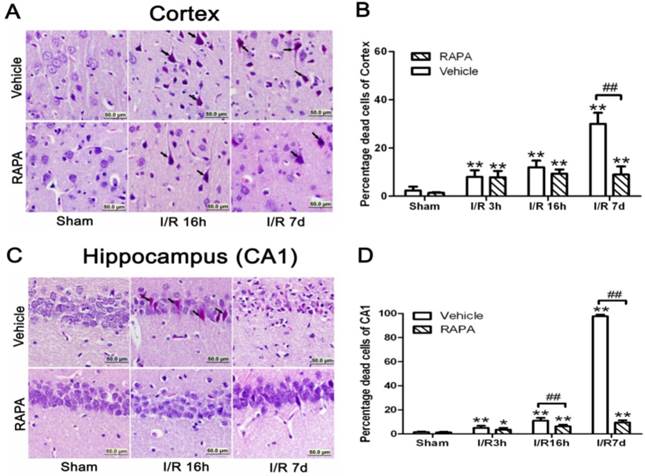
In the hippocampal CA1 area, there were no dead neurons observed in the sham controls. After 3 h of reperfusion, very few scattered dead neurons, less than 5% of the total CA1 population, were detected. The percentage of dead neurons increased to an average of 11% after 16 h of reperfusion, and a drastic increase to close to 100% death was observed after 7 days in all animals. Rapamycin treatment completely blocked the damage in the CA1 area at 3 and 16 h of reperfusion and significantly reduced the damage to less than 10% after the 7 days of recovery. A set of representative photomicrographs are presented in Fig. 1C, and a summarized bar graph is presented in Fig. 1D.
Rapamycin Reduced the Number of TUNEL Positive Neurons
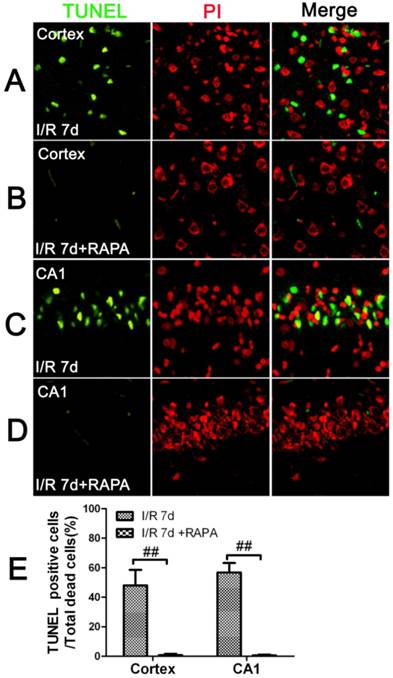
To characterize the nature of the cell death after cerebral ischemia in the vehicle- and rapamycin-treated animals, we performed TUNEL staining and counter labeled the sections with propidium iodide using a separate set of frozen brain sections. The results showed that there were no TUNEL positive neurons detected at 3 and 16 h of reperfusion in the cortical and hippocampal CA1 areas in both the vehicle- and rapamycin-treated animals. After 7 days of reperfusion, the number of TUNEL positive neurons increased significantly in both the cortex and the CA1 areas. Interestingly, not all dead neurons were TUNEL positive. In fact, the TUNEL positive neurons constituted 48% of all the dead neurons in the cortex and 56.8% in the CA1. These results suggest that ischemic neuronal death is both an apoptotic and necrotic feature. Rapamycin treatment significantly reduced TUNEL positive cells in both the cortex and CA1. As shown in Fig. 2, TUNEL positive cells could hardly be found in the rapamycin groups at 7 days of reperfusion in both the cortex and CA1.
TUNEL immunofluorescent staining in the cortex and CA1 at 7 days of reperfusion in vehicle- and rapamycin-treated animals. A-D. Representative images from the cortex and CA1 in vehicle- (A,C) and rapamycin-treated (B,D) animals. E. Summarized percentage of TUNEL positive cells over total number of dead cells in the cortex and CA1. Green color denotes TUNEL positive neurons, red, propidium iodide staining. Bar=50 μm. Data are presented as means ± s.d. ## p<0.01 between rapamycin- (RAPA) and vehicle-treated groups at an identical reperfusion stage.
Ischemia Increased the mRNA and Protein Levels of mTOR
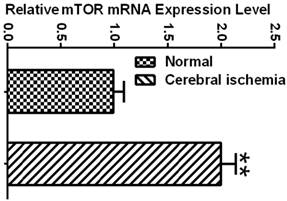
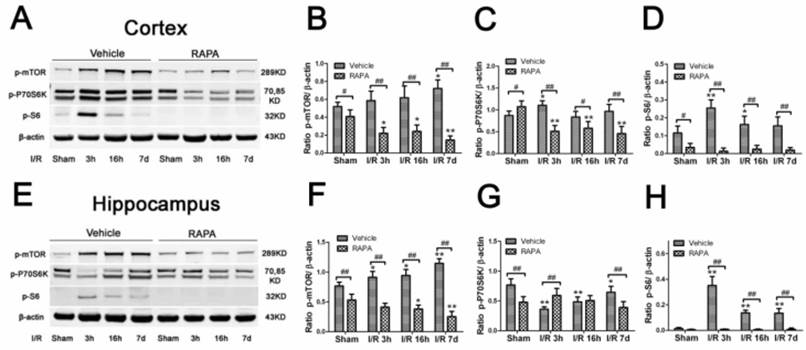
To determine whether the mTOR pathway was activated after ischemic reperfusion, we measured both the mRNA and protein levels of mTOR. Messenger RNA levels of mTOR were detected in cortical tissues by reverse transcription-polymerase chain reaction (RT-PCR). The Ct values (the number of the threshold cycle) of each group were recorded and compared. The results showed that the mTOR mRNA levels were up-regulated after 16 h of reperfusion following 10 min of forebrain ischemia in the rat brain (Fig. 3). To confirm whether ischemia induced the upregulation of mTOR signaling at the translational level, we measured the protein levels of p-mTOR, p-P70S6K, and p-S6 using Western blot analysis and the cytosolic fractions of both the cortical and hippocampal tissues. Consistent with the mRNA data, the protein levels of p-mTOR and its downstream effector molecules p-P70S6K and p-S6 were elevated significantly after reperfusion. The p-mTOR level increased mildly after 3 and 16 h and significantly after 7 day of reperfusion in both the cortex and hippocampus. Phospho-P70S6K was transiently increased after 3 h of reperfusion and then returned to its baseline level after 16 h and 7 days in the cortex. In the hippocampus, p-P70S6K exhibited a decreasing trend after reperfusion. Phospho-S6, which has been used as a major indicator for mTOR pathway activation, significantly increased after 3 h of reperfusion in both the cortex and hippocampus, returning to its baseline level after 16 h and 7 days in the cortex but remaining elevated at 16 h and 7 days in the hippocampus (Fig. 4).
mTOR mRNA expression in normal control and ischemic groups. Relative expression level was calculated by ΔCt = (the Ct values of mTOR in experimental group - the Ct values of GAPDH in experimental group) - (the Ct values of mTOR in sham control group-the Ct values of GAPDH in sham control group). ** p<0.01, Student's t test.
Phosphorylation of the mTOR pathway after ischemia and reperfusion (I/R) in vehicle- and rapamycin-treated animals. A,E. Representative Western blots of p-mTOR, p-P70S6K, p-S6. B-D,F-H semi-quantification of band intensities of the aforementioned proteins. Data are presented as means ± s.d. * p< 0.05, **p<0.01 versus sham in the same treatment group, #p<0.05, ## p<0.01 between rapamycin- (RAPA) and vehicle-treated groups at an identical reperfusion stage.
Rapamycin Inhibited Ischemia-Activated mTOR Signaling Pathway
To determine whether the neuroprotective effects of rapamycin on ischemic brain damage are associated with inhibition of the mTOR pathway, we measured the protein levels of p-mTOR, p-P70S6K, and p-S6 using Western blotting. As shown in Fig. 4, in the cortex, p-mTOR and p-S6 were moderately but significantly reduced in the sham-operated animals by rapamycin. However, p-P70S6K was mildly increased in the rapamycin-treated sham group. After ischemia and reperfusion, all three mTOR pathway markers were significantly suppressed by rapmaycin at all three reperfusion endpoints, with the most pronounced inhibition observed for p-S6 (Fig. 4 A-D). In the hippocampus, mTOR and p-70S6K were decreased in the rapamycin-treated controls, and p-S6 levels were hardly detectable in both the vehicle- and rapamycin-treated controls. After ischemia, the changes in the p-mTOR and p-S6 levels in the hippocampus were similar compared to the cortex, which showed a decreased level of mTOR and a much more pronounced reduction of p-S6 in all three reperfusion endpoints. The changes in p-P70S6K did not show a consistent pattern in the hippocampus: increasing at 3 h, no difference at 16 h and decreasing again at 7 days of reperfusion in the rapamycin-treated animals compared to the vehicle-treated ischemic animals (Fig. 4 E-J). An inhibition of p-S6 was observed in both structures at all reperfusion endpoints.
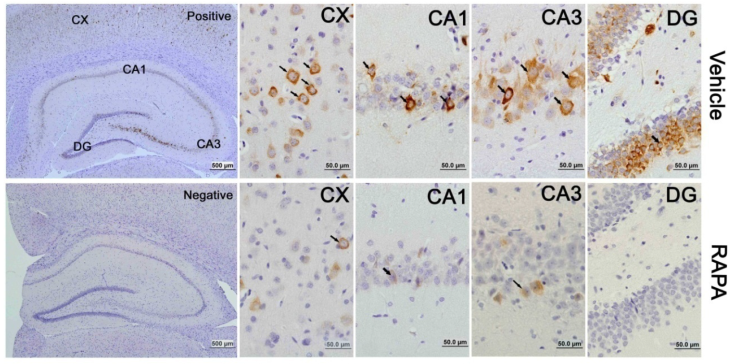
The activation of the mTOR pathway after cerebral ischemia and its inhibition by rapamycin were further confirmed by p-S6 immunohistochemistry. Consistent with the results obtained from the protein blotting, p-S6 positive neurons increased after 3 h of reperfusion and persisted for 7 days of reperfusion in both the cortex and hippocampus. The p-S6 was located in the cytoplasm, and p-S6 positively stained neurons were not only observed in the CA1 area but also in the CA3 and dentate gyrus areas (Fig. 5). Consistent with the protein data, the number of p-S6 positive neurons significantly decreased after rapamycin intervention.
mTOR activity as reflected by phosphorylated p-S6 protein in vehicle- and rapamycin-treated animals with 16 h of reperfusion. Arrows indicate p-S6 positive neurons. CX, cortex; DG, dentate gyrus. Bar= 50 μm.
Rapamycin suppressed p-Akt
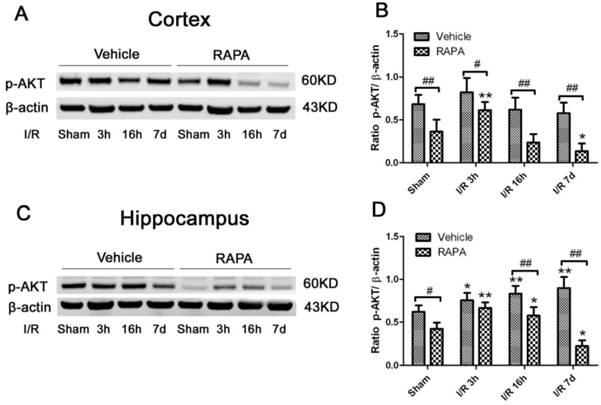
Akt serves as both an upstream regulator and a downstream target of the mTOR pathway [34]. To study the effects of rapmaycin on the phosphorylation of Akt, we measured p-Akt using Western blotting. The results showed that the p-Akt content slightly increased after 3 h and returned to its control level at 16 h and 7 days of recovery in both the cortex and hippocampus. In contrast, rapamycin significantly inhibited the protein level of p-Akt in the sham-operated control and after 3 h, 16 h and 7 days of reperfusion following transient forebrain ischemia in both the cortex and hippocampus (Fig. 6).
Western blot of p-Akt after ischemia and reperfusion (I/R) in vehicle- and rapamycin-treated animals. A,C. Representative Western blots of p-Akt in the cortex (A) and hippocampus (C); B,D. Semi-quantification of band intensities in the cortex (B) and hippocampus (D). Data are presented as means ± s.d. * p< 0.05, **p<0.01 versus sham in the same treatment group, #p<0.05, ## p<0.01 between rapamycin- (RAPA) and vehicle-treated group at an identical reperfusion stage.
Ischemia Increased- and Rapamycin Inhibited-Autophagy Markers
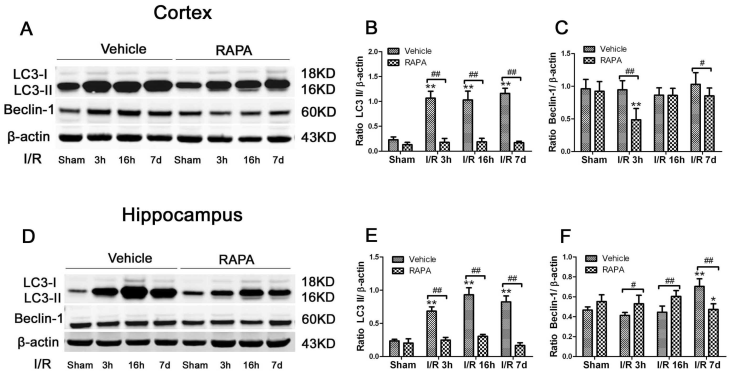
To assess the level of autophagy after ischemia and rapamycin treatment, we measured two commonly used autophagy markers, LC3-II and Beclin-1, in the cytosolic fractions. As demonstrated in Fig. 7, LC3-II increased drastically after 3 h of reperfusion following ischemia and persisted for at least 7 days in both the cortex and hippocampus. However, the Beclin-1 level was not increased after ischemia and reperfusion in the cortex while the level of the hippocampus increased after 7 days of reperfusion. Rapamycin treatment reduced the LC3-II levels to equal or below the baseline levels in both the cortex and hippocampus after ischemia and reperfusion up to 7 days. The influence of rapamycin on Beclin-1 was more complex. In the cortex, the Beclin-1 level decreased at 3 h and 7 days in the rapamycin-treated animals; however, its level in the hippocampus moderately increased after rapamycin treatment at 3 h and 16 h and decreased after 7 days of reperfusion.
LC3-II and Beclin-1 protein levels in the cortex (A-C) and hippocampus (D-F). A,D. Representative Western blots in the cortex (A) and hippocampus (D). B,C. Bar graphs summarize the band intensities of LC3-II (B) and Beclin-1 (C) in the cortex. E,F. Bar graphs summarize the band intensities of LC3-II (E) and Beclin-1 (F) in the hippocampus. Data are presented as means ± s.d. * p< 0.05, **p<0.01 versus sham in the same treatment group, #p<0.05, ## p<0.01 between rapamycin- (RAPA) and vehicle-treated groups at an identical reperfusion stage.
Rapamycin suppressed-ischemia induced Cytochrome c release
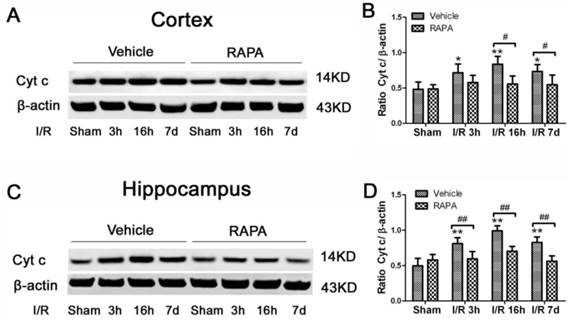
Cytochrome c release is a critical step in activating the caspase-dependent cell death pathway. To examine whether rapamycin has any effect on cell death pathway activation, we detected cytochrome c release in the cytosolic fraction in vehicle- and rapamycin-treated animals. Cytosolic cytochrome c levels increased moderately at 3 h of reperfusion, peaked at 16 h and remained elevated at 7 days of recovery in both the cortex (Fig. 8A, 8B) and hippocampus (Fig. 8C, 8D). Rapamycin blocked ischemia-induced cytochrome c release after 3 and 16 h and 7 days of reperfusion.
Cytochrome c levels in the cytosolic fractions of the vehicle and rapamycin-treated animals. A,C. Representative Western blots in the cortex (A) and hippocampus (C). B,D. Summarized band intensities in the cortex (B) and hippocampus (D). Data are presented as means ± s.d. * p< 0.05, **p<0.01 versus sham in the same treatment group, #p<0.05, ## p<0.01 between rapamycin- (RAPA) and vehicle-treated groups at an identical reperfusion stage.
Discussion
The present study has revealed that transient forebrain ischemia-caused neuronal death (both apopototic and necrotic in nature) is associated with activation of the mTOR signaling pathway, autophagy and cytochrome c release. Inhibition of the mTOR pathway by rapamycin significantly reduced neuronal death. In addition, rapamycin inhibited autophagy and cytochrome c release in ischemic rats. Thus, mOTR plays an injury role in cerebral ischemia.
Transient forebrain ischemia induces delayed selective neuronal death [35]. The neuronal death occurs after 3 days of reperfusion following a transient forebrain ischemic reperfusion injury [36]. It has been controversial whether such delayed neuronal death is apoptotic or necrotic in nature. On the one hand, ischemia causes proapoptotic factors such as cytochrome c, Smac/Diablo, apoptosis-inducing factor (AIF) and endonuclease G to be released from the mitochondria and subsequently activates caspase-dependent and caspase-independent apoptotic cell death pathways [37-39]. On the other hand, not many cells fall into the typical apoptotic morphology, and cell membrane rupture and subcellular organelle damage such as mitochondria swelling are common. As such, a new term, necroptosis, has been used to describe the nature of ischemic cell death due to the fact that ischemic neuronal cell death has both apoptotic and necrotic components [40,41]. In the present study, neuronal death reached to close to 30% in the cortex and close to 100% in the CA1 after 7 days of reperfusion. In contrast, only 48% and 56.8% of the total dead cells in the cortex and CA1 areas, respectively, were TUNEL positive, suggesting that transient forebrain ischemia causes both necrotic and apoptotic neuronal death. Although it has been argued that TUNEL staining is not specific for apoptosis, our results showed that TUNEL did not label all dead cells, suggesting the TUNEL kit we have used is relatively specific for apoptosis.
Rapamycin reduced ischemic brain damage. Rapamycin is an mTOR inhibitor. Although rapamycin has been shown to reduce infarct size and to ameliorate neurological deficits in animal cerebral focal ischemic models [30], in vitro glucose-oxygen deprivation [32] and its effects on global ischemia has not been reported in adult rodents. In the present study, we have shown that pretreatment with rapamycin for 7 days prior to and 7 days after a transient forebrain ischemia significantly reduced cortical and hippocampal CA1 neuronal death. As a result, the damage in the cortex was reduced from 30% in the vehicle- to 9% in the rapamycin-treated animals. More convincingly, the damage in the CA1 was reduced from close to 100% in the vehicle- to less than 10% in the rapamycin-treated rats. Further analyses revealed that rapamycin reduced both apoptotic and necrotic cell death. Therefore, the percentages of TUNEL positive cells out of the total dead neurons were significantly reduced. In fact, TUNEL positive neurons could hardly be found in either the cortex or CA1 at 7 days of reperfusion.
The neuroprotective effect of rapamycin on cerebral ischemia was associated with inhibition of the mTOR pathway. Previous studies have shown that the mTOR pathway is down-regulated after ischemia, which is likely caused by a low cellular energy state during the ischemic period [42-45]. In the present study, we observed an early activation of the mTOR pathway, as reflected by increased levels of p-mTOR, p-P70S6K and p-S6 after transient forebrain ischemia. The activation of the mTOR pathway was observed after 3 h and persisted to 7 days of reperfusion. This finding is consistent with the results of Crozier and colleagues, who reported an increase in PI3K-Akt-mTOR pathway activation after cardiac ischemia and reperfusion [46] and those of Fletcher et al. who demonstrated mTOR activation after in vitro oxygen-glucose deprivation [32]. The activation of the mTOR pathway during reperfusion may be induced by the restoration of blood flow and glucose to the affected region [47,48]. It is not clear whether activation of the mTOR pathway plays a protective or destructive role for ischemic animals. However, it is clear that inhibition of mTOR by rapamycin provides a potent protection to neuronal cells against ischemia and reperfusion injury. It is also worth mentioning that rapamycin is a potent immunosuppressant. Because neuro-inflammation plays an important role in mediating ischemia/reperfusion injury, it remains to be shown whether the neuroprotective effect of rapamycin against ischemia/reperfusion injury is attributed to mTOR inhibition or immunosuppresion. One limitation of this study is that the neurobehavioral evaluation was not performed. Forebrain ischemia causes more cognitive disturbance than motor dysfunction. Another limitation of this study is that rapamycin was given 7 days pre-ischemic and continued till 7days post-ischemically. This might have changed the baseline of mTOR activity in the animals. However, we also had a separate group in which the rapamycin was only be injected 3 h after reperfusion and followed by daily administration for 7 days. The same protective effects were observed in post-treated animals (Hei, Yang, Liu et al., data not shown). This suggest inhibition of mTOR has a therapeutic value against cerebral ischemia.
Rapamycin inhibits Akt. We observed that cerebral ischemia increased the phosphorylation of Akt and rapamycin reduced the p-Akt level, especially after 16 h and 7 days of reperfusion. Akt (a.k.a protein kinase B) serves as both an upstream regulator of mTOR and as a downstream target of mTOR signaling. Akt suppresses tuberous sclerosis complex (TSC) while the latter suppresses mTOR activation through inhibition of Rheb [34]. mTOR consists of mTOR complex 2 (TORC2) and mTOR complex 1 (TORC1). TORC2 causes phosphorylation of serum- and glucocorticoid-regulated kinase (SGK), Akt and protein kinase c (PKC), thereby enhancing cell survival and promoting cytoskeleton assembly. TORC1 phosphorylates P70S6K and eukaryotic initiation factor 4E-BP, resulting in chromatin proliferation, protein synthesis, cell growth, mitochondrial biogenesis, lipogenesis, and changes in ion channels and neurotransmission [49,50,51,52]. Rapamycin is thought to inhibit mTORC1 due to the rapamycin-FKBP12 interaction. However, Sarbassov and colleagues found that prolonged rapamycin treatment also inhibited the assembly of mTORC2 [53]. It is possible that 7 days of rapamycin treatment reduces mTORC2 expression to below the level that is required to maintain Akt signaling.
In the present study, we observed that transient cerebral ischemia and reperfusion activated autophagy. LC3-II and Beclin-1 were dramatically elevated after ischemia and reperfusion in both the cortex and hippocampus. The increases in LC3-II started early (3 h) and lasted up to 7 days, which suggests activation of autophagy in this model. Similar findings have also been reported in neonatal hypoxia-ischemia [54,55] and after middle cerebral artery (MCA) occlusion [56,57]. However, autophagy is a double edged sword. Under physiological conditions, cells maintain a balance between synthesis, degradation, and the recycling of cellular components through an evolutionarily conserved key homeostatic mechanism—autophagy, in which misfolded or aggregated proteins, lipids, and organelles are engulfed by double- membrane vesicles, called autophagosomes, and degraded after fusing with lysosomes [58]. Decreased autophagy is implicated in the pathogenesis of many neurodegenerative diseases such as Parkinson's disease, Alzheimer's disease, and Huntington disease [59]. However, overactivation of autophagy triggers non-apoptotic programmed cell death, termed autophagic cell death, through excessive self-digestion and the degradation of essential cellular constituents [60,61]. It has been previously shown that transient forebrain ischemia activates autophagy, as reflected by increased levels of ATG5 and LC3-II and increased autophagosomes detected by electron microscopy [62]. Whether such an increase in autophagy is a normal cellular response to injury signals to remove damaged cellular constituents or an active cell death mechanism is still under debate. Interestingly, comparing the ischemic- vulnerable CA1 to the ischemic-resistant dentate gyrus, the elevation of autophagy markers is much pronounced in the CA1, implying an association between overly activated autophagy and extensive cell damage [62]. The detrimental role of autophagy in cerebral ischemic neuronal death has been further confirmed by the immuno-colocalization of Beclin-1 with TUNEL staining [63]. In the present study, we observed that rapamycin inhibited autophagy markers in both the cortical and hippocampal structures. It has been reported that inhibition of autophagy mediates the neuroprotective events against cerebral ischemic neuronal death [57]. Furthermore, inhibition of autophagy by 3-methyladenine (3-MA) significantly ameliorated ischemic brain damage [63], suggesting that reduced autophagy is associated with neuroprotection in a cerebral ischemic model.
Under normal conditions, mTOR inhibits autophagy by modulating more than 20 downstream genes that encode proteins that are essential for the execution of autophagy [64]. Chemical inhibition of mTOR is often used as an approach to activate autophagy. Rapamycin has been shown to increase autophagy in neonatal hypoxia-ischemia [55], in ischemic human umbilical vein endothelial cells (HUVEC) [29], and in acute cerebral focal ischemia [65]. Unexpectedly, we observed that administration of rapamycin in rats subjected to transient forebrain ischemia suppressed autophagy. It is not known why in our experimental setting rapamycin inhibits, instead of activates, autophagy. Perhaps the experimental model, rapamycin dosage, plasma concentration and timing of the administration lead to these results. In addition, an effect of rapamycin to influence the cross-talk between the survival kinases is likely another possibility as activation or inhibition of one pathway may affect another elsewhere.
Cytochrome c release from the mitochondria to cytosol plays a pivotal role in activating the apoptotic cell death pathway. The released cytochrome c binds to apf-1 and procaspase-9 to form apoptosomes, which leads to cleavage of caspase-9 and its downstream caspase, caspase-3. Caspase-3 then either activates caspase-activated DNase (CAD) or directly translocates to the nucleus to cause DNA damage [38]. We observed an increased cytochrome c level in the cytosolic fraction after ischemia and reperfusion, which is consistent with previous findings [37,66]. Rapamycin treatment prevented the release of cytochrome c induced by cerebral ischemia. These results suggest that rapamycin prevents the activation of the mitochondria-initiated cell death pathway in addition to inhibiting the mTOR pathway.
In conclusion, the present data have demonstrated that transient forebrain ischemia causes both necrotic and apoptotic cell death in brain tissues; schemia results in the activation of mTOR signaling, enhancement of autophagy and release of cytochrome c. In addition, rapamycin treatment markedly reduced ischemic brain damage, and the protective effects are associated with suppression of mTOR signaling, inhibition of autophagy and prevention of cytochrome c release.
Acknowledgements
This work is supported by grants from American Epileptic Society to PAL, National Natural Science Foundation of China (81560226) and Natural Science Foundation of Ningxia Hui Autonomous (NZ14281) to XY, and Chinese National Science Foundation 973-Preproject (2014CB560711) JN. The Biomanufacturing Research Institute and Technology Enterprise (BRITE) is partially funded by the Golden Leaf Foundation. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The authors appreciate Dr. Robert Onyenwoke for proofreading.
Author Contributions
PAL, FW, JN, ST conceived and designed the experiments. XY, CH, PL, ZS, TT, ST performed the experiments. XY, PL, CH, PAL analyzed the data. PAL, JN, TS contributed reagents/materials/analysis tools. XY, PAL wrote the paper.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Thrift AG, Dewey HM, Macdonell RA, McNeil JJ, Donnan GA. Incidence of the major stroke subtypes: initial findings from the North East Melbourne stroke incidence study (NEMESIS). Stroke. 2001;32:1732-8
2. Dorado L, Millan M, Davalos A. Reperfusion therapies for acute ischemic stroke: an update. Curr Cardiol Rev. 2014;10:327-35
3. Boyle Jr M, Edward M, Pohlman M, Timothy H, Cornejo M, Carol J. et al. Ischemia-reperfusion injury. Ann Thoracic Surg. 1997;64:S24-S30
4. Buja LM. Myocardial ischemia and reperfusion injury. Cardiovas Pathol. 2005;14:170-5
5. Nour M, Scalzo F, Liebeskind DS. Ischemia-reperfusion injury in stroke. Interv Neurol. 2013;1:185-99
6. Pac-Soo CK, Mathew H, Ma D. Ischaemic conditioning strategies reduce ischaemia/reperfusion-induced organ injury. Br J Anaesth. 2015;114:204-16
7. White BC, Sullivan JM, DeGracia DJ, O'Neil BJ, Neumar RW, Grossman LI. et al. Brain ischemia and reperfusion: molecular mechanisms of neuronal injury. J Neurol Sci. 2000;179:1-33
8. Wong M. Mammalian target of rapamycin (mTOR) pathways in neurological diseases. Biomed J. 2013;36:40-50
9. Tee AR, Fingar DC, Manning BD, Kwiatkowski DJ, Cantley LC, Blenis J. Tuberous sclerosis complex-1 and -2 gene products function together to inhibit mammalian target of rapamycin (mTOR)-mediated downstream signaling. Proc Natl Acad Sci U S A. 2002;99:13571-6
10. El-Hashemite N, Zhang H, Henske EP, Kwiatkowski DJ. Mutation in TSC2 and activation of mammalian target of rapamycin signalling pathway in renal angiomyolipoma. Lancet. 2003;361:1348-9
11. Kwon CH, Zhu X, Zhang J, Baker SJ. mTor is required for hypertrophy of Pten-deficient neuronal soma in vivo. Proc Natl Acad Sci U S A. 2003;100:12923-8
12. Zeng LH, Xu L, Gutmann DH, Wong M. Rapamycin prevents epilepsy in a mouse model of tuberous sclerosis complex. Ann Neurol. 2008;63:444-53
13. Sunnen CN, Brewster AL, Lugo JN, Vanegas F, Turcios E, Mukhi S. et al. Inhibition of the mammalian target of rapamycin blocks epilepsy progression in NS-Pten conditional knockout mice. Epilepsia. 2011;52:2065-75
14. McDaniel SS, Wong M. Therapeutic role of mammalian target of rapamycin (mTOR) inhibition in preventing epileptogenesis. Neurosci Lett. 2011;497:231-9
15. Edlich F, Weiwad M, Wildemann D, Jarczowski F, Kilka S, Moutty MC. et al. The specific FKBP38 inhibitor N-(N',N'-dimethylcarboxamidomethyl) cycloheximide has potent neuroprotective and neurotrophic properties in brain ischemia. J Biol Chem. 2006;281:14961-70
16. Jaworski J, Spangler S, Seeburg DP, Hoogenraad CC, Sheng M. Control of dendritic arborization by the phosphoinositide-3'-kinase-Akt-mammalian target of rapamycin pathway. J Neurosci. 2005;25:11300-12
17. Kumar V, Zhang MX, Swank MW, Kunz J, Wu GY. Regulation of dendritic morphogenesis by Ras-PI3K-Akt-mTOR and Ras-MAPK signaling pathways. J Neurosci. 2005;25:11288-99
18. Bove J, Martinez-Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci. 2011;12:437-52
19. Gupta YK, Chauhan A. Potential of immunosuppressive agents in cerebral ischaemia. Indian J Med Res. 2011;133:15-26
20. Vignot S, Faivre S, Aguirre D, Raymond E. mTOR-targeted therapy of cancer with rapamycin derivatives. Annals of oncology. 2005;16:525-37
21. Dowling RJ, Topisirovic I, Fonseca BD, Sonenberg N. Dissecting the role of mTOR: lessons from mTOR inhibitors. Biochim Biophys Acta. 2010;1804:433-9
22. King MA, Hands S, Hafiz F, Mizushima N, Tolkovsky AM, Wyttenbach A. Rapamycin inhibits polyglutamine aggregation independently of autophagy by reducing protein synthesis. Mol Pharmacol. 2008;73:1052-63
23. Koh PO, Cho JH, Won CK, Lee HJ, Sung JH, Kim MO. Estradiol attenuates the focal cerebral ischemic injury through mTOR/p70S6 kinase signaling pathway. Neurosci Lett. 2008;436:62-6
24. Pastor MD, Garcia-Yebenes I, Fradejas N, Perez-Ortiz JM, Mora-Lee S, Tranque P. et al. mTOR/S6 kinase pathway contributes to astrocyte survival during ischemia. J Biol Chem. 2009;284:22067-78
25. You H, Li T, Zhang J, Lei Q, Tao X, Xie P. et al. Reduction in ischemic cerebral infarction is mediated through golgi phosphoprotein 3 and Akt/mTOR signaling following salvianolate administration. Curr Neurovasc Res. 2014;11:107-13
26. Koh P-O. Ferulic acid attenuates focal cerebral ischemia-induced decreases in p70S6 kinase and S6 phosphorylation. Neurosci lett. 2013;555:7-11
27. Zare Mehrjerdi F, Aboutaleb N, Habibey R, Ajami M, Soleimani M, Arabian M. et al. Increased phosphorylation of mTOR is involved in remote ischemic preconditioning of hippocampus in mice. Brain Res. 2013;1526:94-101
28. Xie R, Wang P, Ji X, Zhao H. Ischemic post-conditioning facilitates brain recovery after stroke by promoting Akt/mTOR activity in nude rats. J Neurochem. 2013;127:723-32
29. Urbanek T, Kuczmik W, Basta-Kaim A, Gabryel B. Rapamycin induces of protective autophagy in vascular endothelial cells exposed to oxygen-glucose deprivation. Brain Res. 2014;1553:1-11
30. Yin L, Ye S, Chen Z, Zeng Y. Rapamycin preconditioning attenuates transient focal cerebral ischemia/reperfusion injury in mice. Int J Neurosci. 2012;122:748-56
31. Chauhan A, Sharma U, Jagannathan NR, Reeta KH, Gupta YK. Rapamycin protects against middle cerebral artery occlusion induced focal cerebral ischemia in rats. Behav Brain Res. 2011;225:603-9
32. Fletcher L, Evans TM, Watts LT, Jimenez DF, Digicaylioglu M. Rapamycin treatment improves neuron viability in an in vitro model of stroke. PLoS One. 2013;8:e68281
33. Buckley KM, Hess DL, Sazonova IY, Periyasamy-Thandavan S, Barrett JR, Kirks R. et al. Rapamycin up-regulation of autophagy reduces infarct size and improves outcomes in both permanent MCAL, and embolic MCAO, murine models of stroke. Exp Transl Stroke Med. 2014;6:8
34. Wullschleger S, Loewith R, Hall MN. TOR signaling in growth and metabolism. Cell. 2006;124:471-84
35. Smith M-L, Auer R, Siesjö B. The density and distribution of ischemic brain injury in the rat following 2-10 min of forebrain ischemia. Acta Neuropathol. 1984;64:319-32
36. Abe K, Aoki M, Kawagoe J, Yoshida T, Hattori A, Kogure K. et al. Ischemic delayed neuronal death a mitochondrial hypothesis. Stroke. 1995;26:1478-89
37. Broughton BR, Reutens DC, Sobey CG. Apoptotic mechanisms after cerebral ischemia. Stroke. 2009;40:e331-e9
38. Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495-516
39. Fujimura M, Morita-Fujimura Y, Kawase M, Copin J-C, Calagui B, Epstein CJ. et al. Manganese superoxide dismutase mediates the early release of mitochondrial cytochrome C and subsequent DNA fragmentation after permanent focal cerebral ischemia in mice. J Neurosci. 1999;19:3414-22
40. Galluzzi L, Kroemer G. Necroptosis: a specialized pathway of programmed necrosis. Cell. 2008;135:1161-3
41. Walsh CM. Grand challenges in cell death and survival: apoptosis vs. necroptosis. Cell Death Survival. 2014;2:3
42. Foster KG, Fingar DC. Mammalian target of rapamycin (mTOR): conducting the cellular signaling symphony. J Biol Chem. 2010;285:14071-7
43. Sofer A, Lei K, Johannessen CM, Ellisen LW. Regulation of mTOR and cell growth in response to energy stress by REDD1. Mol Cell Biol. 2005;25:5834-45
44. Koh P-O. Gingko biloba extract (EGb 761) prevents cerebral ischemia-induced p70S6 kinase and S6 phosphorylation. Am J Chin Med. 2010;38:727-34
45. Koh P-O, Cho J-H, Won C-K, Lee H-J, Sung J-H. Estradiol attenuates the focal cerebral ischemic injury through mTOR/p70S6 kinase signaling pathway. Neurosci Lett. 2008;436:62-66
46. Crozier SJ, Zhang X, Wang J, Cheung J, Kimball SR, Jefferson LS. Activation of signaling pathways and regulatory mechanisms of mRNA translation following myocardial ischemia-reperfusion. J Appl Physiol. 2006;101:576-82
47. Liebeskind DS. Reperfusion for acute ischemic stroke: arterial revascularization and collateral therapeutics. Curr Opinion Neurol. 2006;23:36-45
48. Cimarosti H, Henley JM. Investigating the mechanisms underlying neuronal death in ischemia using in vitro oxygen-glucose deprivation: potential involvement of protein SUMOylation. Neuroscientist. 2008;14:626-36
49. Soukas AA, Kane EA, Carr CE, Melo JA, Ruvkun G. Rictor/TORC2 regulates fat metabolism, feeding, growth, and life span in Caenorhabditis elegans. Genes Devel. 2009;23:496-511
50. Sarbassov DD, Ali SM, Kim D-H, Guertin DA, Latek RR, Erdjument-Bromage H. et al. Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol. 2004;14:1296-302
51. Sarbassov DD, Guertin DA, Ali SM, Sabatini DM. Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science. 2005;307:1098-101
52. Garcia-Martinez J, Alessi D. mTOR complex 2 (mTORC2) controls hydrophobic motif phosphorylation and activation of serum-and glucocorticoid-induced protein kinase 1 (SGK1). Biochem J. 2008;416:375-85
53. Sarbassov DD, Ali SM, Sengupta S, Sheen J-H, Hsu PP, Bagley AF. et al. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22:159-68
54. Carloni S, Girelli S, Scopa C, Buonocore G, Longini M, Balduini W. Activation of autophagy and Akt/CREB signaling play an equivalent role in the neuroprotective effect of rapamycin in neonatal hypoxia-ischemia. Autophagy. 2010;6:366-77
55. Carloni S, Buonocore G, Balduini W. Protective role of autophagy in neonatal hypoxia-schemia induced brain injury. Neurobiol Dis. 2008;32:329-39
56. Rami A, Langhagen A, Steiger S. Focal cerebral ischemia induces upregulation of Beclin 1 and autophagy-like cell death. Neurobiol Dis. 2008;29:132-41
57. Gao L, Jiang T, Guo J, Liu Y, Cui G, Gu L. et al. Inhibition of autophagy contributes to ischemic postconditioning-induced neuroprotection against focal cerebral ischemia in rats. PLoS One. 2012;7:e46092
58. Debnath J, Baehrecke EH, Kroemer G. Does autophagy contribute to cell death? Autophagy. 2005;1:66-74
59. Wong E, Cuervo AM. Autophagy gone awry in neurodegenerative diseases. Nat Neurosci. 2005;13:805-11
60. Shintani T, Klionsky DJ. Autophagy in health and disease: a double-edged sword. Science. 2004;306:990-5
61. Uchiyama Y, Shibata M, Koike M, Yoshimura K, Sasaki M. Autophagy-physiology and pathophysiology. Histochem Cell Biol. 2008;129:407-20
62. Liu C, Gao Y, Barrett J, Hu B. Autophagy and protein aggregation after brain ischemia. J Neurochem. 2010;115:68-78
63. Li W-L, Yu S, Chen D, Yu S, Jiang Y-J, Genetta T. et al. The regulatory role of NF-κB in autophagy-like cell death after focal cerebral ischemia in mice. Neuroscience. 2013;244:16-30
64. Mizushima N, Klionsky DJ. Protein turnover via autophagy: Implications for metabolism. Annu Rev Nutr. 2007;27:19-40
65. Viscomi MT, D'Amelio M, Cavallucci V, Latini L, Bisicchia E, Nazio F. et al. Stimulation of autophagy by rapamycin protects neurons from remote degeneration after acute focal brain damage. Autophagy. 2012;8:222-35
66. Fujimura M, Morita-Fujimura Y, Murakami K, Kawase M, Chan PH. Cytosolic redistribution of cytochrome c after transient focal cerebral ischemia in rats. J Cereb Blood Flow Metab. 1998;18:1239-47
Author contact
![]() Corresponding author: P. Andy Li, M.D., Ph.D., Department of Pharmaceutical Sciences, North Carolina Central University, 1801 Fayetteville Street, Durham, NC 27707, Fax: 919-530-6600; E-mail: pliedu
Corresponding author: P. Andy Li, M.D., Ph.D., Department of Pharmaceutical Sciences, North Carolina Central University, 1801 Fayetteville Street, Durham, NC 27707, Fax: 919-530-6600; E-mail: pliedu