Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2015; 11(12):1436-1446. doi:10.7150/ijbs.13371 This issue Cite
Research Paper
A20 Attenuates FFAs-induced Lipid Accumulation in Nonalcoholic Steatohepatitis
Luoyan Ai1,2*, Qingqing Xu1,2*, Changwei Wu1,2, Xiaohan Wang1,2, Zhiwei Chen1,2, Dazhi Su1,2, Xiaoke Jiang1,2, Antao Xu2, Qing Lin 1 ![]() , Zhuping Fan1
, Zhuping Fan1 ![]()
1. Department of Health Care Center, RenJi Hospital, School of Medicine, Shanghai Jiao Tong University, Shang hai, China;
2. Division of Gastroenterology and Hepatology, RenJi Hospital, School of Medicine, Shanghai Jiao Tong University, Shanghai Institute of Digestive Disease; Key Laboratory of Gastroenterology & Hepatology, Ministry of Health (Shanghai Jiao Tong University), Shanghai, China.
*These authors contribute equally to this work.
Received 2015-7-28; Accepted 2015-10-1; Published 2015-11-25
Abstract

A20 is a ubiquitin-editing enzyme that attenuates the activity of proximal signaling complexes at pro-inflammatory receptors. It has been well documented that A20 protein plays an important role in response to liver injury and hepatocytes apoptosis in pro-inflammatory pathways. However, there was little evidence showing that A20 protein was involving in fatty-acid homeostasis except the up-regulation of two fatty acid metabolism regulatory genes at mRNA level (PPARa and CPT1a) by adenovirus-mediated A20 protein overexpression. In this study we found that: 1) the expression level of A20 protein was significantly higher in the steatotic liver from MCD-fed mice than the controls; 2) Overexpression of A20 protein suppressed FFAs-stimulated triglyceride deposition in HepG2 cells while under expression of A20 protein increased FFAs-stimulated triglyceride deposition; 3) Overexpression of A20 protein in HepG2 cells upregulated genes that promote β-oxidation and decreased the mRNA levels of key lipogenic genes such as fatty acid synthase (FAS), indicating A20 function as anti-steatotic factor by the activation of mitochondrial β-oxidation and attenuation of de novo lipogenesis; 4) Nonalcoholic steatohepatitis (NASH) patients showed significantly higher A20 expression level in liver compared with control individuals. Our results demonstrated that A20 protein plays an important role in fatty-acid homeostasis in human as well as animals. In addition, our data suggested that the pathological function of A20 protein in hepatocyte from lipotoxicity to NASH is by the alleviation of triglyceride accumulation in hepatocytes. Elevated expression of A20 protein could be a potential therapeutic strategy for preventing the progression of nonalcoholic steatohepatitis.
Keywords: A20, Nonalcoholic Steatohepatitis, NF-κB, FFAs
Introduction
Nonalcoholic fatty liver disease (NAFLD) is a burgeoning health problem that affects one-third of adults and an increasing number of children[1]. This disorder includes a wide spectrum of liver injury from simple steatosis to nonalcoholic steatohepatitis (NASH). While simple steatosis has few adverse outcomes on liver function, NASH is characterized by hepatocyte injury and inflammation, which can lead to hepatic fibrosis and eventually cirrhosis. It is still unclear why certain patients progress toward inflammation, fibrosis and cirrhosis, whereas others do not[2]. Insights into the molecular mechanisms related to the progression of NASH may be helpful in designing therapeutic strategies for this disease entity.
Recent studies reported that patients with NASH showed a remarkable increasing of NF-κB activation in liver biopsies, correlated with progression of inflammation and fibrosis, indicating a possible role of this transcription factor in the pathogenesis of NASH[3]. This was confirmed by studies in obese foz/foz strain of mice which carried a truncating mutation in the Alström syndrome protein (Alms1) gene and showed significant NF-κB activation when the liver pathology converted from bland steatosis to NASH with high-fat (HF) diet, while reversion to a physiological dietary composition attenuated NASH and showed suppressed NF-κB activation[4]. In the methionine choline-deficient (MCD) dietary model of steatohepatitis, NF-κB was identified as a critical pro-inflammatory mediator of lesion development[5]. These evidences suggested that deprivation of NF-κB-mediated pro-inflammatory effects could be a rational strategy in the treatment of NAFLD. However, the hepatoprotective effect of NF-κB, especially in preventing apoptosis and promoting regeneration of hepatocytes, was supported by other studies[6-8]. Consequences of complete blockade of NF-κB pathway could be complicated. Therefore, molecules can inhibit NF-κB activation-mediated inflammation without affect its hepatoprotective function could be suitable targets for clinical intervention of NASH.
A20 is a cytoplasmic seven zinc finger protein that was upregulated by NF-κB in most cell types, including hepatocytes[9, 10]. The critical role of A20 in the regulation of inflammatory response through inhibition of NF-κB activation pathway has been well established [11-15]. Serial studies by Dr. Ferran and her colleagues suggested that despite inhibition of NF-κB activation, A20 exerted multiple hepatoprotective functions, including anti-apoptotic and pro-proliferative effects[10, 16, 17]. Altogether, A20 inhibited NF-κB-mediated inflammatory response without influencing or even could be taken as the mediator of its hepatoprotective function[10, 11, 16, 17]. Another evidence came from transcriptional analysis of liver with adenovirus-mediated A20 overexpression, A20 promoted expression of two critical fatty acid metabolism regulators, peroxisome proliferator activated receptor alpha (PPARα) and carnitine palmytoyl transferase 1α (CPT1α) in the context of extensive liver resection[18]. Inhibition of PPARα increased susceptibility to high fat-induced NASH in mouse model[19]. However, whether A20 could interfere with the pathogenesis of NASH still remained unclear. Thus, we hypothesized that A20 could be part of physiological response of hepatocytes to lipotoxicity and contributed to the prevention of NASH development through its modulation of lipid metabolism.
Material and Methods
Patients and Biopsies
Human liver tissues were obtained from seven patients with NAFLD and four healthy individuals who underwent surgery or liver biopsy at the Shanghai RenJi Hospital. Informed consent was obtained from each patient included in the study and the study protocol conforms to the ethical guidelines of the 1975 Declaration of Helsinki as reflected in a priori approval by the human research committee of RenJi Hospital, School of Medicine, Shanghai JiaoTong University. The diagnosis of NAFLD was according to conventional clinical and histological criteria, as described by Chalasani, N[20]. i.e.1) There is hepatic steatosis by histology, 2) there is no significant alcohol consumption, 3) there are no competing etiologies for hepatic steatosis, and 4) there are no co-existing causes for chronic liver disease. Liver tissues taken from patients without NAFLD with no histological abnormalities served as controls. Characteristics of patients were summarized in Table 1.
Patient Characteristics
| Normal (n=4) | NAFLD (n=7) | P | |
|---|---|---|---|
| Gender | 1.0 | ||
| Male | 2 | 4 | |
| Female | 2 | 3 | |
| Age, mean ±SD, yr | 45.5 ±5 | 49.2±10.3 | 0.0947 |
| Comordibities | — | ||
| Obesity | — | 6 | |
| Type 2 diabetes mellitus | — | 2 | |
| Dyslipidemia | — | 2 | |
| Medication use | — | ||
| Thiazolidinediones | — | 1 | |
| Insulin Sensitizing Agents | — | 1 | |
| Vitamin E | — | — | |
| Ursodeoxycholic Acid | — | 2 | |
| None | — | 3 |
Tissue culture and treatments
The human hepatocellular carcinoma cell line (HepG2, HB-8065), U937 mononuclear cells (U937, CRL-1593.2™) and the human embryonic kidney cell line (293T) were purchased from American Type Culture Collection (ATCC) (Manassas, VA). HepG2 cells were cultured at 37℃ in a 5% CO2 atmosphere in MEM medium (Invitrogen, Carlsbad, CA) containing 10% fetal bovine serum (Gibco) and 1% MEM non-essential amino acid solution. U937 cells were cultured in RPMI 1640 medium (Invitrogen, Carlsbad, CA). The 293T cells were cultured in high glucose Dulbecco's Modified Eagle's Medium (DMEM; Invitrogen, Carlsbad, CA), both containing 10% FBS. For cell treatment, the FFAs mixture was prepared with oleic acid (OA; Sigma, O1257) and palmitic acid (PA; Sigma, P0500-10G) in a 2:1 ratio and used at a 0.5 mmol/L final concentration.
Construction oflentivirus and plasmids
To obtain stable A20 knockdown cells, we constructed three shRNAs for human A20. The sequence of the most efficient one was 5′-TGCTGTTGACAGTGAGCGCTCCGAGCTGTTCCACTTGTTATAGTGAAGCCACAGATGTATAACAAGTGGAACAGCTCGGA-3′. We then subcloned the shRNA cassettes into the pGIPZ lentiviral vector. Correct insertion of the shRNA cassette was confirmed by restriction mapping and DNA sequencing. Recombinant lentiviruses were produced by co-transfecting 293T cells with the lentiviral expression plasmid and packaging plasmids using the lipofectin method. Lentivirus was purified by ultracentrifugation and titered for use.
The A20-overexpression plasmid was a gift from Dr. Han from the Second Military Medical University, Shanghai, China.
Animal models
Male C57BL/6 mice (6-8 weeks of age) were purchased from the Shanghai SLAC Laboratory Animal Co. Ltd (Shanghai, China) and housed under pathogen-free conditions in the animal facility of Renji Hospital, School of Medicine, Shanghai Jiao Tong University. Mice were fed either a methionine and choline deficient (MCD) diet, or a chow diet for 6 weeks. Animals were euthanized and then the livers were harvested. A20 mRNA and protein levels in the liver tissues of mice were measured by qRT-PCR and immunoblotting, respectively. All animal experiments were performed in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals, with the approval of the Institutional Animal Care and Use Committee (IACUC) of Shanghai Jiaotong University.
RNA extraction and quantitative real-time reverse transcription polymerase chain reaction (qRT-PCR)
Total RNA was extracted from the cultured cells using TRIzol reagent (Invitrogen, USA), and 1 µg of total RNA was reverse-transcribed using the Prime Script PTMP RT Reagent Kit (Perfect Real Time; Takara, Shiga, Japan). Quantitative real-time PCR was performed on an Applied Biosystem 7900 quantitative PCR system (Applied Biosystems, Foster City, CA). The amplified transcript level of each specific gene was normalized to β-actin levels. Primers were designed according to published cDNA or genomic sequences.
Immunoblots
Whole cell lysates from HepG2 and U937 cells were harvested after treatment with the FFAs mixture. Samples were analyzed using immunoblots. The antibodies used included: A20 (Cell Signaling Technology, Boston, MA, 5630s), phospho-I-κBα(Cell Signaling Technology, 2859), phospho-p65 (Cell Signaling Technology, 3033), and GAPDH (Kangcheng, Shanghai, KC-5G5) as a loading control. Immunoblots were scanned and bands were quantified by densitometry using ImageJ 1.41 software (NIH, Bethesda, MD).
Fluorescence activated cell sorting (FACS) analysis
The supernatant from the cells treated and untreated with FFAs were collected and filtered through a 0.22 micron sterile filter (Millipore, Billerica, MA, USA). The concentration of IL-8 from each sample were determined using a Cytometric Bead Array (CBA) Human Inflammatory Cytokine kit (BD Biosciences) following the manufacturer's protocol. These assays were performed in triplicate, and mock untreated cells were used as control. These samples were acquired on a FACS Canto II (BD Biosciences) flow cytometer and analyzed using the FCAP array software (BD Biosciences).
Oil Red O staining
Subconfluent monolayers of HepG2 cells were exposed to the FFAs mixture or BSA as a control for 24 hours. Cells were then stained with Oil Red O to examine the amount of lipid accumulation. Briefly, dishes were washed with cold phosphate-buffered saline and fixed in 4% paraformaldehyde for 30 minutes. Then, Oil Red O was added for 30 minutes, followed by washing in 60% isopropanol. The dishes were then rinsed in distilled water and counterstained with hematoxylin. For each dish, three images were photographed, and a representative image is shown.
Intracellular triglyceride assay
HepG2 whole cell lysates were harvested after FFAs mixture treatment for 24 hours. Intracellular triglyceride (TG) content was determined using an enzymatic kit (Nanjing Jiancheng Bioengineering).
Immunohistochemistry
Expression of A20 was determined by immunohistochemistry using a primary antibody against A20 (Abcam, Cambridge, UK, ab13597). After deparaffination and rehydration the sections were subjected to antigen retrieval (10 mM Citric Acid Buffer, pH 6.0). The endogenous peroxidase activity was blocked using 30% H2O2. The primary antibody in PBS was incubated overnight at 4°C. The secondary antibody (Vector Laboratories) was applied in PBS for 30 min at room temperature. Visualization of the signal was performed using the DAB-Substrate-Kit (Vector Laboratories). Slides were counter stained with hematoxylin (Dako Deutschland GmbH, Hamburg). The number of A20 positive cells per high power field (HPF) was counted using ImageJ automated or manual cell counting.
Statistical analysis
Statistical analysis was performed using SPSS 13.0 software (SPSS Inc., Chicago, IL, USA). Data are presented as mean values ±SD; n refers to the number of independent experiments. Levels of significance for comparisons between two or more independent samples were determined using a two-tailed unpaired Student's t-test. Differences were considered significant at P<0.05.
Results
A20 was part of physiological response to lipotoxicity in NASH
To investigate any potential correlation of A20 expression with metabolic homeostasis in the fatty liver, we examined hepatic A20 expression in MCD-fed mice by real-time PCR and western blot. As depicted in Fig. 1A-C, the expression of A20 mRNA and protein were significantly higher in steatotic livers from MCD-fed mice than chow diet-fed (CD) animals. Consistent with our observation, reanalysis of microarray data from the study results by Duval .C et al[21], also showed A20 mRNA level was much higher in livers of HFD-induced NASH mice than in non-NASH mice (Fig. 1D).
A20 was part of physiological response to lipotoxicity in NASH. (A) Immunoblot analysis of A20 protein in the livers of MCD-fed (n = 10) vs control diet mice (n = 8). (B) Graphs represent the relative intensities of A20/GAPDH determined by densitometry. (C) A20 mRNA levels in the livers of MCD-fed (n=10) vs control diet mice (n=8). (D) Relative A20 mRNA levels in the livers of HFD-induced NASH and non-NASH mice models (Reanalysis of microarray data from the study of Duval .C et al, GSE 24031). (E) and (F) Relative A20 mRNA level was measured by RT-PCR in HepG2 and U937 cells treated with FFAs(OA: PA = 2:1, 0.5mmol/L) or 1% BSA as a control for 24 hours. (G) A20 protein level was evaluated by western blot in HepG2 and U937 cells treated with FFAs (OA: PA = 2:1, 0.5mmol/L) at different time points. Data is shown as means ±SD, n = 3 -5 experiments.
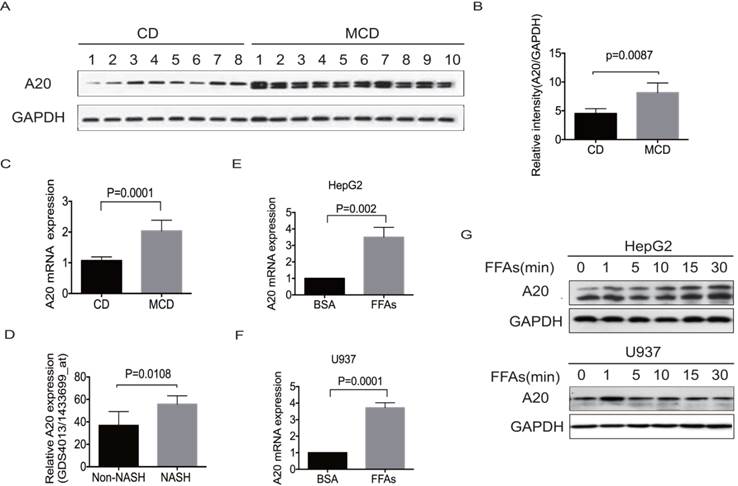
In obese individuals, adipocytes undergo hypertrophy and secrete free fatty acids (FFAs). The overwhelming influx of FFAs may cause hepatotoxicity and stimulate progression from simple steatosis to NASH via several mechanisms beyond direct cytotoxicity. We incubated hepatocyte HepG2 and monocyte U937 cells to pathophysiologically relevant concentration of mixed free fatty acids (0.5mmol/L FFAs; OA: PA = 2:1) for indicated times to simulate the excessive uptake of FAs. Consistent with the upregulation of A20 in steatotic livers in vivo, A20 was also rapidly increased after FFAs stimulation in both HepG2 and U937 cells (Fig.1 E-G). Immunoblot analysis of A20 expression in HepG2 cells and MCD-fed mice showed as a doublet as shown in Fig. 1 A and G, which may be associated with the alternative phosphorylation of A20 protein. These results indicated that A20 was part of physiological response to lipotoxicity in NASH.
A20 regulated inflammatory cytokines
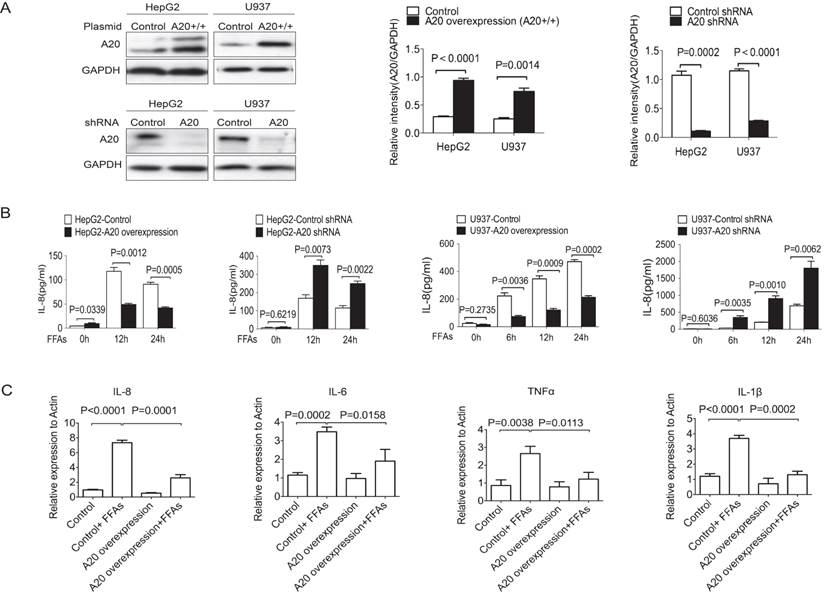
Since chronic inflammation was a key factor in the pathogenesis of NASH and A20 was reported to inhibit inflammation in a variety of diseases, we need to know whether A20 protein could affect the inflammatory response in NASH or not. We then generated four stable cell lines to overexpress or to knockdown A20 expression by using HepG2 and U937 cells (Fig. 2A). We measured the protein levels of IL-8 in the supernatant using FACS. After treatment with 0.5 mmol/L FFAs for the indicated times, overexpression of A20 inhibited IL-8 secretion in both HepG2 and U937 cells, and knockdown of A20 increased IL-8 production in HepG2 and U937 cells (Fig. 2 B). We also found that overexpression of A20 in HepG2 cells reduced the mRNA levels of pro-inflammatory cytokines such as TNF-α, IL-1β, IL-6 and IL-8 (Fig. 2C). These results were in line with the well-established anti-inflammation function of A20, A20 could also inhibit the inflammatory reaction in response to FFAs stimulation too.
A20 regulated inflammatory cytokines (A) Immunoblotting of A20 protein in stable cell lines of HepG2 and U937 that either overexpression or knockdown of A20. Graphs represent quantification of target protein bands relative to GAPDH. (B) Cytokine analysis of IL-8 in the culture supernatant of A20-overexpression and A20-knockdown cells after treatment of 0.5 mmol/L FFAs for the indicated times. Cytokine analysis was measured by FACS. (C) Real-time PCR analysis of IL-8, IL-6, TNF-α and IL-1β in HepG2-A20 overexpression and HepG2-Control cells after treatment of 0.5mmol/L FFAs for 16 hours. Graphs represent means ±SD of relative mRNA levels after normalization with β-actin. Data is shown as means ±SD, n = 3 experiments.
A20 is a negative regulator of FFAs-induced NF-κB activation
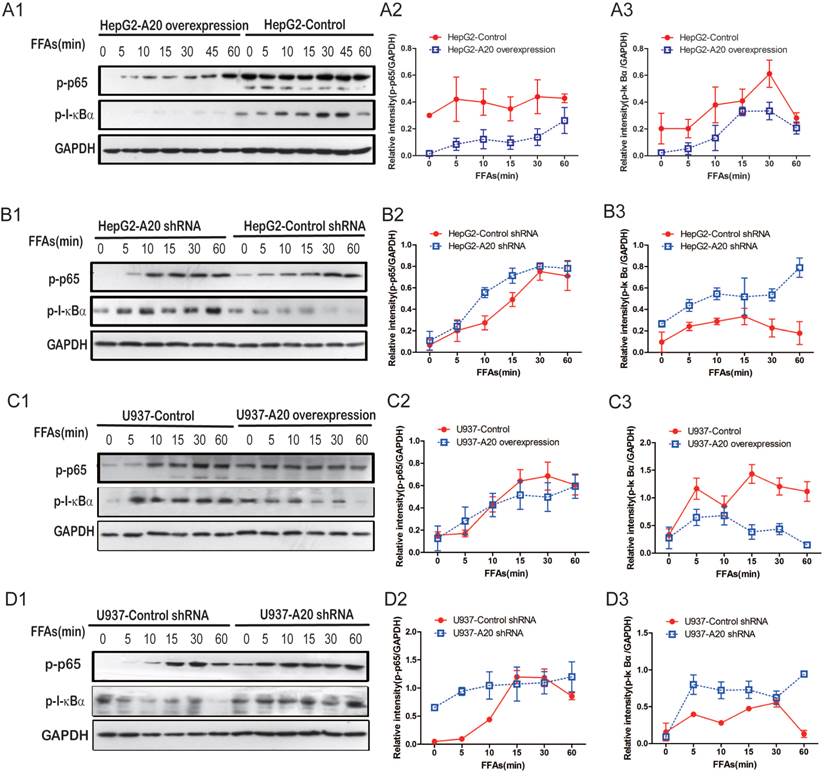
As A20 was a putative inhibitor of the NF-κB pathway[22], we analyzed NF-κB signaling in HepG2 and U937 cells in which A20 was either stably overexpressed or knocked down. A20-overexpression cells showed impaired activation of NF-κB in response to FFAs as indicated by lower phospho-p65 levels and lower phospho-I-κBα levels (Fig. 3A and C). A20-knockdown cells showed enhanced NF-κB activation, which was demonstrated by higher phospho-p65 and phospho-I-κBα levels (Fig. 3B and D). These findings support the canonical role played by A20 as an inhibitor of NF-κB signaling, even in response to FFAs stimulation. Taken together, A20 inhibited FFAs-induced inflammatory response via attenuating NF-κB activation in four cell lines we generated.
A20 negatively regulated FFAs-induced NF-κB activation. (A1) Immunoblot analysis of p-p65, p-I-κBα expression in HepG2-A20 overexpression cells and control cells with 0.5 mmol/L FFAs treatment for the indicated times. (A2) Corrected densitometry of p-p65/GAPDH in (A1). (A3) Corrected densitometry of p-I-κBα/GAPDH in (A1). (B1) Immunoblot analysis of p-p65, p-I-κBα expression in HepG2-A20 shRNA cells and control cells after 0.5 mmol/L FFAs treatment for the indicated times. (B2) and (B3) Corrected densitometry of p-p65/GAPDH, p-I-κBα/GAPDH in (B1), respectively. (C1) Immunoblot analysis of p-p65, p-I-κBα expression in U937-A20 overexpression cells and control cells after 0.5 mmol/L FFAs treatment for the indicated times. (C2) and (C3) Corrected densitometry of p-p65/GAPDH, p-I-κBα/GAPDH in (C1), respectively. (D1) Immunoblot analysis of p-p65, p-I-κBα expression in U937-A20 shRNA cells and control cells after 0.5 mmol/L FFAs treatment for the indicated times. (D2) and (D3) Corrected densitometry of p-p65/GAPDH, p-I-κBα/GAPDH in (D1), respectively. GAPDH was used as loading control. Data is shown as means ±SD of 3-5 independent experiments.
A20 decreased lipids accumulation in HepG2 cells via decreasing lipogenesis and increasing fatty acid oxidation
We then studied whether A20 could decrease lipids accumulation in HepG2 cells or not. After FFAs stimulation for 24 hours, Oil Red O staining was used to measure lipids accumulation. HepG2-A20 overexpression cells were observed to have less lipid droplet accumulation compared with the HepG2 control cells (Fig. 4A). As an increased availability and uptake of FFAs was expected to augment hepatic TG synthesis, we also measured intracellular TG content. Along with their morphological presentation, FFA-treated HepG2-A20 overexpression cells accumulated only half the amount of TG compared with control cells (Fig. 4B). Knockdown of A20 in HepG2 cells increased TG deposition, both qualitatively and quantitatively (Fig. 4 A and B). These findings demonstrated that A20 could suppress lipids deposition in hepatocytes.
A20 decreased lipids accumulation in HepG2 cells (A) HepG2 cells that stably overexpression or knockdown of A20 were treated with 0.5 mmol/L FFAs (OA: PA = 2:1) or 1% BSA as a control (data not shown) for 24 hours. Then, these cells were stained with Oil Red O to measure intracellular lipid accumulation. (B) Quantitative analysis of intracellular TG content in cells treated as in (A). Absorbance readings were normalized by protein concentrations. (C) and (D) HepG2-A20 overexpression and HepG2-control cells were treated with 0.5 mmol/L FFAs for 16 hours, then the expression of genes involved in de novo lipogenesis (C) and FA oxidation (D) were measured by real-time PCR. Data is shown as means ±SD, n = 3 - 5 experiments. SREBP-1c, sterol regulatory element binding protein 1c; ACC1, acetyl-CoA carboxylase 1; FAS, fatty acid synthase; PPARα, peroxisome proliferator-activated receptor alpha; CPT1α, carnitine palmitoyl transferase 1α; MCAD, medium-chain acyl-CoA dehydrogenase; PGC-1α, peroxisome proliferator-activated receptor-gamma coactivator-1 alpha.
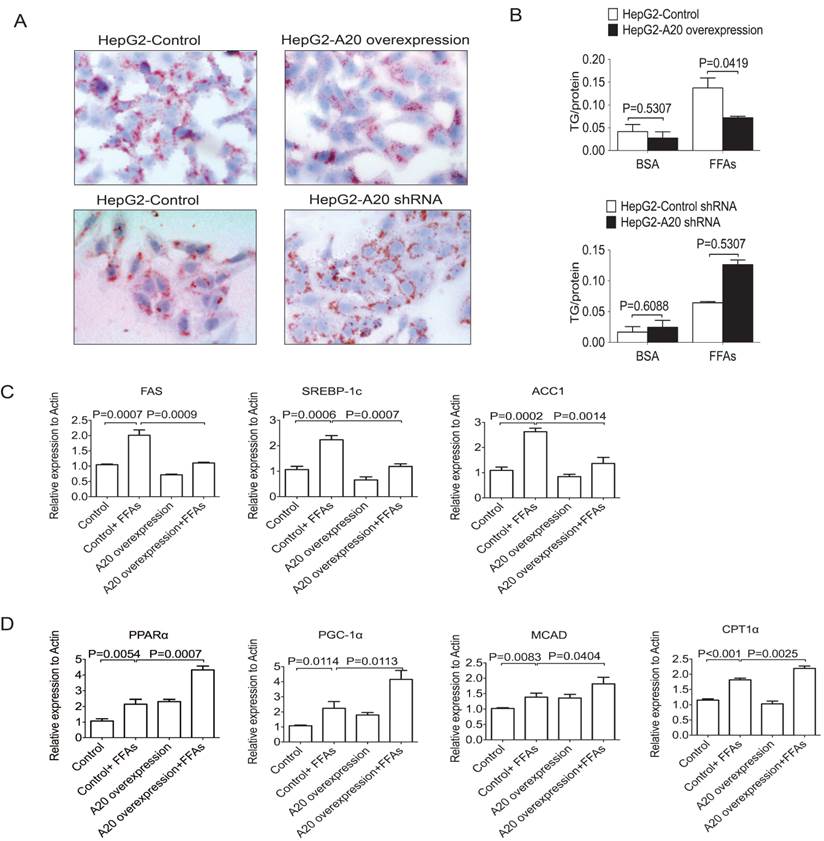
To determine how A20 inhibits lipids deposition, we measured the relative expression of genes involved in de novo lipogenesis and FA oxidation. We found genes that related to de novo lipogenesis and fatty acid oxidation were both upregulated upon FFAs stimulation (Figure 4C&D). Overexpression of A20 in HepG2 cells markedly decreased the mRNA levels of key lipogenic genes such as fatty acid synthase (FAS), sterol regulatory element binding protein1c (SREBP-1c) and acetyl-CoA carboxylase 1 (ACC1) (Fig. 4C), but not sterol regulatory element binding protein 2 (SREBP2) or diacylglycerol acyltransferase-2 (DGAT2) (data not shown), indicating that A20 protein plays a role as negative regulator for lipogenesis in hepatocytes.
In addition, we also observed that A20 overexpression prominently upregulated genes that promote β-oxidation, including peroxisome proliferator-activated receptor alpha (PPARα), peroxisome proliferator-activated receptor-gamma coactivator-1 alpha (PGC-1α), medium-chain acyl-CoA dehydrogenase (MCAD) and the rate-limiting enzyme of mitochondrial β-oxidation, carnitinepalmitoyl transferase 1α (CPT1α; Fig.4D). These findings strongly suggest that, in addition to negatively regulate the de novo lipogenesis, A20 function as anti-steatotic factor by the activation of mitochondrial β-oxidation.
Elevated expression of hepatic A20 in steatotic human liver samples
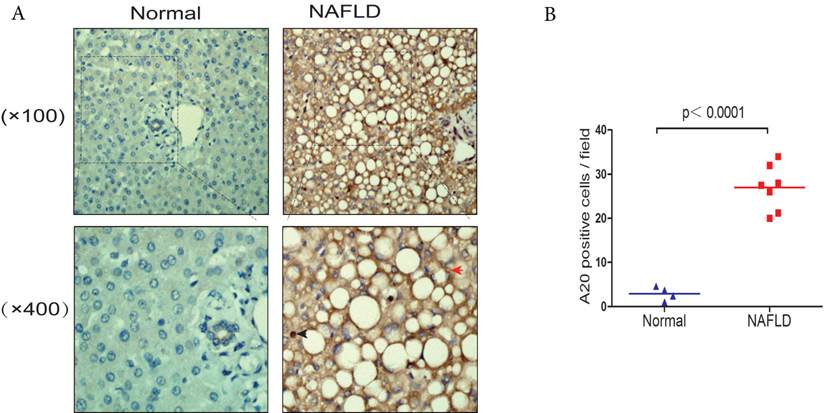
There was very limited information about the expression and location of A20 in human NAFLD patient samples, we compared the expression levels in human liver tissues from NAFLD patients and normal controls by immunohistochemistry. Liver samples from seven NAFLD patients were used in this study, and four samples from normal liver tissue were used as control. We observed that A20 protein levels were increased in the livers from NAFLD patients in comparison to normal controls, especially in hepatocytes and kupffer cells (Fig. 5 A and B). This indicated A20 was indeed involved in the pathogenesis of NASH in human.
Elevated expression of hepatic A20 in steatotic human liver samples (A) A20 protein levels in normal liver tissue (Normal, n = 4) and liver tissue taken from NAFLD patients (NAFLD, n = 7) were analyzed by immunohistochemistry (IHC; brown). The red arrows indicated A20-positive hepatocytes. Blue arrows indicated A20-positive Kupffer cells. (B) Quantification of number of A20-positive cells in each biopsy from patients with NAFLD compared with controls.
Discussion
NF-κB-mediated pro-inflammatory effects had been taken as a major concern in the development of steatosis progression to steatohepatitis. As a downstream negative regulator, A20 mediated hepatoprotective function of NF-κB, but inhibited its role in recruitment of inflammation. Together with the evidence that A20 modulated genes expression related to lipid metabolism, we hypothesized that A20 was also an important mediator of anti-lipotoxic protection, restraining FFAs-promoted NASH development through modulation of lipid metabolism.
Dr. Ferran and her colleagues showed that A20 was significantly upregulated in mouse models following severe liver injuries, including acute toxic hepatitis (D-galactosamine/lipolysaccharide) and extended (78% of total liver mass) hepatectomy[10, 16]. These findings indicated that A20 could be a common protective response of hepatocytes to various insults. However, these physiological responses mediated hepatoprotective function seemed to be insufficient, better prognosis need the higher A20 expression level. This was supported by studies in mouse models showing that, although hepatic expression of A20 increased in control mice, the survival rate of mice suffering from acute toxic hepatitis was only 15% to 20%, while virus-mediated overexpression of A20 yielded a survival rate of 85%[10]. In the present study, significantly higher expression of A20 in liver biopsies was found, especially in hepatocytes and Kupffer cells, from patients with NASH and MCD diet-induced NASH models. Previous studies reported that the elevation of A20 could be results of various stimulations, including TNF-α, ConA and LPS. FFAs were also important mediators of lipotoxicity, both as potential cellular toxins and through leading to lipid over-accumulation[23]. The elevation of circulating FFAs was verified in patients with NAFLD and their levels correlated with disease severity[24, 25]. Our study showed that FFAs alone could rapidly elevate A20 expression in hepatocytes and Kupffer cells. The elevation of A20 in liver with steatosis or steatohepatitis could at least partly due to excessive liver influx of fatty acids. These results supported that A20 was part of physiological response to lipotoxicity during NASH development.
The “two hits” model of NASH pathogenesis was proposed in 1998, the initial hit involved fat accumulation in the livers, which sensitized hepatocytes to subsequent insults, including inflammatory cytokines that promoted hepatocellular damage, inflammation and progressive liver disease[26]. Recent studies reported that FFAs-promoted hepatic fat accumulation induced the release of various “danger signals” from hepatocytes, such as IL-8, IL-1β, CCN1 and TNF-α and these danger signals play critical roles in the development of hepatic inflammation[23, 27-29]. Our results confirmed Joshi-Barve's findings, which showing that FFAs stimulation increased IL-8 gene expression and secretion (both mRNA and protein) from hepatocytes[27]. This should be taken as an important part of the lipotoxicity, because a failure of anti-lipotoxic protection against it would facilitate progression of the possible ensuing inflammation, which promoted the transition from simple steatosis to NASH. In this study, we found that A20 could obviously interfere with FFAs-stimulated production of these 'danger' signals from hepatocytes. Overexpression of A20 significantly downregulated hepatocyte-derived mRNA expression of IL-6, TNF-α and IL-8 in hepG2 cells with FFAs treatment. Furthermore, hepatocyte production of IL-8 was suppressed by A20 overexpression but enhanced by A20 depletion. Together with the evidence that A20 was rapidly induced in hepatocyte following FFAs stimulation, the view that A20 was an important mediator of anti-lipotoxic protection against production of hepatocyte-derived pro-inflammatory cytokines was indicated. Previous studies showed that production of these cytokines, e.g. IL-8 and TNF-α, were NF-κB -dependent, supporting a pathogenic role of NF-κB in progression of NASH. In our study, FFAs-induced activation of NF-κB could weaken by A20 overexpression, because hepatocytes with decreased A20 expression showed amplified activation of NF-κB. The regulatory effects of A20 on FFAs-induced expression of pro-inflammatory cytokines from hepatocyte were probably through modulation of NF-κB activation.
Several mechanisms may lead to lipid accumulation in the liver, including excessive supply of FFAs which accounted for almost two-thirds of the accumulation, decreased very low density lipoprotein-triglyceride export, increased de novo hepatic lipogenesis and decreased FFAs oxidation[2, 30]. Therapies capable of regulating these processes may have a role in the management of NAFLD and associated co-morbidities. In the present study, we induced obvious lipid accumulation in HepG2 cells by exposing them to FFAs, which was shown morphologically by Oil Red O staining and evaluation of intracellular triglyceride levels. Loss- and gain- of -function analysis showed clearly that A20 protected hepatocytes from FFAs-induced fat accumulation. Previous gene chip-based biological analysis showed that A20 optimized fatty acid metabolism, in which accompanied with A20 overexpression, PPARα and its transcriptional target carnitine palmytoyl transferase 1α (CPT1α) were significantly upregulated, both of which were critical regulators of fatty acid metabolism, mainly through activating mitochondrial and peroxisomal fatty acid β-oxidation pathways[18]. Studies showed that inhibition of PPARα increased susceptibility to high fat-induced NAFLD in mouse models, with increased steatosis, oxidative stress and inflammation[19], while agonist-mediated activation of PPARα inhibited lipid accumulation in hepatocytes, as well as liver inflammation[31, 32]. In this study, we also found PPARα mRNA expression was elevated under FFAs treatment; probably it was a functional adaptation, while enforced A20 expression further increased the expression of PPARα. The expression of CPT1α and MCAD were also showed the same pattern as PPARα too.
Similar to previous study, genes involved in de novo lipogenesis, including fatty-acid synthase (FAS) and sterol regulatory element-binding proteins-1c (SREBP-1c), were also induced by FFAs[33]. FAS activity is believed to be a determinant of the maximal capacity of liver to synthesize fatty acids by de novo lipogenesis. Inhibition of FAS by a selective inhibitor, platensimycin[34, 35], was reported to reduce hepatic TG levels and further ameliorate diabetes in db/db mice[36]. In line with it, we demonstrated that higher expression of A20 significantly down regulates the mRNA levels of FAS. Thus, A20 mediated amelioration of lipid accumulation in hepatocytes probably through up regulation of genes involved in fatty acid oxidation and inhibition of genes involved in de novo lipogenesis.
In summary, in this study we reported that A20 could be a physiological factor to FFAs stimulation in the pathogenesis of NASH, and also acted as an important anti-lipotoxic mediator through inhibition of lipid accumulation and pro-inflammatory cytokines releasing. A potential link between A20 and NASH was proposed in this study, more in vivo studies were needed to confirm the therapeutic potential of A20 in NASH development in future.
Abbreviations
MCD, methionine choline-deficient
FA, fatty acid
NAFLD, nonalcoholic fatty liver disease
HCC, hepatocellular carcinoma
NASH, nonalcoholic steatohepatitis
FFAs, free fatty acids
TNFAIP3, TNF-α-induced protein 3
PPARα, peroxisome proliferator-activated receptor alpha
SREBP-1c, sterol regulatory element binding protein1c
ACC1, acetyl-CoA carboxylase 1
FAS, fatty acid synthase
PGC-1α, peroxisome proliferator-activated receptor-gamma coactivator-1 alpha
MCAD, medium-chain acyl-CoA dehydrogenase
OA, oleic acid
PA, palmitic acid
IACUC, Institutional Animal Care and Use Committee
FACS, Fluorescence activated cell sorter analysis
TG, triglyceride
HPF, high power field
HFD, high-fat diets
IHC, immunohistochemistry
H&E, hematoxylin and eosin
CD, control diet
Acknowledgements
This work was supported by grants from the Baoen Wang liver fibrosis research fund, hepatitis prevention and treatment foundation of China (No.20110002), and Key discipline construction plan of Shanghai Public Health projects (No.12GWZX0903). Also thanks for Dr. Jun Wang for giving scientific advice and reviewing this article.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Cohen JC, Horton JD, Hobbs HH. Human fatty liver disease: old questions and new insights. Science. 2011;332:1519-1523
2. Tilg H, Moschen AR. Evolution of inflammation in nonalcoholic fatty liver disease: the multiple parallel hits hypothesis. Hepatology. 2010;52:1836-1846
3. Ribeiro PS, Cortez-Pinto H, Sola S. et al. Hepatocyte apoptosis, expression of death receptors, and activation of NF-kappaB in the liver of nonalcoholic and alcoholic steatohepatitis patients. The American journal of gastroenterology. 2004;99:1708-1717
4. Larter CZ, Yeh MM, Haigh WG. et al. Dietary modification dampens liver inflammation and fibrosis in obesity-related fatty liver disease. Obesity. 2013;21:1189-1199
5. Dela Pena A, Leclercq I, Field J, George J, Jones B, Farrell G. NF-kappaB activation, rather than TNF, mediates hepatic inflammation in a murine dietary model of steatohepatitis. Gastroenterology. 2005;129:1663-1674
6. Brenndorfer ED, Weiland M, Frelin L. et al. Anti-tumor necrosis factor alpha treatment promotes apoptosis and prevents liver regeneration in a transgenic mouse model of chronic hepatitis C. Hepatology. 2010;52:1553-1563
7. Iimuro Y, Nishiura T, Hellerbrand C. et al. NFkappaB prevents apoptosis and liver dysfunction during liver regeneration. The Journal of clinical investigation. 1998;101:802-811
8. Wang CY, Mayo MW, Baldwin AS Jr. TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-kappaB. Science. 1996;274:784-787
9. Cooper JT, Stroka DM, Brostjan C, Palmetshofer A, Bach FH, Ferran C. A20 blocks endothelial cell activation through a NF-kappaB-dependent mechanism. J Biol Chem. 1996;271:18068-18073
10. Arvelo MB, Cooper JT, Longo C. et al. A20 protects mice from D-galactosamine/lipopolysaccharide acute toxic lethal hepatitis. Hepatology. 2002;35:535-543
11. Lee EG, Boone DL, Chai S. et al. Failure to regulate TNF-induced NF-kappaB and cell death responses in A20-deficient mice. Science. 2000;289:2350-2354
12. Lu TT, Onizawa M, Hammer GE. et al. Dimerization and ubiquitin mediated recruitment of A20, a complex deubiquitinating enzyme. Immunity. 2013;38:896-905
13. Xia M, Liu J, Wu X. et al. Histone methyltransferase Ash1l suppresses interleukin-6 production and inflammatory autoimmune diseases by inducing the ubiquitin-editing enzyme A20. Immunity. 2013;39:470-481
14. Coornaert B, Carpentier I, Beyaert R. A20: central gatekeeper in inflammation and immunity. J Biol Chem. 2009;284:8217-8221
15. Enesa K, Moll HP, Luong L, Ferran C, Evans PC. A20 suppresses vascular inflammation by recruiting proinflammatory signaling molecules to intracellular aggresomes. FASEB journal: official publication of the Federation of American Societies for Experimental Biology. 2015;29:1869-1878
16. Longo CR, Patel VI, Shrikhande GV. et al. A20 protects mice from lethal radical hepatectomy by promoting hepatocyte proliferation via a p21waf1-dependent mechanism. Hepatology. 2005;42:156-164
17. da Silva CG, Studer P, Skroch M. et al. A20 promotes liver regeneration by decreasing SOCS3 expression to enhance IL-6/STAT3 proliferative signals. Hepatology. 2013;57:2014-2025
18. Damrauer SM, Studer P, da Silva CG. et al. A20 modulates lipid metabolism and energy production to promote liver regeneration. PloS one. 2011;6:e17715
19. Abdelmegeed MA, Yoo SH, Henderson LE, Gonzalez FJ, Woodcroft KJ, Song BJ. PPARalpha expression protects male mice from high fat-induced nonalcoholic fatty liver. The Journal of nutrition. 2011;141:603-610
20. Chalasani N, Younossi Z, Lavine JE. et al. The diagnosis and management of non-alcoholic fatty liver disease: practice guideline by the American Gastroenterological Association, American Association for the Study of Liver Diseases, and American College of Gastroenterology. Gastroenterology. 2012;142:1592-1609
21. Duval C, Thissen U, Keshtkar S. et al. Adipose tissue dysfunction signals progression of hepatic steatosis towards nonalcoholic steatohepatitis in C57BL/6 mice. Diabetes. 2010;59:3181-3191
22. Hymowitz SG, Wertz IE. A20: from ubiquitin editing to tumour suppression. Nature reviews Cancer. 2010;10:332-341
23. Feldstein AE, Werneburg NW, Canbay A. et al. Free fatty acids promote hepatic lipotoxicity by stimulating TNF-alpha expression via a lysosomal pathway. Hepatology. 2004;40:185-194
24. Nehra V, Angulo P, Buchman AL, Lindor KD. Nutritional and metabolic considerations in the etiology of nonalcoholic steatohepatitis. Digestive diseases and sciences. 2001;46:2347-2352
25. de Almeida IT, Cortez-Pinto H, Fidalgo G, Rodrigues D, Camilo ME. Plasma total and free fatty acids composition in human non-alcoholic steatohepatitis. Clinical nutrition. 2002;21:219-223
26. Day CP, James OF. Steatohepatitis: a tale of two "hits"? Gastroenterology. 1998;114:842-845
27. Joshi-Barve S, Barve SS, Amancherla K. et al. Palmitic acid induces production of proinflammatory cytokine interleukin-8 from hepatocytes. Hepatology. 2007;46:823-830
28. Csak T, Ganz M, Pespisa J, Kodys K, Dolganiuc A, Szabo G. Fatty acid and endotoxin activate inflammasomes in mouse hepatocytes that release danger signals to stimulate immune cells. Hepatology. 2011;54:133-144
29. Bian Z, Peng Y, You Z. et al. CCN1 expression in hepatocytes contributes to macrophage infiltration in nonalcoholic fatty liver disease in mice. Journal of lipid research. 2013;54:44-54
30. Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, Parks EJ. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. The Journal of clinical investigation. 2005;115:1343-1351
31. Nagasawa T, Inada Y, Nakano S. et al. Effects of bezafibrate, PPAR pan-agonist, and GW501516, PPARdelta agonist, on development of steatohepatitis in mice fed a methionine- and choline-deficient diet. European journal of pharmacology. 2006;536:182-191
32. Jia Y, Kim S, Kim J. et al. Ursolic acid improves lipid and glucose metabolism in high-fat-fed C57BL/6J mice by activating peroxisome proliferator-activated receptor alpha and hepatic autophagy. Molecular nutrition & food research. 2015;59:344-354
33. Dorn C, Riener MO, Kirovski G. et al. Expression of fatty acid synthase in nonalcoholic fatty liver disease. International journal of clinical and experimental pathology. 2010;3:505-514
34. Wang J, Soisson SM, Young K. et al. Platensimycin is a selective FabF inhibitor with potent antibiotic properties. Nature. 2006;441:358-361
35. Method of treatment using fatty acid synthesis inhibitors. Singh SB TM, Wang J. http://www.wipo.int/pctdb/en/wo.jsp?WO=2008039327
36. Wu M, Singh SB, Wang J. et al. Antidiabetic and antisteatotic effects of the selective fatty acid synthase (FAS) inhibitor platensimycin in mouse models of diabetes. Proc Natl Acad Sci U S A. 2011;108:5378-5383
Author contact
![]() Corresponding authors: Zhuping Fan, Ph.D., Address: No.160, Pujian Road, Shanghai, China; Tel.: +86-21-68383551; Fax: 86 21 50903024; Email: zhuping_fancom. Qing Lin, Ph.D., Current address: No.381, Fenglin Road, Shanghai, China; Tel.:+86-21-53897598; Email: qing.lincom
Corresponding authors: Zhuping Fan, Ph.D., Address: No.160, Pujian Road, Shanghai, China; Tel.: +86-21-68383551; Fax: 86 21 50903024; Email: zhuping_fancom. Qing Lin, Ph.D., Current address: No.381, Fenglin Road, Shanghai, China; Tel.:+86-21-53897598; Email: qing.lincom