Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Development and clinical...
3. Overcome the limiting...
4. Conclusions
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(6):718-729. doi:10.7150/ijbs.14405 This issue Cite
Review
New Strategies for the Treatment of Solid Tumors with CAR-T Cells
Hao Zhang1*, Zhen-long Ye1*, Zhen-gang Yuan1, Zheng-qiang Luo2, Hua-jun Jin1 ![]() , Qi-jun qian1, 2, 3
, Qi-jun qian1, 2, 3 ![]()
1. Laboratory of Viral and Gene Therapy, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, Shanghai 200438, China;
2. Xinyuan Institute of Medicine and Biotechnology College of Life Science, Zhejiang Sci-Tech University, Hangzhou 310018, China;
3. Ningbo NO.5 Hospital (Ningbo Cancer Hospital), Ningbo 315201, China.
*These authors contributed equally to this manuscript.
Received 2015-11-12; Accepted 2016-1-16; Published 2016-4-28
Abstract

Recent years, we have witnessed significant progresses in both basic and clinical studies regarding novel therapeutic strategies with genetically engineered T cells. Modification with chimeric antigen receptors (CARs) endows T cells with tumor specific cytotoxicity and thus induce anti-tumor immunity against malignancies. However, targeting solid tumors is more challenging than targeting B-cell malignancies with CAR-T cells because of the histopathological structure features, specific antigens shortage and strong immunosuppressive environment of solid tumors. Meanwhile, the on-target/off-tumor toxicity caused by relative expression of target on normal tissues is another issue that should be reckoned. Optimization of the design of CAR vectors, exploration of new targets, addition of safe switches and combination with other treatments bring new vitality to the CAR-T cell based immunotherapy against solid tumors. In this review, we focus on the major obstacles limiting the application of CAR-T cell therapy toward solid tumors and summarize the measures to refine this new cancer therapeutic modality.
Keywords: chimeric antigen receptor, adoptive immunotherapy, cell therapy, solid tumor.
1. Introduction
Advances in our understanding on the interaction between the immune system and tumor cells have contributed to the rapid development of novel therapeutic strategies based on chimeric antigen receptor (CAR) or T cell receptor (TCR) modified T cells. CAR-T cell therapy has achieved outstanding progresses in clinical observations, which makes it even more attractive in the development of cancer adoptive immunotherapy. The CARs endow T cells with antigen-specific recognition, activation and proliferation in a major histocompatibility complex (MHC) independent manner [1], Currently, CARs have considerably evolved to the third generation, containing two co-stimulatory molecules, such as CD28+CD134 (OX40) or CD28+CD137 (4-1BB), which have usually been demonstrated with enhanced cytokine production and tumor lytic activity and reduced activation-induced cell death (AICD) than the second or first generation CARs [2].
The emerging therapeutic approach of CAR-T cell therapy has sparked great interests, extensive studies in preclinical and clinical trials have revealed encouraging therapeutic efficacy in treating a variety of cancers, particularly in treating B-cell hematologic malignancies with CD19 CAR-T cells [3]. Nevertheless, targeting solid tumors is more challenging than targeting hematological malignancies because of tumor histopathological characteristics, shortage of specific antigens and local strong immunosuppressive microenvironment [4]. Furthermore, the on-target/off-tumor toxicity can pose significant risks. Thus, it is imperative to develop more competent and safer immunotherapy approaches by optimizing the design of CAR vectors, exploring new targets, incorporating conditional safe switches and combining with other strategies. And much work remains to be done to improve the efficacy of CAR-T cell therapy for solid tumors. It may be achieved, at least partially, by more extensive basic studies investigating the spatiotemporal dynamics of T cell activation by CARs and unraveling the connection between T cell migration in solid tumors and the effectiveness in eradication of solid tumors and metastases [5]. In this review, we discuss the current status and major obstacles for the treatment of solid tumors with CAR-T cells, thus provide some potential measures to refine this novel therapeutic modality.
2. Development and clinical application of CARs
2.1 Evolving architecture of CARs
It has been exclusively reported that CARs combine the exquisite antigen specificity of antibodies with the poly functionality and potency of cellular immunity. The unique structure of CAR endows T cells with tumor specific cytotoxicity and elevated anti-tumor activity in an MHC independent manner, through applying viral-vector technology or transposon-based system to transfect immune effector cells [6]. The classic CARs consist of an extracellular antigen recognition domain, a hinge domain, a transmembrane (TM) domain and an intracellular domain (Fig 1) [7]. The extracellular antigen-binding moiety in CARs, typically derived from a single chain variable fragment (scFv) that isolated from an antigen-specific monoclonal antibody, renders T cells the ability to bind antigens with retained specificity and affinity [8]. The hinge region mediates CAR flexibility, transduces essential signals, and exerts profound impacts on ensuring the suitable positioning of the binding domain during scFv-antigen interactions (Fig 1A) [9]. The transmembrane domains are derived from CD3-ζ, CD4, CD8, OX40, and H2-Kb [10] , and it has been clearly proved that the transmembrane domain can indeed influence the function of CAR-T cells [11]. Other investigators suggested that CAR-T cells with the CD3-ζ transmembrane domain showed more potent cytolytic activity, while CAR-T cells with CD28 transmembrane domains were more persistent [12]. A transmembrane domain from native CD3-ζ chain induces enhanced T-cell activation in comparison to mutated CD3-ζ transmembrane [13]. The intracellular domain is responsible for signal delivery within CARs, this element has been manipulated extensively in an attempt to optimize functions of engineered T cells. T cell activation relies on the phosphorylation of immune receptor tyrosine based activation motifs (ITAMs) presented in the cytoplasmic CD3-ζ domain of the TCR complex [14] (Fig 1B). The signaling domain is critical for CAR-T cells to fulfill anti-tumor functions, the construct of CARs has seen several incarnations according to the different compositions of signaling domain.
Chimeric antigen receptors (CARs) architecture. (A) CARs consist of an extracellular domain, a hinge, a transmembrane domain, and an intracellular domain. The extracellular domain is typically a scFv fragment that isolated from an antigen-specific monoclonal antibody, with retained specificity and affinity. (B) The intracellular domain, derived from the phosphorylation of immunoreceptor tyrosine based activation motifs (ITAMs) presented in the cytoplasmic CD3-ζ domain of the TCR complex, transmits activation and co-stimulatory signals to T cells. (C) According to the number of signaling molecules, CARs are classified into the 1st generation (one), 2nd generation (two) and 3rd generation (three) CARs. The most applied co-stimulatory signaling molecules are CD28, 4-1BB, ICOS and OX-40.
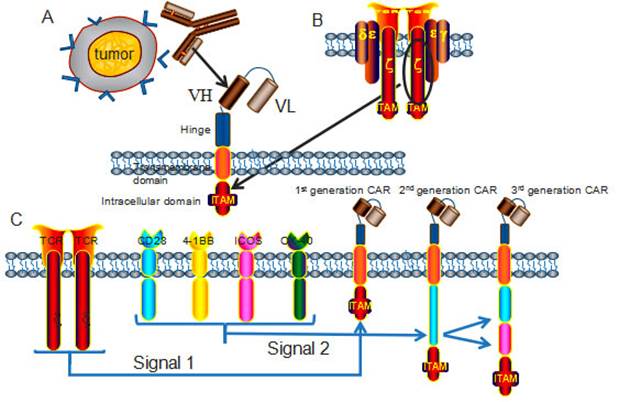
The first generation CARs provided the proof for the concept of the targeting and activation of CAR-T cells, but had very modest clinical activity and poor persistence in vivo [15], to overcome these limitations, the second and third generation CARs have incorporated co-stimulatory molecules, including CD27, CD28, CD134, CD137 and ICOS [16]. CAR-T cells with multiple signaling receptors have been demonstrated with sustained proliferation, enhanced cytokine production, improved tumor lytic activity, and reduced AICD both in vitro and in vivo [17] (Fig 1C). Nowadays, the second-generation CAR-T cells have been more exclusively and ubiquitously applied in clinical trials than the third generation CAR-T cells, because the reduced activation threshold of the third generation CAR-T cells may cause on-target/off-tumor side effects to normal tissues.
2.2 Therapeutic advantages of CARs technology
The use of CARs confers several advantages over TCR transgenes. Based on the MHC independent antigen recognition, CARs are able to bypass the mechanism that employed by tumors to evade immune detection through down-regulating MHC-I molecule [18]. Theoretically, CARs are able to detect almost all antigens that can be recognized by antibodies, including protein antigens, carbohydrate and lipid antigens and so on, that is, CARs are more universally applicable for immunotherapy to treat diseases [19]. In addition, the intracellular signaling domains within CARs are more flexible so that can be designed to compensate the down-regulation of co-stimulatory molecules induced by cancer cells [20]. Therefore, CAR-T cells harness maximal treatment resources for adoptive immunotherapy over TCR-T cells, for which the main hurdle is that the effect is HLA/MHC dependent.
2.3 Clinical trials utilizing CAR-T cells
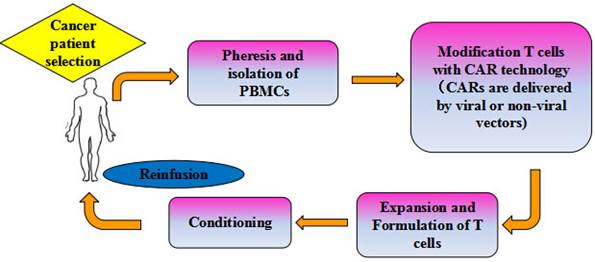
The promising clinical trials have generated remarkable responses in cancer patients, which provided a solid foundation for inspiring the application of CAR-T cell based adoptive cell immunotherapy in multiple oncological settings. To date, clinical trials utilizing the second generation CD19 CAR-T cells to treat hematological malignancies have resulted in the most encouraging clinical responses [21], and the CD19-CAR T cell therapy (CTL019) has been highly appraised and approved as the breakthrough therapy by the FDA. Currently, adoptive CAR-T cells focusing on CD20 are also being evaluated in clinical trials for the treatment of B-cell malignancies, for which other CAR-T cells targeting CD22, CD30 and CD33 are now at the stage of in vitro trials [22]. At present, CAR-T cell therapy has demonstrated success as a novel treatment modality that the commercial manufacture of gene-modified T cells at industrial scale for the treatment of advanced cancers is becoming a hotspot worldwide. Mention worthy, the Juno therapeutics, Novartis and Kite are leading Big Pharmacies in the world due to their pioneering contributions to the development of CAR-T cell therapy. Figure 2 shows a flow chart of adoptive immunotherapy using CAR-T cells in clinical treatment.
Schema of adoptive cellular therapy with CAR-T cells. PBLs harvested from specifically selected patients. T cells were isolated, activated and genetically modified to express a transgene encoding tumor-specific CARs. The genetically modified T cells are then expanded on a large scale using a cell processing center in vitro to a sufficient number, and thus infused back into patients, with or without chemo-radio therapeutic preconditioning.
In contrast to the remarkable clinical responses of CAR-T cell immunotherapy for hematologic malignancies, treating solid tumors with CAR-T cells has been limited by tumor histopathological structure and strong immunosuppressive environment, wherein the lack of ideal target is another crucial deficiency for the treatment of solid tumors. Currently the preferred therapeutic targets to treat ovarian cancer and neuroblastoma with CAR-T cells are FRα and GD2 respectively [23]. The updated statistics of therapeutic targets in solid tumor immunotherapy with CAR-T cells are showed in table 1.
Therapeutic targets in treating solid tumors with CAR-T cells.
| Target | Tumor types | Number of cases | Clinical stage | Results | Citation |
|---|---|---|---|---|---|
| mesothelin FRα | mesothelioma | 4 | I | partial remission | [24] |
| Lung Cancer | 24 | I | Ongoing | NCT02414269 | |
| Breast Cancer | 14 | I | partial remission | [25] | |
| Ovarian Cancer | 15 | I | Ongoing | [26] | |
| L1-CAM | Metastatic neuroblastoma | 6 | I | One case of partial remission; five cases of progress | [27] |
| CAIX | Metastatic renal cell carcinoma | 3 | I | test was forced to stop because of the serious liver toxicity | [28] |
| GD2 | Neuroblastoma | 11 | I | 5 cases of complete remission; 2 cases of partial remission; 2 cases of stable; 2 cases of tumor necrosis | [29] |
| 19 | I | 6 cases of complete remission; 3 cases of sick to survive; 10 cases of death | [30] | ||
| FAP | Malignant pleural mesothelioma | 9 | I | Ongoing | [31] |
| Lewis Y | Bone marrow lymphoma | 5 | I | 2 cases of stable; 1 patient died (in treatment); one case progress | [32] [33] |
| EGFRvIII | Brain tumor | 160 | I/II | Ongoing | NCT01454596 |
| HER2 | Colon Cancer | 1 | I | Death for off-target effects and cytokine storm syndrome | [34] |
| HER2-positive Lung Cancer | 18 | I | Ongoing | NCT00889954 | |
| Malignant gliomas | 18 | I | Ongoing | NCT01109095 | |
| CD20 | Follicular lymphoma; Mantle cell lymphoma | 7 | I | 2 cases of complete remission; 1 case of partial remission; 4 cases of stable | [35] |
| 3 | II | 2 cases of complete remission; 1 case of partial remission | [36] | ||
| PSMA | Prostate Cancer | 18+18 | I | Ongoing | NCT00664196NCT01140373 |
| kLC | B-cell lymphoma, CLL, multiple myeloma | 18 | I | Ongoing | NCT00881920 |
| CD30 | Hodgkin's lymphoma,NHLs | 18 | I | Ongoing | NCT01316146 |
| CEA | Stomach cancer et.al | 14 | I | Ongoing | [37] |
| Metastatic adenocarcinoma | 48 | II | Ongoing | NCT01723306 | |
| Metastatic Breast Cancer | 26 | I | Ongoing | NCT00673829 |
FRα, α-folate receptor; L1-CAM, L1-cell adhesion molecule; CAIX, carboxy-anhydrase-IX; FAP, Fibroblast activation protein; HER2, human epidermal growth factor receptor 2; CEA, carcinoembryonic antigen; PSMA, Prostate Specific Membrane Antigen; CEA, Carcino Embryonie Antigen.
3. Overcome the limiting obstacles of CAR-T cell therapy against solid tumors
CAR-T cells recognize cell surface antigens through scFv structures, which typically contain the variable domains of the light and heavy chains, in non-MHC restricted manner [38]. The membrane protein CD19 is widely expressed by almost all the B cells, and B-cell hematologic malignancies are with relatively uniform structure characteristics and so on [39]. All these properties lead to that most patients with B-cell malignancies exhibited inspiring curative effect after CD19 CAR-T cell therapy. But the application of CAR-T cell therapy in solid tumor treatment is severely limited by heterogeneity characteristics, shortage of tumor specific antigens and immunosuppressive microenvironment. Now we make an analysis on the limiting factors for the application of CAR-T cells in solid tumor treatment and discuss the relevant countermeasures.
3.1 Poor infiltration of T lymphocytes into solid tumors
Most hematologic malignancies are associated with hematopoietic stem cell regeneration dysfunction [40], without forming tissue structure. In contrast, solid tumors have special histopathological features, such as high concentration of blood vessel, wide gap of vessel wall clearance, extensive vascular leakage, poor integrity of issue structure, and so on. And these features cause selectively enhanced permeability and retention of lipid particles and macromolecular substances within solid tumors. The phenomenon of enhanced permeability and retention effect is called the EPR effect [41, 42]. The presence of high number of tumor-infiltrating lymphocytes (TIL) and extensive infiltration have been found as major indicators of favorable patient prognosis and positive therapeutic responses in treating several solid tumors [43], including colorectal cancer [44], lung cancer [45], and ovarian carcinomas [46, 47]. The EPR effect of solid tumor and the suppressive nature of the tumor microenvironment play important roles in impeding the infiltration into tumor tissues of effector T lymphocytes [48, 49]. Understanding and manipulating the factors contributing to the infiltration of T lymphocytes can be helpful to further improve the selective targeting of tumor tissues.
The process of T cells trafficking include rolling, adhesion, extravasation, and chemotaxis [50], and the trafficking of T cells to the tumor microenvironment is essential for the success of T cell based cancer immunotherapy. The clinical curative effect of T cell based immunotherapy against solid tumors has been more moderate than advanced melanoma or hematologic malignancies, overcoming hurdles of the migration of T cells is one of the major challenges in CAR-T cell immunotherapy, mismatching of chemokine-chemokine receptor pairs, down regulation of adhesion molecules, and aberrant vasculature may also contribute to the poor homing of T cells.
Studies have found that the CD3+, CD8+ T cell as well as B lymphocytes infiltrations are significantly correlated with the existence of tumor high endothelial venules (HEVs). Tumor HEVs are specifically located in lymphocytes concentrated areas, high density of tumor HEVs predicts low risk of relapse and metastasis [43, 51]. It is also an important access for lymphocyte infiltration into tumor sites, being associated with clinical prognosis makes it an indicator of tumor diagnosis and therapy. Over-expression of endothelin B receptor (ETBR) in tumor blood vessels is another limiting factor of lymphocyte infiltrating into tumor tissues through impeding the adhesion of lymphocyte to vascular endothelium [52]. And tumor angiogenesis has been found down-regulating endothelial cell-adhesion molecules, such as intercellular adhesion molecule 1 (ICAM-1) [53]. All the above limiting factors block the homing of T cells and thus impact the efficiency of tumor immunotherapy. Theoretically, ETBR blockade and ICAM-1 up-regulation could therapeutically promote T cells homing and enhance immunotherapy efficacy. ETBR inhibitor BQ-788 (a specific ETBR inhibitor peptide) has been revealed increasing T cell adhesion to human endothelium in vitro. Angiogenesis is a prerequisite for the outgrowth and metastasis of cancer cells [54], some angiogenic factors produced by tumor cells are responsible for the down-regulation of ICAM-1. For example, previous evidences have showed the up-regulation of ICAM-1 expression of human umbilical vein endothelial cells (HUVECs) after VEGF stimulation. But in contrast, prolonged stimulation (which occurs during the development of tumor) results in the down-regulation of ICAM-1 expression and leukocyte adhesion [55]. In addition, tumor necrosis factor-α (TNF-α) also induces the up-regulation of ICAM-1 [56]. Some relevant strategies to enhance the trafficking of CAR-T cells into solid tumors are discussed below.
3.1.1 Enhance CAR-T cells trafficking to tumor sites
A potential result of the special histopathological structure of solid tumor may lie in the observed lack of sufficient T cells within tumor tissues. Similarly, the insufficient migration of CAR-T cells to tumor sites also critically limits the efficacy of CAR-T cell immunotherapy against solid tumors. The limiting impact may result from unfavorable chemokine gradients, which means that tumor-specific T cells may lack the appropriate chemokine receptors for chemokines secreted by tumor cells [57].
Tumor-derived chemokines are also attractive targets for CAR-T cell immunotherapy due to their immune-modulatory effects: decrease the immunogenicity of tumors and the desensitization of chemokine receptors on T cells [58]. At the same time, tumor cells can utilize chemokines in this manner to provide autocrine growth signals and signals to enhance angiogenesis [59]. Thereby, once these tumor cells are eliminated, the remaining tumor cells will be more vulnerable. These above mechanisms envision the possibility to redirect T cells to predetermined targets through arming T cells with relevant chemokine receptors. Several studies have verified this principle through arming CAR-T cells with the expression of CXCR2 (CXCL1 receptor) [60], CCR4 (CCL17 receptor), Gro-a, CCL17 [61], and CCL2.
Nevertheless, tumor cells adopt multiple inhibitory strategies, it is challenging to derive CAR-T cells accommodating all the immune-modulatory genes which are required to overcome tumor inhibition while increasing CAR-T cell trafficking, survival, and safety [62]. Some researchers resorted to oncolytic viruses, which selectively infect, lyse, and replicate in malignant cells while sparing normal cells to solve this complex task by arming CAR-T cells with oncolytic virus expressing the chemokine RANTES and the cytokine IL-15 [63].
In addition, the difference of T cell administration also exerts an important impact on CAR-T cells expansion and effector differentiation. Studies have evaluated two different routes of CAR-T cells delivery, regional intra-pleural administration and conventional systemic intravenous of mesothelin-targeted M28z CAR-T cells, the former route presented robust T cells persistence and enhanced anti-tumor efficacy compared to the latter by circumventing obligate circulation and transient pulmonary sequestration [64]. Similarly, the intra-cerebral injection method of CAR-T cells has also been applied to treat glioblastoma with CAR-T cells to avoid the traffic blocking blood brain barrier [65]. The remarkable ability of regional delivery of CAR-T cells provides another approach to enhance functional T cell persistence and improve therapeutic efficacy through choosing favorable traffic route for CAR-T cells.
3.1.2 Cytokines released by engineered CAR-T cells promote tumor elimination
Inflammatory cells have a significant correlation with the growth and metastasis of cancer cells, indicating an improvement measure for CAR-T cell therapy by modulating tumor stroma through engineering CAR-T cells to secrete cytokines, such as interleukin-12 (IL-12) [66, 67]. The mechanism is that once activated by the CAR vector, T cells secrete IL-12, which can further activate innate immune cells response toward tumor cells that are invisible to CAR-T cells and subsequent inaccessible to antigen-directed immunotherapy, the process of T cells redirected for universal cytokine-mediated killing is also known as TRUCKs [68]. The TRUCKs have been demonstrated with remarkable therapeutic efficacy against tumors in pre-clinical models. So design T cells redirected by a tumor-targeting CAR and additionally engineered with a CAR-inducible cytokine cassette upon CAR engagement of cognate antigen (also termed as the fourth generation CARs by some scientists), such as the CAR-inducible IL-12 (iIL-12), which has been revealed with recruiting macrophage effect [69, 70]. This design can supplement the defect that CAR-T cell can't eliminate inaccessible tumor lesions, in a manner associated with reduced systemic toxicity.
3.1.3 Optimizing culture condition for CAR-T cells
Cytokine and stimulation conditions are indispensable ingredients in the process of CAR-T cells manufacturing, and several reports have indicated the influence of cytokines and growth conditions on the expansion and phenotype of immune T cells [71-73]. Thus, determining the choice of cytokines and optimizing the growth conditions are crucial for the expansion and related anti-tumor activity of CAR-T cells. The cytokines of IL-7 and IL-15 or IL-2 are mostly used as growth factors for the culture of CAR-T cells [74]. Studies have shown that IL-7 and IL-15 are superior to IL-2 for preserving CAR-T cell expansion in vitro and in vivo, CAR-T cells fed with IL-7 and IL-15 showed more sustained expansion and superior survival when exposed to serial antigens stimulation, and thus exhibited enhanced persistence and antitumor activity [75, 76]. In conclusion, these approaches lead to better living conditions for CAR-T cells, and can be translated into clinical immunotherapy.
3.2 Scarcity of specific antigen within solid tumors
The solid tumor heterogeneity in biological structure is a preponderant limiting factor of CAR-T cell immunotherapy for solid tumors. Tumor heterogeneity may result from subject factors and individual factors. The subject factors include the differences in cell origin and patient ethnicity, diversity that caused by genetic and epigenetic changes [77]. While the individual factors are mainly caused by tumor physiological heterogeneity among patients, intra-tumor heterogeneity, different distribution of an individual tumor, the presence of cancer stem cells or the direction of evolution [78]. Tumor heterogeneity results in that the immunotherapy target become specific to only a portion of tumor cells, which worsens the prognosis of patient and increases the recurrence and metastasis of cancer.
Therefore, the most advantageous method to treat solid tumors with CAR-T cells is to identify and project the specific cell surface antigens, but this optimal selection is severely hindered by the shortage of tumor specific antigens (TSA) under the circumstances of high heterogeneity. The posterior selection is tumor associated antigens (TAA) that relatively over expressed on the tumor cell surface, but CAR-T cells targeting TAAs may cause collateral damage to normal tissues. Therefore, new strategies improving the safety of clinical practice while maintaining the anti-tumor activity of CAR-T cells, including target tumor cell specific neoantigens that derived from somatic mutations of tumor cells (e.g. mutant EGFR variant III), target intracellular antigens (e.g.WT1, a peptide induced by Wilms' tumor gene 1), optimize CAR system with bi-signal independent pathways, apply suicide gene and other safe switches.
3.2.1 Engineered CARs targeting mutation phenotype of tumor cells
Epidermal growth factor receptor (EGFR) is a member of HER2 family, which frequently overexpressed in cancers and negatively correlated with clinical efficiency of treatment [79], and this makes it an inspiring research target. Researchers have found that 40-70% of brain tumors express mutant EGFR variant III (EGFRvIII) with a deletion of exons 2-7 of EGFR, which causes a defect in the extracellular ligand-binding domain and constitutive activation in a ligand-independent manner [80, 81]. Its specific expression on tumor cells, significant correlation with invasion and angiogenesis of tumors and patients' survival make EGFRvIII a novel promising target [82]. Arming polyclonal CTLs with tumor-specific TCR can avoid many obstacles in cellular immunotherapy, and this is called ''T-body'' approach [80, 83]. So the EGFRvIII targeting CAR system was utilized in the treatment of EGFRvIII expressing gliomas, and the generated T-body approach was able to secrete cytokines and lyse tumor cells in an EGFRvIII-dependent manner. This research brings us a new direction for CAR-T cell therapy, that is, to target specific tumor cell phenotypes induced by mutations of tumor cells.
3.2.2 Modify CARs to better target tumor associated antigens
Recognition of the peptide/MHC class I complexes endues T lymphocytes specific anti-tumor efficacy and enhanced cytokine secretion. Simultaneously, incorporation of the co-stimulatory signals or CD8+ adhesion molecules to the CARs can enhance the activation of T cells [84]. Peptide WT1, presented in the context of HLA-A* 02:01 (RMF/A2) [85], is an important immunologically validated oncogenic target with limited expression in normal tissues, while overexpressed in majority of leukemia and a wide range of solid tumors, especially mesothelioma and ovarian cancer [86, 87]. Therefore, CAR-T cells that targeting WT1 is a good improvement project. The modified CAR-T cells, containing the antigen recognition domain derived from fully human TCR-like ESK1 mAb (Called WT1 28z) have been found cytotoxic to primary AML bone marrow cells through targeting the intracellular oncoprotein WT1 [88]. Compared with targeting other antigens, WT1 28z CAR-T cells showed improved secretion of pro-inflammatory cytokines, such as IFN-γ, IL-2. And the therapeutic potential of TCR-like scFv CAR-T cells was proved able to be further enhanced by affinity maturation of the scFv fragment, such as TCR-like Q2L mAb derived scFv [89]. Hence, these researches indicate that improvement to the anti-tumor activity of CAR-T cells can be made through bidirectional modification of CAR-T cells to target intracellular antigens.
Mesothelin, a kind of cell surface glycoprotein with molecular weight of 40 kDa, is gaining much attention in clinical therapy of advanced solid tumors due to its high expression on the surface of numerous solid tumor cells [90]. As reported on the American Association for Cancer Research, a phase I study from Novartis and the University of Pennsylvania is ongoing, meso-CAR T cells were administered to advanced cancer patients who were no longer responding to multiple lines of prior therapies, the results indicated safety and pretty functionality in treated patients (table 1).
Mesothelin expression is also found in normal tissues, which raises a concern that meso-CAR T cells may damage healthy tissues and organs [91-93]. Researchers found some evidences showing that the meso-CAR T cells were detectable in the fluid around the heart of patients, but there were no related toxicities reported [94]. Although meso-CAR T cells program is still being evaluated, there is no doubt that mesothelin is another promising protein target and opens up the possibility that such an approach can benefit patients with various solid tumors.
3.2.3 Tuning affinity of CARs to selectively target tumor cells
In order to improve the specificity of CAR-T cells, separate dual CAR system was designed to recognize two different tumor antigens and separately transmit the first and second signals that are essential for T cell activation [95]. The co-transduced T cells maintain the therapeutic efficacy on the basis of the second generation and the third generation CARs, which empowers wider use of CAR-T cells and avoids potential safety issues.
Theoretically, the co-transduced T cells only destroy tumor cells that express both antigens (double positive) instead of these that express either antigen (single positive). However, studies have confirmed that co-transduced T cells are cytolytic against both single positive and double positive tumor cells. The most possible explanation is that the CARs transmitting the first signal have too robust stimulatory functions on T cells, and activate T cells even without the second signal. Thus in 2012, Kloss CC and colleagues reduced the affinity of the CARs that are responsible for transmitting the first signal in the dual CAR system and reported that co-transduced CAR-T cells did not exhibit cytolytic activity against single positive tumor cells but double positive tumor cells [96]. This research opened the door of adjusting the affinity to improve CAR-T cell specificity and thus safety performance.
Recently, the validity of the above strategy, through adjusting the affinity of the scFv components of CARs to selectively target tumor cells from normal cells, so as to reduce the off-target effects and improve the safety performance, was further verified. Tuning the functional affinity of CAR was performed to selectively target tumor cells overexpressing EGFR from normal cells based on the disparate density of EGFR expression [97]. The use of affinity-tuned scFv to target HER2 has found that decreasing the affinity of the scFv could significantly increase the therapeutic index of CAR-T cells [98]. Thus, another direction to improve safety performance of CAR-T cell based immunotherapy against solid tumors is tuning the functional affinity of CARs to discriminate the overexpressing tumor cells from normal tissues that express target at physiologic levels.
3.2.4 Applying suicide gene to enhance the safety of CAR-T cell therapy
Clinical application of CAR-T cells suggested that the anti-tumor efficacy is associated with some degree of toxicity [99], especially when targeting TAAs which are also expressed on normal cells. Therefore, strategies are needed to reverse any sign of toxicity effect.
Adding the “ideal” suicide gene to the construct of CARs can lead to selective ablation of gene modified T cells, thus prevent collateral damage to normal tissues or organs [100]. Therefore, CAR-T cell application with transgenic expression of one or two suicide genes is advisable, at the same time, the selective suicide gene would ensure the safety by irreversible elimination of specific part cells that are responsible for the unwanted toxicity. Currently, two validated suicide genes have been successfully used in clinical setting, herpes-simplex-thymidine-kinase (HSV-TK) and inducible-caspase-9 (iCasp9) to enhance the safety of CAR-T cell therapy against hematologic malignancies [101, 102]. So applying suicide gene modification to CAR-T cells may greatly increase the safety performance and clinical therapeutic efficacy.
3.3 Immunosuppressive environment within solid tumor
The clinical therapeutic efficacy of CAR-T cells in the treatment of solid tumors keeps a marginal characteristics compared with hematological malignancies, because the efficacy is significantly impeded by the strong immunosuppressive environment of solid tumors [4, 103]. The relative kinetics of CAR-T cells accumulation versus the rate of inactivation within solid tumors will ultimately determine the overall anti-tumor efficacy, and the balance will likely to be tumor-specific. The limiting factors that hinder T cells efficiency within solid tumor microenvironment are mainly from two aspects: 1) intrinsic microenvironment characteristics, such as hypoxia and low pH, the lack of arginine or tryptophan, inhibitory effects of tumor-derived cytokines, 2) inhibitory pathways against activated T cells, including intrinsic inhibitory pathways mediated by up-regulation of inhibitory receptors [104], intracellular inhibitory pathways to inhibit T cell receptor pathways [105], and effector functions after T cell activation [106]. Advanced generation of CAR-T cell inactivation is reversible within the solid tumor microenvironment by multiple mechanisms. Studies have showed that CAR-T cells undergo rapid loss of functional activity limited their therapeutic efficacy within solid tumor microenvironment, but this hypo-function was reversible when the CAR-T cells were isolated away from the tumor [107]. Whether this reversible characteristics can be attributed to the removal of inhibitory factors that reside in the tumor microenvironment is being investigated. Therefore, it is of great interests to optimize the efficacy of CAR-T cell therapy by combining with other treatments for solid tumors.
3.3.1 Engineer CARs to convert immunoregulatory signaling pathway
As mentioned above, solid tumors employ a variety of countermeasures to impair the function of CAR-T cells. Within the tumor microenvironment, the activated CAR-T cells are exhausted by many negative signal-regulated pathways, including cognate ligands reacting with their up-regulated inhibitory receptors expressed on T cells [108] and the lack of ligands for T cell co-stimulatory receptors [109]. Tumors exploit negative control signals to attenuate CAR-T cell responses, such as programmed death ligand 1 (PD-L1), which interacts with programmed death 1 (PD-1) expressed on activated T cells, and this exhausts CAR-T cells [110].
So developing a new engineering strategy to equip CAR-T cells with the capacity to convert tumor negative signal-regulated pathways into regulating pathways will be helpful, such as constructing a new chimera to convert cognate ligand into a ligand for T cell co-stimulation receptor by exchanging its transmembrane and cytoplasmic tail with that of CD28 or 4-1BB. The validated PD1: CD28 chimera was shown to efficiently convert PD-L1 into a co-stimulation ligand of primary human CD8+ CTL, resulting in enhanced cytokine secretion, increased proliferative capacity and augmented anti-tumor activity [111]. It is reasonable to speculate that genetic modification of CAR-T cells to express the above new type of chimera may greatly enhance the anti-tumor functions and also provide a platform to improve the clinical efficacy of CAR-T cell therapy under the immunosuppressive environment of solid tumors.
3.3.2 Engineer CARs to target stroma cells
With the exploration of solid tumor immunology, the tumor-associated stroma, occupying up to 90% of the tumor volume, has gained increasing attention for its role in initiating and sustaining tumor growth [112]. Cancer associated fibroblasts (CAFs), the principle ingredient of the tumor-associated stroma, play a preponderant influence in the formation of a highly protumorigenic and immunosuppressive microenvironment that mediates therapeutic resistance [113]. Additionally, immunotherapies with CAR-T cells targeting tumor- associated antigens (TAAs) often fail to eradicate CAFs, which support tumor progression directly through paracrine secretion of cytokines and growth factors.
Therefore, CAR-T cells targeting TAAs combine with CAR-T cells targeting CAFs may augment the anti-tumor function. The treatment by co-targeting CAFs in addition to cancer cells has been validated with significantly enhanced anti-tumor effects against solid tumors when compared with the treatment targeting CAFs or tumor cells alone. An immunotherapeutic target expressed on CAFs within a majority of solid tumors is necessary, fibroblast activation protein-α (FAP), a type 2 dipeptidyl peptidase, is a marker of a major subset of stromal cells in virtually carcinomas [114], making it an attractive therapeutic target. Studies have showed that genetically modified T cells with the expression of FAP-specific CAR can effectively recognize and kill FAP-positive tumor cells [115]. When combined with CAR-T cells targeting TAAs, FAP-specific CAR-T cells presented ever more attractive anti-tumor effects [116]. This novel combination provides another direction for solid tumor immunotherapy.
3.3.3 Combine with immune checkpoint inhibitors or cytokine expressing oncolytic virus
The field of cancer immunotherapy has considerably expanded with several new treatment options: immune checkpoint inhibitors, cancer vaccines, and adoptive T-cell immunotherapies. However, many drawbacks are still exist in efficacy, such as, immune checkpoint inhibitors are efficacious for just few patients with high mutation loaded melanoma and lung cancer, the efficacy in treating solid tumors with CAR-T cells is limited due to the unfavorable microenvironment [117, 118]. Thus, combination therapy of CAR-T cells with immune checkpoint inhibitors may be a solution. Recently, CAR-T cell therapy was combined with immune checkpoint inhibitors, which can create more favorable microenvironment by reducing its immunosuppressive effect to improve the efficacy of CAR-T cell therapy, and many clinical trials of combination therapy of CAR-T cells with checkpoint inhibitors are ongoing [19]. In addition to immune checkpoint inhibitors, CAR-T cell therapy has also been combined with oncolytic virus expressing the chemokine RANTES and the cytokine IL-15, and showed enhanced function of CAR-T cells by improving CAR-T cell trafficking and recruiting innate immune cells [119]. In conclusion, combination therapy of CAR-T cells with other treatments holds great potential for treating solid tumors.
4. Conclusions
CAR-T cell based immunotherapy has made a great success in treating B cell malignancies, but targeting solid tumors remains a tough task mainly due to the scarcity of TSAs and suppressive environment of solid tumors. However, researchers have been trying to improve the efficacy of CAR-T cell therapy against various solid tumors from many aspects including: 1) arming with cytokine or chemokine to enhance the infiltration of T cells and recruit other immune effectors; 2) optimizing the culture conditions to derive more potent CAR-T cells; 3) modifying the targeting system by changing the antigen recognition domain toward intracellular antigens or neo-antigens even bi-directional; 4) reducing the on-target/off tumor effect by applying dual targeting system, tuning the affinity of scFv fragments and applying safe switches; 5) combining with other treatments to eliminate tumor cells more thoroughly. Many clinical trials of CAR-T cell therapy targeting different antigens for treating solid tumors are ongoing. Nevertheless, mechanical studies on how CARs activate T cells, comparison between CAR and TCR, and optimization on each element in CARs are still needed to better apply CAR-T cell based immunotherapy in treating solid tumors.
Acknowledgements
This work was supported by the Chinese Key Project for Infectious Diseases (No. 2012ZX0002-014-005, 2013ZX10002-010-007), the Military Youth Project (No. 13QNP101), the State Project for Essential Drug Research and Development (2013ZX09102-060) and the Chinese State 863 projects (No. 2012AA020806).
Author contribution
Zhang H and Ye Z wrote the manuscript. Zhang H, Ye Z, Jin H and Qian Q conceived the study and revised the manuscript. Gang Y and Luo Z assisted searching the literature and provided conceptual input. All the authors participated in the discussion of the study, have read and approved the final manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Bouhassira DC, Thompson JJ, Davila ML. Using gene therapy to manipulate the immune system in the fight against B-cell leukemias. Expert opinion on biological therapy. 2015;15:403-16
2. Han EQ, Li X, Wang C, Li T, Han S. Chimeric antigen receptor-engineered T cells for cancer immunotherapy: progress and challenges. J Hematol Oncol. 2013;6:47
3. Maus MV, Grupp SA, Porter DL, June CH. Antibody-modified T cells: CARs take the front seat for hematologic malignancies. Blood. 2014;123:2625-35
4. Beavis PA, Slaney CY, Kershaw MH, Gyorki D, Neeson PJ, Darcy PK. Reprogramming the tumor microenvironment to enhance adoptive cellular therapy. Seminars in immunology: Elsevier. 2015
5. Chmielewski M, Hombach A, Heuser C, Adams GP, Abken H. T cell activation by antibody-like immunoreceptors: increase in affinity of the single-chain fragment domain above threshold does not increase T cell activation against antigen-positive target cells but decreases selectivity. The Journal of Immunology. 2004;173:7647-53
6. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. cell. 2011;144:646-74
7. Whilding LM, Maher J. CAR T-cell immunotherapy: The path from the by-road to the freeway? Molecular oncology. 2015;9:1994-2018
8. Shi H, Sun M, Liu L, Wang Z. Chimeric antigen receptor for adoptive immunotherapy of cancer: latest research and future prospects. Molecular cancer. 2014;13:219
9. Hudecek M, Sommermeyer D, Kosasih PL, Silva-Benedict A, Liu L, Rader C. et al. The non-signaling extracellular spacer domain of chimeric antigen receptors is decisive for in vivo antitumor activity. Cancer immunology research. 2014 canimm.0127.2014
10. Harris DT, Kranz DM. Adoptive T Cell Therapies: A Comparison of T Cell Receptors and Chimeric Antigen Receptors. Trends in Pharmacological Sciences. 2015
11. Dotti G, Gottschalk S, Savoldo B, Brenner MK. Design and development of therapies using chimeric antigen receptor-expressing T cells. Immunological reviews. 2014;257:107-26
12. Savoldo B, Ramos CA, Liu E, Mims MP, Keating MJ, Carrum G. et al. CD28 costimulation improves expansion and persistence of chimeric antigen receptor-modified T cells in lymphoma patients. The Journal of clinical investigation. 2011;121:1822
13. Bridgeman JS, Hawkins RE, Bagley S, Blaylock M, Holland M, Gilham DE. The optimal antigen response of chimeric antigen receptors harboring the CD3ζ transmembrane domain is dependent upon incorporation of the receptor into the endogenous TCR/CD3 complex. The Journal of Immunology. 2010;184:6938-49
14. Kershaw MH, Westwood JA, Slaney CY, Darcy PK. Clinical application of genetically modified T cells in cancer therapy. Clinical & Translational Immunology. 2014;3:e16
15. Kenderian SS, Ruella M, Gill S, Kalos M. Chimeric antigen receptor T-cell therapy to target hematologic malignancies. Cancer research. 2014;74:6383-9
16. Zhong X-S, Matsushita M, Plotkin J, Riviere I, Sadelain M. Chimeric Antigen Receptors Combining 4-1BB and CD28 Signaling Domains Augment PI3kinase/AKT/Bcl-XL Activation and CD8+ T Cell-mediated Tumor Eradication. Molecular Therapy. 2010;18:413-20
17. Long AH, Haso WM, Shern JF, Wanhainen KM, Murgai M, Ingaramo M. et al. 4-1BB costimulation ameliorates T cell exhaustion induced by tonic signaling of chimeric antigen receptors. Nature medicine. 2015
18. Catalán E, Charni S, Jaime P, Aguiló JI, Enríquez JA, Naval J. et al. MHC-I modulation due to changes in tumor cell metabolism regulates tumor sensitivity to CTL and NK cells. Oncoimmunology. 2015;4:e985924
19. Hillerdal V, Essand M. Chimeric Antigen Receptor-Engineered T Cells for the Treatment of Metastatic Prostate Cancer. BioDrugs. 2015;29:75-89
20. Duong CP, Yong CS, Kershaw MH, Slaney CY, Darcy PK. Cancer immunotherapy utilizing gene-modified T cells: from the bench to the clinic. Molecular immunology. 2015
21. Maude SL, Frey N, Shaw PA, Aplenc R, Barrett DM, Bunin NJ. et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. New England Journal of Medicine. 2014;371:1507-17
22. Boulassel M-R, Galal A. Immunotherapy for B-Cell Neoplasms using T Cells expressing Chimeric Antigen Receptors: From antigen choice to clinical implementation. Sultan Qaboos University medical journal. 2012;12:273
23. Singh N, Liu X, Hulitt J, Jiang S, June CH, Grupp SA. et al. Nature of tumor control by permanently and transiently modified GD2 chimeric antigen receptor T cells in xenograft models of neuroblastoma. Cancer Immunol Res. 2014;2:1059-70
24. Maus MV, Haas AR, Beatty GL, Albelda SM, Levine BL, Liu X. et al. T cells expressing chimeric antigen receptors can cause anaphylaxis in humans. Cancer immunology research. 2013;1:26-31
25. Hasegawa K, Nakamura T, Harvey M, Ikeda Y, Oberg A, Figini M. et al. The use of a tropism-modified measles virus in folate receptor-targeted virotherapy of ovarian cancer. Clinical Cancer Research. 2006;12:6170-8
26. Kandalaft LE, Powell Jr DJ, Coukos G. A phase I clinical trial of adoptive transfer of folate receptor-alpha redirected autologous T cells for recurrent ovarian cancer. J Transl Med. 2012;10:157
27. Park JR, DiGiusto DL, Slovak M, Wright C, Naranjo A, Wagner J. et al. Adoptive transfer of chimeric antigen receptor re-directed cytolytic T lymphocyte clones in patients with neuroblastoma. Molecular Therapy. 2007;15:825-33
28. Lamers CH, Langeveld SC, Groot-van Ruijven CM, Debets R, Sleijfer S, Gratama JW. Gene-modified T cells for adoptive immunotherapy of renal cell cancer maintain transgene-specific immune functions in vivo. Cancer Immunology, Immunotherapy. 2007;56:1875-83
29. Pule MA, Savoldo B, Myers GD, Rossig C, Russell HV, Dotti G. et al. Virus-specific T cells engineered to coexpress tumor-specific receptors: persistence and antitumor activity in individuals with neuroblastoma. Nature medicine. 2008;14:1264-70
30. Louis CU, Savoldo B, Dotti G, Pule M, Yvon E, Myers GD. et al. Antitumor activity and long-term fate of chimeric antigen receptor-positive T cells in patients with neuroblastoma. Blood. 2011;118:6050-6
31. Petrausch U, Schuberth PC, Hagedorn C, Soltermann A, Tomaszek S, Stahel R. et al. Re-directed T cells for the treatment of fibroblast activation protein (FAP)-positive malignant pleural mesothelioma (FAPME-1). BMC cancer. 2012;12:615
32. Peinert S, Prince H, Guru P, Kershaw M, Smyth M, Trapani J. et al. Gene-modified T cells as immunotherapy for multiple myeloma and acute myeloid leukemia expressing the Lewis Y antigen. Gene Ther. 2010;17:678-86
33. Ritchie DS, Neeson PJ, Khot A, Peinert S, Tai T, Tainton K. et al. Persistence and efficacy of second generation CAR T cell against the LeY antigen in acute myeloid leukemia. Molecular Therapy. 2013;21:2122-9
34. Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, Rosenberg SA. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Molecular Therapy. 2010;18:843-51
35. Zhao Y, Moon E, Carpenito C, Paulos CM, Liu X, Brennan AL. et al. Multiple injections of electroporated autologous T cells expressing a chimeric antigen receptor mediate regression of human disseminated tumor. Cancer research. 2010;70:9053-61
36. Till BG, Jensen MC, Wang J, Qian X, Gopal AK, Maloney DG. et al. CD20-specific adoptive immunotherapy for lymphoma using a chimeric antigen receptor with both CD28 and 4-1BB domains: pilot clinical trial results. Blood. 2012;119:3940-50
37. Guest RD, Kirillova N, Mowbray S, Gornall H, Rothwell DG, Cheadle EJ. et al. Definition and application of good manufacturing process-compliant production of CEA-specific chimeric antigen receptor expressing T-cells for phase I/II clinical trial. Cancer immunology, immunotherapy: CII. 2014;63:133-45
38. Sadelain M. T-cell engineering for cancer immunotherapy. The Cancer Journal. 2009;15:451-5
39. Gill S, Porter DL. CAR-modified anti-CD19 T cells for the treatment of B-cell malignancies: rules of the road. Expert opinion on biological therapy. 2014;14:37-49
40. Trifilio S, Bennett C, Yarnold P, McKoy J, Parada J, Mehta J. et al. Breakthrough zygomycosis after voriconazole administration among patients with hematologic malignancies who receive hematopoietic stem-cell transplants or intensive chemotherapy. Bone marrow transplantation. 2007;39:425-9
41. Fang J, Nakamura H, Maeda H. The EPR effect: unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Advanced drug delivery reviews. 2011;63:136-51
42. Maeda H, Wu J, Sawa T, Matsumura Y, Hori K. Tumor vascular permeability and the EPR effect in macromolecular therapeutics: a review. Journal of controlled release. 2000;65:271-84
43. Martinet L, Garrido I, Filleron T, Le Guellec S, Bellard E, Fournie J-J. et al. Human solid tumors contain high endothelial venules: association with T-and B-lymphocyte infiltration and favorable prognosis in breast cancer. Cancer research. 2011;71:5678-87
44. Pagès F, Berger A, Camus M, Sanchez-Cabo F, Costes A, Molidor R. et al. Effector memory T cells, early metastasis, and survival in colorectal cancer. New England Journal of Medicine. 2005;353:2654-66
45. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62-8
46. Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G. et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. New England Journal of Medicine. 2003;348:203-13
47. Hamanishi J, Mandai M, Iwasaki M, Okazaki T, Tanaka Y, Yamaguchi K. et al. Programmed cell death 1 ligand 1 and tumor-infiltrating CD8+ T lymphocytes are prognostic factors of human ovarian cancer. Proceedings of the National Academy of Sciences. 2007;104:3360-5
48. Yin H, Liao L, Fang J. Enhanced permeability and retention (EPR) effect based tumor targeting: the concept, application and prospect. JSM Clin Oncol Res. 2014;2:1010-4
49. Rahir G, Moser M. Tumor microenvironment and lymphocyte infiltration. Cancer Immunology, Immunotherapy. 2012;61:751-9
50. Slaney CY, Kershaw MH, Darcy PK. Trafficking of T Cells into Tumors. Cancer research. 2014;74:7168-74
51. Martinet L, Le Guellec S, Filleron T, Lamant L, Meyer N, Rochaix P. et al. High endothelial venules (HEVs) in human melanoma lesions: major gateways for tumor-infiltrating lymphocytes. Oncoimmunology. 2012;1:829-39
52. Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K. et al. Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy. Nature medicine. 2008;14:28-36
53. Dirkx AE, oude Egbrink MG, Kuijpers MJ, van der Niet ST, Heijnen VV, Bouma-ter Steege JC. et al. Tumor angiogenesis modulates leukocyte-vessel wall interactions in vivo by reducing endothelial adhesion molecule expression. Cancer research. 2003;63:2322-9
54. Griffioen AW, Molema G. Angiogenesis: potentials for pharmacologic intervention in the treatment of cancer, cardiovascular diseases, and chronic inflammation. Pharmacological reviews. 2000;52:237-68
55. Griffioen AW, Damen CA, Martinotti S, Blijham GH, Groenewegen G. Endothelial intercellular adhesion molecule-1 expression is suppressed in human malignancies: the role of angiogenic factors. Cancer research. 1996;56:1111-7
56. J Tuorkey M, K Abdul-Aziz K, A Zidan A-A. Active immunization against tumor necrosis factor-alpha decreases proinflammatory cytokines, oxidative stress mediators and adhesion molecules risk factors in streptozotocin-induced diabetic rats. Endocrine, Metabolic & Immune Disorders-Drug Targets (Formerly Current Drug Targets-Immune, Endocrine & Metabolic Disorders). 2013;13:269-74
57. Karin N, Wildbaum G. The Role of Chemokines in Shaping the Balance Between CD4+ T Cell Subsets and Its Therapeutic Implications in Autoimmune and Cancer Diseases. Frontiers in immunology. 2015:6
58. Springer TA. Traffic signals for lymphocyte recirculation and leukocyte emigration: the multistep paradigm. cell. 1994;76:301-14
59. Kershaw MH, Wang G, Westwood JA, Pachynski RK, Tiffany HL, Marincola FM. et al. Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2. Hum Gene Ther. 2002;13:1971-80
60. Craddock JA, Lu A, Bear A, Pule M, Brenner MK, Rooney CM. et al. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. Journal of immunotherapy (Hagerstown, Md: 1997). 2010;33:780
61. Di Stasi A, De Angelis B, Rooney CM, Zhang L, Mahendravada A, Foster AE. et al. T lymphocytes coexpressing CCR4 and a chimeric antigen receptor targeting CD30 have improved homing and antitumor activity in a Hodgkin tumor model. Blood. 2009;113:6392-402
62. Curran KJ, Pegram HJ, Brentjens RJ. Chimeric antigen receptors for T cell immunotherapy: current understanding and future directions. The journal of gene medicine. 2012;14:405-15
63. Nishio N, Diaconu I, Liu H, Cerullo V, Caruana I, Hoyos V. et al. Armed Oncolytic Virus Enhances Immune Functions of Chimeric Antigen Receptor-Modified T Cells in Solid Tumors. Cancer research. 2014;74:5195-205
64. Adusumilli PS, Cherkassky L, Villena-Vargas J, Colovos C, Servais E, Plotkin J. et al. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Science translational medicine. 2014;6:261ra151-261ra151
65. Choi BD, Suryadevara CM, Gedeon PC, Herndon II JE, Sanchez-Perez L, Bigner DD. et al. Intracerebral delivery of a third generation EGFRvIII-specific chimeric antigen receptor is efficacious against human glioma. Journal of Clinical Neuroscience. 2014;21:189-90
66. Pegram HJ, Lee JC, Hayman EG, Imperato GH, Tedder TF, Sadelain M. et al. Tumor-targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119:4133-41
67. Kerkar SP, Muranski P, Kaiser A, Boni A, Sanchez-Perez L, Yu Z. et al. Tumor-specific CD8+ T cells expressing interleukin-12 eradicate established cancers in lymphodepleted hosts. Cancer research. 2010;70:6725-34
68. Chmielewski M, Hombach AA, Abken H. Of CARs and TRUCKs: chimeric antigen receptor (CAR) T cells engineered with an inducible cytokine to modulate the tumor stroma. Immunological reviews. 2014;257:83-90
69. Chmielewski M, Kopecky C, Hombach AA, Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively Muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer research. 2011;71:5697-706
70. Zhang L, Kerkar SP, Yu Z, Zheng Z, Yang S, Restifo NP. et al. Improving adoptive T cell therapy by targeting and controlling IL-12 expression to the tumor environment. Molecular Therapy. 2011;19:751-9
71. Gargett T, Brown MP. Different cytokine and stimulation conditions influence the expansion and immune phenotype of third-generation chimeric antigen receptor T cells specific for tumor antigen GD2. Cytotherapy. 2015;17:487-95
72. Porter DL, Levine BL, Bunin N, Stadtmauer EA, Luger SM, Goldstein S. et al. A phase 1 trial of donor lymphocyte infusions expanded and activated ex vivo via CD3/CD28 costimulation. Blood. 2006;107:1325-31
73. Hamid O, Robert C, Daud A, Hodi FS, Hwu W-J, Kefford R. et al. Safety and tumor responses with lambrolizumab (anti-PD-1) in melanoma. New England Journal of Medicine. 2013;369:134-44
74. Ma A, Koka R, Burkett P. Diverse functions of IL-2, IL-15, and IL-7 in lymphoid homeostasis. Annu Rev Immunol. 2006;24:657-79
75. Xu Y, Zhang M, Ramos CA, Durett A, Liu E, Dakhova O. et al. Closely related T-memory stem cells correlate with in vivo expansion of CAR. CD19-T cells and are preserved by IL-7 and IL-15. Blood. 2014;123:3750-9
76. Caserta S, Alessi P, Basso V, Mondino A. IL-7 is superior to IL-2 for ex vivo expansion of tumour-specific CD4+ T cells. European journal of immunology. 2010;40:470-9
77. Shen H, Laird PW. Interplay between the cancer genome and epigenome. cell. 2013;153:38-55
78. Roessler S, Budhu A, Wang XW. Deciphering cancer heterogeneity: the biological space. Frontiers in cell and developmental biology. 2014:2
79. Panousis C, Rayzman V, Johns T, Renner C, Liu Z, Cartwright G. et al. Engineering and characterisation of chimeric monoclonal antibody 806 (ch806) for targeted immunotherapy of tumours expressing de2-7 EGFR or amplified EGFR. British journal of cancer. 2005;92:1069-77
80. Ohno M, Natsume A, Ichiro Iwami K, Iwamizu H, Noritake K, Ito D. et al. Retrovirally engineered T-cell-based immunotherapy targeting type III variant epidermal growth factor receptor, a glioma-associated antigen. Cancer science. 2010;101:2518-24
81. Sugawa N, Ekstrand AJ, James CD, Collins VP. Identical splicing of aberrant epidermal growth factor receptor transcripts from amplified rearranged genes in human glioblastomas. Proceedings of the National Academy of Sciences. 1990;87:8602-6
82. Yamazaki H, Ohba Y, Tamaoki N, Shibuya M. A deletion mutation within the ligand binding domain is responsible for activation of epidermal growth factor receptor gene in human brain tumors. Cancer science. 1990;81:773-9
83. Eshhar Z. The T-body approach: redirecting T cells with antibody specificity. Therapeutic Antibodies. Springer. 2008 p. 329-42
84. Watanabe K, Terakura S, Martens AC, van Meerten T, Uchiyama S, Imai M. et al. Target Antigen Density Governs the Efficacy of Anti-CD20-CD28-CD3 ζ Chimeric Antigen Receptor-Modified Effector CD8+ T Cells. The Journal of Immunology. 2015;194:911-20
85. Veomett N, Dao T, Liu H, Xiang J, Pankov D, Dubrovsky L. et al. Therapeutic efficacy of an Fc-enhanced TCR-like antibody to the intracellular WT1 oncoprotein. Clinical Cancer Research. 2014;20:4036-46
86. Krug LM, Dao T, Brown AB, Maslak P, Travis W, Bekele S. et al. WT1 peptide vaccinations induce CD4 and CD8 T cell immune responses in patients with mesothelioma and non-small cell lung cancer. Cancer Immunology, Immunotherapy. 2010;59:1467-79
87. Dohi S, Ohno S, Ohno Y, Takakura M, Kyo S, Soma G-I. et al. WT1 peptide vaccine stabilized intractable ovarian cancer patient for one year: a case report. Anticancer research. 2011;31:2441-5
88. Rafiq S, Dao T, Liu C, Scheinberg DA, Brentjens RJ. Engineered T cell receptor-mimic antibody,(TCRm) chimeric antigen receptor (CAR) T cells against the intracellular protein Wilms tumor-1 (WT1) for treatment of hematologic and solid cancers. Blood. 2014;124:2155 -
89. Zhao Q, Ahmed M, Tassev D, Hasan A, Kuo T, Guo H. et al. Affinity maturation of T-cell receptor-like antibodies for Wilms tumor 1 peptide greatly enhances therapeutic potential. Leukemia. 2015
90. Tchou J, Wang L-C, Selven B, Zhang H, Conejo-Garcia J, Borghaei H. et al. Mesothelin, a novel immunotherapy target for triple negative breast cancer. Breast cancer research and treatment. 2012;133:799-804
91. Servais EL, Colovos C, Rodriguez L, Bograd AJ, Nitadori J-i, Sima C. et al. Mesothelin overexpression promotes mesothelioma cell invasion and MMP-9 secretion in an orthotopic mouse model and in epithelioid pleural mesothelioma patients. Clinical Cancer Research. 2012;18:2478-89
92. Rizk NP, Servais EL, Tang LH, Sima CS, Gerdes H, Fleisher M. et al. Tissue and serum mesothelin are potential markers of neoplastic progression in Barrett's associated esophageal adenocarcinoma. Cancer Epidemiology Biomarkers & Prevention. 2012;21:482-6
93. Kachala SS, Bograd AJ, Villena-Vargas J, Suzuki K, Servais EL, Kadota K. et al. Mesothelin overexpression is a marker of tumor aggressiveness and is associated with reduced recurrence-free and overall survival in early-stage lung adenocarcinoma. Clinical Cancer Research. 2014;20:1020-8
94. Pastan I, Hassan R. Discovery of mesothelin and exploiting it as a target for immunotherapy. Cancer research. 2014;74:2907-12
95. Wilkie S, van Schalkwyk MC, Hobbs S, Davies DM, van der Stegen SJ, Pereira ACP. et al. Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. Journal of clinical immunology. 2012;32:1059-70
96. Kloss CC, Condomines M, Cartellieri M, Bachmann M, Sadelain M. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nature biotechnology. 2013;31:71-5
97. Caruso HG, Hurton LV, Najjar A, Rushworth D, Ang S, Olivares S. et al. Tuning Sensitivity of CAR to EGFR Density Limits Recognition of Normal Tissue While Maintaining Potent Antitumor Activity. Cancer research. 2015;75:3505-18
98. Liu X, Jiang S, Fang C, Yang S, Olalere D, Pequignot EC. et al. Affinity-Tuned ErbB2 or EGFR Chimeric Antigen Receptor T Cells Exhibit an Increased Therapeutic Index against Tumors in Mice. Cancer research. 2015;75:3596-607
99. Casucci M, Bondanza A. Suicide gene therapy to increase the safety of chimeric antigen receptor-redirected T lymphocytes. Journal of Cancer. 2011;2:378
100. Jones BS, Lamb LS, Goldman F, Di Stasi A. Improving the safety of cell therapy products by suicide gene transfer. Frontiers in pharmacology. 2014:5
101. Gargett T, Brown MP. The inducible caspase-9 suicide gene system as a “safety switch” to limit on-target, off-tumor toxicities of chimeric antigen receptor T cells. Frontiers in pharmacology. 2014:5
102. Ciceri F, Bonini C, Stanghellini MTL, Bondanza A, Traversari C, Salomoni M. et al. Infusion of suicide-gene-engineered donor lymphocytes after family haploidentical haemopoietic stem-cell transplantation for leukaemia (the TK007 trial): a non-randomised phase I-II study. The lancet oncology. 2009;10:489-500
103. Shiao SL, Ganesan AP, Rugo HS, Coussens LM. Immune microenvironments in solid tumors: new targets for therapy. Genes & development. 2011;25:2559-72
104. Park HJ, Kusnadi A, Lee E-J, Kim WW, Cho BC, Lee IJ. et al. Tumor-infiltrating regulatory T cells delineated by upregulation of PD-1 and inhibitory receptors. Cellular immunology. 2012;278:76-83
105. Zheng Y, Zha Y, Gajewski TF. Molecular regulation of T-cell anergy. EMBO reports. 2008;9:50-5
106. Moon EK, Wang L-C, Dolfi DV, Wilson CB, Ranganathan R, Sun J. et al. Multifactorial T-cell hypofunction that is reversible can limit the efficacy of chimeric antigen receptor-transduced human T cells in solid tumors. Clinical Cancer Research. 2014;20:4262-73
107. Beatty GL, Moon EK. Chimeric antigen receptor T cells are vulnerable to immunosuppressive mechanisms present within the tumor microenvironment. Oncoimmunology. 2014;3:e970027
108. Gajewski TF, Meng Y, Blank C, Brown I, Kacha A, Kline J. et al. Immune resistance orchestrated by the tumor microenvironment. Immunological reviews. 2006;213:131-45
109. Anderson RC, Anderson DE, Elder JB, Brown MD, Mandigo CE, Parsa AT. et al. Lack of B7 expression, not human leukocyte antigen expression, facilitates immune evasion by human malignant gliomas. Neurosurgery. 2007;60:1129-36
110. Tewalt EF, Cohen JN, Rouhani SJ, Guidi CJ, Qiao H, Fahl SP. et al. Lymphatic endothelial cells induce tolerance via PD-L1 and lack of costimulation leading to high-level PD-1 expression on CD8 T cells. Blood. 2012;120:4772-82
111. Prosser ME, Brown CE, Shami AF, Forman SJ, Jensen MC. Tumor PD-L1 co-stimulates primary human CD8+ cytotoxic T cells modified to express a PD1: CD28 chimeric receptor. Molecular immunology. 2012;51:263-72
112. Santos AM, Jung J, Aziz N, Kissil JL, Puré E. Targeting fibroblast activation protein inhibits tumor stromagenesis and growth in mice. The Journal of clinical investigation. 2009;119:3613
113. Kakarla S, Chow KK, Mata M, Shaffer DR, Song X-T, Wu M-F. et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Molecular Therapy. 2013;21:1611-20
114. Saadi A, Shannon NB, Lao-Sirieix P, O'Donovan M, Walker E, Clemons NJ. et al. Stromal genes discriminate preinvasive from invasive disease, predict outcome, and highlight inflammatory pathways in digestive cancers. Proceedings of the National Academy of Sciences. 2010;107:2177-82
115. Kraman M, Bambrough PJ, Arnold JN, Roberts EW, Magiera L, Jones JO. et al. Suppression of antitumor immunity by stromal cells expressing fibroblast activation protein-α. Science. 2010;330:827-30
116. Lo A, Wang L-CS, Scholler J, Monslow J, Avery D, Newick K. et al. Tumor-promoting desmoplasia is disrupted by depleting FAP-expressing stromal cells. Cancer research. 2015 canres.3041.2014
117. Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science. 2015;348:62-8
118. Schumacher TN, Schreiber RD. Neoantigens in cancer immunotherapy. Science. 2015;348:69-74
119. Nishio N, Dotti G. Oncolytic virus expressing RANTES and IL-15 enhances function of CAR-modified T cells in solid tumors. Oncoimmunology. 2015;4:e988098
Author contact
![]() Corresponding authors: Qi-Jun Qian, Tel: + 86-21-81875371; Fax: + 86-21-65580677; E-mail: qianqjcn; Hua-Jun Jin, Tel: + 86-21-81875372; E-mail: hj-jincom.
Corresponding authors: Qi-Jun Qian, Tel: + 86-21-81875371; Fax: + 86-21-65580677; E-mail: qianqjcn; Hua-Jun Jin, Tel: + 86-21-81875372; E-mail: hj-jincom.