Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Results and Discussion
Materials and Methods
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(8):911-916. doi:10.7150/ijbs.15172 This issue Cite
Research Paper
Generation of a Mouse Full-length Balancer with Versatile Cassette-shuttling Selection Strategy
Zhisheng Ye1*, Lei Sun1*, Rongbo Li1*, Min Han1,2, Yuan Zhuang1,3, Xiaohui Wu1 ![]() , Tian Xu1,4
, Tian Xu1,4 ![]()
1. State Key Laboratory of Genetic Engineering and National Center for International Research of Development and Disease, Collaborative Innovation Center for Genetics and Development, Fudan-Yale Biomedical Research Center, Institute of Developmental Biology and Molecular Medicine, Fudan University, Shanghai 200433, China.
2. Howard Hughes Medical Institute, Department of Molecular, Cellular, Developmental Biology, University of Colorado at Boulder, Boulder, CO 80309, USA.
3. Department of Immunology, Duke University Medical Center, Durham, NC 27710, USA.
4. Howard Hughes Medical Institute, Department of Genetics, Yale University School of Medicine, New Haven, CT 06536, USA.
* These authors contribute equally to this work.
Received 2016-2-2; Accepted 2016-4-20; Published 2016-6-7
Abstract

Balancer chromosomes are important tools for a variety of genetic manipulations in lower model organisms, owing to their ability to suppress recombination. In mouse, however, such effort has not been accomplished, mostly due to the size of the chromosomes and the complexity of multiple step chromosomal engineering. We developed an effective and versatile cassette-shuttling selection (CASS) strategy involving only two selection markers to achieve the sequential production of multiple large inversions along the chromosome. Using this strategy, we successfully generated the first full-length balancer in mice and showed that Balancer 17M-GFP can efficiently suppress recombination. Our study has not only generated a useful genetic resource, but also provided a strategy for constructing mammalian balancer chromosomes.
Keywords: Mouse full-length balancer chromosome, cassette-shuttling selection (CASS) strategy, 17M-GFP.
Introduction
A Drosophila chromosome carrying an inversion was first used by Muller in 1920s as a balancer for genetic manipulation [1]. An efficient balancer, however, carries multiple inversions along its chromosome (full-length balancer), which suppresses recombination between homologous chromosomes. Balancers with multiple inversions had been constructed for each major Drosophila chromosome in 1950s [2]. They have since been continuously used for a variety of manipulations including genetic screens, stock maintenance, phenotypic and epistasis analysis, and constructing strains with specific genetic backgrounds.
In mouse, balancers for chromosome 4 and 11, which each carries a single regional inversion, have been generated using Cre-loxP recombination and successfully employed in genetic screens [3-7]. However, mouse chromosomes are much longer than Drosophila ones. It is not clear how many inversions are needed to sufficiently suppress recombination. Furthermore, a new approach is needed to efficiently accomplish the genetic manipulations and selection for multiple large inversions. For these reasons, a full-length balancer has not been constructed for any mouse chromosome. Here we present an effective and versatile cassette-shuttling selection (CASS) strategy using two drug selection markers to achieve the sequential integration of knock-in constructs and production of multiple large inversions along the chromosome. To demonstrate the feasibility of the approach, we constructed a full-length balancer for mouse chromosome 17 by introducing three continuous large inversions. We showed that this GFP-marked full-length balancer, 17M-GFP, can efficiently suppress recombination.
Mouse chromosome 17 is gene rich, which contains 3% of the sequence and yet nearly 5% of the known genes. Furthermore, it possesses large syntenies homologous to human sequences including the major histocompatibility complex. Thus, it is an attractive target for large-scale mutagenesis screens [8, 9].
Results and Discussion
Though the longest recombination ever generated by Cre-loxP system in mouse cell line is 60 cM [10], the chromosomal inversion known to suppress meiotic recombination in mice spans 30 cM [5]. To generate balancers for all mouse chromosomes including those that are over 100 cM, we designed an approach to generate three inversions per chromosome. For chromosome 17, we divided it into three major regions, Distal (28.8 cM or 32 Mb), Internal (16.5 cM or 32 Mb) and Proximal (12.0 cM or 24 Mb), for generating inversions (Fig. 1A). The full-length balancer with triple inversions was expected to efficiently suppress recombination.
The cassette-shuttling selection (CASS) strategy and flowchart for engineering the full-length mouse chromosome 17 balancer, 17M-GFP. A: Genetic and physical map of three inversions and four selected breakpoints on wild-type chromosome 17. B: The CASS strategy-based multi-step process used for engineering the distal inversion. Each step rendered ES cells a different drug resistance, Neo+ (G418 resistance) or Puro+ (puromycin resistance), as labeled on the left. C: Workflow used for generating full-length balancer. Green mouse indicates GFP visible marker. The loxP sites (red triangles); Cytomegalovirus immediate-early enhancer and promoter (CMV); Phosphoglycerate kinase 1 promoter (PGK); The coding region of neomycin resistance gene (Neo); The coding region of puromycin resistance gene (Puro); The reversed order of letters in abbreviations indicate the inverted orientation of the corresponding gene or regulatory element. Empty boxes indicate inactive elements while solid ones indicate active components in the shuttling cassettes.
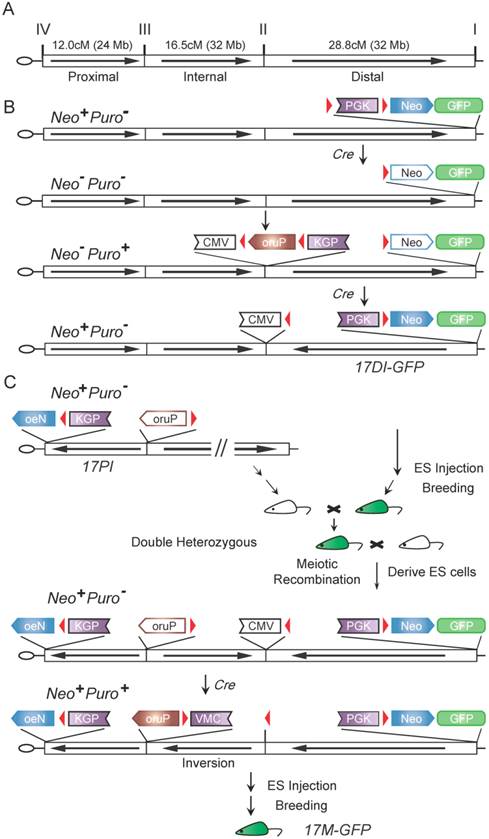
To produce a single inversion, two loxP sites have to be engineered into the chromosome. Targeting two loxP sites and subsequently selecting inversion normally require three different drug selection markers [3]. For generating multiple inversions on the same chromosome, it would become unrealistic as too many different drug selection markers are required. To solve this problem, we developed the cassette-shuttling selection (CASS) strategy, which requires only two drug selection markers. Specifically, either the promoter or the coding region of the neomycin (Neo) or puromycin (Puro) drug resistant gene is flanked by two loxP sites to form four shuttling cassettes (Fig. S2, Fig. 1B-D; >PGK>Neo; PGK>Puro>VMC; PGK>Puro>; oruP>PGK>Neo). Upon Cre expression, recombination between loxP sites could inactivate a drug selection marker in a knock-in shuttling cassette. Subsequently, recombination induced chromosome inversion could reactivate a drug selection marker (Fig. 1B-C; Fig. S2). The strategy is designed such that the process could be repeated for generating the next inversion. Thus, this CASS strategy could accomplish targeting constructs at multiple sites and subsequently generating multiple inversions on the same chromosome with only two drug selection markers.
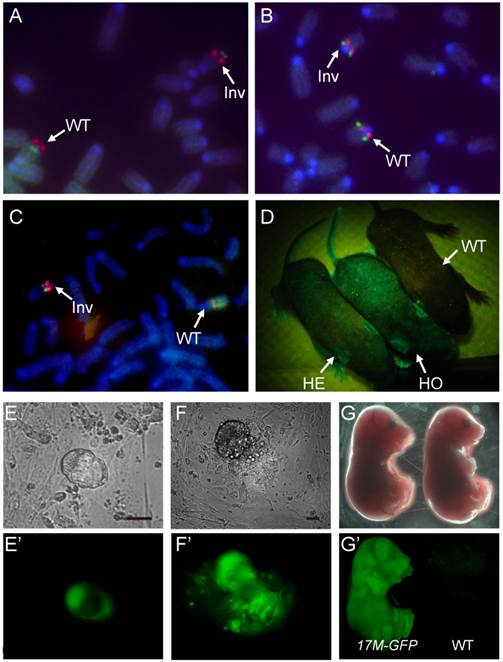
Using the CASS strategy, we first generated the distal inversion for chromosome 17. Drosophila balancers often carry lethal mutations, which permits self-sustainable stock keeping since individuals homozygous for a balancer will be eliminated. However, mouse breeding is expansive. Mouse litter is small in size, and individuals carrying fluorescence or hair markers can be visually identified in the process of changing cages. Mice homozygous for a balancer are useful resource, which can be used for other breeding purposes. Thus, in choosing targeting sites or inversion breaking points, we preferred intergenic regions to avoid potential lethality. On the other hand, we did adopt the dominant marker strategy using a GFP transgene for visually tracking the balancer. Specifically, at site I (93.41 Mb) on chromosome 17, the >PGK>Neo shuttling cassette and a constitutive GFP expression transgene were knocked in 129S1/SvImJ embryonic stem cells (ES cells) with G418/Neo resistance (Neo+) selection (Fig. 1B & Fig. S1). After successful knock-in, a Cre plasmid was then electroporated into the targeted ES cells to excise the PGK promoter, thus inactivating the resistant gene (Neo-). Subsequently, at site II (61.58 Mb), the PGK>Puro>VMC shuttling cassette was knocked in with Puro resistance selection (Puro+) (Fig. 1B & Fig. S1). The loxP sequences on site I and II were designed to be oriented at opposite directions so that inversion could be induced. Upon Cre expression, the long range inversion between loxP sequences at site I and II can be selected with reactivation of the PGK-Neo gene (Neo+, Puro-; Fig. 1B). The Puro shuttling cassette at site II was also inactivated during the process. Together with the inverted CMV promoter at site II, they laid down the ground for future selection of internal inversion (see below). 16 of the 24 Neo+ clones were confirmed as positive for the inversion by PCR. We also performed fluorescence in situ hybridization (FISH) on metaphase chromosomes of two positive clones to confirm the inversion and correct karyotype (Fig. 2A).
ES cells and mouse with Distal, Proximal and internal inversions.A,B,C: FISH analysis of ES cells carrying the distal(A), the proximal(B) and the internal(C) inversions. The probes were described in Methods. D: Mice heterozygous or homozygous for 17M-GFP can be identified by lamination of a ultraviolet lamp. GFP signal can be detected as early as blastocysts stage (E), in all three germ layers outgrown from inner cell mass (F) and late embryos (G),as compared to wild type (E'-G'). Scale bars are 100 µm.
A mouse line carrying the distal inversion on chromosome 17 was established in 129S1/SvImJ background by a standard ES cell injection procedure (17DI-GFP). Both heterozygous and homozygous 17DI-GFP mice are viable and fertile. To determine whether the distal inversion chromosome suppresses crossovers, the 17DI-GFP mice were crossed to C57BL/6 wild-type animals and the F1 heterozygotes were further back crossed to C57BL/6 animals (Fig. S4). Recombination in the inversion region could be detected by the disassociation of the PCR products from the inversion sites in the F2 progenies (Fig. 3A). As the distal region is the longest among the three inversions, we also used a SNP marker to detect possible double crossover events within the region (Fig. 3A). Among 146 F2 progenies, no evidence of recombination in the distal inverted region was detected (Fig. 3A).
Suppression of recombination by inversions on chromosome 17. Expected and detected recombination ratio in heterozygous mice carrying the 17DI-GFP (A), the 17PI (B), and 17M-GFP(C). Recombination was effectively suppressed by the inversions. Circles represent different primer pairs, which were used for detecting recombination (see Supplementary Table S1). Expected recombination ratio was inferred from genetic distance between variant sites (see Materials and Methods).

Using a similar strategy, we generated the proximal inversion (Fig. S2). The oruP>PGK>Neo shuttling cassette was knocked in on site IV (5.77 Mb) with Neo selection (Neo+) (Fig. S1 and S2). The Neo resistant gene was then inactivated (Neo-) by deleting the PGK promoter (Fig. S2). Subsequently, the PGK>Puro> shuttling cassette was knocked in on site III (29.72 Mb) with puromycin selection (Puro+) (Fig. S1 and S2). Again, the loxP sequences of two sites are at opposite directions, which allowed the induction of the proximal inversion with Neo selection (reconstitution of the PGK>Neo cassette, Neo+) (Fig. S2). PCR results indicated that 40 of the 45 Neo+ clones carried the inversions. FISH performed on the two positive clones also confirmed the inversion and correct karyotype (Fig. 2B). During the process of generating the inversion, the Puro shuttling cassette at site III was also inactivated while the Puro coding cassette at site VI was relocated to site III. This laid the ground for future selection of the internal inversion. A mouse line carrying the proximal inversion on chromosome 17 was established in 129S1/SvImJ background (17PI). The 17PI mice were bred to FVB/NJ wild-type animals for detecting possible crossovers between chromosomes (Fig. S4). No crossover event was detected in 150 F2 progenies for the proximal inversion (Fig. 3B).
To bring the distal and proximal inversions onto the same chromosome, the 17DI-GFP and17PI mice were bred together and the double heterozygotes were then mated to wild-type 129S1/SvImJ animals (Fig. 1C). The GFP-positive ES cell lines were established from the F2 blastocysts and two ES cell lines carrying the recombinant chromosome with both inversions were identified by PCR and confirmed by FISH. Finally, the internal inversion was generated in these cells by inducing recombination between site II and III and selected by the reactivated Puro resistant gene (Neo+, Puro+, Fig. 1C). These cells were simultaneously selected for G418 resistance. Although there are loxP sequences at all four sites and recombination could occur at any two of them, the Neo+ and Puro+ double selection ensures that the survived cells have the correct triple inversions (17M-GFP, Fig. S3). Though another unintended double inversion outcome may obtain the same drug resistance (Fig. S3), it would demand a 57.3 cM recombination induced on loxP loci, which would be of low frequency [10]. We did not detect such event in our experiment by P6 and P7 (Fig. 3C and Table S1). The new internal inversion was confirmed by FISH (Fig. 2C). The triple inversions on chromosome 17 were also verified by PCR on all four inversion points (Fig. 1C and Fig. 3). A mouse line carrying the 17M-GFP chromosome was established in 129S1/SvImJ background. Both 17M-GFP heterozygous and homozygous animals are viable and fertile. Organisms carrying the 17M-GFP chromosome can be easily detected from early embryonic stage to adult following the GFP signal (Fig. 2D-G & 2E'-2G'). For the 17M-GFP chromosome, no crossover event was detected in any of the three inversion regions in 187 F2 progenies, indicating that recombination between homologous chromosome 17 has been effectively suppressed (Fig. 3C; Fig. S4).
By constructing 17M-GFP and generating mice carrying this balancer, we have shown that the CASS strategy is an effective and versatile approach for constructing multiple inversions on chromosomes. Furthermore, mice carrying multiple inversions along a major chromosome have no obvious detrimental defect. Finally, three long inversions can effectively suppress crossovers for a large chromosome. Together, our study has not only produced the first full-length mouse chromosome balancer, 17M-GFP, which is a useful resource for a variety of genetic manipulations, but also provided a strategy for constructing full-length balancers for all mouse chromosomes.
Materials and Methods
Construction of the CASS shuttling cassettes and knock-in constructs
>PGK>Neo: a PGK-Neo expression cassette was first cloned from pMSCVneo into pBSKS III backbone; two synthesized loxP sequences were then inserted in the 5' and 3' region of the PGK promoter separately with a same direction.
PGK>Puro>: two synthesized loxP sequences were inserted separately into 5' and 3' of the coding region of a PGK-puro expression cassette cloned from pMSCVpuro into pBSKS III backbone.
The genomic sequences used for targeting were isolated from the BAC library of mouse genome (purchased from Invitrogen, Catalog No. 96051) and the phage clones from Yuan Zhuang's lab. DNA probes used in screen was PCR products containing following regions on mouse chromosome 17 (Site I to Site IV): 93,409,211-93,412,711; 61,583,933-61,587,832; 29,719,881-29,721,481; 5,768,771-5,770,771. All nucleotide co-ordinates for the mouse are those corresponding to NCBI m37.
>PGK>Neo containing knock-in construct for Site I: a BamHI and PstI fragment was cloned from BAC clone, from which an 11kb long arm and a 1.5kb short arm were isolated separately. An additional beta-actin driven GFP cassette was inserted into >PGK>Neo next to PGK-neo before both arms were assembled onto the targeting vector. A PGK driven Diphtheria Toxin alpha chain (PGK-DTA) was added as negative selection marker.
PGK>Puro>VMC containing knock-in construct for Site II: a 2.4kb and a 14 kb BglII fragments were cloned from BAC as the short arm and the long arm. The orientation of short arm and long arm in this vector was arranged in an Ends-in targeting fashion. A CMV promoter was cloned into PGK>Puro> as XhoI /EcoRI fragment before PGK>Puro> was inserted between two arms.
PGK>Puro> containing knock-in construct for Site III: a 2.4kb and a 10 kb EcoRI/XhoI fragments were cloned from BAC as the short arm and the long arm. An intact PGK>Puro> was then inserted. PGK-DTA was added as negative selection marker.
oruP>PGK>Neo containing knock-in construct for Site IV: an EcoRV and NotI fragment was cloned from positive phage clone, from which a 12kb long arm and a 1.6kb short arm were separated and used in targeting vector. A Hind III and XmnI fragment, containing puromycin resistant gene and 3'LTR region was cloned from pMSCVpuro and inserted outside of the >PGK>Neo in the reverse orientation of neomycin resistance cassette. The whole cassette was then inserted between two arms. PGK-DTA was used as negative selection marker.
Culture, derivation, and electroporation of ES cell
The conditions for culture and electroporation of W4/129S6 mouse ES cells were described in the manufacturer-recommended protocols (Taconic). Briefly, ES cells were cultured and passaged on mitomycin-C treated mouse embryonic stem cells as a feeder layer. The growth media contains 1000U/mL Leukemia Inhibitory Factor to maintain ES cells' growth. The de novo isolation of ES cells was performed according to the established protocols[11]. Briefly, 3.5dpc blastocysts were harvested and plated on feeder. The clump derived from inner cell mass was disaggregated with trypsin and plated into new wells. The ES cell like cells emerged from these wells were colonized and examined with PCR. The ES cell lines positive for desired inversions and Y chromosome were expanded for further experiments.
Thirty micrograms of linearized knock-in constructs, or fifteen micrograms PGK-Cre expression plasmids were used for electroporation of 10 million cells. Immediately after electroporation, cells were seeded onto three 10 cm plates containing feeder cells. When necessary, drug selection was initiated 48 hrs after electroporation with medium containing 200 g/ml G418 or 1-1.5 μg/ml puromycin. Drug selection continued for two weeks. Diphtheria toxin expressed from the PGK-DTA cassette should be expressed only in cells with random integration of the targeting vector, providing a negative selection. Positive ES clones were karyotyped and prepared for blastocysts injection according to standard procedures.
Animals
Adult wild-type male and female mice were group housed up to five in a cage and kept on a reverse 12-h light/dark cycle with ad libitum food and water. All mouse inversion strains in this study were generated in our laboratory and maintained on a 129S1/SvImJ background. All animal-related procedures were reviewed and approved by the Institute of Developmental Biology and Molecular Medicine Institutional Animal Care and Use Committee.
Recombination analysis
The genetic distances on the wild-type chromosome were derived from chromosomal positions (in cM) in the Chromosome Committee Reports (http://www.informatics.jax.org). Site I was mapped to marker Gm9665 (60.69 cM). Site II was mapped between D17Dcr18 (31.94 cM) and D17Dcr29 (31.97 cM). Site III was mapped between Gm20161 (15.46 cM) and Ccdc167 (15.48 cM). Site IV was mapped to marker 3300005D01Rik (3.47 cM). The expected recombination ratios were inferred from the genetic distance between sites.
To determine the actual recombination frequencies, we examine possible recombinants in F2 test cross (see also Fig. S4). For example, 17DI-GFP carriers were crossed to wild-type C57BL/6 animals and the F1 heterozygotes were further back crossed to C57BL/6 strains. In the F2 offspring, any possible crossover event would result in the separation of markers at the terminal of inversion or SNP markers within the region. For each regional inversion, we used primer pairs across the breakpoints to distinguish the inverted allele and wild type allele. SNP markers polymorphic between 129S1/SvImJ and indicated strains were also used to determine recombination ratio at selected loci. For 17M-GFP, all four breakpoints were examined by PCR to detect any possible double crossovers. Primers used to detect crossovers were listed in Supplementary Table S1.
FISH
For each inversion, two BAC clones that mapped within inversion interval were selected. BAC DNA was probed with either biotin or digoxin by nick translation (Roche). For the proximal inversion, BACs RP24-264D8 (9.0Mb-9.2Mb) and RP24-319F22 (25.8Mb-26.0Mb) were used, while for the distal inversion BACs RP24-247F10 (65.9Mb-66.1Mb) and RP24-374P20 (87.6Mb-87.8Mb) were used. And BACs RP24-157E7 (37.8Mb-37.9Mb2) and RP24-401F21 (57.0Mb-57.1Mb) were used for internal inversion.
Spreads of metaphase chromosomes were prepared from ES cells and FISH was performed as described [8]. Briefly, slides were first digested with pepsin, fixed and dehydrated in ethanol. After applying two probes, slides were sealed and denature in 85 oC and hybridized in 37 oC overnight. The slides were then washed and incubated with Avidin-FITC and Digoxin-Rhodamine (Roche). DAPI was used as counterstain. Photographs were taken with a Leica DMRXA2 camera and images prepared with Leica FW4000 and Adobe Photoshop. The strong DAPI signal on each chromosome indicated the location of centromeres region. The arrangement of the red and green signals relative to the centromere was used to distinguish the chromosome carrying inversion.
Supplementary Material
Supplementary table and figures.
Acknowledgements
We thank Yanling Yang, Yanfeng Tan, and Boying Tan for technical assistance, Yueying Chen, Chunyan Gao, and Xiaoping Huang for mouse husbandry, Ling Sun and Wufan Tao for discussion. This work was supported by the Hi-tech Research and Development Project of China (863) (2014AA021104); National Natural Science Foundation of China (NSFC) (81170789); the Rising-Star Program (10QH1400100), the 'Shu Guang' project (09SG04), and the STCSM grant (12431900100) of Shanghai Municipal Government to X.W., and the Chinese Key Projects for Basic Research (973) grant (2013CB945301 and 2012AA022401) to T.X. and H.M., respectively. T.X. and H.M are Howard Hughes Medical Institute investigators. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Muller HJ. The Measurement of Gene Mutation Rate in Drosophila, Its High Variability, and Its Dependence upon Temperature. Genetics. 1928;13:279-357
2. Lindsley DL, Zimm GG. The Genome of Drosophila melanogaster; 1992.
3. Zheng B, Sage M, Cai WW, Thompson DM, Tavsanli BC, Cheah YC. et al. Engineering a mouse balancer chromosome. Nat Genet. 1999;22:375-8
4. Nishijima I, Mills A, Qi Y, Mills M, Bradley A. Two new balancer chromosomes on mouse chromosome 4 to facilitate functional annotation of human chromosome 1p. Genesis. 2003;36:142-8
5. Klysik J, Dinh C, Bradley A. Two new mouse chromosome 11 balancers. Genomics. 2004;83:303-10
6. Boles MK, Wilkinson BM, Maxwell A, Lai L, Mills AA, Nishijima I. et al. A mouse chromosome 4 balancer ENU-mutagenesis screen isolates eleven lethal lines. BMC Genet. 2009;10:12
7. Boles MK, Wilkinson BM, Wilming LG, Liu B, Probst FJ, Harrow J. et al. Discovery of Candidate Disease Genes in ENU-Induced Mouse Mutants by Large-Scale Sequencing, Including a Splice-Site Mutation in Nucleoredoxin. PLoS genetics. 2009;5:e1000759
8. Sun L, Wu X, Han M, Xu T, Zhuang Y. A mitotic recombination system for mouse chromosome 17. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:4237-41
9. Vacik T, Ort M, Gregorova S, Strnad P, Blatny R, Conte N. et al. Segmental trisomy of chromosome 17: a mouse model of human aneuploidy syndromes. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:4500-5
10. Zheng B, Sage M, Sheppeard EA, Jurecic V, Bradley A. Engineering mouse chromosomes with Cre-loxP: range, efficiency, and somatic applications. Molecular and cellular biology. 2000;20:648-55
11. A N. Manipulating the Mouse Embryo: A Laboratory Manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. 2003
Author contact
![]() Corresponding authors: T.X. (tian.xuedu) and X.W. (xiaohui_wuedu.cn).
Corresponding authors: T.X. (tian.xuedu) and X.W. (xiaohui_wuedu.cn).