Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(8):979-989. doi:10.7150/ijbs.15071 This issue Cite
Research Paper
BDNF-mediates Down-regulation of MicroRNA-195 Inhibits Ischemic Cardiac Apoptosis in Rats
Pengzhou Hang1, Chuan Sun1, Jing Guo1, Jing Zhao2, Zhimin Du1 ![]()
1. Institute of Clinical Pharmacology of the Second Affiliated Hospital (Key Laboratory of Drug Research, Heilongjiang Higher Education Institutions), Harbin Medical University, Harbin 150086, Heilongjiang Province, P. R. China;
2. Department of Cardiology, the First Affiliated Hospital (Key Laboratory of Cardiac Diseases and Heart Failure), Harbin Medical University, Harbin 150001, Heilongjiang Province, P. R. China.
Received 2016-1-24; Accepted 2016-6-7; Published 2016-7-9
Abstract

Background: Our previous studies suggested that brain-derived neurotrophic factor (BDNF)/tropomyosin-related kinase B (TrkB) axis inhibited cardiomyocyte apoptosis in myocardial infarction (MI). However, the relationship between BDNF and microRNA (miRNA) in cardiomyocytes are unclear. The present study was performed to investigate the role of miR-195 and the interplay between BDNF and miR-195 in ischemic cardiomyocyte apoptosis.
Methods: Male Wistar rats were subjected to coronary artery ligation, and primary neonatal rat ventricular myocytes were treated with hypoxia or hydrogen peroxide (H2O2). BDNF level in rat ventricles was measured by enzyme linked immunosorbent assay (ELISA). miR-195 mimic, inhibitor or negative control was transfected into the cardiomyocytes. Cell viability and apoptosis were detected by MTT assay and TdT-mediated dUTP nick end labeling (TUNEL) staining, respectively. Cardiac function and apoptosis were detected in MI rats intravenously injected with antagomiR-195. Luciferase assay, Western blot and Real-time RT-PCR were employed to clarify the interplay between miR-195 and BDNF.
Results: miR-195 level was dynamically regulated in response to MI and significantly increased in ischemic regions 24 h post-MI as well as in hypoxic or H2O2-treated cardiomyocytes. Meanwhile, BDNF protein level was rapidly increased in MI rats and H2O2-treated cardiomyocytes. Apoptosis in both hypoxic and H2O2-treated cardiomyocytes were markedly reduced and cell viability was increased by miR-195 inhibitor. Moreover, inhibition of miR-195 significantly improved cardiac function of MI rats. Bcl-2 but not BDNF was validated as the direct target of miR-195. Furthermore, BDNF abolished the pro-apoptotic role of miR-195, which was reversed by its scavenger TrkB-Fc.
Conclusion: Up-regulation of miR-195 in ischemic cardiomyocytes promotes ischemic apoptosis by targeting Bcl-2. BDNF mitigated the pro-apoptotic effect of miR-195 in rat cardiomyocytes. These findings may provide better understanding of the pro-apoptotic role of miR-195 in MI and suggest that BDNF/miR-195/Bcl-2 axis may be beneficial for limiting myocardial ischemic injury.
Keywords: Brain-derived neurotrophic factor, miR-195, Myocardial ischemia, Apoptosis, Bcl-2.
Introduction
Acute myocardial infarction (MI) can lead to sudden cardiac death, which is one of the leading causes of death and disability worldwide [1]. Numerous studies have documented that cardiomyocyte apoptosis occurs in border zone of infarct scars and in the remote zone of non-infarcted myocardium [2-4], which exacerbates the post-MI remodeling and aggravates cardiac dysfunction [5]. Studies have also suggested that both acute substantial cardiomyocyte loss and chronic low levels of apoptosis contributed to the development of heart failure [6, 7]. Thus, successfully reversal of cardiomyocyte apoptosis during early stage of MI is crucial for repairing the injured heart and ameliorating cardiac function [8].
MicroRNAs (miRNAs or miRs) refer to a group of small noncoding RNA molecules, typically 18~25 nucleotides in length, that function in RNA silencing and post-transcriptional regulation of gene expression [9]. miR-195 is one of the members of miR-15 family including miR-15a, miR-15b, miR-16, miR-195 and miR-497 [10]. Recent studies elucidate that miR-195 plays a critical role in many cardiovascular diseases such as cardiac hypertrophy/heart failure, interstitial fibrosis and diabetic cardiomyopathy [11-13]. On the other hand, studies also suggested that miR-195 promoted apoptosis in a variety of cell types, e.g. hepatocellular carcinoma cells, neural progenitor cells, chondrocytes and cardiomyocytes [14-17]. At the same time, several apoptotic genes were screened as direct targets of miR-195, including Bcl-2, Sirtuin (Sirt1) and hypoxia-inducible factor (HIF)-1α according to bioinformatical prediction and experimental validation [16-18]. However, the role of miR-195 in cardiac ischemia-induced apoptosis and underlying mechanism is not fully understood.
Brain-derived neurotrophic factor (BDNF) is a member of neurotrophin factors, which is known to play key regulatory roles in neuronal activity and cognitive functions [19, 20]. Recently, BDNF is recognized to protect the heart by regulating the central nervous system [21] and promote neovascularization of ischemic tissue by recruiting the endothelial cells [22]. Our recent findings suggested that BDNF/tropomyosin-related kinase B (TrkB) axis possessed anti-ischemic apoptosis effect by increasing Bcl-2 expression and inhibiting caspase-3 activity [23]. Besides, BDNF was reported as a potential target of miR-195 based on previous studies [24]. Moreover, studies also revealed the link between BDNF and miRNAs in retinal ganglion cells and podocytes [25, 26]. Therefore, our present study focused on the role of miR-195, and the association between miR-195 and BDNF in ischemic cardiomyocyte apoptosis.
Materials and methods
Ethics statement
This study was approved by the Experimental Animal Ethic Committee of Harbin Medical University. All experimental procedures conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (Publication, 8th Edition, 2011).
Rat MI model
Male Wistar rats (weight 220 ± 20 g, provided by Animal Center of the Second Affiliated Hospital of Harbin Medical University, Harbin, China) were raised under standard animal room conditions (temperature 23 ± 1 °C; humidity 55 ± 5 %) with free access of food and water. Rats were randomly divided into four groups: sham and MI for 1 h, 6 h or 24 h. MI model was established by ligating the left anterior descending coronary artery as we previously described [23]. Sham-operated animals subjected to the same procedure but the coronary ligature was left untied. Electrocardiograms before, after ligation and at the end of ligation were recorded to confirm the ischemic condition. The surgery procedures were performed under sodium pentobarbital anesthesia. Remote, border and infarcted zones were quickly dissected for subsequent Real-time PCR analysis. To investigate the effect of miR-195 on ischemic heart function, antagomiR-195 (5 mg/kg, RiboBio Co., China) or negative control (NC) were injected via tail vein in the rats after the left anterior descending coronary artery was ligated. AntagomiR-195 is a single-stranded RNA analog complementary to the sequence of mature miR-195 (5'-UAGCAGCACAGAAAUAUUGGC-3') that is chemically modified and cholesterol-conjugated from a hydroxyprolinol-linked cholesterol solid support and 2'-OMe phosphoramidites. 24 h later, they were injected again at the same dose. 7 days post-MI, cardiac function was detected by echocardiography. To explore the role of BDNF on miR-195 in ischemic rat heart, recombinant human BDNF (10 μg in 25 μl PBS, R&D Systems, USA) and TrkB-Fc (5 μg in 25 μl PBS, 688-TK-100, TrkB-Fc chimera, R&D Systems, USA) were intramuscular injected into the left ventricles according to previous studies [27]. 24 h post-MI border zones were collected.
Echocardiography measurement and histological analysis
Left ventricular function was measured by Acuson Sequoia 512 Ultrasound System (Siemens Medical Solutions USA, Inc., Mountain View, CA, USA). Left ventricular systolic diameter (LVDs), left ventricular diastolic diameter (LVDd) was measured, and left ventricular ejection fraction (LVEF) and fractional shortening (FS) were calculated. For histological analysis, border zones were embedded with paraffin, sectioned into 5 μm slices. Then, they were stained with standard hematoxylin and eosin (HE).
Cell culture and transfection
Neonatal rat ventricular myocytes (NRVMs) were isolate and cultured according to previous study [23, 28]. The isolated cells were cultured with DMEM (Corning, USA) and 10% fetal bovine serum (ExCell Bio, China). The cardiomyocytes were cultured at 37°C with 5% CO2 and 95% air. Cells were starved in serum-free medium for 12 h, and then transiently transfected with miR-195 (50 nM), miR-195 inhibitor (100 nM) or negative control (NC), using X-treme GENE siRNA transfection reagent (Roche, USA) according to the manufacturer's instructions. miR-195 mimic, inhibitor and NC were synthesized by RiboBio Co., Ltd (China). The sequence of miR-195 mimic is 5'-UAGCAGCACAGAAAUAUUGGC-3', and mimic NC is 5'-UUUGUACUACACAAAAGUACUG-3'. The sequence of miR-195 inhibitor is 5'-GCCAAUAUUUCUGUGCUGCUA-3' and inhibitor NC is 5'-CAGUACUUUUGUGUAGUACAAA-3', which contains 2'-O-methyl modifications. 6h later, the culture medium were removed and DMEM containing serum was added to the cells. Cy3-labeled transfection control (Ribo Bio Co., Ltd, China) were used to validate the transfection efficiency. For hypoxic treatment, cells were placed in a hypoxic incubator (Thermo Scientific Series WJ 8000, USA) and were kept at 37°C for 12 h with a constant stream of water-saturated 94% N2, 5% CO2, and 1% O2. Then, the cells were subsequently treated with different concentrations of hydrogen peroxide (H2O2, 50 and 100 μM) for 4 h or 24 h. H2O2 (100 μM, 4 h) was selected. Moreover, recombinant human BDNF (200 ng/ml, R&D Systems, USA) and/or 4 μg/ml BDNF scavenger TrkB-Fc was treated to the cells as previously described [23]. Forty-eight hours after transfection, cardiomyocytes were collected for total RNA isolation or protein extraction.
RNA extraction and Real-time PCR
Total RNA were extracted from left ventricles of rats or NRVMs using Trizol reagent (Invitrogen, USA) according to manufacturer's protocols. RNA quantity was assessed using the NanoDropTM 8000 spectrophotometer (Thermo Scientific, France). cDNA was synthesized using reverse transcriptase kit (Roche, USA). Sequences of gene-specific PCR primers (Shanghai Generay Biotech Co. Ltd, China) used were as follow: miR-195 RT: 5'-GTCGTATCCAGTGCGTGTCGTGGAGTCGGCAATTGCACTGGATACGACGCCAAT-3'; F: 5'-GGGGTAGCAGCAGCACAGAAAT-3'; R: 5'-TCCAGTGCGTGTCGTGGA-3'; U6 RT: 5'-CGCTTCACGAATTTGCGTGTCAT-3'; F: 5'-GCTTCGGCACATATACTAAAAT-3'; R: 5'-CGCTTCACGAATTTGCGTGTCAT-3'. Primes of miR-15a, -15b, -16, -497 were listed in Supplementary Material: Table S1. Quantitative real-time PCR was performed in 20 μl volumes with SYBR Green PCR Master Mix (Roche, USA) at 95°C for 10 min and 40 cycles at 95°C for 15 s, 60°C for 30 s and 72°C for 30 s, using LightCycler 480 (Roche, USA). miR-195 levels were quantified with the 2-ΔΔCt relative quantification method that was normalized to U6.
BDNF level measurement
BDNF level in ischemic zone of rat left ventricles was examined by enzyme linked immunosorbent assay (ELISA) according to the manufacturer's instructions (Promega, USA).
TdT-mediated dUTP nick end labeling (TUNEL) staining
TUNEL staining was employed to detect the apoptosis in left ventricles (border zones) and NRVMs as described in our previous study [23] using a TUNEL fluorescence FITC kit (Roche, USA) according to the manufacturer's instruction. After TUNEL staining, the cardiomyocytes were immerged into DAPI (1:30, Beyotime Biotechnology, China) solution to stain nuclei. Fluorescence staining was viewed by microscope (Olympus, BX-60, Japan) or a Laser Scanning Confocal Microscope (FV1000, Olympus, Japan). The apoptotic rate was calculated as TUNEL-positive cells per field.
Cell viability assay
MTT (3-[4, 5-dimethylthiazol-2-yl]-2, 5 diphenyl tetrazolium bromide) assay was used to examine the viability of cardiomyocytes. The cells were treated as designated and were subsequently incubated with 20 μl MTT (0.5 mg/ml) for 4 h. Then, 200 μl Dimethyl sulphoxide (DMSO) was added to each well to dissolve the formazan after removing the culture medium. The absorbance values were examined at 490 nm wavelength using an Infinite M200 microplate reader (Tecan, Salzburg, Austria) after rocking the cells for 10 min.
Luciferase activity assay
The luciferase reporter assay was performed as previously described [29]. Briefly, luciferase reporters containing full-length wild-type 3'UTR of BDNF were constructed using pmiR-RB-REPORTTM vector. Then, 293T cells were seeded in a 24-well plate and co-transfected with 0.5 mg plasmid and miR-195 mimic or NC by riboFectTM (Guangzhou Ribo Bio Co., Ltd). Renilla luciferase was used as an internal control. Forty-eight hours later, the cells were collected, and firefly and Renilla luciferase activities were measured in Dual-Glo® Luciferase Assay System (Promega, Madison, WI, USA).
Western blot
Total protein was extracted from the cardiomyocytes as described in previous studies [23, 29]. Briefly, proteins were separated by electrophoresis on SDS-polyacrylamide gels and transferred moist to nitrocellulose filter membranes. Membranes were incubated with anti-BDNF (A1307, 1:500, Abclonal, China), anti-TrkB (A2099, 1:500, Abclonal, China) or anti-Bcl-2 (ab7973, 1:500, Abcam, USA) antibody overnight at 4°C. After washing, the membrane was incubated with IRDye secondary antibodies (LI-COR) for 1 h. The images were captured by the Odyssey CLx Infrared Imaging System (LI-COR Biosciences, Lincoln, NE, USA). Western blot bands were quantified by measuring intensity in each group using Odyssey CLx version 2.1 with anti-GAPDH antibody (TA-09, 1:1000, ZSGB-Bio, China) as an internal control.
Annexin V-FITC/propidium iodide (PI) staining assay
An Annexin V-FITC Apoptosis Detection kit was used to examine early apoptosis (Annexin V-FITC+/PI-, Q4), late apoptosis (Annexin V-FITC+/PI+, Q2), and necrosis (Annexin V-FITC-/PI+, Q1) according to the manufacturer's instructions (Vazyme, Nanjing, China). The cells were digested with 0.25% trypsin and collected by centrifugation after treatment. Then, the cells were washed twice with PBS and stained with Annexin V-FITC for 15 min and PI for 5 min. The apoptotic cells were detected by flow cytometry (BD Bioscience, USA).
Statistical analysis
All data are expressed as mean ± SEM and analyzed by GraphPad Prism 5.0 software. Two-group comparisons were performed by Student's t test. Multiple-group comparisons were carried out using one-way ANOVA followed by Dunnet's t-test. A two-tailed P < 0.05 was considered to be significant.
Results
miR-195 and BDNF were dynamically regulated in response to myocardial ischemic injury
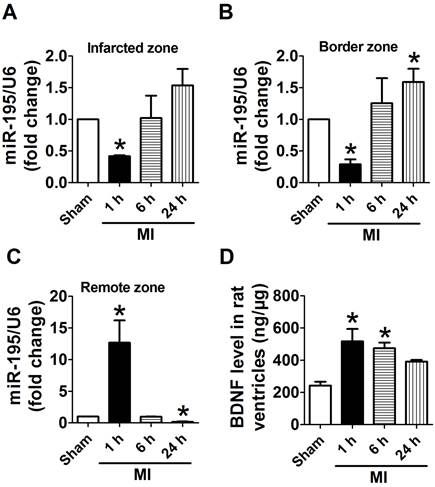
Because infarct healing is a dynamic process involving specific regional and temporal changes in cardiomyocytes, we first examined the expression of miR-195 in different zones after MI for 1 h, 6 h and 24 h, respectively. Real-time PCR analysis revealed that miR-195 level was rapidly decreased in infarcted and border zones (Fig. 1A, B) whereas dramatically increased in remote zone 1 h after ischemic injury (Fig. 1C). Interestingly, miR-195 level was found to be elevated in infarcted and border zone while reduced in remote zone during the ischemic process. And miR-195 level was markedly higher in both infarcted and border zones 24 h after ischemic injury than sham rats (Fig. 1A, B). In contrast, it was significantly lower in remote zone of MI rats compared with sham rats (Fig. 1C). The expression of other miR-15 family members including miR-15a, miR-15b, miR-16, and miR-497 were also examined in infarcted, border and remote zones, respectively. As shown in Supplementary Material: Fig. S1, miR-15a, -15b, -16 and -497 were all increased in 24 h post-MI compared with sham rats. Thus, the expression of these four miRNAs together with miR-195 had different amplitudes and characteristics in response to MI although belonging to the same family. Meanwhile, BDNF level in ischemic myocardium was found rapidly increased at 1 h and 6 h post-MI and recovered 24 h post-MI (Fig. 1D). Thus, both miR-195 and BDNF are dynamically regulated in response to MI.
miR-195 and BDNF levels were dynamically regulated in different regions of rat myocardium in response to ischemic injury. (A, B) Real-time PCR analysis indicates that miR-195 is increased in the infarcted and border zones 24 h after myocardial infarction (MI). (C) miR-195 is decreased in remote zone in rat myocardium 24 h after MI. (D) BDNF level in ischemic zone of rat ventricles. *p<0.05, **p<0.01 vs. sham, n = 3.
miR-195 mimic and inhibitor transfection validation
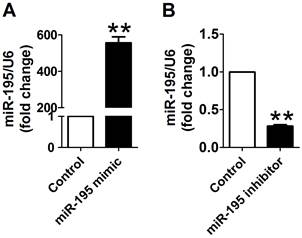
Cy3-labeled transfection control was used to examine the transfection efficiency of miR-195 mimic and inhibitor. As displayed in Supplementary Material: Fig. S2, Cy3-labeled transfection control (red color) was widely distributed around the nuclear (stained in blue with DAPI), which suggested that the transfection efficiency satisfied with experimental requirements. Then, to overexpress or knockdown miR-195 expression, miR-195 mimic or inhibitor was transfected into the cardiomyocytes. We further validate the miR-195 level using Real-time PCR. We found that miR-195 level was significantly increased after transfecting with miR-195 mimic (~557 folds, p<0.01, Fig. 2A). Moreover, miR-195 was markedly decreased in miR-195 inhibitor transfection group compared with non-transfection group (~0.28 fold, p<0.001, Fig. 2B). These findings strongly supported that miR-195 mimic or inhibitor were successfully transfected into the cardiomyocytes to upregulate or downregulate miR-195, respectively.
miR-195 mimic and inhibitor transfection validation. (A) Real-time PCR analysis indicated that miR-195 level was dramatically increased after transfecting with miR-195 mimic compared with control group. (B) miR-195 level was markedly decreased after transfecting with miR-195 inhibitor compared with control group. **p<0.01 vs. control, ***p<0.001 vs. control, n = 3.
miR-195 inhibitor prevented hypoxia-induced cardiomyocyte apoptosis
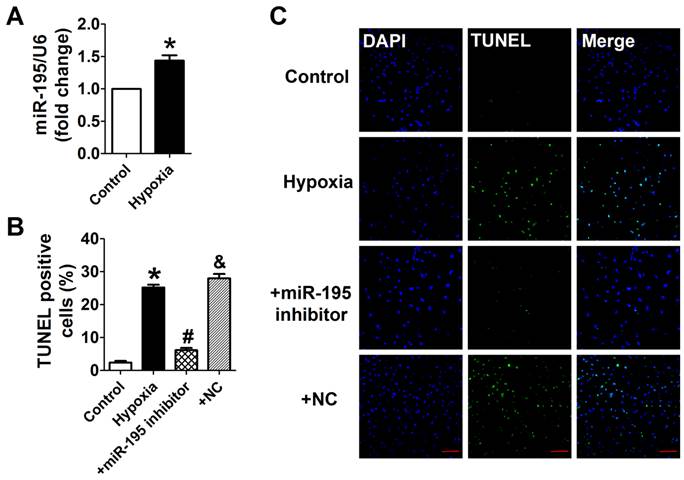
Cardiomyocytes were subjected to hypoxia to simulate ischemic status in vitro. Consistently, we found that the miR-195 level was significantly higher in hypoxic cells than normoxia cells (Fig. 3A). In addition, TUNEL assay was used to detect the role of miR-195 in hypoxia-induced cardiomyocyte apoptosis. It was observed that the TUNEL-positive cells were significantly increased in hypoxia group, which was diminished by miR-195 inhibitor but not by miR-NC (Fig. 3B, C). These data implied that miR-195 inhibitor prevented hypoxia-induced cardiomyocyte apoptosis.
miR-195 inhibitor attenuated hypoxia-induced cardiomyocyte apoptosis. (A) Real-time PCR analysis indicates that miR-195 is increased in hypoxia-treated cardiomyocytes. (B) Statistical results of TUNEL-positive cells per field. (C) Representative images of TUNEL staining of cardiomyocyte showing the apoptotic cells (apoptotic cells stained in green and nucleus stained in blue with DAPI). *p<0.05 vs. control, #p<0.05 vs. hypoxia, n = 5, scale bar: 100 μm.
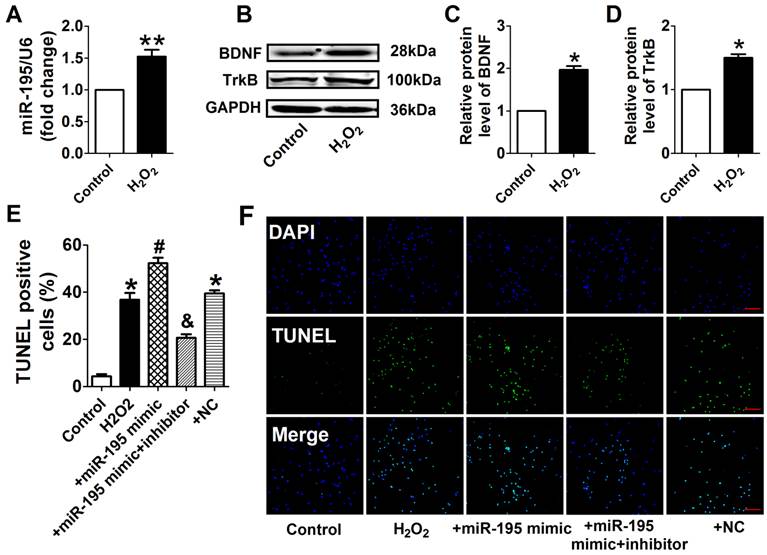
miR-195 inhibitor prevented H2O2-induced cardiomyocyte apoptosis
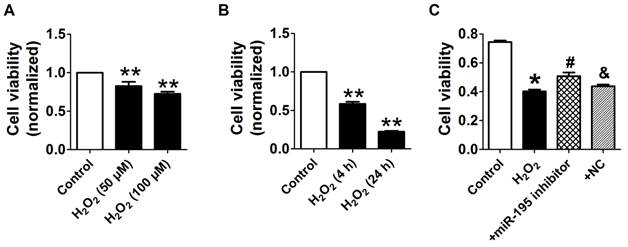
Consistent with previous study [28], cell viability was significantly decreased by 50 μM and 100 μM H2O2 for 4 h, meanwhile markedly decreased after H2O2 treatment for 4 h and 24 h at 100 μM (Fig. 4A, B). Accordingly, H2O2 reduced cardiomyocyte viability in a concentration and time-dependent manner and 100 μM H2O2 treated for 4 h was chosen for subsequent experiments. We found that cell viability treated with H2O2 was restored by miR-195 inhibitor but not by miR-NC (Fig. 4C). In agreement with hypoxia, miR-195 level was found to be increased in H2O2-treated cardiomyocytes compared with control cells (Fig. 5A). Western blot analysis indicated that BDNF and TrkB were significantly increased in H2O2-treated cardiomyocytes (Fig. 5B-D). Furthermore, we found that the TUNEL-positive cells were significantly increased by miR-195 mimic in H2O2-treated group, which was reversed by miR-195 inhibitor but not by miR-NC (Fig. 5E, F). These results together suggested that miR-195 is pro-apoptotic in H2O2-treated cardiomyocytes. Meanwhile, knockdown of endogenous miR-195 prevented H2O2-induced apoptosis of cardiomyocytes.
miR-195 inhibitor protected against cardiomyocyte injury induced by hydrogen peroxide (H2O2). (A, B) Cultured neonatal rat cardiomyocytes were exposed to different concentration of of H2O2 (50, 100 μM) for 4 h, or different timepoints (4 h, 24 h) at 100 μM. (C) MTT assay suggested that miR-195 inhibitor restored cell viability after H2O2 treatment. *p<0.05, **p<0.01 vs Control, #p<0.05 vs H2O2, &p<0.05 vs miR-195 inhibitor, n = 5.
miR-195 inhibitor attenuated H2O2-induced cardiomyocyte apoptosis. (A) Real-time PCR analysis indicates that miR-195 is increased in H2O2-treated cardiomyocytes. (B) Western blot bands of BDNF and TrkB in control and H2O2-treated cardiomyocytes. (C, D) Statistical results of protein level of BDNF and TrkB, *p<0.05, n = 5. (E) Statistical results of TUNEL-positive cells per field. (F) Representative images of TUNEL staining of cardiomyocyte showing the apoptotic cells (apoptotic cells stained in green and nucleus stained in blue with DAPI). *p<0.05 vs. control, #p<0.05 vs. H2O2, &p<0.05 vs. +miR-195 mimic, n = 5, scale bar: 100 μM.
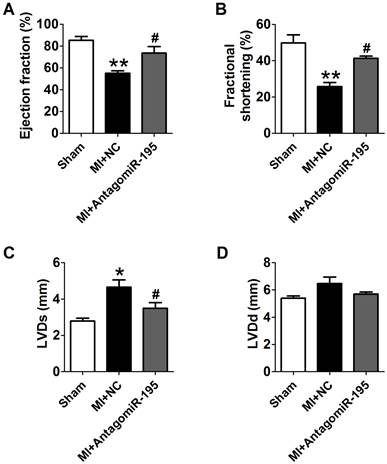
AntagomiR-195 improved cardiac function in MI rats
EF and FS were markedly decreased in MI+NC rats compared with sham rats, which was significantly restored by antagomiR-195 (Fig. 6A, B). Meanwhile, MI-induced increment of LVDs and LVDd were reduced in antagomiR-195 rats (Fig. 6C, D). HE staining displayed that cardiac injury induced by MI was markedly attenuated by antagomiR-195 but not by NC (Fig. 7A). Moreover, the role of miR-195 in MI-induced cardiomyocyte apoptosis was also validated in rats. As shown in Fig. 7B, C, TUNEL positive cells were markedly decreased in antogomiR-195-treated MI rats compared with MI+NC rats. Consistently, Bcl-2 protein expression was found upregulated by antagomiR-195 (Fig. 7D, E). So, these data together supported that inhibition of miR-195 improved cardiac function in MI.
Improvement of cardiac function by antagomiR-195 in MI rats. (A) Ejection fractions. (B) Fractional shortening. (C) Left ventricular systolic diameter (LVDs). (D) Left ventricular diastolic diameter (LVDd). *p<0.05, **p<0.01 vs. sham, #p<0.05 vs. MI+NC, n = 5.
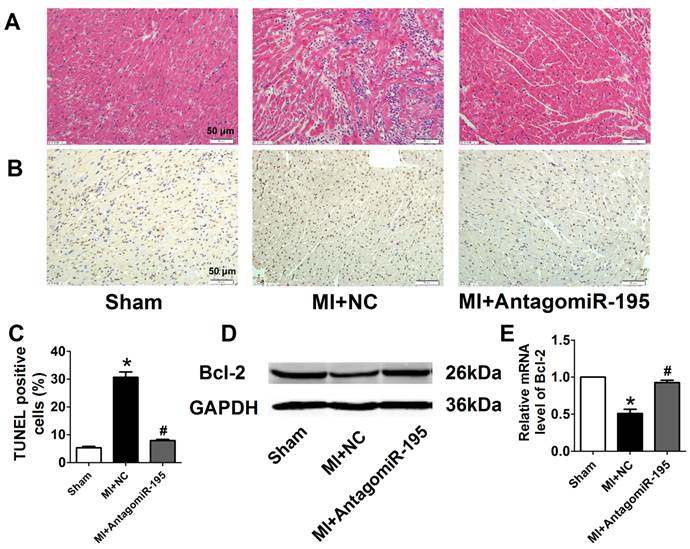
AntagomiR-195 inhibited cardiac injury and apoptosis by upregulating Bcl-2 in MI rats. (A) Representative HE staining pictures, scale bar: 50 μM. (B) Representative images of TUNEL staining in rat myocardium (apoptotic cells stained in brown), scale bar: 50 μM. (C) Statistical results of TUNEL-positive cells per field. (D) Western blot bands of Bcl-2 in sham, MI+NC and MI+antagomiR-195 rat hearts. (E) Statistical results of protein level of Bcl-2. *p<0.05 vs. sham, #p<0.05 vs. MI+NC, n = 5.
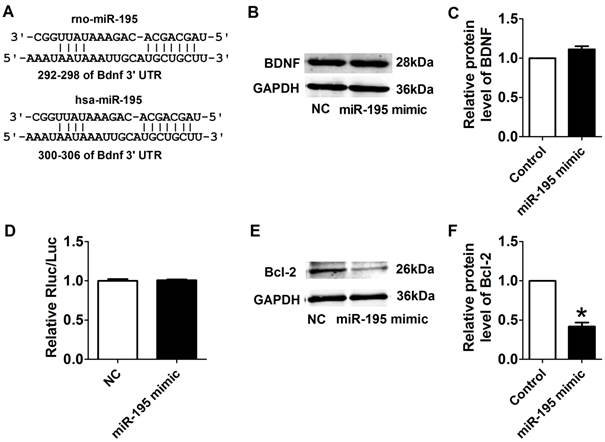
Interplay between miR-195 and BDNF
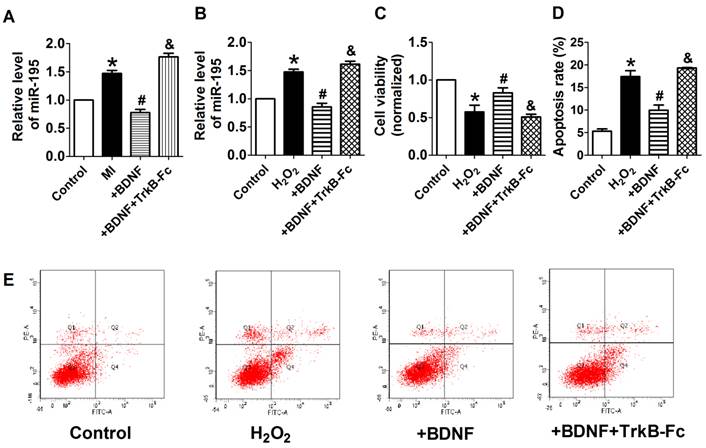
We further examined the possible interaction between miR-195 and BDNF. As illustrated in Fig. 8A, the 3'UTR of Bdnf has one binding site of miR-195. We detected the protein expression of BDNF after transfected with miR-195 mimic or inhibitor. And no significant change was observed among control, miR-195 mimic and miR-195 inhibitor groups (Fig. 8B, C). Then, luciferase assay was further employed to validate the regulatory effect of miR-195 on BDNF. Consistent to western blot results, no significant change of luciferase reporter activity was found between miR-195 mimic and NC (Fig. 8D). These findings demonstrated that BDNF is not a direct target of miR-195. Besides, previous bioinformatical analysis and experimental studies have proved the anti-apoptotic factor Bcl-2 was a direct target of miR-195 [18]. In our present study, we found that protein expression of Bcl-2 was significantly inhibited by miR-195 mimic (Fig. 8E, F), and validated the relatioship between miR-195 and Bcl-2. On the other hand, we tried to clarify the effect of BDNF on miR-195 by detecting miR-195 level after administering with BDNF or its blocker TrkB-Fc in both rats and NRVMs. We found that miR-195 level was obviously repressed by BDNF, which could be antagonized by TrkB-Fc both in vivo and in vitro (Fig. 9A, B). Next, we found that BDNF increased cell viability H2O2 treatment and was reversed by TrkB-Fc (Fig. 9C). Finally, flow cytometry was utilized to validate the protective role of BDNF. We found that the apoptosis rate was increased by H2O2 and diminished by BDNF, which was reversed by TrkB-Fc (Fig. 9D, E). Taken together, these findings suggested that BDNF inhibited miR-195 expression and prevented cardiomyocyte apoptosis.
Target validation of miR-195. (A) Sequence alignment show between miR-195 and the binding sites in the 3'UTR of the Bdnf gene. (B) Representative western blot bands of BDNF. (C) Statistical results of protein level of BDNF in miR-195 mimic and NC group, n = 3. (D) The interaction between miR-195 and its binding sites in the 3'UTR of Bdnf was examined by luciferase assay in HEK293 cells, n = 3. (E) Representative western blot bands of Bcl-2. (F) Statistical results of protein level of Bcl-2 in miR-195 mimic and NC group, *p<0.05, vs. control, n = 3.
BDNF inhibited miR-195 expression and protected cardiomyocytes against H2O2-induced apoptosis. (A) Real-time PCR analysis indicates that miR-195 level is reduced by BDNF and restored by TrkB-Fc, *p<0.05, vs. control, #p<0.05 vs H2O2, &p<0.05 vs +BDNF, n = 5. (B) MTT assay showed that BDNF improved cell viability in H2O2-treated cardiomyocytes and was reversed by TrkB-Fc, *p<0.05, vs. control, #p<0.05 vs H2O2, &p<0.05 vs +BDNF, n = 5. (C) The quantitative presentation of apoptotic cells by Annexin V-FITC/propidium iodide (PI) staining, *p<0.05, vs. control, #p<0.05 vs H2O2, &p<0.05 vs +BDNF, n = 3. (D) Representative Annexin V-FITC/PI staining pictures.
Discussion
The present study demonstrated that miR-195 was up-regulated in both ischemic myocardium and hypoxia/H2O2-induced cardiomyocytes. Up-regulation of miR-195 in ischemic cardiomyocytes promotes ischemic apoptosis by targeting Bcl-2. BDNF mitigated the pro-apoptotic effect of miR-195 in cardiomyocytes. These findings suggest that BDNF/miR-195/bcl-2 axis may be beneficial for repairing the ischemic cardiomyocytes.
The regulation of miR-195 is spatial, temoral and dynamic, and its expression profile depends on species and pathological stress [30-32]. In an earlier study by Hullinger et al. [30], miR-195 was found to be up-regulated in the infarcted zone 24 h after ischemic-reperfusion (IR) injury in the porcine MI model but did not significantly increased in the border zone. Another study reported that miR-195 was downregulated in both border and infarct zone of mice in response to MI [31]. In contrast, we noted that miR-195 expression elevated in both infarcted and border zones and reduced in remote zone in response to MI. Moreover, it has also been reported that only miR15b but no other miR-15 family members was upregulated in mouse heart subjected to IR for 2 or 7 days [32]. These different results may at least partly be explained by distinct species and animal models.
We focused on miR-195, as this miR has been previously reported as a miR with potential pro-apoptotic properties [14-17], but to the best of our knowledge, the present study firstly demonstrated the critical role of miR-195 in ischemic apoptosis and heart function. In previous studies, miR-195 mimic inhibited the protein level of Bcl-2 and contributed to podocyte apoptosis by increasing caspase-3 in high-glucose culture podocytes [33]. In cardiomyocytes, up-regulation of miR-195 expression promoted apoptosis and ROS production after palmitate-stimulation [17]. In line with these studies, our data demonstrated that knockdown of miR-195 alleviated cardiomyocyte apoptosis. Considering several previous studies have proved that Bcl-2 was an important direct target of miR-195 [17, 18]. Only western blot was employed to validate the effect of miR-195 mimic on Bcl-2 protein expression in the cardiomyocytes.
There is controversy in regarding of whether BDNF is the direct target of miR-195. Mellios et al. proposed that miR-195 potentially target 3'-untranslated region of BDNF using luciferase assays [24]. But they did not validate the protein expression of BDNF after transfection of miR-195 mimic. On the contrary, recent studies pointed out that BDNF is not the target of miR-195 according to luciferase assay results [34-36]. To clarify this question, both luciferase assay and western blot experiments were employed in the present study. And our data suggested that transfection with miR-195 mimic could not reduce luciferase report activity. Consistently, neither miR-195 mimic nor miR-195 inhibitor changed the protein expression of BDNF. Therefore, our findings supported that BDNF is not a direct target of miR-195. On the other hand, protein level of BDNF was also dynamically regulated (rapidly increased after 1 h ischemia and gradually decreased after 24 h ischemia) in response to ischemic injury. Interestingly, we noted an inverse relationship between BDNF and miR-195 expression. So we examined the effect of BDNF on miR-195 and found that miR-195 was negatively regulated by BDNF. Our previous study has proved that BDNF treatment up-regulated Bcl-2 expression [23], which is widely accepted as a direct target of miR-195 [18]. Incorporating with these studies, we speculated the protective role of BDNF/miR-195/Bcl-2 axis against ischemic apoptosis. Likewise, we noted that previous studies by Iekushi et al. demonstrated that bone marrow-derived mononuclear cells released insulin-like growth factor-1 (IGF-1), and prevented cardiomyocyte apoptosis by blocking expression of the precursor and the mature miR-34a [37]. This study indicates that the effect of cytokines/growth factor on miRNAs maybe a general phenomenon. Such possible interactions may provide further evidence for exploring upstream of miRNAs in cardiomyocytes.
In conclusion, this study suggested that BDNF-mediated down-regulation of miR-195 inhibited ischemic cardiomyocyte apoptosis, which may provide novel idea for treatment of ischemic heart diseases.
Supplementary Material
Supplementary tables and figures.
Acknowledgements
This work was supported in part by Major Program of National Natural Science Foundation of China (81230081), National Natural Science Foundation of China (81300080, 81200077), Natural Science Foundation of Heilongjiang Province (QC2013C087), and Postgraduate Innovation Foundation of Harbin Medical University (YJSCX2015-9HYD).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M. et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133:e38-e360
2. Saraste A, Pulkki K, Kallajoki M, Henriksen K, Parvinen M, Voipio-Pulkki LM. Apoptosis in human acute myocardial infarction. Circulation. 1997;95:320-323
3. Olivetti G, Quaini F, Sala R, Lagrasta C, Corradi D, Bonacina E, Gambert SR, Cigola E, Anversa P. Acute myocardial infarction in humans is associated with activation of programmed myocyte cell death in the surviving portion of the heart. J Mol Cell Cardiol. 1996;28:2005-2016
4. Piro FR, di Gioia CR, Gallo P, Giordano C, d'Amati G. Is apoptosis a diagnostic marker of acute myocardial infarction? Arch Pathol Lab Med. 2000;124:827-831
5. Palojoki E, Saraste A, Eriksson A, Pulkki K, Kallajoki M, Voipio-Pulkki LM, Tikkanen I. Cardiomyocyte apoptosis and ventricular remodeling after myocardial infarction in rats. Am J Physiol Heart Circ Physiol. 2001;280:H2726-H2731
6. Narula J, Haider N, Virmani R, DiSalvo TG, Kolodgie FD, Hajjar RJ, Schmidt U, Semigran MJ, Dec GW, Khaw BA. Apoptosis in myocytes in end-stage heart failure. N Engl J Med. 1996;335:1182-1189
7. Wencker D, Chandra M, Nguyen K, Miao W, Garantziotis S, Factor SM, Shirani J, Armstrong RC, Kitsis RN. A mechanistic role for cardiac myocyte apoptosis in heart failure. J Clin Invest. 2003;111:1497-1504
8. Eltzschig HK, Eckle T. Ischemia and reperfusion-from mechanism to translation. Nat Med. 2011;17:1391-1401
9. Boon RA, Dimmeler S. MicroRNAs in myocardial infarction. Nat Rev Cardiol. 2015;12:135-142
10. Finnerty JR, Wang WX, Hébert SS, Wilfred BR, Mao G, Nelson PT. The miR-15/107 group of microRNA genes: evolutionary biology, cellular functions, and roles in human diseases. J Mol Biol. 2010;402:491-509
11. van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci U S A. 2006;103:18255-18260
12. Tijsen AJ, van der Made I, van den Hoogenhof MM, Wijnen WJ, van Deel ED, de Groot NE, Alekseev S, Fluiter K, Schroen B, Goumans MJ, van der Velden J, Duncker DJ, Pinto YM, Creemers EE. The microRNA-15 family inhibits the TGFβ-pathway in the heart. Cardiovasc Res. 2014;104:61-71
13. Zheng D, Ma J, Yu Y, Li M, Ni R, Wang G, Chen R, Li J, Fan GC, Lacefield JC, Peng T. Silencing of miR-195 reduces diabetic cardiomyopathy in C57BL/6 mice. Diabetologia. 2015;58:1949-1958
14. Yang X, Yin J, Yu J, Xiang Q, Liu Y, Tang S, Liao D, Zhu B, Zu X, Tang H, Lei X. miRNA-195 sensitizes human hepatocellular carcinoma cells to 5-FU by targeting BCL-w. Oncol Rep. 2012;27:250-257
15. Zhou Y, Jiang H, Gu J, Tang Y, Shen N, Jin Y. MicroRNA-195 targets ADP-ribosylation factor-like protein 2 to induce apoptosis in human embryonic stem cell-derived neural progenitor cells. Cell Death Dis. 2013;4:e695
16. Bai R, Zhao AQ, Zhao ZQ, Liu WL, Jian DM. MicroRNA-195 induced apoptosis in hypoxic chondrocytes by targeting hypoxia-inducible factor 1 alpha. Eur Rev Med Pharmacol Sci. 2015;19:545-551
17. Zhu H, Yang Y, Wang Y, Li J, Schiller PW, Peng T. MicroRNA-195 promotes palmitate-induced apoptosis in cardiomyocytes by down-regulating Sirt1. Cardiovasc Res. 2011;92:75-84
18. Liu L, Chen L, Xu Y, Li R, Du X. microRNA-195 promotes apoptosis and suppresses tumorigenicity of human colorectal cancer cells. Biochem Biophys Res Commun. 2010;400:236-240
19. Wong YH, Lee CM, Xie W, Cui B, Poo MM. Activity-dependent BDNF release via endocytic pathways is regulated by synaptotagmin-6 and complexin. Proc Natl Acad Sci U S A. 2015;112:E4475-E4484
20. Ma JC, Duan MJ, Sun LL, Yan ML, Liu T, Wang Q, Liu CD, Wang X, Kang XH, Pei SC, Zong DK, Chen X, Wang N, Ai J. Cardiac over-expression of microRNA-1 induces impairment of cognition in mice. Neuroscience. 2015;299:66-78
21. Okada S, Yokoyama M, Toko H, Tateno K, Moriya J, Shimizu I, Nojima A, Ito T, Yoshida Y, Kobayashi Y, Katagiri H, Minamino T, Komuro I. Brain-derived neurotrophic factor protects against cardiac dysfunction after myocardial infarction via a central nervous system-mediated pathway. Arterioscler Thromb Vasc Biol. 2012;32:1902-1909
22. Kermani P, Rafii D, Jin DK, Whitlock P, Schaffer W, Chiang A, Vincent L, Friedrich M, Shido K, Hackett NR, Crystal RG, Rafii S, Hempstead BL. Neurotrophins promote revascularization by local recruitment of TrkB+ endothelial cells and systemic mobilization of hematopoietic progenitors. J Clin Invest. 2005;115:653-663
23. Hang P, Zhao J, Cai B, Tian S, Huang W, Guo J, Sun C, Li Y, Du Z. Brain-derived neurotrophic factor regulates TRPC3/6 channels and protects against myocardial infarction in rodents. Int J Biol Sci. 2015;11:536-545
24. Mellios N, Huang HS, Grigorenko A, Rogaev E, Akbarian S. A set of differentially expressed miRNAs, including miR-30a-5p, act as post-transcriptional inhibitors of BDNF in prefrontal cortex. Hum Mol Genet. 2008;17:3030-3042
25. Marler KJ, Suetterlin P, Dopplapudi A, Rubikaite A, Adnan J, Maiorano NA, Lowe AS, Thompson ID, Pathania M, Bordey A, Fulga T, Van Vactor DL, Hindges R, Drescher U. BDNF promotes axon branching of retinal ganglion cells via miRNA-132 and p250GAP. J Neurosci. 2014;34:969-979
26. Li M, Armelloni S, Zennaro C, Wei C, Corbelli A, Ikehata M, Berra S, Giardino L, Mattinzoli D, Watanabe S, Agostoni C, Edefonti A, Reiser J, Messa P, Rastaldi MP. BDNF repairs podocyte damage by microRNA-mediated increase of actin polymerization. J Pathol. 2015;235:731-744
27. Zhang X, Wang J, Zhou Q, Xu Y, Pu S, Wu J, Xue Y, Tian Y, Lu J, Jiang W, Du D. Brain-derived neurotrophic factor-activated astrocytes produce mechanical allodynia in neuropathic pain. Neuroscience. 2011;199:452-460
28. Xu C, Hu Y, Hou L, Ju J, Li X, Du N, Guan X, Liu Z, Zhang T, Qin W, Shen N, Bilal MU, Lu Y, Zhang Y, Shan H. β-Blocker carvedilol protects cardiomyocytes against oxidative stress-induced apoptosis by up-regulating miR-133 expression. J Mol Cell Cardiol. 2014;75:111-121
29. Dong DL, Chen C, Huo R, Wang N, Li Z, Tu YJ, Hu JT, Chu X, Huang W, Yang BF. Reciprocal repression between microRNA-133 and calcineurin regulates cardiac hypertrophy: a novel mechanism for progressive cardiac hypertrophy. Hypertension. 2010;55:946-952
30. Hullinger TG, Montgomery RL, Seto AG, Dickinson BA, Semus HM, Lynch JM, Dalby CM, Robinson K, Stack C, Latimer PA, Hare JM, Olson EN, van Rooij E. Inhibition of miR-15 protects against cardiac ischemic injury. Circ Res. 2012;110:71-81
31. Porrello ER, Mahmoud AI, Simpson E, Johnson BA, Grinsfelder D, Canseco D, Mammen PP, Rothermel BA, Olson EN, Sadek HA. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proc Natl Acad Sci U S A. 2013;110:187-192
32. Roy S, Khanna S, Hussain SR, Biswas S, Azad A, Rink C, Gnyawali S, Shilo S, Nuovo GJ, Sen CK. MicroRNA expression in response to murine myocardial infarction: miR-21 regulates fibroblast metalloprotease-2 via phosphatase and tensin homologue. Cardiovasc Res. 2009;82:21-29
33. Chen YQ, Wang XX, Yao XM, Zhang DL, Yang XF, Tian SF, Wang NS. MicroRNA-195 promotes apoptosis in mouse podocytes via enhanced caspase activity driven by BCL2 insufficiency. Am J Nephrol. 2011;34:549-559
34. Varendi K, Kumar A, Härma MA, Andressoo JO. miR-1, miR-10b, miR-155, and miR-191 are novel regulators of BDNF. Cell Mol Life Sci. 2014;71:4443-4456
35. Tian N, Cao Z, Zhang Y. MiR-206 decreases brain-derived neurotrophic factor levels in a transgenic mouse model of Alzheimer's disease. Neurosci Bull. 2014;30:191-197
36. Varendi K, Mätlik K, Andressoo JO. From microRNA target validation to therapy: lessons learned from studies on BDNF. Cell Mol Life Sci. 2015;72:1779-1794
37. Iekushi K, Seeger F, Assmus B, Zeiher AM, Dimmeler S. Regulation of cardiac microRNAs by bone marrow mononuclear cell therapy in myocardial infarction. Circulation. 2012;125:1765-1773 S1-S7
Author contact
![]() Corresponding author: Zhimin Du, PhD, Institute of Clinical Pharmacology of the Second Affiliated Hospital, Harbin Medical University, Xuefu Road 246, Nangang District, Harbin 150086, Heilongjiang Province, P. R. China. Fax: +86 451 86665559, Tel: +86 451 86605353, Email: dzm1956com.
Corresponding author: Zhimin Du, PhD, Institute of Clinical Pharmacology of the Second Affiliated Hospital, Harbin Medical University, Xuefu Road 246, Nangang District, Harbin 150086, Heilongjiang Province, P. R. China. Fax: +86 451 86665559, Tel: +86 451 86605353, Email: dzm1956com.