Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Materials and methods
3. Results
4. Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(8):1000-1009. doi:10.7150/ijbs.13222 This issue Cite
Research Paper
Carbon Monoxide Improves Neurologic Outcomes by Mitochondrial Biogenesis after Global Cerebral Ischemia Induced by Cardiac Arrest in Rats
Peng Wang1,2#, Lan Yao1,2,3#, Li-li Zhou1,2, Yuan-shan Liu1,2, Ming-di Chen1,2, Hai-dong Wu1,2, Rui-ming Chang1,2, Yi Li1,2, Ming-gen Zhou1,2, Xiang-shao Fang1,2, Tao Yu1,2, Long-yuan Jiang1,2, Zi-tong Huang1,2 ![]()
1. Department of Emergency Medicine, Sun Yat-sen Memorial Hospital of Sun Yat-sen University, Guangzhou, China
2. Institute of Cardiopulmonary Cerebral Resuscitation, Sun Yat-sen University, Guangzhou, China
3. Department of Emergency Medicine, The fifth affiliated hospital, Sun Yat-sen University, Zhuhai, China
# These authors contributed equally to this study.
Received 2015-7-11; Accepted 2016-6-20; Published 2016-7-17
Abstract

Mitochondrial dysfunction contributes to brain injury following global cerebral ischemia after cardiac arrest. Carbon monoxide treatment has shown potent cytoprotective effects in ischemia/reperfusion injury. This study aimed to investigate the effects of carbon monoxide-releasing molecules on brain mitochondrial dysfunction and brain injury following resuscitation after cardiac arrest in rats. A rat model of cardiac arrest was established by asphyxia. The animals were randomly divided into the following 3 groups: cardiac arrest and resuscitation group, cardiac arrest and resuscitation plus carbon monoxide intervention group, and sham control group (no cardiac arrest). After the return of spontaneous circulation, neurologic deficit scores (NDS) and S-100B levels were significantly decreased at 24, 48, and 72 h, but carbon monoxide treatment improved the NDS and S-100B levels at 24 h and the 3-day survival rates of the rats. This treatment also decreased the number of damaged neurons in the hippocampus CA1 area and increased the brain mitochondrial activity. In addition, it increased mitochondrial biogenesis by increasing the expression of biogenesis factors including peroxisome proliferator-activated receptor-γ coactivator-1α, nuclear respiratory factor-1, nuclear respiratory factor-2 and mitochondrial transcription factor A. Thus, this study showed that carbon monoxide treatment alleviated brain injury after cardiac arrest in rats by increased brain mitochondrial biogenesis.
Keywords: carbon monoxide, brain injury, cardiac arrest, mitochondria biogenesis
1. Introduction
Global cerebral ischemia is a clinical outcome of cardiac arrest. Cardiopulmonary cerebral resuscitation is essential for the treatment of cardiac arrest. However, patients experiencing the return of spontaneous circulation (ROSC) after cardiac arrest have high morbidity and mortality rates owing to a systemic ischemia/reperfusion response involving multiple organs. Brain injury is a major cause of morbidity and mortality and is responsible for approximately two-thirds of the deaths in the post-cardiac arrest period [1]. Global cerebral ischemia during cardiac arrest and reperfusion after ROSC result in heterogeneous injury to the brain, and the hippocampus and brain cortex are the most vulnerable areas [2]. Mild therapeutic hypothermia has been proven to be effective for neurological protection after cardiac arrest in both experimental and clinical studies [3], but some other current treatments are limited. Therefore, many pharmacological neuroprotective drugs that focus on specific brain injury mechanisms after cardiac arrest have been developed and have shown benefits in preclinical studies.
Mitochondria are vital organelles in cells that not only produce adenosine triphosphate (ATP) for cellular functions but also participate in a number of intracellular processes such as cell division, oxidative response, metabolic signaling pathways, regulation of cytosolic Ca2+ concentration, and determination of cell death or survival [4]. Mitochondrial dysfunction is central in the pathogenesis of numerous human diseases including neurodegenerative diseases, cardiovascular diseases, diabetes, and cancer. In recent years, mitochondrial abnormalities have been identified in many ischemia/reperfusion injuries such as mitochondrial ultrastructure, mitochondrial respiration, redox state, Ca2+ overload, fusion/fission, and biogenesis [4-6]. It is evident that knowledge regarding mitochondrial dysfunction in pathological states is crucial not only for our understanding of the basic cause of disease but also for developing therapeutic strategies by alleviating mitochondrial dysfunction to attenuate a given disease process. For example, inhibiting mitochondrial Ca2+ overload, preventing mitochondrial permeability transition pore opening, and maintaining normal mitochondrial energy metabolism are potential ways to attenuate cardiac and vascular dysfunction [7-10].
Mitochondrial biogenesis is a highly regulated process by which new mitochondria are formed in cells, where it is controlled by the nuclear genome. Alteration in mitochondrial biogenesis is usually triggered by mitochondrial dysfunction or high-energy demands during some pathophysiological conditions. Several transcriptional regulators are involved in the process of mitochondrial biogenesis. Nuclear respiratory factor (NRF)-1 and NRF-2 are important contributors leading to the increase in the transcription of key mitochondrial enzymes and have been shown to interact with mitochondrial transcriptional factor A (TFAM), which drives the transcription and replication of mitochondrial DNA [11, 12]. Peroxisome proliferator-activated receptor-γ coactivator-1α (PGC-1α) is a major regulator of mitochondrial biogenesis that induces biogenesis by activating different transcription factors including NRF-1 and NRF-2, which promote the expression of TFAM [11, 12].
Carbon monoxide (CO) is best known as a toxic gas, which binds to hemoglobin and forms carboxyhemoglobin and severely impairs the respiratory system. However, CO is also generated physiologically in cells through the breakdown of heme by heme oxygenase enzymes and has potent antioxidant and anti-inflammatory activities. Therapeutic application of exogenous CO can be achieved by direct inhalation of CO gas, which has shown potent cytoprotective effects in liver transplantation, lung injury, and stroke. However, the applicability of gaseous CO is limited owing to its toxic effects with prolonged exposure and the difficulty in controlling the gas [13, 14]. Therefore, CO-releasing molecules (CORMs) were developed to allow selective delivery and local release of CO from a nontoxic prodrug, which has been shown to be a valid alternative to the use of gaseous CO. A metal carbonyl complex tricarbonyldichlororuthenium(II) dimer known as CORM-2 can efficiently release defined amounts of CO and shows biological activities, including protection of cardiac function, attenuation of liver inflammation, and improvement in ischemia/reperfusion outcomes, without significant toxicity [15-17]. Importantly, CORM-2 treatment is associated with minimal formation of carboxyhemoglobin [13], suggesting that it is a safer alternative to CO gas inhalation.
The role of CO in the modulation of mitochondrial dysfunction and regulation of mitochondrial biogenesis-associated brain injury after cardiac arrest has not yet been investigated. In the present study, we investigated whether exogenous CO has a therapeutic effect on brain injury and improves mitochondrial activity by inducing mitochondrial biogenesis after global cerebral ischemia in a rat model of cardiac arrest.
2. Materials and methods
Animals and drugs
Male Sprague-Dawley rats (body weight, 370-420 g) were purchased from the Medical Experimental Animal Center of Guangdong Province (Guangzhou, China). Animals were maintained on laboratory chow and housed in a specific pathogen-free room at a constant temperature (20-22°C) with 10 h of light and 14 h of dark exposure. All animal studies were approved by the Institutional Animal Care and Use Committee of Sun Yat-sen University, and all experiments were performed in accordance with the National Institutes of Health guidelines for the care and use of laboratory animals.
The tricarbonyldichlororuthenium(II) dimer (CORM-2) was obtained from Sigma Aldrich (St. Louis, MO, USA) and was dissolved in dimethyl sulfoxide as a stock solution. The inactive form of CORM-2 (iCORM-2) was prepared as described [18]. Vecuronium was purchased from ShiQiao BioPharm (Shanghai, China). Pentobarbital sodium was purchased from Sigma Aldrich (St. Louis, MO, USA).
Animal preparation and drug treatment
Cardiac arrest was induced by asphyxia and this procedure has been described in detail previously [19]. Briefly, after an overnight fast except for free access to water, the rats were anesthetized by intraperitoneal injection of pentobarbital sodium (45 mg/kg). The trachea was orally intubated with a 14-gauge cannula (Abbocath-T, USA). A 23-gauge polyethylene-50 (PE-50) catheter (Abbocath-T, USA) was advanced through the left femoral artery for measuring blood pressure. An additional 23-gauge PE-50 catheter was advanced through the left femoral vein into the inferior vena cava for infusing the CORM-2 or placebo. Mean arterial pressure (MAP) was measured using a pressure transducer (BD, Germany). A conventional electrocardiogram lead II was continuously recorded. Hemodynamic data were continuously measured and recorded in a computer-based data acquisition system (WinDaq acquisition system, USA).
Asphyxia was induced by arterial injection of vecuronium (1 mg/kg) and discontinuation of ventilation. Cardiac arrest was defined as a MAP of ≤20 mmHg, which occurred approximately 4.5 min after asphyxia. After 6 min of cardiac arrest, cardiopulmonary resuscitation (CPR) was started. ROSC was defined as the return of supraventricular rhythm with a MAP of ≥60 mmHg for a minimum of 5 min. Mechanical ventilation was continued for 1 h after ROSC, during which FiO2 was 100% at 30 min, 50% at 20 min, and 30% at 10 min.
After successful resuscitation, the animals were randomly divided into 3 groups: (1) CPR group, cardiac arrest was induced and CPR was performed in the rats; (2) CPR+CO group, cardiac arrest was induced and CPR was performed in the rats, after which CORM-2 (or iCORM-2) treatment was administered; (3) Control group, the same operation was performed, but cardiac arrest was not induced and CPR was not performed in the rats.
Thirty minutes after ROSC, the CPR+CO group rats received a continuous intravenous infusion of CORM-2 (or iCORM-2) at a rate of 10 mg/kg/h for 30 min (5 mg/kg) according to the dose as previously described [20]. Meanwhile, the control and CPR rats received a placebo (vehicle dimethyl sulfoxide with 0.9% NaCl solution) at a rate of 4 mL/kg/h.
Neurological functional examination
Neurologic deficit scores (NDS) were measured by 2 investigators who were unaware of the group identities, as previously described [21]. NDS assessment involves the scoring of 19 items including arousal, reflex, motor, sensory, and balance responses, resulting in scores ranging from 0 (brain death) to 80 (normal brain function). In this study, dead rats were excluded from the NDS assessment.
Serum S-100B concentrations
Intravenous blood was obtained from the inferior vena cava. After whole blood collection, the blood was allowed to clot by leaving it undisturbed at room temperature. The clot was removed by centrifugation at 1500 g for 15 min and the serum was stored at -80°C until use. Serum S-100B concentrations were determined using a rat S-100B enzyme-linked immunosorbent assay kit (Cusabio, China) according to the manufacturer instructions.
Measurement of ATP levels
ATP levels in brain tissues were measured using an ATP assay kit (Beyotime, China). Briefly, brain tissues were harvested and homogenized in an ice-cold ATP-releasing buffer and then centrifuged at 12,000 g for 5 min. After centrifugation, the supernatant was transferred to a new tube for ATP testing. Luminescence from a 100 μL sample was assayed in a luminometer (Molecular Devices, SpectraMax M5) together with 100 μL of ATP detection buffer and was then normalized by protein concentration.
Measurement of mitochondrial respiration and mitochondrial membrane potential (ΔΨm) levels
Rat brain mitochondria were isolated using a Qproteome mitochondria isolation kit (Qiagen, Germany) according to the manufacturer instructions. Mitochondria were added to the respiration buffer (GenMed Scientifics, USA), and respiratory function was measured using an oxygen electrode (Oxygraph, Hansatech Instruments, UK). State III mitochondrial respiration was stimulated by the addition of adenosine 5´-diphosphate (ADP, 0.5 mmol/L). On ADP depletion, state IV respiration was measured. The mitochondrial respiratory control ratio (RCR) was calculated as the ratio of the respiratory rate in state III to that in state IV. The mitochondrial membrane potential (ΔΨm) levels were measured using a JC-1 mitochondrial membrane potential detection kit (Beyotime, China) according to the manufacturer instructions. Mitochondrial fluorescence was measured using a microplate reader (Molecular Devices, SpectraMax M5). The RCR and ΔΨm levels were normalized by mitochondrial protein concentration.
Nissl staining and terminal deoxyribonucleotide transferase-mediated dUTP nick-end labeling (TUNEL) assay
Brain tissues were harvested and immediately fixed in 4% formaldehyde in 0.1 mol/L phosphate buffer. The paraffin-embedded brain tissues were cut into 5-μm sections. Nissl staining (Beyotime, China) and TUNEL assay (Roche, USA) were performed for in situ detection of neuronal injury, according to the manufacturer instructions. Seven fields of vision (40× magnification) in the hippocampal CA1 area were randomly selected from each brain tissue sample, and the Nissl- or TUNEL-stained neuronal cells were counted.
Specimen preparation for transmission electron microscopy (TEM)
The brain hippocampus area was isolated and immediately fixed in cold 2.5% glutaraldehyde with 0.1 mol/L cacodylate buffer (pH 7.4), post-fixed in 1% osmium tetroxide, dehydrated, and embedded in Epon. According to the standard stereological principle of randomization, ultrathin sections (60-80 μm) were randomly cut regardless of the orientation and mounted on copper grids, stained with lead citrate and uranyl acetate, and then viewed using a transmission electron microscope (FEI, Tecnai G2).
Real-time reverse transcription-polymerase chain reaction (RT-PCR) analysis
Total RNA was extracted from the hippocampus and brain cortex using TRIzol solution (Invitrogen, USA) according to the manufacturer instructions. RNase-free DNase I was used to remove DNA contamination. Reverse transcription was performed using PrimeScript RT master mix (TaKaRa, China). Next, cDNA amplification was performed by real-time PCR (Bio-Rad, USA) using SYBR Premix Ex Taq mix (TaKaRa, China). The specific primers for PGC-1α, NRF-1, NRF-2, TFAM, and actin (an internal control) were as follows: PGC-1α, 5´-AATGCAGCGGTCTTAGCACT-3´ (forward), 5´-TTTCTGTGGGTTTGGTGTGA-3´ (reverse); NRF-1, 5´-TGGTCCAGAGAGTGCTTGTG-3´ (forward), 5´-TTCCTGGGAAGGGAGAAGAT-3´ (reverse); NRF-2, 5´-CTCGCTGGAAAAAGAAGTGG-3´ (forward), 5´-CCGTCCAGGAGTTCAGAGAG-3´ (reverse); TFAM, 5´-GTTTGAAGGTGGTGGAAGGA-3´ (forward), 5´-TTGCTCTGAAGCACATGGTC-3´ (reverse); actin, 5´-AACTCCATCATGAAGTGTGACG-3´ (forward), 5´-GATCCACATCTGCTGGAAGG-3´ (reverse). Real-time PCR was performed at 95°C for 2 min, followed by 40 cycles at 95°C for 3 s and at 60°C for 30 s. The relative mRNA expression levels of the target genes were measured using the 2-ΔΔCt method normalized to mRNA levels for actin.
Western blot analysis
The brain tissues were homogenized in radioimmunoprecipitation assay buffer, and the lysates were cleared by centrifugation (14,000 rpm) at 4°C for 30 min. Protein samples (approximately 40 μg each) were separated on a 10% sodium dodecyl sulfate-polyacrylamide gel, and the fractionated proteins were transferred to polyvinylidene difluoride membranes. After blocking the nonspecific binding sites with 5% nonfat milk for 60 min, the membranes were incubated overnight at 4°C with primary monoclonal antibodies against PGC-1α (Cell Signaling Technology, 1:1000 dilution), NRF-2 (Santa Cruz, 1:1000 dilution), and α-tubulin (Cell Signaling Technology, 1:5000 dilution). Next, the membranes were subjected to three 15-min washes with Tris-buffered saline and Tween 20 (TBST) and then incubated with horseradish peroxidase-conjugated secondary antibody (1:2000 dilution) for 45 min at room temperature. The membranes were washed 3 more times with TBST and developed using an enhanced chemiluminescence system (Cell Signaling Technology).
Mitochondrial DNA copy number determination
Total DNA (genomic and mitochondrial) was extracted from brain tissues by using the QIAamp DNA mini kit (Qiagen, Germany). The relative mitochondrial DNA (mtDNA) copy number was measured by real-time PCR using SYBR and corrected by simultaneous measurement of nuclear DNA. The forward and reverse primers for mtDNA, which are complementary to the sequence of the mitochondrial cytochrome c oxidase subunit II gene, were 5´-TTGGTCTACAAGACGCCACA-3´ (forward) and 5´-CGGTTAATACGGGGTTGTTG-3´ (reverse), respectively. The 5´-GAGGATGAGGTGGAACGTGT-3´ (forward) and 5´-GGACCTGGCTGTATTTTCCA-3´ (reverse) sequences, complementary to the 18S gene, were the primers used for the detection of nuclear DNA. The relative mtDNA copy number was normalized against the nuclear-encoded 18S gene. The relative fold change was then calculated using the 2-ΔΔCt method.
Statistical analysis
All measurement data were presented as mean ± standard deviation (SD), and statistical analyses were performed using SPSS version 19.0 for Windows. An unpaired t test was used to compare the same parameters between 2 groups, and one-way analysis of variance was used to compare the same parameters among 3 groups. Differences between pairs of means were assessed post hoc using Tukey's test. Fisher's exact test was employed to compare the NDS between the groups. Differences in the survival rates between the groups were compared using Kaplan-Meier survival analysis. A P value of < 0.05 was considered statistically significant.
3. Results
Significant decrease in neurological and brain mitochondrial functions after cardiac arrest in rats
NDS were measured to estimate the neurological outcomes after cardiac arrest in the animals. These scores ranged from 0 (brain death) to 80 (normal brain function) and were significantly decreased at 24, 48, and 72 h after ROSC (Fig. 1A). Serum S-100 is a prognostic marker of neurological outcomes after cardiac arrest and is therefore used to predict neurological injuries [22, 23]. The S-100B levels increased significantly at 24, 48, and 72 h after ROSC (Fig. 1B). Therefore, the NDS and S-100B levels indicate that neurological injuries occurred after cardiac arrest and were more severe at 24 h after ROSC.
Neurological and brain mitochondrial functions significantly decreased after cardiac arrest in rats. (A) Neurologic deficit scores (NDS) were measured at 24, 48, and 72 h after the return of spontaneous circulation (ROSC). The NDS ranged from 0 (brain death) to 80 (normal brain function) and significantly decreased within 3 days after ROSC. Horizontal bars present mean values. (B) Serum S-100B levels were measured using enzyme-linked immunosorbent assay at 24, 48, and 72 h after ROSC. The S-100B levels significantly increased within 3 days after ROSC. (C) Brain ATP levels significantly decreased at 24, 48, and 72 h after ROSC. (D) Brain mitochondrial respiratory control ratios (RCR) significantly decreased at 24 h, 48, and 72 h after ROSC. Results are presented as mean ± standard deviation (SD). n = 5-10, *P < 0.05, **P < 0.01.
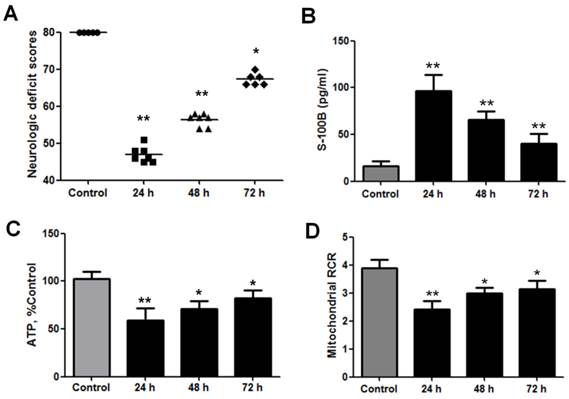
Since mitochondrial energy metabolism is vital for brain injury after cardiac arrest, ATP production and brain mitochondrial respiration were studied to evaluate mitochondrial activity after ROSC. The results revealed that brain ATP levels significantly decreased at 24, 48, and 72 h after ROSC (Fig. 1C). The mitochondrial RCR indicates the tightness of the coupling between mitochondrial respiration and phosphorylation. The results also showed that brain mitochondrial RCRs significantly decreased at 24, 48, and 72 h after ROSC (Fig. 1D). Taken together, these data indicate that mitochondrial activities significantly decreased after ROSC, especially at 24 h.
CO treatment improved neurological outcomes after cardiac arrest
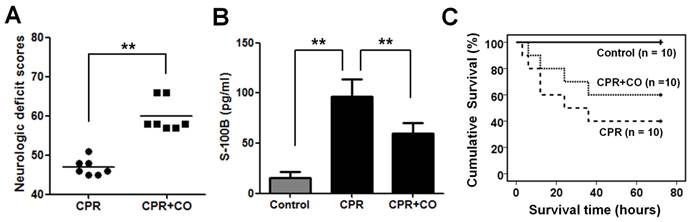
CORM-2 treatment was used to explore the effects of CO on neurological functions after cardiac arrest in rats. To exclude direct effect of CORM-2 to the brain function in the rats, we first used iCORM-2 to treat the rats and detected its effects to the rats. The results demonstrated that the iCORM-2 treatment has no effect on brain NDS, S-100B levels, ATP levels and mitochondrial RCRs (Fig. S1A-D). However, The CORM-2 treatment results showed that CO increased NDS (Fig. 2A) and decreased S-100B levels (Fig. 2B) significantly at 24 h after ROSC. Kaplan-Meier survival analysis showed that CO treatment significantly increased the 3-day survival rates of the rats after ROSC (Fig. 2C). Taken together, these data suggest that CO treatment improves neurological outcomes after cardiac arrest.
CO treatment alleviated neurological injury after cardiac arrest and increased the survival time of rats. (A) CO treatment significantly increased the NDS of the rats at 24 h after ROSC. Results are presented as mean ± SD. n = 7, **P < 0.01. Horizontal bars present mean values. (B) CO treatment significantly decreased the serum S-100B levels in the rats at 24 h after ROSC. Results are presented as mean ± SD. n = 5, **P < 0.01. (C) CO treatment significantly increased the survival time of the rats after ROSC. CPR+CO vs. CPR group, P < 0.05, by Kaplan-Meier survival analysis. Control indicates the control group; CPR indicates the cardiopulmonary resuscitation (CPR) group; CPR+CO indicates the CPR plus CO treatment group.
CO treatment protected brain neurons from injuries after ROSC
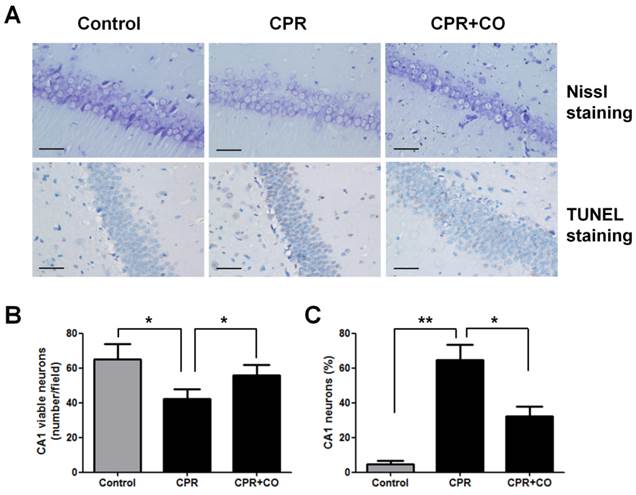
To evaluate the effects of CO on brain neurons, Nissl staining and TUNEL assay were performed to detect neuronal damage in the hippocampal CA1 area at 24 h after ROSC. Nissl staining revealed that there were more Nissl bodies in the cytoplasm of neurons in the CPR+CO group than in the CPR group (Fig. 3A). The number of viable neurons significantly decreased in the CPR group compared with the control group (P < 0.05) and significantly increased in the CPR+CO group compared with the CPR group (P < 0.05) (Fig. 3B). TUNEL staining showed a large number of strongly TUNEL-positive apoptotic cells in the CPR group and a few weakly positive cells in the CPR+CO group (Fig. 3A). The number of TUNEL-positive apoptotic cells significantly increased in the CPR group compared with the control group (P < 0.01); moreover, the number of these cells was significantly lower in the CPR+CO group than in the CPR group (P < 0.05) (Fig. 3C).
CO treatment protected the hippocampal CA1 neurons at 24 h after ROSC. (A) Representative images of Nissl and TUNEL staining of the hippocampal CA1 region of the 3 groups (×400). Scale bar = 10 μm. (B) Quantification of the viable hippocampal CA1 neurons by Nissl staining. (C) Quantification of the apoptotic hippocampal CA1 neurons by TUNEL staining. Data in (B) and (C) are shown as mean ± SD. For each group, n = 6 and 7 photomicrographs were counted per animal. *P < 0.05, **P < 0.01.
CO treatment increased brain mitochondrial activities after ROSC
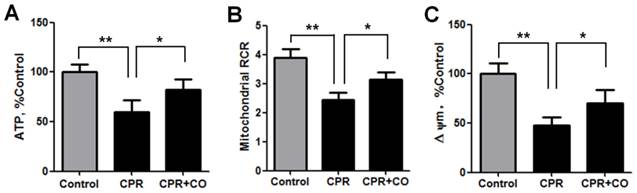
To explore the effects of CO on brain mitochondrial activities, the ATP levels and mitochondrial RCR were measured at 24 h after ROSC. The data showed that CO treatment significantly increased the ATP levels (Fig. 4A) and mitochondrial RCR (Fig. 4B). Furthermore, CO treatment significantly increased the ΔΨm, another indicator of mitochondrial function, at 24 h after ROSC (Fig. 4C). Taken together, these data show that CO treatment alleviated the mitochondrial dysfunction occurring after ROSC.
CO treatment increased brain mitochondrial activity after ROSC. (A) CO treatment increased brain ATP levels at 24 h after ROSC. (B) CO treatment increased the brain mitochondrial RCR at 24 h after ROSC. (C) CO treatment increased the brain mitochondrial membrane potential at 24 h after ROSC. Data are shown as mean ± SD. n = 5, *P < 0.05, **P < 0.01.
CO treatment increased brain mitochondrial biogenesis after ROSC
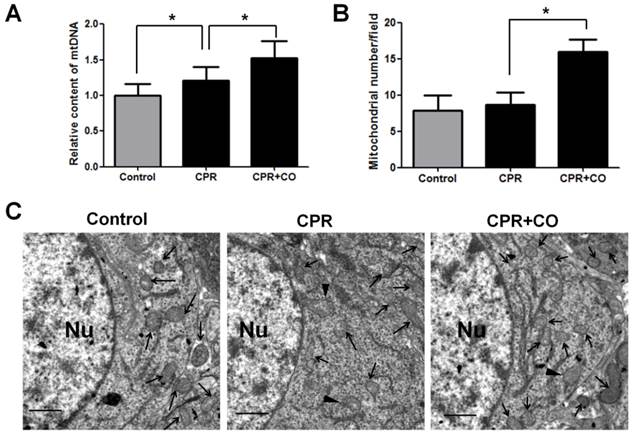
To further determine the effects of CO on mitochondrial function, the mitochondrial amount was measured because CO treatment caused an increase in the ATP levels, mitochondrial RCR, and ΔΨm. Mitochondrial DNA (mtDNA) copy number analysis showed that CO treatment significantly increased brain mtDNA copy number at 24 h after ROSC (Fig. 5A). Moreover, TEM analysis showed that CO treatment significantly increased the brain mitochondrial amount at 24 h after ROSC (Fig. 5B-C). Further, a few damaged mitochondria with broken or disrupted cristae were observed in the CPR group compared with the control group, whereas fewer damaged mitochondria were observed in the CPR+CO group (Fig. 5C). Therefore, these data suggest that CO treatment increased brain mitochondrial biogenesis and alleviated mitochondrial damage after ROSC.
CO treatment increased brain mitochondrial biogenesis after ROSC. (A) The brain mitochondrial DNA (mtDNA) copy number was measured using real-time polymerase chain reaction (PCR). CO treatment increased the brain mtDNA copy number at 24 h after ROSC. (B) Quantification of the brain mitochondrial number per photograph by transmission electron microscopy (TEM) at 24 h after ROSC. For each group, n = 3 and 7 photomicrographs were counted per animal. (C) Representative TEM micrographs of brain mitochondria at 24 h after ROSC. The neuronal nucleus (Nu) is surrounded by relatively uniform and compact mitochondria. Arrows indicate mitochondria; arrowheads indicate damaged mitochondria. Scale bar = 1 μm. Data are shown as mean ± SD. n = 3, *P < 0.05.
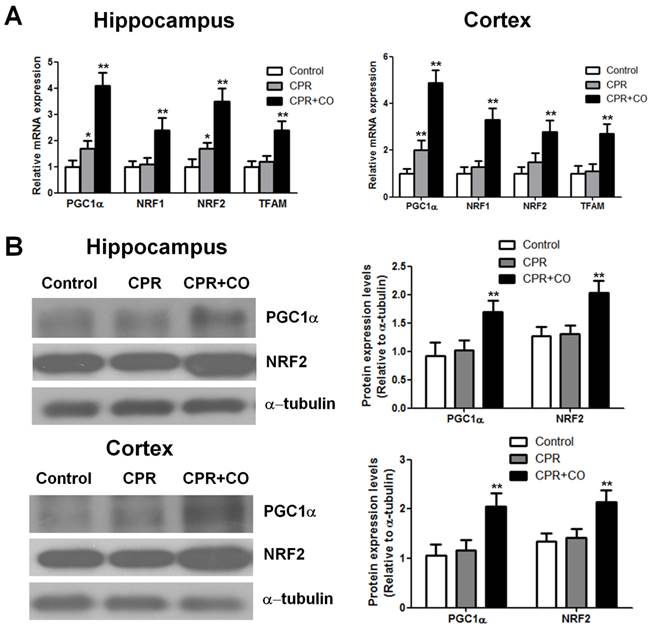
CO treatment increased mitochondrial biogenesis factor expression after ROSC
Transcription and replication of mtDNA are regulated by some nucleus-encoded transcription factors and coactivators. To investigate the effects of CO on mtDNA transcription, some important biogenesis factors were detected at 24 h after ROSC. Real-time PCR analysis showed that CO treatment significantly increased the PGC-1α, NRF-1, NRF-2, and TFAM mRNA expression levels in the hippocampus and brain cortex at 24 h after ROSC (P < 0.01) (Fig. 6A). Furthermore, western blot analysis revealed that CO treatment significantly increased the PGC-1α and NRF-2 protein expression levels in the hippocampus and brain cortex at 24 h after ROSC (Fig. 6B). Thus, these data showed that CO treatment increased brain mitochondrial biogenesis after ROSC by regulating the mitochondrial biogenesis factor mRNA and protein expression levels.
CO treatment increased mitochondrial biogenesis factor expression in the hippocampus and brain cortex at 24 h after ROSC. (A) CO treatment increased the mRNA expression of mitochondrial biogenesis factors (PGC-1α, NRF-1, NRF-2, and TFAM) in the hippocampus and brain cortex at 24 h after ROSC. The PGC-1α, NRF-1, NRF-2, and TFAM mRNA expression levels were assessed using real-time reverse transcription-PCR. Data are shown as mean ± SD. n = 5, *P < 0.05, **P < 0.01. (B) CO treatment increased PGC-1α and NRF-2 protein expression levels in the hippocampus and brain cortex at 24 h after ROSC. Representative results of western blot analysis of PGC-1α and NRF-2 protein expression in the hippocampus and brain cortex after CO treatment at 24 h after ROSC (left). α-tubulin was used as the loading control. PGC-1α and NRF-2 protein expressions were quantified by western blot band intensity measurement (right). Data are shown as mean ± SD. n = 3, **P < 0.01.
4. Discussion
In the present study, we found that brain neurologic and mitochondrial functions decreased after cardiac arrest and ROSC, and the neurologic functions were consistent with brain mitochondrial activity at 24, 48, and 72 h after ROSC. The brain neurologic and mitochondrial functions were severely damaged at 24 h after ROSC, whereas these functions were restored to some extent at 72 h after ROSC. Therefore, our results suggest that there is a close relationship between mitochondrial function and brain function after cardiac arrest and ROSC. Indeed, mitochondrial homeostasis is necessary to maintain neuronal function because neurons have a high energy requirement. Disruptions in mitochondrial function are associated with neurological diseases. Some studies have reported that mitochondrial dysfunction occurs in both acute and chronic neurologic disorders [24-26]. Mild mitochondrial injury, with maintenance of near-normal cellular ATP, results in mainly programmed cell death, while more extensive injury that causes ATP depletion results in cell necrosis [27]. Therefore, mitochondria have become increasingly important targets for neuroprotective interventions.
Brain injury after cardiac arrest and resuscitation is a common cause of morbidity and mortality. The most common mechanism of brain injury after cardiac arrest and resuscitation is global cerebral ischemia caused by systemic hypoperfusion. Some mechanisms involved in mitochondrial dysfunction after brain injury triggered by cardiac arrest and resuscitation include inhibition of oxidative phosphorylation, disruption of calcium homeostasis, formation of free radicals, and activation of cell death signaling pathways [2]. In recent years, some therapeutic interventions that target mitochondrial dysfunction have been proven to be effective for post-cardiac arrest brain injury in animal models. For example, nitrite therapy after cardiac arrest reduces reactive oxygen species generation, improves neurological function, and enhances survival via reversible inhibition of mitochondrial complex I [28]. Therapeutic hypothermia is thought to prevent brain injury through several mechanisms including protection of brain mitochondrial activity [29]. In the present study, our results demonstrated that CO treatment improved brain function after cardiac arrest by protecting mitochondrial activity in a rat model.
Mitochondrial biogenesis is important for cell homeostasis and is essential for cells and tissues to survive and recover from mitochondrial damage, for example, ischemic injury, as well as from other damage that activates energy-requiring repair processes. Some studies have proven that enhancing mitochondrial biogenesis reduces ischemic cerebral injury. A study showed that the glycogen synthase kinase-3 (GSK-3) inhibitor SB216763 activated an efficient mitochondrial biogenesis program in cortical neurons and counteracted oxygen-glucose deprivation-mediated neuronal damage [30]. Sodium hydrosulfide treatment improved neurological outcomes at 24 h after ROSC by allowing maintenance of mitochondrial function by improving mitochondrial biogenesis in rats [31]. In the present study, CO administration increased mitochondrial ATP production, respiration, and membrane potential at 24 h after ROSC via mitochondrial biogenesis and therefore decreased brain injury after cardiac arrest. However, mitochondrial biogenesis can also be induced by ischemic or hypoxic damage and can help in maintaining aerobic metabolism by increasing the abundance of mitochondria [32-34]. A previous study showed that brain mtDNA content was markedly increased at 6 h after neonatal hypoxic/ischemic brain injury and continued to increase up to 24 h [34]. Similarly, in our rat cardiac arrest model, we found that brain mtDNA content was increased at 24 h after ROSC.
Our data showed that the PGC-1α, NRF-1, NRF-2, and TFAM mRNA and protein expression levels in the hippocampus and brain cortex increased after CO treatment at 24 h after ROSC. Thus, CO treatment increased brain mitochondrial biogenesis after ROSC by regulating the mitochondrial biogenesis factor expression. CO activation of mitochondrial biogenesis involves the prosurvival kinase Akt/PKB [35, 36]. Akt then phosphorylates NRF-1 and promotes its nuclear localization and inactivates GSK-3β, which improves the nuclear translocation of NRF-2. The accumulation of NRF-1 and NRF-2 leads to gene activation for mitochondrial biogenesis [37]. CO may also activate guanylate cyclase, which produces cyclic guanosine monophosphate (cGMP) as the second messenger effector. Mitochondrial biogenesis is regulated by cGMP by increasing the expression of the PGC-1α, NRF-1, and TFAM genes [38, 39].
In conclusion, our study demonstrated that CO treatment improved neurological outcomes after cardiac arrest and resuscitation by maintaining mitochondrial function by improving mitochondrial biogenesis. We consider that mitochondria-targeted therapy is a new intervention for brain injury during resuscitation and that CO may be a promising candidate for the treatment of brain injury after cardiac arrest and resuscitation. However, determination of the optimal dose of CO is important to achieve the therapeutic benefits effects of CO. At low concentrations, CO shows cytoprotective on effects and can activate distinct endogenous cell defense pathways, whereas at high concentrations, it may show cytotoxicity. Thus, further studies are needed to optimize the therapeutic dose of CO for brain injury after cardiac arrest to achieve more potent therapeutic effects.
Supplementary Material
Figure S1.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (81501137) and the Natural Science Foundation of Guangdong Province (2015A030310042).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Stub D, Bernard S, Duffy SJ, Kaye DM. Post cardiac arrest syndrome: a review of therapeutic strategies. Circulation. 2011;123:1428-35
2. Harukuni I, Bhardwaj A. Mechanisms of brain injury after global cerebral ischemia. Neurol Clin. 2006;24:1-21
3. Bernard SA, Gray TW, Buist MD, Jones BM, Silvester W, Gutteridge G. et al. Treatment of comatose survivors of out-of-hospital cardiac arrest with induced hypothermia. N Engl J Med. 2002;346:557-63
4. Camara AK, Bienengraeber M, Stowe DF. Mitochondrial approaches to protect against cardiac ischemia and reperfusion injury. Front Physiol. 2011;2:13
5. Honda HM, Korge P, Weiss JN. Mitochondria and ischemia/reperfusion injury. Ann N Y Acad Sci. 2005;1047:248-58
6. Perez-Pinzon MA, Stetler RA, Fiskum G. Novel mitochondrial targets for neuroprotection. J Cereb Blood Flow Metab. 2012;32:1362-76
7. Cour M, Loufouat J, Paillard M, Augeul L, Goudable J, Ovize M. et al. Inhibition of mitochondrial permeability transition to prevent the post-cardiac arrest syndrome: a pre-clinical study. Eur Heart J. 2011;32:226-35
8. Ayoub IM, Kolarova JD, Kantola RL, Radhakrishnan J, Wang S, Gazmuri RJ. Zoniporide preserves left ventricular compliance during ventricular fibrillation and minimizes postresuscitation myocardial dysfunction through benefits on energy metabolism. Crit Care Med. 2007;35:2329-36
9. Ayoub IM, Radhakrishnan J, Gazmuri RJ. Targeting mitochondria for resuscitation from cardiac arrest. Crit Care Med. 2008;36:S440-6
10. Gazmuri RJ, Ayoub IM, Kolarova JD, Karmazyn M. Myocardial protection during ventricular fibrillation by inhibition of the sodium-hydrogen exchanger isoform-1. Crit Care Med. 2002;30:S166-71
11. Scarpulla RC, Vega RB, Kelly DP. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab. 2012;23:459-66
12. Wenz T. Regulation of mitochondrial biogenesis and PGC-1alpha under cellular stress. Mitochondrion. 2013;13:134-42
13. Motterlini R, Otterbein LE. The therapeutic potential of carbon monoxide. Nat Rev Drug Discov. 2010;9:728-43
14. Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006;86:583-650
15. Wang X, Qin W, Qiu X, Cao J, Liu D, Sun B. A novel role of exogenous carbon monoxide on protecting cardiac function and improving survival against sepsis via mitochondrial energetic metabolism pathway. Int J Bio Sci. 2014;10:777-88
16. Wei Y, Chen P, de Bruyn M, Zhang W, Bremer E, Helfrich W. Carbon monoxide-releasing molecule-2 (CORM-2) attenuates acute hepatic ischemia reperfusion injury in rats. BMC Gastroenterol. 2010;10:42
17. Bolognesi M, Sacerdoti D, Piva A, Di Pascoli M, Zampieri F, Quarta S. et al. Carbon monoxide-mediated activation of large-conductance calcium-activated potassium channels contributes to mesenteric vasodilatation in cirrhotic rats. J Pharmacol Exp Ther. 2007;321:187-94
18. Sawle P, Foresti R, Mann BE, Johnson TR, Green CJ, Motterlini R. Carbon monoxide-releasing molecules (CO-RMs) attenuate the inflammatory response elicited by lipopolysaccharide in RAW 264.7 murine macrophages. Br J Pharmacol. 2005;145:800-10
19. Fink EL, Alexander H, Marco CD, Dixon CE, Kochanek PM, Jenkins LW. et al. Experimental model of pediatric asphyxial cardiopulmonary arrest in rats. Pediatr Crit Care Med. 2004;5:139-44
20. Van Phan T, Sul OJ, Ke K, Lee MH, Kim WK, Cho YS. et al. Carbon monoxide protects against ovariectomy-induced bone loss by inhibiting osteoclastogenesis. Biochem Pharmacol. 2013;85:1145-52
21. Geocadin RG, Ghodadra R, Kimura T, Lei H, Sherman DL, Hanley DF. et al. A novel quantitative EEG injury measure of global cerebral ischemia. Clin Neurophysiol. 2000;111:1779-87
22. Bottiger BW, Mobes S, Glatzer R, Bauer H, Gries A, Bartsch P. et al. Astroglial protein S-100 is an early and sensitive marker of hypoxic brain damage and outcome after cardiac arrest in humans. Circulation. 2001;103:2694-8
23. Rosen H, Sunnerhagen KS, Herlitz J, Blomstrand C, Rosengren L. Serum levels of the brain-derived proteins S-100 and NSE predict long-term outcome after cardiac arrest. Resuscitation. 2001;49:183-91
24. Bayir H, Kochanek PM, Kagan VE. Oxidative stress in immature brain after traumatic brain injury. Dev Neurosci. 2006;28:420-31
25. Calkins MJ, Manczak M, Mao P, Shirendeb U, Reddy PH. Impaired mitochondrial biogenesis, defective axonal transport of mitochondria, abnormal mitochondrial dynamics and synaptic degeneration in a mouse model of Alzheimer's disease. Hum Mol Genet. 2011;20:4515-29
26. Cui L, Jeong H, Borovecki F, Parkhurst CN, Tanese N, Krainc D. Transcriptional repression of PGC-1alpha by mutant huntingtin leads to mitochondrial dysfunction and neurodegeneration. Cell. 2006;127:59-69
27. Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA. et al. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961-73
28. Dezfulian C, Shiva S, Alekseyenko A, Pendyal A, Beiser DG, Munasinghe JP. et al. Nitrite therapy after cardiac arrest reduces reactive oxygen species generation, improves cardiac and neurological function, and enhances survival via reversible inhibition of mitochondrial complex I. Circulation. 2009;120:897-905
29. Gong P, Li CS, Hua R, Zhao H, Tang ZR, Mei X. et al. Mild hypothermia attenuates mitochondrial oxidative stress by protecting respiratory enzymes and upregulating MnSOD in a pig model of cardiac arrest. PloS One. 2012;7:e35313
30. Valerio A, Bertolotti P, Delbarba A, Perego C, Dossena M, Ragni M. et al. Glycogen synthase kinase-3 inhibition reduces ischemic cerebral damage, restores impaired mitochondrial biogenesis and prevents ROS production. J Neurochem. 2011;116:1148-59
31. Pan H, Xie X, Chen D, Zhang J, Zhou Y, Yang G. Protective and biogenesis effects of sodium hydrosulfide on brain mitochondria after cardiac arrest and resuscitation. Eur J Pharmacol. 2014;741C:74-82
32. Gutsaeva DR, Carraway MS, Suliman HB, Demchenko IT, Shitara H, Yonekawa H. et al. Transient hypoxia stimulates mitochondrial biogenesis in brain subcortex by a neuronal nitric oxide synthase-dependent mechanism. J Neurosci. 2008;28:2015-24
33. Xie Y, Li J, Fan G, Qi S, Li B. Reperfusion promotes mitochondrial biogenesis following focal cerebral ischemia in rats. PloS One. 2014;9:e92443
34. Yin W, Signore AP, Iwai M, Cao G, Gao Y, Chen J. Rapidly increased neuronal mitochondrial biogenesis after hypoxic-ischemic brain injury. Stroke. 2008;39:3057-63
35. Suliman HB, Carraway MS, Tatro LG, Piantadosi CA. A new activating role for CO in cardiac mitochondrial biogenesis. J Cell Sci. 2007;120:299-308
36. Zhang X, Shan P, Alam J, Fu XY, Lee PJ. Carbon monoxide differentially modulates STAT1 and STAT3 and inhibits apoptosis via a phosphatidylinositol 3-kinase/Akt and p38 kinase-dependent STAT3 pathway during anoxia-reoxygenation injury. J Biol Chem. 2005;280:8714-21
37. Piantadosi CA, Carraway MS, Babiker A, Suliman HB. Heme oxygenase-1 regulates cardiac mitochondrial biogenesis via Nrf2-mediated transcriptional control of nuclear respiratory factor-1. Circ Res. 2008;103:1232-40
38. Liu XM, Chapman GB, Peyton KJ, Schafer AI, Durante W. Carbon monoxide inhibits apoptosis in vascular smooth muscle cells. Cardiovasc Res. 2002;55:396-405
39. Nisoli E, Clementi E, Paolucci C, Cozzi V, Tonello C, Sciorati C. et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 2003;299:896-9
Author contact
![]() Corresponding author: Zi-tong Huang, Sun Yat-sen Memorial Hospital of Sun Yat-sen University, 107 Yan Jiang Xi Road, Guangzhou 510120, China. Email: syxhztcom; Tel : +86-20-81332084; Fax : +86-20-81332410
Corresponding author: Zi-tong Huang, Sun Yat-sen Memorial Hospital of Sun Yat-sen University, 107 Yan Jiang Xi Road, Guangzhou 510120, China. Email: syxhztcom; Tel : +86-20-81332084; Fax : +86-20-81332410