Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(9):1041-1051. doi:10.7150/ijbs.16134 This issue Cite
Research Paper
Multiple Growth Factors, But Not VEGF, Stimulate Glycosaminoglycan Hyperelongation in Retinal Choroidal Endothelial Cells
Othman Al Gwairi1*, Narin Osman1,2*, Robel Getachew, Wenhua Zheng3,4, X-L. Liang4, Danielle Kamato1, Lyna Thach5, Peter J. Little1,5 ![]()
1. School of Health and Biomedical Sciences, RMIT University, Bundoora, VIC 3083 Australia;
2. Department of Immunology, Monash University, Melbourne 3004 VIC, Australia.
3. Faculty of Health Sciences, University of Macau, Taipa, Macau, China;
4. State Key Laboratory of Ophthalmology, Zhongshan Ophthalmic Center, Sun Yat-sen University, Guangzhou 510006, China.
5. School of Pharmacy. The University of Queensland, Wooloongabba, QLD 4102, Australia.
*These two authors contributed equally to this work.
Received 2016-5-11; Accepted 2016-6-30; Published 2016-7-18
Abstract

A major feature of early age-related macular degeneration (AMD) is the thickening of Bruch's membrane in the retina and an alteration in its composition with increased lipid deposition. In certain pathological conditions proteoglycans are responsible for lipid retention in tissues. Growth factors are known to increase the length of glycosaminoglycan chains and this can lead to a large increase in the interaction between proteoglycans and lipids. Using choroidal endothelial cells, we investigated the effects of a number of AMD relevant growth factors TGFβ, thrombin, PDGF, IGF and VEGF on proteoglycan synthesis. Cells were characterized as of endothelial origin using the specific cell markers endothelial nitric oxide synthesis and von Willebrand factor and imaged using confocal microscopy. Cells were treated with growth factors in the presence and absence of the appropriate inhibitors and were radiolabeled with [35S]-SO4. Proteoglycans were isolated by ion exchange chromatography and sized using SDS-PAGE. Radiosulfate incorporation was determined by the cetylpyridinium chloride (CPC) precipitation technique. To measure cellular glycosaminoglycan synthesizing capacity we added xyloside and assessed the xyloside-GAGs by SDS-PAGE. TGFβ, thrombin, PDGF & IGF dose-dependently stimulated radiosulfate incorporation and GAG elongation as well as xyloside-GAG synthesis, however VEGF treatment did not stimulate any changes in proteoglycan synthesis. VEGF did not increase pAKT but caused a large increase in pERK relative to the response to PDGF. Thus, AMD relevant agonists cause glycosaminoglycan hyperelongation of proteoglycans synthesised and secreted by retinal choroidal endothelial cells. The absence of a response to VEGF is intriguing and identifies proteoglycans as a novel potential target in AMD. Future studies will examine the relevance of these changes to enhanced lipid binding and the development of AMD.
Keywords: Age related macular degeneration, proteoglycans, growth factors, Bruch's membrane, VEGF, xyloside.
Introduction
Age-related macular degeneration (AMD) is one of the most important eye diseases with a high prevalence and serious health and quality of life implications [1, 2]. In its simplest manifestation, AMD has two presentations representing potentially its natural history [3]. In the earliest “dry” stage there are changes in endothelial cells, epithelial cells and photo receptor cells leading to thickening of Bruch's Membrane (BM) and a loss of central vision [2, 4]. The thickening of Bruch's membrane is associated with the deposition of serum-derived lipoproteins in the tissue [4-6]. As the disease progresses, there is a breakdown of the barrier function of BM allowing migration and proliferation of endothelial cells and the penetration of micro vessels into the retina in the “wet” phase of the disease. The wet phase of the disease is the most aggressive and the target of therapies but the dry phase is more prevalent, most likely representing the chronological progression of the disease [2]. The pathophysiology underlying the dry phase of AMD is not well understood and there are currently no therapies targeted at this stage of AMD.
There is an increasing recognition that a number of major diseases are associated with the disposition of lipids in tissues [7-10]. Apolipoproteins on LDL particles become trapped and deposited into tissues where subsequent biochemical modifications generate immunogenic particles which initiate an inflammatory reaction [7, 8]. This inflammatory response often becomes chronic or unresolving and is the underlying pathology of diseases including atherosclerosis [11]. The biochemical events underlying the trapping of lipids in tissues involve a primary role of proteoglycans [12, 13]. Proteoglycans are protein carbohydrate adducts which are ubiquitously expressed. Proteoglycans play a role in normal physiology such as in the function of joints but modified proteoglycans are associated with disease processes [6, 14-17]. Some small leucine-rich proteoglycans such a biglcyan have chondroitin and dermatan sulfate glycosaminoglycan (GAG) chains which are sulfated and highly negatively charged and they avidly bind to positively charged amino acid residues of apolipoproteins on LDL particles [6, 18-22]. A wide range of growth factors and hormones stimulate the expression of genes for GAG synthesising enzymes leading to hyperelongation of GAG chains [21-28]. The relationship between the size (length) of a GAG chain and propensity to bind lipid particles is such that increasing the size of the natural GAG chains leads to a marked and proportionate increase in the binding to lipids [5, 29-34]. Thus, initial tissue insults leading to increased expression and availability of growth factors, hormones and cytokines can lead to an increase in GAG length with increased lipid binding and the initiation of a pathological cascade ultimately setting off the inflammatory response which can lead to the more serious and acute manifestation of the disease. There is strong clinical evidence for the role of GAG elongation in early human atherosclerosis [9, 16, 35, 36]. There are as yet no clinical therapies which target this early stage of GAG modification although experimentally there have been studies showing an association of GAG elongation with the development of atherosclerosis [37, 38] and also successful proof of concept studies in animal models showing that blocking GAG elongation can prevent lipid deposition and atherosclerosis [39].
We have investigated the actions of multiple growth factors and hormones on a retinal endothelial cell model to address the question if the phenomena of GAG elongation occurs in these cells. We investigated the actions of protein tyrosine kinase and protein serine/threonine kinase growth factor receptor agonists as well as a GPCR agonist, all of which have been shown to stimulate hyperelongation of GAG chains on biglycan produced by vascular smooth muscle cells (VSMCs) [17, 25, 28, 30, 33, 40]. The process of GAG hyperelongation can be investigated in more depth by supplying cells with exogenous xyloside which serves as a false acceptor for the initiation of the synthesis of small free GAG chians known as “xyloside GAGs” and several agonists were evaluated in this assay to assess the existence and activity of this process in retinal cells [36, 41, 42]. In other contexts we have identified the genes which are rate limiting in the process of GAG hyperelongation - increased expression of several genes correlates with GAG hyperelongation and we have assessed the effect of various agents on the expression of these genes as a contributor to the underlying pathological process, as a potential marker for early disease and a potential therapeutic target for early AMD [43].
We found that retinal endothelial cells expressed a proteoglycan with a molecular weight similar to that of lipid-binding PGs secreted by other cells. We found that representative agonists at protein tyrosine kinase, serine/threonine kinase and GPCRs all stimulated GAG hyperelongation on a secreted proteoglycan from endothelial cells but with the notable exception that vascular endothelial cell growth factor (VEGF) was without effect on GAG hyperelongation, the size of xyloside GAGs or on the expression of GAG synthesizing enzymes. We showed surprisingly that VEGF relatively strongly stimulated Mitogen Activated Protein (MAP) kinase activity in these cells as assessed by an increased level of phosphoErk (pERK) [8, 44-48]. The data in the study demonstrates that the phenomenon of GAG hyperelongation occurs in retinal endothelial cells and lays the foundation for a later complete analysis of the properties of the proteoglycan (PG) and its lipid binding capacity.
Materials and Methods
Materials
Dulbecco's Modified Eagle medium (DMEM) (0 and 25 mM glucose), trypsin‑versene, antibiotics (10,000 U/ml penicillin, 10,000μg/ml streptomycin) were purchased from GIBCO, (Grand Island, USA). Foetal bovine serum (FBS) was from In Vitro Technologies, Pty. Ltd. (VIC, Australia). Platelet-derived growth factor (PDGF‑BB) (Human recombinant BB isoform), thrombin, SB431542, Dulbecco's phosphate buffered saline (PBS) (10X), sodium dodecyl sulfate (SDS), 2-mercaptoethanol, dimethyl sulfoxide (DMSO) and chondroitin sulphate were purchased from Sigma-Aldrich (St Louis, MO, USA). UO126 was from Promega (Madison, WI, USA). Human transforming growth factor beta-1, anti-pErk1/2 (Thr202/Tyr204), anti-GAPDH and vascular endothelial growth factor were from Cell Signalling Technology (Danvers, MA, USA). Imatinib was obtained from (Alfred Hospital Pharmacy, Australia). SCH79797 was from Tocris Biosciences. Human recombinant IGF-1 was obtained as a gift from Genentech Inc. (San Francisco, CA). Real-time polymerase chain reaction (RT-PCR) primers for glycosaminoglycan (GAG) synthesising enzymes were based on their respective GenBank sequences and were purchased from GeneWorks Pty Ltd. (SA, Australia). Housekeeping gene 18S, the RNeasy Mini Kit, the QuantiTect Reverse Transcription Kit, and the RT-PCR and two-step RT-PCR kit were purchased from Qiagen (VIC, Australia).Carrier-free [35S]-SO4 and [35S] methionine/cysteine were obtained from MP Biomedicals. Cetylpyridinium chloride was from Unilab Chemicals Pharmaceuticals (India); Whatman 3MM chromatography paper was from Biolab (Mulgrave, Australia); Insta-Gel Plus scintillation fluid was from PerkinElmer Life Sciences. SN30978 was provided by Dr Graeme Lancaster (Baker IDI Heart & Diabetes Institute, Melbourne, VIC, Australia).
Methods
Cell Culture
Monkey Retinal Choroidal Endothelium Cells (RF/6A) were purchased from the American Type Culture Collection (ATCC). Samples were thawed by agitating in a water bath at 37°C. Cells were maintained in DMEM (5mM glucose, 10% heat inactivated serum and 1 % antibiotics at 37°C in 5% CO2). RF/6A cells were seeded on 60 mm dishes and 24 well-plates. Cells were grown to confluence then rendered quiescent by serum deprivation for 48 h.
Cell Characterization
Cells were grown on 1.5 grade glass coverslips and then rendered quiescent via serum deprivation. After treatments, cells were fixed in 2% paraformaldehyde in 1 N phosphate buffer and then permeabilized and blocked in 0.1% Triton X-100, 1% horse serum for 30 min. Cells were incubated, with anti‑smooth muscle α‑actin, Endothelial nitric oxide synthase or von-Willebrand factor overnight followed by Alexa-Flour 488 or 594 and Hoechst stain for 30 mins. Coverslips were then mounted on slides and cells were imaged using a Nikon D-eclipse C1confocal microscope.
Quantitation of radiolabel incorporation into proteoglycans
Quiescent cells were changed to fresh medium containing 50 μCi/ml [35S]-SO4 in the presence and absence of thrombin, TGF-β, VEGF, IGF, or PDGF for 24 h. Media from the cell cultures were harvested and protease inhibitors (5mM bezamidine in 0.1 M 60 aminocaproic acid) were added to prevent degradation. Incorporation of the radiolabel into proteoglycans was measured by cetylpyridinium chloride precipitation assay, as described previously [49].
Synthesis of xyloside-initiated GAG chains
Cells rendered quiescent were treated in 0.5ml DMEM and 0.1% FBS supplemented with xyloside (0.5mM) under basal conditions and in the presence of agonists before the addition of [35S]-SO4 (50μCi/ml) for a further 24 hours. Secreted proteoglycans were harvested, isolated, concentrated and SDS-PAGE was used for the quantification.
SDS-PAGE analysis of proteoglycan size
Proteoglycans labelled with [35S]-SO4 were prepared for SDS-PAGE by isolation through DEAE-Sephacel anionic exchange mini columns. Samples were added to pre-equilibrated columns and then washed extensively with low salt buffer (8M urea, 0.25M NaCl, 2mM disodium EDTA, 0.5% Triton X-100). Proteoglycans were eluted with high salt buffer (8M urea, 3M NaCl, 2mM disodium EDTA 0.5% Triton X-100) and fractions containing the highest number of [35S]-SO4 cpm were pooled. Equal counts of proteoglycans were precipitated by ethanol solution (1.3% potassium acetate in 95% ethanol) and chondroitin sulphate then added to serve as a “cold carrier”. Samples were resuspended in buffer (8 M urea, 2 mM disodium EDTA, at pH 7.5), to which and equal volume of sample buffer was added. Radiolabelled samples were separated on 4-13% (PGs) or 4-20% (for xyloside‑GAGs) acrylamide gels and 3% acrylamide stacking gel at 60 V overnight. Gels were fixed then dried and were exposed to an imaging plate (Fujifilm BAS-MS 2040 imaging plate) for approximately 4-5 days. Images were developed on a Cyclone Plus Phosphor Imager (Perkin Elmer).
Western blotting for signalling phosphoproteins
Total cell lysates were resolved by SDS-PAGE on 10% gels and transferred onto PVDF membranes. Membranes were blocked with 5% bovine serum albumin and incubated with anti-phospho-Akt(Ser473) and anti-phospho-Erk1/2(Thr202/Tyr204) rabbit monoclonal antibody followed by HRP anti-rabbit IgG and ECL detection. Blots were imaged using the Bio-Rad gel documentation system and densitometry analysis was performed with Image Lab imaging software.
Assessing mRNA gene expression
The mRNA level of the GAG enzymes was determined by real-time polymerase chain reaction (RT-PCR). Total RNA was isolated from treated RF/6A cells. Total RNA was extracted from 5×105 cells using RNeasy Mini kit (Qiagen) according to the manufacturers' instructions. RNA concentration and purity was checked by spectrophotometry (260/280 nm) using a Nanodrop2000 spectrophotometer (Thermo Fisher Scientific). First strand cDNA was synthesized for 1μg RNA using Quantitect reverse transcriptase kit (Qiagen) according to the manufacturers' instructions. Quantitative RT-PCR was performed using Qiagen Rotor Gene Q and QuantiFast SYBR Green PCR Master mix kit (Qiagen). The data was normalised to 18S as the house keeping gene. Relative expression of the mRNA levels was quantified using comparative delta delta Ct method.
Statistical analysis
Data was normalised and expressed as the mean ± standard error of the mean (SEM) of three independent experiments unless stated otherwise. A one-way ANOVA was used to calculate statistical significance of normalised data, followed by least significant difference post-hoc analysis. Results were considered significant when the probability was less than 0.05 (*p<0.05) and 0.01 (**p<0.01).
Results
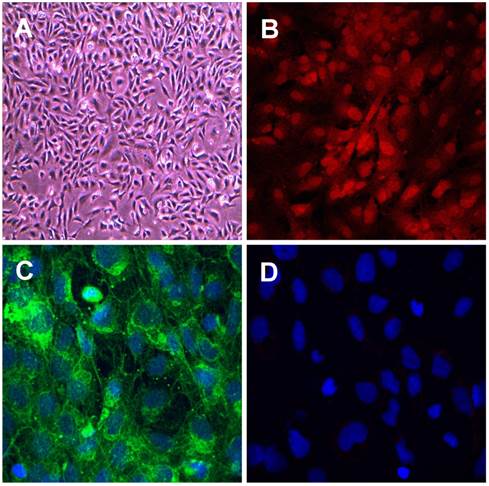
We characterised the RF/6A choroidal retinal endothelial cells using confocal microscopy and staining with two markers for endothelial and as a negative control, one marker for vascular smooth muscle cells (VSMCs). The RF/6A choroidal endothelial cells showed the typical cobblestone pattern and stained positively for the endothelial cell markers, endothelial cell nitric oxide synthase (ecNOS) and von Willebrand factor (vWF) and were negative for the VSMC marker smooth muscle α‑actin (see Fig. 1 A - D). The data confirms the endothelial cell lineage of the cells used in this project.
Characterisation of retinal choroidal endothelial cells. Photomicrographs showing retinal choroidal endothelial cells (A) Phase contrast image of RF/6A cells. (B-D) Confocal images of cells stained for expression of specific endothelial markers (B) eNOS (C) von Willebrand factor and (D) vascular smooth muscle cell marker smooth muscle α‑actin. Primary antibodies 1:1000, secondary antibodies 1:400 green Alexa Fluor 488 donkey anti- rabbit IgG, red Alexa Fluor 594 donkey anti-mouse IgG, and nuclear stain blue Hoechst 408 1:500. Magnification (A) x200, (B), (C) and (D) x400.
We then commenced a systematic study of the effect of multiple growth factors and hormones on the synthesis and size of PGs secreted by these cells. PGs are heavily sulfated and their synthesis can be monitored by the metabolic incorporation of radiosulfate in an assay which assesses the expression of core proteins on which GAG chains extend and also the length of the GAG chains. Thus, PG synthesis in choroidal endothelial cells was assessed by radiosulfate incorporation into material secreted into the cell culture medium which was analysed by the CPC precipitation method which is specific for the analysis of very large anionic molecules over 95 per cent of which are PGs. Choroidal endothelial cells were passaged and grown to confluency then serum‑deprived for 24 h before the addition of radiosulfate and growth factors. The media was harvested, spotted on Whatman chromatography paper for quantitative analysis and the bulk of the media was processed through ion exchanges columns and concentrated for size analysis by SDS‑PAGE.
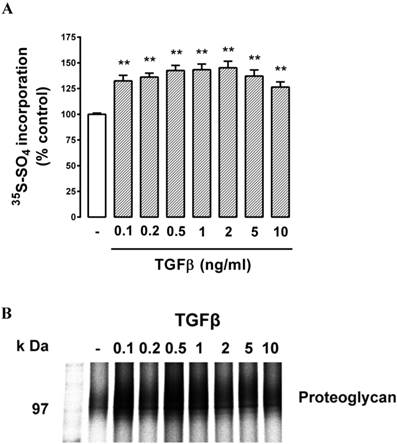
Cells were treated with TGFβ (0.1 - 10 ng/ml, 24 h) which resulted in a modest but statistically significant (P<0.05) increase in radiosulfate incorporation at the lowest concentration tested, followed by a concentration-dependent increase up to a peak response at 2 ng/ml and a lower response at the two highest concentrations tested (Fig. 2A). The parabolic dose response relationship is typical of responses to TGFβ. The peak response represented an increase in radiosulfate incorporation of almost 50% which is similar to the response observed in VSMCs [25, 36, 41, 50]. The SDS‑PAGE showed a weak band in the region of 100 kDa which is the expected area for lipid binding chondroitin/dermatan sulfate PGs (Fig. 2B) with no other substantial bands visible on the gels. Although the bands were weak, it was possible to observe reduced electrophoretic mobility meaning an increase in the size of the bands (and a decrease at the highest concentrations of TGFβ) consistent with the data obtained in the quantitation (Fig. 2A). Thus, the data indicated that there was a potentially lipid-binding PG expressed by these retinal endothelial cells and the synthesis and expression of this band responded to TGFβ stimulation of the cells by increasing the size of the GAG chains.
Effect of TGFβ on proteoglycan synthesis and apparent molecular size in choroidal endothelial cells. Cells were stimulated with TGFβ at 0.1, 0.2, 0.5, 1, 2, 5, 10 ng/ml for 24 hours as indicated. (A) Histogram showing the radiosulfate incorporation for cells across a range of TGFβ concentrations. (B) SDS-PAGE of the isolated proteoglycans shows a broad band 95-120 kDa. **p<0.01.
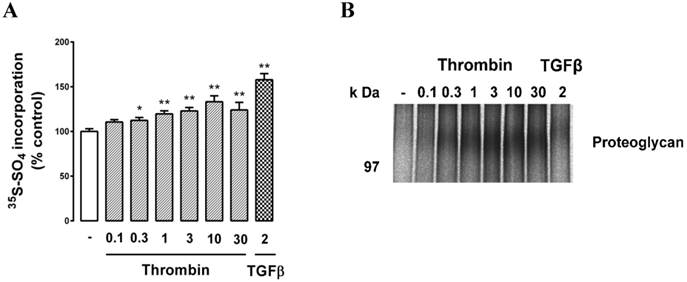
We then treated cells with the GPCR agonist thrombin and assessed PG synthesis. Cells were treated with thrombin (0.1, 0.3, 1, 3, 10, 30 U/ml) which resulted in a dose-dependent increase in radiosulfate with a maximum response of almost 40% (Fig. 3A). TGFβ was used as a positive control and it gave a similar result to that reported in Fig. 2. SDS‑PAGE analysis again revealed a weak band at around 100 kDa and the size of this band responded to thrombin treatment as observed earlier for the response to TGFβ (Fig. 3B).
Effect of thrombin on proteoglycan synthesis and apparent molecular size in choroidal endothelial cells. RF/6A cells were treated with the agonists thrombin (0.1, 0.3, 1, 3, 10, & 30 U/ml) or treated with TGFβ (2ng/ml) as a positive control for 24 hours and proteoglycans were collected and analysed as described in the text. (A) Histogram shows the radiosulfate incorporation for cells across a range of thrombin concentrations and (B) SDS-PAGE gel of the proteoglycans. The experiments were repeated at least 3 times with similar results. **p<0.01 and *p<0.05
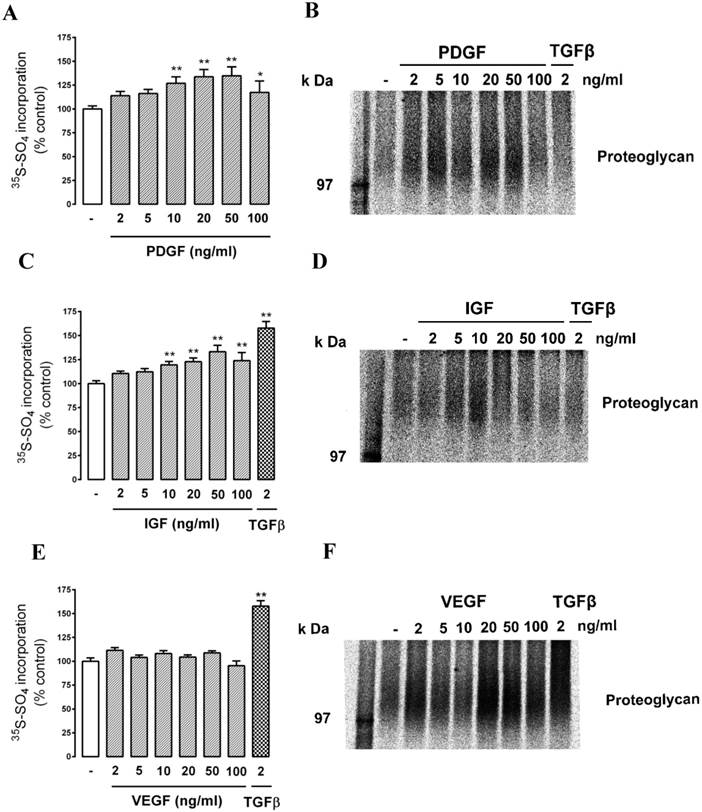
We then assessed the response to a series of protein tyrosine receptor agonists - PDGF (2 - 100 ng/ml), IGF (2 - 100 ng/ml) and VEGF. (2 - 100 ng/ml). The cells responded to PDGF and IGF in a similar manner to the response to thrombin (Fig. 4 A (PDGF) and C (IGF)). Intriguingly, there was no response to VEGF as either radiosulfate incorporation (Fig. 4E) or a change in electrophoretic mobility (Fig. 4F) but in the same experiment the cells responded to TGFβ as a positive control. This experiment was repeated four times and there was no response to VEGF on any occasion. This is a surprising result in consideration of the association of VEGF with endothelial cells and retinal diseases although it can be noted that the effect of VEGF on PG synthesis does not appear to have been reported for any cell type.
Effect of tyrosine kinase receptor agonists PDGF, IGF and VEGF on proteoglycan synthesis and GAG elongation in choroidal endothelial cells. Cells were treated with the agonists (A, B) PDGF (1, 2, 5, 10, 20, 50 & 100 ng/ml), (C, D) IGF (2 , 5, 10, 20, 50 & 100 ng/ml), (E, F) VEGF (2 , 5, 10, 20, 50 & 100 ng/ml) or with TGFβ (2ng/ml) as a positive control, for 24 hours and analysed as described in text. Histograms show the radiosulfate incorporation for cells across a range of agonist concentrations. SDS-PAGE gels for PDGF, IGF and VEGF treatments (B, D, F) respectively and show the size of the isolated proteoglycan band. The experiments were repeated at least 3 times with similar results. **p<0.01 and *p<0.05 using a one-way ANOVA.
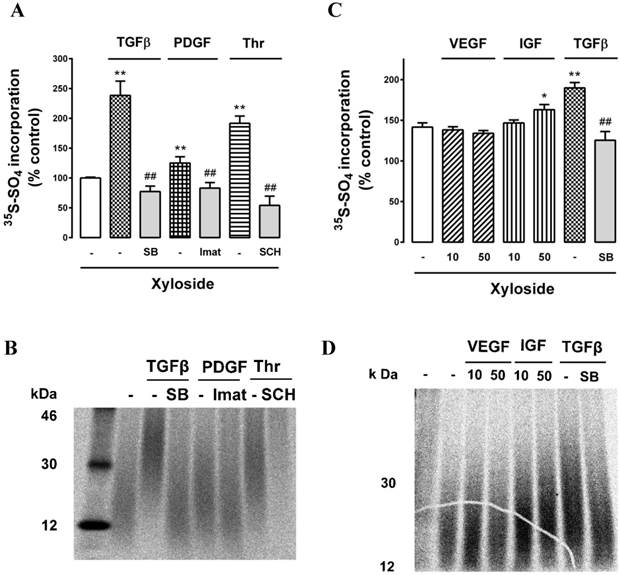
With the weak bands on SDS‑PAGE making visualization of the critical band difficult, we employed an additional technique to evaluate the effect of growth factors on the GAG synthesizing capacity of the retinal endothelial cells. The first sugar added to the serine residue on a core protein of a PG is xylose and adding exogenous xyloside to cell cultures induces the cells to engage the xyloside as a “false acceptor” [42] and synthesise short GAG chains which are termed “xyloside GAGs” [17, 42]. We have previously used this technique to evaluate the effects of agonists and antagonists in VSMCs [50-52]. Cells were prepared and incubated as previously described and in the additional presence of β‑D-xyloside (0.5 mM). We treated cells with TGFβ, PDGF and thrombin and their antagonists, SB431542, imatinib and SCH55555, respectively,.in the presence of xyloside and analysed the culture media quantitatively and by SDS‑PAGE for the production and size of xyloside GAGs. All three growth factors stimulated an increase in radiosulfate incorporation into CPC precipitable material and the responses were completely blocked by the respective antagonists (Fig. 5A). Analysis of the size of the xyloside GAGs by SDS‑PAGE showed that all three agonists increased the size of the xyloside GAGs and that the responses were blocked by the respective antagonists (Fig. 5B). The effect to increase the size of the xyloside GAGs correlated with the size of the increase in radiosulfate incorporation (compare Fig. 5A and 5B). We then evaluated further protein tyrosine kinase receptor agonists, VEGF and IGF, noting that VEGF did not activate GAG synthesis when assessed as intact PGs. VEGF did not increase radiosulfate into PGs and xyloside GAGs synthesized in the presence of xyloside (Fig. 5C) whereas IGF caused a small stimulation and TGFβ used as a positive control increased radiosulfate incorporation (Fig. 5C). We then examined the effect on the size of the xyloside GAGs and the results correlated with the effects on radiosulfate incorporation and notably VEGF had no effect on the size of the xyloside GAGs (Fig. 5D). These results confirm that multiple agonists can activate the GAG synthesizing capacity of retinal endothelial cells but VEGF, uniquely thus far, is without effect.
Effect of TGFβ, PDGF, IGF, VEGF and thrombin on GAG synthesizing capacity of choroidal endothelial cells using xyloside as a false acceptor. Cells were untreated (-) or treated with (A) TGFβ (2 ng/ml) and in the presence of SB431542 (SB 10μM), PDGF (50 ng/ml) and in the presence of imatinib (Imat 10μM) and thrombin (10 U/ml) in and in the presence of SCH79797 (SCH 10μM) or (B) VEGF (10 and 50 ng/ml), IGF (10 and 50 ng/ml) or TGFβ (2 ng/ml) and in the presence of SB431542 (SB 10μM). The cell culture media was supplemented with xyloside (0.5mM) to enable the production of xyloside GAGs (see text for explanation) and radiosulfate (50mCi/well) was added for a further 24 hours. Histograms show the radiosulfate incorporation for the different treatments. Samples were run on SDS-PAGE (4-20% gradient gel) and xyloside GAGs showed molecular weights in the range of 15 - 30 kDa. Experiments were performed at least three times, **p<0.01 for agonists or ##p<0.01 for antagonists.
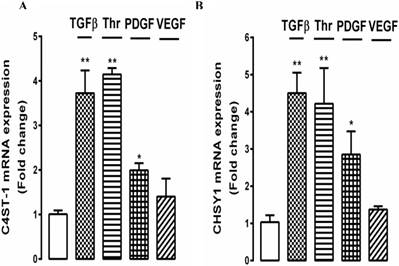
To further explore the effect of growth factors on GAG hyperelongation by choroidal endothelial cells, we investigated the underlying cellular mechanisms. In VSMCs we have identified several genes for GAG synthesizing enzymes, the expression of which correlates with the effects on GAG hyperelongation and we are working on the hypothesis that these genes represent the proteins which are rate limiting for the process of GAG hyperelongation [43]. The two most prominent genes are chondroitin‑4‑sulfotransferase‑1 (C4ST‑1) and chondroitin synthase‑1 (CHSy‑1). We established that several growth factors gave a peak response for the stimulation of the expression of these genes at 4h (results not shown) and we then evaluated the effects of several growth factors targeting different classes of receptors as used in the above experiments on the expression of these genes. Choroidal endothelial cells were treated with TGFβ, thrombin, PDGF and VEGF for 4 h and the expression of C4ST‑1 and CHSy‑1 was determined relative to untreated cells. TGFβ, thrombin and PDGF stimulated the expression of both C4ST‑1 and CHSy‑1 by 2 - 4 fold with a similar but not identical response for both gene products (Fig. 6A and 6B). The extent of stimulation of gene expression correlated with the size of the effects of the three growth factors on radiosulfate incorporation and the increase in the size of the xyloside GAGs (see Fig. 5). In contrast, but consistent with the date reported above, VEGF did not stimulate the expression of either of the target genes (Fig. 6A and B).
Effect of multiple growth factors TGFβ, thrombin, PDGF, and VEGF on the expression of genes mediating glycosaminoglycan (GAG) chain elongation. RF/6A cells were prepared and treated with TGFβ (2 ng/ml), Thrombin (10 U/ml), PDGF (50 ng/ml), and VEGF (50 ng/ml) for 4 h. Total RNA was harvested and the mRNA expression of C4ST-1 and ChSy-1 were analysed using qRT-PCR. 18s was used as a housekeeping gene. **p<0.01 and *p<0.05.
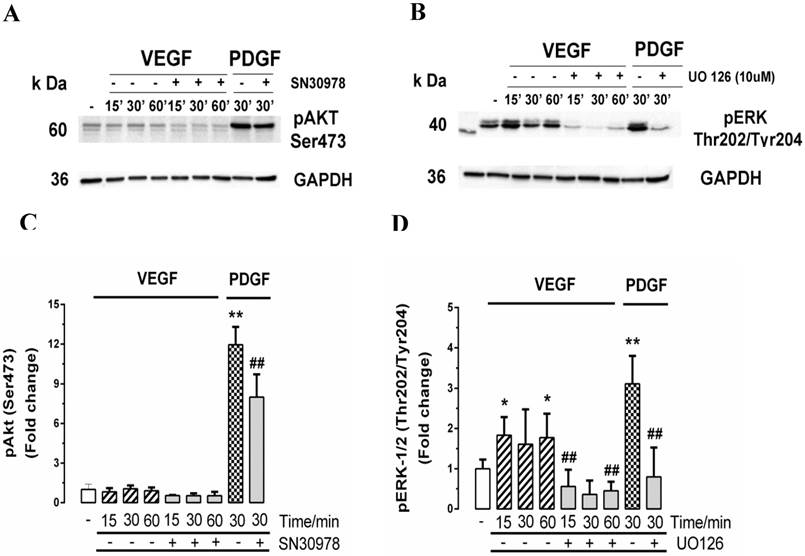
In order to further explore the absence of a response of the retinal endothelial cells to VEGF, we explored two of the major signalling pathways that have been reported to be activated by VEGF, being the phosphorylation of the ubiquitous and poly functional kinase Akt, which has been implicated in many disease states [53] and the phosphorylation of the Mitogen Activated Protein (MAP) kinase pathway, ERK [54, 55]. We have previously explored these pathways in the signalling pathways controlling PG synthesis in VSMCs [34, 56]. Cells were treated for 0 - 60 mins with VEGF and for 30 mins with PDGF as a positive control and we assessed pAKT (Fig. 7A and 7C) and pERK (Fig. 7B and 7D). In response to VEGF, the cells showed a very small early increase in pAKT (Fig. 7A and 7C) but more substantial and statistically significant increase in pERK (Fig. 7B and 7D). In both cases, PDGF used as a positive control very substantially activated pAKT and pERK and the respective inhibitors, SN30978 (an AKT blocker) and UO126 (an ERK blocker) attenuated the response to PDGF. PDGF is an efficacious agonist of pERK and the VEGF response was almost 50% of the response to PDGF indicating that there is an active VEGF activated signalling pathway in these cells.
Effect of VEGF on the ERK and Akt signalling pathways in choroidal endothelial cells. Cells were untreated (-) or pre-treated for 30 min with the (A) Akt1/2 inhibitor SN30978 (2 μM) or (B) with MEK inhibitor U0126 (10 µM) and then stimulated with VEGF (50ng/ml) for 15, 30 or 60 mins or with PDGF (50ng/ml) for 30 mins as a positive control. Western blots of lysates from these treatments were probed for phospho-Akt (Ser473) or (B) phospho-ERK (Thr202/Tyr204). GAPDH was used as the loading control. Histograms depict the fold-change of phosphorylated proteins. Experiments were repeated three times. **p<0.01 or *p<0.05 for agonists and ##p<0.01 for antagonists.
Discussion
We are experimentally evaluating the general hypothesis that modifications in the GAG chains on PGs, specifically hyperelongation, has the potential to lead to increased binding of plasma-derived lipids and initiate pathological responses in tissues. As there is evidence for the deposition of plasma lipids in early AMD, we have addressed the question if growth factors potentially associated with retinal disease activate GAG hyperelongation in PGs produced by retinal endothelial cells. We observed that retinal endothelial cells secreted a PG of the size associated with lipid binding in other tissues and that a range of protein tyrosine kinase, protein serine/threonine kinase and a GPCR agonist all increased the synthesis of PGs and increased the size of the PGs indicating GAG hyperelongation. Several of the agonists stimulated an increase in the size of xyloside GAGs, used as a measure of cellular GAG synthesising capacity and also increased the expression of two of the genes for the enzymes which are considered to be rate limiting in the process of GAG elongation. However and notably, the protein tyrosine kinase receptor agonist, VEGF, which is associated with retinal pathologies, did not stimulate PG synthesis or size, did not stimulate the synthesis or size of xyloside GAGs, did not stimulate the expression of the two GAG elongation genes but VEGF weakly increased pAKT and relatively strongly increased pERK levels in the cells indicating that the signalling pathway for VEGF was present and active in these cells. These results confirm the hypothesis that retinal endothelial cells respond to multiple growth factors with an activation of GAG synthesis and that the cells synthesize and secrete PGs with elongated GAG chains which have the potential to increase binding of plasma‑derived lipids and to initiate a pathological process in the macular.
The results obtained in this work are consistent with our extensive earlier work in VSMCs and confirm that the process of growth factor initiated GAG hyperelongation occurs in these retinal endothelial cells. The interaction of lipids with the GAG chains on PGs or even free GAG chains released from PGs occurs over a very wide range of GAG sizes but notably the increases above the basal or natural size, which we have termed hyperelongation [35] immediately lead to increases in binding to cationic residues of apolipoproteins on lipoproteins [57]. There is much more work to do in analysing the exact molecular entity observed and studied in these experiments and most importantly to assess if it is a lipid-binding PG. However, these studies demonstrating that GAG hyperelongation occurs in response to growth factors relevant to retinal disease strongly supports and validates this line of research in early or dry phase AMD where there are few other current options being explored. There is detailed information available of the multiple and distinct signalling pathways that regulate the expression of the genes for GAG hyperelongation and also a demonstration that an agent, the tyrosine kinase inhibitor imatinib, which inhibits GAG elongation in vitro prevents lipid deposition in an animal model of atherosclerosis so all the pathways are present to explore the role of the process of GAG hyperelongation as a target for the treatment of early AMD [32, 50].
The absence of a response to VEGF is very interesting from the perspective of both cell biology and therapeutics. From a cell biology perspective this is the first agent that we have identified that does not stimulate GAG hyperelongation in any cell. We showed that VEGF quite strongly stimulates the pERK pathway in these cells. We have previously shown that there are several pathways involving ERK and leading to GAG hyperelongation [58]. In particular, in response to TGFβ, pERK is upstream of the phosphorylation of the transcription factor Smad2 and phosphorylation of Smad2 in the linker region correlates with hyperelongation of GAG chains on biglycan in VSMCs [34, 46, 58]. Both PDGF and thrombin stimulate an increase in cellular pERK with thrombin acting via transactivation of the EGF receptor and PDGF directly via the kinase mediated signalling pathway and both agonists stimulate GAG hyperelongation so why VEGF stimulates ERK phosphorylation but does not stimulate GAG hyperelongation is unknown at this time. The signalling for PG core protein expression is distinct from the pathways leading to GAG hyperelongation and have more similarities with the pathways controlling the cell cycle including stimulation of the pAKT pathway [56, 59]. Both TGFβ and PDGF stimulate pAKT as the pathway to stimulate the expression of biglycan in VSMCs [56, 59]. VEGF did not stimulate pAKT levels in the retinal endothelial cells so it is not surprising that it did not increase the expression of PG core proteins sufficiently to be observed as an increase radiosulfate expression (see Fig. 7).
The therapeutic implications of VEGF not stimulating GAG elongation are intriguing. The current meaning would be that VEGF does not contribute to GAG elongation on PGs and that anti-VEGF therapies do not have any actions on PG synthesis in retinal cells. Thus, there is no impact of VEGF or VEGF therapies on PG synthesis and structure in the macula and there is nothing that impacts on the hypothesis that GAG elongation might be contributing to the pathology of AMD. It remains valid to explore the role of GAG hyperelongation as a potential therapeutic target for the treatment of early AMD.
Conclusions
We observed that representative agonists at protein tyrosine kinase, serine/threonine kinase and GPCRs all stimulated GAG hyperelongation of a secreted PG from retinal endothelial cells but with the notable exception that VEGF had no effect. Surprisingly, the tyrosine kinase growth factor agonist VEGF did not stimulate GAG hyperelongation notwithstanding that the cells responded to VEGF with a modest increase in pERK which has previously been shown to be a signalling pathways for GAG hyperelongation. These results raise the possibility that growth factor mediated hyperelongation of GAG chains on PGs may be playing a role in early AMD and it may therefore represent a potential target for the development of new therapeutic agents. VEGF did not stimulate proteoglycan synthesis, creating an opportunity for a therapeutic area completely distinct from that which is most prominent for the present therapy of AMD.
The findings from these studies demonstrate that AMD relevant agonists impact the elongation of glycosaminoglycan chains of PGs synthesised by retinal choroidal endothelial cells. Future studies will examine the relevance of these changes to the development of AMD.
Acknowledgements
OA was supported by the Saudi Arabia Ministry of Higher Education. We thank the Ministry of Foreign Experts of the Government of the People's Republic of China for support of PJL by way of a High End Professor (Education) Award through Zhongshan (Sun Yat-sen) University to Prof. Zheng.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Cook HL, Patel PJ, Tufail A. Age-related macular degeneration: diagnosis and management. British Medical Bulletin. 2008;85:127-49
2. Schmidt-Erfurth U, Chong V, Loewenstein A, Larsen M, Souied E, Schlingemann R. et al. Guidelines for the management of neovascular age-related macular degeneration by the European Society of Retina Specialists (EURETINA). The British journal of ophthalmology. 2014;98:1144-67
3. Alten F, Eter N. Current knowledge on reticular pseudodrusen in age-related macular degeneration. The British journal of ophthalmology. 2015;99(6):717-722
4. Fletcher EL, Jobling AI, Greferath U, Mills SA, Waugh M, Ho T. et al. Studying Age-Related Macular Degeneration Using Animal Models. Optometry & Vision Science. 2014;91:878-86
5. Pikuleva IA, Curcio CA. Cholesterol in the retina: the best is yet to come. Prog Retin Eye Res. 2014;41:64-89
6. Nakashima Y, Wight TN, Sueishi K. Early atherosclerosis in humans: role of diffuse intimal thickening and extracellular matrix proteoglycans. Cardiovascular Research. 2008;79(1):14-23
7. Bays HE, Toth PP, Kris-Etherton PM, Abate N, Aronne LJ, Brown WV. et al. Obesity, adiposity, and dyslipidemia: A consensus statement from the National Lipid Association. Journal of Clinical Lipidology. 2013;7:304-83
8. Delgado-Roche L. The response-to-retention hypothesis: From theory to the potential therapeutic approaches. Biomedicine & Aging Pathology. 2014;4:291-5
9. Ballinger ML, Nigro J, Frontanilla KV, Dart AM, Little PJ. Regulation of glycosaminoglycan structure and atherogenesis. Cell Mol Life Sci. 2004;61:1296-306
10. Burch ML, Osman N, Getachew R, Al-Aryahi S, Poronnik P, Zheng W. et al. G protein coupled receptor transactivation: extending the paradigm to include serine/threonine kinase receptors. Int J Biochem Cell Biol. 2012;44:722-7
11. Little PJ, Chait A, Bobik A. Cellular and cytokine-based inflammatory processes as novel therapeutic targets for the prevention and treatment of atherosclerosis. Pharmacol Ther. 2011;131:255-68
12. Little PJ, Drennon KD, Tannock LR. Glucosamine inhibits the synthesis of glycosaminoglycan chains on vascular smooth muscle cell proteoglycans by depletion of ATP. Arch Physiol Biochem. 2008;114:120-6
13. Osman N, Little PJ. The Prolyl Peptidyl Isomerase Pin1 as a Potential Therapeutic Target in Atherosclerosis. Clinical & Experimental Pharmacology. 2012;2:3
14. Nakata S, Tsutsui M, Shimokawa H, Tamura M, Tasaki H, Morishita T. et al. Vascular neuronal NO synthase is selectively upregulated by platelet-derived growth factor: involvement of the MEK/ERK pathway. Arteriosclerosis, thrombosis, and vascular biology. 2005;25:2502-8
15. Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994;14:133-40
16. Nakashima Y, Fujii H, Sumiyoshi S, Wight TN, Sueishi K. Early human atherosclerosis: accumulation of lipid and proteoglycans in intimal thickenings followed by macrophage infiltration. Arterioscler Thromb Vasc Biol. 2007;27:1159-65
17. Getachew R, Ballinger ML, Burch ML, Little PJ, Osman N. Characterisation of Ki11502 as a potent inhibitor of PDGF beta receptor-mediated proteoglycan synthesis in vascular smooth muscle cells. Eur J Pharmacol. 2010;626:186-92
18. Olofsson SO, Wiklund O, Boren J. Apolipoproteins A-I and B: biosynthesis, role in the development of atherosclerosis and targets for intervention against cardiovascular disease. Vascular health and risk management. 2007;3:491-502
19. Stillemark-Billton P, Beck C, Boren J, Olofsson SO. Relation of the size and intracellular sorting of apoB to the formation of VLDL 1 and VLDL 2. Journal of lipid research. 2005;46:104-14
20. Flood C, Gustafsson M, Richardson PE, Harvey SC, Segrest JP, Boren J. Identification of the proteoglycan binding site in apolipoprotein B48. J Biol Chem. 2002;277:32228-33
21. Burch ML, Yang SN, Ballinger ML, Getachew R, Osman N, Little PJ. TGF-beta stimulates biglycan synthesis via p38 and ERK phosphorylation of the linker region of Smad2. Cellular and molecular life sciences: CMLS. 2010;67:2077-90
22. Kamato D, Babaahmadi Rezaei H, Getachew R, Thach L, Guidone D, Osman N. et al. (S)-[6]-Gingerol inhibits TGF-beta-stimulated biglycan synthesis but not glycosaminoglycan hyperelongation in human vascular smooth muscle cells. J Pharm Pharmacol. 2013;65:1026-36
23. Al-aryahi S, Kamato D, Getachew R, Zheng W, Potocnik SJ, Cohen N. et al. Atherogenic, fibrotic and glucose utilising actions of glucokinase activators on vascular endothelium and smooth muscle. Cardiovasc Diabetol. 2014;13:80
24. Rezaei HB, Kamato D, Ansari G, Osman N, Little PJ. Cell biology of Smad2/3 linker region phosphorylation in vascular smooth muscle. Clin Exp Pharmacol Physiol. 2012;39:661-7
25. Ballinger ML, Ivey ME, Osman N, Thomas WG, Little PJ. Endothelin-1 activates ETA receptors on human vascular smooth muscle cells to yield proteoglycans with increased binding to LDL. Atherosclerosis. 2009;205:451-7
26. Ballinger ML, Thomas MC, Nigro J, Ivey ME, Dilley RJ, Little PJ. Glycated and carboxy-methylated proteins do not directly activate human vascular smooth muscle cells. Kidney Int. 2005;68:2756-65
27. Rostam MA, Piva TJ, Rezaei HB, Kamato D, Little PJ, Zheng W. et al. Peptidyl-prolyl isomerases: functionality and potential therapeutic targets in cardiovascular disease. Clinical and experimental pharmacology & physiology. 2015;42(2):117-124
28. Osman N, Ballinger ML, Dadlani HM, Getachew R, Burch ML, Little PJ. p38 MAP kinase mediated proteoglycan synthesis as a target for the prevention of atherosclerosis. Cardiovasc Hematol Disord Drug Targets. 2008;8:287-92
29. Ballinger ML, Osman N, Wilks AF, Su S, Burns CJ, Bu X. et al. Pyrido-pyrimidine derivative CYC10424 inhibits glycosaminoglycan changes on vascular smooth muscle-derived proteoglycans and reduces lipoprotein binding. J Cardiovasc Pharmacol. 2008;52:403-12
30. Burch ML, Getachew R, Osman N, Febbraio MA, Little PJ. Thrombin-mediated proteoglycan synthesis utilizes both protein-tyrosine kinase and serine/threonine kinase receptor transactivation in vascular smooth muscle cells. J Biol Chem. 2013;288:7410-9
31. Osman N, Getachew R, Little AM P. Aortic smooth muscle cells from ApoE(-/-) mice secrete biglycan with hyperelongated glycosaminoglycan chains. Clinical and Experimental Pharmacology. 2013;3:6
32. Ballinger ML, Osman N, Hashimura K, de Hann J, Jandeleit-Dahm K, Allen TJ. et al. Imatinib inhibits vascular smooth muscle proteoglycan synthesis and reduces LDL binding in vitro and aortic lipid deposition in vivo. Journal of Cellular and Molecular Medicine. 2010;14:1408-18
33. Burch ML, Ballinger ML, Yang SN, Getachew R, Itman C, Loveland K. et al. Thrombin stimulation of proteoglycan synthesis in vascular smooth muscle is mediated by protease-activated receptor-1 transactivation of the transforming growth factor beta type I receptor. J Biol Chem. 2010;285:26798-805
34. Burch ML, Yang SN, Ballinger ML, Getachew R, Osman N, Little PJ. TGF-β stimulates biglycan synthesis via p38 and ERK phosphorylation of the linker region of Smad 2. Cell Mol Life Sci. 2010;67:2077-90
35. Little PJ, Osman N, O'Brien KD. Hyperelongated biglycan: the surreptitious initiator of atherosclerosis. Current Opinion in Lipidology. 2008;19:448-54
36. Ivey ME, Little PJ. Thrombin regulates vascular smooth muscle cell proteoglycan synthesis via PAR-1 and multiple downstream signalling pathways. Thromb Res. 2008;123:288-97
37. Grande-Allen KJ, Osman N, Ballinger ML, Dadlani H, Marasco S, Little PJ. Glycosaminoglycan synthesis and structure as targets for the prevention of calcific aortic valve disease. Cardiovasc Res. 2007Oct1;76(1):19-28
38. Anggraeni VY, Emoto N, Yagi K, Mayasari DS, Nakayama K, Izumikawa T. et al. Correlation of C4ST-1 and ChGn-2 expression with chondroitin sulfate chain elongation in atherosclerosis. Biochem Biophys Res Commun. 2011;406:36-41
39. Ballinger ML, Osman N, Hashimura K, de Haan JB, Jandeleit-Dahm K, Allen T. et al. Imatinib inhibits vascular smooth muscle proteoglycan synthesis and reduces LDL binding in vitro and aortic lipid deposition in vivo. Journal of cellular and molecular medicine. 2010;14:1408-18
40. Kamato D, Burch ML, Piva TJ, Rezaei HB, Rostam MA, Xu S. et al. Transforming growth factor-beta signalling: Role and consequences of Smad linker region phosphorylation. Cell Signal. 2013;25:2017-24
41. Little PJ, Tannock L, Olin KL, Chait A, Wight TN. Proteoglycans synthesized by arterial smooth muscle cells in the presence of transforming growth factor-beta1 exhibit increased binding to LDLs. Arterioscler Thromb Vasc Biol. 2002;22:55-60
42. Potter-Perigo S, Braun KR, Schonherr E, Wight TN. Altered proteoglycan synthesis via the false acceptor pathway can be dissociated from beta-D-xyloside inhibition of proliferation. Arch Biochem Biophys. 1992;297:101-9
43. Kamato D, Thach L, Getachew R, Burch M, Hollenberg MD, Zheng W. et al. Protease activated receptor-1 mediated dual kinase receptor transactivation stimulates the expression of glycosaminoglycan synthesizing genes. Cell Signal. 2016;28:110-9
44. Bullard LE, Qi X, Penn JS. Role for extracellular signal-responsive kinase-1 and -2 in retinal angiogenesis. Investigative ophthalmology & visual science. 2003;44:1722-31
45. Rebekah Bernard LT, Danielle Kamato, Narin Osman, Peter J Little. Assessing the Role of Gαq/11 in Cellular Responses: An Analysis of Investigative Tools. Clinical & Experimental Pharmacology. 2014;4:164
46. Burch ML, Zheng W, Little PJ. Smad linker region phosphorylation in the regulation of extracellular matrix synthesis. Cell Mol Life Sci. 2011;68:97-107
47. Getachew R, Ballinger ML, Burch ML, Reid JJ, Khachigian LM, Wight TN. et al. PDGF beta-receptor kinase activity and ERK1/2 mediate glycosaminoglycan elongation on biglycan and increases binding to LDL. Endocrinology. 2010;151:4356-67
48. Kamato D, Rostam MA, Piva TJ, Babaahmadi Rezaei H, Getachew R, Thach L. et al. Transforming growth factor beta-mediated site-specific Smad linker region phosphorylation in vascular endothelial cells. J Pharm Pharmacol. 2014;66:1722-33
49. Wasteson A, Uthne K, Westermark B. A novel assay for the biosynthesis of sulphated polysaccharide and its application to studies on the effects of somatomedin on cultured cells. Biochem J. 1973;136:1069-74
50. Getachew R, Ballinger ML, Burch ML, Reid JJ, Khachigian LM, Wight TN. et al. Platelet-Derived Growth Factor {beta}-Receptor Kinase Activity and ERK1/2 Mediate Glycosaminoglycan Elongation on Biglycan and Increases Binding to Low-Density Lipoprotein. Endocrinology. 2010;151:4356-67
51. Nigro J, Ballinger M, Dilley R, Jennings G, Wight T, Little P. Fenofibrate modifies human vascular smooth muscle proteoglycans and reduces LDL binding. Diabetologia. 2004;47:2105-13
52. Nigro J, Dilley RJ, Little PJ. Differential effects of gemfibrozil on migration, proliferation and proteoglycan production in human vascular smooth muscle cells. Atherosclerosis. 2002;162:119-29
53. Zheng W, Wang H, Zeng Z, Lin J, Little PJ, Srivastava LK. et al. The possible role of the Akt signaling pathway in schizophrenia. Brain Res. 2012;1470:145-58
54. Byeon SH, Lee SC, Choi SH, Lee HK, Lee JH, Chu YK. et al. Vascular endothelial growth factor as an autocrine survival factor for retinal pigment epithelial cells under oxidative stress via the VEGF-R2/PI3K/Akt. Invest Ophthalmol Vis Sci. 2010;51:1190-7
55. Goetze S, Eilers F, Bungenstock A, Kintscher U, Stawowy P, Blaschke F. et al. PPAR activators inhibit endothelial cell migration by targeting Akt. Biochem Biophys Res Commun. 2002;293:1431-7
56. Osman N, Getachew R, Burch M, Lancaster G, Wang R, Wang H. et al. TGF-beta stimulates biglycan core protein synthesis but not glycosaminoglycan chain elongation via Akt phosphorylation in vascular smooth muscle. Growth Factors. 2011;29:203-10
57. Boren J, Olin K, Lee I, Chait A, Wight TN, Innerarity TL. Identification of the principal proteoglycan-binding site in LDL. A single-point mutation in apo-B100 severely affects proteoglycan interaction without affecting LDL receptor binding. J Clin Invest. 1998;101:2658-64
58. Dadlani H, Ballinger ML, Osman N, Getachew R, Little PJ. Smad and p38 MAP kinase-mediated signaling of proteoglycan synthesis in vascular smooth muscle. J Biol Chem. 2008;283:7844-52
59. Osman N, Getachew R, Thach L, Wang H, Su X, Zheng W. et al. Platelet-derived growth factor-stimulated versican synthesis but not glycosaminoglycan elongation in vascular smooth muscle is mediated via Akt phosphorylation. Cell Signal. 2014;26:912-6
Author contact
![]() Corresponding author: Prof Peter J. Little, School of Pharmacy, The University of Queensland, Pharmacy Australia Centre of Excellence, 20 Cornwall Street, Woolloongabba, QLD 4102, Australia. Tel: +61 7 3346 1701 Email: p.littleedu.au.
Corresponding author: Prof Peter J. Little, School of Pharmacy, The University of Queensland, Pharmacy Australia Centre of Excellence, 20 Cornwall Street, Woolloongabba, QLD 4102, Australia. Tel: +61 7 3346 1701 Email: p.littleedu.au.