Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2016; 12(10):1236-1246. doi:10.7150/ijbs.15195 This issue Cite
Research Paper
Indoxyl Sulfate Enhance the Hypermethylation of Klotho and Promote the Process of Vascular Calcification in Chronic Kidney Disease
Jing Chen1,2,3*, Xiaoyan Zhang1,2,3 ![]() *, Han Zhang1, Tongqiang Liu1, Hui Zhang1, Jie Teng2, Jun Ji2, Xiaoqiang Ding1,2,3
*, Han Zhang1, Tongqiang Liu1, Hui Zhang1, Jie Teng2, Jun Ji2, Xiaoqiang Ding1,2,3 ![]()
1. Department of Nephrology, Zhongshan Hospital, Fudan University, Shanghai, China.
2. Kidney and Dialysis Institute of Shanghai, Shanghai, China.
3. Kidney and Blood Purification Laboratory of Shanghai, Shanghai, China.
*These authors contributed equally.
Received 2016-2-3; Accepted 2016-7-29; Published 2016-9-15
Abstract

Chronic kidney disease (CKD) is a state of Klotho deficiency. The Klotho expression may be suppressed due to DNA hypermethylation in cancer cells so we have investigated the effects and possible mechanisms by which Klotho expression is regulated in human aortic smooth muscle cells (HASMCs). The vascular Klotho hypermethylation in radial arteries of patients with end-stage renal disease was described. Cultured HASMCs and 5/6-nephrectomized Sprague Dawley (SD) rats treated with indoxyl sulfate (IS) were used as in vitro and in vivo models, respectively. IS increased CpG hypermethylation of the Klotho gene and decreased Klotho expression in HASMCs, and potentiated HASMCs calcification. The expression of DNA methyltransferase (DNMT) 1 and 3a in HASMCs treated with IS was significantly increased and specific inhibition of DNA methyltransferase 1 by 5-aza-2'-deoxycytidine(5Aza-2dc) caused demethylation of the Klotho gene and increased Klotho expression. In rats, injection of IS potentiated vascular calcification, increased CpG hypermethylation of the Klotho gene and decreased Klotho expression in the aortic medial layer and all of these changes could be reverted by 5Aza-2dc treatment. Transcriptional suppression of vascular Klotho gene expression by IS and epigenetic modification of Klotho by IS may be an important pathological mechanism of vascular calcification in CKD.
Keywords: DNA methylation, DNA methyltransferase, Klotho, Indoxyl sulfate, Vascular calcification, Chronic kidney disease.
Introduction
Chronic kidney disease (CKD) is the gradual and usually permanent loss of kidney function. CKD consists of five stages of increasing severity. The fifth stage is called as end-stage renal disease (ESRD). The mortality of cardiovascular disease (CVD) in the patients with chronic kidney disease (CKD) is significantly higher than in the general population [1, 2]. CKD-related mineral and bone disorders (MBDs) include abnormal mineral metabolism, abnormal bone, and extra-skeletal calcification. Accelerated vascular calcification (VC) has been considered to be one of the major causes of this elevated mortality [3].
Several mechanisms contribute to uremic VC, including systemic calcium/phosphate imbalances, decreased expression of calcification inhibitors, vascular smooth muscle cell (VSMC) osteogenic differentiation, and elastic remodeling. The VSMC phenotype change is particularly striking, and can be triggered by elevated extracellular phosphate. Large observational studies have correlated elevated serum phosphate with increased cardiovascular mortality in end-stage renal disease (ESRD), CKD and the general population. Of note, phosphate loading occurs early in CKD stage 3, as evidenced by increased serum levels of fibroblast growth factor 23 (FGF23) which precedes overt hyperphosphatemia [4].
It has been demonstrated that indoxyl sulfate (IS), a protein-bound uremic toxin, derives from the metabolism of dietary tryptophan[5]. Generally, intestinal bacteria metabolize tryptophan into indole which is further converted into IS in the liver after intestinal absorption. Kidney is the main organ that excretes IS via proximal tubular secretion. Thus, IS is usually accumulated in the blood of patients with CKD[5]. Moreover, conventional hemodialysis cannot efficiently remove IS due to its high binding affinity to albumin[6]. Recently, it has been reported that IS may act as a vascular toxin [4]. It is demonstrated IS can directly stimulate proliferation of rat vascular smooth muscle cells in a concentration-dependent manner [7]. In Dahl salt-sensitive hypertensive rats treated with IS, increased aortic wall thickness and severe aortic calcification accompanied with increased expression of osteoblast-specific proteins (core binding factor α 1, osteopontin, and alkaline phosphatase) [8]. These findings all suggest that IS may be one of the major contributors of vascular calcification in the patients with CKD. At present, it is still unclear how IS may contribute to vascular calcification.
Klotho expression is decreased in uremic patients [9]. Klotho knockout mice (Klotho-/-) have shortened lifespan, and develop hyperphosphatemia and multiple accelerated age-related disorders, including extensive and diffuse VC which is also seen in patients with CKD [10]. Studies have demonstrated that Klotho comprises anti-aging and cardiovascular-protective properties [11, 12]. Cleavage of the extracellular domain of Klotho generates soluble Klotho found in blood and urine [13]. Whether vascular smooth muscle cells express endogenous Klotho is currently a matter of debate [14, 15]. This matter has now been addressed again in an interesting article by Ritter [16, 17, 18], and it remains an important topic for future investigations. CKD is generally accepted as a state of renal [12, 19] and therefore systemic state of Klotho deficiency, which will in itself readily induce vascular calcification [20, 21]. Lim and colleagues described for the first time that endogenous Klotho is expressed in human arteries as well as human aortic smooth muscle cells (HASMCs) in culture [22]. Donate-Correa found human vascular tissue expresses Klotho [23]. Recently, Chang found intermedin (IMD) 1-53 protects against vascular calcification by up regulating α-Klotho [24]. However, claims of absence of Klotho expression have also been reported [25]. Until now, the possible cause of vascular Klotho deficiency in CKD remains unclear. Indoxyl sulfate (IS) can reduce renal Klotho expression through CpG hypermethylation of the Klotho gene, suggesting that epigenetic modification of Klotho gene may be an important mechanism of renal Klotho deficiency [19]. Based on these observations, we hypothesized that IS could increase DNA methyltransferase expression in vascular cells and suppress Klotho expression by DNA hypermethylation. We tested this hypothesis in human aortic smooth muscle cells (HASMCs) treated with IS, as well as in an animal model of uremia.
Materials and methods
Human samples
Human radial arteries were collected from ESRD patients undergoing arteriovenostomy for hemodialysis. The radial arteries harvested as grafts from age- and sex-matched coronary heart disease patients undergoing coronary artery bypass graft (CABG) were included as control. Human artery collection was performed at Zhongshan Hospital, Shanghai Medical College, Fudan University, Shanghai, China. Ethical approval was obtained from the Clinical Research Ethical Committee of Zhongshan Hospital, Shanghai Medical College, Fudan University and the methods were carried out in accordance with the approved guidelines. Each patient provided written informed consent to the use of their tissues for research purposes. All of the demographic and clinical data of patients were summarized in table S1.
Cell culture
HASMCs (Lot. 6001) were obtained from ScienCell Research Laboratory, Carlsbad, CA. HASMCs were cultured in 5 % CO2 at 37°C incubator with smooth muscle cell medium containing 0.5 mmol/L phosphate and 1.6 mmol/L calcium (Catalog No. 1101; ScienCell Research Laboratory, Carlsbad, CA). DMEM (Catalog No.1013, Invitrogen, Carlsbad, CA) with 0.5% fetal bovine serum was used as starvation medium for short-term experiments to assess the role of IS. HASMCs cultures at ~70% confluence were synchronized under serum-free conditions for 48 h and then treated with IS at concentrations of 0, 200, 500, and 1000 μmol/L for 6 d. For inhibition experiments, 5-aza-2'-deoxycytidine (5Aza-2dc) at concentrations of 0, 0.1, 1, and 10 μmol/L were added to HASMCs treated with IS at a concentration of 1000 μmol/L for 6d. Each treatment was done in triplicates.
Study animals
Eight-week-old SD rats with 5/6-nephrectomy were used in this study [19]. The experimental rats received intraperitoneal injection with IS (Alfa Aesar, Lancashire, England; n=5) at a dosage of 100 mg / kg / 48 h [8] for 24 weeks. The control rats (n=5) received same volume of phosphate-buffered saline injection every 48 h for 24 weeks. At the end of study, the body weight of study animal was recorded and the serum levels of blood urea nitrogen (BUN) and creatinine (Cr) were analyzed. The aortic tissue was micro dissected for further analysis. In the 5Aza-2dc treatment study, the IS -injected rats received intraperitoneal injection with 5Aza-2dc (Sigma, St Louis, MO, USA) simultaneously at a dosage of 1 mg/kg /48 h for 24 weeks (n=5 for each group). All animal experiments were approved by the experimental animal ethics committee at the Zhongshan Hospital, Shanghai Medical College, Fudan University and all experiments were performed in accordance with relevant guidelines and regulations. The flow diagram for animal study is summarized in Figure S1 in the online-only Data Supplement.
Quantitative bisulfate pyrosequencing
DNA methylation analyses were performed on bisulfate-treated DNA using a quantitative assay based on polymerase chain reaction (PCR)-pyrosequencing described in previous reports [26, 27]. Briefly, DNA was treated with sodium bisulfate using EZ DNA Methylation-Gold kit (Zymo Research, CA, USA) per manufacturer's instructions. The bisulfate-treated DNA was amplified using Qiagen PyroMark PCR Kit (Qiagen, Hilden, Germany). The methylation percentage of 6 CpG sites in humans (-432 to -401) and 5 CpG sites in rats (-387 to -351) was measured (Figure S2 in the online-only Data Supplement). Thermal cycling program and PCR primers are listed in Table S2 in the online-only Data Supplement. The specific PCR products were then subjected to quantitative pyrosequencing analysis using a PyroMark™ Q96 MD Pyrosequencing System (Qiagen, Germany). The degree of methylation in the CpG sites tested is expressed herein as the percentage of methylated cytosine (mC) over the sum of methylated and unmethylated cytosine residues. The reported methylation levels are the averages of all CpG sites in Klotho. Each marker was pyrosequenced in duplicates, and averaged of the results were considered.
Indoxyl sulfate measurement
Serum IS was determined by ultra-performance liquid chromatography-mass spectrometry / mass spectrometry (UPLC-MS/MS). Agilent 1100 system (Agilent, Waldbronn, Germany) consisting of G1312A quaternary pump, G1379A vacuum degasser, G1316A thermostattedcolumn oven and HTS PAL autosampler was used. Mass spectrometric detection was performed on API 3000 triple quadrupole instrument (Applied Biosystems, Ontario, Canada) in multiple reactions monitoring (MRM) mode. Turbo Ion Spray ionization interface in negative ionization mode was used. The samples were stored at -20ºC and allowed to thaw at room temperature before processing. All the parameters of LC-MS were controlled by Analyst software version 1.4.1 (Applied Biosystems).
Statistical analysis
All data were expressed as mean ± standard error. The one-way analysis of variance was performed with Bonferroni corrections for analyzing the data of cell culture study. Data from different study animal groups were compared using the Wilcoxon-Mann-Whitney test and P-values of <0.05 were considered statistically significant.
Supplemental methods
Detailed description for quantitative real-time PCR, western blotting analysis, calcification staining, HE and immunohistochemistry staining protocols used can be found in the online-only Data Supplement.
Results
Vascular calcification and Klotho hypermethylation in patients with ESRD
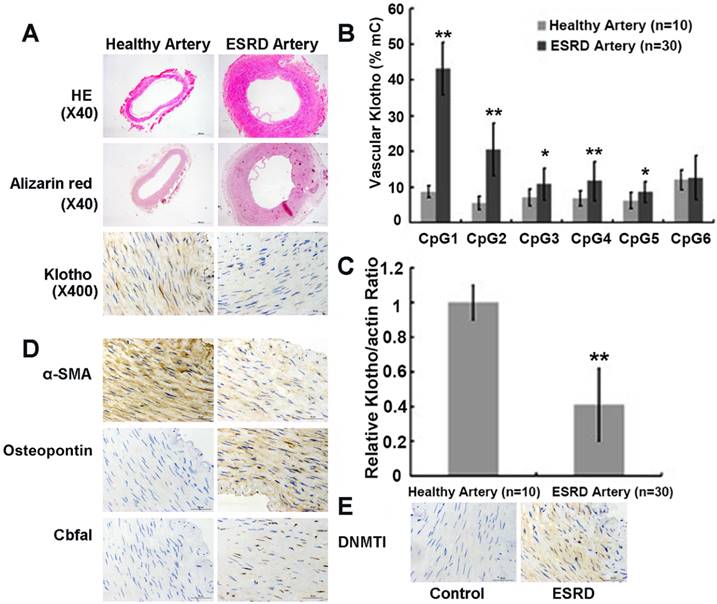
A great reduction in Klotho protein expression in the medial layer of radial arteries from ESRD patients were observed compared to healthy controls by immunohistochemistry analysis (Figure 1A top panels). This reduction in vascular Klotho protein expression in ESRD patients was associated with extensive medial calcification assessed by Alizarin red staining (Figure 1A mid panels). In addition, Figure 1D showed that the protein level of osteopontin (OPN) and core binding factor 1 (Cbfα1) increased in ESRD group while α-smooth muscle actin (α-SMA) decreased, which suggested vascular calcification in the ESRD group. We found that 0 of 10 (0%) of healthy arteries from control group and 21 of 30 (70%) of ESRD arteries having calcification. We also performed quantitative bisulfate-pyrosequencing analysis in these artery samples to check the levels of vascular Klotho promoter methylation (Figure S2 in the online-only Data Supplement for pyrosequencing positions). Arteries from ESRD patients exhibited increased levels of vascular Klotho promoter methylation compared to control group (7.7 ± 0.7 vs. 18.0 ± 3.8%, P<0.001), suggesting the existence of hypermethylation in ESRD group. Figure 1B summarizes the comparison of methylation level between ESRD arteries and healthy arteries at 6 individual CpG islands of Klotho gene, of which 5 sites showed difference with statistical significance. Consistent with promoter hypermethylation, Klotho mRNA and protein expression was significantly down-regulated in ESRD arteries (Figure 1C and 1A bottom panels). We also noted that DNMT1 increased in ESRD group (Figure 1E). These findings suggest that vascular Klotho hypermethylation may be involved in the vascular Klotho deficiency and vascular dysfunction of ESRD patients.
Arteries from ESRD patients have increased calcification and reduced Klotho expression due to its promoter hypermethylation. (A) Hematoxylin-eosin, Alizarin red calcium and immunohistochemistry staining. Histologically, medial layer of the arterial wall was observed much thicker in ESRD group than the control group. Alizarin Red calcium staining showed no calcium nodule formation in the control group. However, various degrees of calcium nodule formation were found in radial medial layer of ESRD group. Immunohistochemical staining showed that Klotho was weakly and sporadically positive in radial medial layer of ESRD patients. (B) Vascular methylation level for 6 CpG islands of Klotho. ESRD Patients had higher vascular methylation level for 5 CpG islands of Klothothan control group. (C) Real-time PCR analysis of artery Klotho mRNA expression. Vascular Klotho mRNA expression in ESRD patients (n=30) was significantly lower than in healthy individuals (n=10). mC%, percentage of methylated cytosines. Compared with control group, *P<0.05, ** P<0.001. (D) and (E) Immunohistochemical staining showed that α-SMA, Cbfα1, OPN and DNMT1 in ESRD patients and control.
IS suppresses Klotho expression and potentiates the development of calcification in HASMCs
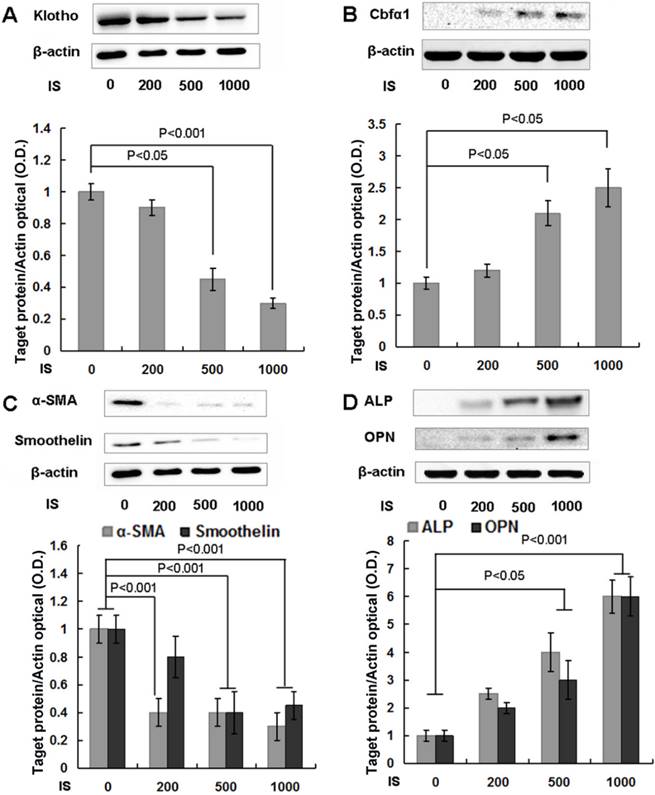
Klotho expression was significantly reduced after treatment with IS of 500 μmol/L for 6 days (Figure 2A) and cells exhibited remarkable calcification (Figure 4C). IS has been shown to induce osteogenic transformation of HASMCs, an important process that orchestrates the development of calcification [28]. In parallel with calcification, Cbfα1 expression was up-regulated, indicating that HASMCs underwent transformation to the calcifying phenotype (Figure 2B). The changes of key cellular regulatory/marker proteins further confirmed this transformation, such as the down-regulation of α-SMA and smoothelin, as well as the up-regulation of alkaline phosphatase (ALP) and OPN (Figure 2C, D).
IS reduces Klotho expression and accelerates osteogenic transformation of HASMCs. (A) HASMCs were treated with IS at concentrations of 0, 200, 500, and 1000 μmol/L for 6 d, and cellular protein was analyzed with Western blotting. Klotho protein expression was suppressed by 500 and 1000 μmol/L IS. (B, C, D) Cbfα1 protein expression, the master regulator towards to a calcifying osteogenic phenotype, increased dramatically, together with decreased expression of α-SMA and smoothelin, and increased ALP and OPN.
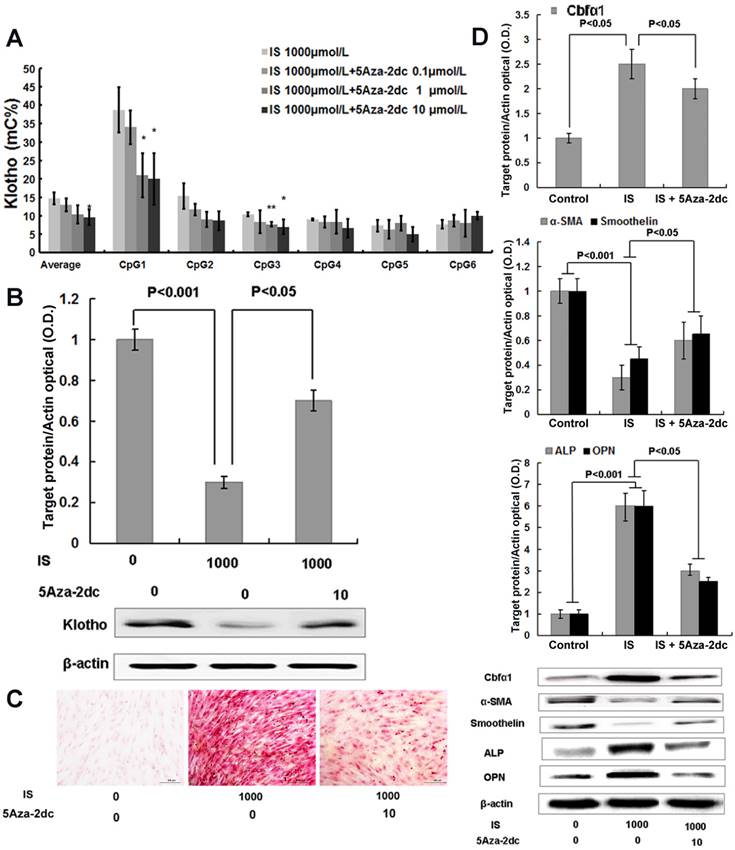
DNA methyltransferase 1 inhibitor demethylates the Klotho gene, increases Klotho expression and mitigates osteogenic transformation of HASMCs induced by IS. (A) Quantitative bisulfite pyrosequencing showed that 10 μmol/L 5-Aza-2dc inhibited Klotho gene hypermethylation in cultured HASMCs treated with 1000 μmol/L IS. Compared with lane 1, *P<0.05, ** P<0.01. (B) Western blotting showed that 10 μmol/L 5Aza-2dc significantly increased the Klotho protein expression in HASMCs treated with 1000 μmol/L IS. (C)Qualitative assessment of calcification by Alizarin red staining in HASMCs treated with IS at concentrations of 1000 μmol/L for 6 d revealed calcium deposition. These effects were mitigated by10 μmol/L 5Aza-2dc. (D) 5Aza-2dc dramatically down-regulated Cbfα1 protein expression, up-regulated α-SMA and smoothelin expression, and down-regulated ALP and OPN expression.
IS increases DNMT1 and 3a expression and DNA methylation of the Klotho gene in HASMCs, which can be reverted by DNMT1 inhibitor, 5Aza-2dc
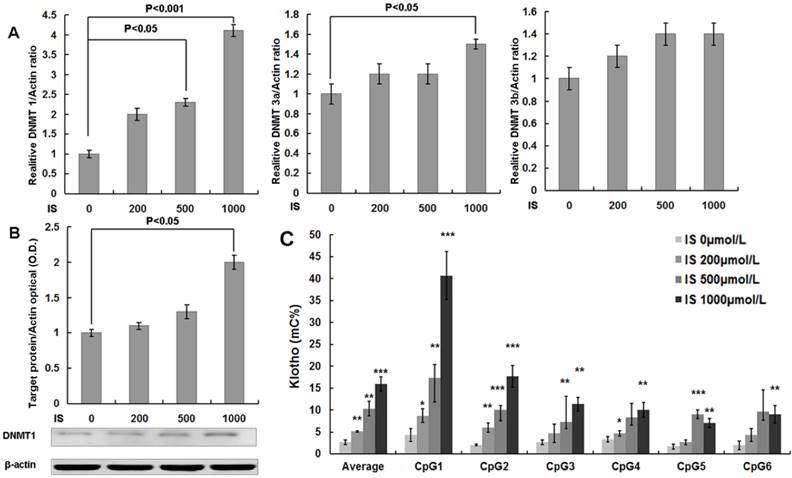
Quantitative RT-PCR analysis showed that either 500 or 1000 μmol/L of IS treatments significantly increased DNMT1 mRNA expression in cultured HASMCs. The mRNA expression of DNMT3a was also increased in HASMCs culture in the presence of 1000 μmol/L of IS (Figure 3A). Western blotting analysis further confirmed the up-regulation of DNMT1 expression by IS at the concentration of 1000 μmol/L (Figure 3B). Furthermore, quantitative bisulfate-pyrosequencing analysis indicated that HASMCs treated with IS had significantly increased CpG methylation levels of the Klotho gene (Figure 3C).
IS increases DNA methyltransferase 1, 3a, and 3b expression and DNA methylation of the Klotho gene in HASMCs. (A) Real-time PCR analysis showed that IS significantly increased DNMT 1 and 3a expression. (B) Western blotting analysis showed that IS at 1000 μmol/L significantly increased DNMT 1 protein expression. (C) Chromatin DNA samples from cultured HASMCs treated with IS for 6 d were analyzed by quantitative bisulfite pyrosequencing. It revealed that Klotho methylation levels of HASMCs treated with IS were significantly increased at concentrations of 200, 500, and 1000 μmol/L. HASMCs treated with IS of 1000 μmol/L had higher methylation level for 6 CpG islands of Klotho. Compared with lane 1, *P<0.05, ** P<0.01, ***P<0.001. mC%, percentage of methylated cytosines.
We next investigated whether 5Aza-2dc, a DNMT1 inhibitor, could suppress Klotho gene hypermethylation and restore its expression. As shown in Figure 4A, 5Aza-2dc decreased the enhanced CpG hypermethylation of the Klotho gene, which was induced by 1000μmol/L of IS treatment, in cultured HASMCs by quantitative bisulfate-pyrosequencing analysis. Moreover, the differences detected in CpG1 and CpG3 reached statistical significance. The effect of 5Aza-2dc on the CpG hypermethylation showed a dose-dependent response, and 10 μmol/L concentration was used in the following project. Western blotting results showed that 5Aza-2dc, at the concentration of 10 μmol/L, largely restored the Klotho protein expression in HASMCs (Figure 4B). 5Aza-2dc also significantly mitigated HASMCs calcification treated with IS of 1000 μmol/L (Figure 4C), consistent with the observation of Cbfα1 down-regulation and the changes of key cellular regulatory proteins, including α-SMA, smoothelin, ALP and OPN (Figure 4D).
Chronic IS injection reduces aortic Klotho expression and causes aortic calcification in 5/6-nephrectomized rats
To model the high IS concentration in ESRD patients, we continuously injected rats with IS for 24 weeks (Figure S1 in the online-only Data Supplement). The IS concentration and other biochemical indicators are listed in Table 1. The serum IS concentration in IS-injected rats was significantly higher than the control rats (14.3±3μM vs. 0.9±0.13μM, P<0.001), which is in the same range of with the IS concentration in ESRD patients [8, 29]. However, the body weight of IS-injected rats was not significantly different from those of the control rats (334±14 vs. 323±12g, P>0.05). HE and Alizarin red calcium staining results showed that IS-injected rats had significantly increased aortic calcification compared to the control rats (Figure 5C). The increased Cbfα1 expression also indicated vascular cells transformation to the calcifying phenotype, together with changes of the key cellular regulatory proteins α-SMA, smoothelin, and OPN (Figure 5D).
Laboratory parameters in Control, IS and IS+ 5-Aza-2dc rats.
| Control(n=5) | IS(n=5) | IS+5-Aza-2dc(n=5) | |
|---|---|---|---|
| BW(g) | 334±14 | 323±12 | 341±17 |
| BUN(mg/dL) | 10.1±5.4 | 20.4±6.4* | 19.2±5.9# |
| Scr(μmol/L) | 45±9.7 | 179.3±16.9* | 140.2±10.6# |
| U-protein(μg/mL) | 134.4±23.1 | 165.7±34.5* | 150.6±36.8# |
| IS(pg/mL) | 0.21±0.03 | 3.3±0.7* | 3.4 ± 0.6* |
| Ca (mg/dL) | 9.3±0.3 | 9.1±0.2 | 9.4±0.4 |
| P (mg/mL) | 7.1±0.26 | 6.8±0.43 | 7.3±0.36 |
* Compared with control group, P<0.01, # Compared with IS group, P<0.01.
DNMT 1 inhibitor demethylates the Klotho gene, increases Klotho expression, and mitigated aortic calcification induced by IS in5/6-nephrectomized rats. (A) Quantitative bisulfite pyrosequencing revealed that Klotho methylation levels of IS-injected rats were significantly increased. IS-injected rats had higher methylation level for 6 CpG islands of Klotho than control rats, while simultaneous treatment with 5Aza-2dc in IS-injected mice significantly demethylated the Klotho gene. mC%, percentage of methylated cytosines. (B) Western blotting showed that 5Aza-2dc significantly increased the Klotho expression in IS-injected rats. (C) Hematoxylin-eosin and Alizarin red staining in IS -injected 5/6-nephrectomized rats revealed increased aortic wall thickness and severe aortic calcification. These effects were mitigated by 5-aza-2dc. (D) 5Aza-2dc dramatically down-regulated Cbfα1 protein expression, up-regulated expression of α-SMA and smoothelin, and suppressed ALP and OPN expression. (E) Immunohistochemical staining revealed that Klotho protein expression was significantly lower in aorta of the rats treated with IS. (F) Western blot analysis showed that IS significantly increased the DNMT 1 expression.
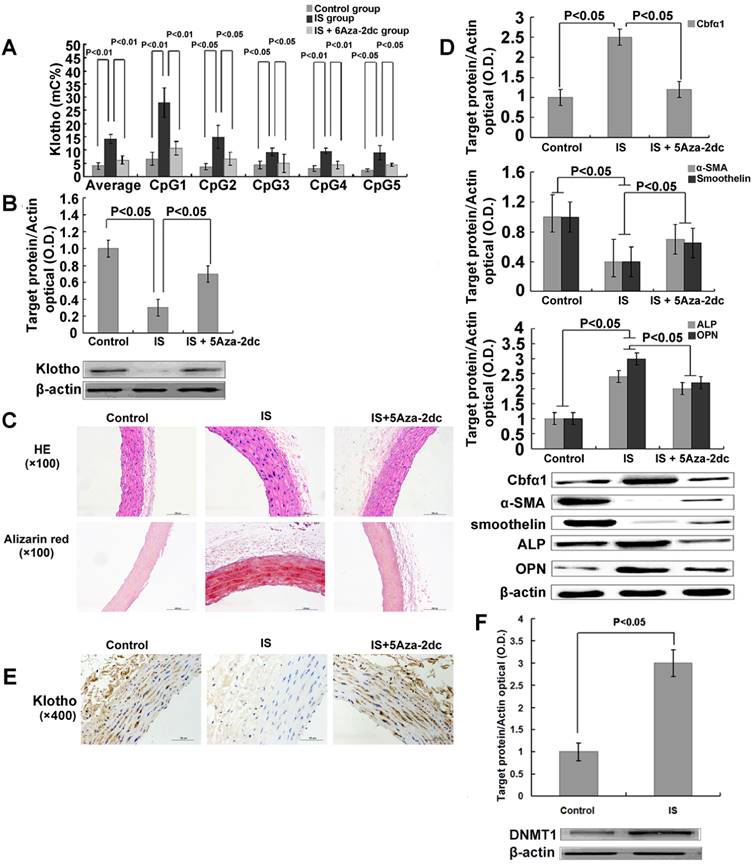
Consistent with the HASMCs data again, both western blotting analysis and immunohistochemistry stain showed that Klotho protein expression was significantly lower in the aorta of IS-injected rats when than in the 5/6-nephrectomized rats without IS injection (Figure 5B). Quantitative bisulfate-pyrosequencing analysis indicated that IS-injected rats had significantly increased aortic CpG methylation levels of the Klotho gene (Figure 5A, Figure 2 in the online-only Data Supplement for pyrosequencing positions), which was probably due to the upregulation of DNMT1 in these cells (Figure 5F).
Lastly, we tested if 5Aza-2dc could revert the DNA hypermethylation of the Klotho gene and restore its expression in these IS treated experimental animals. We gave 5Aza-2dc every 48 h together with IS injection throughout the 24-week treatment period (Figure S1 in the online-only Data Supplement for experiment design). 5Aza-2dc did not change the serum IS level itself (14.7 ± 2.6μM for 5Aza-2dc group vs.14.3±3μM for IS group) but indeed, efficiently prevented DNA hypermethylation of Klotho gene in IS- injected rats as shown by quantitative bisulfate-pyrosequencing analysis (Figure 5A). As a result, aortic Klotho expression in these rats was largely restored (Figure 5B). Furthermore, 5Aza-2dc significantly inhibited aortic calcification in IS-injected rats (Figure 5C). These observations were again confirmed by examining protein expression of Cbfα1, α-SMA, smoothelin, ALP and OPN (Figure 5D).
Discussion
The relation between chronic kidney disease (CKD) and cardiovascular disease (CVD) has been extensively studied in the past years. CKD is considered to be one of the major risk factors that lead to accelerated vascular calcification. Klotho, a gene functions as a cofactor for FGF23 regulated urinary phosphate excretion, is thought to link CKD and vascular calcification [15]. It has been shown that severe Klotho deficiency is usually found in CKD patients, however underlying mechanisms remain elusive [15]. In the current study, we have compared Klotho gene methylation status in human arteries from CKD patients and from healthy individuals, and found that Klotho gene hypermethylation could be one of the mechanisms for uremia induced Klotho silencing. Studies using HASMCs and 5/6 nephrectomized rats further demonstrated that IS, a protein-bound uremic toxin, decreased vascular Klotho expression through epigenetically silencing its promoter region.
IS is usually accumulated in the serum of CKD patients. IS induces the production of free radicals in both renal cells and vascular smooth muscle cells and oxidative stress is one of the most important mechanisms for the tissue injury caused by IS [30, 31]. Oxidative stress results in increased expression of DNMT during carcinogenesis [32]. It has been suggested that IS could increase DNMT expression in kidney tubular cells [19]. DNMTs are the key enzymes that are responsible for the regulation of DNA methylation [33] and DNMT1 is the most abundant DNMT in mammals and is considered to be the key methyltransferase responsible for maintaining promoter methylation [34, 35]. Exactly, as DNMT inhibition may have effects on other genes, we could not conclude that 5-Aza2dc treatment inhibits vascular calcification via Klotho, although this may partially be the case. Previous studies have shown that SM22a promoter methylation was an important event in vascular smooth muscle cells calcification and that high phosphate could induce this epigenetic modification via increased DNMT expression [36]. Several other studies have shown that Klotho hypermethylation is associated with DNMT1 expression in kidney. Our study in cultured HASMCs suggested that administration of IS increased DNMT1 and 3a expression. Inhibiting DNMT1 activity with 5Aza-2dc reduced CpG methylation of the Klotho gene and increased Klotho expression in HASMCs treated with IS.
Previous studies have shown that administration of IS to hypertensive rats promote aortic calcification through activation of Cbfα1 and promote cell senescence with expression of senescence-related proteins such as p16, p21 and p53 in the aorta of hypertensive rats [28, 37]. Inhibition of Klotho gene expression by RNA interference up-regulated p53/p21 pathway and induced premature senescence in human cells [38]. Our results showed that epigenetic silencing of the Klotho gene by IS partially explained the possible mechanism of cell senescence process caused by uremic toxins.
It has been demonstrated that osteogenic transformation of vascular smooth muscle cells can render them mineralization competent to secrete an osteoid-like extracellular matrix over a defined time course [39]. Our results showed that suppression of Klotho is associated with up-regulation of Cbfα1 and vascular smooth muscle cells phenotype transformation. Up-regulation of Cbfα1 orchestrates with osteoblastic differentiation of vascular smooth muscle cells by regulating downstream bone-related proteins, including alkaline phosphatase and OPN. Our data suggested that phenotypic adaptation of HASMCs may be involved in the pathogenesis of endogenous vascular Klotho loss. Inhibiting DNMT1 activity with 5Aza-2dc demethylated CpG of the Klotho gene, and increased α-SMA and smoothelin expression and suppressed ALP and OPN expression in HASMCs treated with IS.
The levels of serum IS are different at different stages of CKD [4, 40]. The IS level induced in our experimental rats (14.3 μM) was similar to the level of patients with CKD stage 4, although serum IS level can be as high as 500-600 μM when severe kidney function loss was developed in ESRD patients. Thus, our animal model may only represent the pathology of relatively end-stage CKD patients. However, the development of vascular calcification in CKD patients is a very slow process and it may be caused in vivo by other risk factors such as hyperphosphatemia. HASMC in vitro culture system only allowed us to test the effect of IS on Klotho gene hypermethylation and other cellular changes within 7 days. Under this condition, it required at least 200-500 μM IS treatment to visualize the changes (Figure 2). We are currently developing a prolonged primary cell culture system for studying vascular calcification with pathologically relevant concentration of IS.
Transcriptional suppression of vascular Klotho gene expression by IS is associated with CpG hypermethylation of Klotho and epigenetic modification of Klotho by IS may be an important pathological mechanism of vascular calcification in chronic kidney disease. A more comprehensive animal study with different serum IS levels may lead to a better understanding of the role of serum IS and Klotho hypermethylation in the progression of chronic kidney disease.
Supplementary Material
Supplementary Table and Figures.
Acknowledgements
We would like to thank Professor Qing Wu (School of Public Health, Fudan University), for help on primer designing.
Funding
This study was supported by research grants from the Ministry of Science and Technology of People's Republic of China (2011BAI10B03), Science and Technology Commission of Shanghai Municipality (14ZR1406400), and Young Physician Training Program from Shanghai Health and Family Planning Commission. The funders had no roles in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Author Contributions
Conceived and designed the experiments: XZ XD. Performed the experiments: JC HZ HZ. Analyzed the data: JC XZ. Contributed reagents/materials/analysis tools: XD TL JT JJ. Wrote the manuscript: JC XZ XD.
Competing Interests
The authors have declared that no competing interest exists.
References
1. London GM, Guerin AP, Marchais SJ. et al. Arterial media calcification in end-stage renal disease: impact on all-cause and cardiovascular mortality. Nephrol Dial Transplant. 2003;18:1731-40
2. Shanahan CM, Crouthamel MH, Kapustin A. et al. Arterial calcification in chronic kidney disease: key roles for calcium and phosphate. Circ Res. 2011;109:697-711
3. Blacher J, Guerin AP, Pannier B. et al. Arterial calcifications, arterial stiffness, and cardiovascular risk in end-stage renal disease. Hypertension. 2001;38:938-42
4. Lau WL, Leaf EM, Hu MC. et al. Vitamin D receptor agonists increase klotho and osteopontin while decreasing aortic calcification in mice with chronic kidney disease fed a high phosphate diet. Kidney Int. 2012;82:1261-70
5. Liabeuf S, Drueke TB, Massy ZA. Protein-bound uremic toxins: new insight from clinical studies. Toxins (Basel). 2011;3:911-9
6. Niwa T, Takeda N, Tatematsu A. et al. Accumulation of indoxyl sulfate, an inhibitor of drug-binding, in uremic serum as demonstrated by internal-surface reversed-phase liquid chromatography. Clin Chem. 1988;34:2264-7
7. Yamamoto H, Tsuruoka S, Ioka T. et al. Indoxyl sulfate stimulates proliferation of rat vascular smooth muscle cells. Kidney Int. 2006;69:1780-5
8. Adijiang A, Goto S, Uramoto S. et al. Indoxyl sulphate promotes aortic calcification with expression of osteoblast-specific proteins in hypertensive rats. Nephrol Dial Transplant. 2008;23:1892-901
9. Sugiura H, Yoshida T, Tsuchiya K. et al. Klotho reduces apoptosis in experimental ischaemic acute renal failure. Nephrol Dial Transplant. 2005;20:2636-2645
10. Kuro-o M, Matsumura Y, Aizawa H. et al. Mutation of the mouse klotho gene leads to a syndrome resembling ageing. Nature. 1997;390:45-51
11. Kurosu H, Yamamoto M, Clark JD. et al. Suppression of aging in mice by the hormone Klotho. Science. 2005;309:1829-33
12. Hu MC, Shi M, Zhang J. et al. Klotho deficiency causes vascular calcification in chronic kidney disease. J Am Soc Nephrol. 2011;22:124-36
13. Imura A, Iwano A, Tohyama O. et al. Secreted Klotho protein in sera and CSF: implication for post-translational cleavage in release of Klotho protein from cell membrane. FEBS Lett. 2004;565:143-7
14. Hu MC. Klotho connects intermedin1-53 to suppression of vascular calcification in chronic kidney disease. Kidney Int. 2016;89:534-537
15. Hu MC, Kuro-o M, Moe OW. αKlotho and vascular calcification: an evolving paradigm. Curr Opin Nephrol Hypertens. 2014;23:331-9
16. C. Ritter, S. Zhang, J. Delmez, et al. Differential expression and regulation of Klotho by paricalcitol in the kidney, parathyroid, and aorta of uremic rats. Kidney Int. 2015;87:1089-1091
17. Faul C, Wolf M. Hunt for the culprit of cardiovascular injury in kidney disease. Cardiovasc Res. 2015;108:209-11
18. Lewin E, Olgaard K. The vascular secret of Klotho. Kidney Int. 2015;87:1089-91
19. Sun CY, Chang SC, Wu MS. Suppression of Klotho expression by protein-bound uremic toxins is associated with increased DNA methyltransferase expression and DNA hypermethylation. Kidney Int. 2012;81:640-50
20. Hu MC, Shi M, Cho HJ. Klotho and phosphate are modulators of pathologic uremic cardiac remodeling. J Am Soc Nephrol. 2015;26:1290-302
21. Lindberg K, Amin R, Moe OW. The kidney is the principal organ mediating klotho effects. J Am Soc Nephrol. 2014;25:2169-75
22. Lim K, Lu TS, Molostvov G. et al. Vascular Klotho deficiency potentiates the development of human artery calcification and mediates resistance to fibroblast growth factor 23. Circulation. 2012;125:2243-55
23. Donate-Correa J, Mora-Fernández C, Martínez-Sanz R. et al. Expression of FGF23/KLOTHO system in human vascular tissue. Int J Cardiol. 2013;165:179-83
24. Chang JR, Guo J, Wang Y. et al. Intermedin1-53 attenuates vascular calcification in rats with chronic kidney disease by upregulation of α-Klotho. Kidney Int. 2016;89:586-600
25. Lindberg K, Olauson H, Amin R. Arterial klotho expression and FGF23 effects on vascular calcification and function. PLoS One. 2013;8:e60658
26. Zhang C, Liang Y, Lei L. et al. Hypermethylations of RASAL1 and KLOTHO is associated with renal dysfunction in a Chinese population environmentally exposed to cadmium. Toxicol Appl Pharmacol. 2013;271:78-85
27. Chen J, Zhang X, Zhang H. et al. Elevated Klotho promoter methylation is associated with severity of chronic kidney disease. PLoS One. 2013;8:e79856
28. Muteliefu G, Enomoto A, Jiang P. et al. Indoxyl sulphate induces oxidative stress and the expression of osteoblast-specific proteins in vascular smooth muscle cells. Nephrol Dial Transplant. 2009;24:2051-8
29. Boelaert J, Lynen F, Glorieux G. et al. A novel UPLC-MS-MS method for simultaneous determination of seven uremic retention toxins with cardiovascular relevance in chronic kidney disease patients. Anal Bioanal Chem. 2013;405:1937-47
30. Tumur Z, Niwa T. Indoxyl sulfate inhibits nitric oxide production and cell viability by inducing oxidative stress in vascular endothelial cells. Am J Nephrol. 2009;29:551-7
31. Palm F, Nangaku M, Fasching A. et al. Uremia induces abnormal oxygen consumption in tubules and aggravates chronic hypoxia of the kidney via oxidative stress. Am J Physiol Renal Physiol. 2010;299:F380-6
32. Campos AC, Molognoni F, Melo FH. et al. Oxidative stress modulates DNA methylation during melanocyte anchorage blockade associated with malignant transformation. Neoplasia. 2007;9:1111-21
33. Turek-Plewa J, Jagodzinski PP. The role of mammalian DNA methyltransferases in the regulation of gene expression. Cell Mol Biol Lett. 2005;10:631-47
34. Ting AH, Jair KW, Schuebel KE. et al. Differential requirement for DNA methyltransferase 1 in maintaining human cancer cell gene promoter hypermethylation. Cancer Res. 2006;66:729-35
35. Fandy TE. Development of DNA methyltransferase inhibitors for the treatment of neoplastic diseases. Curr Med Chem. 2009;16:2075-85
36. Montes de Oca A, Madueno JA, Martinez-Moreno JM. et al. High-phosphate-induced calcification is related to SM22alpha promoter methylation in vascular smooth muscle cells. J Bone Miner Res. 2010;25:1996-2005
37. Muteliefu G, Shimizu H, Enomoto A. et al. Indoxyl sulfate promotes vascular smooth muscle cell senescence with upregulation of p53, p21, and prelamin A through oxidative stress. Am J Physiol Cell Physiol. 2012;303:C126-34
38. de Oliveira RM. Klotho RNAi induces premature senescence of human cells via a p53/p21 dependent pathway. FEBS Lett. 2006;580:5753-8
39. Reynolds JL, Joannides AJ, Skepper JN. et al. Human vascular smooth muscle cells undergo vesicle-mediated calcification in response to changes in extracellular calcium and phosphate concentrations: a potential mechanism for accelerated vascular calcification in ESRD. J Am Soc Nephrol. 2004;15:2857-67
40. Wu IW, Hsu KH, Lee CC. et al. p-Cresyl sulphate and indoxyl sulphate predict progression of chronic kidney disease. Nephrol Dial Transplant. 2011;26:938-47
Author contact
![]() Corresponding authors: Xiaoqiang Ding, Department of Nephrology, Zhongshan Hospital, Fudan University, No 180 Fenglin Road, Shanghai 200032, China. E-mail: ding.xiaoqiangsh.cn; Tel. / fax: +86-21-64038472. Xiaoyan Zhang, Kidney and Dialysis Institute of Shanghai, No 136 Medical College Road, Shanghai 200032, China. E-mail: zhang.xiaoyansh.cn; Tel. / fax: +86-21-64038472.
Corresponding authors: Xiaoqiang Ding, Department of Nephrology, Zhongshan Hospital, Fudan University, No 180 Fenglin Road, Shanghai 200032, China. E-mail: ding.xiaoqiangsh.cn; Tel. / fax: +86-21-64038472. Xiaoyan Zhang, Kidney and Dialysis Institute of Shanghai, No 136 Medical College Road, Shanghai 200032, China. E-mail: zhang.xiaoyansh.cn; Tel. / fax: +86-21-64038472.