Impact Factor ISSN: 1449-2288

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2017; 13(3):391-400. doi:10.7150/ijbs.17318 This issue Cite
Research Paper
Novel approaches to vitiligo treatment via modulation of mTOR and NF-κB pathways in human skin melanocytes
Jerry Wan1*, Fuquan Lin2, Wei Zhang3, Aie Xu2, Joseph DeGiorgis1, Hongguang Lu3, Yinsheng Wan1 ![]()
1. Department of Biology, Providence College, Providence, RI 02918, USA.
2. Department of Dermatology, the 3 rd Hospital of Hangzhou, Hangzhou 310000, Zhejiang Province, China.
3. Department of Dermatology, Guiyang Medical University, Guizhou 550004, China.
* Current address: Tandon School of Engineering, New York University, Brooklyn, NY 11201, USA.
Received 2016-8-23; Accepted 2017-1-9; Published 2017-2-25
Abstract

Vitiligo is a skin depigmentation disorder with an increasing prevalence. Among recognized mechanisms is the oxidative stress that affects melanocytes which are responsible for skin pigmentation. Studies have shown that high concentration of hydrogen peroxide, or H2O2, induces apoptotic activities. Few studies have been done with lower doses of H2O2. Using human skin melanocytes, we investigated the effect of moderate concentration of H2O2 on melanocyte dendrites. Confocal data show that H2O2 at 250 µM induces loss of dendrites, as indicated by cytoskeletal proteins. α-melanocyte stimulating hormone or α-MSH pretreatment protects against H2O2-induced loss of dendrites, while α-MSH alone enhances dendrites. PI3K/AKT inhibitor LY294002 and mTORC1 inhibitor Rapamycin inhibit α-MSH-induced dendrites. In this study, we also investigated the effect of TNFα on cultured human skin melanocytes, since TNFα plays important roles in vitiligo. Confocal data demonstrate that TNFα induces NFκB activation. Western blot analysis shows that TNFα induces IκB phosphorylation and degradation. Interestingly, α-MSH does not have any effect of TNFα-induced IκB degradation and NF-κB activation. α-MSH, however, activates mTORC1 pathway. TNFα induces p38 but not AMPKα activation. Collectively, our data suggest that modulation of mTOR and NF-κB pathways may be a novel approach for better clinical management of vitiligo.
Keywords: α-MSH, mTORC1, TNFα, vitiligo
Introduction
Vitiligo is a depigmentation disorder with an estimated worldwide prevalence from 0.5 to 2%[1, 2]. Vitiligo is not life threatening, but affects people cosmetically and even emotionally as a result of a negative social stigma. With the increasing prevalence of this skin disorder, more and more people are interested in understanding the cellular and molecular mechanism in hopes of overcoming and better yet preventing this disorder.
Vitiligo occurs when functioning melanocytes disappear from the epidermis and the production of pigments decreases, leaving white patches on the skin. Vitiligo is the result of complex interactions of biochemical, environmental and immunological events in a permissive genetic milieu, while the precise mechanism of vitiligo pathogenesis has remained elusive[3, 4]. It has been recognized that this unique type of skin disorder is associated with melanocyte and possibly other skin cell apoptosis[5, 6]. Melanocyte death is related to reactive oxygen species, cytokines[7, 8], and more complicatedly, autoimmunity[3, 9-13].
Reactive oxygen species or ROS, has been shown to be related to a variety of human diseases including vitiligo[12]. Hydrogen peroxide or H2O2, is one of the ROS that induces apoptosis. Excess ROS has been documented in active vitiligo skin. The removal of H2O2 by antioxidants has been proven to be beneficial to patients with vitiligo[14]. And minocycline, an antibiotic possessing antioxidant activity, has been shown to protect melanocytes against H2O2-induced apoptosis in vitro and may be used for the treatment in the early stages of vitiligo[15].
Melanocytes are responsible for melanin production and transport to surrounding keratinocytes. Melanin transfer takes place at the junction between dendrites of melanocytes and membrane of keratinocytes. Dendrites play critical roles of melanosomal transfer in melanocytes[16, 17]. In addition to apoptosis of melanocytes, the damage of dendrites may also affect melanin transport leading to vitiligo. Our preliminary study showed that oxidative stress such as H2O2 treatment induces loss of dendrites. However, the mechanism of the loss of dendrites remains to be unraveled.
In addition to oxidative stress, there also is evidence for altered immunological processes in vitiligo, particularly in chronic and progressive conditions. While both innate and adaptive immunities are proposed to be involved as a primary event or as a consequence in vitiligo, there is an interplay between ROS and the immune system in the pathogenesis of vitiligo. There is evidence linking oxidative stress and autoimmunity to vitiligo pathogenesis, supporting the notion of a convergent terminal pathway of oxidative stress-autoimmunity-mediated melanocyte loss in vitiligo[12, 18, 19].
α-MSH activates putative cell surface receptor MC1R and stimulates melanogenesis and proliferation of human melanocytes. α-MSH also promotes human melanocyte survival by inhibiting UV-induced apoptosis[15, 20]. Studies predict that the survival effect of α-MSH is caused by reduction of UV-induced DNA damage and contributes to the prevention of melanoma[20-22]. Our preliminary studies have shown that H2O2 incudes loss of dendrites in human melanocytes, and α-MSH protects this loss of dendrites. However, the molecular mechanism of this process remains unknown.
PI3K/AKT and mTOR pathway has been suggested to be associated with cell survival in response to UV radiation and oxidative stress[23]. Growth factors protect against UV and oxidative stress-induced apoptosis via activation of AKT and mTOR pathways[24-26]. While indirect data suggest that α-MSH-stimulated melanogenesis through the activation of MEK/ERK or PI3K/AKT[27], the question of whether α-MSH protects against oxidative stress-induced cell damage either apoptosis or loss of dendrites via AKT/mTOR pathway activation remains to be addressed.
Autoimmunity remains a complex issue, and vitiligo specific antigens are yet to be identified. Studies thus far have shown that UV radiation induces generation of reactive oxidative species and cytokines, which may have deleterious effects on melanocytes and may partially account for an increased rate of vitiligo[28]. Cytokines such as tumor necrosis factor (TNF)-alpha or TNFα, a paracrine inhibitor of melanocytes, play critical roles in the pathogenesis of several autoimmune diseases including vitiligo[29]. Anti-TNFα has been tested for the treatment of vitiligo[30-32]. And yet, the cellular and molecular mechanisms of the actions of cytokines have not been thoroughly studied in cultured human melanocytes.
Given that H2O2 induces cell damages, either apoptosis or loss of dendrites or both, oxidative stress may be associated with vitiligo, and cytokines such as TNFα play important roles in vitiligo, we undertook this study to investigate the effect of H2O2 on melanocyte dendrites, to study cell signaling pathway leading to α-MSH's protective activity, to investigate the effect of cytokines on melanocytes, and to study the cell signaling pathways of cytokine actions. We, for the first time, found that oxidative stress reduces dendrites and α-MSH protects against oxidative stress-induced loss of dendrites via activation of mTORC1 pathway. We also, for the first time, found that TNFα, but not IL1β, activates NFκB pathway and α-MSH does not crosstalk with TNFα cell signaling pathway. Our novel findings support the notion that modulation of mTORC1 pathway and NF-κB pathway may offer better clinical management of vitiligo.
Materials and Methods
Cell Culture
Primary human skin melanocytes were purchased from American Type Culture Collection (ATCC, Manassas, VA) and cultured in 254 medium (Invitrogen, Carlsbad, CA) with growth factor supplements and Penicillin/Streptomycin (1:100, Sigma-Aldrich, St. Louis, MO) as described previously[15, 20]. Cells were cultured in a CO2 incubator at 37oC. For confocal microscopy, cells were seeded in eight well chamber slides and grew to eighty percent of confluence for treatment. For Western blot analysis, cells were cultured in six well slides and grew to ninety percent confluence for treatment.
Reagents and Antibodies
Recombinant human TNFα and IL1β were purchased from R & D Systems (Minneapolis, MN). α-MSH was from Sigma-Aldrich (St. Louis, MO). LY294002 and Rapamycin were from EMD/Calbiochem (San Diego, CA). Rabbit Anti-p65, Rabbit anti-IκB, Rabbit-anti α-tubulin were purchased from Santa-Cruz Biotechnology (Santa-Cruz, CA). Rabbit anti-phosphor IκB, rabbit anti-phosphor p38, and rabbit anti-phosphor-S6 ribosomal protein were purchased from Cell Signaling Technology (Bevery, MA). Mouse anti-β-actin was purchased from Sigma-Aldrich (St. Louis, MO). Alexa Fluor 488 goat anti-mouse IgG or anti-rabbit IgG, Alexa Fuor 594 goat anti-mouse IgG or anti-rabbit IgG, and Alexa Fluor 680 goat anti-rabbit IgG were from Life Technologies (Grand Island, NY). IRDye Goat anti-mouse and anti-rabbit secondary antibodies were from LI-COR (Lincoln, NE).
Confocal microscopy
As previously reported[33], cells were cultured in eight well chamber slides to 90% confluence and treated with various reagents and fixed with 4% formaldehyde in PBS for 10 min. After PBS wash, the cells were permeabilized with cold methanol for 15 min. After PBS wash, cells were blocked with goat serum for 30 min. After PBS wash, cells were stained with various antibodies for 1 hour. After PBS wash, cells were stained with Alexa fluor labeled secondary antibodies for 1 hour. After PBS wash, cells were stained with Hoechst dye for 15 min. After PBS wash, slides were disassembled, covered by anti-fade (Invitrogen, Carlsbad, CA), and slides were sealed for confocal microscopic observation (Carl Zeiss LSM 700). Images were captured by Zen 2009 Light Edition and exported to Photoshop and processed and assembled in Adobe Illustrator CS6.
Quantification of dendricity
Images of cultured melanocytes were captured under confocal microscope, with one cell under one microscope field. The length of the dendrite was measured using a ruler. A total of six cells were used for quantification of dendricity.
Western blot analysis
As previously reported[34], cells were cultured in six-well plates to 80% confluence. Cells were treated with various reagents. Cells were collected at different time points by scraping them into 120 µl of RIPA cell lysis buffer (50 mM Tris-HCL, pH 7.4, 150 mM NaCl, 1% NP40, 1 mM EDTA, 0.25% sodium deoxycholate, with 1 mM NaF, 10 µM Na3VO4, 1mM phenylmethylsulfonyl fluoride, and protease inhibitor cocktail) in a microfuge tube. Cell lysates were incubated in 4°C for 30 min on a shaker. After centrifugation at 12000rpm for 10 min, supernatant was collected. Twenty micrograms of proteins were denatured in 5x SDS-PAGE sample buffer for 5 min at 95°C. The proteins were separated by 10% SDS-PAGE and transferred onto PVDF membrane (EMD/Millipore, Bedford, MA). Nonspecific binding was blocked with 10% dry milk in TBS for 1 h at room temperature. After blocking, membranes were incubated with specific antibodies in dilution buffer (2% BSA in TBS) overnight at 4oC on the shaker. The blots were incubated with secondary antibodies at 1:10000 dilution for 1 h. Antibody binding was detected using Odyssey Infrared Imaging System (LI-COR, Lincoln, NE) following manufacturer's instructions. The images were exported to and processed in Photoshop and assembled in Adobe Illustrator CS6.
Statistical analysis
Statistical comparisons were performed using an unpaired Student's t test in Microsoft Excel.
Results
H2O2 induces apoptosis and loss of dendrites in cultured human skin melanocytes
To test the effect of H2O2 on melanocytes, we first cultured human melanocytes in eight well chamber slides and treated with a different does of H2O2 (ranging from 10 µM to 500 µM). The results showed that as expected, melanocytes died 24 hours later at high concentrations, as previously reported[15]. This is one of the causes of the loss of melanocytes leading to vitiligo[15, 35]. Existing data have indicated that dendrites of melanocytes play critical roles in melanin transfer from melanocytes to surrounding keratinocytes. The loss or reduction of dendrites may also affect the capability of melanin transport. Interestingly, we found that, at the concentration of 250 µM, melanocytes lose dendrites within four hours of the treatment, as shown in Fig. 1 A and B. α-melanocyte stimulating hormone or α-MSH, has been shown to have protective effect on melanocytes[20]. To test whether α-MSH could protect melanocytes from the loss of dendrites under H2O2 treatment, we treated the cells with α-MSH first for one hour and then H2O2 for two hours. We found that melanocytes pre-treated with α-MSH maintained the integrity of dendrites, while H2O2 alone induced loss of dendrites (Fig. 1C, 1D). We also observed that α-MSH induced S6 phosphorylation in melanocytes (Fig. 1C). S6, a ribosomal protein, downstream of mTORC1, plays an important role in protein synthesis, thus is critical for cell survival and maintaining dendrites of melanocytes in response to oxidative stress.
H2O2 induces loss of dendrites and α-MSH protects against it in cultured human skin melanocytes. Cultured human skin melanocytes in eight well chamber slides were treated with 250 µM of H2O2 and fixed and stained with Hoechst for nucleus, stained with anti β-actin and α-tubulin as shown in (A), anti β-actin and vimentin as shown in (B) for cytoskeletal proteins. And cells were pretreated with α-MSH (10-8M) for 1 hour and then treated with H2O2 for two hours. The cells were then fixed and stained with Hoechst for nucleus, stained with anti β-actin for cytoskeletal actin and stained with anti-phosphor-ribosomal protein S6 antibody for S6 phosphorylation as shown in (C). Dendricity was quantified based on β-actin staining in C, mean±SD, *p<0.05 (D). Scale bar=50µm.
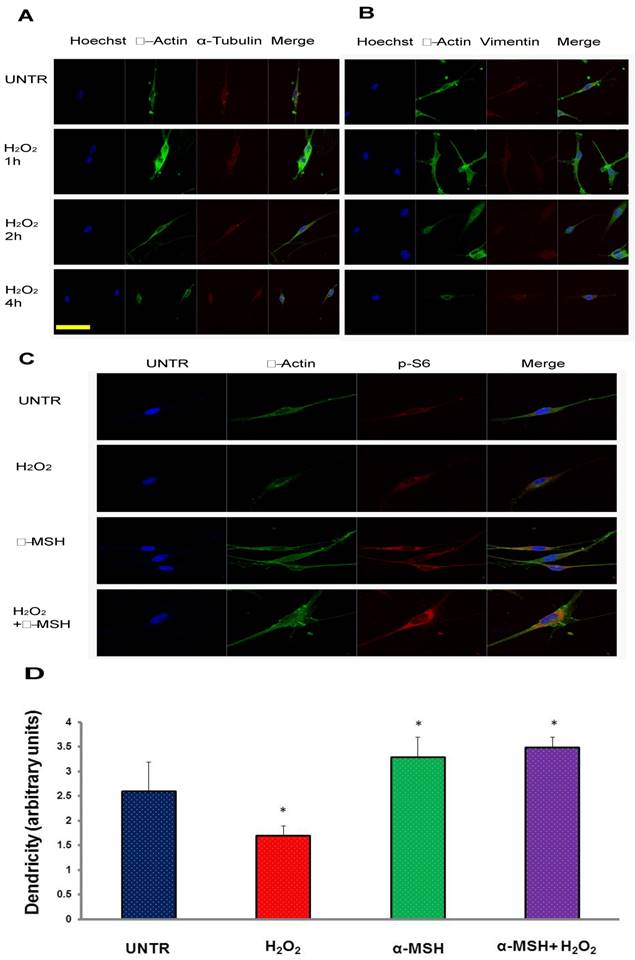
mTORC1 plays important roles in maintaining dendrites under oxidative stress
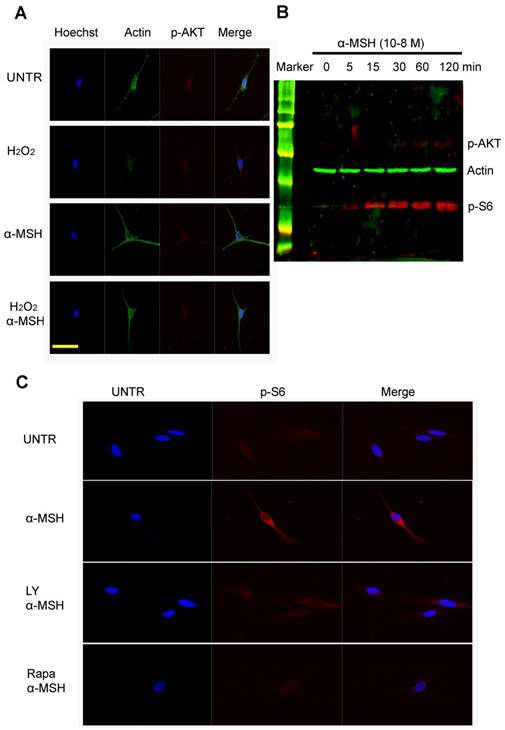
To investigate the cellular signaling pathways that are important in dendrites, we treated cells with H2O2, α-MSH alone, or a combination. Confocal data showed that α-MSH slightly induced AKT phosphorylation (Fig. 2A), however, remarkably induced S6 phosphorylation (Fig. 1C). To confirm this observation, we performed Western blot analysis. Cells were cultured in six well plates and treated with α-MSH and cell lysates were collected at different time points after treatment. The data showed that α-MSH significantly induced S6 phosphorylation in a time dependent manner (Fig. 2B). Confocal data also showed that LY294002, an inhibitor of PI3K/AKT slightly reduced α-MSH-induced S6 phosphorylation, and Rapamycin, an inhibitor of mTORC1, almost completely abolished α-MSH-induced S6 activation (Fig. 2C). These data suggest that mTORC1 plays an important role in α-MSH-induced S6 phosphorylation which may bypass PI3K/AKT and mTORC2.
α-MSH induces mTORC1 as measured as S6 phosphorylation but moderately AKT activation in cultured human skin melanocytes. Cells were cultured in eight well slides and pretreated with or without α-MSH for 1 hour and then treated with H2O2 for two hours. Cells were then fixed and stained with Hoechst for nucleus, stained with anti β-actin for cytoskeletal proteins and anti-phosphor-AKT for AKT activation as shown in (A). Cells were cultured in six well plates and treated with α-MSH (10-8M). Cell lysates were collected at different time points post treatment for Western blot analysis, probed by anti-phosphor AKT and anti-phosphor S6 for mTORC1 activation, by anti-β-actin as loading control, as shown in (B). And cells were pretreated with PI3K/AKT inhibitor LY294002 (10µM), or mTORC1 inhibitor Rapamycin (10µM) for 1h, and then treated with α-MSH (10-8M) for 1h. Cells were fixed and stained with Hoechst for nucleus and anti-phosphor-S6 for S6 activation as shown in (C). Scale bar=50µm.
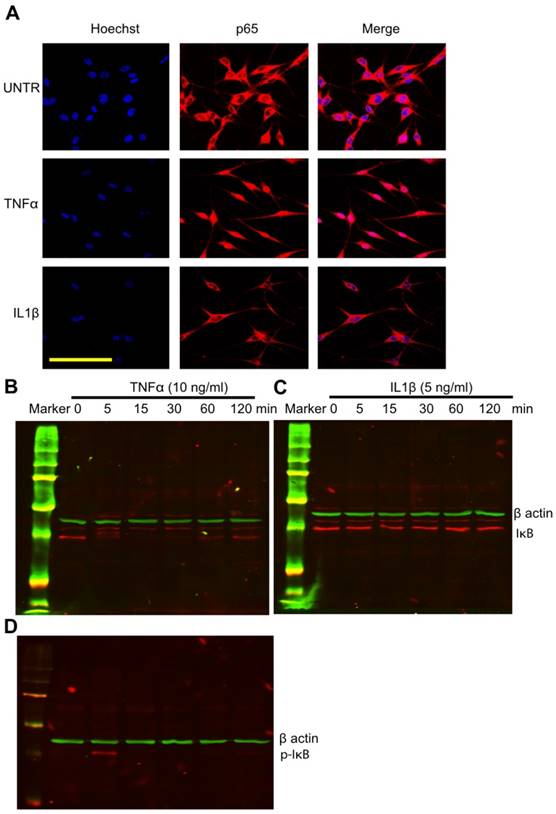
TNFα but not IL1β, induces NF-κB p65 translocation from the cytoplasm to the nucleus in human skin melanocytes
Accumulating data have shown that cytokines, especially TNFα, play an important role in vitiligo. It has been suggested that anti-TNFα antibody could be used to treat vitiligo[7]. However, the molecular mechanism of TNFα in vitiligo remains unclear. To study whether TNFα activates NF-κB (with subunits of p65 and p50) in human skin melanocytes, cells were treated with TNFα at various time points (0, 5, 15, 30 min, 1 and 2 hour) and fixed and stained with anti-p65 antibody. The confocal microscopy data showed that NF-κB subunit p65 translocated from the cytoplasm to the nucleus in a time-dependent manner. Nuclear staining was most visible at 30 min after treatment (Fig. 3A). The results demonstrate that TNFα activates NF-κB in cultured human melanocytes. To our knowledge, this is the first report of TNFα-induced NF-κB activation in cultured human skin melanocytes. Interestingly, however, IL1β does not have any effect on p65 translocation, since in human skin keratinocytes IL1β does activate NF-κB. To further investigate the mechanism of NF-κB activation, cells were treated with TNFα at different time points (0, 5, 15, 30 min, 1 and 2 hour). Collected cell lysates were subject to SDS-PAGE Western blot analysis. The results showed that TNFα induced IκB degradation in a time-dependent manner. IκB disappeared 5 min after TNFα treatment, remained invisible, and reappeared 30 min post-TNFα treatment (Fig. 3B). There is a band above IκB which might be the phosphorylated form of IκB. It is equally interesting but expected that IL1β did not induce IκB degradation (Fig. 3C), since IL1β did not induce p65 translocation in melanocytes (Fig. 3A). To further investigate the activation and degradation of IκB, cells were treated with TNFα at different time points (0, 5, 15, 30 min, 1 and 2 hour). Cell lysates were subject to SDS-PAGE and Western blot by anti-phosphor IκB. The results showed that TNFα induced phosphorylation of IκB dramatically at 5 min (Fig. 3D), which was corresponding to the degradation of IκB (Fig. 3A).
TNFα but not IL1β induces NFκB activation and TNFα induces IκB phosphorylation and degradation in cultured human skin melanocytes. Cells were cultured in eight well chamber slides and treated with TNFα (10ng/ml) or IL1β (5ng/ml) at different time points. Cells were fixed with 4% formaldehyde in PBS and stained with anti-p65, then observed by confocal microscopy as shown in A. Cells were cultured in six well plates and treated with TNFα (10ng/ml) or IL1β (5ng/ml) at different time points. Cells were collected for SDS-PAGE/Western blot analysis using anti-IκB for IκB degradation by TNFα as shown in (B) or IL1β as shown in (C), or using anti-phosphor-IκB for IκB phosphorylation by TNFα as shown in (D). β-actin was probed as loading control. Scale bar=50µm.
α-MSH does not inhibit TNFα-induced NF-κB activation in cultured melanocytes
Existing data from other cells indicate that α-MSH inhibits TNFα-induced NF-κB activation[36, 37]. To study whether α-MSH has any effect on TNFα-induced NF-κB activation in cultured human skin melanocytes, cells were treated with α-MSH 1 hour prior to TNFα treatment. Western blot analysis showed that α-MSH pre-treatment did not affect TNFα-induced IκB degradation (Fig. 4A), and NF-κB p65 translocation (data not shown). These results are different from previous publication in keratinocyte[37]. It is likely that melanocytes are respondent to α-MSH and TNFα differently from keratinocytes. And our data showed that α-MSH induced mTORC1 activation as indicated by phosphorylation of S6 ribosomal protein (Fig. 1C, Fig. 2B, 2C, Fig. 4B).
α-MSH does not inhibit TNFα-induced IκB degradation but induces mTORC1 activation and TNFα induces p38 activation in cultured human skin melanocytes. Cells were cultured in six well plates, pre-treated with or without α-MSH (10-8M) for 1 hour and treated with TNFα (10ng/ml) at different time points. Cells were collected for SDS-PAGE/Western blot analysis using anti-IκB or phosphor-S6 as shown in (A). Cells were cultured in six well plates and treated with TNFα (10ng/ml) at different time points. Cells were collected for SDS-PAGE/Western blot analysis using anti-phosphor p38 as shown in (B). β-actin was probed as a loading control.
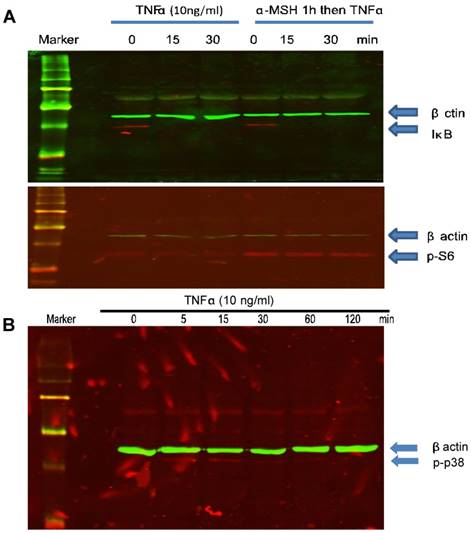
TNFα induces p38 but not AMPKα activation in culture human skin melanocytes
To further study the effect of TNFα on apoptotic pathways, cells were treated with TNFα at different time points (0, 5, 15, 30 min, 1 and 2 hour). Cell lysates were collected for SDS-PAGE and Western blot analysis. The results showed that TNFα induced p38 phosphorylation in a time dependent manner. p38 phosphorylation started a few minutes after treatment and peaked at about 15 minutes and the activity returned to basal level in one hour (Fig. 4B). AMPKα activation has been shown to be related apoptosis and mTOR pathways. Our data did not indicate that TNFα induced AMPKα activation in cultured human skin melanocytes (data not shown).
Discussion
Vitiligo is a common pigmentary skin disorder, characterized by the appearance of white macules on the skin, mucosal or hair that may spread over the entire body skin. Depigmentation arises from the loss of functioning melanocytes[38]. Treatment is often a tough challenge and involves a wide range of therapies[39] including UV radiation, laser[40-42], transplant[43, 44], and complementary medicine[45-47]. The most efficient therapy remains autologous skin graft, performed successfully daily in some of the dermatology clinics.
While the cellular and molecular mechanisms of vitiligo are yet to be fully elucidated, in the field of dermatology, three dominant theories regarding the etiology of vitiligo, namely, reactive oxygen species, cytokines and autoimmunity, have been proposed and supported by experimental and clinical data. However, due to the complexity of this disease, and the likely interconnections of those three possibilities, the studies on vitiligo remain an unparalleled challenge.
It has been proposed that melanocytes are under oxidative stress in vitiligo with a variety of causes including UV radiation and overproduction of cytokines. H2O2 levels are elevated locally in the skin of vitiligo patients. Increased ROS contributes to melanocyte apoptosis and the development of cutaneous diseases or disorders via autoimmunity[12]. However, mechanisms and inter-relationships between ROS and autoimmunity are unknown. Most recent data demonstrate that ER protein calreticulin or CRT exposure via H2O2-induced oxidative stress plays a significant role in melanocyte apoptosis, suggesting a relationship between apoptosis and immune reactions during melanocyte destruction[48, 49].
In addition to apoptosis, we found in this study that H2O2 induced the loss of dendrites in melanocytes within hours of treatment (Fig. 1, 2). Dendrites are critically important in melanin transfer. The loss of dendrites would result in the reduced melanin levels in keratinocytes that account for the skin pigmentation. We found that α-MSH, that has been used in the treatment of vitiligo[50] protects against H2O2-induced loss of dendrites in melanocytes (Fig. 1). This novel discovery supports the notion that α-MSH could be one of the options for the management of vitiligo.
α-MSH plays important roles in melanocyte growth and melanin production via cell surface receptor activation[51]. α-MSH has been also identified as a potent anti-inflammatory in various tissues including the skin. We found that α-MSH activates S6 ribosomal protein and enhances dendrites of melanocytes (Fig. 1) and protects against H2O2-induced loss of dendrites. To further understand the molecular mechanism through which MSH protects against H2O2-induced loss of dendrites, we investigated mammalian target of rapamycin or mTOR pathways that are related to protein synthesis, metabolism, cell survival and other cellular activities.
mTOR signaling pathway couples energy and nutrient abundance to the execution of cell growth and division. Mammalian TOR complex 1 (mTORC1) and mTORC2 exert their actions by regulating other important kinases, such as S6 kinase (S6K) and AKT[52] respectively, or collectively. mTORC1 triggers cell growth and proliferation by promoting protein synthesis and metabolism, and by reducing autophagy or enhancing survival[53]. Our data suggest that α-MSH-induced activation of mTORC1 pathway, inhibited by rapamycin (Fig. 2), help maintain dendrites of melanocytes under oxidative stress (Fig. 1, Fig. 2).
Studies have shown that levels of H2O2 and IL-6, a pro-inflammatory cytokine and a key factor in the pathogenesis of autoimmune diseases are elevated in vitiligo lesions, suggesting that H2O2-induced overexpression of IL-6 by melanocytes via p38 and NF-κB pathways may be a molecular linkage for the oxidative stress and inflammatory/autoimmune reactions in vitiligo and may provide a novel target for the treatment of vitiligo[54, 55]. However, the mechanism through which cytokines affect pigmentation and dendrites is not fully understood[56] and warrants further investigation.
TNFα has been suggested to play important roles in vitiligo. Anti-TNFα antibody which has been proposed and used, albeit in only one case in vivo, appears promising[7]. And yet, thus far, no reports have been published on the cellular signaling pathways of cytokine actions in human skin melanocytes. We, for the first time, have shown that TNFα, but not IL1β, induces NF-κB pathway activation through IκB phosphorylation and degradation (Fig. 3). We have provided insights into the understanding of the molecular actions of cytokines in human skin melanocytes. Our data also support the notion and TNFα antibody could be used in human skin in vivo in vitiligo patients.
It has previously been shown in skin cell keratinocytes and melanocytes/melanoma cells that α-MSH inhibits TNFα-stimulated NF-κB activity[37]. α-MSH has an anti-inflammatory action on dermal fibroblast signaling by inhibiting the pro-inflammatory activity of TNFα in vitro[55]. Interestingly, we found in this study that in human skin melanocytes, α-MSH activates mTOC1 pathway, and does not cross talk with TNFα-induced NF-κB pathway (Fig. 4), which is also a novel observation. Since α-MSH plays such an important role in melanocytes, crosstalk with other pathways would introduce more complications to the complex cellular responses.
In conclusion, our data demonstrate for the first time that oxidative stress reduces dendrites of melanocytes. α-MSH protects against H2O2-induced loss of dendrites via activation of mTORC1 pathway. Our data also for the first time show that TNFα induces NF-κB activation via IκB phosphorylation and degradation. α-MSH, does not inhibit TNFα-induced NF-κB activation in cultured human skin melanocytes (Fig. 5). Collectively, our data suggest that modulation of mTORC1 and NF-κB pathway may offer better approaches for clinical management of vitiligo.
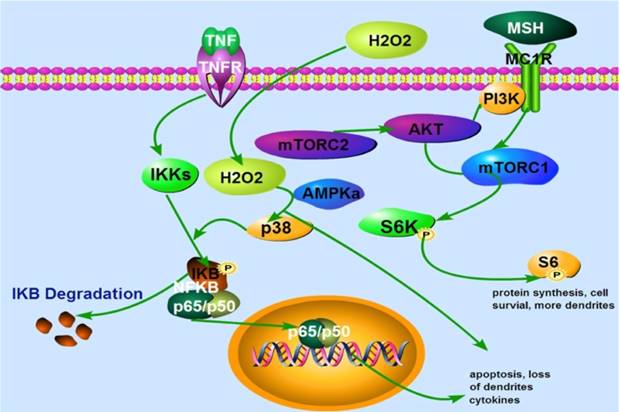
A proposed model that α-MSH activates mTORC1 and protects against H2O2-Induced loss of dendrites in human skin melanocytes. Extracellular H2O2 activates p38 and NFκB leading to apoptosis and loss of dendrites. α-MSH activates mTORC1 pathway via cell surface receptor MC1R which is blocked by rapamycin, leading to protein synthesis, cell survival and more dendrites. TNFα activates cell surface receptor which induces IκB phosphorylation and degradation. This results in translocation of NF-κB subunits p65 and p50 from cytoplasm to nucleus, and expression of more cytokines, apoptosis, and loss of dendrites. TNFα may also induce intracellular H2O2 production that activates NF-κB pathway.
Abbreviations
mTOR: mammalian target of rapamycin; MSH: melanocyte stimulating hormone;
Acknowledgements
This work was supported by National Natural Science Foundation of China (81071294, A Xu) and Natural Science Foundation of Zhejiang Province, China (Z2100973, A XU); and a grant from NIH (P20 RR016457 from INBRE program of the National Center for Research Resources, Y Wan).
Author contributions
All authors contributed significantly to this research project and the preparation of the manuscript. JW, AX, JD and YW conceived and designed the experiments and analyzed the data. FL cultured the cells and JW treated the cells and ran Western blot and confocal microscopy. JW and YW wrote the draft of the manuscript. WZ and HL were involved in the revision process. All authors revised the manuscript and approved the final version for publication. All authors are accountable for all aspects pertinent to the project including accuracy and integrity.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Kruger C, Schallreuter KU. Cumulative life course impairment in vitiligo. Curr Probl Dermatol. 2013;44:102-17
2. Kruger C, Panske A, Schallreuter KU. Disease-related behavioral patterns and experiences affect quality of life in children and adolescents with vitiligo. Int J Dermatol. 2013
3. Malhotra N, Dytoc M. The pathogenesis of vitiligo. J Cutan Med Surg. 2013;17:153-72
4. Alikhan A, Griffin J, Nguyen N, Davis DM, Gibson LE. Pediatric follicular mucinosis: presentation, histopathology, molecular genetics, treatment, and outcomes over an 11-year period at the Mayo Clinic. Pediatr Dermatol. 2013;30:192-8
5. Le Poole C, Boissy RE. Vitiligo. Semin Cutan Med Surg. 1997;16:3-14
6. Bhawan J, Bhutani LK. Keratinocyte damage in vitiligo. J Cutan Pathol. 1983;10:207-12
7. Lv Y, Li Q, Wang L, Gao T. Use of anti-tumor necrosis factor agents: a possible therapy for vitiligo. Med Hypotheses. 2009;72:546-7
8. Yazici AC, Erdal ME, Kaya TI, Ikizoglu G, Savasoglu K. et al. Lack of association with TNF-alpha-308 promoter polymorphism in patients with vitiligo. Arch Dermatol Res. 2006;298:46-9
9. Bowcock AM, Fernandez-Vina M. Targeting skin: vitiligo and autoimmunity. J Invest Dermatol. 2012;132:13-5
10. Boissy RE, Dell'Anna ML, Picardo M. On the pathophysiology of vitiligo: possible treatment options. Indian J Dermatol Venereol Leprol. 2012;78:24-9
11. Zhou H, Zhao J, Tang X, Zhang X, He D. Autoimmune Hyperthyroidism, Vitiligo, Halo Nevus and Lupus. Am J Med Sci. 2015
12. Xie H, Zhou F, Liu L, Zhu G, Li Q. et al. Vitiligo: How do oxidative stress-induced autoantigens trigger autoimmunity? J Dermatol Sci. 2016;81:3-9
13. Shi Q, Zhang W, Guo S, Jian Z, Li S. et al. Oxidative stress-induced overexpression of miR-25: the mechanism underlying the degeneration of melanocytes in vitiligo. Cell Death Differ. 2016;23:496-508
14. Liu B, Jian Z, Li Q, Li K, Wang Z. et al. Baicalein protects human melanocytes from H(2)O(2)-induced apoptosis via inhibiting mitochondria-dependent caspase activation and the p38 MAPK pathway. Free Radic Biol Med. 2012;53:183-93
15. Song X, Xu A, Pan W, Wallin B, Kivlin R. et al. Minocycline protects melanocytes against H2O2-induced cell death via JNK and p38 MAPK pathways. Int J Mol Med. 2008;22:9-16
16. Scott G. Rac and rho: the story behind melanocyte dendrite formation. Pigment Cell Res. 2002;15:322-30
17. Scott G, Leopardi S, Printup S, Madden BC. Filopodia are conduits for melanosome transfer to keratinocytes. J Cell Sci. 2002;115:1441-51
18. Laddha NC, Dwivedi M, Mansuri MS, Gani AR, Ansarullah M. et al. Vitiligo: interplay between oxidative stress and immune system. Exp Dermatol. 2013;22:245-50
19. Glassman SJ. Vitiligo, reactive oxygen species and T-cells. Clin Sci (Lond). 2011;120:99-120
20. Song X, Mosby N, Yang J, Xu A, Abdel-Malek Z. et al. alpha-MSH activates immediate defense responses to UV-induced oxidative stress in human melanocytes. Pigment Cell Melanoma Res. 2009;22:809-18
21. Abdel-Malek ZA, Kadekaro AL, Kavanagh RJ, Todorovic A, Koikov LN. et al. Melanoma prevention strategy based on using tetrapeptide alpha-MSH analogs that protect human melanocytes from UV-induced DNA damage and cytotoxicity. Faseb J. 2006;20:1561-3
22. Bohm M, Wolff I, Scholzen TE, Robinson SJ, Healy E. et al. alpha-Melanocyte-stimulating hormone protects from ultraviolet radiation-induced apoptosis and DNA damage. J Biol Chem. 2005;280:5795-802
23. Cao C, Wan Y. Parameters of protection against ultraviolet radiation-induced skin cell damage. J Cell Physiol. 2009;220:277-84
24. Cao C, Huang X, Han Y, Wan Y, Birnbaumer L. et al. Galpha(i1) and Galpha(i3) are required for epidermal growth factor-mediated activation of the Akt-mTORC1 pathway. Sci Signal. 2009;2:ra17
25. Cao C, Lu S, Jiang Q, Wang WJ, Song X. et al. EGFR activation confers protections against UV-induced apoptosis in cultured mouse skin dendritic cells. Cell Signal. 2008;20:1830-8
26. Cheng LB, Cheng L, Bi HE, Zhang ZQ, Yao J. et al. Alpha-melanocyte stimulating hormone protects retinal pigment epithelium cells from oxidative stress through activation of melanocortin 1 receptor-Akt-mTOR signaling. Biochem Biophys Res Commun. 2014;443:447-52
27. Kadekaro AL, Kavanagh R, Kanto H, Terzieva S, Hauser J. et al. alpha-Melanocortin and endothelin-1 activate antiapoptotic pathways and reduce DNA damage in human melanocytes. Cancer Res. 2005;65:4292-9
28. Moretti S, Spallanzani A, Amato L, Hautmann G, Gallerani I. et al. New insights into the pathogenesis of vitiligo: imbalance of epidermal cytokines at sites of lesions. Pigment Cell Res. 2002;15:87-92
29. Moretti S, Fabbri P, Baroni G, Berti S, Bani D. et al. Keratinocyte dysfunction in vitiligo epidermis: cytokine microenvironment and correlation to keratinocyte apoptosis. Histol Histopathol. 2009;24:849-57
30. Grimes PE. White patches and bruised souls: advances in the pathogenesis and treatment of vitiligo. J Am Acad Dermatol. 2004;51:S5-7
31. Grimes PE, Morris R, Avaniss-Aghajani E, Soriano T, Meraz M. et al. Topical tacrolimus therapy for vitiligo: therapeutic responses and skin messenger RNA expression of proinflammatory cytokines. J Am Acad Dermatol. 2004;51:52-61
32. Simon JA, Burgos-Vargas R. Vitiligo improvement in a patient with ankylosing spondylitis treated with infliximab. Dermatology. 2008;216:234-5
33. Liu Y, Cao GF, Xue J, Wan J, Wan Y. et al. Tumor necrosis factor-alpha (TNF-alpha)-mediated in vitro human retinal pigment epithelial (RPE) cell migration mainly requires Akt/mTOR complex 1 (mTORC1), but not mTOR complex 2 (mTORC2) signaling. Eur J Cell Biol. 2012;91:728-37
34. Cao GF, Liu Y, Yang W, Wan J, Yao J. et al. Rapamycin sensitive mTOR activation mediates nerve growth factor (NGF) induced cell migration and pro-survival effects against hydrogen peroxide in retinal pigment epithelial cells. Biochem Biophys Res Commun. 2011;414:499-505
35. Song X, Xu A, Pan W, Wallin B, Kivlin R. et al. Nicotinamide attenuates aquaporin 3 overexpression induced by retinoic acid through inhibition of EGFR/ERK in cultured human skin keratinocytes. Int J Mol Med. 2008;22:229-36
36. Luger TA, Scholzen TE, Brzoska T, Bohm M. New insights into the functions of alpha-MSH and related peptides in the immune system. Ann N Y Acad Sci. 2003;994:133-40
37. Moustafa M, Szabo M, Ghanem GE, Morandini R, Kemp EH. et al. Inhibition of tumor necrosis factor-alpha stimulated NFkappaB/p65 in human keratinocytes by alpha-melanocyte stimulating hormone and adrenocorticotropic hormone peptides. J Invest Dermatol. 2002;119:1244-53
38. Guerra L, Dellambra E, Brescia S, Raskovic D. Vitiligo: pathogenetic hypotheses and targets for current therapies. Curr Drug Metab. 2010;11:451-67
39. Colucci R, Lotti T, Moretti S. Vitiligo: an update on current pharmacotherapy and future directions. Expert Opin Pharmacother. 2012;13:1885-99
40. Hegyi V, Petrovajova M, Novotny M. An objective assessment of melanin in vitiligo skin treated with Balneo PUVA therapy. Skin Res Technol. 2013
41. Hamzavi IH, Lim HW, Syed ZU. Ultraviolet-based therapy for vitiligo: what's new? Indian J Dermatol Venereol Leprol. 2012;78:42-8
42. Hossani-Madani A, Halder R. Treatment of vitiligo: advantages and disadvantages, indications for use and outcomes. G Ital Dermatol Venereol. 2011;146:373-95
43. Hong WS, Hu DN, Qian GP, McCormick SA, Xu AE. Ratio of size of recipient and donor areas in treatment of vitiligo by autologous cultured melanocyte transplantation. Br J Dermatol. 2011;165:520-5
44. Huggins RH, Henderson MD, Mulekar SV, Ozog DM, Kerr HA. et al. Melanocyte-keratinocyte transplantation procedure in the treatment of vitiligo: the experience of an academic medical center in the United States. J Am Acad Dermatol. 2012;66:785-93
45. Abd El-Samad Z, Shaaban D. Treatment of localized non-segmental vitiligo with intradermal 5-flurouracil injection combined with narrow-band ultraviolet B: a preliminary study. J Dermatolog Treat. 2012;23:443-8
46. Abu Tahir M, Pramod K, Ansari SH, Ali J. Current remedies for vitiligo. Autoimmun Rev. 2010;9:516-20
47. Pacifico A, Leone G. Photo(chemo)therapy for vitiligo. Photodermatol Photoimmunol Photomed. 2011;27:261-77
48. Zhang Y, Liu L, Jin L, Yi X, Dang E. et al. Oxidative Stress-Induced Calreticulin Expression And Translocation: New Insights Into The Destruction Of Melanocytes. J Invest Dermatol. 2013
49. Zheng X, Li Y, Zhao R, Yan F, Ma Y. et al. xCT deficiency induces autophagy via endoplasmic reticulum stress activated p38-mitogen-activated protein kinase and mTOR in sut melanocytes. Eur J Cell Biol. 2016;95:175-81
50. Fabrikant J, Touloei K, Brown SM. A review and update on melanocyte stimulating hormone therapy: afamelanotide. J Drugs Dermatol. 2013;12:775-9
51. Wilson S, Ginger RS, Dadd T, Gunn D, Lim FL. et al. NCKX5, a natural regulator of human skin colour variation, regulates the expression of key pigment genes MC1R and alpha-MSH and alters cholesterol homeostasis in normal human melanocytes. Adv Exp Med Biol. 2013;961:95-107
52. Zoncu R, Efeyan A, Sabatini DM. mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol. 2011;12:21-35
53. Laplante M, Sabatini DM. Regulation of mTORC1 and its impact on gene expression at a glance. J Cell Sci. 2013;126:1713-9
54. Yao L, Hu DN, Chen M, Li SS. Subtoxic levels hydrogen peroxide-induced expression of interleukin-6 by epidermal melanocytes. Arch Dermatol Res. 2012;304:831-8
55. Shah AA, Sinha AA. Oxidative stress and autoimmune skin disease. Eur J Dermatol. 2013;23:5-13
56. Kim NH, Jeon S, Lee HJ, Lee AY. Impaired PI3K/Akt activation-mediated NF-kappaB inactivation under elevated TNF-alpha is more vulnerable to apoptosis in vitiliginous keratinocytes. J Invest Dermatol. 2007;127:2612-7
Author contact
![]() Corresponding author: Prof. Yinsheng Wan, Department of Biology, Providence College, 1 Cunningham Sq., Providence, RI 02918, USA. E-mail address: yswanedu
Corresponding author: Prof. Yinsheng Wan, Department of Biology, Providence College, 1 Cunningham Sq., Providence, RI 02918, USA. E-mail address: yswanedu