Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(2):124-136. doi:10.7150/ijbs.22619 This issue Cite
Research Paper
Ten hub genes associated with progression and prognosis of pancreatic carcinoma identified by co-expression analysis
Zhou Zhou1, Yian Cheng2, Yinan Jiang3, Shi Liu1, Meng Zhang1, Jing Liu1, Qiu Zhao1 ![]()
1. Department of Gastroenterology, Hubei Clinical Center and Key Lab of Intestinal and Colorectal Diseases, Zhongnan Hospital of Wuhan University;
2. Department of Gastroenterology, Renming Hospital of Wuhan University;
3. Department of Gastrointestinal Surgery, Zhongnan Hospital of Wuhan University.
Received 2017-8-31; Accepted 2017-12-21; Published 2018-1-12
Abstract

Since the five-year survival rate is less than 5%, pancreatic ductal adenocarcinoma (PDAC) remains the 4th cause of cancer-related death. Although PDAC has been repeatedly researched in recent years, it is still predicted to be the second leading cause of cancer death by year 2030. In our study, the differentially expressed genes in dataset GSE62452 were used to construct a co-expression network by WGCNA. The yellow module related to grade of PDAC was screened. Combined with co-expression network and PPI network, 36 candidates were screened. After survival and regression analysis by using GSE62452 and TCGA dataset, we identified 10 real hub genes (CCNA2, CCNB1, CENPF, DLGAP5, KIF14, KIF23, NEK2, RACGAP1, TPX2 and UBE2C) tightly related to progression of PDAC. According to Oncomine database and The Human Protein Atlas (HPA), we found that all real hub genes were overexpressed in pancreatic carcinoma compared with normal tissues on transcriptional and translational level. ROC curve was plotted and AUC was calculated to distinguish recurrent and non-recurrent PDAC and every AUC of the real hub gene was greater than 0.5. Finally, functional enrichment analysis and gene set enrichment (GSEA) was performed and both of them showed the cell cycle played a vital role in PDAC.
Keywords: pancreatic ductal adenocarcinoma, prognosis, progression, co-expression, bioinformatics analysis.
Introduction
Since the five-year survival rate is less than 5%, pancreatic ductal adenocarcinoma (PDAC) remains the 4th cause of cancer-related death [1]. Rapid progress, later presentation of symptoms and lack of early specific diagnostic method may lead to this awful prognosis [2-4]. Although PDAC has been repeatedly researched in recent years and numerous biomarkers have been proved to be effectual for diagnosis and treatment for PDAC [5-7]. It is still predicted to be the second leading cause of cancer death by year 2030 [8]. Therefore, further exploration of PDAC about the mechanism of tumorigenesis and progression is inevitable. Davies et al. reported that impaired JNK signaling dramatically accelerated the appearance of KrasG12D-induced acinar-to-ductal metaplasia and pancreatic intraepithelial neoplasias, which rapidly progressed to invasive PDAC within 10 weeks of age [9]. Another analysis showed that aberrant DNA methylation altered the expression of SLIT-ROBO, MET and ITGA2 thus impacting the progress of PDAC [10]. Several studies also proved that immune system played an irreplaceable role in the development of PDAC [5, 7]. Thus, it can be seen that the mechanism of PDAC is intricate and more studies is urgently needed.
Due to the limitation of experiment, the development of microarray and sequencing technology provided an excellent tool and platform for cancer research [11-13]. By associating clinical data with molecular mechanism, new biomarkers for diagnosis, treatment and prognosis might be recovered. Omer et al. released that the homeostasis of cholesterol greatly contributed to the development of cancers by using TCGA database [14]. Frank et al. emphasized the value of cancer network on learning about the connection and difference in various types of cancers [15]. The weighted correlation network analysis (WGCNA) is an R package for weighted correlation network analysis and can be used as a data exploratory tool or a gene screening (ranking) method to find clusters (modules) of highly correlated genes [16]. It was used widely to finding hub genes in various cancers. For example, Colin et al. identified 11 gene co-expression clusters from a large-scale breast cancer data using WGCNA and UBE2S might indicate a poor prognosis for breast cancer [17]. In order to further explore the progress of PDAC, we used this algorithm to identify network-centric genes associated with clinical features.
Materials and methods
Data collection and preprocessing
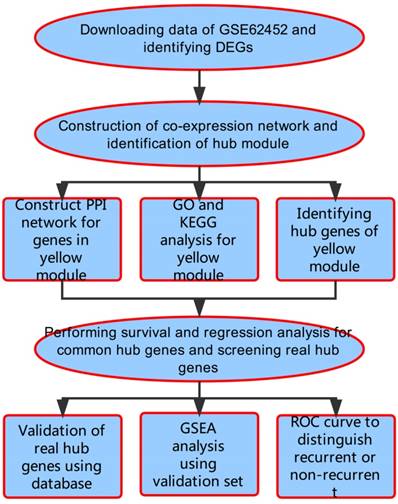
A workflow of this study is shown in Figure 1. We downloaded expressing profiles of mRNA of human pancreatic cancer from Gene Expression Omnibus (GEO) database (http://www.ncbi.nlm.nih.gov/geo/). Dataset GSE62452 performed on Affymetrix Human Gene 1.0 ST Array (Affymetrix, Santa Clara, CA, USA), including 69 pancreatic tumors and 61 adjacent non-tumor tissues from patients with pancreatic ductal adenocarcinoma (PDAC), was used as the training set to construct co-expression networks and identify hub genes in our study [18]. Meanwhile, we also downloaded RNA-sequencing data of pancreatic carcinoma from The Cancer Genome Atlas (TCGA) database (https://genome-cancer.ucsc.edu/), screening 146 samples of pancreas adenocarcinoma ductal type (PDAC) with complete expression profile and clinic information to further validate our results. Moreover, the additional independent dataset GSE62165 performed on Affymetrix Human Gene 1.0 ST Array (Affymetrix, Santa Clara, CA, USA) consisting of 118 PDAC samples and 13 control samples was used as a validation set to perform gene set enrichment analysis (GSEA) in order to identify potential function of PDAC [19].
Flow diagram of the analysis procedure: data collection, preprocessing, analysis and validation.
For the next analyses, raw expression data in dataset GSE62452 was firstly subjected to Robust Multiarray Averaging (RMA) background correction and their processed signals were log2 transformed and normalized by quantile normalization. Then we used the “affy” R package to summarize median-polish probesets. Microarray quality was assessed by sample clustering according to the distance between different samples in average linkage.
Differentially expressed genes (DEGs) screening
The “limma” R package was used to screen the DEGs between non-malignant pancreas samples and PDAC tissues in the expressing data. The significance analysis of microarrays (SAM) with false discovery rate (FDR) < 0.05 and |log2 fold change (FC)| ≥ 0.585 was applied to select genes further considered in the network construction.
Co-expression network construction
We kept the expression data profile of DEGs qualified and then constructed the co-expression network by “WGCNA” package in R for the DEGs [16, 20]. Next, Pearson's correlation matrices were performed for pair-wise genes. We constructed a weighted adjacency matrix using a power function amn = |cmn|β (cmn = Pearson's correlation between gene m and gene n; amn = adjacency between gene m and gene n). Parameter β could emphasize strong correlations between genes and penalize weak correlations. After choosing the appropriate β, the adjacency was transformed into topological overlap matrix (TOM), which could measure the network connectivity of a gene defined as the sum of its adjacency with all other genes for network generation [21]. To classify genes with similar expression profiles into gene modules, average linkage hierarchical clustering was conducted according to the TOM-based dissimilarity measure with a minimum size (gene group) of 50 for the genes dendrogram [22]. To further analyze the module, we calculated the dissimilarity of module eigengenes, chose a cut line for module dendrogram and merged some module.
Identification of clinical significant modules
Two approaches were used to identify modules related to clinical information. Module eigengenes (MEs) were considered as the major component in the principal component analysis for each gene module and the expression patterns of all genes could be summarized into a single characteristic expression profile within a given module. In addition, we calculated the correlation between MEs and clinical trait to identify the relevant module. Afterwards, gene significance (GS) was defined as the log10 transformation of the P value (GS = lgP) in the linear regression between gene expression and clinical information. In addition, module significance (MS) was defined as the average GS for all the genes in a module. In general, the module with the absolute MS ranked first among all the selected modules was considered as the one related with clinical trait.
Candidate genes selection
Hub genes, highly interconnected with nodes in a module, have been considered functionally significant. In our study, an interesting module was chosen, and hub genes were defined by module connectivity and clinical traits significance. Furthermore, we uploaded all genes in the hub module to the Search Tool for the Retrieval of Interacting Genes (STRING) database to construct protein-protein interaction (PPI) to screen hub nodes in PPI network [23, 24]. We defined genes with the node connectivity > 2 (total edges / total nodes) as the hub nodes in PPI network and using Cytoscape to perform the network [25]. Hub genes common in both networks were chosen as the candidates to be further analyzed and validated.
Hub genes identification and efficacy evaluation
We performed survival analysis and regression analysis for common hub genes using dataset GSE62452 and TCGA data. The genes with p value significantly less than 0.05 were identified as real hub genes. Venn diagram was performed based on the survival and regression analysis using TCGA data and training set. Genes with a significant p value in all tests were real hub genes. Next, the database Oncomine (https://www.oncomine.org/) and The Human Protein Atlas (HPA) (http://www.proteinatlas.org/) were used to validate the expression of the real hub genes on transcriptional and translational level [26]. ROC curve was plotted and AUC was calculated with “ROCR” package to evaluate the capability of distinguishing tumor and normal tissue as well as recurrent and non-recurrent PDAC [27].
Functional enrichment analysis
To obtain further insight into the function of DEGs in hub module, DEGs in hub module were uploaded to the Database for Annotation, Visualization and Integrated Discovery (DAVID) (https://david.ncifcrf.gov/home.jsp/) to screen enriched biological themes, particularly GO terms and KEGG pathways [28, 29]. P < 0.05 was set as the cut-off criterion.
Gene set enrichment analysis (GSEA)
In validation set GSE62165, samples of PDAC were divided into two groups according to the expression level of the real hub genes respectively. To identify potential function of the hub genes, GSEA (http://software.broadinstitute.org/gsea/index.jsp) was conducted to detect whether a series of priori defined biological processes were enriched in the gene rank derived from DEGs between the two groups [30]. Terms with FDR < 0.05 and enriched in all real hub genes were identified.
Results
Training set quality assessment and clinical data
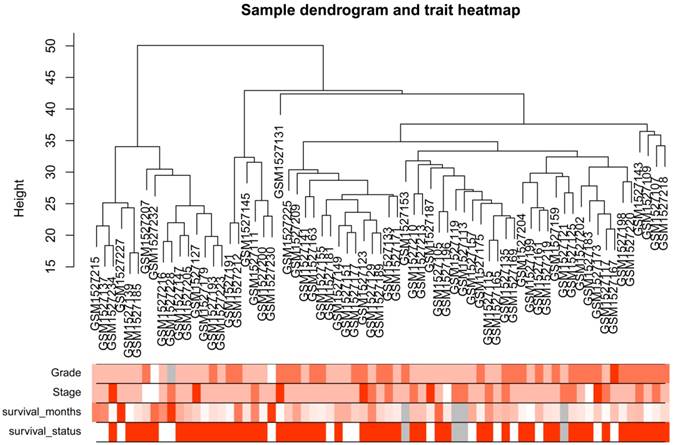
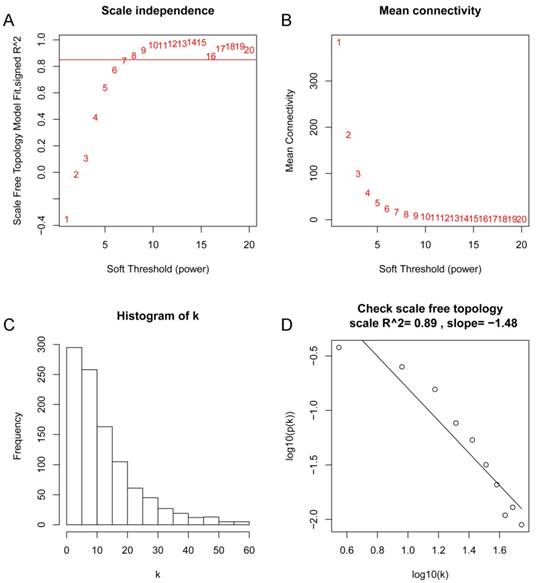
After the first quality check by WGCNA R package, no sample was removed from subsequent analysis in GSE62452 (Figure 2). And in Figure 2, we could find 4 types of clinical data including histological grade, tumor pathological stage, survival months and survival status of PDAC patients. To ensure a scale-free network, here, the power of β = 8 (scale free R2 = 0.89) was selected (Figure 3).
Samples clustering to detect outliers (GSE62452): sample dendrogram and trait indicator. The clustering was based on the expression data of differentially expressed genes between tumor samples and non-tumor samples in PDAC (only the tumor samples, n = 69). The color intensity was proportional to older age, higher histological grade and pathological stage, longer survival month. In survival status, white means patient alive and red means patient dead.
Determination of soft-thresholding power in the weighted gene co-expression network analysis (WGCNA). (A) Analysis of the scale-free fit index for various soft-thresholding powers (β). (B) Analysis of the mean connectivity for various soft-thresholding powers. (C) Histogram of connectivity distribution when β = 8. (D) Checking the scale free topology when β = 8.
DEGs screening
After data preprocessing and quality assessment, we obtained the expression matrices from the 69 samples in training set GSE62452. Under the threshold of FDR < 0.05 and |log2FC| ≥ 0.585, a total of 1008 DEGs (699 up-regulated and 309 down-regulated) were chosen for subsequent analysis. The heatmap and volcano plot of DEGs were shown in Figure S1.
Weighted co-expression network construction and key modules identification
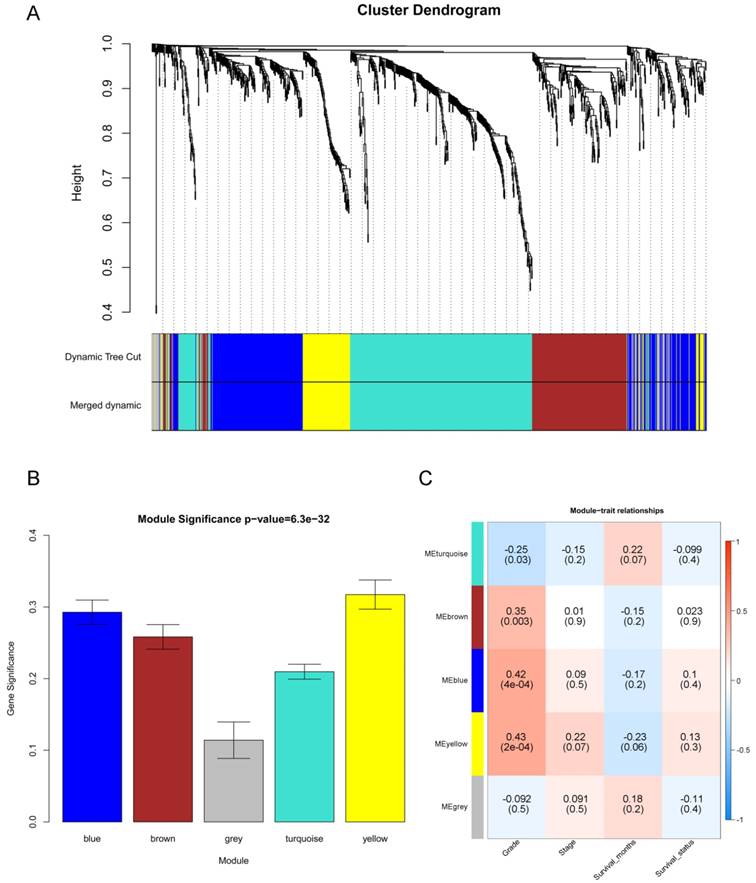
“WGCNA” package in R was used to put the DEGs with similar expression patterns into modules by average linkage clustering, and a total of 5 modules were identified based on the histological grade of PDAC (Figure 4A). We used 2 methods to test the relevance between each module and the PDAC progression. Modules with a greater MS were considered to have more connection with the disease progression, and we found that the MS of the yellow module was higher than those of any other modules (Figure 4B). Afterwards, the ME of the yellow module showed a higher correlation with disease progression than other modules (Figure 4C). Based on the two methods, we identified the yellow module was the module most relevant to the disease progression of PDAC.
Identification of modules associated with the progression of PDAC. (A) Dendrogram of all differentially expressed genes clustered based on a dissimilarity measure (1- TOM). (B) Distribution of average gene significance and errors in the modules associated with the progression of PDAC. (C) Heatmap of the correlation between module eigengenes and the disease progression of PDAC.
Hub gene identification for grade in the yellow module
A total of 43 genes which were highly connective to yellow module were identified as candidate hub genes (Figure 5A). Moreover, we also constructed a network of protein-protein interaction (PPI) for all genes in yellow module by Cytoscape consisted of 80 nodes and 930 edges according to the STRING database, and 42 genes connected with more than 23 nodes were taken as hub nodes in the PPI network (Figure 5B). 36 common network genes were screened as the candidates to be further analyzed and validated (Figure 5C-D).
Hub genes detection and protein-protein network (PPI). (A) Scatter plot of module eigengenes in yellow module. (B) Protein-protein interaction network of genes in the yellow module. The color intensity in each node was proportional to the degree of connectivity in the weighted gene co-expression network (positive correlation in red and negative correlation in green). The size of circles was equal to the Fold Change (FC). The nodes with bold circle represented network hub genes identified by WGCNA. The edge width was proportional to the score of protein- protein interaction based on the STRING database. (C) Selection of hub genes in PPI network and co-expression network. (D) Identification of real hub genes.
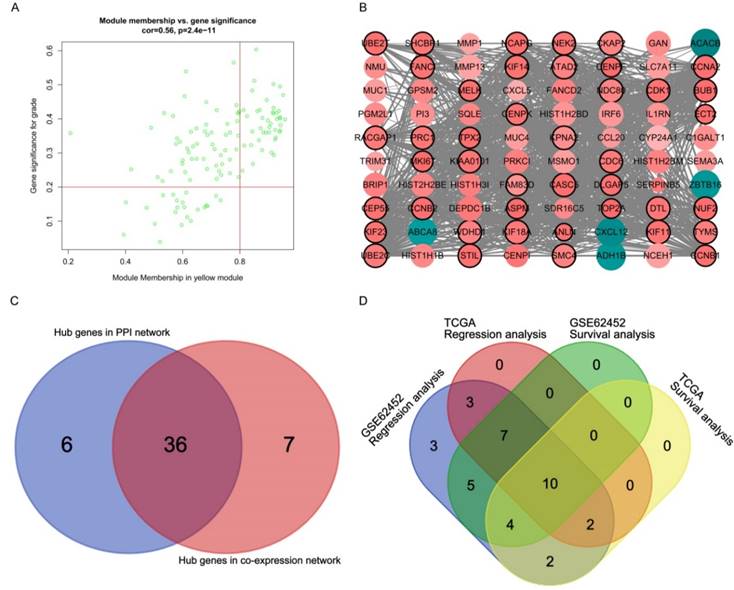
Survival analysis and regression analysis for common hub genes
The dataset GSE62452 containing 69 pancreatic tumors and mRNASeq data followed information of 146 PDAC patients in TCGA were separately subjected to survival analysis and regression analysis. And 10 real hub genes (CCNA2, CCNB1, CENPF, DLGAP5, KIF14, KIF23, NEK2, RACGAP1, TPX2 and UBE2C) among 36 common network genes were highlighted with p value significant less than 0.05 (Figure 6-9). The remaining 26 genes were shown in Figure S2-5.
Survival analysis of real hub genes in the dataset GSE62452. (A) CCNA2. (B) CCNB1. (C) CENPF. (D) DLGAP5. (E) KIF14. (F) KIF23. (G) NEK2. (H) RACGAP1. (I) TPX2. (J) UBE2C. Red lines represent high expression of the real hub genes and blue lines represent low expression.
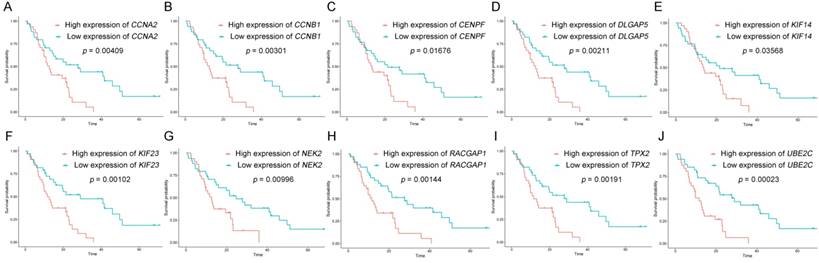
Survival analysis of real hub genes in the TCGA dataset. (A) CCNA2. (B) CCNB1. (C)CENPF. (D) DLGAP5. (E) KIF14. (F) KIF23. (G) NEK2. (H) RACGAP1. (I) TPX2. (J) UBE2C. Red lines represent high expression of the real hub genes and blue lines represent low expression.
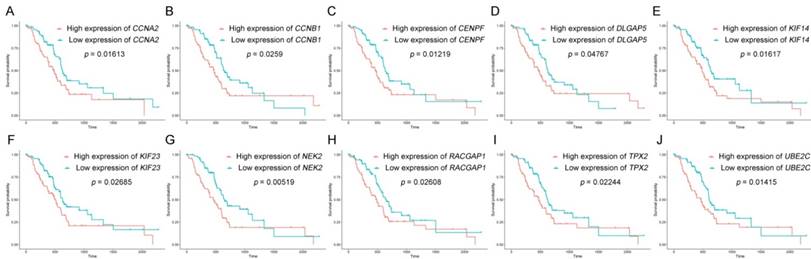
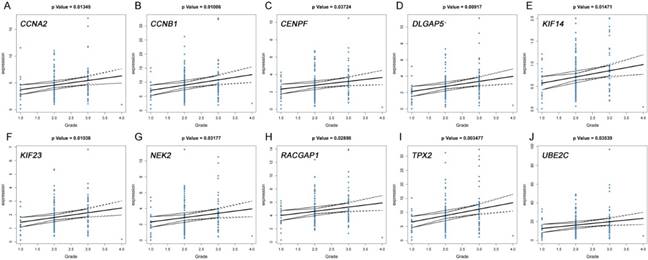
The correlation between the expression levels of real hub genes in the dataset GSE62452 and the disease progression of PDAC. (A) CCNA2. (B) CCNB1. (C) CENPF. (D) DLGAP5. (E) KIF14. (F) KIF23. (G) NEK2. (H) RACGAP1. (I) TPX2. (J) UBE2C. Grade means histological grade.
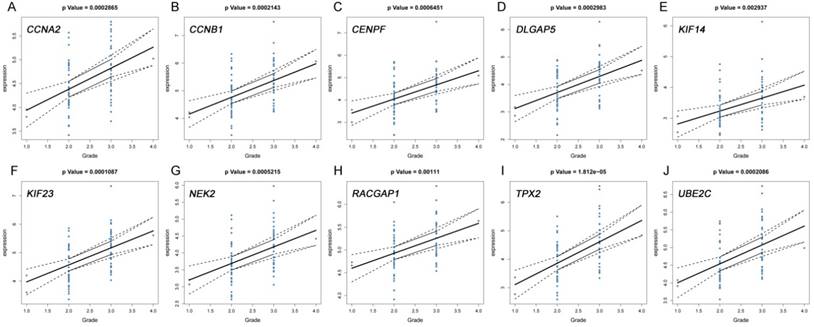
The correlation between the expression levels of real hub genes in the TCGA dataset and the disease progression of PDAC. (A) CCNA2. (B) CCNB1. (C) CENPF. (D) DLGAP5. (E) KIF14. (F) KIF23. (G) NEK2. (H) RACGAP1. (I) TPX2. (J) UBE2C. Grade means histological grade.
Real hub genes validation and efficacy evaluation
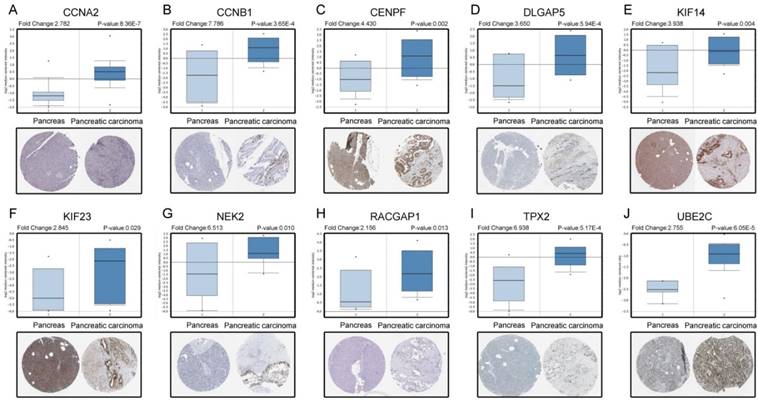
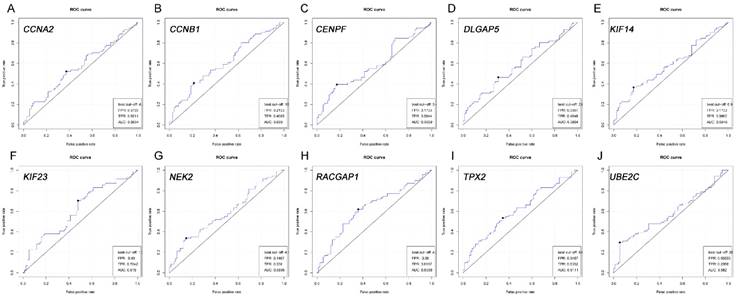
Based on the Oncomine database, we could find that the expression of real hub genes was significantly elevated in pancreatic carcinoma compared with normal tissues. Moreover, immunohistochemistry staining obtained from The Human Protein Atlas database also demonstrated the deregulations of real hub genes expression (Figure 10), and the patient data of the IHC was list in Table S1. In addition, ROC curve analysis was implemented to evaluate the capacity of real hub genes to distinguish recurrent and non-recurrent PDAC as well as tumor and normal tissues. AUC values for 10 genes were greater than 0.5 (Figure 11 and Figure S6).
Validation the expression of real hub genes on transcriptional and translational level by Oncomine database and The Human Protein Atlas database (immunohistochemistry). (A) CCNA2. (B) CCNB1. (C) CENPF. (D) DLGAP5. (E) KIF14. (F) KIF23. (G) NEK2. (H) RACGAP1. (I) TPX2. (J) UBE2C.
ROC analysis of real hub genes in TCGA database. (A) CCNA2. (B) CCNB1. (C) CENPF. (D) DLGAP5. (E) KIF14. (F) KIF23. (G) NEK2. (H) RACGAP1. (I) TPX2. (J) UBE2C.Receiver operating characteristic (ROC) curves and area under the curve (AUC) statistics is to evaluate the capacity of distinguishing recurrent and non-recurrent PDAC.
Functional enrichment analysis
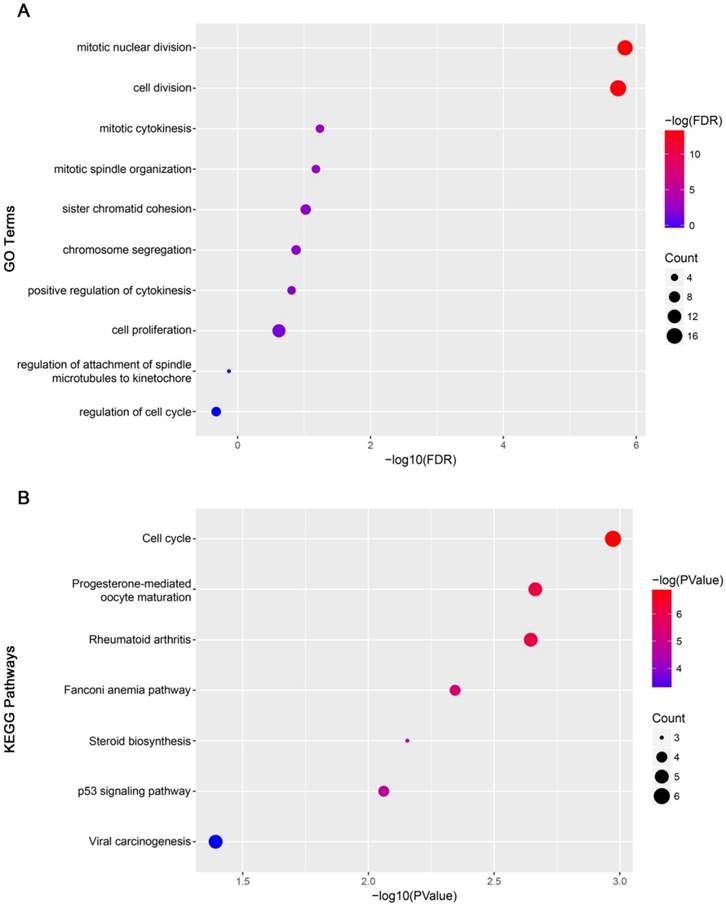
GO analysis results showed that mitotic nuclear division and cell division were significantly enriched (Figure 12A). Cell cycle was most enriched in KEGG pathways analysis (Figure 12B).
Functional analysis. (A) Gene Ontology analysis for genes in yellow module. (B) KEGG pathway enrichment analysis for genes in yellow module. The x-axis shows the -log10 (P- value) of each term and the y-axis shows the GO and KEGG pathway terms.
Gene set enrichment analysis (GSEA)
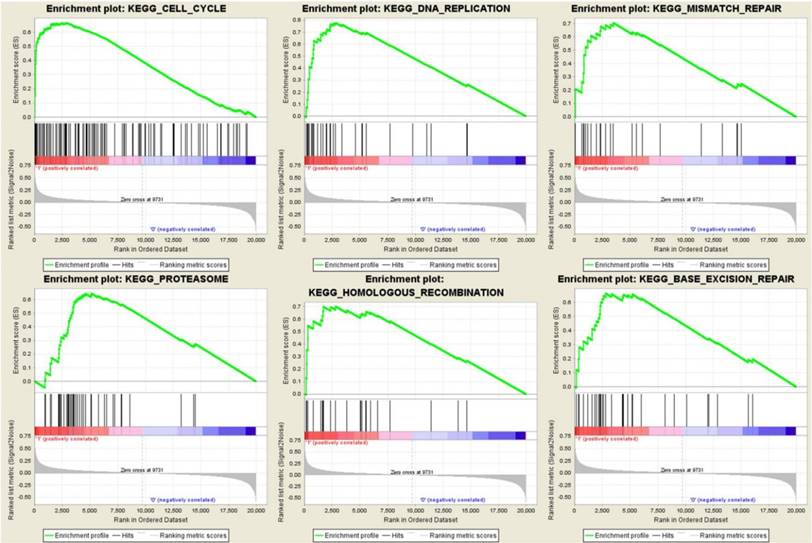
To identify the potential function of the real hub genes in PDAC, GSEA was conducted to search KEGG pathways enriched in the highly-expressed samples. Six gene sets (n = 118), “Cell cycle”, “DNA replication”, “mismatch repair”, “proteasome”, “homologous recombination” and “base excision repair” were enriched (FDR < 0.05) (Figure 13).
Gene set enrichment analysis (GSEA) using GSE62165. Only listed the six most common functional gene sets enriched in PDAC samples with real hub genes highly expressed.
Discussion
In the present study, first, we screened yellow module related to grade of PDAC by using WGCNA. And then a total of 36 candidates, common hub genes both in co-expression network and PPI network, were distinguished. After a series of bioinformatics analysis, 10 real hub genes tightly associated with progression and prognosis of PDAC were identified. The findings may contribute to the improvement of therapeutic decision-making, risk stratification and prognosis prediction for PDAC patients.
To a great extent, tumor grade has a close correlation with prognosis. Auvinen et al. demonstrated that hyaluronan synthases in stromal and malignant cells was correlated with breast cancer grade and predicted patient survival [31]. Azimi et al. also stated that tumor-infiltrating lymphocyte (TIL) grade is an independent predictor of survival and sentinel lymph node (SLN) status in patients with melanoma [32]. Here, we performed survival analysis and regression analysis to screen real hub genes with a significant p value. A total of 10 genes (CCNA2, CCNB1, CENPF, DLGAP5, KIF14, KIF23, NEK2, RACGAP1, TPX2 and UBE2C) were especially outstanding. They were not only highly correlated with PDAC grade, but also may be potential biomarkers for prognosis.
Many researchers found that those 10 real hub genes were involved in the process of cell cycle, participating in the tumorigenesis and tumor proliferation. It was showed that CCNA2 and its associated kinase activity are important for progestin-induced activation of endogenous progesterone receptor target genes in breast cancer cells [33]. Down-regulation of CCNB1 impaired proliferation and induced apoptotic death in colorectal cancer [34]. Aytes et al. reported that FOXM1 and CENPF function synergistically promoted tumor growth by coordinating regulation of target gene expression and activation of key signaling pathways associated with prostate cancer malignancy [35]. KIF14 was proved to be overexpressed in most primary hepatocellular carcinoma (HCC) tissues compared with the adjacent normal liver tissues and closely related to tumor grade, stage and poor survival [36]. KIF23, another number in kinesins family, whose elevated levels was correlate with ANCCA in the tumors and with poor relapse-free survival of patients with ER-positive breast cancer [37]. TPX2 was confirmed to affect spindle formation and microtubule nucleation around the chromosomes [38]. High expression of TPX2 may serve as a prognostic marker and promotes tumorigenesis and metastasis of HCC [39]. In addition, DLGAP5 combined expression with BUB1B and PINK1 may be a predictor of poor outcome in adrenocortical tumors [40]. High expression of RACGAP1 was significantly associated with progression and prognosis of colorectal cancer (CRC) and its biological and clinical significance provided sufficient evidence as a potential biomarker for identifying patients with lymph node metastasis and poor prognosis [41]. And dysregulation of UBE2C was associated with proliferative marker Ki-67, accumulation of cyclin A and B1, and a poor overall survival in colorectal cancer [42]. Moreover, recent studies have revealed that CCNB1 and CENPF were biomarkers of PDAC by bioinformatic analysis [43, 44]; meanwhile, TPX2 was proved to be a candidate target in PDAC for designing improved treatments [45]. As for the rest real hub genes, they were not reported to participate in PDAC progression. More study was needed. In order to further verify our results, we used the Oncomine database and The Human Protein Atlas to validate the expression of the real hub genes on transcriptional and translational level. All these gens were significantly up-regulated in pancreatic carcinoma compared with normal tissue in mRNA level and immunohistochemistry staining, proving their vital role in carcinogenesis.
According to the statistics, about 80% of surgically resected PDAC recurred. As the tumor recurrence played a critical role in prognosis, we then performed ROC curve and calculated AUC, showing that the real hub genes, especially CCNB1, KIF23, RACGAP1 and TPX2, were capable of distinguishing recurrent and non-recurrent PDAC with excellent diagnostic efficacy [46]. Several researchers were committed to finding indicators that could be used to detect recurrence of PDAC, like CT or magnetic resonance imaging for routine follow-up, serum levels of CA19-9 and CD133 [47, 48]. CCNB1, KIF23 and RACGAP1 were all stated to be potential candidate biomarkers for HCC recurrence after surgery [49, 50]. It was reported that co-expression of TPX2 and PD-L1 was significantly higher in CIN persistence/recurrence group than the group without CIN persistence/recurrence [51]. Our findings might serve as the promising biomarkers for diagnosis of disease progression, poor prognosis and recurrence.
The functional and pathway enrichment analysis showed that three inseparable biological processes, mitotic nuclear division, cell division and cell cycle, were significantly enriched. Moreover, the gene set enrichment analysis also found that the function of those real hub genes were enriched in terms related to cell cycle regulation. Cell cycle has been proved to regulate various tumors progression and prognosis, including PDAC [52-55]. Chen et al. reported that down-regulation of FAM83B in PDAC cells contributed to G0/G1 phase arrest and inhibition of cell proliferation [56]; Liu et al. discovered YY1 suppressed proliferation and migration of PDAC by regulating the CDKN3/MdM2/P53/P21 signaling pathway [57]. Numerous biomarkers were reported to impact PDAC via cell cycle pathway, including the 10 hub real genes mentioned above [58-60]. Therefore, further exploration of cell cycle and associated genes was of enormous significance.
In summary, by using a series of bioinformatics analysis, we illustrated ten real hub genes which might be involved in the prognosis and capable of distinguishing recurrent and non-recurrent PDAC. The crucial pathway enriched in the real hub genes was cell cycle. These findings would greatly contribute to the reveal the progress of PDAC.
Abbreviations
PDAC: pancreatic ductal adenocarcinoma; HPA: Human Protein Atlas; GSEA: enrichment analysis and gene set enrichment; WGCNA: weighted gene co-expression network analysis; TCGA: The Cancer Genome Atlas; DEGs: Differentially expressed genes; TOM: topological overlap matrix; MEs: Module eigengenes; GS: gene significance; MS: module significance; STRING: Search Tool for the Retrieval of Interacting Genes; PPI: protein-protein interaction; DAVID: Database for Annotation, Visualization and Integrated Discovery.
Supplementary Material
Supplementary table and figures.
Acknowledgements
We are grateful to Lushun Yuan for valuable discussion and advice. This work was supported by The National Natural Science Foundation of China (CN), (No. 81472735).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Gifford JB, Huang W, Zeleniak AE, Hindoyan A, Wu H, Donahue TR, Hill R. Expression of GRP78, Master Regulator of the Unfolded Protein Response, Increases Chemoresistance in Pancreatic Ductal Adenocarcinoma. Mol Cancer Ther. 2016;15:1043-52
2. Costa-Silva B, Aiello NM, Ocean AJ. et al. Pancreatic cancer exosomes initiate pre-metastatic niche formation in the liver. Nat Cell Biol. 2015;17:816-26
3. Chang JC, Kundranda M. Novel Diagnostic and Predictive Biomarkers in Pancreatic Adenocarcinoma. Int J Mol Sci. 2017:18
4. Lancet O. Pancreatic cancer: a neglected killer? Lancet Oncol. 2010;11:1107
5. Zheng L, Xue J, Jaffee EM, Habtezion A. Role of immune cells and immune-based therapies in pancreatitis and pancreatic ductal adenocarcinoma. Gastroenterology. 2013;144:1230-40
6. Poruk KE, Firpo MA, Scaife CL, Adler DG, Emerson LL, Boucher KM, Mulvihill SJ. Serum osteopontin and tissue inhibitor of metalloproteinase 1 as diagnostic and prognostic biomarkers for pancreatic adenocarcinoma. Pancreas. 2013;42:193-7
7. Yoshikawa K, Mitsunaga S, Kinoshita T, Konishi M, Takahashi S, Gotohda N, Kato Y, Aizawa M, Ochiai A. Impact of tumor-associated macrophages on invasive ductal carcinoma of the pancreas head. Cancer Sci. 2012;103:2012-20
8. Rahib L, Smith BD, Aizenberg R, Rosenzweig AB, Fleshman JM, Matrisian LM. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res. 2014;74:2913-21
9. Davies CC, Harvey E, McMahon RF, Finegan KG, Connor F, Davis RJ, Tuveson DA, Tournier C. Impaired JNK signaling cooperates with KrasG12D expression to accelerate pancreatic ductal adenocarcinoma. Cancer Res. 2014;74:3344-56
10. Nones K, Waddell N, Song S. et al. Genome-wide DNA methylation patterns in pancreatic ductal adenocarcinoma reveal epigenetic deregulation of SLIT-ROBO, ITGA2 and MET signaling. Int J Cancer. 2014;135:1110-8
11. Srivastava P, Mangal M, Agarwal SM. Understanding the transcriptional regulation of cervix cancer using microarray gene expression data and promoter sequence analysis of a curated gene set. Gene. 2014;535:233-8
12. Mancini-DiNardo D, Judkins T, Woolstenhulme N. et al. Design and validation of an oligonucleotide microarray for the detection of genomic rearrangements associated with common hereditary cancer syndromes. Journal of experimental & clinical cancer research: CR. 2014;33:74
13. Kandoth C, McLellan MD, Vandin F. et al. Mutational landscape and significance across 12 major cancer types. Nature. 2013;502:333-9
14. Kuzu OF, Noory MA, Robertson GP. The Role of Cholesterol in Cancer. Cancer research. 2016;76:2063-70
15. Emmert-Streib F, de Matos Simoes R, Glazko G, McDade S, Haibe-Kains B, Holzinger A, Dehmer M, Campbell F. Functional and genetic analysis of the colon cancer network. BMC bioinformatics. 2014;15(Suppl 6):S6
16. Langfelder P, Horvath S. WGCNA: an R package for weighted correlation network analysis. BMC bioinformatics. 2008;9:559
17. Clarke C, Madden SF, Doolan P. et al. Correlating transcriptional networks to breast cancer survival: a large-scale coexpression analysis. Carcinogenesis. 2013;34:2300-8
18. Yang S, He P, Wang J. et al. A Novel MIF Signaling Pathway Drives the Malignant Character of Pancreatic Cancer by Targeting NR3C2. Cancer research. 2016;76:3838-50
19. Janky R, Binda MM, Allemeersch J, Van den Broeck A, Govaere O, Swinnen JV, Roskams T, Aerts S, Topal B. Prognostic relevance of molecular subtypes and master regulators in pancreatic ductal adenocarcinoma. BMC cancer. 2016;16:632
20. Zhou Z, Liu S, Zhang M, Zhou R, Liu J, Chang Y, Zhao Q. Overexpression of Topoisomerase 2-Alpha Confers a Poor Prognosis in Pancreatic Adenocarcinoma Identified by Co-Expression Analysis. Digestive diseases and sciences. 2017
21. Botia JA, Vandrovcova J, Forabosco P. et al. An additional k-means clustering step improves the biological features of WGCNA gene co-expression networks. BMC systems biology. 2017;11:47
22. Rahmani B, Zimmermann MT, Grill DE, Kennedy RB, Oberg AL, White BC, Poland GA, McKinney BA. Recursive Indirect-Paths Modularity (RIP-M) for Detecting Community Structure in RNA-Seq Co-expression Networks. Frontiers in genetics. 2016;7:80
23. Szklarczyk D, Morris JH, Cook H. et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic acids research. 2017;45:D362-D8
24. Szklarczyk D, Franceschini A, Wyder S. et al. STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic acids research. 2015;43:D447-52
25. Su G, Morris JH, Demchak B, Bader GD. Biological network exploration with Cytoscape 3. Current protocols in bioinformatics. 2014;47:8 13 1-24
26. Uhlen M, Fagerberg L, Hallstrom BM. et al. Proteomics. Tissue-based map of the human proteome. Science. 2015;347:1260419
27. Sing T, Sander O, Beerenwinkel N, Lengauer T. ROCR: visualizing classifier performance in R. Bioinformatics. 2005;21:3940-1
28. Huang da W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nature protocols. 2009;4:44-57
29. Huang da W, Sherman BT, Lempicki RA. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic acids research. 2009;37:1-13
30. Subramanian A, Kuehn H, Gould J, Tamayo P, Mesirov JP. GSEA-P: a desktop application for Gene Set Enrichment Analysis. Bioinformatics. 2007;23:3251-3
31. Auvinen P, Rilla K, Tumelius R. et al. Hyaluronan synthases (HAS1-3) in stromal and malignant cells correlate with breast cancer grade and predict patient survival. Breast Cancer Res Treat. 2014;143:277-86
32. Azimi F, Scolyer RA, Rumcheva P, Moncrieff M, Murali R, McCarthy SW, Saw RP, Thompson JF. Tumor-infiltrating lymphocyte grade is an independent predictor of sentinel lymph node status and survival in patients with cutaneous melanoma. J Clin Oncol. 2012;30:2678-83
33. Moore NL, Edwards DP, Weigel NL. Cyclin A2 and its associated kinase activity are required for optimal induction of progesterone receptor target genes in breast cancer cells. J Steroid Biochem Mol Biol. 2014:144 Pt B: 471-82
34. Fang Y, Yu H, Liang X, Xu J, Cai X. Chk1-induced CCNB1 overexpression promotes cell proliferation and tumor growth in human colorectal cancer. Cancer Biol Ther. 2014;15:1268-79
35. Aytes A, Mitrofanova A, Lefebvre C. et al. Cross-species regulatory network analysis identifies a synergistic interaction between FOXM1 and CENPF that drives prostate cancer malignancy. Cancer Cell. 2014;25:638-51
36. Yang T, Zhang XB, Zheng ZM. Suppression of KIF14 expression inhibits hepatocellular carcinoma progression and predicts favorable outcome. Cancer Sci. 2013;104:552-7
37. Zou JX, Duan Z, Wang J, Sokolov A, Xu J, Chen CZ, Li JJ, Chen HW. Kinesin family deregulation coordinated by bromodomain protein ANCCA and histone methyltransferase MLL for breast cancer cell growth, survival, and tamoxifen resistance. Mol Cancer Res. 2014;12:539-49
38. Wittmann T, Wilm M, Karsenti E, Vernos I. TPX2, A novel xenopus MAP involved in spindle pole organization. J Cell Biol. 2000;149:1405-18
39. Huang Y, Guo W, Kan H. TPX2 is a prognostic marker and contributes to growth and metastasis of human hepatocellular carcinoma. Int J Mol Sci. 2014;15:18148-61
40. Fragoso MC, Almeida MQ, Mazzuco TL. et al. Combined expression of BUB1B, DLGAP5, and PINK1 as predictors of poor outcome in adrenocortical tumors: validation in a Brazilian cohort of adult and pediatric patients. European journal of endocrinology. 2012;166:61-7
41. Imaoka H, Toiyama Y, Saigusa S. et al. RacGAP1 expression, increasing tumor malignant potential, as a predictive biomarker for lymph node metastasis and poor prognosis in colorectal cancer. Carcinogenesis. 2015;36:346-54
42. Bavi P, Uddin S, Ahmed M. et al. Bortezomib stabilizes mitotic cyclins and prevents cell cycle progression via inhibition of UBE2C in colorectal carcinoma. Am J Pathol. 2011;178:2109-20
43. Fiorini C, Cordani M, Padroni C, Blandino G, Di Agostino S, Donadelli M. Mutant p53 stimulates chemoresistance of pancreatic adenocarcinoma cells to gemcitabine. Biochimica et biophysica acta. 2015;1853:89-100
44. Grutzmann R, Pilarsky C, Ammerpohl O. et al. Gene expression profiling of microdissected pancreatic ductal carcinomas using high-density DNA microarrays. Neoplasia. 2004;6:611-22
45. Zhang G, Schetter A, He P. et al. DPEP1 inhibits tumor cell invasiveness, enhances chemosensitivity and predicts clinical outcome in pancreatic ductal adenocarcinoma. PloS one. 2012;7:e31507
46. Kyriazanos ID, Tsoukalos GG, Papageorgiou G, Verigos KE, Miliadis L, Stoidis CN. Local recurrence of pancreatic cancer after primary surgical intervention: how to deal with this devastating scenario? Surg Oncol. 2011;20:e133-42
47. Katz MH, Wang H, Fleming JB. et al. Long-term survival after multidisciplinary management of resected pancreatic adenocarcinoma. Ann Surg Oncol. 2009;16:836-47
48. Welsch T, Keleg S, Bergmann F, Degrate L, Bauer S, Schmidt J. Comparative analysis of tumorbiology and CD133 positivity in primary and recurrent pancreatic ductal adenocarcinoma. Clin Exp Metastasis. 2009;26:701-11
49. Weng L, Du J, Zhou Q, Cheng B, Li J, Zhang D, Ling C. Identification of cyclin B1 and Sec62 as biomarkers for recurrence in patients with HBV-related hepatocellular carcinoma after surgical resection. Mol Cancer. 2012;11:39
50. Wang SM, Ooi LL, Hui KM. Upregulation of Rac GTPase-activating protein 1 is significantly associated with the early recurrence of human hepatocellular carcinoma. Clin Cancer Res. 2011;17:6040-51
51. Zhang H, Zhang T, You Z, Zhang Y. Positive Surgical Margin, HPV Persistence, and Expression of Both TPX2 and PD-L1 Are Associated with Persistence/Recurrence of Cervical Intraepithelial Neoplasia after Cervical Conization. PLoS One. 2015;10:e0142868
52. Angus L, van der Watt PJ, Leaner VD. Inhibition of the nuclear transporter, Kpnbeta1, results in prolonged mitotic arrest and activation of the intrinsic apoptotic pathway in cervical cancer cells. Carcinogenesis. 2014;35:1121-31
53. Phan L, Chou PC, Velazquez-Torres G. et al. The cell cycle regulator 14-3-3sigma opposes and reverses cancer metabolic reprogramming. Nat Commun. 2015;6:7530
54. Cuzick J, Swanson GP, Fisher G. et al. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: a retrospective study. Lancet Oncol. 2011;12:245-55
55. Baan B, Dihal AA, Hoff E. et al. 5-Aminosalicylic acid inhibits cell cycle progression in a phospholipase D dependent manner in colorectal cancer. Gut. 2012;61:1708-15
56. Shen CQ, Yan TT, Liu W. et al. High Expression of FAM83B Predicts Poor Prognosis in Patients with Pancreatic Ductal Adenocarcinoma and Correlates with Cell Cycle and Cell Proliferation. Journal of Cancer. 2017;8:3154-65
57. Liu D, Zhang J, Wu Y. et al. YY1 suppresses proliferation and migration of pancreatic ductal adenocarcinoma by regulating the CDKN3/MdM2/P53/P21 signaling pathway. International journal of cancer. 2017
58. Kaistha BP, Krattenmacher A, Fredebohm J. et al. The deubiquitinating enzyme USP5 promotes pancreatic cancer via modulating cell cycle regulators. Oncotarget. 2017;8:66215-25
59. Zhu Y, Gu J, Li Y. et al. MiR-17-5p enhances pancreatic cancer proliferation by altering cell cycle profiles via disruption of RBL2/E2F4-repressing complexes. Cancer Lett. 2018;412:59-68
60. Loc WS, Linton SS, Wilczynski ZR. et al. Effective encapsulation and biological activity of phosphorylated chemotherapeutics in calcium phosphosilicate nanoparticles for the treatment of pancreatic cancer. Nanomedicine: nanotechnology, biology, and medicine. 2017;13:2313-24
Author contact
![]() Corresponding author: Qiu Zhao, Email: qiuzhaoedu.cn, Tel: +8615972116991; Fax: +86-27-6781-2892.
Corresponding author: Qiu Zhao, Email: qiuzhaoedu.cn, Tel: +8615972116991; Fax: +86-27-6781-2892.