Impact Factor ISSN: 1449-2288
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Issue 6; 2026
- Issue 5; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Methods and materials
Results
Discussion
Conclusions
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(3):253-265. doi:10.7150/ijbs.23489 This issue Cite
Research Paper
Instillation of particulate matter 2.5 induced acute lung injury and attenuated the injury recovery in ACE2 knockout mice
Chung-I Lin1,2*, Chin-Hung Tsai1,3*, Yu-Ling Sun4, Wen-Yeh Hsieh 5,6, Yi-Chang Lin1, Cheng-Yi Chen1,7, Chih-Sheng Lin1 ![]()
1. Department of Biological Science and Technology, National Chiao Tung University, Hsinchu, Taiwan
2. Bioresource Collection and Research Center, Food Industry Research and Development Institute, Hsinchu, Taiwan
3. Division of Pulmonary Medicine, Department of Internal Medicine, Tungs' Taichung Metro Harbor Hospital, Taichung, Taiwan
4. Aquatic Technology Laboratories, Agricultural Technology Research Institute, Hsinchu, Taiwan
5. Division of Chest Medicine, Department of Internal Medicine, Hsinchu Mackay Memorial Hospital, Hsinchu, Taiwan
6. Department of Senior Citizen Service Management, Minghsin University of Science and Technology, Hsinchu, Taiwan
7. Division of Nephrology, Department of Internal Medicine, Hsinchu Mackay Memorial Hospital, Hsinchu, Taiwan
*These authors contributed equally to the manuscript.
Received 2017-10-25; Accepted 2018-1-9; Published 2018-2-12
Abstract

Inhaled particulate matter 2.5 (PM2.5) can cause lung injury by inducing serious inflammation in lung tissue. Renin-angiotensin system (RAS) is involved in the pathogenesis of inflammatory lung diseases and regulates inflammatory response. Angiotensin-converting enzyme II (ACE2), which is produced through the angiotensin-converting enzyme (ACE)/angiotensin II (Ang II) axis, protects against lung disease. However, few studies have focused on the relationships between PM2.5 and ACE2. Therefore, we aimed to explore the role of ACE2 in PM2.5-induced acute lung injury (ALI). An animal model of PM2.5-induced ALI was established with wild type (C57BL/6, WT) and ACE2 gene knockout (ACE2 KO) mice. The mice were exposed to PM2.5 through intratracheal instillation once a day for 3 days (6.25 mg/kg/day) and then sacrificed at 2 days and 5 days after PM2.5 instillation. The results show that resting respiratory rate (RRR), levels of inflammatory cytokines, ACE and MMPs in the lungs of WT and ACE2 KO mice were significantly increased at 2 days postinstillation. At 5 days postinstillation, the PM2.5-induced ALI significantly recovered in the WT mice, but only partially recovered in the ACE2 KO mice. The results hint that PM2.5 could induce severe ALI through pulmonary inflammation, and the repair after acute PM2.5 postinstillation could be attenuated in the absence of ACE2. Additionally, our results show that PM2.5-induced ALI is associated with signaling p-ERK1/2 and p-STAT3 pathways and ACE2 knockdown could increase pulmonary p-STAT3 and p-ERK1/2 levels in the PM2.5-induced ALI.
Keywords: particulate matter 2.5, renin-angiotensin system, angiotensin-converting enzyme II, inflammation, acute lung injury
Introduction
Air pollution has worsened in recent years, particularly anthropogenic sources due to the development of heavy industry in recent years. Particulate matter 2.5 (PM2.5), an index of air pollution, is defined as particulate matter with an aerodynamic diameter of less than 2.5 μm and can adversely affect the respiratory and circulatory systems of humans [1, 2]. Approximately 96% of PM2.5 were retained in the lungs given its size effect [3]. In addition, PM2.5 contains different components, such as polycyclic aromatic hydrocarbons, oxygenated volatile organic compounds and heavy metals, which exert toxic effects [4, 5].
The relationships between PM2.5 and inflammation have been mentioned in many pulmonary diseases, such as acute lung injury (ALI), asthma and chronic obstructive pulmonary disease (COPD) [6-8]. PM2.5-induced inflammation is associated with the release of numerous cytokines and chemokines, such as interleukin-6 (IL-6), tumor necrosis factor alpha (TNF-α) and tumor growth factor beta 1 (TGF-β1). These cytokines activate the mitogen-activated protein kinase (MAPK) and Janus-activated kinase (JAKs) pathways [9-11]. Furthermore, matrix metalloproteinases (MMPs) are important factors in lung diseases and are regulated via the pathways of signal transducer and activator of transcription 3 (STAT3) and MAPK signaling transducer [12, 13]. Therefore, the fundamental mechanism of PM2.5-induced ALI must be understood for the effective prevention of PM2.5-related pulmonary diseases.
Renin-angiotensin system (RAS) is an important endocrine system that regulates cardiovascular physiology and participates in pulmonary injury by activating inflammatory factors in the lung [14, 15]. The angiotensin-converting enzyme (ACE)/angiotensin II (Ang II)/Ang II type I receptor (AT1R) (ACE/Ang II/AT1R) axis and angiotensin-converting enzyme II (ACE2)/angiotensin 1-7 (Ang-(1-7))/Mas receptor (ACE2/Ang-(1-7)/Mas) axis are two pathways in RAS with opposing effects [16]. These two pathways counter-regulate the expression of pro-inflammatory factors. Increased ACE levels cause the activation of AT1R by Ang II, consequently inducing the expression of the inflammatory cytokines IL-6, TNF-α and TGF-β1 [17]. ACE2 counter-regulates the effects produced by Ang II by converting Ang II to Ang-(1-7), activates Mas to repress the signaling pathways of STAT3 and extracellular signal-regulated kinases (ERK) [18], and acts as an anti-inflammatory factor [19].
As mentioned above, PM2.5 is associated with various respiratory and cardiovascular diseases. Inflammation is one of its mechanisms. Interestingly, RAS is strongly related with the cardiopulmonary system and inflammation. Therefore, we hypothesized that PM2.5-induced ALI is regulated by RAS, and ACE2/Ang-(1-7)/Mas axis has a crucial role in the pathogenesis of lung injury. To test this hypothesis, we exposed C57BL/6 and ACE2 knockout (ACE2 KO) mice to PM2.5 collected from urban traffic. PM2.5 was administered to the mice through intratracheal instillation for 3 consecutive days. The mice were then sacrificed at 2 and 5 days after PM2.5 instillation. We then measured physiological parameters, expression levels and activities of ACE, ACE2, and MMPs in lung tissue. Furthermore, we detected the phosphorylation of ERK1/2 to p-ERK1/2 and of STAT3 to p-STAT3 to explore the potential mechanisms and the role of ACE2 in PM2.5-induced ALI.
Methods and materials
Source and analysis of PM2.5
Urban traffic is the major source of PM2.5 [20, 21]. We collected PM2.5 from traffic by continuous pumping air in accordance with the protocol of previous study with slight modifications [22, 23]. Particulates with diameters of 2 μm that attached to PTFE paper (TaiwanGrace International Co., Taichung, Taiwan) were carefully scraped to another piece of filter paper (Toyo Roshi Kaisha, Tokyo, Japan). Then, water was added to wash out particulates by suction filtration. The liquid was then collected and sonicated for 5 minutes, and this step was repeated thrice. The particulates were freeze-dried until excess water was completely removed. The particulates were sterilized and dissolved in DMSO (Sigma-Aldrich, Louis, MO, USA) to 10 mg/mL as stock for later use.
The collected PM2.5 were diluted to 10 μg/mL in acetone (Sigma-Aldrich) and dropped onto copper mesh for analysis through transmission electron microscopy (TEM) (Ted Pella, Redding, CA, USA). The TEM grids were incubated in an oven for 5 min. Finally, the configuration and elemental composition of PM2.5 were characterized using TECNAI 20 instrument (Philips, Netherlands) and a scanning electron microscope (Hitachi SU8010; Hitachi High-Technologies Co., Tokyo, Japan).
Animal treatment with PM2.5
Six-week-old wild type (WT, C57BL/6, male) and ACE2 gene knockout (ACE2 KO, male) mice were respectively divided to the sham, 2 days and 5 days postinstillation group (n = 7 for each group; a total of 21 mice used) in this study. The WT mice were obtained from National Laboratory Animal Center (NLAC), Taiwan. The first ACE2 KO (B6, 129S5-Ace2tm1 Lex/Mmcd) mice were purchased from Mutant Mouse Regional Resource Centers (MMRRC) and breed in NLAC [15]. All of the animal experiments conformed to the “Guide for the Care and Use of Laboratory Animals published by National Institutes of Health” (NIH Publication No. 85-23, revised 1996) and was approved by the Animal Welfare Committee of National Chiao Tung University (NCTU-IACUC-104026).
The mice were treated once daily with PM2.5 in 100 μL saline solution (6.25 mg/kg/day) via intratracheal instillation for 3 consecutive days and sacrificed at 2 and 5 days postinstillation. The sham group was treated with saline by intratracheal instillation. The body weight and resting respiratory rate (RRR) of the mice were measured daily. The RRR (BPM, breaths per minute) of the mice was measured in accordance with the protocol of a previous study with some modifications [24]. The RRR of mice after 15 min of exercise was detected daily before saline or PM2.5 treatment by using whole-body unrestrained chambers (Emka, Middletown, PA, USA). RRR data were analyzed by BIOPAC Student Lab Systems software (Biopac System, Goleta, CA, USA) [24].
Enzyme-linked immunosorbent assay (ELISA) and Western blot
Pulmonary TNF-α, TGF-β1 and IL-6 were detected by sandwich ELISA (Abcam, Cambridge, MA, USA). Proteins were incubated in 96-well ELISA plates with primary antibodies. After the addition of biotinylated antibodies, the plates were washed and reacted with HRP-conjugated streptavidin. Tetramethylbenzidine (TMB) one-step substrate tablets for the detection of TNF-α, TGF-β1 or IL-6, and the results were measured at 450 nm using a micro-plate reader (Thermo Scientific, Waltham, MA, USA).
Lung homogenates with an equivalent protein content of 25 μg protein were electrophoresed on 12% SDS-PAGE gels and then transferred to polyvinylidene fluoride membranes (Immobilon-PTM; Millipore, Bedford, MA, USA). The primary ACE, ACE2, phosphor-STAT3, phosphor-ERK1/2, STAT3, ERK1/2 and β-actin antibodies were obtained from Genetex (Irvine, CA, USA) or Cell Signaling Technology (Beverly, MA, USA). Immunoreactivity was detected through enhanced chemiluminescence (Immobilon Western Chemiluminescent HRP Substrate; Millipore, Billerica, MA, USA). PAGE membranes were exposed in Lumi-Film Chemiluminescent Detection Film (Roche, Indianapolis, IN, USA). Band density was measured with Scion Image software (Scion, Frederick, MD, USA). The levels of ACE and ACE2 were normalized to that of β-actin, and those of p-ERK1/2 and p-STAT3 were normalized to that of total ERK1/2 and STAT3, respectively.
Gelatin zymography assay
Lung MMP-2 and MMP-9 activities were detected through gelatin zymography with gelatin-containing gels in accordance with our previously reported protocol [26]. Lung homogenates were mixed with 6× zymography sample buffer (0.375 M Tris-HCl, pH 6.8, 20% (v/v) glycerol, 12% (w/v) SDS, and 0.015% bromophenol blue), incubated for 10 min at 37℃, and loaded onto 10% acrylamide gels with 0.1% (w/v) gelatin (Sigma-Aldrich) for SDS-PAGE. After electrophoresis, the gel was washed twice with zymography renaturing buffer (2.5% Triton X-100) for 30 min and incubated in developing buffer (50 mM Tris-HCl, pH 7.4, 200 mM NaCl, 5 mM CaCl2, and 0.02% Brij35) for 20 h at 37°C. The gels were then stained with Coomassie blue for 30 min and further distained with destaining buffer (50% methanol, 10% acetic acid, and 40% ddH2O). MMP-2 and MMP-9 activities were determined on the basis of clear or unstained areas, which are indicative of enzyme activity on the gelatin substrate. The unstained areas were quantified by using Scion Image software (Scion). Each gel contained MMP-2 or MMP-9 positive controls (Chemicon, Temecula, CA, USA) for the standardization of intensity value to sample intensity and expressed in arbitrary units [27].
In situ zymography
Tissue treatment and in situ zymography were performed in accordance with our previous report [15]. Lung tissues were collected and embedded in O.C.T. (Thermo, Waltham, MA, USA), immediately frozen, and stored at -80℃. Frozen tissue was cut into 7-10 μm thick sections on a slide. Then, 20 μL substrate gel solution was placed on the surface of the tissue sample. Another slide was then placed on top of the first slide to form a sandwich. The slides were placed in a box that contained Tris buffer (pH 7.4) at 37℃ and protected from light for 20 h. Substrate that was broken down by gelatinase emitted green fluorescent signal. Fluorescence images were taken using a Leica SP5X confocal laser-scanning microscope (Leica, Wetzlar, Hesse, Germany).
Histological determination
Lung tissues were collected from C57BL/6 and ACE2 KO mice, soaked in 10% formaldehyde overnight, embedded in paraffin, and cut into 4 μm-thick sections on acid-pretreated slides for Hematoxylin/ Eosin (H&E) and immunohistochemical (IHC) staining. The tissue samples were incubated with primary IHC antibody against TGF-β1 (1:2000; Genetex) at 4°C overnight and then with HRP-labeled Goat Anti-Mouse IgG (H + L) as a secondary antibody (1:100 dilution) at 37°C for 30 min. The stained pathological sections were photographed by a digital camera mounted on a microscope. A computerized microscope equipped with a high-resolution video camera (BX 51; Olympus, Tokyo, Japan) was used for morphometric analysis.
Statistical analysis
All values were expressed as the mean ± standard deviation. Student's t-test was performed for comparisons between two groups. One-way analysis of variance (ANOVA) test was performed to evaluate differences among multiple groups. A value of p < 0.05 was considered statistically significant.
Results
PM2.5 characterization
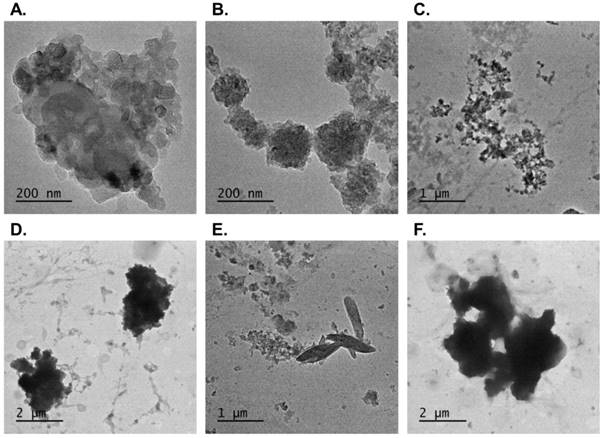
Configurations of the PM2.5 collected in the present study were shown in Fig. 1. The PM2.5 comprised numerous small particulates with diameters of 500 nm (Fig. 1A). These small particulates may be aggregated or dispersed (Fig. 1B and 1C) and form large particles with diameters of approximate 2.5 μm (i.e., PM2.5) (Fig. 1D). The elemental composition of the particulates was characterized. Figs. 1E and 1F show the silicon crystal and sulfur components of the collected PM2.5, respectively.
TEM images of PM2.5 configuration. Configurations of the particulates collected from traffic were shown under transmission electron microscopy. The small particles which were 500 nm in size (A) distributed in many types (B) (C) and formed PM2.5 (D). Few particles showed other different composition such as silicon (E) or sulfur (F).
PM2.5-induced ALI associated with inflammation
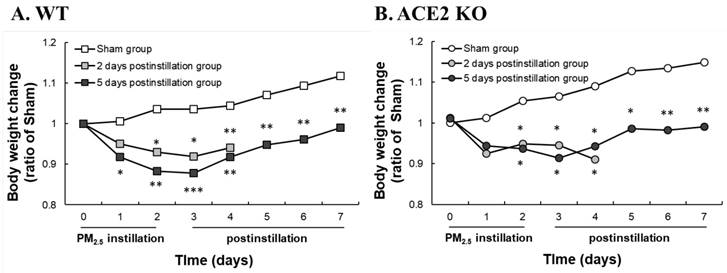
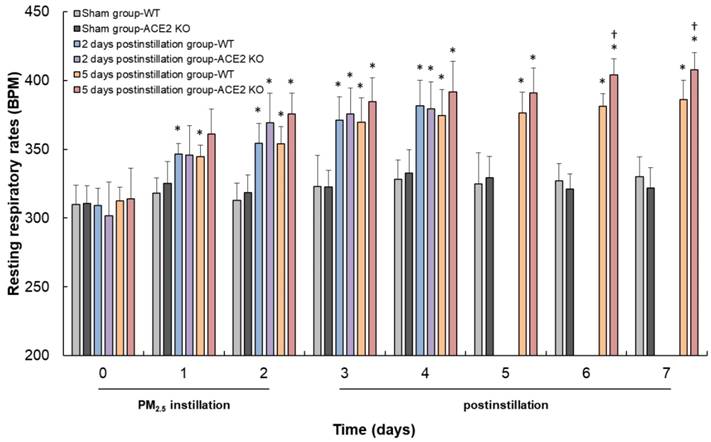
WT and ACE2 KO mice were treated with PM2.5 via intratracheal instillation once a day for 3 days and sacrificed at 2 and 5 days postinstillation. Body weight and RRR of the animals were monitored daily until sacrifice. The body weight of WT and ACE2 KO mice were markedly decreased throughout the entire PM2.5 treatment period and slightly recovered to almost their initial body weight at 5 days postinstillation (Fig. 2). However, the WT and ACE2 KO mice in the sham (saline treatment) control steadily gained body weight during the experimental period. RRR in the WT and ACE2 KO mice significantly increased during PM2.5 instillation period and remained at a high level at 5 days postinstillation (Fig. 3). Compared with those of the mice of PM2.5 treatment group, RRR of the WT and ACE2 KO mice in the sham group remained below 330 BPM throughout the experimental period.
Changes in the body weight of WT and ACE2 KO mice. WT (C57BL/6, n = 7 for each group) and ACE2 KO mice (n = 7 for each group) treated with PM2.5 or saline by intratracheal administration for consecutive 3 days and sacrificed at 2 and 5 days postinstillation. Body weight of WT (A) and ACE2 KO mice (B) were detected every day until sacrifice. All values are expressed as the mean from each group; * p < 0.05, ** p < 0.01 and *** p < 0.001 compared with the body weight in the same day.
Changes in the RRR of WT and ACE2 KO mice. The RRR of WT and ACE2 KO mice were measured every day until sacrifice. The RRR increased after PM2.5 administration, and RRR of ACE2 KO mice was higher than RRR of WT mice at 6 days and 7 days. All values are expressed as the mean ± SD from each group; * p < 0.05 compared with the initial (i.e., 0 day) RRR in the same group; † p < 0.05 compared with RRR of ACE2 KO mice to RRR of WT mice in the same day.
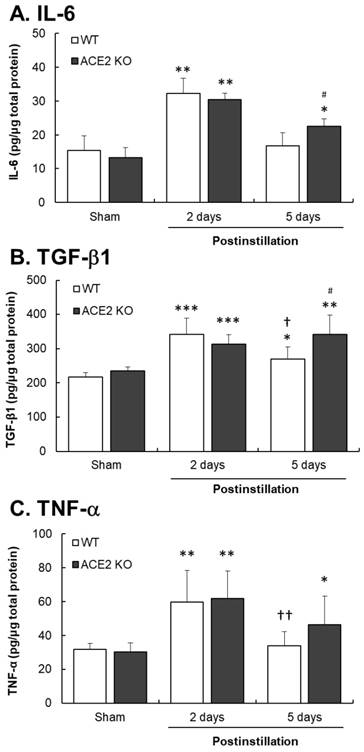
Inflammatory cytokine levels were determined to confirm that whether PM2.5 instillation could induce pulmonary inflammation in mice. At 2 days postinstillation, IL-6, TGF-β1 and TNF-α levels in the lungs significantly increased by 2.1-, 1.5- and 1.9-fold in WT mice, and 2.3-, 1.5- and 2.0-fold in ACE2 KO mice, respectively (Fig. 4). At 5 days postinstillation, the levels of IL-6 and TNF-α in the lungs of WT mice returned to almost their basal levels, whereas that of TGF-β1 still increased by 1.3-fold. IL-6, TGF-β1 and TNF-α levels in the lungs of ACE2 KO mice increased by 1.7-, 1.5-, 1.6-fold at 5 days postinstillation, respectively. In particular, IL-6 and TGF-β1 levels in ACE2 KO mice were significantly higher than those in WT mice (Fig. 4). The expression and distribution of TGF-β1 in the lungs were also detected by IHC staining (Fig. 5). TGF-β1 level in the lung tissues, especially in the bronchus, significantly increased at 2 and 5 days postinstillation.
Cytokines expression in the lungs of ACE2 KO mice after PM2.5 treatment. WT mice and ACE2 KO mice treated with PM2.5 for consecutive 3 days and sacrificed at 2 and 5 days postinstillation. The expressions of IL-6 (A), TGF-β1 (B) and TNF-α (C) in isolated lung tissue were determined by ELISA. All values are expressed as the mean ± SD from each group; * p < 0.05, ** p < 0.01 and *** p < 0.001 compared with the Sham value in the same group; † p < 0.05 and †† p < 0.01 compared with the value of 5 days to 2 days postinstillation in same group; # p < 0.05 compared with the value of ACE2 KO mice to WT mice in the same day.
Immunohistochemistry staining for TGF-β1 expression in bronchial tissue from ACE2 KO mice after PM2.5 treatment. The TGF-β1 (dark brown) of WT (A) and ACE2 KO mice (B) distributed only on a thin layer in bronchus and around blood cells in the sham group. Compared to sham group, the pulmonary TGF-β1 accumulated in bronchus at 2 days postinstillation, and numerous white blood cells (blue) infiltrated. However, the pulmonary TGF-β1 reduced but still obviously accumulated in bronchus at 5 days postinstillation. Images were captured at medium magnification (Scale = 50 μm).
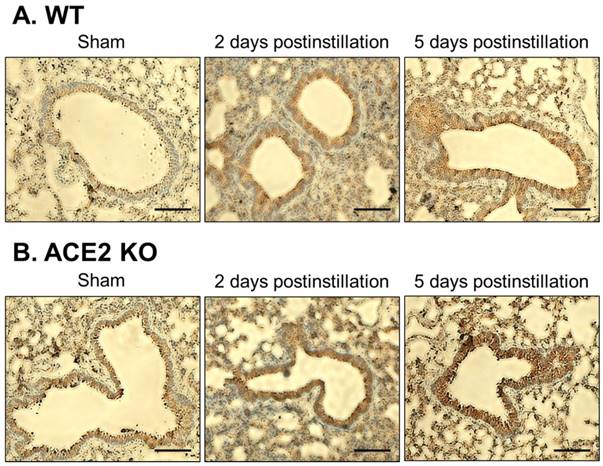
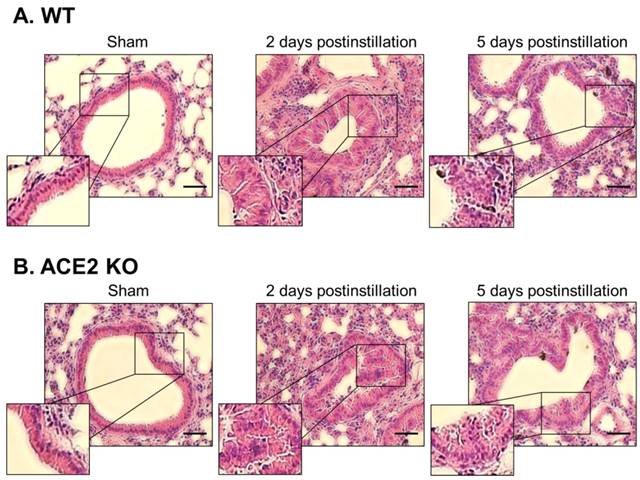
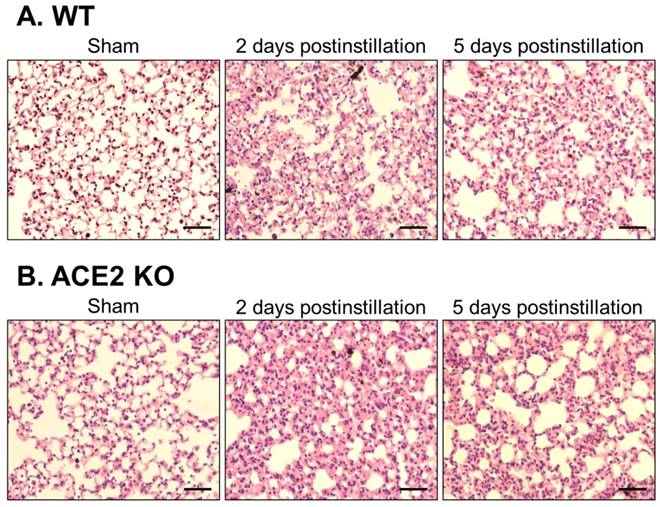
Lung sections from the WT and ACE2 KO mice were subjected to H&E staining, which revealed extensive white blood cells infiltration around the bronchus and airway epithelial thickening in WT and ACE2 KO mice at 2 days postinstillation (Fig. 6). The extent of infiltration and airway epithelial thickening in the WT and ACE2 KO mice decreased at 5 days postinstillation, but still more severe than that of sham group. Alveolar damage and fusion were also found in the mice at 5 days postinstillation (Fig. 7). Moreover, infiltration and airway epithelial thickening were more extensive in the ACE2 KO mice than those in WT mice after PM2.5 treatment.
Pathology of bronchial tissue from WT and ACE2 KO mice after PM2.5 treatment. The lung bronchus sections of WT (A) and ACE2 KO mice (B) after PM2.5 instillation were stained with haematoxylin-eosin (H&E). Relative to initiation of sham mice, the white blood cells markedly infiltrated after PM2.5 treatment, especially at 2 days postinstillation. Besides, the bronchus wall was significantly thickened at 2 days postinstillation but the airway epithelial thickening reduced at 5 days postinstillation. Images were captured at medium magnification (Scale = 50 μm).
Pathology of alveolar tissue from WT and ACE2 KO mice after PM2.5 treatment. The lung alveolar sections of WT (A) and ACE2 KO mice (B) after PM2.5 treatment were stained with haematoxylin-eosin (H&E). Relative to initiation of sham mice, the white blood cells markedly infiltrated and airway epithelial was thickening after PM2.5 treatment. Besides, the ruptured alveoli were revealed at 5 days postinstillation. Images were captured at medium magnification (Scale = 100 μm).
Effects of PM2.5 on ACE and ACE2 expression
To identify the RAS mediators associated with PM2.5-induced ALI, the protein expression of ACE and ACE2 in the pathogenesis of lung injury induced by PM2.5 were investigated.
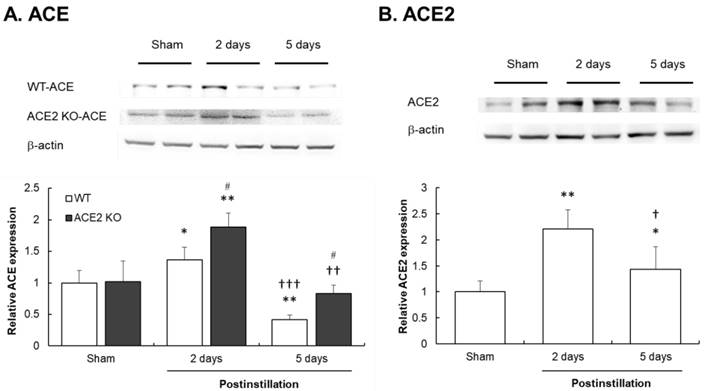
Fig. 8A shows that ACE expression in the lungs of WT and ACE2 KO mice was significantly increased by 1.3- and 1.8-fold, respectively, compared with those in the lungs of sham group mice at 2 days postinstillation. Notably, ACE expression in the lungs of WT and ACE2 KO mice significantly decreased to 0.4- and 0.8-fold which was a level lower than that in the sham group at 5 days postinstillation. The ACE expression level in WT mice was only one half compared with that in ACE2 KO mice. ACE2 expression in the lungs of WT mice significantly increased at both 2 and 5 days postinstillation, and markedly increased ACE2 expression level at 2 days postinstillation reduced at 5 days postinstillation (Fig. 8B).
Change in ACE and ACE2 expressions in the lungs of mice after PM2.5 treatment. The pulmonary ACE and ACE2 expression were determined by western blot. β-actin was used as the internal control for ACE and ACE2. The value of ACE and ACE2 expression in non-PM2.5 treatment was calculated in 100% as the sham value. The relative changes of pulmonary ACE expression in WT and ACE2 KO mice at 2 and 5 days postinstillation (A). The relative changes of pulmonary ACE2 expression in ACE2 KO mice at 2 and 5 days postinstillation (B). All values are expressed as the mean ± SD from each group; * p < 0.05 and ** p < 0.01 compared with the Sham value in the same group; † p < 0.05, †† p < 0.01 and ††† p < 0.001 compared with 5 days to 2 days postinstillation in same group; # p < 0.05 compared with the ACE2 KO mice to WT mice in the same day.
Effects of PM2.5 on lung gelatinase activity
Gelatinase containing MMP-2 and MMP-9 was subjected to in situ zymography assay to compare the gelatinase localization and activity in the lungs of WT and ACE2 KO mice.
Gelatinase activity mainly localized near the bronchus (Fig. 9). Gelatinase activity significantly increased in the lung of WT and ACE2 KO mice at 2 days postinstillation, as shown by thickening area in the mouse bronchus (Fig. 9B). At 5 days postinstillation, pulmonary gelatinase activity of the WT and ACE2 KO mice decreased, but remained higher than that in samples from the sham group.
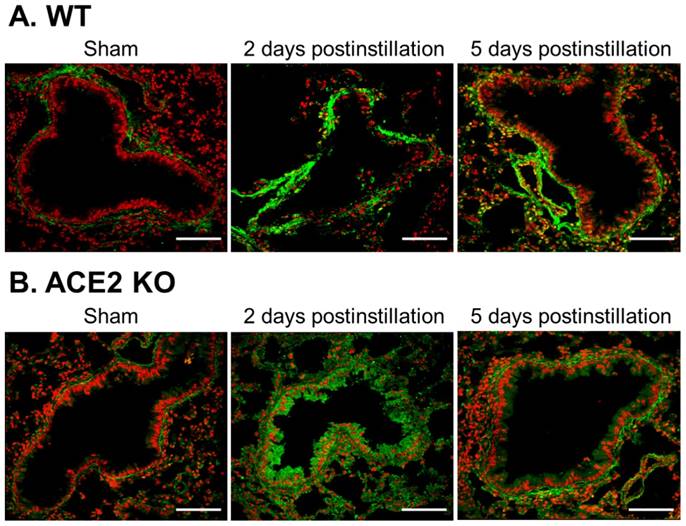
Change and distribution of gelatinase activity in the bronchus of WT and ACE2 KO mice after PM2.5 treatment. The location and gelatinase activity in the lungs of WT (A) and ACE2 KO mice (B) were detected by in situ zymography. The gelatin substrate was digested by MMPs and was shown in green, the nuclear signal was stained and was shown in red. Gelatinase activity around the bronchus was significantly increased after PM2.5 treatment. Images were captured at medium magnification (Scale = 50 μm).
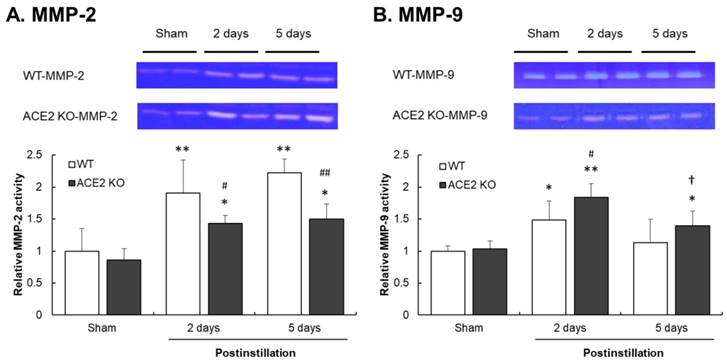
To confirm that ACE2 deficiency affects lung gelatinase activity, the activity of MMP-2 and MMP-9 determined by gelatin zymography assay was performed. MMP-2 activity significantly increased in the lungs of WT mice, but only slightly increased in the lungs of ACE2 KO mice after PM2.5 treatment (Fig. 10A). Furthermore, MMP-9 activity in the lung increased in both WT and ACE2 KO mice at 2 days postinstillation. MMP-9 activity was also increased at 5 days postinstillation in the lungs of ACE2 KO mice, but not in those of WT mice (Fig. 10B).
Relative MMP-2 and MMP-9 activities in the lungs of mice after PM2.5 treatment. WT and ACE2 KO mice treated with PM2.5 or saline by intratracheal administration for consecutive 3 days and sacrificed at 2 and 5 days postinstillation. The pulmonary MMP-2 and MMP-9 activity was determined by gelatin zymography. The value of MMP-2 and MMP-9 activity in non-PM2.5 treatment was calculated in 100% as the sham value. The pulmonary MMP-2 activity (A) increased in the lungs of WT and ACE2 KO mice after PM2.5 treatment. The MMP-9 activity (B) increased in the lungs of WT and ACE2 KO mice at 2 days postinstillation but only remained at higher level in those of ACE2 KO mice at 5 days postinstillation. All values are expressed as the mean ± SD from each group; * p < 0.05 and ** p < 0.01 compared with the sham value in the same group; † p < 0.05 compared with 5 days to 2 days postinstillation in same group; # p < 0.05 and ## p < 0.01 compared with the ACE2 KO mice to WT mice in the same day.
ACE2 knockdown increases p-STAT3 and p-ERK1/2 in PM2.5-induced ALI
The above results show that the levels of inflammatory cytokines and inflammation in pulmonary system were significantly induced after the mice treated with PM2.5. IL-6, TGF-β1 and TNF-α participate in inflammatory responses by activating the MAPK and JAK-STAT3 signaling pathways. To further explore whether the ACE2 knockdown would also influence the mediators of MAPK and JAK-STAT3 signaling pathways in PM2.5-induced ALI, we detected ERK1/2 and STAT-3 phosphorylation in the lung tissues by Western blotting.
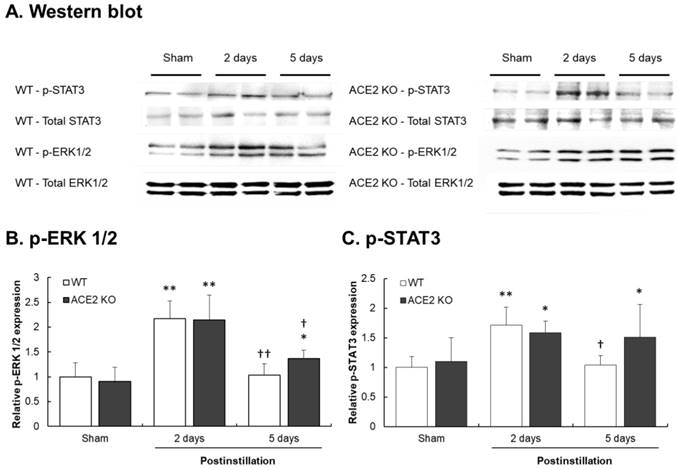
The results show that in the lungs of WT mice, pulmonary p-ERK1/2 and p-STAT3 levels in the lungs of WT mice significantly increased by 2.2- and 1.7-fold at 2 days postinstillation, respectively, but recovered to normal levels at 5 days postinstillation. Similarly, the pulmonary p-ERK1/2 and p-STAT3 levels in ACE2 KO mice significantly increased by 2.3- and 1.5-fold at 2 days postinstillation. However, increased p-ERK1/2 and p-STAT3 levels in the lungs of ACE2 KO mice remained at a relative higher level at 5 days postinstillation compared those in the WT mice (Fig. 11). Additionally, the recovery effect on the pulmonary p-STAT3 in ACE2 KO mice was not observed.
Elevation of p-ERK1/2 and p-STAT3 levels after PM2.5 treatment. WT and ACE2 KO mice treated PM2.5 or saline by intratracheal administration for consecutive 3 days and sacrificed at 2 and 5 days postinstillation. The expression of p-ERK1/2 and p-STAT3 in the lungs was determined by western blot (A). ERK1/2 and STAT3 were used as the internal controls for p-ERK1/2 and p-STAT3, respectively. The expression of p-ERK1/2 and p-STAT3 in the non-PM2.5 treatment mice were calculated in 100% as the sham. The relative pulmonary p-ERK1/2 (B) and p-STAT3 (C) expression increased in both WT and ACE2 KO mice at 2 days postinstillation, but p-ERK1/2 (B) and p-STAT3 (C) only returned to normal level in WT mice at 5 days postinstillation. All values are expressed as the mean ± SD from each group; * p < 0.05 and ** p < 0.01 compared with the Sham value in the same group; † p < 0.05 and †† p < 0.01 compared with the 5 days to 2 days postinstillation in same group.
Discussion
The results of this study show that PM2.5 collected from urban traffic can induce lung injury through mechanisms that are similar, but a little different from those reported in another study due to the elements composition of PM2.5 [28]. Carbon and oxygen are the major constituents of PM2.5 in our collection and account for 47.1% and 40.8%, respectively, of the total weight of the particulates. The minor constituent elements silicon, sulfate and copper contributed 9.1%, 0.5% and 2.6%, to the total weight of the particulates, respectively. The heavy metal and transition metal contents of PM2.5 would cause injury through their size effect and toxicity [4, 5]. However, given the lack of harmful elements, such as arsenic, cadmium and mercury in the PM2.5 collected in this study, the size effect might be the major mechanism that causes injuries in mice.
In this study, ACE2 KO mice were utilized to investigate the role of ACE2 in PM2.5-induced ALI. In previous studies, PM2.5 dosage used to induce ALI was usually under 100 µg [29, 30]. We used higher dosage in this study to induce ALI as to close to reality. The PM2.5 concentration in developed countries is usually low, but much higher in developing countries such as China. In the winter, the level of PM2.5 can exceed more than 200 µg/m3 and maintain for several days [31, 32]. In our study, PM2.5 was mainly administered to experimental animals through intratracheal instillation [29, 30] or through aerosol inhalation in an exposure chamber that contains PM2.5 [33, 34] for consecutive 3 days, to imitate the reality of how PM2.5 invades our body. Furthermore, we selected intratracheal administration given its accurate and rapid delivery of PM2.5 to the tracheae. The effect after administration of PM2.5 through aerosol inhalation is affected by many factors including mouse physiological stability and RRR, and may not have stable effects [33]. Compared with aerosol inhalation, intratracheal administration can decrease dose loss and better represent the relationship between dosage and pathological changes [33]. However, one of limitations in the use of intratracheal instillation in animal models is that particulates may be unevenly distributed and accumulate in some sections of the lungs after PM2.5 instillation [34, 35]. Another limitation is that intractracheally instilled PM2.5 may not reproduce the pathological reactions induced by inhaled PM2.5. Despite these limitations, intratracheal instillation still reveals the adverse effects caused by PM2.5.
Recent studies indicated that PM2.5 can induce inflammatory response and can recruit numerous inflammatory cytokines, such as TGF-β1, TNF-α and IL-6 [9-11]. Several pulmonary diseases, e.g., asthma, ALI, COPD and acute respiratory distress syndrome (ARDS), are associated with abnormal TGF-β1, TNF-α and IL-6 expression [38-40]. Our results accordingly show that TGF-β1, TNF-α and IL-6 levels in the lungs of WT and ACE2 KO mice significantly increased at 2 days postinstillation (Fig. 4). Moreover, the lung sections also showed serious infiltration and injury (Fig. 5 and Fig. 6). The experimental results indicated that severe inflammation was induced at 2 days postinstillation. However, inflammation decreased at 5 days postinstillation (Fig. 4). The impact of PM2.5 may be very strong at the beginning of exposure, especially as the dosage use in this study was larger than that used in other studies [41, 42]. Few days after PM2.5 exposure, the function of self-repair in the body may attenuate the injury [43]. TGF-β1 and IL-6 levels in the lungs of ACE2 KO mice were higher than in those of WT mice at 5 days postinstillation. Pathogenic lesions and inflammation were also more severe in the lungs of ACE2 KO mice than those in WT mice. Lung injury in ACE2 KO mice was more severe than that in WT mice at 5 days postinstillation. The results imply that a massive dose of PM2.5 exposure could induce severe pulmonary inflammation and tissue injury, and the injury could recover when mice are no longer exposed to PM2.5. Moreover, the existence of ACE2 in the lung might be crucial in the repair of PM2.5-induced ALI given that ACE2 KO mice exhibited more severe inflammation than WT mice.
Furthermore, ACE and ACE2 levels changed after PM2.5 treatment. The results show that ACE expression in the lung increased at 2 days postinstillation with a trend that opposed that at 5 days postinstillation (Fig. 8A). In the previous study, ACE expression in lung injury is controversial as it had been reported to exhibit either increasing or decreasing trend. For instance, hypertensive rats instilled with PM2.5 showed that ACE levels increased in the heart but decreased in the lung at 1 and 3 days postinstillation [44]. Similarly, when Sprague-Dawley (SD) rats were acutely exposed to particulates through aerosol inhalation for 3 days, ACE levels in the lung decreased. However, when the SD rats were subchronically (8 weeks) exposed to particulates using a particulate concentrator, lung ACE protein level and mRNA levels increased [45, 46]. We speculated that ACE increased in the lung because the lung is a major organ of ACE production and the initiation of PM2.5 treatment induced ACE expression [47]. However, ACE expression in the lung gradually normalized after instillation, and ACE in the lung might enter the circulatory system, thus causing ACE levels in the lung to decrease. Additionally, ACE expression in the lungs of ACE2 KO mice was higher than that in WT mice at 2 and 5 days postinstillation (Fig. 8A). A possible reason for this result is that ACE2 KO mice got more injuries and recovered slower induced by PM2.5 than WT mice after PM2.5 treatment.
Compared with ACE, the importance of ACE2 in PM2.5-induced ALI is seldom discussed. Imai et al. [48] reported that ACE2 has protective functions in murine lungs and can attenuate the acute injury induced by ARDS and SARS. In addition, ACE2 acts as a central role in lung endothelial function and pathophysiology [19, 49]. For example, lung injury induced by cigarette smoke exposure, and pulmonary fibrosis induced by bleomycin are associated with ACE2 activity [15, 50]. In our study, ACE2 expression in the lung increased at 2 and 5 days postinstillation (Fig. 8B), and PM2.5-induced ALI in ACE2 KO mice was more severe than that in WT mice. We speculated that the activation of ACE2/Ang-(1-7)/Mas receptor axis is absent in ACE2 KO mice. Thus, ACE2 KO mice sustained severe injuries and inflammation from PM2.5 exposure. On the basis of these findings, we hypothesized that ACE2 has a protective role against lung injury induced by PM2.5.
The members of MMP family, particularly gelatinase MMP-2 and MMP-9, are highly related with pulmonary inflammation and lung disease, such as asthma, ALI, and pulmonary fibrosis [51, 52]. Although MMP-9 and MMP-2 are the target enzymes of tissue remodeling, they have different roles in lung injury [51]. MMP-2 is synthesized by structural cells, including fibroblasts and endothelial and epithelial cells; MMP-2 is associated with impaired tissue remodeling, thus leading to pathological collagen deposition and pulmonary fibrosis [53, 54]. By contrast, MMP-9 is mainly secreted by the neutrophils, eosinophils, mast cells, alveolar macrophages, and is associated with acute inflammation [55]. Our results show that MMP-2 significantly increased in the lungs of WT and ACE2 KO mice at 2 and 5 days postinstillation (Fig. 10A). This result indicates that lung was undergoing repair with time after PM2.5 treatment. These results, combined with that for inflammation, confirm that PM2.5 -induced ALI can be repaired. Furthermore, MMP-2 activity in WT mice is higher than that in ACE2 KO mice at 2 and 5 days postinstillation, which indicated that WT mice exhibited more extensive tissue remodeling and might have faster recovery than ACE2 KO mice. On the other hand, MMP-9 increased in the lungs of WT and ACE2 KO mice at 2 days postinstillation but only remained at higher level in those of ACE2 KO mice at 5 days postinstillation (Fig. 10B). Under severe inflammation at 2 days postinstillation, MMP-9 activity elevated more significantly in the lungs of ACE2 KO mice than in WT mice. At 5 days postinstillation, inflammation in the lungs of WT mice decreased as MMP-9 normalized, but activation of inflammatory response persisted in ACE2 KO mice as MMP-9 activity remained in slightly increased level (as compared with the sham group). The lack of ACE2 would increase MMP-9 activity in lungs through inflammation, thus affecting recovery after PM2.5 instillation. Therefore, ACE2 likely plays an important role in the repair of PM2.5-induced injury.
The results of our study show that PM2.5 induces severe inflammatory response and unbalances the RAS system. The RAS system is involved in inflammatory response in several lung diseases [14, 15]. Inflammatory cytokines, such as TNF-α, IL-6 and TGF-β1, are associated with MAPK pathways [9-11]. ACE2 is associated with the regulation of the ERK-p38 and JAK-STAT signaling pathways [15, 56]. Moreover, MMPs participate in tissue repair through the mediation of transcription factors, e.g., STAT-3, that might indirectly activate MAPK signal pathways [57, 58]. Therefore, the changes in the phosphorylation of ERK1/2 and STAT3 in the lung after PM2.5 treatment were investigated in this study. The result shows that PM2.5-induced ALI is associated with the MAPK pathway. p-ERK1/2 and p-STAT3 levels in the lungs significantly increased at 2 days postinstillation (Fig. 11). The results demonstrate that PM2.5 causes lung injury via ERK1/2 and STAT3 signaling pathways and are consistent with the conclusions of previous reports that p38, ERK1/2 and JNK phosphorylation regulate key inflammatory, proteolytic and apoptotic responses in the lungs [59, 60]. Interestingly, p-ERK1/2 and p-STAT3 levels decreased to normal levels in the lungs of WT mice but remained at significantly increased levels in ACE2 KO mice at 5 days postinstillation (Fig. 11). The result suggests that inflammation and RAS were alleviated and PM2.5-induced ALI was also attenuated in WT mice. However, the ACE2 KO mice sustained severe injuries and exhibited slow recovery, inflammatory responses and RAS change, which caused the ERK1/2 and STAT3 signaling pathway to remain to be activated at 5 days postinstillation. These observations are consistent with the results mentioned above: the absence of ACE2 would affect and regulate the protective function that retards PM2.5-induced ALI.
Conclusions
The present study is the first to use a mouse model of PM2.5-induced ALI to investigate the effects of ACE2 deficiency. ACE2 deficiency would attenuate injury repair, inflammatory response and tissue remodeling in response to PM2.5 instillation. Therefore, ACE2 may have protected the respiratory system from PM2.5-induced injuries. Further studies, however, are necessary to validate this conclusion. Moreover, circulating or lung Ang II and Ang-(1-7) and the actions of both peptides on PM2.5-induced ALI remain to be further explored.
Abbreviations
ACE: Angiotensin converting enzyme; ACE2: Angiotensin converting enzyme II; Ang II: Angiotensin II; Ang-(1-7): Angiotensin 1-7; ARDS: Acute respiratory distress syndrome; AT1R: Ang II type I receptor; COPD: Chronic obstructive pulmonary disease; ERK1/2: Extracellular signal-regulated kinases 1 and 2; JAKs: Janus-activated kinases; KO: Knockout; IL-6: Interleukin-6; MAPKs: Mitogen-activated protein kinases; MMP-2: Matrix metalloproteinase 2; MMP-9: Matrix metalloproteinase 9; MMPs: Matrix metalloproteinases; OVOCs: Oxygenated volatile organic compounds; p-ERK1/2: Phosphorylated ERK1/2; p-STAT3: Phosphorylated STAT3; PAHs: Polycyclic aromatic hydrocarbons; PM2.5: Particulate matter 2.5; RAS: Renin-angiotensin system; RRR: Resting respiratory rate; SD: Sprague Dawley; STAT3: Signal transducer and activator of transcription 3; TEM: Transmission electron microscope; TGF-β1: Transforming growth factor beta 1; TNF-α: Tumour necrosis factor alpha; WT: Wild-type.
Acknowledgements
This work was supported by the grants of MOST 104-2313-B-009 -001-MY3 from the Ministry of Science and Technology and the grants of MMH-CT-10503 and MMH-CT-10507 from the Hsinchu Mackay Memorial Hospital, Taiwan.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Pun VC, Kazemiparkouhi F, Manjourides J. et al. Long-term PM2.5 exposures and respiratory, cancer and cardiovascular mortality in American older adults. Am J Epidemiol. 2017;186:961-969
2. Song C, He J, Wu L. et al. Health burden attributable to ambient PM2.5 in China. Environ Pollut. 2017;223:575-586
3. Churg A, Brauer M. Human lung parenchyma retains PM2.5. Am J Respir Crit Care Med. 1997;155:2109-2111
4. Li YY, Lin T, Wang FW. et al. Seasonal variation of polybrominated diphenyl ethers in PM 2.5 aerosols over the East China Sea. Chemosphere. 2015;119:675-681
5. Zhang Y, Ji X, Ku T. et al. Heavy metals bound to fine particulate matter from northern China induce season-dependent health risks: A study based on myocardial toxicity. Environ Pollut. 2016;216:380-390
6. Gent JF, Triche EW, Holford TR. et al. Association of low-level ozone and fine particles with respiratory symptoms in children with asthma. JAMA. 2003;290:1859-1867
7. Gong HJ, Linn WS, Clark KW. et al. Respiratory responses to exposures with fine particulates and nitrogen dioxide in the elderly with and without COPD. Inhal Toxicol. 2005;17:123-132
8. Tong Y, Zhang G, Li Y. et al. Synchrotron microradiography study on acute lung injury of mouse caused by PM(2.5) aerosols. Eur J Radiol. 2006;58:266-272
9. Monn C, Becker S. Cytotoxicity and induction of proinflammatory cytokines from human monocytes exposed to fine (PM2.5) and coarse particles (PM10-2.5) in outdoor and indoor air. Toxicol Appl Pharmacol. 1999;155:245-252
10. Dagher Z, Garçon G, Gosset P. et al. Pro-inflammatory effects of Dunkerque city air pollution particulate matter 2.5 in human epithelial lung cells (L132) in culture. J Appl Toxicol. 2005;25:166-175
11. Baulig A, Blanchet S, Rumelhard M. et al. Fine urban atmospheric particulate matter modulates inflammatory gene and protein expression in human bronchial epithelial cells. Front Biosci. 2007;12:771-782
12. Geraghty P, Wyman AE, Garcia-Arcos I. et al. STAT3 modulates cigarette smoke-induced inflammation and protease expression. Front Physiol. 2013;4:267
13. Pan T, Xiao ZH. Expression of P38 MAPK and MMP-2 mRNA in neonatal rats with hyperoxia-induced lung injury. Zhongguo Dang Dai Er Ke Za Zhi. 2013;15:383-386
14. Marshall RP. The pulmonary renin-angiotensin system. Curr Pharm Des. 2003;9:715-722
15. Hung YH, Hsieh WY, Hsieh JS. et al. Alternative Roles of STAT3 and MAPK Signaling Pathways in the MMPs Activation and Progression of Lung Injury Induced by Cigarette Smoke Exposure in ACE2 Knockout Mice. Int J Biol Sci. 2016;12:454-465
16. Parajuli N, Ramprasath T, Patel V.B. et al. Targeting angiotensin-converting enzyme 2 as a new therapeutic target for cardiovascular diseases. Can J Physiol Pharmacol. 2014;92:558-565
17. Lee YB, Nagai A, Kim SU. Cytokines, chemokines and cytokine receptors in human microglia. J Neurosci Res. 2002;69:94-103
18. Dagenais NJ, Jamali F. Protective effects of angiotensin II interruption: evidence for antiinflammatory actions. Pharmacotherapy. 2005;25:1213-1229
19. Patel VB, Basu R, Oudit GY. ACE2/Ang 1-7 axis: A critical regulator of epicardial adipose tissue inflammation and cardiac dysfunction in obesity. Adipocyte. 2016;5:306-311
20. Feng J, Zhong M, Xu B. et al. Concentrations, seasonal and diurnal variations of black carbon in PM 2.5 in Shanghai, China. Atmos Res. 2014;147:1-9
21. Song J, Wu Y, Zheng X. et al. Chemical characterization of roadside PM 2.5 and black carbon in Macao during a summer campaign. Atmos Pollut Res. 2014;5:381-387
22. Kenny LC, Bowry A, Crook B. et al. Field testing of a personal size-selective bioaerosol sampler. Ann Occup Hyg. 1999;43:393-404
23. Chao MW, Kozlosky J, Po IP. et al. Diesel exhaust particle exposure causes redistribution of endothelial tube VE-cadherin. Toxicology. 2011;279(1-3):73-84
24. Kulkarni AA, Thatcher TH, Hsiao HM. et al. The triterpenoid CDDO-Me inhibits bleomycin-induced lung inflammation and fibrosis. PLoS One. 2013;8:e63798
25. Vickers C, Hales P, Kaushik V. et al. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem. 2002;277:14838-14843
26. Chen CL, Huang SK, Lin JL. et al. Upregulation of matrix metalloproteinase-9 and tissue inhibitors of metalloproteinases in rapid atrial pacing-induced atrial fibrillation. J Mol Cell Cardiol. 2008;45:742-753
27. Lu LC, Yang CW, Hsieh WY. et al. Decreases in plasma MMP-2/TIMP-2 and MMP-9/TIMP-1 ratios in uremic patients during hemodialysis. Clin Exp Nephrol. 2016;20:934-942
28. Yan J, Lai CH, Lung SC. et al. Industrial PM2.5 cause pulmonary adverse effect through RhoA/ROCK pathway. Sci Total Environ. 2017;599-600:1658-1666
29. Mantecca P, Farina F, Moschini E. et al. Comparative acute lung inflammation induced by atmospheric PM and size-fractionated tire particles. Toxicol Lett. 2010;198:244-254
30. Farina F, Sancini G, Mantecca P. et al. The acute toxic effects of particulate matter in mouse lung are related to size and season of collection. Toxicol Lett. 2011;202:209-217
31. Gautam S, Yadav A, Tsai CJ. et al. A review on recent progress in observations, sources, classification and regulations of PM2.5 in Asian environments. Environ Sci Pollut Res Int. 2016;23(21):21165-21175
32. Zheng G, Duan F, Ma Y. et al. Episode-Based Evolution Pattern Analysis of Haze Pollution: Method Development and Results from Beijing, China. Environ Sci Technol. 2016;50(9):4632-41
33. Park JH, Han KT, Eu KJ. et al. Diffusion flame-derived fine particulate matters doped with iron caused genotoxicity in B6C3F1 mice. Toxicol Ind Health. 2005;21:57-65
34. Pereira FA, Lemos M, Mauad T. et al. Urban, traffic-related particles and lung tumors in urethane treated mice. Clinics. 2011;66:1051-1054
35. Molina RM, Konduru NV, Hirano H. et al. Pulmonary distribution of nanoceria: comparison of intratracheal, microspray instillation and dry powder insufflation. Inhal Toxicol. 2016;28:550-560
36. Li W, Hu Y, Yuan W. et al. Comparison of two mouse models of lung fibrosis induced by intratracheal instillation and intratracheal aerosol administration of bleomycin. Nan Fang Yi Ke Da Xue Xue Bao. 2012;32:221-225
37. Liu F, Li W, Pauluhn J. et al. Lipopolysaccharide-induced acute lung injury in rats: comparative assessment of intratracheal instillation and aerosol inhalation. Toxicology. 2013;304:158-166
38. Mak JC, Chan-Yeung MM, Ho SP. et al. Elevated plasma TGF-beta1 levels in patients with chronic obstructive pulmonary disease. Respir Med. 2009;103(7):1083-9
39. Wei J, Xiong XF, Lin YH. et al. Association between serum interleukin-6 concentrations and chronic obstructive pulmonary disease: a systematic review and meta-analysis. PeerJ. 2015;3:e1199
40. Rincon M, Irvin CG. Role of IL-6 in Asthma and Other Inflammatory Pulmonary Diseases. Int J Biol Sci. 2012;8(9):1281-1290
41. Wegesser TC, Last JA. Mouse lung inflammation after instillation of particulate matter collected from a working dairy barn. Toxicol Appl Pharmacol. 2009;236:348-357
42. Riva DR, Magalhães CB, Lopes AA. et al. Low dose of fine particulate matter (PM2.5) can induce acute oxidative stress, inflammation and pulmonary impairment in healthy mice. Inhal Toxicol. 2011;23:257-267
43. Upadhyay S, Stoeger T, Harder V. et al. Exposure to ultrafine carbon particles at levels below detectable pulmonary inflammation affects cardiovascular performance in spontaneously hypertensive rats. Part Fibre Toxicol. 2008;5:19
44. Upadhyay S, Ganguly K, Stoeger T. et al. Cardiovascular and inflammatory effects of intratracheally instilled ambient dust from Augsburg, Germany, in spontaneously hypertensive rats (SHRs). Part Fibre Toxicol. 2010;7:27
45. Aztatzi-Aguilar OG, Uribe-Ramírez M, Arias-Montaño JA. et al. Acute and subchronic exposure to air particulate matter induces expression of angiotensin and bradykinin-related genes in the lungs and heart: Angiotensin-II type-I receptor as a molecular target of particulate matter exposure. Part Fibre Toxicol. 2015;12:17
46. Hasegawa G, Hirano M, Ishihara Y. Differential gene expression associated with inflammation and blood pressure regulation induced by concentrated ambient particle exposure. Inhal Toxicol. 2011;23:897-905
47. Gaddam RR, Chambers S, Bhatia M. ACE and ACE2 in inflammation: a tale of two enzymes. Inflamm Allergy Drug Targets. 2014;13(4):224-34
48. Imai Y, Kuba K, Rao S. et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature. 2005;436:112-116
49. Olkowicz M, Chlopicki S, Smolenski RT. Perspectives for angiotensin profiling with liquid chromatography/mass spectrometry to evaluate ACE/ACE2 balance in endothelial dysfunction and vascular pathologies. Pharmacol Rep. 2015;67:778-785
50. Li X, Molina-Molina M, Abdul-Hafez A. et al. Angiotensin converting enzyme-2 is protective but downregulated in human and experimental lung fibrosis. Am J Physiol Lung Cell Mol Physiol. 2008;295:L178-185
51. Corbel M, Belleguic C, Boichot E. et al. Involvement of gelatinases (MMP-2 and MMP-9) in the development of airway inflammation and pulmonary fibrosis. Cell Biol Toxicol. 2002;18:51-61
52. Smigiel KS, Parks WC. Matrix Metalloproteinases and Leukocyte Activation. Prog Mol Biol Transl Sci. 2017;147:167-195
53. Iwazu Y, Muto S, Hirahara I. et al. Matrix metalloproteinase 2 induces epithelial-mesenchymal transition in proximal tubules from the luminal side and progresses fibrosis in mineralocorticoid/salt-induced hypertensive rats. J Hypertens. 2011;29:2440-2453
54. Eldred JA, Hodgkinson LM, Dawes LJ. et al. MMP2 activity is critical for TGFβ2-induced matrix contraction implications for fibrosis. Invest Ophthalmol Vis Sci. 2012;53:4085-4098
55. Yoon HK, Cho HY, Kleeberger SR. Protective role of matrix metalloproteinase-9 in ozone-induced airway inflammation. Environ Health Perspect. 2007;115:1557-1563
56. Zhang C, Zhao YX, Zhang YH. et al. Angiotensin-converting enzyme 2 attenuates atherosclerotic lesions by targeting vascular cells. Proc Natl Acad Sci USA. 2010;107:15886-15891
57. Sun J. Matrix metalloproteinases and tissue inhibitor of metalloproteinases are essential for the inflammatory response in cancer cells. J Signal Transduct. 2010;2010:985132
58. Balakumar P, Jagadeesh G. A century old renin-angiotensin system still grows with endless possibilities: AT1 receptor signaling cascades in cardiovascular physiopathology. Cell Signal. 2014;26:2147-2160
59. Deng X, Rui W, Zhang F. et al. PM2.5 induces Nrf2-mediated defense mechanisms against oxidative stress by activating PIK3/AKT signaling pathway in human lung alveolar epithelial A549 cells. Cell Biol Toxicol. 2013;29:143-157
60. Xu Z, Zhang Z, Ma X. et al. Effect of PM2.5 on oxidative stress-JAK/STAT signaling pathway of human bronchial epithelial cells. Wei Sheng Yan Jiu. 2015;44:451-455
Author contact
![]() Corresponding author: Chih-Sheng Lin, Ph.D., Department of Biological Science and Technology. National Chiao Tung University, No.75 Po-Ai Street, Hsinchu 30068, Taiwan. Tel.: +886-3-5131338; Fax: +886-3-5729288; E-mail: lincsnctu.edu.tw
Corresponding author: Chih-Sheng Lin, Ph.D., Department of Biological Science and Technology. National Chiao Tung University, No.75 Po-Ai Street, Hsinchu 30068, Taiwan. Tel.: +886-3-5131338; Fax: +886-3-5729288; E-mail: lincsnctu.edu.tw