Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and methods
Results and Discussion
Conclusion
Abbreviation
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(4):369-380. doi:10.7150/ijbs.24117 This issue Cite
Research Paper
Splicing Factor RBM20 Regulates Transcriptional Network of Titin Associated and Calcium Handling Genes in The Heart
Wei Guo1,2 ![]() , Chaoqun Zhu1,2, Zhiyong Yin1,3, Qiurong Wang1,2, Mingming Sun1,2, Huojun Cao4,5, Marion L. Greaser6
, Chaoqun Zhu1,2, Zhiyong Yin1,3, Qiurong Wang1,2, Mingming Sun1,2, Huojun Cao4,5, Marion L. Greaser6
1. Animal Science, University of Wyoming, Laramie, WY 82071, USA
2. Center for Cardiovascular Research and Alternative Medicine, University of Wyoming, Laramie, WY 82071, USA
3. Department of Cardiology, Xi Jing Hospital, Fourth Military Medical University, Xi'an 710032, China
4. Iowa Institute for Oral Health Research, College of Dentistry,
5. Craniofacial Anomalies Research Center, Carver College of Medicine, The University of Iowa, Iowa City, IA 52242, USA
6. Animal Science, University of Wisconsin-Madison, Madison, WI 53705, USA
Received 2017-11-30; Accepted 2018-2-27; Published 2018-3-9
Abstract

RNA binding motif 20 (RBM20) regulates pre-mRNA splicing of over thirty genes, among which titin is a major target. With RBM20 expression, titin expresses a larger isoform at fetal stage to a smaller isoform at adult resulting from alternative splicing, while, without RBM20, titin expresses exclusively a larger isoform throughout all ages. In addition to splicing regulation, it is unknown whether RBM20 also regulates gene expression. In this study, we employed Rbm20 knockout rats to investigate gene expression profile using Affymetrix expression array. We compared wild type to Rbm20 knockout at day1, 20 and 49. Bioinformatics analysis showed RBM20 regulates fewer genes expression at younger age and more at older age and commonly expressed genes have the same trends. GSEA indicated up-regulated genes are associated with heart failure. We examined titin binding partners. All titin direct binding partners are up-regulated and their increased expression is associated with dilated cardiomyopathy. Particularly, we found that genes involving calcium handling and muscle contraction are changed by RBM20. Intracellular calcium level measurement with individual cardiomyocytes further confirmed that changes of these proteins impact calcium handling. Selected genes from titin binding partners and calcium handling were validated with QPCR and western blotting. These data demonstrate that RBM20 regulates gene splicing as well as gene expression. Altered gene expression by RBM20 influences protein-protein interaction, calcium releasing and thus muscle contraction. Our results first reported gene expression impacted by RBM20 with heart maturation, and provided new insights into the role of RBM20 in the progression of heart failure.
Keywords: Titin, RBM20, Titin binding partners, Calcium handling, Intracellular calcium concentration
Introduction
Pre-mRNA splicing is an important biological process that contributes to protein diversity in eukaryotic organisms [1]. This process is determined by RNA-binding splicing factors to produce protein isoforms in a tissue-specific and developmental-regulated manner [2]. Tissue-specific alternative splicing is usually regulated by tissue-specific splicing factors or a combination with ubiquitously expressed splicing factors to influence spliceosome assembly at splice sites [3]. Therefore, identification of tissue-specific splicing factors and their targets is essential to decipher the mechanism of pre-mRNA splicing. In muscle tissues, only a few splicing factors have been reported [1], and RNA binding motif 20 (RBM20) is one of the muscle tissue specific splicing factors that has been identified recently [4]. Protein RBM20 contains two highly conserved functional domains, an RNA-recognition domain 1 (RRM1) and a serine (S)/arginine (R) rich domain (SR-domain) [5]. These structural features defined it as a serine/arginine (SR) protein that is essential for assembly in the spliceosome of pre-mRNA splicing [6, 7]. RBM20 is highly expressed in striated muscles, with the highest expression levels in cardiac muscle [4]. Rbm20 deficiency in animal models leads to dilated cardiomyopathy, a major cause of heart failure and a significant source of mortality and morbidity worldwide [4, 8], and mutations in RBM20 have been identified in human patients with familial dilated cardiomyopathy [4, 5, 9, 10, 11, 12, 13]. Given the role of RBM20 in the regulation of gene splicing, efforts toward the identification of its RNA targets have been made [4, 14] with a goal that these findings would shed light on human heart failure progression and provide strategies for molecular therapy. With these efforts, more than thirty RNA targets of RBM20 have been identified with a major target being titin [4, 14].
Titin is a giant multifunctional protein in the sarcomere that plays a critical role in elastic recoil of cardiac myocytes and contributes to passive stiffness of the myocardium [15-16, 17-18]. It has been increasingly recognized as a major disease-causing gene [16, 19]. Titin runs through distinct sarcomere regions: the Z-band, the I-band, the A-band and the M-band regions [20, 21]. Over twenty proteins have been found directly bound to these regions of titin, and over fifty proteins are indirectly bound to titin, linking it to multiple potential mechanic signaling pathways [22, 23]. Cardiac titin is encoded by a single gene, and it has two major classes of isoforms resulting from alternative splicing. These isoforms have been termed “N2B” and “N2BA” [24]. The ratio changes of these two isoforms are a major determinant for diastolic passive stiffness during left ventricular filling [15-18, 25]. Aberrant ratio changes of titin isoforms have been found in both systolic and diastolic heart failure [1, 26-32]. Therefore, the role of RBM20 in the regulation of titin alternative splicing may be critical for correction of abnormal titin isoform changes, and thereby, the treatment of diastolic dysfunction. In addition to its major role of the regulation in titin splicing, it is unknown whether RBM20 also regulates gene expression profiles in the heart. In this study, we compared the gene expression profiles of wild type and the Rbm20 knockout rats, and demonstrated that calcium handling channel proteins are affected and regulated by RBM20.
Materials and methods
Animals and Sample preparation
Sprague-Dawley strain rats were crossed with Fisher 344 inbreeds. Both strains were obtained from Harlan Sprague Dawley, Indianapolis, IN. Animals were maintained on standard rodent chow using protocols approved by the University of Wisconsin-Madison and University of Wyoming Animal Use and Care Committees. Hearts were removed immediately after euthanasia and the left ventricle, right ventricle, and atria were separated. Samples were obtained from animals ranging from 16 days fetuses (E16) to 1 year after birth. Tissues were snap-frozen in liquid nitrogen and stored in -80°C freezer prior to gel sample and RNA preparation. All samples for protein gel electrophoresis were prepared as described previously [33].
Gel electrophoresis
Titin isoforms were resolved using a previously published method using a vertical sodium dodecyl sulfate (SDS)-1% agarose gel electrophoresis (VAGE) system [33]. The frozen tissue samples from E16, E18 (18 days fetuses), day (d) 1, 10, 20, 30, 60, 180, and 1 year after birth were dissolved in urea-thiourea-SDS-dithiothreitol sample buffer using a small Dounce homogenizer or a MiniBeadBeater (Biospec Products, Bartlesville, OK) and were heated for 10min at ~65℃. Silver stained agarose gels were dried between sheets of mylar and dried gels were scanned. Human cardiac titin N2B (or the identically sized rat N2B) were used as standards to estimate molecular weights of the rat N2BA titin isoform. Full details have been discussed in a previous published report [33].
Microarray hybridization and data analysis
Total RNA was extracted in 1ml TRIzol (Invitrogen, Frederick, MD), following manufacturer instructions. RNA was precipitated from the TRIzol reagent with isopropanol and resuspended at 0.5-1.0mg/ml using RNA secure solution (Ambion, Austin, TX). RNA samples were treated with DNaseI (DNA-free; Ambion, Austin, TX). After extraction, all samples were stored at -80 degree. RNA concentration was measured with a NanoDrop ND 1000 spectrometer (NanoDrop Technologies, Wilmington, DE), and RNA integrity was assessed on an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA). RNA was further purified by the RNeasy mini kit (Qiagen, Valencia, CA). Double stranded cDNA was synthesized from total RNA (SuperScript II system; Invitrogen). An in vitro transcription reaction was then performed to obtain biotin-labeled cRNA from the double-stranded cDNA (Enzo BioArray High Yield RNA Transcript Labeling kit; Enzo Diagnostics, Farmingdale, NY). The cRNA was fragmented before hybridization, and then mixed in a hybridization mixture containing probe array controls, BSA, and herring sperm DNA [34]. A cleanup procedure was performed on the hybridization cocktail using an RNeasy spin column (Qiagen), after which it was applied to the Affymetrix Rat 230 2.0 probe array. A total of eighteen hybridization experiments were performed in which each stage (d1, d20 and d49) was represented by three wild type (Wt) and three RBM20 homozygous knockout (Hm) individual ventricular RNA extracts (Fig. 2A). Hybridization was allowed to continue for 16 h at 45°C in a GeneChip 640 hybridization oven, after which the arrays were washed and stained with phycoerythrin-conjugated streptavidin (Molecular Probes, Eugene, OR). Images were scanned using a GeneArray scanner (Agilent Technologies, Palo Alto, CA) and GeneChip cel files were subsequently processed by the log scale robust multi-array analysis (RMA). The RMA estimates were based upon a robust average of log2 (B (PM)), where B (PM) is background corrected perfect match intensities. Compared with the GCOS method the RMA method has better precision, provided more consistent estimates of fold change and provided higher specificity and sensitivity when using fold change analysis to detect differential expression [35].
Gene expression profile analysis with gene-chip data. (A), Biological replicates of heart tissues of Wt and Hm with development; (B), Venn Diagram analysis of differentially expressed genes at three ages; (C), Percentage of DEG with up- and down-regulated genes at three ages; The Heatmap indicated the up- and down-regulated genes at three ages between Wt and Hm. Wt, wild type; Hm, homozygous Rbm20 knockout; D, day; DEG, differentially expressed genes; Up, up-regulation; Down, down-regulation.
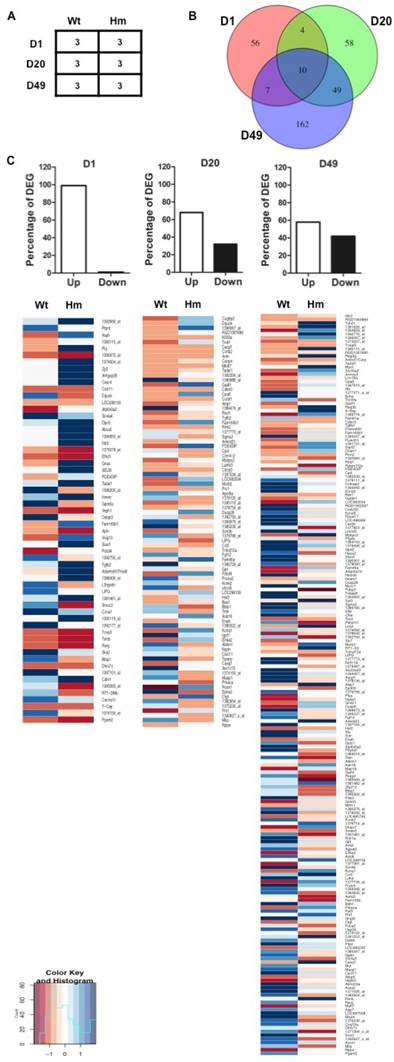
The complete data set is publicly available in the NCBI Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/; accession numbers GSE11137). Differential gene expression analysis was performed with Limma (R/Bioconductor) Gene set and pathway enrichment analysis was performed using DAVID [36] (http://david.niaid.nih.gov./david/beta/index.htm) and GSEA (http://software.broadinstitute.org/gsea/index.jsp)
Cardiomyocyte isolation and intracellular calcium measurements
Rats were injected with heparin (1000U/kg) 20mins before anesthetized with 60mg/kg ketamine and 8mg/kg xylazine (i.p.). Myocytes were isolated as described previously [11]. In brief, under anesthesia, the heart was dissected and cannulated via the aorta. The heart was perfused at 37°C for 4 min with perfusion buffer (in mM: 113 NaCl, 4.7 KCl, 0.6 Na2HPO4, 1.2 MgSO4, 12 NaHCO3, 10 KHCO3, 10 HEPES, and 30 taurine, pH 7.4) equilibrated with 5%CO2-95% O2, followed by perfusion with digestion buffer (perfusion buffer plus 0.45 mg/ml collagenase II [240 U/mg; Worthington Biochemical Corporation], 0.13 mg/ml trypsin, and 25 µM CaCl2) for 8-10 min. When the heart was flaccid, digestion was halted and the heart was placed in myocyte stopping buffer 1 (perfusion buffer plus 0.04 ml bovine calf serum [BCS]/ml buffer and 5 µM CaCl2). The left ventricle (LV) was cut into small pieces, and the rest of the heart was discarded. The small pieces were then triturated several times with a transfer pipette and then filtered through a 300µm nylon mesh filter. After this, the cells were gravity pelleted and the supernatant was discarded. Next, 10 ml of myocyte stopping buffer 2 was added (perfusion buffer plus 0.05 ml BCS/ml buffer and 12.5 µM CaCl2), and Ca2+ was reintroduced to a final concentration of 1.8 mM. Then, myocytes were loaded with Fura-2 AM (0.5 μM) for 10 min, and fluorescence measurements were recorded with a dual-excitation fluorescence photomultiplier system (IonOptix). Myocytes were placed on an Olympus IX-70 inverted microscope stage and imaged through a Fluor 40× oil objective. Myocytes were exposed to light emitted by a 75-W lamp and passed through either a 360- or a 380-nm filter while being stimulated to contract at 0.5 Hz. Fluorescence emissions were detected between 480 and 520 nm by a photomultiplier tube after first illuminating the myocytes at 360 nm for 0.5 sec and then at 380 nm for the duration of the recording protocol (333Hz sampling rate). The excitation scan at 360 nm was repeated at the end of the protocol and qualitative changes in intracellular Ca2+ concentration were inferred from the ratio of Fura-2 fluorescence intensity (FFI) at the two wavelengths (360/380). Fluorescence decay time was measured as an indication of the intracellular Ca2+ clearing rate. Single exponential curve fit programs were used to derive the intracellular Ca2+ decay constant [37].
Quantitative real time PCR (QPCR)
Total RNA was extracted using TRIzol, as described above. For reverse transcription, 60 ng of RNA were mixed with 5 uM random hexamers, 1 mM each dNTP, 7.5 mM MgCl2, 40 U RNasin (Promega, Madison, WI), 1X PCR buffer II (Applied Biosystems, Foster City, CA) and 250 U of SuperScript II reverse transcriptase (Invitrogen). The reaction mixture was incubated at 25°C for 10 min, 48°C for 45 min, and 95°C for 5 min, then cooled down to 4°C. Two volumes of ethanol were added to the cDNA and evenly mixed, and then stored at -20°C for 30 minutes, centrifuging for 15 minutes at 13,000 rpm, washed with 75% ethanol and dried. The cDNA was resuspended in distilled water and used as a template. For SYBR Green quantitative real-time PCR, all primers were designed from the selected genes (Supplement table S1). All primer pairs produced a single PCR product as determined by the dissociation curve and gel analysis. Real-time PCR was performed in a 20ul reaction, 96-well format and 1 X SYBR PCR Master Mix (Biorad). The reaction was incubated in a CFX96 real-time PCR system (Biorad) for 40 cycles consisting of denaturation at 95°C for 15 s and annealing/extension at 58-60°C for 1 min. Three samples were measured in each experimental group in quadruplicate, with a minimum of two independent experiments. The relative amount of target mRNA normalized to GAPDH was calculated according to the method described previously [38].
Western Blot
Total protein was separated by SDS-PAGE gel, and transferred onto a PVDF membrane. The membrane was probed with antibodies against FHL (Santa Cruz), CRP2 (Santa Cruz), IGFBP8/CTGF (Abcam), KCNQ1 (Santa Cruz), TGF β2 (Cell Signaling), RyR2 (Abcam), SERCA2A (Santa Cruz), TRDN (Santa Cruz), CASQ1 (Santa Cruz), PLB (Abcam), Rabbit anti-mouse IgG-conjugated with horseradish peroxidase (Fisher Scientific) was served as the secondary antibody. GAPDH (Santa Cruz) was served as the protein loading control. The detailed procedure was described previously [34].
Statistical analysis
GraphPad prism software was used for statistical analysis. Results were expressed as means ± SEM. Statistical significance between groups was determined using one way ANOVA with a Tukey post test, or a 2-tailed t-test for comparison of two groups. The significance level was P<0.05, P<0.01 and P<0.001.
Results and Discussion
RBM20 knockout Rat model characterization
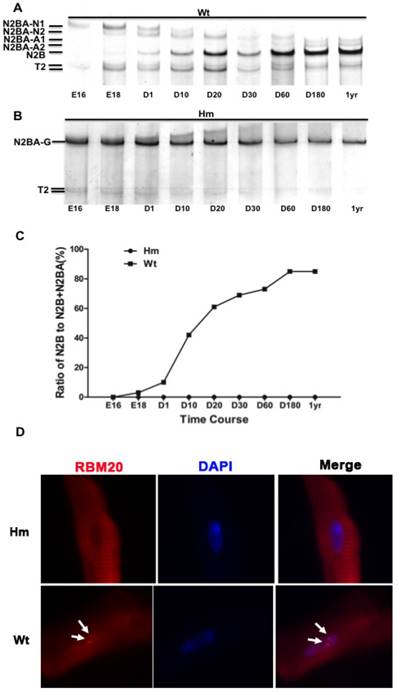
A major RBM20 target, titin is a striated muscle-specific giant elastic protein and largely responsible for the generation of the diastolic force in the cardiac myocyte [15-18]. Titin has two classes of isoforms with one smaller isoform N2B that is stiffer, and the larger isoforms N2BAs that are more compliant [24-25]. Cardiac titin undergoes a series of isoform transitions during maturation in wild type rat (Wt) heart as observed by SDS agarose electrophoresis (embryonic day (E) 16, E18, day (d) 1, d10, d20, d30, d60, d180 and year (yr) 1) (Fig. 1A). Larger fetal titin N2BA isoform is predominantly expressed before E18, and decreases in sizes and amounts with maturation, while smaller N2B isoform starts to be present after E18 and dominant after d10 (Fig. 1A and 1C). Percentage of N2B to total titin (N2B+N2BA) changes from about 2% at E18 to approximately 85% at adult (Fig 1C). However, in Rbm20 knockout rats (Hm), titin is expressed as a constant size that is similar to the larger fetal titin N2BA isoform through all ages when compared to Wt rats (Fig. 1B and 1C). This constantly expressed larger titin isoform in Rbm20 Hm rats leads to reduced ventricular wall stiffness and has been associated with heart failure progression as described previously [20]. Immunocytochemistry staining of cardiomyocytes isolated from adult rat hearts with RBM20 antibody indicated that RBM20 is located in splicing speckles in the nucleus with two spots in Wt cardiomyocytes and completely depleted in Hm cardiomyocytes (Fig 1D). Splicing speckles are nuclear bodies that are important for splicing factor storage and modification and contain components of the splicing machinery and polyA RNA [39-40].
Rbm20 deficient rat model. (A and B), Agarose gel showing titin protein isoform changes with development in wild type (Wt) and homozygous Rbm20 knockout (Hm); (C), Percentage of N2B with development in Wt and Hm; (D), Rbm20 location and expression in Wt and Hm rat adult cardiomyocytes. The arrows indicate the Rbm20 localization in the nucleus. Wt, wild type; Hm, homozygous Rbm20 knockout; E, embryonic day; D, day; yr, year.
Our previous study has shown that Rbm20 deficiency in the rat model leads to heart failure with reduced ejection fraction, enlarged ventricular chamber and thinner ventricular walls [4]. Additionally, Ours and other studies have identified over thirty RBM20 target genes that undergo alternative splicing [4, 14]. However, it is unknown whether Rbm20 deficiency leads to gene expression changes in the heart. Therefore, this study employs the Rbm20 knockout rat model to investigate gene expression profile using Affymetrix gene expression microarray and determine which genes or signaling pathways are associated with heart failure progression in Rbm20 knockout rats.
Gene expression profile indicated that developmental changes of gene expression are consistent with titin splicing pattern regulated by RBM20
In order to examine the effect of RBM20 on gene differential expression in heart muscle at distinct developmental stages, we selected d1, d20 and d49 as representative maturation time courses according to titin isoform expression pattern as indicated in Fig. 1. The experimental design is shown in Fig 2A. Gene expression array data were analyzed based on student t test between Wt and Hm with p value cutoff less than 0.05 and fold change more than 1.5. Out of a total 31099 probes on the Affymetrix Rat 230 2.0 Chip, 418 probes representing 381 genes including 21 unannotated genes were differentially changed when comparing Hm to Wt rats (Supplement table S2). Among 381 genes, 77 of them are differentially expressed at d1 (Supplement table S3), 121 at d20 (Supplement table S4), and 227 at d49 (Supplement table S5). Venn diagram analysis of differentially expressed genes at three ages demonstrated that four genes were commonly expressed at d1 and 20, seven at d1 and 49, forty-nine at d20 and 49, and ten at all three ages (Fig. 2B). These data hint that most of differentially expressed genes regulated by RBM20 are development dependent. RBM20 regulates the expression of fewer genes at early developmental stages when compared to late stages, which matches the pattern of titin size changes from the larger ones to smaller during maturation. Interestingly, we found that almost 99% of differentially expressed genes are up-regulated at early maturation stage, and percentage of up-regulated genes decreases at d20 to approximately 68% and at d49 to 57% (Fig. 2C). These data suggest that genes regulated by RBM20 are fewer at d1 with mostly elevated expression by comparing to late maturation stages d20 and d49 with more down-regulated genes. However, gene expression pattern regulated by RBM20 at d20 and d49 are similar, which is consistent with titin splicing pattern with maturation (Fig. 1A).
Gene functional analysis showed that up-regulated genes are associated with heart failure and gene expression pattern is similar at later maturation stages
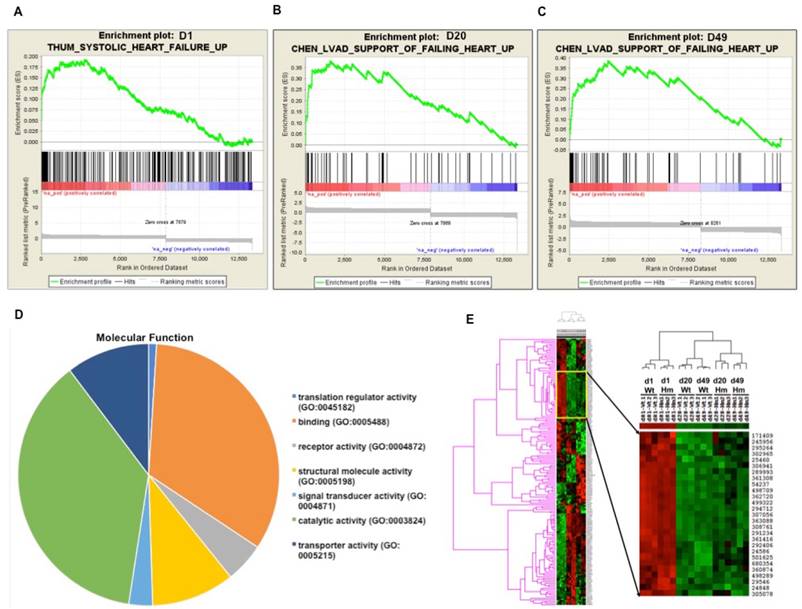
Next we did further data mining for the relevant function of these differentially expressed genes. Gene Set Enrichment Analysis (GSEA) is a computational method that determines whether a priori defined set of genes shows statistically significant, concordant differences between two biological states [41]. In order to interpret gene expression data by analyzing gene sets, GSEA analysis indicated that gene sets associated with failing heart were up-regulated at all three ages (Fig. 3A, 3B and 3C). Gene Ontology (GO) analysis indicated that most of differentially expressed genes were associated with protein binding and cell signal transduction (Fig. 3D). Gene cluster analysis suggested that the expression pattern of d20 and d49 between Wt and Hm was similar, but it was different from d1. GO and gene cluster analysis further confirmed that gene expression pattern regulated by RBM20 was consistent with titin splicing pattern changes in the heart of Wt rats which was also modulated by RBM20 (Fig. 3E, 1A and 1B).
Gene functional annotation and heatmap of pairwise correlations between Wt and Hm. (A, B and C), Gene Set Enrichment analysis indicated that gene sets associated with heart failure are up-regulated at all three ages; (D), A cluster of differentially expressed gene between Wt and Hm at three ages; (E), Molecular function of differentially expressed genes with Gene Ontology (GO) analysis. D, day; Wt, wild type; Hm, homozygous Rbm20 knockout; GO, Gene Ontology.
Differentially expressed genes associated with titin direct and indirect binding and these binding partners play diverse roles that may together contribute to heart failure progression induced by RBM20
Titin binds over 70 proteins directly and indirectly [23]. It is unknown whether differentially expressed genes regulated by RBM20 contain titin binding partners. We searched the list of differentially expressed genes with known titin partners, and found that among these genes, proteins encoded by five of them directly bound to titin and twenty-two of them were indirectly associated with titin (Fig. 4). Five direct binding proteins were MyBP-C, Fhl1, Carp, Darp, and β-myosin. MyBPC has three isoforms: slow skeletal, fast skeletal and cardiac isoform that are encoded by three different genes [42-45]. Gene MyBPC2 encodes a fast skeletal isoform (fMyBP-C) that is expressed primarily in fast skeletal muscles [45]. However, it can also be expressed in cardiac muscle, and its increased expression in the heart is associated with severe dilated cardiomyopathy [46]. Mammalian cardiac muscle contains two myosin isoforms: α-myosin and β-myosin encoded by two distinct genes [47]. In adult human cardiac muscle, the major isoform is β-myosin [48]. Elevated β-myosin level has been found in human failing heart myocardium, cardiomyopathy, aging and pressure overload [49-51]. Four-and-a-half Lim domain protein-1 (Fhl1) is a member of Fhl family structurally characterized by four complete LIM domain and a N-terminal half LIM domain [52], which has been found associated with titin N2B domain [53] and the mitogen-activated protein kinase signaling cascade (Raf1/Mek1/2/Erk2) [54]. Fhl1 plays an important role in sensing biomechanical hypertrophic stress responses in the sarcomere via titin N2B, which in turn regulates MAPK/hypertrophic signaling and muscle compliance [54-55]. Carp and Darp are two members of the three homologous muscle-ankyrin-repeat proteins (MARPs) that interact with titin I-band N2A region [56-57]. They are associated with muscle stress signaling and up-regulated when muscles undergo mechanical or metabolic challenge [56]. Carp and Darp exist both in the nucleus and in the sarcomeric I-band, and show increased expression levels in end-stage failing human dilated cardiomyopathy hearts [32]. All of these five direct titin binding partners were up-regulated in Rbm20 knockout rats. According to previous studies described above, up-regulation of these five genes in human heart or animal models leads to heart failure with preserved ejection fraction [46,49, 50, 55, 56], which was consistent with the phenotype of Rbm20 knockout rats and human patients with Rbm20 loss-of-function mutations [4].
Titin direct and indirect binding partners. The central rectangle depicts the sarcomere positions of different titin segments. The direct titin binding partners are orange ovals; the indirect titin binding partners are yellow rectangles; the direct and indirect titin binding partners with differential expression are red ovals and rectangles. sANK1, small ankyrin 1 isoform; p94, calpain-3; FHL1/2, four-and-a-half-LIM-domain-protein; MyBPC, myosin binding protein C; Nbr1, neighbor-of-BRCA1-gene-1; MURF, muscle-specific-RING-finger-protein; MyHC, myosin-heavy-chain; MyLC, myosin-light-chain; TPM, tropomyosin; CARP, cardiac-ankyrin-repeat-protein; DARP, diabetes-related-ankyrin-repeat-protein; ARPP, ankyrin-repeat-domain-protein-2; TMOD, tropomodulin; MYOT, myotilin; FLNC, Υ-filamin; T-cap, telethonin; minK, potassium-channel subunit; MLP (CRP3), muscle-LIM-protein (cysteine-rich-protein-3); CRP2, muscle-LIM-protein (cysteine-rich-protein-2); ENAH, protein enabled homolog; ANP, atrial natriuretic peptide; BNP, brain natriuretic peptide; KCND2/3, potassium voltage-gated channel subfamily D member 2/3; KCNK2, potassium channel subfamily K member 2; KCNQ1, potassium voltage-gated channel subfamily Q member 1; TNFa-CE (ADAM-17), tumor-necrosisfactor-alpha-converting-enzyme (A-disintegrin-and metalloprotease-17 protein); ADAM33, disintegrin-and metalloprotease-33 protein; IGFBP, insulin growth factor binding protein; ZNF, zinc finger protein; ERK2, extracellular-signal-regulated-kinase-2; ANK2, ankyrin 2; YB-1, Y-box-binding-protein-1; ISOT-3 (USP13), isopeptidase-T-3; USP24, ubiquitin specific peptidase 24; UBC9, SUMO-conjugating enzyme UBC9; UBCH10, SUMO-conjugating enzyme E2C.
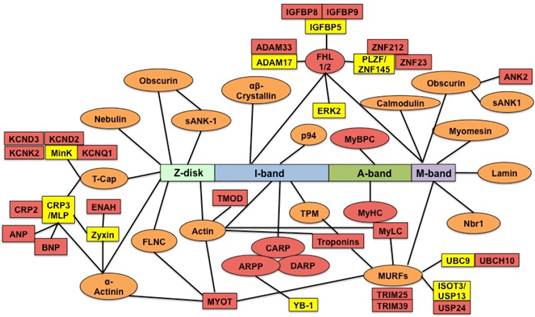
In addition to titin direct binding partners, we found more titin indirect binding partners that are regulated by RBM20. These indirect binding partners play diverse roles in different physiological function. For example, Kcnq1, Kcnd2, Kcnd3 and Kcnk2 are potassium voltage-gated channels required for cardiac action potential, cellular excitability and heart function [58-61]. The cardiac gene expression and plasma level of ANP and BNP are in response to changes of diuresis, natriuresis and systemic blood pressure. Increased levels of ANP and BNP are associated with chronic heart failure, and can be predictors for left ventricular ejection fraction, mortality and morbidity in human patients [62-63]. Another example is Myl4 that encodes a protein called atrial light chain 1 (ALC-1). Increased expression of ALC-1 was found in patients with hypertrophic obstructive cardiomyopathy, and in ischemic and idiopathic cardiomyopathy with larger individual differences [51, 64-65]. RBM20-regulated genes involving diverse functions indicate that Rbm20 deficiency leads to heart failure through cooperation of multiple biological processes instead of single gene effect such as titin, though they may play minor roles when compared to titin.
Validation of titin binding partners and non-titin binding partners by QPCR and western blotting
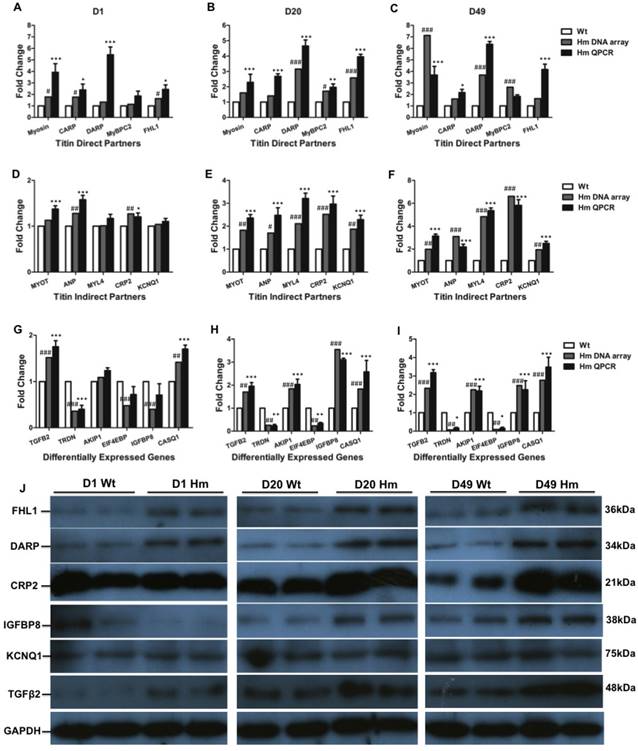
We next validated gene expression resulting from DNA microarray by QPCR and western blotting. We selected all five genes (Myosin, Carp, Darp, Mybpc2 and Fhl1) from direct titin binding partners and five genes (Myot, Anp, Myl4, Crp2 and Kcnq1) from indirect titin binding partners. We also selected six genes (Tgfβ2, Triadin/Trdn, Akip1, Eif4ebp, Igfbp8/Ctgf and Casq1) that haven't been identified associated with titin binding for validation at d1, d20 and d49. Comparisons were made between gene expression levels from DNA microarray and QPCR at individual ages. For titin direct binding partners, we found that all five genes were up-regulated and validated by QPCR at three ages which is consistent with DNA microarray data (Fig. 5A, 5B and 5C). For titin indirect binding partners, DNA microarray data indicated that all five genes were not increased by comparing to Wt at d1, while one gene Anp was validated to increase by QPCR at d1 (Fig. 5D). All five genes were significantly increased at d20 and d49 from both DNA microarray and QPCR (Fig. 5E and 5F). For other genes which are not related to titin binding, the expression level of genes Akip1 and Eif4ebp was similar to Wt at d1 from both DNA microarray and QPCR data. Expression of Igfbp8 was slightly lower by comparing to Wt at d1 from DNA microarray data, but it had no significant difference validated from QPCR. Gene Casq1 was slightly increased, but no significant difference at d1 from DNA microarray data, while QPCR results indicated significant increase. The expression level of Tgfβ2 and Trdn validated by QPCR was consistent with DNA microarray data. Interestingly, Genes Trdn and Eif4ebp were significantly decreased and Tgfβ2, Akip1, Igfbp8 and Casq1 were significantly increased by comparing to Wt at d20 and d49 from DNA microarray, which were confirmed by QPCR (Fig. 5G, 5H and 5I). We then selected total six proteins (Fhl1, Darp, Crp2, Igfbp8, Kcnq1 and Tgfβ2) from direct and indirect titin binding partners and non-titin binding partners to validate in protein level with western blotting at three ages (Fig. 5J). We found that proteins Fhl1, Darp, Crp2 and Tgfβ2 were consistent with DNA microarray and QPCR data. Protein Igfbp8 was slightly reduced in d1 Hm which was consistent with DNA microarray data and significantly increased at d20 and 49 that was consistent with both DNA microarray and QPCR data. Protein level of Kcnq1 was not changed at three ages which was consistent with DNA microarray and QPCR data at d1, but not d20 and 49 (Fig. 6J). These data suggest that most of genes validated by QPCR and western blotting have consistency between mRNA level and protein level, and they are consistent with DNA microarray data too, confirming the gene expression changes obtained from DNA microarray data are reliable.
Validation of differentially expressed genes with QPCR and western blot. (A, B and C), Expression level of selected titin direct binding partners with DNA microarray and QPCR; (D, E and F), Expression level of selected titin indirect binding partners with DNA microarray and QPCR; (G, H and I), Expression level of selected genes that are not titin binding partners with DNA microarray and QPCR; (J), Validation of selected proteins from titin direct and indirect partners and non-titin binding partners with western blot. D, day; Wt, wild type; Hm, homozygous Rbm20 knockout; GAPDH, protein loading control. Mean±SEM (n=3); *P<0.05; **P<0.01; ***P<0.001; #P<0.05; ##P<0.01; ###P<0.001.
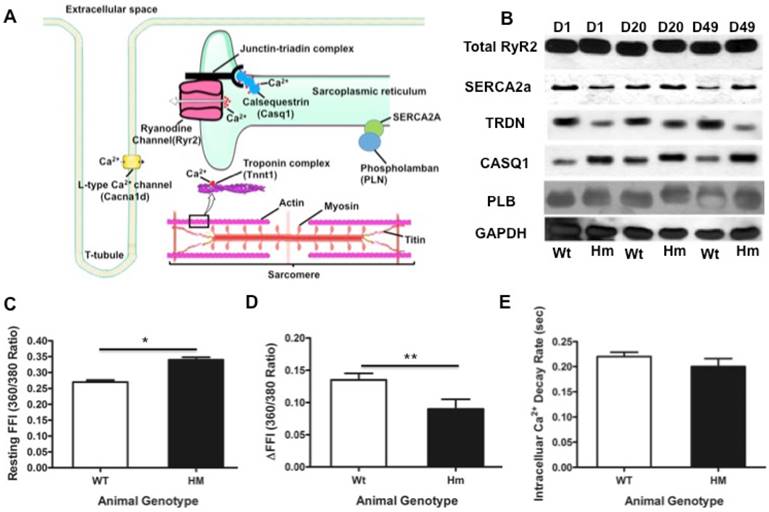
Transcriptional network of calcium handling genes and intracellular calcium changes. (A), Schematic diagram of calcium release signaling and sarcomere; (B), validation of key calcium handling proteins with western blotting; (C), Intracellular calcium concentration changes with cardiomyocytes isolated from the rat heart at d49. Resting FFI, basal intracellular calcium level; (D), ΔFFI, Rise in intracellular calcium in response to electrical stimulus shown as changes in Fura-2 fluorescence intensity; (E), D, day; Wt, wild type; Hm, Rbm20 homozygous knockout; GAPDH, protein loading control. Mean ± SEM (n=30), *P < 0.05, **P<0.01.
RBM20 regulates a transcriptional network of calcium handling genes in the heart and alters intracellular calcium levels during contraction
We next studied and validated genes involving in calcium release signaling. From our DNA microarray data, we found that genes Trdn, Casq1, Cacna1c, Tnnt1, MyHC involved in calcium handling and sarcomere contractility were differentially expressed. These genes were displayed in schematic diagram of calcium handling and sarcomeric structure (Fig. 6A). We validated calcium handling genes Trdn and Casq1 with fold changes, while other genes (RyR2, Serca2a and phospholamban/PLN) that have no fold changes in calcium handling from DNA microarray. Western blotting analysis demonstrated that Trdn was down-regulated and casq1 was up-regulated in Hm at d1, d20 and d49, while other genes (RyR2, Serca2a and PLB) had no changes (Fig. 6B). These results are consistent with the DNA microarray data. Interestingly, western blot indicated that Trdn and casq1 protein bands had migration difference between Wt and Hm. This could be caused by alternative splicing that is regulated by RBM20 as reported previously [4]. Given that expression profile changes of some calcium handling genes, we assumed that intracellular calcium concentration may be subjected to changes during muscle contraction. Thus, we measured the calcium transient with cardiomyocytes isolated from the heart at d49 using a calcium contractility system (IonOptix). The results indicated that basal level of the intracellular calcium concentration was significant higher in Hm cardiomyocytes than that in Wt (Fig. 6C), while, in response to electrical stimulus, intracellular calcium change was lower in Hm cardiomyocytes than that in Wt (Fig. 6D). However, intracellular calcium decay rate had no significant difference between Wt and Hm cardiomyocytes (Fig. 6E). These data suggest that calcium releasing signaling could be impaired, leading to calcium leaking, while calcium ATPase pump system was physiologically functional and could normally pump calcium from cytosol to sarcoplasmic reticulum (SR) for calcium uptake. Calcium releasing from SR is through a multi-molecular signaling complex channel includeing RyR, Trdn, Junctin and Casq that plays a key role in excitation-contraction coupling (EC-coupling). The calcium enters the cytosol through plasmalemmal voltage-gated calcium channels and activates RyR channel to release more calcium from SR for muscle contraction [66-68]. However, the RyR channel is also positively controlled by the luminal side of the channel [66, 69-70]. Auxiliary proteins (Trdn, Junctin and Casq) with luminal localization mediate the effect of luminal calcium on RyR [71-72]. Casq is a major calcium binding protein and changes conformation upon calcium binding, resulting in interaction with RyR to sense luminal calcium [73-76]. Trdn and Junctin mediate the inhibitory interactions between Casq and the RyR channel [73]. Therefore, down-regulated Trdn and up-regulated Casq1 may cooperate for calcium releasing. Since RyR2 is not changed based on our data, it still remains unknown how calcium leaking occurs in Rbm20 Hm rats. Previous study indicated that RBM20 regulates alternative splicing of RyR2, leading to a 24bp insertion in Rbm20 Hm rats [14], which could be a cause for calcium leaking. However, further study is needed to confirm this hypothesis. Nevertheless, expression changes of Trdn and Casq1 may play a role in the impaired calcium release signaling in Rbm20 Hm rats. On the other hand, SR calcium uptake is mediated by a calcium ATPase Serca2, whose activity is reversibly regulated by PLN [77]. Since no changes of these two proteins were found in Rbm20 Hm rats, calcium uptake appears normal, which was consistent with the measurement of intracellular calcium decay rate (Fig. 6E).
Conclusion
Our results reveal that the muscle tissue specific splicing factor RBM20 not only regulates gene alternative splicing but also gene expression in the heart. Previous studies have identified approximately thirty genes that are undergoing alternative splicing regulated by RBM20. In this study, we have demonstrated that RBM20 also modulates gene expression of about four hundred genes with heart maturation. Intriguingly, we found that for certain genes such as Trdn and MyHC, RBM20 regulates their splicing as well as their expression level. Rbm20 deficiency in animal models leads to dilated cardiomyopathy and heart failure, and mutations found in RBM20 are also associated with dilated cardiomyopathy in human patients. However, the detailed mechanisms of how RBM20 causes heart dysfunction remain unclear. Our data suggest that the mechanisms of both gene splicing and gene expression regulated by RBM20 will require more detailed study in the future. Particularly, whether gene expression is directly regulated by RBM20 or is a secondary effect remains unknown. At least, previous studies have shown that pre-mRNA splicing can be coupled to transcription and crosstalk and impact each other [78-79], and trans- and cis-acting factors can regulate RNA polymerase II elongation [80, 81]. However, whether RBM20-mediated transcription is involved in coupling to RBM20-mediated splicing is very interesting to be further studied. In conclusion, our results provide new insights into the mechanism of RBM20-induced heart disease.
Abbreviation
RBM20: RNA binding motif 20; RRM: RNA-recognition domain; SR: serine /arginine; E16: 16 days fetuses; SDS-VAGE: sodium dodecyl sulfate agarose gel electrophoresis; Wt: wild type; Hm: homozygous knockout; RMA: robust multi-array analysis; LV: left ventricule; FFI: Fura-2 fluorescence intensity; d: day; yr: year; GO: Gene Ontology; GSEA: Gene Set Enrichment Analysis; Fhl1: four-and-a-half Lim domain protein-1; MARPs: muscle-ankyrin-repeat proteins; SR: sarcoplasmic reticulum; EC-coupling: excitation-contraction coupling; sANK1: small ankyrin 1 isoform; p94: calpain-3; MyBPC: myosin binding protein C; Nbr1: neighbor-of-BRCA1-gene-1; MURF: muscle-specific-RING-finger-protein; MyHC: myosin-heavy-chain; MyLC: myosin-light-chain; TPM: tropomyosin; CARP: cardiac-ankyrin-repeat-protein; DARP: diabetes-related-ankyrin-repeat-protein; ARPP: ankyrin-repeat-domain-protein-2; TMOD: tropomodulin; MYOT: myotilin; FLNC: Υ-filamin; T-cap: telethonin; mink: potassium-channel subunit; MLP (CRP3): muscle-LIM-protein (cysteine-rich-protein-3); CRP2: muscle-LIM-protein (cysteine-rich-protein-2); ENAH: protein enabled homolog; ANP: atrial natriuretic peptide; BNP: brain natriuretic peptide; KCND2/3: potassium voltage-gated channel subfamily D member 2/3; KCNK2: potassium channel subfamily K member 2; KCNQ1: potassium voltage-gated channel subfamily Q member 1; TNFa-CE (ADAM-17): tumor-necrosisfactor-alpha-converting-enzyme (A-disintegrin-and metalloprotease-17 protein); ADAM33: disintegrin-and metalloprotease-33 protein; IGFBP: insulin growth factor binding protein; ZNF: zinc finger protein; ERK2: extracellular-signal-regulated-kinase-2; ANK2: ankyrin 2; YB-1: Y-box-binding-protein-1; ISOT-3 (USP13): isopeptidase-T-3; USP24: ubiquitin specific peptidase 24; UBC9: SUMO-conjugating enzyme UBC9; UBCH10: SUMO-conjugating enzyme E2C.
Supplementary Material
Supplementary tables.
Acknowledgements
This work was supported by the National Institute of Health/National Institute of General Medical Sciences [NIGMSP20GM103432]; the BGIA from the American Heart Association [16BGIA27790136 to WG]; the USDA National Institute of Food and Agriculture [Hatch project 1009266 to WG] and the National Institutes of Health [grant HL77196 to MLG]. The authors are grateful to Mark D. Hanna for his technical assistance, animal care and laboratory management.
Author contribution
W.G., and M.G., conceived and designed the experiments; Z.C., Y.Z., W.Q., S.M., C.H. and W.G. performed the experiments and interpreted the data; W.G. wrote the paper; M.G., revised the paper.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Zhu C, Chen Z, Guo W. Pre-mRNA mis-splicing of sarcomeric genes in heart failure. Bba-Biomembranes. 2016;16:30290-30293 doi:10.1016/j.bbadis.2016.11.008
2. Chen M, Manley JL. Mechanisms of alternative splicing regulation: insights from molecular and genomics approaches. Nat Rev Mol Cell Biol. 2009;10:741-754 doi:10.1038/nrm2777
3. Black DL. Mechanisms of alternative pre-messenger RNA splicing. Annu Rev Biochem. 2003;72:291-336 doi:10.1146/annurev.biochem.72.121801.161720
4. Guo W, Schafer S, Greaser ML. et al. RBM20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nat Med. 2012;18:766-773 doi:10.1038/nm.2693
5. Brauch KM, Karst ML, Herron KJ, de Andrade M, Pellikka PA, Rodeheffer RJ, Michels VV, Olson TM. Mutations in ribonucleic acid binding protein gene cause familial dilated cardiomyopathy. Annu Rev Biochem. 2009;54:930-941 doi:10.1016/j.jacc.2009.05.038
6. Lin S, Fu XD. SR proteins and related factors in alternative splicing. Adv Exp Med Biol. 2007;623:107-122
7. Zhong XY, Ding JH, Adams JA, Ghosh G, Fu XD. Regulation of SR protein phosphorylation and alternative splicing by modulating kinetic interactions of SRPK1 with molecular chaperones. Genes Dev. 2009;23:482-495 doi:10.1101/gad.1752109
8. Jefferies JL, Towbin JA. Dilated cardiomyopathy. Lancet. 2010;375:752-762 doi:10.1016/S0140-6736(09)62023-7
9. Li D, Morales A, Gonzalez Quintana J, Norton N, Siegfried JD, Hofmeyer M, Hershberger RE. Identification of novel mutations in RBM20 in patients with dilated cardiomyopathy. Cts-Clin Transl Sci. 2010;3:90-97 doi:10.1111/j.1752-8062.2010.00198.x
10. Millat G, Bouvagnet P, Chevalier P. et al. Clinical and mutational spectrum in a cohort of 105 unrelated patients with dilated cardiomyopathy. Eur J Med Genet. 2011;54:e570-e575 doi:10.1016/j.ejmg.2011.07.005
11. Rampersaud E, Siegfried JD, Norton N, Li D, Martin E, Hershberger RE. Rare variant mutations identified in pediatric patients with dilated cardiomyopathy. Prog Pediatr Cardiol. 2011;31:39-47 doi:10.1016/j.ppedcard.2010.11.008
12. Refaat MM, Lubitz SA, Makino S. et al. Genetic variation in the alternative splicing regulator RBM20 is associated with dilated cardiomyopathy. Heart Rhythm. 2012;9:390-396 doi:10.1016/j.hrthm.2011.10.016
13. Wells QS, Becker JR, Su YR. et al. Whole exome sequencing identifies a causal RBM20 mutation in a large pedigree with familial dilated cardiomyopathy. Circulation. 2013;6:317-326 doi:10.1161/CIRCGENETICS.113.000011
14. Maatz H, Jens M, Liss M. et al. RNA-binding protein RBM20 represses splicing to orchestrate cardiac pre-mRNA processing. The J Clin Invest. 2014;124:3419-3430 doi:10.1172/JCI74523
15. Granzier HL, Irving TC. Passive tension in cardiac muscle: contribution of collagen, titin, microtubules, and intermediate filaments. Biophys J. 1995;68:1027-1044 doi:10.1016/S0006-3495(95)80278-X
16. Granzier HL, Labeit S. The giant protein titin. Circ Res. 2004;94:284-295 doi:10.1161/01.RES.0000117769.88862.F8
17. LeWinter MM, Granzier HL. Cardiac titin. Circulation. 2010;121:2137-2145 doi:10.1161/circulationaha.109.860171
18. LeWinter MM, Granzier HL. Cardiac titin and heart disease. J Cardiovasc Pharm. 2014;63:207-212 doi:10.1097/FJC.0000000000000007
19. Guo W, Sun M. RBM20, a potential target for treatment of cardiomyopathy via titin isoform switching. Biophysical Reviews. 2018;10:15-25 doi:10.1007/s12551 -017-0267-5
20. Bang ML, Centner T, Fornoff F. et al. The complete gene sequence of titin, expression of an unusual≈ 700-kDa titin isoform, and its interaction with obscurin identify a novel Z-line to I-band linking system. Circ Res. 2001;89:1065-1072 doi:10.1161/hh2301.100981
21. Labeit S, Barlow DP, Gautel M. et al. A regular pattern of two types of 100-residue motif in the sequence of titin. Nature. 1990;345:273-276 doi:10.1038/345273a0
22. Krüger M, Linke WA. The giant protein titin: a regulatory node that integrates myocyte signaling pathways. J Biol Chem. 2011;286:9905-9912 doi:10.1074/jbc.R110.173260
23. Linke WA. Sense and stretchability: the role of titin and titin-associated proteins in myocardial stress-sensing and mechanical dysfunction. Cardiovasc Res. 2008;77:637-648 doi:10.1016/j.cardiores.2007.03.029
24. Freiburg A, Trombitas K, Hell W. et al. Series of exon-skipping events in the elastic spring region of titin as the structural basis for myofibrillar elastic diversity. Circ Res. 2000;86:1114-1121 doi:10.1161/01.RES.86.11.1114
25. Taylor M, Graw S, Sinagra G, Barnes C, Slavov D, Brun F, Pinamonti B, Salcedo EE, Sauer W, Pyxaras S. Genetic variation in titin in arrhythmogenic right ventricular cardiomyopathy-overlap syndromes. Circulation. 2011;124:876-885 doi:10.1161/circulationaha.110.005405
26. Borbély A, Falcao-Pires I, Van Heerebeek L. et al. Hypophosphorylation of the Stiff N2B titin isoform raises cardiomyocyte resting tension in failing human myocardium. Circ Res. 2009;104:780-786 doi:10.1161/ circresaha.108.193326
27. Borbély A, Van der Velden J, Papp Z, Bronzwaer JG, Edes I, Stienen GJ, Paulus WJ. Cardiomyocyte stiffness in diastolic heart failure. Circulation. 2005;111:774-781 doi:10.1161/01.CIR.0000155257.33485.6D
28. Katz AM, Zile MR. New molecular mechanism in diastolic heart failure. Circ Res. 2006;113:1922-1925 doi:10.1161/circulationaha.106.620765
29. Makarenko I, Opitz CA, Leake MC, Neagoe C, Kulke M, Gwathmey JK, Del MF, Hajjar RJ, Linke WA. Passive stiffness changes caused by upregulation of compliant titin isoforms in human dilated cardiomyopathy hearts. Circ Res. 2004;95:708-716 doi:10.1161/01.RES.0000143901.37063.2f
30. Neagoe C, Kulke M, Del Monte F, Gwathmey JK, de Tombe PP, Hajjar RJ, Linke WA. Titin isoform switch in ischemic human heart disease. Circulation. 2002;106:1333-1341 doi:10.1161/01.CIR.0000029803.93022.93
31. Van Heerebeek L, Borbély A, Niessen HW, Bronzwaer JG, Van der Velden J, Stienen GJ, Linke WA, Laarman GJ, Paulus WJ. Myocardial structure and function differ in systolic and diastolic heart failure. Circulation. 2006;113:1966-1973 doi:10.1161/circulationaha.105.587519
32. Nagueh SF, Shah G, Wu Y, Torre-Amione G, King NM, Lahmers S, Witt CC, Becker K, Labeit S, Granzier HL. Altered titin expression, myocardial stiffness, and left ventricular function in patients with dilated cardiomyopathy. Circulation. 2004;110:155-162 doi:110.1161/01.CIR.0000135591.37759.AF
33. Warren CM, Krzesinski PR, Greaser ML. Vertical agarose gel electrophoresis and electroblotting of high-molecular-weight proteins. Electrophoresis. 2003;24:1695-1702 doi:10.1002/elps.200305392
34. Guo W, Pleitner JM, Saupe KW, Greaser ML. Pathophysiological defects and transcriptional profiling in the RBM20-/-rat model. PLoS One. 2013;8:e84281. doi:10.1371/journal.pone.0084281
35. Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31:e15. doi:10.1093/nar/gng015
36. Dennis G, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 2003;4:P3. doi:10.1186/gb-2003-4-9-r60
37. Doser TA, Turdi S, Thomas DP, Epstein PN, Li S, Ren J. Transgenic overexpression of aldehyde dehydrogenase-2 rescues chronic alcohol intake-induced myocardial hypertrophy and contractile dysfunction. Circulation. 2009;119:1941-1949 doi:10.1161/circulationaha.108.823799
38. Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi:10.1093/nar/29.9.e45
39. Spector DL, Lamond AI. Nuclear Speckles. Cold Spring Harbor Perspectives in Biol. 2011;3:a000646. doi:10.1101/cshperspect.a000646
40. Xie SQ, Martin S, Guillot PV, Bentley DL, Pombo A. Splicing Speckles Are Not Reservoirs of RNA Polymerase II, but Contain an Inactive Form, Phosphorylated on Serine2 Residues of the C-Terminal Domain. Mol Biol Cell. 2006;17:1723-1733 doi:10.1091/mbc.E05-08-0726
41. Subramanian A, Tamayo P, Mootha VK. et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545-15550 doi:10.1073/pnas.0506580102
42. Carrier L, Bonne G, Bahrend E. et al. Organization and sequence of human cardiac myosin binding protein C gene (MYBPC3) and identification of mutations predicted to produce truncated proteins in familial hypertrophic cardiomyopathy. Circ Res. 1997;80:427-434 doi:10.1161/01.res.0000435859.24609.b3
43. Dhoot GK, Hales MC, Grail BM, Perry SV. The isoforms of C protein and their distribution in mammalian skeletal muscle. J Muscle Res Cell M. 1985;6:487-505 doi:10.1007/BF00712585
44. Einheber S, Fischman DA. Isolation and characterization of a cDNA clone encoding avian skeletal muscle C-protein: an intracellular member of the immunoglobulin superfamily. Proc Natl Acad Sci U S A. 1990;87:2157-2161 doi:10.1073/pnas.87.6.2157
45. Weber Fe, Vaughan Kt, Reinach Fc, Fischman Da. Complete sequence of human fast-type and slow-type muscle myosin-binding-protein C (MyBP-C). Febs Lett. 1993;216:661-669 doi:10.1111/j.14321033.1993.tb18186.x
46. Lin B, Govindan S, Lee K, Zhao P, Han R, Runte KE, Craig R, Palmer BM, Sadayappan S. Cardiac myosin binding protein-C plays no regulatory role in skeletal muscle structure and function. PLoS One. 2013;8:e69671. doi:10.1371/journal.pone.0069671
47. Yamauchi-Takihara K, Sole MJ, Liew J, Ing D, Liew C. Characterization of human cardiac myosin heavy chain genes. P Natl A Sci U S A. 1989;86:3504-3508 doi:10.1073/pnas.86.10.3504
48. Bouvagnet P, Mairhofer H, Leger J, Puech P, Leger JJ. Distribution pattern of α and β myosin in normal and diseased human ventricular myocardium. Basic Res Cardiol. 1989;84:91-102 doi:10.1007/BF01907006
49. Miyata S, Minobe W, Bristow MR, Leinwand LA. Myosin heavy chain isoform expression in the failing and nonfailing human heart. Circ Res. 2000;86:386-390 doi:10.1161/01.RES.86.4.386
50. Lompre A, Schwartz K, D'Albis A, Lacombe G, Van Thiem N, Swynghedauw B. Myosin isoenzyme redistribution in chronic heart overload. Nature. 1979;282:105-107 doi:10.1038/282105a0
51. Yin Z, Ren J, Guo W. Sarcomeric protein isoform transitions in cardiac muscle: a journey to heart failure. Bba-Biomembranes. 2015;1852:47-52 doi:10.1016/j.bbadis.2014.11.003
52. Lee SM, Tsui SK, Chan KK, Garcia-Barcelo M, Waye MM, Fung KP, Liew CC, Lee CY. Chromosomal mapping, tissue distribution and cDNA sequence of four-and-a-half LIM domain protein 1 (FHL1). Gene. 1998;216:163-170 doi:10.1016/S0378-1119(98)00302-3
53. LeWinter MM, Wu Y, Labeit S, Granzier H. Cardiac titin: structure, functions and role in disease. Clin Chim Acta. 2007;375:1-9 doi:10.1016/j.cca.2006.06.035
54. Sheikh F, Raskin A, Chu PH. et al. An FHL1-containing complex within the cardiomyocyte sarcomere mediates hypertrophic biomechanical stress responses in mice. J Clin Invest. 2008;118:3870-3880 doi:10.1172/JCI34472
55. Raskin A, Lange S, Banares K. et al. A novel mechanism involving four-and-a-half LIM domain protein-1 and extracellular signal-regulated kinase-2 regulates titin phosphorylation and mechanics. J Biol Chem. 2012;287:29273-29284 doi:10.1074/jbc.M112.37283
56. Miller MK, Bang M, Witt CC, Labeit D, Trombitas C, Watanabe K, Granzier H, McElhinny AS, Gregorio CC, Labeit S. The muscle ankyrin repeat proteins: CARP, ankrd2/Arpp and DARP as a family of titin filament-based stress response molecules. J Mol Biol. 2003;333:951-964 doi:10.1016/j.jmb.2003.09.012
57. Witt CC, Ono Y, Puschmann E. et al. Induction and myofibrillar targeting of CARP, and suppression of the Nkx2.5 pathway in the MDM mouse with impaired titin-based signaling. J Mol Biol. 2004;336:145-154 doi:10.1016/j.jmb.2003.12.021
58. Jespersen T, Grunnet M, Olesen S. The KCNQ1 potassium channel: from gene to physiological function. Physiology. 2005;20:408-416 doi:10.1152/physiol.00031.2005
59. Oudit GY, Kassiri Z, Sah R, Ramirez RJ, Zobel C, Backx PH. The molecular physiology of the cardiac transient outward potassium current (I to) in normal and diseased myocardium. J Cell Mol Med. 2001;33:851-872 doi:10.1006/jmcc.2001.1376
60. Postma AV, Bezzina CR, De Vries JF, Wilde AA, Moorman AF, Mannens MM. Genomic organisation and chromosomal localisation of two members of the KCND ion channel family, KCND2 and KCND3. Hum Mol Genet. 2000;106:614-619 doi:10.1007/s0043900003
61. Schmidt C, Wiedmann F, Kallenberger SM. et al. Stretch-activated two-pore-domain (K2P) potassium channels in the heart: Focus on atrial fibrillation and heart failure. Prog Biophys Mol Bio. 2017;130:233-243 doi:10.1016/j.pbiomolbio.2017.05.004
62. Braunwald E. Biomarkers in heart failure. New Engl J Med. 2008;358:2148-2159 doi:10.1056/NEJMra0800239
63. Nagaya N, Nishikimi T, Okano Y. et al. Plasma brain natriuretic peptide levels increase in proportion to the extent of right ventricular dysfunction in pulmonary hypertension. J Am Coll Cardio. 1998;31:202-208 doi:10.1016/S0735-1097(97)00452-X
64. Morano I. Tuning the human heart molecular motors by myosin light chains. J Mol Med. 1999;77:544-555 doi:10.1007/s001099900031
65. Ritter O, Luther HP, Haase H, Baltas LG, Baumann G, Schulte HD, Morano I. Expression of atrial myosin light chains but not a-myosin heavy chains is correlated in vivo with increased ventricular function in patients with hypertrophic obstructive cardiomyopathy. J Mol Med. 1999;77:677-685 doi:10.1007/s0010999000
66. Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198-205 doi:10.1161/hh2301.100981
67. Li P, Chen SW. Molecular basis of Ca 2+ activation of the mouse cardiac Ca2+ release channel (ryanodine receptor). J Gen Physiol. 2001;118:33-44 doi:10.1085/jgp.118.1.33
68. Meissner G, Rios E, Tripathy A, Pasek DA. Regulation of skeletal muscle Ca2+ release channel (ryanodine receptor) by Ca2+ and monovalent cations and anions. J Biol Chem. 1997;272:1628-1638 doi:10.1074/jbc.272.3.1628
69. Ching LL, Williams AJ, Sitsapesan R. Evidence for Ca2+ activation and inactivation sites on the luminal side of the cardiac ryanodine receptor complex. Circ Res. 2000;87:201-206 doi:10.1161/01.RES.87.3.201
70. Györke I, Györke S. Regulation of the cardiac ryanodine receptor channel by luminal Ca 2+ involves luminal Ca 2+ sensing sites. Biophys J. 1998;75:2801-2810 doi:10.1016/S0006-3495(98)77723-9
71. Shin DW, Ma J, Kim DH. The asp-rich region at the carboxyl-terminus of calsequestrin binds to Ca2+ and interacts with triadin. Febs Lett. 2000;486:178-182 doi:10.1016/S0014-5793(00)02246-8
72. Zhang L, Kelley J, Schmeisser G, Kobayashi YM, Jones LR. Complex formation between junctin, triadin, calsequestrin, and the ryanodine receptor proteins of the cardiac junctional sarcoplasmic reticulum membrane. J Biol Chem. 1997;272:23389-23397 doi:10.1074/jbc.272.37.23389
73. Györke I, Hester N, Jones LR, Györke S. The role of calsequestrin, triadin, and junctin in conferring cardiac ryanodine receptor responsiveness to luminal calcium. Biophys J. 2004;86:2121-2128 doi:10.1016/S0006-3495(04)74271-X
74. Herzog A, Szegedi C, Jona I, Herberg FW, Varsanyi M. Surface plasmon resonance studies prove the interaction of skeletal muscle sarcoplasmic reticular Ca2+ release channel/ryanodine receptor with calsequestrin. Febs Lett. 2000;472:73-77 doi:10.1016/S0014-5793(00)01431-9
75. Jones LR, Suzuki YJ, Wang W, Kobayashi YM, Ramesh V, Franzini-Armstrong C, Cleemann L, Morad M. Regulation of Ca2+ signaling in transgenic mouse cardiac myocytes overexpressing calsequestrin. J Clin Invest. 1998;101:1385. doi:10.1172/JCI1362
76. Mitchell RD, Simmerman HK, Jones LR. Ca2+ binding effects on protein conformation and protein interactions of canine cardiac calsequestrin. J Biol Chem. 1988;263:1376-1381
77. Kranias EG, Hajjar RJ. Modulation of cardiac contractility by the phopholamban/SERCA2a regulatome. Circ Res. 2012;110:1646-1660 doi:10.1161/Circresaha.111.259754
78. Kornblihtt AR, Schor IE, Allo M, Dujardin G, Petrillo E, Munoz MJ. Alternative splicing: a pivotal step between eukaryotic transcription and translation. Nat Rev Mol Cell Biol. 2013;14:153-165 doi: 10.1038/nrm3525
79. Lazarev D, Manley JL. Concurrent splicing and transcription are not sufficient to enhance splicing efficiency. RNA. 2007;13:1546-1557 doi:10.1261/rna.595907
80. Nojima T, Gomes T, Grosso ARF, Kimura H, Dye MJ, Dhir S, Carmo-Fonseca M, Proudfoot NJ. Mammalian NET-Seq Reveals Genome-wide Nascent Transcription Coupled to RNA Processing. Cell. 2015;161:526-540 doi:10.1016/j.cell.2015.03.027
81. Shukla S, Oberdoerffer S. Co-transcriptional regulation of alternative pre-mRNA splicing. Biochimica et Biophysica Acta. 2012;1819:673-683 doi:10.1016/j.bbagrm.2012.01.014
Author contact
![]() Corresponding author: Dr. Wei Guo, Tel: +1 307-766-3429; Fax: +1 307-766-2355; Email: wguo3edu
Corresponding author: Dr. Wei Guo, Tel: +1 307-766-3429; Fax: +1 307-766-2355; Email: wguo3edu