Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results and Discussions
Conclusions
Acknowledgements
Supplementary Material
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(8):901-906. doi:10.7150/ijbs.24327 This issue Cite
Research Paper
A Sequential Segment Based Alpha-Helical Transmembrane Protein Alignment Method
Han Wang1,2, Jingru Wang1,2, Li Zhang3, Pingping Sun1,2, Ning Du4, Yanwen Li1,2, ![]()
1. School of Information Science and Technology, Northeast Normal University, Changchun, 130117, China.
2. Institute of Computational Biology, Northeast Normal University, Changchun, 130117, China.
3. School of Computer Science and Engineering, Changchun University of Technology, Changchun, China.
4. School of Mathematics and Statistics, Northeast Normal University, Changchun, 130024, China
Received 2017-12-12; Accepted 2018-2-2; Published 2018-5-22
Abstract

Alpha-helical transmembrane protein (αTMP) is one of the two major categories of transmembrane protein (TMP). They are abundant existing in eukaryotic cells and involved in many biological processes. The special physicochemical properties, the structures of αTMP are hard to be experimentally solved, but αTMP's sequential segments are important to determine their conformations, so that TM-specific alignment is necessary to benefit their structure prediction. We used segment information extracted from topology structure and evolutionary information as features to implement a αTMP Segment Alignment method (TMSA). The method was trained using one non-redundant dataset and tested using another non-redundant dataset. Comparing the results to a general alignment method HHalign, TMSA achieved higher alignment accuracy, and easier to recognize the fold of αTMPs.
Keywords: Transmembrane Protein, Segment Alignment, Topology
Introduction
Alpha-helical transmembrane proteins (αTMPs) play vital roles in many biological processes as transporters or receptors, they respond for transporting materials and signals between both sides of biological membrane. Therefore, they are major drug target currently[1], and related with many serious diseases[2], or exist as key nodes in their pathways. For the reason, αTMPs become one of prime factor in disease treatments, they are the targets of most drugs currently on market[3].
Further understanding of αTMPs structures is necessary to study their functions and biological mechanisms for drug design. But high-resolution αTMP structures are hard to derive, the amount of known αTMP structures in Protein Data Bank (PDB) are not more than 2%[4]. Facing the rapidly increasing sequences, alignment-based structure prediction methods are widely used to reduce the gap of amounts between the structures and sequences[5]. However, the methods applied to globular proteins perform poorly on αTMPs due to their special physicochemical properties. It was reported that the structure prediction is estimated to obtain the accuracy as high as that of globular proteins if the αTMP alignment achieves the accuracy as it counterpart[6]. Due to the absence of such methods, a high accuracy αTMP alignment is urgently needed.
Traditional sequence-to-sequence alignment methods, such as BLAST[7], cannot satisfy the high accuracy required, so that the sequence-to-structure alignment methods[8,9], also called threading, are widely used to model the protein structures. These state-of-the-art methods abstract many major structure-based features into the profile-to-profile alignments[10,11] to improve the accuracy, including the evolution information, solvent accessibility and the secondary structure. For the purpose to improve the alignment accuracy for αTMP, these features must be taken reconsiderations, and more αTMP -specific features are necessarily involved.
The αTMPs are obviously different from the globular proteins in their conformation characteristics, they have one or more helices forming helical bundles to cross biological membrane. These transmembrane helixes (TMHs) are more hydrophobic than those helixes in globular proteins. Notably, αTMPs in the same fold always have similar TMH numbers and the conformations. Here, TMH is the TM-segment on the protein sequence, the rest parts, namely, non-TM segments of sequence are divided to inside segment (existing in cytoplasmic) and outside segment (existing in extracellular) according to their locations relative to the biological membrane. Thus, the segments are the specific pattern of αTMPs, which alternatively locate on the sequence regularly.
Topology prediction can be used to identify those segments for a αTMP using its sequence as input. Various methods have been developed for the purpose, where, Hidden Markov Model (HMM) based methods achieved big success[12,13,14,15], while some other machine learning methods also showed the good performance[16,17,18,19,20]. To date, αTMP topology prediction methods achieve high accuracy, which makes it possible applying segment types to the alignment as a TM-specific feature.
In this study, we firstly introduce the segment alignment to αTMPs and implement the method αTMP Segment Alignment (TMSA). Differing to the methods for globular proteins, our method employs the topology structure instead of secondary structure, so that the segments alignment can be applied. Since the solvent accessibility feature is used by topology prediction, and the alignment profile is composed of only two features: the evolution information and the segment information. Correspondingly, the scoring function is tailored for the segment alignment, in which an additional segment broken penalty is added to guarantee the completeness of the segments. Our method was compared to a general top-leading alignment method HHalign against a non-redundant dataset and derived higher alignment accuracy.
Materials and Methods
Training and Testing datasets
To test the method, we use a complete αTMP dataset from Protein Data Bank of Transmembrane Proteins (PDBTM)[21], where totally 1366 αTMP are embodied. The bitopic αTMPs or those αTMPs shorter than 50 amino acids were removed, the left entries were clustered using BlastClust with identity less than 30 mutually. The top two non-redundant clusters were selected as training dataset and testing dataset (see in Supplementary Table S1), respectively compose of 58 and 72 non-redundant entries, these two sets have no overlaps.
Selected Features
The selected features are utilized by the scoring function to determine the compatibility between any two residues which come respectively from target and template. Of course, the more features compatible, the more likely they are aligned. As mentioned, segment and evolution information are selected, the two features are compact and complementary with each other. The details about them are described as following.
Segment Information: The segment information are derived by MEMSAT-SVM[18], one of the best topology predictors using a support vector machine (SVM) which has been widely used in bioinformatics[22,23,24,25] to identify segments on the input sequence. MEMSAT-SVM is a stand-alone tool to predict αTMP's topology structure, a protein sequence is required as input, and an isometric sequence is output as predicted topology structure, in which only three characters are used to label the topology structure type for each sequential position corresponding the input protein sequence, e.g. 'M' is TM-segment residue, 'i' is inside segment, and 'o' is outside segment. Thus, each residue i on the sequence is assigned to an integer value which represents the segment type:
(1)
Evlution Information: Position Specific Scoring Matrix (PSSM) profile generated by PSI-BLAST[7] derives the evolutionary conservation of sequence positions based on large-scale sequence alignment, which has had a significant impact on protein fold recognition[26]. The evolution information is useful to the alignment both for TM and non-TM segments. A PSSM profile pm[i, j] is a n×20 matrix, where the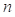 represents the sequence length. Each element in pm[i, j] negatively represents the frequency of the residue type j at position i.
represents the sequence length. Each element in pm[i, j] negatively represents the frequency of the residue type j at position i.
TMSA Method
The dynamic programming (DP) is the most popular paradigm in computational biology[27], also the heart of many well-known programs is DP[28]. As known, the scoring function is the kernel of the dynamic programming, which drives the DP forwarding the global optimal result. In this study, the scoring function is tailored for the αTMP segment alignment, especially the scoring for gap penalty. As the result, the optimal path of the DP will be slightly different with that of other general methods.
1) Fitness Scoring: As a part of scoring function, the fitness score 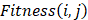 evaluates the compatibility between target sequence position
evaluates the compatibility between target sequence position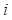 and template sequence position j, is given by the equation,
and template sequence position j, is given by the equation,
(2)
where the SEG(i, j) is the segment fitness score of the two positions, and the EV(i, j) is their fitness score of evolution conservation, w1 and w2 are the weights of two fitness scores, while wshift is a to-be-determinate constant that avoids the unrelated residues aligned[29]. The segment fitness score is defined below:
(3)
The evolution fitness score is calculated according to the equation
(4)
where pmtarget[i, k] is PSSM value of residue type k at position i on the target sequence, as well pmtemplate[i, k] follows the similar meaning.
2) Gap Penalty Scoring: A segment-dependent gap penalty is employed. Satisfying the segment alignment, the TM segments and non-TM segments are respectively denoted the different open gap penalties optm, opnon-tm and extended gap penalties eptm, epnon-tm, so that the TM segments are harder to be broken during the aligning.
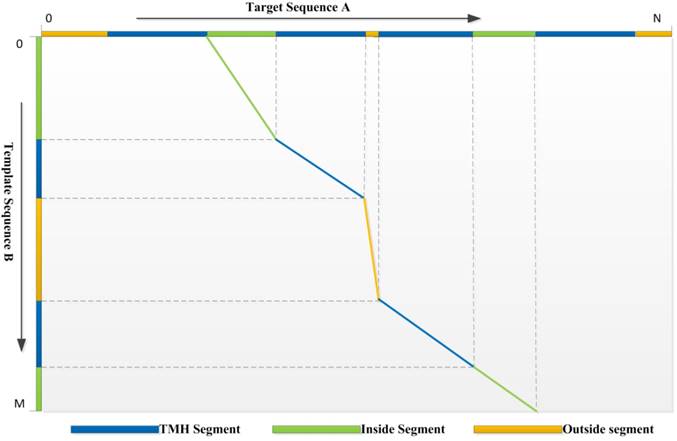
3) Dynamic Programming: We use a local-global algorithm to optimize the alignment path for the requirement of segment alignment. With the scoring function above, the DP procedure of TMSA can find the reasonable alignment path. The segments with the same type are aligned preferentially, while different segment types are hard to match unless they are extremely compatible with the evolutionary conservation. Assuming a target protein A has a topology structure shown as the top in the Fig. 1, while the topology of template protein B shows in the left, it is hard to align correctly without the guiding of segment types, because the properties of TM segments are very similar, such as sequence patterns and solvent accessibilities. But our DP algorithm conquers the problem which can be found by the alignment path shown in the figure. The second TMH of target, but not the first one, aligns to the first TMH of template due the mismatch of their previous non-TM segments. Therefore, our DP algorithm is designed for the αTMP segment alignment.
The optimized alignment path derived by the dynamic programming algorithm. The aligned segments are marked using the polylines with the corresponding colors.
4) Parameterizations: All the parameters used in the scoring function (w1, w2, wshift, optm, opnon-tm, eptm, epnon-tm,) are trained using the same method as on our non-redundant training dataset[30], but the parameters are optimized according to the best TMscore[31].
Results and Discussions
We pairwise aligned all the protein pairs in the testing dataset, by which the performance of the TMSA is evaluated, due to the absence of αTMP alignment methods, the results are compared to a general alignment method HHalign. And then the correlation between the output rawscore of TMSA and the protein structures will be further discussed to show its ability for the final purpose.
Alignment Accuracy
The alignment accuracy can be evaluated by two different benchmarks, the accuracy rate and structure similarity, we adopted the both benchmarks to show the experimental results. The first one (ACC) uses the golden standard structure alignment to count the rate of correctly aligned residues, here TMalign[32] is employed as the golden standard. The second one simply calculates the structure similarity between the aligned pair, and various methods can be applied for the purpose. We use the TMscore due to its high performance and accuracy.
To present the performance of TMSA, the alignment accuracies of both TM segments and non-TM segments are calculated respectively, as well the overall performance is exhibited aside. As shown in Table 1, the comparison is made using the average alignment accuracy derived by all the testing pairs. The results show that, TMSA achieves higher accuracies than HHalign with all the three segment types, whatever which evaluation method is used, so the two criterions are consistent to present the alignment accuracy. It is almost 10 percent that TMSA over-performs the HHalign with TMscore, and the margin enlarges to 11 percent with ACC. In addition, TMSA achieves the even higher accuracy aligning TM segments than non-TM segments, while the gap shown in the results of HHalign is inconspicuous. The reason mainly because our method adopts the stricter strategies to guarantee the TM segments to be better aligned, while the non-TM segments are easier to be broken inserting gaps. Nevertheless, the alignment for non-TM segments is still more accurate of TMSA comparing to its counterpart.
Alignment accuracies comparing with HHalign
| Methods | ACC (%) | TMscore | ||||
|---|---|---|---|---|---|---|
| T | N | O | T | N | O | |
| TMSA | 64.5 | 59.6 | 62.1 | 0.463 | 0.414 | 0.436 |
| HHalign | 51.9 | 52.6 | 51.7 | 0.355 | 0.338 | 0.342 |
T: TM segments, N: Non-TN segments, O: Overall
Comparing the features used in the two alignment methods, the major difference is the TMSA employs the topology structures instead of secondary structure and solvent accessibility. For αTMPs, topology structure more clearly presents the overall structural properties, and indicates their conformations. Furthermore, the sequence pattern of αTMP is much especial and regular. Therefore, the segment alignment method can enormously improve the accuracy. With the increasing quantity of αTMPs, the topology prediction will be more accurate, which can further derive the segment alignment methods.
Alignment Scores and Structure Similarity
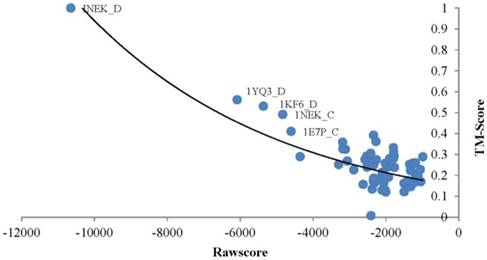
TMSA generates a rawscore for each alignment which negatively relates to the structure similarity of the target and template. The proteins obtained smaller rawscore are more possible to have similar conformations. As an example, the Fig. 2 points out the rawscores derived by chain D of query protein Succinate Dehydrogenase (PDB_ID: 1NEK:D)[33] aligned with all proteins in the testing dataset. The most left top point responses to the query protein aligned to itself, so that it obtained the best TMscore value of 1.0, which means the aligned proteins are completely matched with the tertiary structures. As well it obtained the smallest rawscore. The rest templates basically ordered regularly by their rawscores and TMscores, where the lower TMscore responds to bigger rawscore. However, the points distribute in the right-bottom of the diagram are too intensive to present such correlations, because the corresponding proteins are much different in conformation with the query protein. With this case, rawscores derived by TMSA are showed negatively correlated with the structure similarities, thus the protein fold can be recognized by ranking the rawscores of the templates.
Correlation between the rawscore and structure similarity of 1NEK_D.
Fold Recognition in Superfamily level
Since the superfamily indicates the closer structures than the fold, to better present the alignment accuracy, we adopted the classifications found in Orientations of Proteins in Membranes (OPM) database[34]. Because several αTMPs have not been classified to any superfamily, only a few super families exist in the testing set. The TMSA can recognize the entries which belong to the same superfamily by ranking the alignment rawscores. It is most important to select the high-quality templates for protein structure modeling, where those templates must have the similar structures to the target proteins, so that structure modeling step could compose the structure-unknown target protein by those structure-known template proteins.
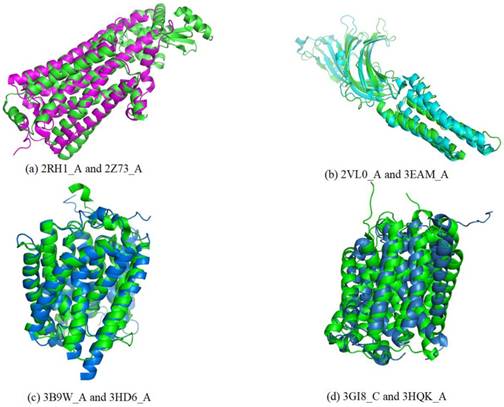
For a given target, the smaller rawscore indicates the responding template is more similar to the target in the structure. Therefore, the templates that have significant smaller rawscores than the average for a target could be the same fold. As shown in Figure 3, several entries were found in the same superfamily using the TMSA. The Figure 3 (a) shows the entries come from superfamily G-protein coupled receptors (ID: 1.1.01.02); the entries in Fig. 3 (b) belong to superfamily pentameric ligand-gated ion channels (ID: 1.1.26); the entries in Fig. 3 (c) and (d) are respectively from superfamily ammonia and urea transporters (ID: 1.1.19) and ligand/cation symporters (ID: 1.1.18). The superposed parts between the proteins are the aligned parts. It can be found that some non-TM segment are not matched in the four pairs, or even the TMHs, but they have overall similar conformations, which is the most important criteria to classify the TMPs.
Entries found in the same superfamily using the TMSA.
Conclusions
This article describes a novel sequence-to-structure alignment method tailoring for αTMP, TMSA, in which the segment alignment is firstly introduced to the study. To the end, the topology structures are employed in the method to describe the proteins' conformations instead of the secondary structures and solvent accessibilities that widely used for globular proteins alignment. Evolutionary information derived from sequence is used with the segment information to scoring the compatibility between two sequence positions, and a segment-dependent gap penalty has been applied in scoring function. We trained the method using a non-redundant dataset, and then pairwise aligned all pairs of protein in our testing dataset, which has no overlaps with training dataset. The testing results shows the method performs well for αTMP comparing to a top-leading alignment tool HHalign, and high structure similarity have been shown between the aligned pairs. The raw score generated by the method is proved negatively correlating to the structure similarity of the templates, which indicates that the method can be used in fold recognition of αTMP.
Acknowledgements
This work is supported by National Key R&D Program of China (No. 2017YFC0909200), and partially supported by National Natural Science Foundation of China (No. 41671379, 81502291, 81773171, 61502093). Jilin Scientific and Technological Development Program of China (No. 20180414006GH, 20180520028JH, 20170520051J, 20170520058JH).
Supplementary Material
Table S1.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Hang Y, Flynn AD. Drugging Membrane Protein Interactions. Annu Rev Biom Eng. 2016;18:51
2. Ng DP, Poulsen BE, Deber CM. Membrane protein misassembly in disease. Biochim Biophys Acta. 2012;1818:1115-22
3. Klabunde T, Hessler G. Drug design strategies for targeting G-protein-coupled receptors. Chembiochem. 2002;33:928-44
4. Berman HM, Bhat TN, Bourne PE. et al. The Protein Data Bank and the challenge of structural genomics. Nat Struct Biol. 2000;7(Suppl):957-9
5. Zou Q, Hu Q, Guo M. et al. HAlign: Fast multiple similar DNA/RNA sequence alignment based on the centre star strategy. Bioinformatics. 2015;31:2475-81
6. Forrest L R, Tang C L, Honig B. On the Accuracy of Homology Modeling and Sequence Alignment Methods Applied to Membrane Proteins. Biophysical J. 2006;91:508-17
7. Altschul SF, Madden TL, Schaffer AA. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389-402
8. Liu S, Zhang C, Liang S. et al. Fold recognition by concurrent use of solvent accessibility and residue depth. Proteins. 2007;68:636-45
9. Ellrott K, Guo JT, Olman V. et al. Improvement in protein sequence-structure alignment using insertion/deletion frequency arrays. Comput Syst Bioinformatics Conf. 2007;6:335-42
10. Mittelman D, Sadreyev R, Grishin N. Probabilistic scoring measures for profile-profile comparison yield more accurate short seed alignments. Bioinformatics. 2003;19:1531-9
11. Wang G Jr DR. Scoring profile-to-profile sequence alignments. Protein Sci. 2004;13:1612-26
12. Krogh A, Larsson B, Heijne G V. et al. Predicting transmembrane protein topology with a hidden markov model: application to complete genomes. J Mol Biol. 2001;305:567-80
13. Tusnady GE, Simon I. The HMMTOP transmembrane topology prediction server. Bioinformatics. 2001;17:849-50
14. Kahsay RY, Gao G, Liao L. An improved hidden Markov model for transmembrane protein detection and topology prediction and its applications to complete genomes. Bioinformatics. 2005;21:1853-8
15. Viklund H, Elofsson A. Best alpha-helical transmembrane protein topology predictions are achieved using hidden Markov models and evolutionary information. Protein Sci. 2004;13:1908-17
16. Shen H, Chou JJ. MemBrain: Improving the Accuracy of Predicting Transmembrane Helices. Plos One. 2008;3:e2399
17. Jones DT. Improving the accuracy of transmembrane protein topology prediction using evolutionary information. Bioinformatics. 2007;23:538-44
18. Nugent T, Jones DT. Transmembrane protein topology prediction using support vector machines. BMC Bioinformatics. 2009;10:159
19. Wang G, Zhao Y, Jiang Y. et al. BinMemPredict: a Web Server and Software for Predicting Membrane Protein Types. Curr Proteomics. 2013;10:2-9
20. Wan S, Duan Y, Zou Q. HPSLPred: An ensemble multi-label classifier for human protein subcellular location prediction with imbalanced source. Proteomics. 2017;17:17-8
21. Berman HM, Westbrook J, Feng Z. et al. The Protein Data Bank. Nucleic Acids Res. 2000;28:235-42
22. Tang Q, Nie F, Kang J. et al. NIEluter: Predicting peptides eluted from HLA class I molecules. J Immunol Methods. 2015;422:22-7
23. Chen XX, Tang H, Li WC. et al. Identification of Bacterial Cell Wall Lyases via Pseudo Amino Acid Composition. Biomed Res Int. 2016;2016:1654623
24. Yang H, Tang H, Chen XX. et al. Identification of Secretory Proteins in Mycobacterium tuberculosis Using Pseudo Amino Acid Composition. BioMed Res Int. 2016;2016:5413903
25. Chen W, Yang H, Feng P. et al. iDNA4mC: identifying DNA N4-methylcytosine sites based on nucleotide chemical properties. Bioinformatics. 2017;33:3518-23
26. Yona G, Levitt M. Within the twilight zone: a sensitive profile-profile comparison tool based on information theory. J Mol Biol. 2002;315:1257
27. Giegerich R. A systematic approach to dynamic programming in bioinformatics. Bioinformatics. 2000;16:665-77
28. Eddy SR. What is dynamic programming? Nature Biotechnol. 2004;22:909-10
29. Wu S, Zhang Y. MUSTER: Improving protein sequence profile-profile alignments by using multiple sources of structure information. Proteins. 2008;72:547-56
30. Zhou H, Zhou Y. Fold Recognition by Combining Sequence Profiles Derived From Evolution and From Depth-Dependent Structural Alignment of Fragments. Proteins. 2005;58:321-8
31. Zhang Y, Skolnick J. Scoring function for automated assessment of protein structure template quality. Proteins. 2007;68:702-10
32. Zhang J, Skolnick Y. TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res. 2005;33:2302-9
33. Yankovskaya V, Horsefield R, Törnroth S. et al. Architecture of Succinate Dehydrogenase and Reactive Oxygen Species Generation. Science. 2003;299:700-4
34. Lomize MA, Lomize AL, Pogozheva ID. et al. OPM: Orientations of Proteins in Membranes database. Bioinformatics. 2006;22:623-25
Author contact
![]() Corresponding author: Yanwen Li: liyw085edu.cn
Corresponding author: Yanwen Li: liyw085edu.cn