Impact Factor ISSN: 1449-2288
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
1. Introduction
2. Materials and Methods
3. Results
4. Discussion
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(10):1221-1231. doi:10.7150/ijbs.25488 This issue Cite
Research Paper
Simultaneously targeting DNA damage repair pathway and mTORC1/2 results in small cell lung cancer growth arrest via ER stress-induced apoptosis
Bin Fang1,2,3, Aarthi Kannan1, Tao Guo2,3, ![]() , Ling Gao1,
, Ling Gao1, ![]()
1. Department of Dermatology, University of Arkansas for Medical Sciences, Little Rock, AR, 72205
2. Department of Hematology, Union Hospital, Tongji Medical College, Huazhong University of Science and Technology, Wuhan, Hubei, China
3. Collaborative Innovation Center of Hematology, Huazhong University of Science and Technology, Wuhan, Hubei, China
Received 2018-2-10; Accepted 2018-4-9; Published 2018-7-13
Abstract

Purpose: Small cell lung cancer (SCLC) is highly lethal with no effective therapy. Wee1 kinase inhibitor AZD1775 (MK-1775) and mTOR kinase inhibitor MLN0128 (TAK228) are in clinical trials for relapsed SCLC and recurrent lung cancer, respectively. However, there is no preclinical data combining these two drugs in human cancers.
Methods: In this study, we set to investigate the combinatorial anti-tumor effects of AZD1775 and MLN0128 on two human SCLC cell lines H69 and H82 in vitro and in vivo.
Results: We have found that AZD1775 or MLN0128 treatment results in remarkably suppressed cell proliferation and increased cell death in vitro, what's more, the salient finding here is the potent anti-tumor effect observed in combinatorial treatment in H82 xenograft tumor. Importantly, we have first observed marked induction of ER stress and CHOP-dependent SCLC cell apoptosis in MLN0128 and AZD1775-primed cells.
Conclusion: Our study has first provided preclinical evidence that combination of AZD1775 and MLN0128 could be a novel effective therapy for advanced SCLC patients.
Keywords: Small Cell Lung Cancer, AZD1775, MLN0128, CHOP, ER Stress, Apoptosis
1. Introduction
Small cell lung cancer (SCLC) is the most lethal form of lung cancer[1]. SCLC patients carry a poor prognosis with a dismal 5-year survival rate of 5%[2]. Genomic studies have revealed that SCLC is highly heterogeneic with near-ubiquitous inactivation of TP53 and RB1; inactivating mutation in the NOTCH family; pathologic amplification of MYC family genes and aberrant activation of PI3K/mTOR pathway[3-5]. Despite these notable discoveries, there is no effective therapy at present for local and metastatic diseases and targeted therapies for SCLC have lagged behind.
Modulation of cell cycle checkpoint machinery is often proposed as a therapeutic strategy to potentiate anticancer therapy[6]. Wee1 is a protein kinase that regulates G2 checkpoint and prevents premature entry into mitosis in response to DNA damage. Recently, Wee1 kinase has received considerable attention as a potential target in cancer therapy[7, 8]. AZD1775 is a potent ATP-competitive inhibitor of the Wee1 kinase, and it abrogates cell cycle checkpoint, and allows premature entry into cell division with unrepaired DNA lesions. Independent of its ability to be a chemotherapy and radiation sensitizer, AZD1775 has demonstrated anti-tumor activities alone or in combination with other drugs in preclinical studies of several human cancers[9-24].
Mammalian target of rapamycin (mTOR) is one of the main growth regulatory pathways in cells[25]. A recent study examining PI3K/Akt/mTOR pathway across more than 11, 000 human cancers representing 32 major types in the TCGA database has shown that a substantial fraction of cancers demonstrating high mTOR pathway activity[26]. In SCLC, 36% of tumors harbor genetic alterations in the PI3K/Akt/mTOR pathway, indicating that this pathway is indeed an attractive therapeutic target[27]. Moreover, PI3K/mTOR pathway inhibitors are identified as potential treatments when screening over 1,000 compounds in SCLC cell lines[28].
There is a growing appreciation for combinatorial therapies to treat human cancers, targeting key pathways and reducing potential drug resistance[29]. A phase II study of AZD1775 monotherapy is underway on relapsed SCLC patients (NCT02688907) and SCLC patients with MYC gene amplification or CDKN2A mutation combined with TP53 mutation (NCT02593019). Recently, an allosteric mTOR inhibitor ridaforolimus is identified as a novel synergistic combination with AZD1775 using an unbiased oncology compound screen in a panel of 39 cancer cell lines[30]. Sen et al have shown that AZD1775 is effective against several SCLC cell lines, whereas SCLC cell lines with high mTOR activity are sensitive to the allosteric mTOR inhibitor RAD001[31]. However, RAD001 has limited clinical efficacy in a phase II clinical trial in previously treated SCLC patients[32]. Importantly, mTOR kinase inhibitor MLN0128 blocks both mTOR complexes and has led to therapeutic benefits in many preclinical models of human cancers[25]. Thus, we hypothesize that defective DNA repair induced by AZD1775 will potentiate the cytotoxic effects of mTOR inhibition and accelerate tumor regression in SCLC.
To this end, we employ AZD1775 and mTOR inhibitor MLN0128 in our experimental models, including the human SCLC cell lines NCI-H69, NCI-H82, and their respective xenograft mouse models. In vitro treatment of H69 and H82 cells with AZD1775 and/or MLN0128 result in suppressed tumor cell proliferation and increased cell death. Treatment with AZD1775 not only causes increased premature mitotic entries but also augmented DNA damage in both cells. ADZ1775 potentiates inhibitory effects of MLN0128 on its downstream pathways. The salient finding in our study is the potent anti-tumor effect observed in combinatorial treatment in H82 xenograft tumor. Interestingly, MLN0128 alone is as effective as the combination treatment in suppressing H69 xenograft tumor growth. Importantly, we have first observed marked induction of ER stress, activation of unfolded protein response (UPR) and the C/EBP homologous protein (CHOP) in MLN0128 and AZD1775-primed cells, leading to CHOP-dependent up-regulation of pro-apoptotic proteins and SCLC cell apoptosis. Taken together, anti-tumor effects of these drugs involves diminished PI3K/mTOR pathway; disrupted cell cycle regualtion; activation of ER-stress pathways and its downstream signaling leading to increased apoptosis in SCLC cells. Our data have provided evidence of combination treatment of AZD1775 and MLN0128 in the management of SCLC.
2. Materials and Methods
Compounds and cell lines
MLN0128 and AZD1775 were purchased from Selleck Chemicals. Human SCLC cell lines NCI-H69 (H69), NCI-H82 (H82) and human embryonic kidney (293T/17) cells were purchased from ATCC. H69 and H82 were maintained in RPMI-1640 (ATCC) and 293T/17 cells were cultured in DMEM (Life Technologies). All media was supplemented with 10% FBS (Atlanta Biologicals), penicillin-streptomycin and L-glutamine (Sigma-Aldrich).
Antibodies
Primary and secondary antibodies for immunoblotting and immunohistochemistry: Mcl-1 (Santa Cruz Biotechnology); Ki67 and GRP78 (Abcam); α-tubulin and β-actin (Sigma-Aldrich); all other primary antibodies purchased from Cell Signaling Technology, all secondary antibodies purchased from Santa Cruz Biotechnology.
Reagents and in vitro analyses
Other kits, supplies, and reagents for in vitro analyses included: Cell Counting Kit-8 (Sigma-Aldrich); MethoCult™ methylcellulose medium (Stem Cell Technologies); Annexin V-FITC, BrdU flow cytometry kits, propidium iodide (BD Biosciences); CHOP shRNA (Sigma-Aldrich); TransIT-LT1™ (Mirus Bio); Lenti-X™ Concentrator (Clontech Laboratories). All reagents used per manufacturer's protocol.
Please see supplementary materials and methods for detailed information regarding in vitro analyses.
Xenograft studies
Six-week-old female NOD/SCID/IL2rgnull (NSG) mice (Jackson Laboratory) were used to generate SCLC xenograft models. The animal protocol was approved by the IACUC at the University of Arkansas for Medical Sciences. Tumor cells were inoculated in NSG mice as previously described[33, 34]. MLN0128 was formulated in 5% N-methyl-2-pyrrolidone (NMP) / 15% polyvinyl pyrrolidone (PVP) in water, administered via oral gavage at 1mg/kg/day. AZD1775 was formulated in 0.5% methylcellulose, administered via oral gavage at 40 mg/kg Q2D[22, 23]. Tumor volume was calculated as L/2 x W2, where length (L) is the longer dimension. Antitumor efficacy was determined as a function of tumor growth inhibition (TGI), calculated by the equation [1-(T-T0/C-T0)] x 100, where T and C represent mean size of tumors in treatment (T) group and control (C) groups, respectively, and T0 refers to tumor size at randomization[35].
Statistical analysis
Data analysis were performed either in the ANOVA framework or using two-tailed Student's t-test, as appropriate, with GraphPad Prism software. P < 0.05 was considered statistically significant.
3. Results
3.1 AZD1775 and MLN0128 inhibit SCLC cell growth in vitro
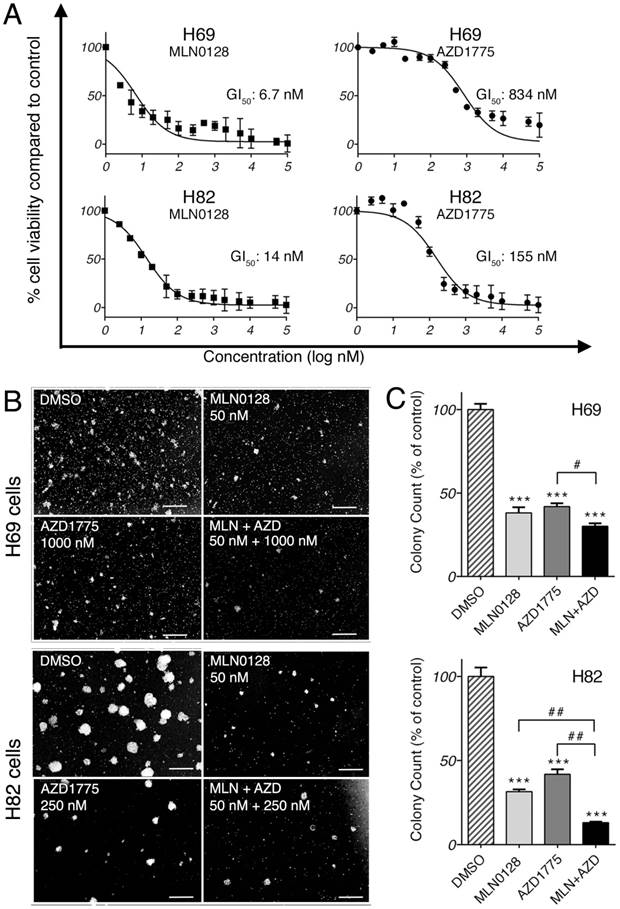
We first investigated the anti-proliferative effect of AZD1775 and MLN0128 by testing 15 serial concentrations of both inhibitors. Cell viability was measured at 72 hours by CCK-8 assay, an assay for the assessment of cell viability and proliferation[34]. As expected, both inhibitors exhibited strong growth inhibitions in both cell lines in a dose-dependent manner (Figure 1A). The half-maximal growth inhibitory concentration (GI50) for MLN0128 was 6.7nM in H69 cells and 14nM in H82 cells, whereas GI50 for AZD1775 was 834nM in H69 cells and 155nM in H82 cells (Figure 1A). GI50 was referred for the treatment concentrations in the following experiments. Furthermore, we examined the effects of AZD1775 and MLN0128 combination treatment on SCLC cells viability using CCK-8 as readout, which has more profound inhibitory effect on SCLC cell survival(see Supplemental Figure 1). To complement the results of short-term treatment, we investigated colony growth as a functional readout of long term in vitro effects. As described previously, H69 and H82 cells were plated and grown in methylcellulose media with respective inhibitors, and colonies were counted on day 14. Compared to DMSO-treated plates, single-agent treated plates caused >50% reduction in colony count (Figure 1B and 1C). With combinatorial treatment, the reduction reached 70% and 87% in H69 and H82 cells, respectively (Figure 1C). However, in H69 cells, there was no significant difference in colony growth reduction between MLN0128 alone and MLN0128 in combination with AZD1775. Taken together, these results confirmed that MLN0128 and AZD1775 inhibit H69 and H82 cell growth in both short- and long-term cell culture.
MLN0128 and AZD1775 inhibit SCLC cell proliferation in vitro. A. Decreased SCLC cell proliferation upon AZD1775 or MLN0128 treatment. H69 and H82 cells were incubated with serial concentrations of MLN0128 or AZD1775 for 72 hours and cell viability was assessed by CCK-8 assay. Average viability of DMSO-treated samples was taken as 100%. Data points represent the mean ± SEM of triplicate experiments. B. Decreased colony formation in SCLC cells treated with AZD1775 or MLN0128 alone or in combination. H69 and H82 cells were plated in methylcellulose medium with DMSO, MLN0128 (50nM), or AZD1775 (1000nM for H69, 250nM for H82), or combination of both inhibitors and cultured for 14 days at 37°C; colonies were counted on Day 14. Left panels (C) show representative images at 40x magnification from different microscopic fields. Right bar graphs (D) indicate the number of colonies at each plate. Data are presented as the mean ± SEM of triplicate experiments. ****p < 0.001 compared with DMSO-treated cells. # p< 0.05, ## p< 0.01 single treatment with combination treatment.
3.2 Augmented SCLC cell death and suppressed cell proliferation upon treatment
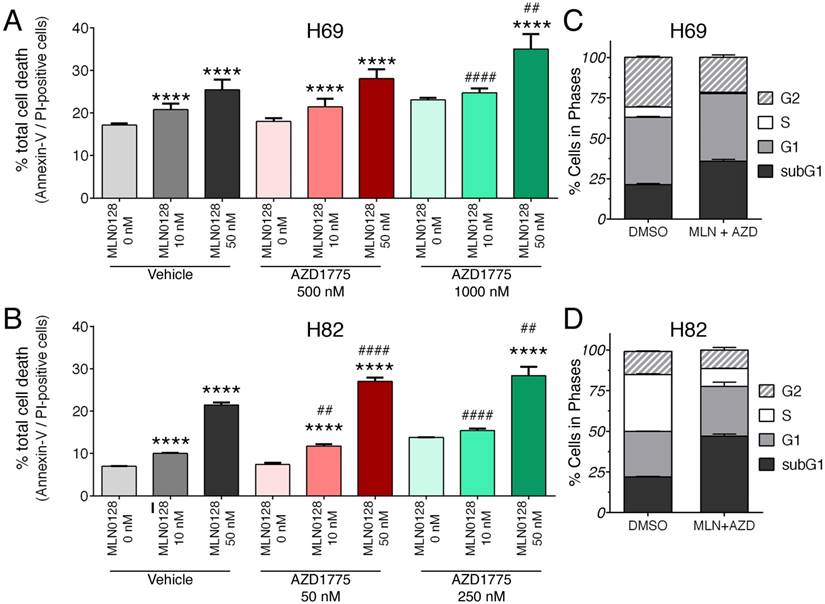
We further examined the effects of inhibitors on SCLC cell death and proliferation. As described above, H69 and H82 cells were treated with inhibitors at indicated concentrations for 48 hours, cell death was monitored by Annexin-V and PI staining. Dose-dependent induction of apoptosis was evident in both SCLC cells treated with MLN0128 or AZD1775, albeit in varying degrees (Figure 2A, 2B and supplementary Figure 3A). While H82 cells responded to lower concentrations of AZD1775 (250nM), H69 cells were sensitive at higher concentrations (1000nM). Then, we evaluated the combinatorial effect on SCLC cell death and cell cycle progression. In both H69 and H89 cells, a more potent inhibitory effect was observed with additional AZD1755 in cells treated with MLN0128 10nM and 50nM, respectively (Figure 2A, 2B and supplementary Figure 3A). By BrdU and 7-AAD staining, there were significant decreases of SCLC cells in S-phase (cell proliferation) and increases of cells in subG1 populations (cell death) (Figure 2C, 2D, and supplementary Figure 3B). The effects of single inhibitor treatments on SCLC cell cycles were shown as supplemental Figure 2. Taken together, our data indicate that MLN0128 and AZD1775 treatment increased apoptosis and decreased cell proliferation in SCLC cells.
Augmented cell death and suppressed cell proliferation in SCLC. A, B: H69 and H82 cells were treated with AZD1775 or MLN0128 alone or combination for 48 hours at indicated concentrations, followed by Annexin-V and PI staining. Cells were analyzed by flow cytometry and the percentage of Annexin V+, PI- (early apoptotic) and Annexin-V+, PI+ (late apoptotic) cells were calculated. Data represent mean ± SEM of total apoptotic cell population in quadruple experiments. ****p < 0.0001 compared treatments among the same group; ## p < 0.01, #### p < 0.0001 compared MLN0128-treated cells to combination-treated cells. C, D: H69 or H82 cells were treated with combination of AZD1775 (1000 nM for H69, 250 nM for H82) and MLN0128 (50nM) for 48 hours, followed by BrdU and 7-AAD staining and flow cytometry analysis. Stacked bars show percentage of SCLC cell population in sub-G1, G1, S, and G2 cell cycle phases. Data are presented as the mean ± SEM of quadruple experiments.
3.3 mTOR and Wee-1 inhibition suppresses PI3K/mTOR pathway and induces DNA damage in SCLC cells
We next investigated the effects of MLN0128 and AZD1775 on their respective downstream targets. Cell lysates were collected followed by western blot with indicated antibodies. Consistent with published studies[34], MLN0128 suppresses phosphorylation of mTOR and its downstream molecules 4E-BP1 and S6K in both H69 and H82 cells (Figure 3A). Interestingly, addition of AZD1775 significantly suppressed p-mTOR and p-S6K activities in H69 cells, whereas additional AZD1775 attenuated p-Akt activities in both Ser473 in H82 cells (Figure 3A).
AZD1775 and MLN0128 treatment suppressed downstream molecules, increased DNA damage, induced ER stress and apoptosis in SCLC cells. A. Suppressed PI3K/mTOR pathway activities in H69 and H82 cells upon treatment with AZD1775 (1000nM for H69, 250nM for H82), or MLN0128 (50nM), or combination of both, for 48 hours. Western blot of SCLC cell lysates with indicated antibodies; tubulin was used as loading control. B. Increased DNA damage and apoptosis in H69 and H82 cells upon treatment with AZD1775 (1000nM for H69, 250nM for H82), or MLN0128 (50nM), or combination of both, for 48 hours. Cell lysates were collected followed by western blot using indicated antibodies; tubulin was used as loading control. C. Induction of ER stress and activation of UPR and CHOP activation in H69 and H82 cells upon treatment with AZD1775 (1000nM for H69, 250nM for H82), or MLN0128 (50nM), or combination of both, for 24 hours. Cell lysates were collected followed by western blot using indicated antibodies; tubulin was used as loading control. D. Up-regulation of pro-apoptotic proteins in H69 and H82 cells upon treatment with AZD1775 (1000nM for H69, 250nM for H82), MLN0128 (50nM), or combination of both, for 48 hours. Western blot was performed with indicated antibodies; tubulin was used as loading control.
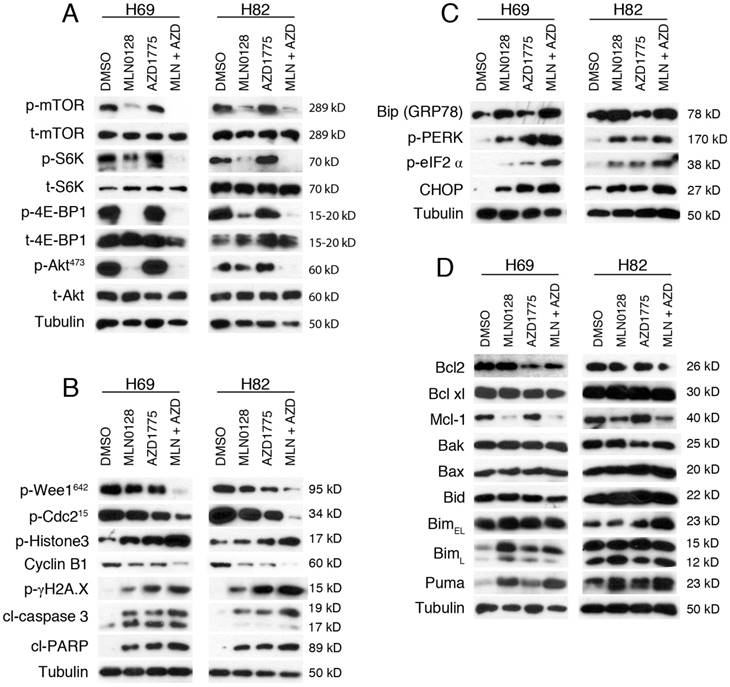
To test the effects of AZD1775 and MLN0128 on cell cycle progression and DNA damage, we examined p-Cdc2. Wee1 kinase phosphorylates Cdc2 to initiate G2 checkpoint in response to DNA damage and Y15 phosphorylation of Cdc2 is a marker of Wee-1 kinase activity. As expected, diminished p-Cdc2 was observed upon treatment with AZD1775 in both cell lines (Figure 3B). Moreover, additional MLN0128 treatment profoundly attenuated Wee-1 and Cdc2 phosphorylation, and significantly increased the level of p-γH2AX, indicative of persistent unrepaired DNA damage in both cells (Figure 3B). A maximal stress response with increased expression of p-Histone-3 (ser10), a marker of mitosis, was observed in combinatorial treatment, suggesting increased premature mitotic entries (Fig. 3B). Progression from G2 to mitosis is controlled by the Cdc2/cyclin B complex and decreased protein level of cyclin B is indicative of effective engagement of Wee-1 inhibition as shown in Figure 3B.
To further confirm our findings, we measured the cleavage products of caspase-3 and PARP as markers of apoptosis. Increased induction of cl-caspase-3 and cl-PARP was detected in H69 and H82 cells upon treatment (Figure 3B). Consistently, the combination therapy caused higher levels of cleaved products of caspase-3 and PARP, suggesting an additive effect in causing SCLC cell apoptosis (Figure 3B). Taken together, these data underscore that combinatorial therapies targeting Wee1 and mTOR significantly enhance DNA damage, increase premature mitotic entry and augment apoptosis in H69 and H82 cells.
3.4 CHOP activation and up-regulation of pro-apoptotic proteins upon treatment
Cellular adaption to ER stress is achieved by the activation of the UPR[36, 37]. Under steady-state conditions, glucose-related protein 78 (GRP78, also known as Bip) is rendered inactive by bounding to the luminal domains of IRE1, ATF6, and PERK. During ER stress, GRP78/Bip is disassociated and recruited to misfolded proteins, resulting in UPR activation. CHOP is a major pro-apoptotic transcription factor trigged by UPR activation, and CHOP-dependent induction of pro-apoptotic proteins Bim and/or Puma are required for ER stress mediated apoptosis in a diverse range of cell types[38]. We have shown previously that MLN0128 induces Merkel cell carcinoma cell death via up-regulation of Bim. Therefore, it prompted us to examine whether treatment with MLN0128 and/or AZD1775 would induce ER stress and its related changes. Increased GRP78/Bip expression was detected upon single agent treatment and reached to a higher level in combination treatment, indicating an induction of ER stress in SCLC cells (Figure 3C). Increased CHOP expression was also evident upon treatment (Figure 3C). It has been shown that PI3K and mTOR inhibition activates PERK and translation initiation factor eIF2α in mammalian cells[39]. Consistently, we observed increased p-PERK and p-eIF2α upon treatment, suggesting UPR activation (Figure 3C). Similar to our previous findings, increased expressions of Bim and Puma were detected upon treatment in SCLC cells (Figure 3D). Interestingly, an increased expression of BimL was detected in H69 cells as compared to increased level of BimEL in H82 cells. Additionally, down-regulated anti-apoptotic proteins Bcl2 and Mcl-1 were also detected (Figure 3D). No changes of expressions in Bak, Bax and Bid protein were detected (Figure 3D). Taken together, both mTOR and Wee-1 inhibition induces ER stress, activates UPR and CHOP, and up-regulates pro-apoptotic proteins Bim and Puma.
3.5 CHOP-mediated SCLC cell apoptosis upon treatment with MLN0128 and AZD1775
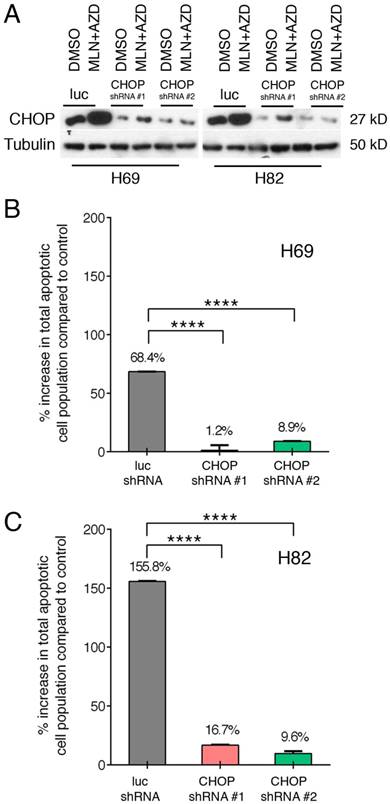
Taking advantage of our well-established lentinvirus mediated shRNA knockdown assay, we silenced CHOP expression using two shRNA followed by incubation with both inhibitors for 48 hours. Transfection efficiency was more than 80% in both cell lines (Figure 4A). Next, by Annexin-V study, we have found that AZD1775+MLN0128 treatment increased 68.4% cell apoptosis in LUC group of H69 cells, while AZD1775+MLN0128 augmented 1.2% cell apoptosis in CHOP shRNA#1 group, the increased rate is 8.9% in CHOP shRNA#2 group. In H82 cells, the increased percentage of apoptosis in LUC group is 155.8%, while the number decreased to 16.7% and 9.6% in CHOP shRNA#1 or CHOP shRNA#2 groups. The above results indicate that CHOP plays a key role in AZD1775+MLN0128-induced cell apoptosis. To our knowledge, this is the first report on ER stress-induced CHOP activation and its role in SCLC cell death upon mTOR and Wee-1 inhibition.
ER stress-induced SCLC cell apoptosis is CHOP dependent. A. H69 and H82 cells were transduced with recombinant lenti-viruses expressing a control shRNA targeting fire fly luciferase (LUC-sh) or shRNA-CHOP #1 or #2 and incubated for 48 hours. Transfected cells were treated with AZD1775 (1000nM for H69, 250nM for H82) and MLN0128 (50nM) for 48 hours, expression of CHOP was determined by Western blot analysis; Tubulin was used as a loading control. B, C. Transfected H69 and H82 cells were treated with MLN0128 (50nM) and AZD1775 (1000nM for H69, 250nM for H82) followed by Annexin-V and PI staining and flow cytometry analysis. Data are presented as the mean ± SEM of quadruple experiments. ****p < 0.0001 compared to cells transfected with control shRNA.
3.6 Treatment of MLN0128 and AZD1775 demonstrated potent in vivo anti-tumor activity
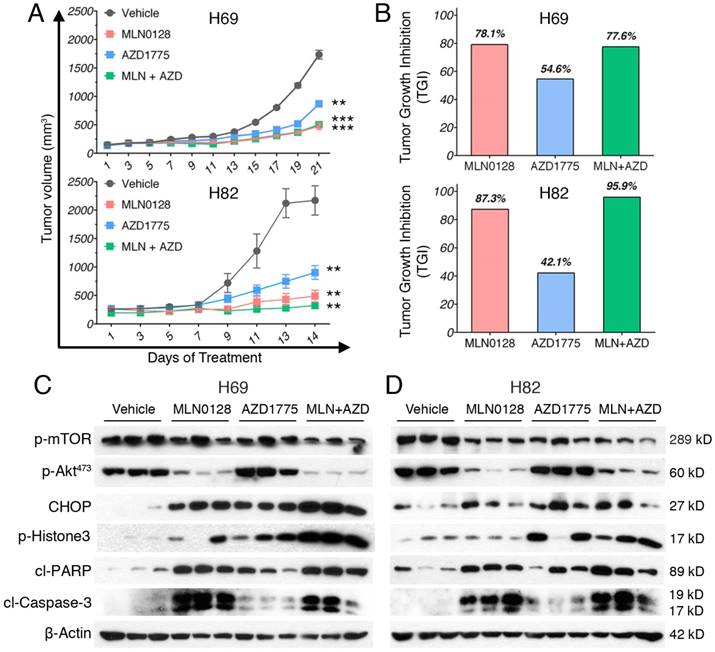
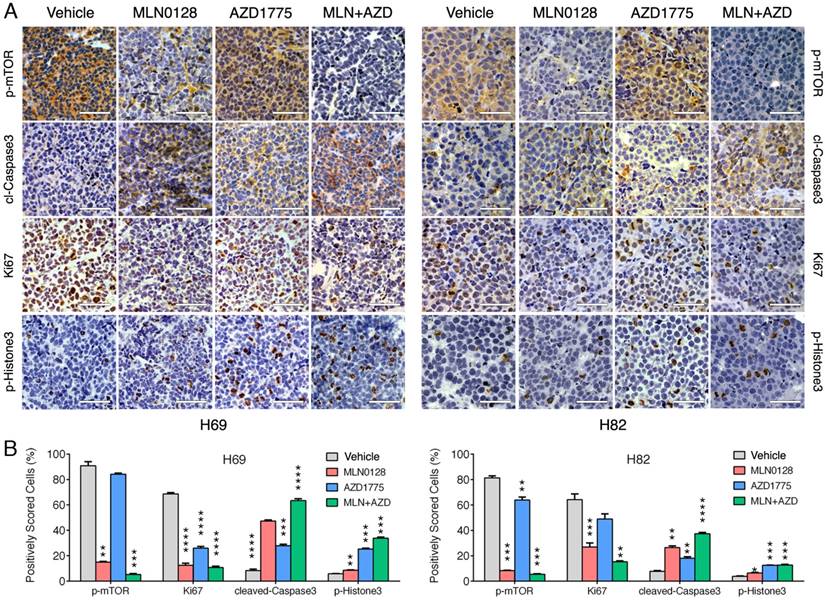
To test the in vivo efficacy of AZD1775 and MLN0128, we have established xenograft tumor using H82 and H69 cells in immunodeficient NSG mice (Jackson Laboratory, strain# 5557). In brief, H82 cells and H69 cells were inoculated subcutaneously on the flanks of NSG mice as described previously[33]. When xenograft tumors approached 7 mm in diameter or 100 mm3 in volume, tumor bearing mice were treated with MLN0128, AZD1775 alone or in combination. Animal receiving treatment showed no obvious signs of toxicity based on body weight, food, water intake, activities, and general exam. Antitumor efficacy was determined by tumor growth inhibition (TGI) as described [35]. By the end of treatment, significant tumor repression was evident in both H69 and H82 xenograft tumors (Figure 5A), Representative pictures of xenograft tumors were shown in Supplementary Fig.4. Although AZD1775 alone was able to suppress both xenograft tumors, it is less potent than MLN0128. TGI of 54.6% and 42.1% was observed in H69 and H82 xenograft tumors treated with AZD1775, respectively (Figure 5B). Interestingly, H69 xenograft tumors treated with MLN0128 alone exhibited similar TGI as H69 xenografts receiving combination therapy (Figure 5B, 78.1% vs. 77.6%). In H82 xenograft tumors, both MLN0128 as a single agent and in combination with AZD1775 significantly suppressed tumor growth (Figure 5B, 87.3% vs. 95.9%). By western blot analysis, there was a significant reduction of p-Akt and p-mTOR activity in mice treated with MLN0128 alone and in combination (Figure 5C and 5D). Significantly increased levels of cl-caspase 3 and cl-PARP were detected in both treated xenograft tumors (Figure 5C and 5D). Increased premature mitotic entry as evident by increased p-Histone 3, a marker for mitosis was detected in both treated xenograft tumors, especially in tumors treated with combinatorial therapy (Figure 5C and 5D). Immunohistochemistry using H69 and H82 xenograft tumors have further confirmed suppressed p-mTOR activity, increased mitotic entries (p-Histone 3 positive cells), decreased cell proliferation (Ki67 positive cells) and increased cell apoptosis (cl-caspase -3 positive cells) upon single or combinatorial treatment (Figure 6A and 6B). These results were very well correlated with tumor regression results obtained with H69 and H82 xenograft mice.
MLN0128 and AZD1775 is effective in repression of SCLC xenograft tumors. A. Mice bearing H69 or H82 xenograft tumors were treated with vehicle, MLN0128 (1mg/kg/day), or AZD1775 (40mg/kg every other day), or MLN0128 + AZD1775 combination therapy by oral gavage. Data are presented as the mean volume ± SEM. B. Tumor growth inhibition (TGI) was greater in MLN0128-treated mice than AZD1775-treated mice, and particularly pronounced in mice receiving AZD1775 + MLN0128 combination therapy. C. Tissue lysates were prepared from H69 xenograft tumors and were analyzed by western blot using the indicated antibodies. β-actin was used as loading control. D. Tissue lysates were prepared from H82 xenograft tumors and were analyzed by western blot using the indicated antibodies. β-actin was used as loading control.
Diminished PI3K/mTOR pathway and increased premature mitotic entry and apoptosis in SCLC xenograft tumors. A. Immunohistochemical analysis of p-mTOR, Ki67, cleaved caspase-3, and p-histone 3 was performed in H69 and H82 xenograft tumors treated with vehicle, MLN0128, or AZD1775, or combination of both. Scale bars, 50μm (inset). Significant reduction of Ki67 positive cells (proliferation), increased cl-caspase-3 positive cells (cell death), increased p-Histone 3 (cellular stress) were observed in MLN0128, or AZD1775, or combination-treated xenograft tumors. Significant reduction of p-mTOR activity was observed in MLN0128-treated xenograft tumors. B. Quantitative cell image analysis was carried out on tissue samples viewed at 400x magnification. Percentage of positivity (brown nuclear/cytoplasmic staining) cells were scored. Vehicle-treated xenograft tumors were compared with MLN0128-, or AZD1775-, or combination-treated tumors. **p < 0.01, ***p < 0.001, ****p < 0.0001 compared with vehicle-treated xenograft tumors.
4. Discussion
SCLC is often metastatic at diagnosis and it is considered the deadliest lung cancer subtypes[40]. It accounts for approximately 15% of all lung cancers. Prognosis is extremely poor for SCLC patient due to lack of effective therapy. Genetic studies have shown that SCLC is highly heterogeneic[3]. Given this clinical scenario, the most effective therapeutic strategies for SCLC will likely be combinatorial in nature targeting multiple key pathways[29]. In this study, we have undertaken such an approach using AZD1775 and MLN0128 that are currently in advanced clinical trials. We have first demonstrated that combinatorial therapy of AZD1775 and MLN0128 is safe and is responsible for significant anti-tumor effect both in vivo and in vitro. Upon treatment, significantly suppressed downstream molecules in the PI3K/Akt/mTOR pathway and in the cell cycle progression were detected in both cell lines and in respective xenograft tumors. Mechanistically, AZD1775 and MLN0128 treatment leads to ER stress, UPR and CHOP activation and subsequent up-regulation of pro-apoptotic proteins in SCLC cells. Collectively, our study has provided preclinical evidence of effective combination therapy of MLN0128 and AZD1775 for SCLC.
Inhibition of Wee-1 kinase activities overrides G2 cell-cycle arrest, causing an accumulation of cells with extensive DNA damage and leading to cell death. Further studies have shown that AZD1775 acts not only as a chemotherapy and radiation sensitizer by allowing increased mitotic entries, but also directly causes DNA damage alone or in combination with other anti-tumor drugs. Consistent with published studies, we have detected increased premature mitotic entry, DNA damage and apoptosis upon treatment with AZD1775 as a single agent in both SCLC cell lines and xenograft tumors. Recently, Sakre et al have demonstrated that SCLC cells with RICTOR copy number gain are sensitive to mTOR kinase inhibition[41]. In that study, H82 cells are used as control cells with normal RICTOR copy number gain and it is less sensitive to mTOR kinase inhibitor, including MLN0128 (formerly named INK128). In contrast, we have shown that H82 cells are sensitive to MLN0128 with a GI50 of 14 nM. It is unclear what underlies the discrepancy between these two studies.
Although AZD1775 and mTOR inhibitor combination treatment has been reported in non-small lung cancer treatment[42]. There is no data to show the efficacy of combination of AZD1775 and MLN0128 in small cell lung cancer. An unbiased oncology screen suggests that AZD1775 and an allosteric mTOR inhibitor ridaforolimus is a novel synergistic combination in a panel of 39 human cancer cell lines[30]. However, a phase II study of RAD001, an allosteric mTOR inhibitor, has shown limited single-agent anti-tumor activity in previously treated SCLC[32]. RAD001 (also named Everolimus) and ridaforolimus, has limited clinical efficacy due to its inability to inhibit mTORC2[25]. This current study builds upon these findings and employs mTOR kinase inhibitor MLN0128 that blocks both mTOR complexes. We here demonstrate for the first time that the combinatorial dose regimen of AZD1775 and MLN0128 is tolerated well in mice. Addition of AZD1775 potentiates MLN0128 in suppressing Akt activities in H82 cells and a potent anti-tumor effect is observed upon combinatorial treatment in repressing H82 xenograft tumor growth. MLN0128 abolishes p-Akt activities in Ser473 in H69 cells and xenograft tumors, and MLN0128 alone is almost as effective as the combination treatment, suggesting Akt activities might be a marker for a subset of SCLC patients who are potentially good responders to MLN0128 treatment. Our finding merits further study to delineate the biomarkers and identify patients who will be sensitive to MLN0128 in combination with AZD1775.
Anticancer drugs can induce potent ER stress responses, and ER stress can facilitate anticancer drug efficacy or the development of chemoresistance, depend on context and tumor types[43, 44]. Vemurafenib (BRAF inhibitor) and epidermal growth factor receptor inhibitor cetuximab potently induce PERK and eIF2α phosphorylation[39]. Moreover, carfilzomib inhibits the chymotrypsin-like activity of the proteasome and it is found to trigger chronic lymphocytic leukemia cell death via CHOP[45]. Furthermore, rapamycin has been shown to induce apoptosis in colon cancer cells via CHOP-dependent DR5 induction[46]. In contrast, little is known about the consequences of ER dysfunction in SCLC, here we report that H69 and H82 cells activate UPR machinery and CHOP in response to MLN0128 and AZD1775 treatment, As ER stress was initiated by unfolded protein response (UPR) that's consists of a complex interplay between three individual signaling “arms”, each of which is from a different ER stress sensor including PERK (pancreatic ER kinase (PKR)-like ER kinase), IRE1 (inositol requiring enzyme 1) and ATF6 (activating transcription factor 6), each sensor can induce relatively individual downstream pathway, for example, PERK is able to induce ER stress by PERK-eIF2α- ATF4-CHOP pathway, while CHOP is a shared target gene of all three arms of the UPR, as it can be induced by ATF4, cleaved ATF6 as well as XBP1[37]. To blocked ER stress by inhibiting the expression of CHOP is a widely used method to address the importance of ER stress in recently published papers [43], that's why we adopt the same method to silence CHOP expression to represent the importance of ER stress. What's more, although AZD1775 or mTOR inhibitor combination has been reported anti-proliferative effect in tumors[31, 42], it's the first time to demonstrate that CHOP is one of the key mechanism to mediate AZD1775/MLN018-induced cell apoptosis. Our data provide the first evidence of ER stress and CHOP-mediated apoptosis in SCLC.
In conclusion, we have proposed a rational combination therapy using AZD1775 and MLN0128 targeting Wee1 kinase and mTORC complexes to arrest SCLC cell growth. Our results show that combinatorial treatment is safe and it attenuates tumor growth in the SCLC mouse models. Further studies are warranted to identify biomarkers for patients who potentially respond to MLN0128 and/or AZD1775. For the first time, we describe an active role of ER stress and CHOP activation in SCLC cells, which has opened new avenues for future studies in this area. These data have immediate clinical implications given the recent advancement in the clinical trials with AZD1775 and MLN0128.
Supplementary Material
Supplementary materials and methods, figures.
Acknowledgements
This work was supported by funds provided by the Department of Dermatology at UAMS, the Winthrop C. Rockefeller Cancer Institute, and the UAMS Clinical Scientist Program. Authors also wish to thank Dr. Krishna Kannan for assisting us with the manuscript preparation.
Author Contributions
Fang performed most of the in vitro experiments and Kannan carried out animal experiments; Fang, Kannan, and Gao contributed to data analysis; Gao and Guo designed, and directed this study.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA: a cancer journal for clinicians. 2017;67:7-30
2. Gardner EE, Lok BH, Schneeberger VE, Desmeules P, Miles LA, Arnold PK. et al. Chemosensitive Relapse in Small Cell Lung Cancer Proceeds through an EZH2-SLFN11 Axis. Cancer cell. 2017;31:286-99
3. George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G. et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524:47-53
4. Semenova EA, Nagel R, Berns A. Origins, genetic landscape, and emerging therapies of small cell lung cancer. Genes & development. 2015;29:1447-62
5. Pietanza MC, Ladanyi M. Bringing the genomic landscape of small-cell lung cancer into focus. Nature genetics. 2012;44:1074-5
6. Geenen JJJ, Schellens JHM. Molecular Pathways: Targeting the Protein Kinase Wee1 in Cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2017;23:4540-4
7. Mahajan K, Mahajan NP. WEE1 tyrosine kinase, a novel epigenetic modifier. Trends in genetics: TIG. 2013;29:394-402
8. Matheson CJ, Backos DS, Reigan P. Targeting WEE1 Kinase in Cancer. Trends in pharmacological sciences. 2016;37:872-81
9. De Witt Hamer PC, Mir SE, Noske D, Van Noorden CJ, Wurdinger T. WEE1 kinase targeting combined with DNA-damaging cancer therapy catalyzes mitotic catastrophe. Clinical cancer research: an official journal of the American Association for Cancer Research. 2011;17:4200-7
10. Hirai H, Iwasawa Y, Okada M, Arai T, Nishibata T, Kobayashi M. et al. Small-molecule inhibition of Wee1 kinase by MK-1775 selectively sensitizes p53-deficient tumor cells to DNA-damaging agents. Molecular cancer therapeutics. 2009;8:2992-3000
11. Kreahling JM, Gemmer JY, Reed D, Letson D, Bui M, Altiok S. MK1775, a selective Wee1 inhibitor, shows single-agent antitumor activity against sarcoma cells. Molecular cancer therapeutics. 2012;11:174-82
12. Zheng H, Shao F, Martin S, Xu X, Deng CX. WEE1 inhibition targets cell cycle checkpoints for triple negative breast cancers to overcome cisplatin resistance. Scientific reports. 2017;7:43517
13. Guertin AD, Li J, Liu Y, Hurd MS, Schuller AG, Long B. et al. Preclinical evaluation of the WEE1 inhibitor MK-1775 as single-agent anticancer therapy. Molecular cancer therapeutics. 2013;12:1442-52
14. Heijink AM, Blomen VA, Bisteau X, Degener F, Matsushita FY, Kaldis P. et al. A haploid genetic screen identifies the G1/S regulatory machinery as a determinant of Wee1 inhibitor sensitivity. Proceedings of the National Academy of Sciences of the United States of America. 2015;112:15160-5
15. Karnak D, Engelke CG, Parsels LA, Kausar T, Wei D, Robertson JR. et al. Combined inhibition of Wee1 and PARP1/2 for radiosensitization in pancreatic cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2014;20:5085-96
16. Tanaka N, Patel AA, Wang J, Frederick MJ, Kalu NN, Zhao M. et al. Wee-1 Kinase Inhibition Sensitizes High-Risk HPV+ HNSCC to Apoptosis Accompanied by Downregulation of MCl-1 and XIAP Antiapoptotic Proteins. Clinical cancer research: an official journal of the American Association for Cancer Research. 2015;21:4831-44
17. Osman AA, Monroe MM, Ortega Alves MV, Patel AA, Katsonis P, Fitzgerald AL. et al. Wee-1 kinase inhibition overcomes cisplatin resistance associated with high-risk TP53 mutations in head and neck cancer through mitotic arrest followed by senescence. Molecular cancer therapeutics. 2015;14:608-19
18. Lal S, Zarei M, Chand SN, Dylgjeri E, Mambelli-Lisboa NC, Pishvaian MJ. et al. WEE1 inhibition in pancreatic cancer cells is dependent on DNA repair status in a context dependent manner. Scientific reports. 2016;6:33323
19. Van Linden AA, Baturin D, Ford JB, Fosmire SP, Gardner L, Korch C. et al. Inhibition of Wee1 sensitizes cancer cells to antimetabolite chemotherapeutics in vitro and in vivo, independent of p53 functionality. Molecular cancer therapeutics. 2013;12:2675-84
20. Porter CC, Kim J, Fosmire S, Gearheart CM, van Linden A, Baturin D. et al. Integrated genomic analyses identify WEE1 as a critical mediator of cell fate and a novel therapeutic target in acute myeloid leukemia. Leukemia. 2012;26:1266-76
21. Lescarbeau RS, Lei L, Bakken KK, Sims PA, Sarkaria JN, Canoll P. et al. Quantitative Phosphoproteomics Reveals Wee1 Kinase as a Therapeutic Target in a Model of Proneural Glioblastoma. Molecular cancer therapeutics. 2016;15:1332-43
22. Zhou L, Zhang Y, Chen S, Kmieciak M, Leng Y, Lin H. et al. A regimen combining the Wee1 inhibitor AZD1775 with HDAC inhibitors targets human acute myeloid leukemia cells harboring various genetic mutations. Leukemia. 2015;29:807-18
23. Ford JB, Baturin D, Burleson TM, Van Linden AA, Kim YM, Porter CC. AZD1775 sensitizes T cell acute lymphoblastic leukemia cells to cytarabine by promoting apoptosis over DNA repair. Oncotarget. 2015;6:28001-10
24. Richer AL, Cala JM, O'Brien K, Carson VM, Inge LJ, Whitsett TG. WEE1 Kinase Inhibitor AZD1775 Has Preclinical Efficacy in LKB1-Deficient Non-Small Cell Lung Cancer. Cancer research. 2017;77:4663-72
25. Saxton RA, Sabatini DM. mTOR Signaling in Growth, Metabolism, and Disease. Cell. 2017;169:361-71
26. Zhang Y, Kwok-Shing Ng P, Kucherlapati M, Chen F, Liu Y, Tsang YH. et al. A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/mTOR Pathway Alterations. Cancer cell. 2017;31:820-32 e3
27. Umemura S, Mimaki S, Makinoshima H, Tada S, Ishii G, Ohmatsu H. et al. Therapeutic priority of the PI3K/AKT/mTOR pathway in small cell lung cancers as revealed by a comprehensive genomic analysis. Journal of thoracic oncology: official publication of the International Association for the Study of Lung Cancer. 2014;9:1324-31
28. Christensen CL, Kwiatkowski N, Abraham BJ, Carretero J, Al-Shahrour F, Zhang T. et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer cell. 2014;26:909-22
29. Bayat Mokhtari R, Homayouni TS, Baluch N, Morgatskaya E, Kumar S, Das B. et al. Combination therapy in combating cancer. Oncotarget. 2017;8:38022-43
30. O'Neil J, Benita Y, Feldman I, Chenard M, Roberts B, Liu Y. et al. An Unbiased Oncology Compound Screen to Identify Novel Combination Strategies. Molecular cancer therapeutics. 2016;15:1155-62
31. Sen T, Tong P, Diao L, Li L, Fan Y, Hoff J. et al. Targeting AXL and mTOR Pathway Overcomes Primary and Acquired Resistance to WEE1 Inhibition in Small-Cell Lung Cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2017;23:6239-53
32. Tarhini A, Kotsakis A, Gooding W, Shuai Y, Petro D, Friedland D. et al. Phase II study of everolimus (RAD001) in previously treated small cell lung cancer. Clinical cancer research: an official journal of the American Association for Cancer Research. 2010;16:5900-7
33. Shao Q, Kannan A, Lin Z, Stack BC Jr, Suen JY, Gao L. BET protein inhibitor JQ1 attenuates Myc-amplified MCC tumor growth in vivo. Cancer research. 2014;74:7090-102
34. Kannan A, Lin Z, Shao Q, Zhao S, Fang B, Moreno MA. et al. Dual mTOR inhibitor MLN0128 suppresses Merkel cell carcinoma (MCC) xenograft tumor growth. Oncotarget. 2016;7:6576-92
35. Paul J, Soujon M, Wengner AM, Zitzmann-Kolbe S, Sturz A, Haike K. et al. Simultaneous Inhibition of PI3Kdelta and PI3Kalpha Induces ABC-DLBCL Regression by Blocking BCR-Dependent and -Independent Activation of NF-kappaB and AKT. Cancer cell. 2017;31:64-78
36. Obacz J, Avril T, Le Reste PJ, Urra H, Quillien V, Hetz C. et al. Endoplasmic reticulum proteostasis in glioblastoma-From molecular mechanisms to therapeutic perspectives. Science signaling. 2017:10
37. Verfaillie T, Garg AD, Agostinis P. Targeting ER stress induced apoptosis and inflammation in cancer. Cancer letters. 2013;332:249-64
38. Puthalakath H, O'Reilly LA, Gunn P, Lee L, Kelly PN, Huntington ND. et al. ER stress triggers apoptosis by activating BH3-only protein Bim. Cell. 2007;129:1337-49
39. Mounir Z, Krishnamoorthy JL, Wang S, Papadopoulou B, Campbell S, Muller WJ. et al. Akt determines cell fate through inhibition of the PERK-eIF2alpha phosphorylation pathway. Science signaling. 2011;4:ra62
40. Augert A, MacPherson D. Treating transcriptional addiction in small cell lung cancer. Cancer cell. 2014;26:783-4
41. Sakre N, Wildey G, Behtaj M, Kresak A, Yang M, Fu P. et al. RICTOR amplification identifies a subgroup in small cell lung cancer and predicts response to drugs targeting mTOR. Oncotarget. 2017;8:5992-6002
42. Hai J, Liu S, Bufe L, Do K, Chen T, Wang X. et al. Synergy of WEE1 and mTOR Inhibition in Mutant KRAS-Driven Lung Cancers. Clinical cancer research: an official journal of the American Association for Cancer Research. 2017;23:6993-7005
43. Cerezo M, Lehraiki A, Millet A, Rouaud F, Plaisant M, Jaune E. et al. Compounds Triggering ER Stress Exert Anti-Melanoma Effects and Overcome BRAF Inhibitor Resistance. Cancer cell. 2016;29:805-19
44. Rodvold JJ, Chiu KT, Hiramatsu N, Nussbacher JK, Galimberti V, Mahadevan NR. et al. Intercellular transmission of the unfolded protein response promotes survival and drug resistance in cancer cells. Science signaling. 2017:10
45. Lamothe B, Wierda WG, Keating MJ, Gandhi V. Carfilzomib Triggers Cell Death in Chronic Lymphocytic Leukemia by Inducing Proapoptotic and Endoplasmic Reticulum Stress Responses. Clinical cancer research: an official journal of the American Association for Cancer Research. 2016;22:4712-26
46. He K, Zheng X, Li M, Zhang L, Yu J. mTOR inhibitors induce apoptosis in colon cancer cells via CHOP-dependent DR5 induction on 4E-BP1 dephosphorylation. Oncogene. 2016;35:148-57
Author contact
![]() Corresponding authors: Ling Gao MD, PhD, Department of Dermatology, University of Arkansas for Medical Sciences, Little Rock, AR 72205. Phone: 501-526-4861; E-mail: LGaoedu and Tao Guo MD, PhD, Department of Hematology, Huazhong University of Science and Technology, Wuhan, Hubei, China. Email: guotao1968com
Corresponding authors: Ling Gao MD, PhD, Department of Dermatology, University of Arkansas for Medical Sciences, Little Rock, AR 72205. Phone: 501-526-4861; E-mail: LGaoedu and Tao Guo MD, PhD, Department of Hematology, Huazhong University of Science and Technology, Wuhan, Hubei, China. Email: guotao1968com