Impact Factor ISSN: 1449-2288
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Approaches to drug repositioning
Text mining-based approaches
Semantics-based approaches
Resources
Challenges and opportunities
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(10):1232-1244. doi:10.7150/ijbs.24612 This issue Cite
Review
Review of Drug Repositioning Approaches and Resources
Hanqing Xue, Jie Li*, ![]() , Haozhe Xie, Yadong Wang
, Haozhe Xie, Yadong Wang
School of Computer Science and Technology, Harbin Institute of Technology, 150001, Harbin, China
*Co-first author
Received 2017-12-28; Accepted 2018-6-12; Published 2018-7-13
Abstract

Drug discovery is a time-consuming, high-investment, and high-risk process in traditional drug development. Drug repositioning has become a popular strategy in recent years. Different from traditional drug development strategies, the strategy is efficient, economical and riskless. There are usually three kinds of approaches: computational approaches, biological experimental approaches, and mixed approaches, all of which are widely used in drug repositioning. In this paper, we reviewed computational approaches and highlighted their characteristics to provide references for researchers to develop more powerful approaches. At the same time, the important findings obtained using these approaches are listed. Furthermore, we summarized 76 important resources about drug repositioning. Finally, challenges and opportunities in drug repositioning are discussed from multiple perspectives, including technology, commercial models, patents and investment.
Introduction
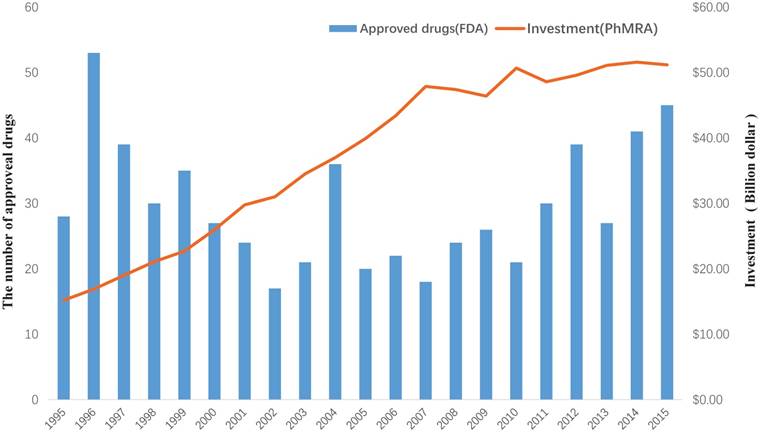
Drug discovery is a time-consuming, laborious, costly and high-risk process. According to a report by the Eastern Research Group (ERG) [1], it usually takes 10-15 years to develop a new drug. However, the success rate of developing a new molecular entity is only 2.01% [2], on average. As demonstrated in a report by the Food and Drug Administration (FDA), the number of drugs approved by the FDA has been declining since 1995 [3]. Moreover, investment in drug development has been gradually increasing, as reported by Pharmaceutical Research and Manufacturers of America (PhRMA) [4] (Figure 1). This indicates that the cost of new drug development will continue to grow. Hence, it is urgent to find a new strategy to discover drugs.
The investment in drug development by PhRMA member companies and the number of approved drugs by the FDA from 1995 to 2015.
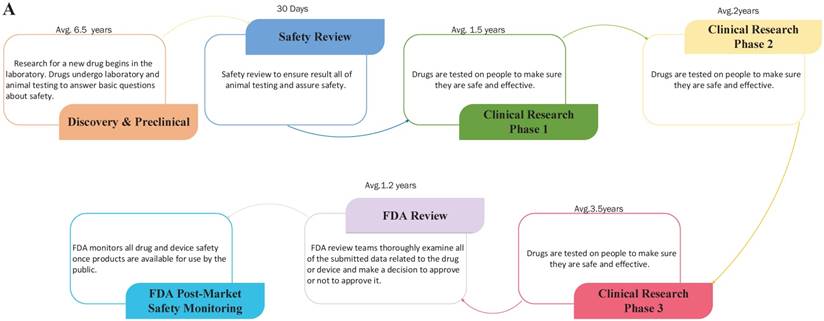
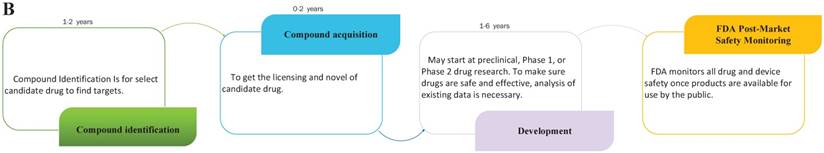
Drug repositioning, also known as old drugs for new uses, is an effective strategy to find new indications for existing drugs and is highly efficient, low-cost and riskless. Traditional drug development strategies usually include five stages: discovery and preclinical, safety review, clinical research, FDA review, and FDA post-market safety monitoring [4, 5]. However, there are only four steps in drug repositioning: compound identification, compound acquisition, development, and FDA post-market safety monitoring (Figure 2). Due to the fast growth of bioinformatics knowledge and biology big data, drug repositioning decreases the time cost of the drug development process significantly. Researchers only need 1-2 years to identify new drug targets and 8 years to develop a repositioned drug, on average [1]. Furthermore, the research and development investment required for drug repositioning is lower than that for traditional strategies. Drug repositioning breaks the bottlenecks of cost for many countries. It only costs $1.6 billion to develop a new drug using a drug repositioning strategy, while the cost of the traditional strategy is $12 billion [6]. Thus, drug repositioning offers an opportunity for many countries to develop drugs with lower investments.
The contrast of traditional drug development and drug repositioning. A) Flowchart of the traditional drug development process. B) Flowchart of drug repositioning.
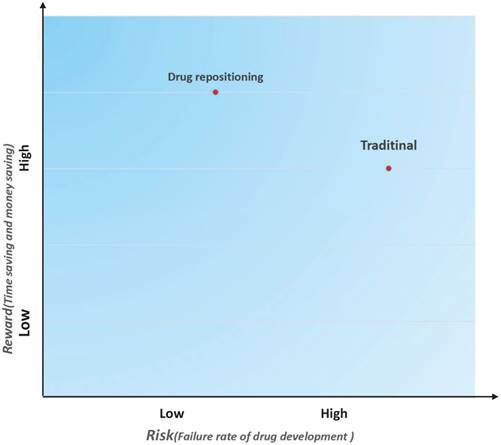
In addition to reducing the time cost and investment, drug repositioning is also a low-risk strategy. A risk-reward diagram is often used to describe the relationship between a risk and the reward on investment [7]. We drew a risk-reward diagram to compare repositioning and traditional drug development strategies (Figure 3). As shown in Figure 3, drug repositioning holds a higher reward with a lower risk. Because repositioned drugs have passed all clinical tests in Phase I, Phase II, and Phase III, their safety has been confirmed. In addition, some repositioned drugs may be marketed as molecular entities and have more opportunities to be pushed into the market once a new indication is discovered.
Risk and reward in two different drug development strategies
Approaches to drug repositioning
The main issue in drug repositioning is the detection of novel drug-disease relationships. To address this issue, a variety of approaches have been developed including computational approaches, biological experimental approaches and mixed approaches.
With the fast development of biology microarray techniques, various drug and disease knowledge databases such as DrugBank [8], ChemBank [9], OMIM [10], KEGG [11], and Pubmed [12] have appeared, and massive genomic databases such as MIPS[13], PDB [14], GEO [15], and GenBank [16] have been built (see Resource section for details). This knowledge and data further promoted the rapid development of a variety of novel computational approaches. Compared to biological experimental approaches, computational approaches have much lower costs and much fewer barriers [17]. In this review, we mainly introduce computational approaches.
Most existing computational approaches are based on the gene expression response of cell lines after treatment or merging several types of information about disease-drug relationships [18] that can be divided into different types from different viewpoints [19-21]. For instance, some researchers grouped drug repositioning methods according to the biological networks used [19], and others divided drug repositioning methods into two types: data-driven and hypothesis-driven [21]. However, the above studies did not focus on methodology. In this paper, we emphasized the core methodologies of drug repositioning approaches, so we divided them into three categories: network-based approaches [22-32], text-mining approaches [33-45] and semantic approaches [46-49].
Network-based approaches
Network-based approaches are widely used in drug repositioning due to the associated ability to integrate multiple data sources. These approaches have been proposed in the past few decades and became a hot topic approximately ten years ago. In this section, two types of network-based approaches are reviewed: network-based cluster approaches [22-24, 26] and network-based propagation approaches [27, 29, 31, 32, 50].
Network-based cluster approaches
Inspired by the fact that biologic entities (disease, drug, protein, etc.) in the same module of biological networks share similar characteristics, network-based cluster approaches have been proposed to discover novel drug-disease relationships or drug-target relationships. These approaches aim to find several modules (also known as subnetworks, groups or cliques) using cluster algorithms according to the topology structures of networks. These modules include various relationships such as drug-disease, drug-drug or drug-target relationships. The most common network-based cluster approaches, including DBSCAN [51], CLIQUE [52], STING [53], and OPTICS [54], cannot detect overlapping clusters. To address this problem, Lu et al. [55] studied the drug repositioning of SCLC (small-cell lung cancer) using a k-means-based network cluster algorithm. Chemical-chemical interactions and chemical-protein interactions were utilized to select candidate drug compounds that had close associations with approved lung cancer drugs and lung cancer-related genes. The experimental results revealed that the proposed algorithm predicted some drugs for treating SCLC, indications which were verified by retrieving references.
Tamas´ et al. [22] proposed a greedy network cluster approach named ClusterONE with two kernel components: cohesiveness score and greedy growth process. There are three steps in the approach: (i) generating growth groups from the greedy growth process using high-cohesiveness seed nodes, (ii) merging highly overlapping group pairs, (iii) discarding some complex groups according to the threshold. The advantage of the approach is its generality, and it can accurately predict not only disease-drug relationships in disease-drug networks but also protein interactions in protein-protein networks. Yu et al. [23] proposed an approach to construct disease-protein-drug networks based on a symmetrical conditional probability and detection of modules on the network via the ClusterONE algorithm. As a result, potential disease-drug interactions were found—for example, the authors discovered that iloperidone could be used to treat hypertension. Wu et al. [24] developed a novel approach through combining ClusterONE and Louvain [25] to detect modules in a heterogeneous network built from KEGG [11] disease-drug and drug-target data. They found 98 clusters and 1160 pairs of disease-drug interactions—for instance, vismodegib was predicted to treat Gorlin syndrome, while its original indication was basal cell carcinoma. Luo et al. [26] presented an approach named MBiRW with three steps: (i) calculating a comprehensive similarity between drugs and diseases; (ii) obtaining drug-drug subnetworks, disease-disease subnetworks and drug-disease subnetworks; and (iii) finding drug-disease relationships using a bi-random walk algorithm. Some novel disease-drug relationships such as Alzheimer's-levodopa were found (see Table 1).
Networks-based drug repositioning
| Name | Method | Network | Description | Key Findings | Advantage | Disadvantage | Ref. |
|---|---|---|---|---|---|---|---|
| RNSC | Cluster | PPI | A global network algorithm to identify protein clusters on PPI networks | Some complex proteins | This method considers both local and global information from networks. Overlap clusters can be detected as well. | Some information may be dropped because the cluster size is small. | [60] |
| RRW | Cluster | PPI | An effective network cluster approach to identify protein clusters on a PPI network | Some complex proteins | This is a general method with a high prediction accuracy. | It is a time-costly and memory-costly method that cannot detect overlap clusters. | [61] |
| ClusterONE | Cluster | PPI | A global network algorithm to identify node clusters on networks. | Some complex proteins | This approach outperformed the other approaches including MCL, RRW, etc., both on weighted and unweighted PPI networks. | There is no a gold standard to evaluate clusters. | [22] |
| - | Cluster | Drug-protein-disease | A variant of ClusterONE algorithm to cluster nodes on heterogeneous networks | (Iloperidone, schizophrenia) → Hypertension | This is an efficient cluster approach that integrates multiple databases. | It is difficult to distinguish between positive associations and negative associations on the network. | [23] |
| - | Cluster | Drug-target-disease | An algorithm to detect clusters on the network | (Vismodegib, Basal cell carcinoma) → Gorlin syndrome | This is a general and highly robust approach. | This approach loses weakly associate genes of diseases and drugs. | [24] |
| MBiRW | Cluster | Drug-disease | A bi-random walk- based algorithm to predict disease-drugs relationships. | (Levodopa, Parkinsonian disorder) → Alzheimer's (Cabergoline, Hyperprolactinemia) →Migraine | Predictions of this approach are reliable. | The approach needs to adopt more biological information to improve the confidence of the similarity metric. | [26] |
| - | Cluster | Drug-protein-chemical | A k-means-based network cluster algorithm on heterogeneous networks. | (Canertinib, Acute lymphoblastic leukemia) → SCLC | This approach is easy to implement. Predictions of this approach are reliable. | This approach needs to integrate multiple databases. | [55] |
| - | Propagation | Drug-target | An algorithm that combines four network-based approaches to predict drug-target relationships. | Melanoma's target cMyc was predicted | This approach is easy to implement. Predictions of this approach are reliable. | This approach needs to integrate multiple databases. | [55] |
| - | Propagation | Disease-protein-gene | A random walk-based network algorithm with a diffusion kernel to predict disease-gene relationships. | Some disease-gene relationships | This is a global efficient method that can be applied on other networkssuch disease-drug networks. | This approach can only be used for genes whose protein-protein relations are known. It does not perform well on small disease-gene family data. | [29] |
| PRINCE | Propagation | Disease-gene | A global propagation algorithm to predict disease-gene relationships. | Some disease-gene relationships | This is a global network approach combined with a novel normalization of protein-proteininteraction weightsand disease-diseasesimilarities. | This approach relies on phenotype data, so some diseases that lack phenotype information are excluded. The performance of this approach relies on data quality. | [31] |
| DrugNet | Propagation | Disease-drug-protein | A comprehensive propagation method to predict different propagation strategies in different subnets. | (Methotrexate, antimetabolite and antifolate)→ cancer (Gabapentin, epilepsy)→neuropathic pain | This method is robust and efficient. | The performance of this approach relies on the quality of disease data. | [31] |
Note: In key findings field, some records are organized as the form: (drug, origin indication) → new indication. For example: (Canertinib, Acute lymphoblastic leukemia) → SCLC
Network-based propagation approaches
Network-based propagation approaches are another important type of network-based approach. The workflow of these approaches is that prior information propagates from the source node to all network nodes and some subnetwork nodes. According to the different propagation ways, these approaches can be divided into two types: local approaches and global approaches. Several studies have proven that these methods perform well in finding disease-targets, disease-genes and disease-drug relationships [27]. Local propagation approaches only take the limited information of the network into account and may fail to make correct predictions [28] in some cases. By contrast, global approaches containing information from the entire network perform better than local approaches. Most current researchers concentrate on global approaches to achieve outstanding performance. For example, Kohle et al. [29] developed a network propagation approach based on the global information of a network to find novel disease-gene interactions. The approach included three phases: (i) extracting drug-disease relationships and constructing a disease-gene network; (ii) obtaining the global information of the network using a random walk propagation algorithm [56] in the network; and (iii) defining global metrics to predict novel disease-gene relationships. The proposed approach performed better than other approaches, including the diffusion kernel approach, PROSPECTR [30]. In addition, cross-validation showed that the accuracy of disease-gene predictions is 98%. Vanunu et al. [31] also proposed a global approach for finding disease-gene and disease-protein relationships via a network propagation approach called PRINCE. The method is based on formulating constraints on a score function related to the smoothness of the disease-gene network. In the proposed method, gene nodes adopt prior information as input and then pump this information to their neighbor node until convergence. The score function gives a confidence level for each predicted disease-gene pair. PRINCE was evaluated on 1369 disease data points from OMIM and could predict unknown causal genes of some diseases such as type 2 diabetes, Alzheimer's disease and prostate cancer. Martinez et al. [32] presented a disease-gene-drug network propagation approach wherein two different propagation approaches were defined: propagation in homogeneous subnetworks (such as gene subnetworks) and propagation between subnetworks. They used a prioritization function to measure the correlation between drugs and diseases. As a consequence, a list of drugs was produced for a queried disease. Novel indications of some drugs such as methotrexate, gabapentin, cisplatin, donepezil, and risperidone were obtained using this approach. In addition, Emig et al. [27] proposed a comprehensive approach combining 4 local and global network approaches through a logistic regression model. The approach was evaluated on 30 different diseases with known drug targets and yielded an AUC (area under the curve) above 80%. Furthermore, melanoma's drug target c-Myc was successfully predicted, and this finding was also confirmed by two other experiential studies. [57, 58]
Network-based approaches are vital for drug repositioning. Researchers often need to make a decision in selecting appropriate approaches, and we summarized these approaches in Table 1 and listed their benefits, bottlenecks, key findings, databases and other information. The networks employed in these approaches can be divided into two classes: homogeneous and heterogeneous. As disease pathways can be constructed from protein-protein interaction (PPI) network analysis [59], a protein-protein interaction (PPI) network, as a homogeneous network, is often employed in some approaches [22, 60, 61] used to identify drug targets involved with multiple pathways. Along with the deeper research associated with the network, the accuracy of PPI networks has been enhanced because numerous PPI databases have been established and updated by experiments. However, PPI networks are limited since they only include protein information without considering additional information. With the advent of the era of big data, the accumulation of various medical data (such as drugs, diseases, and targets data, among others) has made it possible to construct complex heterogeneous networks. Heterogeneous networks, which integrate multiple sources of information including genomes, proteomes and metabolic pathways [62], typically contain two-layer (i.e., disease-drug) or three-layer (i.e., disease-drug-gene) relationships and have attracted researchers' attention. Different biological entities included in the heterogeneous networks not only provide an opportunity to improve the performance of existing methods but also offer a tool to design more efficient and stable approaches [24, 26, 27, 29, 31, 55, 58] (see table 1).
From the method perspective, network-based cluster algorithms are frequently used to find interesting modules, and network-based propagation algorithms are often used to infer new relationships between biological entities. Network-based cluster approaches are general because most network-based cluster algorithms can be employed for detecting biological modules. For example, some cluster algorithms in the social network analysis field can be employed for detecting modules in biological networks [25, 63]. However, there is a challenge of network-based cluster approaches in that there exists no gold standard to test associations among biological modules. Network-based propagation approaches are easy to implement and can make accurate predictions. Researchers can obtain an AUC value and estimate the prediction results. In addition, network-based propagation approaches use not only information from the selected components but also information from expanding components.
Text mining-based approaches
Along with the exploration of drug repositioning, a great deal of medical and biological literature containing fruitful novel biological entity relationships have been published. There is a large challenge in extracting novel and valuable biological entity relationships from the literature. Text mining (TM) techniques have been widely used to address this problem and have been increasingly developed to mine new knowledge from scientific literature and identify connections between biological concepts or biological entities.
Marti Hearst gave a general definition for text mining as 'text mining is the discovery by computer of new, previously unknown information, by automatically extracting information from different written resources' [64]. The main pipeline of biological text mining includes four phrases: information retrieval (IR), biological name entity recognition (BNER), biological information extraction (BIE) and biological knowledge discovery (BKD) (Figure 4). In the IR step, relevant documents are extracted from the literature. These relevant documents need to be filtered because there are some useless concepts in documents. In the BNER step, valuable biological concepts are identified with controlled vocabularies. In the BIE and BKD steps, useful information is extracted to discover knowledge about biological concepts and build a knowledge graph. At the same time, potential associations between knowledge, such as drug-disease and drug-target relationships, can also be detected.
The workflow of text mining.
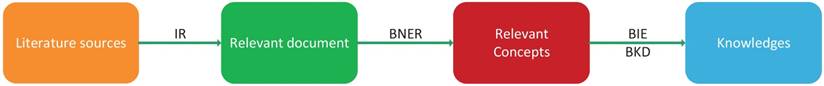
The origin of text mining methods in the medical field is the Swanson 'ABC' model, which states that if concept A is connected with concept B, and concept B is involved with concept C, then concept A may have a novel connection with concept C [65]. Based on the 'ABC' model, various text mining methods have been proposed to find potential disease-drug relationships in the literature. A number of studies have been devoted to applying text mining techniques in drug repositioning. Li et al. [33] developed an approach to building disease-specific drug-protein connectivity maps combining network mining and text mining. In the proposed method, they first extracted disease-protein relationships from molecular interaction networks using network mining. Then, they searched for drug terms indirectly associated with certain diseases such as Alzheimer's disease (AD) in PubMed abstracts through text mining techniques. Finally, drugs and proteins could be linked through drug-disease or disease-protein relationships.
As a result, diltiazem and quinidine are hypertension and arrhythmia drugs, respectively, but authors have also found that the two drugs could be used to cure Alzheimer's disease, which has been confirmed by clinical evidence. Ruggero et al. [34] proposed an approach to building sentence graph networks using text mining techniques. The proposed network can be used to discover relationships between any drug and any disease. These relationships are specific paths among the biomedical entities in the graph network. A novel disease target of sarcoidosis was identified using this approach. Rastegar et al [66] extracted drug-gene and gene-disease relationships from medical abstracts to obtain drug-disease relationships using a rank score. To evaluate the performance of the proposed approach, the obtained drug-disease relationships were validated in the Comparative Toxicogenomics Database (CTD). Experimental results indicated that the discovered relationships confirmed in CTD were highly confident. Jang et al. [35] developed an approach to building dependency graph networks through extracting sentences with genes, drugs and phenotypes from biomedical literature. They calculated the possibility that a drug treats a phenotype based on drug-phenotype associations. The authors compared the predicted drug-phenotype associations using known drug-phenotype associations in databases and proved the good performance of their method. Kuusisto et al [36] proposed another text mining method named KinderMiner to identify potential indications of some old drugs. The method is based on co-occurrence statistics between drugs and diseases in the literature. As a result, new indications of some drugs such as Zestoretic, Zebeta, and Tiazac were found. Zhang et al. [37] reported an algorithm to prioritize anti-AD (Alzheimer's disease) targets. The authors extracted 224 genetic variations, 14 epigenetic modifications, 98 proteins and 86 metabolites associated with AD using text mining and integrated these interactions to construct a weighted sum model to prioritize potential anti-AD drug targets.
With the development of natural language processing (NLP) techniques, increasing numbers of text mining tools have been developed and used to discover repositionable drugs (see [38, 39]). Here, we summarized the inputs, outputs and characteristics of these tools in Table 2. The inputs of these methods are usually biological terms extracted from existing literature, and the corresponding outputs are lists of relationships of biological terms. The confidence levels of the relationships are generally evaluated using computational approaches or biological experiments. These tools can be divided into two categories: static tools and dynamic tools.
Text mining tools for drug repositioning.
| Name | Class | Input | Output | Description | Web Site | Ref |
|---|---|---|---|---|---|---|
| Biovista | Static | Biological knowledge | Gene-protein relationships | A mining framework to extract gene-protein relationships. | http://www.biovista.com/ | [68] |
| BioWisdom | Static | Ontology | Drug-disease, drug-target relationships | A platform to discover novel biological entity relationships. | http://www.biowisdom.com | [39] |
| FACTA+ | Static | Tekst | Abstracts and linked concepts | A system to find associated concepts based on a user query | http://www.nactem.ac.uk/facta/cepts based on a user query | [102] |
| EDGAR | Static | UMLS terms | Drug-gene relationships | A system to extract relationships between drugs and genes involved in cancer using syntactic analysis | https://www.sec.gov/ | [103] |
| PolySearch | Dynamic | Bio-entities | Drug-disease, Drug-gene relationships | A web service to extract links between biological terms | http://wishart.biology.ualberta.ca | [41] |
| TextFlow | Dynamic | Document | Knowledge | A web-based text mining and natural language processing platform | http://textflows.org/ | [42] |
| EXTRACT2 | Static | Bio-entities | Entity relationships | A text mining-based tool to map biological entities to ontology/taxonomy entries | http://extract.jensenlab.org/ | [104] |
| Anni 2.0 | Static | Bio-entities | Linked concepts | An ontology interface of a text mining tool to extract conceptsrelationships | http://biosemantics.org/anni | [69] |
| DrugQuest | Static | Drugs | Drug-drug relations | A knowledge discovery tool todetect drug-drug relationships | http://bioinformatics.med.uoc.gr | [40] |
| MaNER | Dynamic | Medical Document | Relevant entities | A rule-based system to mine relevant entities in medical documents | - | [43] |
| BEST | Dynamic | Biomedical Literature | Relevant bio-entities | A knowledge discovery system to extract relevant bio-entities. | http://best.korea.ac.kr. | [44] |
| Alibaba | Dynamic | Bio-entities | Linked concepts | A tool to fit a PubMed query as a graphical network | - | [45] |
Static tools were built on steady databases or documents with large size. Due to the large data size, the time performance of these tools was poor. To address this problem, indexes for documents or records were created to accelerate the query process in static tools. For example, DrugQuest [40] is a type of query tool for detecting drug-drug relationships. The workflow of this tool includes five stages. (i) Query, in which users provide a query term to retrieve related documents. (ii) Name entity recognition, which identifies proteins, chemicals and pathway terms in related documents using a biomedical concept recognition service named BeCAS [67] and identifies significant terms closely associated with the query by calculating the TF-IDF score (Term Frequency - Inverse Document Frequency) to measure the importance of terms. (iii) Building document network, which uses the similarity of documents. (iv) Clustering, in which various clustering algorithms (MCL, K-means, hierarchical clusters) are employed to cluster documents on the network. (v) Visualization, in which the 'tag cloud' technique is used for representing cluster results. The DrugQuest tool is promising for knowledge discovery and drug-drug relation prediction. However, the proposed tool only supports the DrugBank database, which leads to limitations of the query results. Other query tools [39, 40, 68, 69] were also designed in a similar way (see table 2).
Static tools usually cause outdated result problems. To address this issue, many dynamic tools [41-45] that update their document databases daily were developed. However, these tools also need more time to handle user queries. To reduce the time cost of queries, cache and index techniques were employed in dynamic tools. For example, PolySearch2 [41] used a cache technique to reduce the response time, and BEST [44] used an indexing technique to reduce the computation time. BEST is a biomedical search tool that returns a list of 10 different types of biomedical entities including genes, diseases, drugs, targets, transcription factors, miRNAs, and mutations for a query. The proposed tool consists of two parts: an indexing subsystem and a search subsystem. In the indexing subsystem, the authors used a dictionary-based approach to extract entities from the text and create a document-entity to list paired indexes. To avoid the outdated results problem, the tool automatically downloads abstracts newly indexed from the PubMed system and updates the document-entity indexes every day. In the search subsystem, the proposed tool utilizes the inverted index to obtain matched query terms. All entities obtained from the query are ranked according to their integrated entity scores involving entities and query terms included in all documents. BEST is a real-time and constantly updated tool, for which the time performance and output quality are both considered.
Text mining tools reduce the time complexity of drug repositioning and assist researchers in verifying their experimental results by returning massive amounts of biological entity relationships. However, there are still some issues that need to be addressed. For example, the limited coverage problem is one limitation of text mining tools, which means that partially important biomedical entities or relationships such as mutations, targets, and drugs are not considered. Therefore, there is an urgent need to improve the performance of existing text mining tools.
Semantics-based approaches
Semantics-based approaches are widely used in information retrieval, image retrieval and other fields. Recently, these methods have been applied to drug repositioning. The workflow of these methods mainly includes three steps (Figure 5). First, biological entity relationships are extracted from prior information in massive medical databases to build the semantic network. Then, semantics networks based on existing ontology networks are constructed by adding the prior information obtained in the previous step. Finally, mining algorithms are designed to predict novel relationships in the semantic network.
The workflow of a semantic network inference.
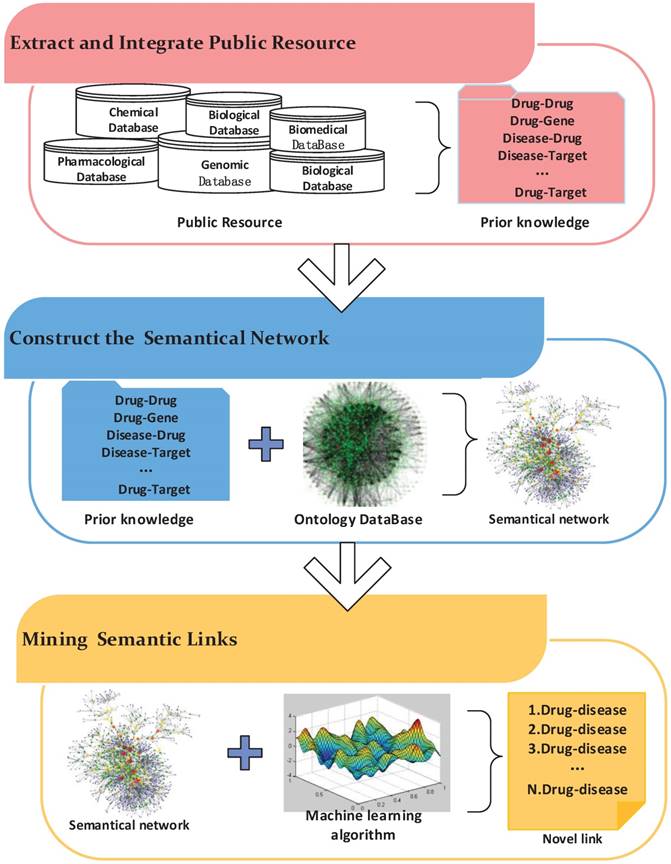
Based on a hypothesis in which similar drugs are correlated with similar targets and similar targets are connected to similar drugs, Guillermo et al. [46] proposed an unsupervised algorithm to predict drug-target relationships. The authors constructed a semantic network including drug-drug, target-target, and drug-target relationships. The proposed approach, which combines semantics link prediction methods and edge partition methods, was evaluated on a network. Due to substantial semantics knowledge being used, the proposed method made accurate predictions about drug-target relationships. Mullell et al. [47] presented a semantics data-driven algorithm for drug repositioning. The authors used a Bayesian statistics approach to rank drug-disease relationships according to prior knowledge. Then, they integrated ranked relationships with other biological entity associations to construct a semantical drug discovery network. To infer drug-disease relationships, the author applied an algorithm for detecting semantic subgraphs. As a result, nitrendipine, a potent blocker of the calcium channel (CACNA1S) used to treat hypokalemic periodic paralysis, was found.
Chen et al. [48] built a semantic linked network consisting of over 290,000 nodes and 720,000 edges with multisource data including drugs, targets, proteins, and disease pathways. Then, the authors applied a statistical model to predict drug-target relationships. Consequently, the proposed model identified some drug-targets pairs and drugs for repurposing. For example, barbiturate, a drug used for treating migraines, was predicted for use in curing insomnia with literature support. Zhu et al. [49] proposed an automatic reasoning approach for heterogeneous semantics networks. Biological entities (such as drugs) are converted to labels in a semantics network. Then, disease-drug relationships are obtained from automatic reasoning techniques. As a demonstration, the authors reported that tamoxifen, a drug used for treating breast cancer, can treat ovarian cancer, which was confirmed by the literature [70].
Semantics-based approaches take full advantage of semantics information included in massive amounts of literature. Therefore, the precision of predicting biological entity relationships was improved. However, there is a still challenge in constructing a semantic network by integrating multisource data. It is urgent to construct semantic networks that contain fruitful medical data.
Resources
Along with the development of biological technology and the accumulation of various omics data (genomics, proteomics or metabolomics, etc.), more databases from chemical, medical, pharmacological and biological fields have been established. We summarized 80 widely used databases or resources that can be used for designing drug repositioning approaches.
Pharmacological databases [8, 71-76] are crucial resources for drug repositioning. These databases collect not only drug property data but also data on interactions between drugs and other biological entities. Pharmacological data lays the foundation of various computational approaches. For instance, DrugBank [8] is a unique bioinformatics and cheminformatics resource that combines detailed drug data with comprehensive drug target information. Many computational approaches, especially network-based approaches [23-26], were designed based on this database, which achieved excellent results (see table 1).
Proteomics databases [13, 14, 76-78] are another type of the data resource for drug repositioning. Most important protein-protein interaction (PPI) networks from proteomics databases are the basis of network-based drug repositioning approaches. In addition, proteomics databases are also important resources for building heterogeneous networks such as drug-protein-disease networks. One of the famous proteomics databases is MIPS [13], which includes manual high-quality PPI data from scientific literature. MIPS can provide PPI information for some network approaches [61, 77]. Moreover, it is a good resource for evaluating the experimental results of some computational approaches.
Chemical features of drugs provide important information for designing chemical-based approaches. Publicly available databases [9] [79-84] of chemical structures contain massive amounts of useful information such as 2D topological fingerprints and 3D conformations. Chemical information on drugs is usually employed for predicting novel drug structures to find new indications for drugs having similar structures. PubChem [85] is a famous database for chemical molecules structures. The database contains a massive amount of 2D data that can be used to measure the similarity of drugs and to construct chemical networks.
With further studies of drug repositioning, enormous amounts of medical and biological literature that contain novel biological entities relations have been published. Collecting massive amounts of literature is a necessary task for researchers; hence, many medical literature databases [10, 12, 86] have been built. Based on these valuable medical textual data, text mining approaches were proposed for drug repositioning (see Section Text mining-based approaches). PubMed [12] is the most widely used literature database. It comprises more than 27 million citations for biomedical literature from MEDLINE, life science journals, and online books. Citations may include links to full-text content from PubMed Central and publisher websites. Many text mining tools and search engines have been built using these databases (Table 2).
Although increasingly more databases are being established, choosing proper approaches to mine novel knowledge is still a large challenge. It is necessary to discover the potential value of the increasing databases and medical literature for designing efficient drug repositioning approaches. We collected these resources in supplementary materials Tables S2 to help researchers choose proper approaches.
Challenges and opportunities
Traditional drug development strategies are costly, failure-prone, and expensive ventures. Therefore, drug repositioning has recently drawn attention and brings drugs out faster for clinical use. However, drug repositioning is a complex process involving multiple factors such as technology, commercial models, patents, investment and market demands. Although many medical databases have been established, selecting the appropriate approach to make full use of massive amounts of medical data is still a challenge. It is urgent to develop new approaches for drug repositioning. The intellectual property (IP) issue is another highlighted issue to be solved. For repositioning drugs, IP protection is limited [87]. For example, some novel drug-target-disease associations found by repositioning researchers were confirmed by publications or online databases; however, it is difficult to seek IP protection for such associations because of the law. The IP issue prevents some repositioned drugs from entering the market. Moreover, some repositioning projects are forced to be abandoned, which is a waste of time and money [88]. It is necessary to develop a new commercial model because the traditional commercial model is a serial model and causes overlapping investment issues.
Opportunities come with challenges. The first example of drug repositioning was an accidental discovery in the 1920s. After about a century of development, more approaches have been proposed for accelerating the process of drug repositioning. For this reason, drug repositioning has acquired great achievements. In supplementary materials Table S1, we list 75 drug repositioning examples collected from the comprehensive literature. Massive machine learning algorithms were introduced to improve the performance of drug repositioning in this scenario. In addition to computational approaches, experimental approaches that give direct evidence of links between drugs and diseases were developed, such as target screening approaches [87-91], cell assay approaches [92-95], animal model approaches [96-99] and clinical approaches [49]. These approaches are reliable and credible. In recent years, increasing numbers of researchers have combined computational approaches and experimental approaches to find new indications for drugs, called mixed approaches [59, 100, 101], wherein the result of computational methods was validated by biological experiments and clinical tests. Mixed approaches offer opportunities for developing repositioned drug effectively and rapidly.
Generating secondary patents provides an opportunity for researchers to find new indications for existing drugs. With the IP problem solved, many repositioning projects have been conducted smoothly with low cost, which has aroused concern from many countries. With regard to the commercial model, parallel strategies bring significant improvement in the efficiency of drug repositioning. For example, multiple tests or studies are conducted for a candidate drug, which reduces the time cost for drug repositioning.
From the market perspective, a large number of diseases require new drugs to be treated, which brings potential economic benefits. Taking rare diseases as an example, there are over 6000 rare diseases that need to be studied. However, only 5% of them are being researched [79]. Rare diseases are a large potential market to explore.
Supplementary Material
Supplementary tables.
Acknowledgements
This work is partially supported by the National Natural Science Foundation of China (61471147, 61371179), the Natural Science Foundation of Heilongjiang Province (F2016016), the Fundamental Research Funds for the Central Universities (HIT.NSRIF.2017037), and the National Key Research and Development Program of China (2016YFC0901905).
Author contributions
Jie Li designed the paper and is the first coauthor, Hanqing Xue and Jie Li analyzed and wrote the manuscript, Haozhe Xie reviewed the manuscript, and Yadong Wang participated in the preparation of the manuscript.
Data availability statement
The data supporting the findings of this work are contained within the manuscript.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Sertkaya A, Birkenbach A, Berlind A, Eyraud J. Examination of clinical trial costs and barriers for drug development. US Department of health and human services, office of the assistant secretary for planning and evaluation report. 2014;1:1-92
2. Yeu Y, Yoon Y, Park S. Protein localization vector propagation: a method for improving the accuracy of drug repositioning. Mol Biosyst. 2015;11(7):2096-102
3. Pharmaceutical Research and Manufacturers of America. http://www.phrma.org/sites/default/files/pdf/2015_phrma_profile.pdf.2015
4. Drug development process. https://www.fda.gov/Drugs/default.htm
5. Drug approval process. https://www.fda.gov/Drugs/DevelopmentApprovalProcess/default.htm
6. Deotarse P. P.1, Jain A. S.1, Baile. M. B, et al. Drug repositioning: a review. Int J Pharma Res Rev. 2015;4:51-58
7. Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discov. 2004;3:673-83
8. Wishart DS, Knox C, Guo AC. et al. DrugBank: a comprehensive resource for in silico drug discovery and exploration. Nucleic Acids Res. 2006;34:668-72
9. Seiler KP, George GA, Happ MP. et al. ChemBank: a small-molecule screening and cheminformatics resource database. Nucleic Acids Res. 2008;36:351-9
10. Hamosh A, Scott AF, Amberger JS, Bocchini CA, Mckusick VA. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005;33:D514-7
11. Ogata H, Goto S, Sato K, Fujibuchi W, Bono H, Kanehisa M. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 1999;27:29-34
12. PubMed: US National Library of Medicine National Institutes of Health. https://www.ncbi.nlm.nih.gov/pubmed/
13. Mewes HW, Hani J, Pfeiffer F, Frishman D. MIPS: a database for protein sequences and complete genomes. Nucleic Acids Res. 1998;26:33-7
14. Bernstein FC, Koetzle TF, Williams GJ. et al. The Protein Data Bank: a computer-based archival file for macromolecular structures. J Mol Biol. 1977;112:535-42
15. Barrett T, Wilhite SE, Ledoux P. et al. NCBI GEO: archive for functional genomics data sets-update. Nucleic Acids Res. 2013;41:D991-5
16. Benson DA, Cavanaugh M, Clark K, Karsch-Mizrachi I, Lipman DJ, Ostell J. et al. GenBank. Nucleic Acids Res. 2012;41:36-42
17. Oprea TI, Overington JP. Computational and Practical Aspects of Drug Repositioning. Assay Drug Dev Technol. 2015;13:299-306
18. Napolitano F, Zhao Y, Moreira VM. et al. Drug repositioning: a machine-learning approach through data integration. J Cheminform. 2013;5:30
19. Lotfi shahreza M, Ghadiri N, Mousavi SR, Varshosaz J, Green JR. A review of network-based approaches to drug repositioning. Brief Bioinformatics. 2017
20. Zou J, Zheng MW, Li G, Su ZG. Advanced systems biology methods in drug discovery and translational biomedicine. Biomed Res Int. 2013;2013:742835
21. Gönen M. Predicting drug-target interactions from chemical and genomic kernels using Bayesian matrix factorization. Bioinformatics. 2012;28:2304-10
22. Wu H, Gao L, Dong J, Yang X. Detecting overlapping protein complexes by rough-fuzzy clustering in protein-protein interaction networks. PLoS ONE. 2014;9(3):e91856
23. Yu L, Huang J, Ma Z, Zhang J, Zou Y, Gao L. Inferring drug-disease associations based on known protein complexes. BMC Med Genomics. 2015;8(Suppl 2):S2
24. Wu C, Gudivada RC, Aronow BJ, Jegga AG. Computational drug repositioning through heterogeneous network clustering. BMC Syst Biol. 2013;7(Suppl 5):S6
25. Subelj L, Bajec M. Unfolding communities in large complex networks: combining defensive and offensive label propagation for core extraction. Phys Rev E Stat Nonlin Soft Matter Phys. 2011;83:036103
26. Luo H, Wang J, Li M. et al. Drug repositioning based on comprehensive similarity measures and Bi-Random walk algorithm. Bioinformatics. 2016;32:2664-71
27. Emig D, Ivliev A, Pustovalova O. et al. Drug target prediction and repositioning using an integrated network-based approach. PLoS ONE. 2013;8(4):e60618
28. Mei J-P, Kwoh C-K, Yang P. et al. Drug-target interaction prediction by learning from local information and neighbors. Bioinformatics. 2012;29:238-45
29. Köhler S, Bauer S, Horn D, Robinson PN. Walking the interactome for prioritization of candidate disease genes. Am J Hum Genet. 2008;82(4):949-58
30. Adie EA, Adams RR, Evans KL, Porteous DJ, Pickard BS. Speeding disease gene discovery by sequence based candidate prioritization. BMC bioinformatics. 2005;6:55
31. Vanunu O, Magger O, Ruppin E, Shlomi T, Sharan R. Associating genes and protein complexes with disease via network propagation. PLoS Comput Biol. 2010;6(1):e1000641
32. Martínez V, Navarro C, Cano C, Fajardo W, Blanco A. DrugNet: network-based drug-disease prioritization by integrating heterogeneous data. Artif Intell Med. 2015;63(1):41-9
33. Li J, Zhu X, Chen JY. Building disease-specific drug-protein connectivity maps from molecular interaction networks and PubMed abstracts. PLoS Comput Biol. 2009;5:e1000450
34. Gramatica R, Di Matteo T, Giorgetti S, Barbiani M, Bevec D, Aste T. Graph theory enables drug repurposing-how a mathematical model can drive the discovery of hidden mechanisms of action. PloS one. 2014;9:e84912
35. Jang G, Lee T, Lee BM, Yoon Y. Literature-based prediction of novel drug indications considering relationships between entities. Mol Biosyst. 2017;13:1399-405
36. Kuusisto F, Steill J, Kuang Z, Thomson J, Page D, Stewart R. A Simple Text Mining Approach for Ranking Pairwise Associations in Biomedical Applications. AMIA Jt Summits Transl Sci Proc. 2017;2017:166-174
37. Zhang M, Schmitt-ulms G, Sato C. et al. Drug Repositioning for Alzheimer's Disease Based on Systematic 'omics' Data Mining. PLoS ONE. 2016;11(12):e0168812
38. Fleuren WW, Alkema W. Application of text mining in the biomedical domain. Methods. 2015;74:97-106
39. Krallinger M, Erhardt RA-A, Valencia A. Text-mining approaches in molecular biology and biomedicine. Drug discovery today. 2005;10:439-45
40. Papanikolaou N, Pavlopoulos GA, Theodosiou T, Vizirianakis IS, Iliopoulos I. DrugQuest - a text mining workflow for drug association discovery. BMC Bioinformatics. 2016;17(Suppl 5):182
41. Cheng D, Knox C, Young N. et al. PolySearch: a web-based text mining system for extracting relationships between human diseases, genes, mutations, drugs and metabolites. Nucleic Acids Res. 2008;36:W399-W405
42. Perovšek M, Kranjc J, Erjavec T, Cestnik B, Lavrač N. TextFlows: A visual programming platform for text mining and natural language processing. Science of Computer Programming. 2016;121:128-52
43. Moreno I, Moreda P, Romá-Ferri MT. MaNER: a MedicAl named entity recogniser. Applications of Natural Language to Information Systems. 2015:418-23
44. Lee S, Kim D, Lee K, Choi J, Kim S, Jeon M. et al. BEST: next-generation biomedical entity search tool for knowledge discovery from biomedical literature. PloS one. 2016;11:e0164680
45. Plake C, Schiemann T, Pankalla M, Hakenberg J, Leser U. AliBaba: PubMed as a graph. Bioinformatics. 2006;22:2444-5
46. Palma G, Vidal M-E, Raschid L. Drug-target interaction prediction using semantic similarity and edge partitioning. ISWC. 2014;1:131-46
47. Mullen J, Cockell SJ, Woollard P, Wipat A. An integrated data driven approach to drug repositioning using gene-disease associations. PloS ONE. 2016;11:e0155811
48. Chen B, Ding Y, Wild DJ. Assessing drug target association using semantic linked data. PLoS Comput Biol. 2012;8:e1002574
49. Zhu Q, Tao C, Shen F, Chute CG. Exploring the pharmacogenomics knowledge base (PharmGKB) for repositioning breast cancer drugs by leveraging Web ontology language (OWL) and cheminformatics approaches. Pac Symp Biocomput. 2014;1:172-82
50. Sun Y, Sheng Z, Ma C. et al. Combining genomic and network characteristics for extended capability in predicting synergistic drugs for cancer. Nat Commun. 2015;6:8481
51. Sander Jr, Ester M, Kriegel H-P, Xu X. Density-based clustering in spatial databases: the algorithm gdbscan and its applications. Data mining and knowledge discovery. 1998;2:169-94
52. Agrawal R, Gehrke J, Gunopulos et al. Automatic subspace clustering of high dimensional data for data mining applications. ACM. 1998;27:94-105
53. Wang W, Yang J, Muntz R. STING: A statistical information grid approach to spatial data mining. VLDB. 1997;1:186-95
54. Ankerst M, Breunig M. M, Kriegel, et al. OPTICS: ordering points to identify the clustering structure. ACM. 1999;28:49-60
55. Lu Jing. et al. Identification of new candidate drugs for lung cancer using chemical-chemical interactions, chemical-protein interactions and a K-means clustering algorithm. J Biomol Struct Dyn. 2016;34:906-917
56. Lovasz Ls. Random walks on graphs. Combinatorics, Paul erdos is eighty. 1993;2:4
57. Hermeking H. The MYC oncogene as a cancer drug target. Curr Cancer Drug Targets. 2003;3:163-75
58. Wang W, Yang S, Zhang X, Li J. Drug repositioning by integrating target information through a heterogeneous network model. Bioinformatics. 2014;30:2923-30
59. Ai N, Wood RD, Welsh WJ. Identification of nitazoxanide as a group imetabotropic glutamate receptor negative modulator for the treatment of neuropathic pain: an in silico drug repositioning study. Pharm Res. 2015;32:2798-807
60. King AD, Przulj N, Jurisica I. Protein complex prediction via cost-based clustering. Bioinformatics. 2004;20:3013-20
61. Macropol K, Can T, Singh AK. RRW: repeated random walks on genome-scale protein networks for local cluster discovery. BMC Bioinformatics. 2009;10:283
62. Wu Z, Wang Y, Chen L. Network-based drug repositioning. Mol Biosyst. 2013;9:1268-81
63. Frey BJ, Dueck D. Clustering by passing messages between data points. Science. 2007;315:972-6
64. Hearst MA. Untangling text data mining. Proceedings of the 37th annual meeting of the association for computational linguistics on computational linguistics: association for computational linguistics. 1999;1:3-10
65. Weeber Marc. et al. Using concepts in literature-based discovery: simulating swanson's raynaud-fish oil and migraine-magnesium discoveries. J Am Soc Inf Sci Technol. 2001;52:972-6
66. Rastegar-Mojarad M, Elayavilli RK, Li D, Prasad R, Liu H. A new method for prioritizing drug repositioning candidates extracted by literature-based discovery. Bioinformatics and Biomedicine (BIBM). 2015;1:669-74
67. Nunes T, Campos D, Matos S, Oliveira JL. BeCAS: biomedical concept recognition services and visualization. Bioinformatics. 2013;29:1915-6
68. Lekka E, Deftereos SN, Persidis A, Persidis A, Andronis C. Literature analysis for systematic drug repurposing: a case study from Biovista. Drug Discov Today Biosilico. 2011;8:103-8
69. Jelier R, Schuemie MJ, Veldhoven A, Dorssers LC, Jenster G, Kors JA. Anni 2.0: a multipurpose text-mining tool for the life sciences. Genome Biol. 2008;9(6):R96
70. Lee JY, Shin JY, Kim HS. et al. Effect of combined treatment with progesterone and tamoxifen on the growth and apoptosis of human ovarian cancer cells. Oncol Rep. 2012;27:87-93
71. Pacini C, Iorio F, Gonçalves E. et al. DvD: An R/Cytoscape pipeline for drug repurposing using public repositories of gene expression data. Bioinformatics. 2013;29(1):132-4
72. Liu Y, Hu B, Fu C, Chen X. DCDB: drug combination database. Bioinformatics. 2009;26:587-8
73. Ekins S, Bunin BA. The Collaborative Drug Discovery (CDD) database. Methods Mol Biol. 2013;993:139-54
74. Fu C, Jin G, Gao J, Zhu R, Ballesteros-villagrana E, Wong ST. DrugMap Central: an on-line query and visualization tool to facilitate drug repositioning studies. Bioinformatics. 2013;29:1834-6
75. Barabási AL, Gulbahce N, Loscalzo J. Network medicine: a network-based approach to human disease. Nat Rev Genet. 2011;12:56-68
76. Thorn CF, Klein TE, Altman RB. PharmGKB: the Pharmacogenomics Knowledge Base. Methods Mol Biol. 2013;1015:311-20
77. Goel R, Muthusamy B, Pandey A, Prasad TS. Human protein reference database and human proteinpedia as discovery resources for molecular biotechnology. Mol Biotechnol. 2011;48:87-95
78. Hodis E, Prilusky J, Martz E, Silman I, Moult J, Sussman JL. Proteopedia - a scientific 'wiki' bridging the rift between three-dimensional structure and function of biomacromolecules. Genome Biol. 2008;9:R121
79. Sardana D, Zhu C, Zhang M, Gudivada RC, Yang L, Jegga AG. Drug repositioning for orphan diseases. Brief Bioinformatics. 2011;12:346-56
80. Gaulton A, Bellis LJ, Bento AP. et al. ChEMBL: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40:D1100-7
81. Chen B, Dong X, Jiao D. et al. Chem2Bio2RDF: a semantic framework for linking and data mining chemogenomic and systems chemical biology data. BMC Bioinformatics. 2010;11:255
82. Chen J, Swamidass SJ, Dou Y. et al. ChemDB: a public database of small molecules and related chemoinformatics resources. Bioinformatics. 2005;21:4133-9
83. Wang Y, Xiao J, Suzek TO, Zhang J, Wang J, Bryant SH. PubChem: a public information system for analyzing bioactivities of small molecules. Nucleic Acids Res. 2009;37:623-33
84. Ma DL, Chan DS, Leung CH. Drug repositioning by structure-based virtual screening. Chem Soc Rev. 2013;42(5):2130-41
85. Bolton Evan E. et al. PubChem: integrated platform of small molecules and biological activities. Annual reports in computational chemistry. 2008;4:217-41
86. Szklarczyk D, Franceschini A, Kuhn M. et al. The STRING database in 2011: functional interaction networks of proteins, globally integrated and scored. Nucleic Acids Res. 2011;39:D561-8
87. Oprea TI, Bauman JE, Bologa CG. et al. Drug Repurposing from an Academic Perspective. Drug Discov Today Ther Strateg. 2011;8(3-4):61-69
88. Roin BN. Unpatentable drugs and the standards of patentability. Tex L Rev. 2008;87:503
89. Swinney DC, Anthony J. How were new medicines discovered? Nat Rev Drug Discov. 2011;10:507-19
90. Fields JD, Bhardwaj A. Non-peptide arginine-vasopressin antagonists (vaptans) for the treatment of hyponatremia in neurocritical care: a new alternative? Neurocrit Care. 2009;11:1-4
91. Kuter DJ. New thrombopoietic growth factors. Blood. 2007;109:4607-16
92. Selby P, Buick RN, Tannock I. A critical appraisal of the "human tumor stem-cell assay". N Engl J Med. 1983;308:129-34
93. Shah ET, Upadhyaya A, Philp LK. et al. Repositioning "old" drugs for new causes: identifying new inhibitors of prostate cancer cell migration and invasion. Clin Exp Metastasis. 2016;33:385-99
94. Eriksson A, Österroos A, Hassan S. et al. Drug screen in patient cells suggests quinacrine to be repositioned for treatment of acute myeloid leukemia. Blood Cancer J. 2015;5:e307
95. Sant'anna R, Gallego P, Robinson LZ. et al. Repositioning tolcapone as a potent inhibitor of transthyretin amyloidogenesis and associated cellular toxicity. Nat Commun. 2016;7:10787
96. Camus S, Quevedo C, Menéndez S. et al. Identification of phosphorylase kinase as a novel therapeutic target through high-throughput screening for anti-angiogenesis compounds in zebrafish. Oncogene. 2012;31:4333-42
97. Ridges S, Heaton WL, Joshi D. et al. Zebrafish screen identifies novel compound with selective toxicity against leukemia. Blood. 2012;119:5621-31
98. Tomlinson ML, Hendry AE, Wheeler GN. Chemical genetics and drug discovery in Xenopus. Methods Mol Biol. 2012;917:155-66
99. Tat J, Liu M, Wen XY. Zebrafish cancer and metastasis models for in vivo drug discovery. Drug Discov Today Technol. 2013;10:e83-9
100. Amelio I, Gostev M, Knight RA, Willis AE, Melino G, Antonov AV. DRUGSURV: a resource for repositioning of approved and experimental drugs in oncology based on patient survival information. Cell Death Dis. 2014;5:e1051
101. Lee H, Kang S, Kim W. Drug repositioning for cancer therapy based on large-scale drug-induced transcriptional signatures. PloS ONE. 2016;11:e0150460
102. Tsuruoka Y, Miwa M, Hamamoto K, Tsujii Ji, Ananiadou S. Discovering and visualizing indirect associations between biomedical concepts. Bioinformatics. 2011;27:111-9
103. Rindflesch TC, Tanabe L, Weinstein JN, Hunter L. EDGAR: extraction of drugs, genes and relations from the biomedical literature. Pac Symp Biocomput. 2000;1:517-28
104. Pafilis Evangelos, Rūdolfs Bērziņš Lars Juhl Jensen. EXTRACT 2.0: text-mining-assisted interactive annotation of biomedical named entities and ontology terms. bioRxiv. 2017:111088
Author contact
![]() Corresponding author: Jie Li, jieliedu.cn
Corresponding author: Jie Li, jieliedu.cn