Impact Factor ISSN: 1449-2288
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Issue 8; 2026
- Issue 7; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Neutrophils production, release...
Neutrophil granules
Neutrophil serine protease...
Endogenous neutrophil serine...
Exogenous and therapeutic...
Conclusions and perspectives
Abbreviations
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(10):1343-1360. doi:10.7150/ijbs.26111 This issue Cite
Review
Neutrophil Elastase Inhibitors and Chronic Kidney Disease
Elsa Bronze-da-Rocha ![]() , Alice Santos-Silva
, Alice Santos-Silva
UCIBIO/REQUIMTE, Laboratório de Bioquímica, Departamento de Ciências Biológicas, Faculdade de Farmácia, Universidade do Porto, 4050-313 Porto, Portugal.
Received 2018-3-16; Accepted 2018-5-18; Published 2018-7-27
Abstract

End-stage renal disease (ESRD), the last stage of chronic kidney disease (CKD), is characterized by chronic inflammation and oxidative stress. Neutrophils are the front line cells that mediate an inflammatory response against microorganisms as they can migrate, produce reactive oxygen species (ROS), secrete neutrophil serine proteases (NSPs), and release neutrophil extracellular traps (NETs). Serine proteases inhibitors regulate the activity of serine proteases and reduce neutrophil accumulation at inflammatory sites. This review intends to relate the role of neutrophil elastase in CKD and the effects of neutrophil elastase inhibitors in predicting or preventing inflammation.
Keywords: End-stage renal disease (ESRD), chronic kidney disease (CKD), neutrophil elastase, neutrophil elastase inhibitors, inflammation
Introduction
Chronic kidney disease (CKD) is a long-term condition characterized by loss of renal function over time leading to kidney failure and to end-stage renal disease (ESRD), the last stage of CKD. The main causes of CKD are diabetes, hypertension, vascular disease, glomerular disease (primary or secondary), cystic kidney disease, tubule interstitial disease, urinary tract obstruction or dysfunction, recurrent kidney stone disease, congenital defects of the kidney or bladder, unrecovered acute kidney injury (AKI) and elderly [1, 2]. The growing prevalence of CKD is associated with the increasing aging of populations. These patients have high morbidity and mortality rates that are mainly due to cardiovascular disease (CVD) events [3, 4]. ESRD patients under hemodialysis (HD) have even higher morbidity and mortality rates. Inflammation, dyslipidemia, as well as disturbances in erythropoiesis, iron metabolism, endothelial function and in nutritional status, are common features in ESRD patients [5]. The chronic inflammation in patients with ESRD under hemodialysis has been associated with an imbalance between oxidant and anti-oxidant mechanisms, neutrophil activation with release of reactive oxygen species (ROS) and release of granule content, namely, neutrophil serine proteases (NSPs), such as neutrophil elastase (NE); and to activation of inflammatory cells through stimulation of cytokines by the released inflammatory products [6-10]. The inflammatory process and the development of oxidative stress are two main factors for worsening of anemia and to increase the risk for cardiovascular events [11-13]. The uremic toxins, including inflammatory cytokines (TNF-α, IL-1β, IL-6, IL-18), chemokines (IL-8), adipokines (adiponectin, resistin and leptin) and anti-inflammatory cytokines (IL-10), are important mediators for the injury and progression of renal tissue damage [11].
Neutrophils are the main immune cell population in adult human blood and the primary mediators of inflammatory response against invading microorganisms and foreign materials [14]. Neutrophils can adhere to endothelium, transmigrate to the infection/inflammatory site, produce oxygen radicals and release the internal components of neutrophil granules outside and within the cell, fusing with the phagosome. Neutrophil serine proteases are key enzymes in inflammation, immune response and coagulation, acting as a host defense mechanism [15-17]. These proteases have specific endogenous and exogenous inhibitors that antagonize their effects, to prevent several inflammatory diseases with severe impact on organ tissue integrity [14, 17, 18]. NSPs are required for the regulation of chemokine, cytokine bioavailability and for the production of neutrophil extracellular traps (NETs) [19]. Neutrophils undergo NETosis, a form of cell death, in response to a variety of activating stimuli, such as bacteria, viruses, parasites, fungi, phorbolmyristate acetate (PMA), oxidized low-density lipoprotein (oxLDL), interleukin-8 (IL-8), lipopolysaccharide (LPS), monosodium urate (MSU) crystals and Leishmania amazonensis, that leads to mitogen-activated protein kinase (MAPK) signaling and NADPH oxidase activation, which finally induces NET release [19-21].
NSPs deficiency is associated with several disorders due to mutations, anti-neutrophil cytoplasmic antibodies (ANCA) and to interference in processing of proteins [17]. NET is associated to some pathologies, namely autoimmune diseases, small vessel vasculitis, auto-inflammatory diseases, chronic inflammatory lung disease, metabolic disease, cancer, nephritis and chronic kidney disease [19, 22, 23].
This review will focus on the role of neutrophil elastase, a serine protease released by neutrophils during inflammation, in CKD; and on the potential value of endogenous and exogenous neutrophil elastase inhibitors to restrict or prevent the inflammation associated with CKD progression and with several CKD-associated complications.
Neutrophils production, release and elimination
Human neutrophils are the result of a process of differentiation and proliferation of myeloid hematopoietic stem cells, in the bone marrow (BM). The granulopoietic cells include undifferentiated hematopoietic stem cells (HSCs) and pluripotent myeloid progenitors that are CD34+ cells, and granulocyte-monocyte progenitor cells (GMPs); the granulocytic precursor cells include myeloblasts, promyelocytes, myelocytes (mitotic pool) and metamyelocytes that mature into band cells and, finally, into mature neutrophils (post-mitotic pool) (Figure 1A) [15, 24-26]. Bone marrow releases 5-10 x 1010 neutrophils per day. By stable isotope labeling, the half-life time of circulating human neutrophils was shown to be less than one day and around 5 days in BM transit [27]. There are also neutrophil pools within the vascular network of spleen, liver and bone marrow, that may be enhanced in case of pathogenic invasion or tissue damage [15, 25]. Neutrophil homeostasis is preserved by granulopoiesis, BM storage and release, intravascular margination, transmigration and destruction [25].
Granulopoietic cells in the bone marrow. (A) From hematopoietic stem and pluripotent progenitor cells, to the mitotic pool of granulocyte precursors (myeloblasts, pro-mielocytes and myelocytes) and the post-mitotic pool of metamyelocytes, band cells and mature granulocytes. (B) Neutrophils are found in the bone marrow, blood (circulating pool), spleen, liver (marginated pool) and in tissues (transmigrated pool). Granulocyte colony-stimulating factor (G-CSF) induces the proliferation of granulocytic progenitors. CXCL1 and CXCL2 are constitutively expressed on endothelial cells of the BM, whereas osteoblasts are the major source of CXCL12. G-CSF regulate the traffic of neutrophils: CXCR4 and its ligand CXCL12 (SDF-1) mediate neutrophil retention in the bone marrow, while CXCR2 and their ligands CXCL1 e CXCL2 promote neutrophil release, contributing for the circulating neutrophil pool. G-CSF enhances the release of neutrophils by inhibiting CXCR4/CXCL12. In physiological conditions, neutrophils in the circulating pool and in the marginated pool are in almost equivalent proportions. Neutrophils in the peripheral blood can be recruited into peripheral tissues (transmigrating pool). During inflammation, the inflammatory mediators released in peripheral tissues can act locally, inducing neutrophil recruitment into peripheral tissue; and, at distance, inducing neutrophil mobilization from the bone marrow, where the concentration of CXCR2 ligands increases, while CXCL12 expression decreases, allowing increased neutrophil migration.
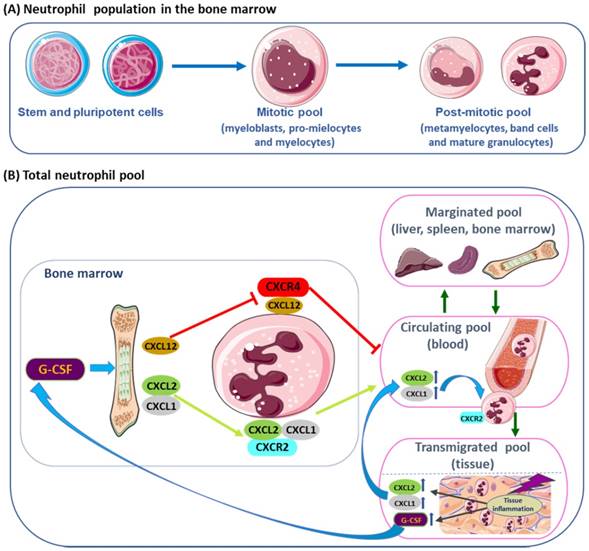
Neutrophil life cycle is mainly regulated by granulocyte colony-stimulating factor (G-CSF) that promotes granulocyte precursor proliferation, differentiation, survival and traffic/mobilization. G-CSF regulates the expression of chemokines, which control the balance between neutrophil release and retention [15, 25, 28]. Granulocyte monocyte colony-stimulating factor (GM-CSF) stimulates granulopoiesis and neutrophil release into the circulation [25, 29]. It is a vital survival signal for neutrophils by activating the janus kinase/signal transducers and activators of transcription (Jak/STAT), phosphoinositide 3-kinase (PI3K) and MAPK pathways [29, 30]. Both PI3K and MAPK signaling are critical for the phosphorylation of protein kinase B (PKB), which is needed for neutrophil chemotaxis [31]. The Jak/STAT pathway is able to delay and rescue GS-CSF induced neutrophils apoptosis; however, the activation of this pathway, by GM-CSF, is altered in neutrophils of aged subjects [29]. The modifications in the activation of Jak/STAT pathway in elderly may also contribute to change the immune response, since this pathway regulates cytokine production [29, 32]. The inhibition of Jak/STAT pathway prevents the secretion of inflammatory cytokines, chemokines, proteases and growth factors and might be used to extend longevity in ageing [32]. In elderly, the activity of PI3K in neutrophils is increased, leading to aberrant neutrophil migration and to an increase of neutrophil proteinase-specific fibrinogen cleavage product Aa-VAL360, a specific marker of neutrophil elastase activity in vivo [33]. Blocking PIK3 activity could contribute to prevent inflammation and increase neutrophil bactericidal and phagocytic functions during infections in older people [33].
Neutrophil release from the bone marrow is regulated by chemokines expressed on neutrophils, namely by the CXC chemokine receptor 2 (CXCR2) that has different ligands, the CXCL1, macrophage inflammatory protein-2 (MIP-2 or CXCL2), epithelial-derived neutrophil-activating peptide 78 (ENA78 or CXCL5) and granulocyte chemotactic protein-2 (GCP-2 or CXCL6); and by the receptor CXCR4 and its major ligand, the stromal-derived factor-1 (SDF-1 or CXCL12), mediates neutrophil retention and signalizes senescent cells for destruction (Figure 1 B) [15, 25, 26, 28].
In the BM, the endothelial cells constitutively express CXCL1 and CXCL2, while osteoblasts are the major source of CXCL12 [28, 34]. CXCR2 signaling interacts antagonistically with CXCR4 to regulate neutrophil release from the BM into the systemic circulation [26, 35]. During neutrophil maturation, CXCR4 and adhesive interaction α4 integrin very late antigen-4 (VLA-4) are downregulated allowing the release of neutrophils into the peripheral blood [36]. Mixed bone marrow Cxcr2-/- chimera mouse showed that the Cxcr2-/- neutrophils, in the BM, were not mobilized into circulating blood, due to transient CXCR4 inhibition, when compared to the wild type. Treatment with CXCR2 agonist increased wild type neutrophils in circulating blood, compared to the Cxcr2-/- neutrophils, suggesting that CXCR2 activation is partially dependent on CXCR4 [35]. Moreover, in double deficient Cxcr2-/-/Cxcr4-/- neutrophils were constitutively mobilized from the BM, indicating that CRCX2 signal was not required in the absence of CXCR4, while CRCX4 regulated neutrophil traffic [35]. The dominance of SDF-1 (CXCL12) over CXCR2 was demonstrated by mutations in CXCR4 that lead to neutrophil retention [37]. CXCR4 autosomal dominant mutations are associated with WHIM syndrome, characterized by hypogammaglobulinemia, immunodeficiency and accumulation of mature, often hypersegmented or dysplasic neutrophils in the BM (myelokathexis) [12, 37]. In murine myeloid cells, deletions of Crcx4 induced increased neutrophil release [38], while CXCR4 antagonists inhibited neutrophil mobilization from bone marrow [39, 40]. Plerixafor (AMD3100, Genzyme Corporation) is an antagonist of the binding of SDF-1, that is used in combined therapy with G-CSF to mobilize hematopoietic stem cells to peripheral blood, for collection and autologous transplantation of patients with some hematopoietic malignancies, as non-Hodgkin's lymphoma and multiple myeloma [40, 41]. CTCE-0021, a cyclized CXCR4 agonist peptide, mobilizes synergistically both neutrophils and hematopoietic progenitor cells when combined with G-CSF, in mice and in Jurkat human lymphoblastoid cell line [42]. CXCR4 receptor agonists (CTCE-0021 and ATI-2341) employed alone or with other agents as mobilizing drugs, may be an alternative for patients that are poor mobilizers [42, 43]. Moreover, the modulation by SDF-1, of (VLA-4) and of its ligand, vascular cell adhesion molecule-1 (VCAM-1), retains neutrophils in the bone marrow [36]. According to in vivo evidences in mouse models, dendritic cells (DCs) control the production of G-CSF and some chemokines that interferes with neutrophil mobilization, recruitment and apoptosis [26, 44].
Neutrophil cell death occurs in homeostasis and is enhanced in inflammation. Old circulating neutrophils are removed by macrophages in the bone marrow, spleen and liver; they undergo spontaneous or constitutive apoptosis, followed by removal of apoptotic neutrophils [24, 30]. Apoptosis is induced and/or controlled by low expression levels of anti-apoptotic proteins; caspase activity; reactive oxygen species (ROS), inhibitors of cyclin-dependent kinases (CDKs); proliferating cell nuclear antigen (PCNA); myeloid nuclear differentiation antigen (MNDA); granular proteases; pathogens that could enhance or restrict signaling pathways; and exposition to pro-survival molecules that delay apoptosis, such as G-CSF, GM-CSF, and interferon-gamma (INF-γ) [30, 45].
Neutrophil death takes place by spontaneous apoptosis or by the extrinsic, intrinsic and phagocytosis-induced apoptotic (PICD) pathways, at inflammatory sites. Tumor necrosis factor-alfa (TNF-α), TNF-related apoptosis-inducing ligand (TRAIL) or Fas-ligand stimulate the extrinsic pathway; the release of cytochrome C into the cytosol is required for the activation of the intrinsic pathway; and the PICD pathway is activated by particle uptake and NADPH oxidase-derived ROS [30, 45]. Other forms of neutrophil death include: autophagy, an intracellular catabolic process activated by high levels of ROS, by which cellular components and pathogens are degraded through the lysosomal machinery [20]; NETosis that takes place by three different models (suicidal NETosis, vital NETosis and vital NETosis with release of mitochondrial DNA), which can be dependent of ROS, NADPH oxidase and on the rapidly accelerated fibrosarcoma-mitogen-activated kinase-extracellular signal-regulated kinase (Raf-MEK-ERK) pathway [19, 20, 22]; necroptosis, which is activated by specific protein kinases, the receptor-interacting protein kinase-3 (RIPK3) and by mixed lineage kinase domain like (MLKL) [24, 45, 46], although NET formation can occur independently of MLKL and RIPK3 signaling [47]; pyroptosis that requires the proteolytic activation of the inflammatory cytokines, IL-1β and IL-18, and is initiated by pyroptosis-mediating caspases; efferocytosis, a process of elimination of dead cells by phagocytes or neighboring cells; and phagocytosis of apoptotic neutrophils by macrophages [24, 30, 45, 46]. Regulation of neutrophil turnover is essential to control cell death and clearance of apoptotic neutrophils in homeostasis and in inflammation. Delayed neutrophil apoptosis may contribute to the pathology of several inflammatory and autoimmune diseases [45].
Neutrophil granules
Mature neutrophils have a segmented nucleus, primary and neutrophil secondary cytoplasmic granules. The main components of primary neutrophil granules, are neutrophil serine proteases that are expressed during the promyelocytic stage of granulopoiesis and present in further myeloid stages and mature granulocytes; they are also expressed in monocytes and mast cells, and become active after a N-terminal modification by dipeptidyl peptidase I (DPPI), also called cathepsin C [24, 30, 45, 46, 48, 49]. There are four types of neutrophil granules, containing approximately 300 proteins, which differs in its content, signaling for granule secretion and place of secretion: primary or azurophilic granules, containing myeloperoxidase (MPO) and the serine proteases; secondary or specific granules, rich in lactoferrin and cathelicidin; tertiary or gelatinase granules, with gelatinase and lysozyme; and secretory granules, with complement and chemotaxis receptors [17, 26, 48-51]. The main components of the four types of neutrophil granules and membrane markers are described in table 1.
Types of neutrophil granules: main components and membrane markers.
| Prymary or azurophilic | Secondary or specific | Tertiary or gelatinase | Secretory | ||||
|---|---|---|---|---|---|---|---|
| Protein | Membrane marker | Protein | Membrane marker | Protein | Membrane marker | Protein | Membrane marker |
| Neutrophil elastase | CD63 | Lactoferrin | CB11b/CD18 | MMPs | CB11b/CD18 | CR1 | CB11b/CD18 |
| Proteinase 3 | CD68 | NGAL | CD67 | Lysozyme | V-type H+-ATPase | FcgRIIIb | CD-10 |
| Cathepsin G | V-type H+-ATPase | hCAP-18 | CD66 | NRAMP1 | Azurocidin | CD-13 | |
| NSP 4 | Presenilin | Cathelicidin | CD15 | Gelatinase B (MMP-9) | Proteinase 3 | CD-14 | |
| Azurocidin | Lysozyme | Leukolysin | AP | CD-16 | |||
| α-defensins | CRISP3 | Arginase 1 | Albumin | CD-35 | |||
| Myeloperoxidase | Pentraxin 3 | β2-microglobulin | Haptoglobulin | CD-45 | |||
| BP1 | Haptoglobin | FicolinI | NOX2 | ||||
| Sialidase | β2-microglobulin | Acetyltransferase | |||||
| Lysozyme | hParanase | DAG deacyliting enzyme | |||||
| Cathepsin C | AP | Lactoferrin | |||||
| Arginase | Aminopeptidase | hCAP-18 | |||||
| Colagenase | Collagenenase (MMP-8) | CRISP3 | |||||
| CAP37 | Haptoglobin | ||||||
| β-galactosidase | SLPI | ||||||
| β-glucoronidase | OLFM4 | ||||||
| Colagenase | Heparinase | ||||||
| β-glicerofosfatase | Histaminase | ||||||
AP: alkaline phosphatase; BP1: bactericidal/permeability-increasing protein 1; CAP-37: cationic antimicrobial protein 37kDa CD: cluster of differentiation; CR1: complement receptor 1;CRISP3: cysteine-rich secretory protein 3; hCAP-18: human cationic antimicrobial; DAG: Diacylglycerol deacyliting enzyme; FcγRIIIb: Fc gamma receptor IIIb; MMPs: matrix metalloproteases; NGAL: neutrophil gelatinase associated lipocain; NRAMP1: natural resistance associated macrophage protein 1, NOX: NADPH oxidase; NSP4: neutrophil serine protease 4; OLFM4: granule protein olfactomedin; SLPI: secretory leukocyte protease inhibitor.
The active serine proteases that belong to the chymotrypsin serine proteases group are neutrophil elastase (NE), proteinase 3 (PR3), cathepsin G (CG) and neutrophil serine protease 4 (NSP4). These four serine proteases are small molecules with low molecular weight (29 kDa) that can form large protein complexes (2000 kDa), with a conserved serine in their catalytic site, and protected of degradation by DPPI [17, 49, 52]. NSPs perform their functions at different compartments, where they have several substrates, and control biological events, namely, the survival of regulatory neutrophil proteins, inflammation and NET formation [49]. NSPs digest phagocytized microorganisms, together with myeloperoxidase and ROS generated by NAPH oxidase complex [17, 48, 52-54]. NSPs are associated with several human pathologies, such as infections, cardiovascular diseases, tumors, congenital neutropenia, and autoimmune syndromes [49].
NE and PR3 enzymes are also able to degrade extracellular matrix (ECM) components, namely, fibrin, fibronectin, collagen, the glycoprotein IIb/IIIa receptor, elastin and cadherins [55]. NE is quickly released from neutrophil granules, and is highly concentrated at inflammation sites. NE translocation to the nucleus is a process requiring ROS and MPO [56]. NE activates matrix metalloproteinases (MMPs) and inactivates their inhibitors; has the ability to degrade components of the coagulation and fibrinolytic pathways. With PR3, NE activates and processes inflammatory cytokines (TNF-α, IL-1β, IL-8, IL-18 and IL-6) and promotes stimulation of protease activated receptors (PAR) that increase the epithelial permeability and neutrophil migration, as well as the selective activation of MAP kinase [55]. NE and CG process and stimulate members of IL-1, namely IL-1α, IL-33, and IL-36 cytokines, while IL-1β and IL-18 promote the release of TNF-α and IL-6 [57]. Thus, NSPs play a key role in the innate immune response against infectious agents (bacteria, fungus and virus), in tissue remodeling processes, and in the initiation/resolution/amplification of inflammation [17, 18, 57]. In this context, key components associated to the role of neutrophils in innate immunity include the inflammatory cytokine TNF-α, signaling through TNF receptors, signaling pathways stimulated by GM-CSF, pathogen associated molecular patterns (PAMPs), damage associated molecular pattern molecules (DAMPs), calpains, receptors on neutrophil surface and autophagy [24, 25].
Neutrophils strategies to combat infections include phagocytosis, the release of granule components into the extracellular space or into the phagosome, the establishment of neutrophil extracellular traps and autophagy [15, 20, 22, 58]. Phagocytosis is the cellular uptake, by which neutrophil engulfs particles or pathogens into the phagosome, a process enhanced by the complement system, IgG antibodies and cell pre-activation [15]. NET is used by neutrophils to capture and kill extracellular pathogens at sites of infection [20] and has coupled antimicrobial peptides released by neutrophils [59]. PR3, CG, NSP4 and NE contribute to the proteolytic activity of NETs (Figure 2); however, NE has a primordial role in this process, as demonstrated by depletion studies [60].
Neutrophil functions. Phagocytosis: neutrophil surrounds the pathogen (bacteria, virus and fungus) engulfing it, so that it winds up enclosed in a phagosome for destruction, a process requiring ROS production and the release of serine proteases and antimicrobial peptides. Degranulation: release of neutrophil granule components, as serine proteases, myeloperoxidase and matrix metalloproteases, required for activation of inflammatory cytokines, degradation of ECM components and digestion of phagocyted pathogens. ROS production: ROS are produced by neutrophils, via NADPH oxidase; ROS scavengers with antioxidant properties are required to counteract extracellular ROS. NET release: ROS and antimicrobial peptides are associated to NET, promoting chromatin decondensation and cell rupture; DNases cleave NETs and macrophages remove NETs.
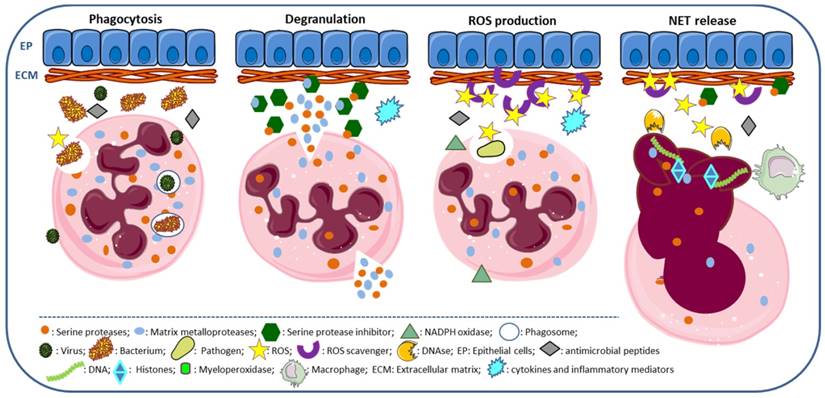
NET can also lead to tissue damage and is a potential source of antigens. The membrane-associated NADPH oxidase system produces ROS, required to initiate NET formation, and is followed by the release of NE and MPO from azurophilic granules that cooperatively enhance chromatin decondensation and cell rupture [19-21, 61]. NETs are associated with the release of cell-free DNA (cfDNA), DNA strands associated with histones (H1, H2A, H2B, H3, and H4) and of several proteins, such as NE, PR3, CG, MPO, high mobility group protein B1 (HMGB1), and LL37, all contributing to the pro-inflammatory process [15, 20, 22, 58]. Pentraxin 3 (PTX3) is expressed in neutrophil precursors (absent in mature neutrophils), mostly stored in specific granules, and moderately in azurophilic and gelatinase granules. It is involved in NET formation, attenuates histone-mediated endothelial cell cytotoxicity through aggregation and limits tissue damage [62].
NETosis is accepted as a specific form of cell death undergone by granulocytes, although different from apoptosis and necrosis [20, 22]. There are three patterns of NETosis: suicidal NETosis, vital NETosis and vital NETosis with release of mitochondrial DNA [19, 58]. In suicidal NETosis, there are loss of nuclear membrane; transport of NE and MPO from the granules into the nucleus; activation of NADPH oxidase complex through protein kinase C (PKC) activation, along with Raf/MEK)/ERK signaling pathway; increase in cytosolic Ca2+ [24, 30, 45, 46, 48]; and chromatin decondensation, which requires ROS for histone cytrulation by the peptidyl arginase desaminase 4 (PAD4) [19, 61, 63]. Decondensation of DNA is required to mix DNA and intracytoplasmic proteins. The pharmacological inhibition of PAD4 suppresses chromatin decondensation and NET formation [64]. In vital NETosis, neutrophils release NETs without loss of nuclear or plasma membrane, is independent of ROS and of the Raf/MEK/ERK pathway, and does not affect neutrophil functions and half-life, even after DNA loss [19, 58]. The vital NETosis with release of mitochondrial DNA, instead of nuclear DNA, is dependent on ROS and does not lead to cell death [19, 65]. These different pathways involved in NET formation can be triggered in different ways, the suicidal pathway is mostly induced by PMA, whereas the vital necrosis is stimulated by LPS and Toll-like receptor-4 (TLR4) on platelets [22]; and the vital NETosis with release of mitochondrial DNA, requires stimulation by GM-CSF and LPS [19, 65].
Neutrophil serine protease inhibitors
The serine protease inhibitors are able to regulate the proteolytic activities of the serine proteases and are important to regenerate the protease/anti-protease balance. Both therapeutic and endogenous serine protease inhibitors regulate NSP activity, preventing excessive elastin proteolysis, as occurs in several inflammatory diseases, with severe impact on organ tissue integrity [55].
Endogenous neutrophil serine protease inhibitors
The most important endogenous biological serine proteases inhibitors (serpins) have a tertiary structure complementary to elastase and play an important role in regulating the immune response. Endogenous inhibitors with anti-neutrophil elastase activity include alfa1-proteinase inhibitor (α1-PI) or alfa1-anti-tripsin (AAT), elafin, secretory leukocyte protease inhibitor (SLPI), α1-anti-chymiotrypsin (ACT), α2-macroglobulin, and monocyte neutrophil elastase inhibitor (MNEI or SerpinB1). The activity of NE is primarily regulated by α-1-PI, Serpin B1, and SLPI [17, 19, 52, 55]. The reduction of elastase activity contributes to the resolution of inflammation and prevents tissue damage. Imbalances between NE and its endogenous inhibitors are implicated in the pathogenesis of a wide range of disorders characterized by severe, progressive or chronic inflammation, such as cystic fibrosis (CF), chronic obstructive pulmonary disease (COPD), acute respiratory distress syndrome (ARDS), pulmonary fibrosis, asthmatic exacerbations, systemic lupus erythematosus (SLE), psoriasis, and rheumatoid arthritis [18, 19, 21].
The endogenous alpha1-anti-trypsin, produced in the liver, is the major circulating serpin. AAT inhibits PR3, CG and plasminogen activator, and has NE as the main target, protecting tissues from damage [66]. AAT has anti-oxidant and anti-protease effects, blocks the activity of TNF, and downregulates neutrophil chemotactic response, abrogating inflammation. In CF and COPD patients, AAT levels rise to equilibrate the increased elastase activity, reducing inflammation; a decrease of neutrophil innate skill to fight infections was also observed, however without great benefits in lung function [21]. AAT can be inactivated by ROS, leading to degradation of extracellular matrix components, associated to exacerbation of elastase-dependent tissue damage, cell death and release of autoantigens, worsening inflammation [18, 21].
SLPI and elafin are serpins produced at the sites of inflammation, in response to inflammatory stimulus, like TNF-α and NE [55]. The primary substrate of elafin is NE [66, 67] and proteinase-3, acting through a competitive tight-binding mechanism [67]. Imbalances between NE and elafin levels have been associated with ARDS mortality [68, 69]. SLPI, a conserved inhibitor produced by epithelial cells, mast cells, neutrophils and macrophages, is an important inhibitor of NE and CG from human neutrophils [66, 70]. SLPI controls inflammation by inhibiting serine proteases, attenuating the monocyte/macrophage response to LPS, stopping the transcription factor NF-kB, preventing bacterial, fungal and viral infections [66, 70]. SLPI is also important for proliferation, differentiation and cell cycle of myeloid cells, as demonstrated by transduction and knockdown studies [71].
SerpinB1, produced by neutrophils and macrophages, is one of the most effective inhibitor of NE. It is not secreted into the extracellular environment, remaining within the cytoplasm and primary granules [53]. SerpinB1 neutralizes the proteinase activity of PR3 in the cytosol and plays a pro-survival function in neutrophils. This was demonstrated in an infectious peritonitis model, in mice with a deficiency in PR3, where neutrophils persisted at inflammation sites, due to a delayed neutrophil death [72]; another study showed that neutrophils lacking SerpinB1 presented an accelerated death process [53]. PR3-mediates caspase-3 activation and controls neutrophil spontaneous death [72]. The inhibition of protease CG by SerpinB1 is essential for neutrophil survival [73].
After neutrophil activation, SerpinB1 and SLPI translocate to the nucleus, where SLPI controls the cleavage of histones by NE and SerpinB1 limits chromatin decondensation, partially preventing NET formation. In plasmocytic dendritc cells (pDCs), at NETs, SLPI, RNA, self-nuclear and mitochondrial DNA stimulates interferon type I (INFI) production through TLR-9. The pDCs and INFI are implicated in the pathogenesis of psoriasis and SLE [53, 74, 75].
Exogenous and therapeutic neutrophil serine protease inhibitors
Therapeutic inhibitors for NSPs are used to decrease intracellular levels of active NE, PR3 and CG, and to reduce neutrophil accumulation at inflammatory sites [14]. They are administrated orally, by aerosol or intravenously, depending on the size and nature of the inhibitor. According to Korkmaz et al. an ideal neutrophil serine inhibitor would: 1) be able to inhibit HNE, PR3 and CG, with similar efficiency, and to control their proteolytic activities within given limits; 2) remain chemically resistant to oxidation and proteolysis; 3) be of small size, thereby affording better access to NSPs when they are bound to extracellular matrices and to molecular or cellular components; 4) be available as aerosol; 5) resist to in vivo degradation [14, 18].
The NE inhibitors available for clinical practice are classified in five groups (Table 2). The 1st generation of inhibitors are biological and some present low stability; α1-anti-tripsin (Prolastin, Zemaira, Aralast) and elafin (Tiprelestat) are some of the NE inhibitors of this group. The 2nd generation of NE inhibitors use a mechanism-based suicide small molecule (SMOL) that inhibits released and membrane-bound elastase (e.g. sivelestat or ONO-5046; freselestat or ONO-6818). The 3rd and 4th generation of NE inhibitors, modern and non-mechanism-based SMOLs, are nonreactive and reversible inhibitors originated from pyridine and dihydropirimidone lead structure; AZD9668 (Alvestat) and BAY-678 are NE inhibitors of the 3rd and 4th generation, respectively. The 5th generation inhibitors have pre-adaptive pharmacophores derived from 4th generation NE inhibitors (e.g. BAY 85-8501) [18, 76].
Classification of neutrophil elastase inhibitors.
| Classification | Origin | Examples |
|---|---|---|
| 1st generation | Biological | AAT (Aralast), Elafin (Tiprelestat) |
| 2sd generation | SMOLs, suicide inhibitors | ONO-5046 (Sivelestat), ONO-6818 (Freselestat) |
| 3rd generation | Modern, non-mechanism-based SMOLs | AZD9668 (Alvestat)* |
| 4th generation | Modern, non-mechanism-based SMOLs | BAY-678* |
| 5th generation | Pre-adaptive pharmacophores derived from 4th generation | BAY 85-8501* |
(*) Under clinical trials (Phase II).
The 3rd generation NE inhibitor AZD9668 (alvelestat) displays a dose-dependent NE inhibition [18, 77, 78]. In patients with CF, AZD9668 was well tolerated, reduced inflammation and lung injury, as showed by the reduction in IL-6, and in urinary desmosine, respectively; however, it seems to have little impact on NE activity or lung function [77]. Moreover, studies using a rat experimental model reported a beneficial effect for AZD9668 in the treatment abdominal aortic aneurysm, reducing the levels of MPO, NE and cfDNA that contribute to progression of the disease [79].
The 5th generation molecules have additional substituents that not directly interact with the target. BAY 85-8501 is one of these inhibitors that display anti-inflammatory effects in animal models [18, 76]. AZD9668 and BAY 85-8501 are in clinical trials (Phase II) for the treatment of patients with CF, COPD, bronchiectasis and pulmonary disease [18, 76, 80]. BAY-8040 showed excellent potency, selectivity, and improved the cardiac function in a rat model [81].
Still, there are other exogenous inhibitors of NE that have been developed and are under study, such as peptide and non-peptide derivatives [82, 83]. The 4-oxo-β-lactams are selective acylating irreversible NE inhibitors. A new 4-oxo-β-lactam containing a N-(4-(phenylsulphonylmethyl)phenyl) group was proposed as a potent slow-tight binding NE inhibitor, showing also selectivity for proteinase 3 and cathepsin G; however, it presents weak stability, due to off-target reactions with plasma and liver enzymes [84]. The design of small-molecule activity-based probes by click chemistry approach on 4-oxo-β-lactams was also reported as an efficient tool for the development of new potent NE inhibitors; these probes showed adequate fluorescence properties, and neutrophil internalization, suitable for detection of NE, making them a promise as diagnostic tools [85]. MK0339, a potent human β-lactam-based inhibitor, was tested in clinical trials, stimulating cell survival and increasing the production of mature neutrophils; data suggested MK0339 as a potential agent to be used in ELANE-associated neutropenia [86]. Derivatives of O3-acyl kojic acid, selected by virtual screening, are also under study as they presented good selectivity for NE and low cytotoxicity [87]. Cinnoline derivatives are reversible competitive inhibitors of human NE that seem to have potential, but further studies are required [88]. The design and synthesis of small molecules based on different bicyclic scaffold allowed the isolation of isoxazol-5(2H)-one scaffold, as good and reversible competitive inhibitor of NE [80], while thiazol-2-(3H)-one nucleus was not a good scaffold for NE inhibitor [89].
Camostat mesilate, a synthetic serine protease inhibitor with antioxidant properties mainly used to treat chronic pancreatitis [90] and reduce the incidence of pancreatic metastasis [91], was proposed as an antihypertensive drug with renoprotective effects in patients with salt-sensitive hypertension [92].
Several serine protease inhibitors have been identified in different organisms. NSPs from Musca domestica (MDSPI16) [93] and from spider Araneus ventricosus (AvCI) [94] showed to be active in inhibiting elastase activity. The fungus Ganoderma mastoporum, found in several Asian countries, has inhibitory effects on elastase release and on superoxide anion generation, due to their content in triterpnoid, which are under studies as potential NE inhibitors [95] [96].
Exogenous NE inhibitors are also able to prevent NET formation, which is advantageous for host defense, but they also induce endothelial cell damage and the release of autoantigens that may contribute to the development of autoimmune diseases [19, 22, 23]. In this context, lactoferrin, a constituent of the specific neutrophil granules, might act as an intrinsic inhibitor of NET, and be useful in the control NET and granule release, in inflammatory and autoimmune diseases [97].
Different treatments for neutrophil-mediated diseases, based on the inhibition of neutrophil infiltration, NE activity, ROS activity, and NET prevention and degradation [21], have been tested in pre-clinical and clinical studies. Currently, Sivelestat is the only non-peptide drug available for the treatment of acute lung injury (ALI) and ARDS [82, 98]. Clinical trials using small molecules ligands, as β2-integrins or ICAM-1, failed to block neutrophil infiltration in response to shock or ischemia-reperfusion (IR) injury [21].
Studies with chemokines and chemokine receptors have shown promising data. An antagonist of CXCR2, SB-656933, was well tolerated in adult patients with cystic fibrosis, acting by preventing neutrophil infiltration and modulating airway inflammation in these patients [99]. MK-7123 (also known as SCH 527123) is an antagonist of the human CXC receptors, with substantially higher affinity for CXCR2 that might selectively reduce neutrophil recruitment to sites of inflammation in COPD patients [100]. Azithromycin therapy, an antagonist of IL-8, reduced the number of worsening episodes in stable COPD, by reducing the levels of neutrophilic airway inflammatory markers [101]. Roflumilast, a selective phosphodiesterase-4 inhibitor, is a therapeutic agent with anti-inflammatory effects used in the treatment of COPD that acts by interfering with the acetylated proline-glycine-proline (AcPGP) pathway [102]. Transhinone IIA was functionally characterized as a pro-resolution molecule that promotes reverse migration of neutrophils and increases the levels of neutrophil apoptosis in human, rats and zebrafish, contributing to inflammation resolution [103]. The origin, mode of action and therapeutic applications of endogenous and exogenous inhibitors are summarized in tables 3 and 4.
Mode of action and therapeutic application of endogenous/biological and exogenous/biological NSPs inhibitors.
| Inhibitor | Origin | Action | Therapeutic use | References |
|---|---|---|---|---|
| AAT | endogenous/biological | - anti-oxidant and anti- protease | ARDS | [21] |
| Elafin | endogenous/biological | - lung tissue destruction protection | ARDS | [68, 69] |
| - biomarker for monitoring early development of ARDS | ||||
| SLPI | endogenous/biological | - inhibition of neutrophil phagocytosis - protection of tissue damage by proteolytic enzymes - inhibition of NFkB | COPD, ARDS | [53, 66, 68, 70, 104] |
| - regulation of granulopoiesis | ||||
| - inhibitor of apoptosis of circulating neutrophils | ||||
| - inhibition of neutrophil phagocytosis | ||||
| SerpinB1 | endogenous/biological | - inhibition of apoptosis of circulating neutrophils | CF | [53, 72, 73] |
| - inhibition of CG - promotion of PMN survival | inflammatory and autoimmune diseases | |||
| - control of neutrophil expontaneous death in PR3 deficient mice | ||||
| Lactoferrin | exogenous/biological | - inhibition of NET formation and prevention of NET spread | inflammatory and autoimmune diseases | [97] |
| Transhinone IIA | exogenous/biological | - block of pro-inflammatory signals in vivo | [103] | |
| - inhibition of survival signals in human neutrophils - modulation of neutrophil reverse migration | ||||
| MDSPI16 (Musca domestica) | exogenous/biological | - inhibition of elastase, chymotrypsin, and microbial serine proteases | [93] | |
| AvCI (Araneus ventricosus) | exogenous/biological | - inhibition of human NE and porcine pancreatic elastase - block potassium channel activity | [94] | |
| Triterpenoids (Ganoderma mastoporum) | exogenous/ biological | - inhibition of superoxide anion generation and elastase release | inflammatory diseases | [96] |
AAT: alfa1-anti-tripsin; ARDS: acute respiratory distress syndrome CF: cystic fibrosis; CG: cathepsin G; COPD: chronic obstructive pulmonary disease; NE: neutrophil elastase; NET: neutrophil extracellular trap: Nf-kB: nuclear factor kappa-light-chain enhancer of activated B cells; NET: neutrophil extracellular trap; PMN: polymorphonuclear; PR3: proteinase 3; SLPI: secretory leucocyte protease inhibitor.
Mode of action and therapeutic application of exogenous/synthetic NSPs inhibitors.
| Inhibitor | Origin | Action | Therapeutic use | References |
|---|---|---|---|---|
| ONO-5046 (Sivelestat) | exogenous/synthetic | - inhibition of released and membrane bound elastase | AATD, COPD, ARSD; CKD | [18, 82] |
| ONO-6818 (Freselestat) | exogenous/synthetic | - inhibition of released and membrane bound elastase | ALI, SIRS | [18, 98] |
| AZD9668 (Alvestat) | exogenous/synthetic | - reversible inhibitor with high selectivity and target specificity | ALI, lung emphysema, COPD | [18] |
| BAY- 678 | exogenous/synthetic | - reversible inhibitor with high selectivity and target specificity | ALI, lung emphysema | [18] |
| BAY-85-8501 | exogenous/synthetic | - improvement in potency and inhibitory capacity for NE | anti-inflammatory in pulmonary diseases | [76] |
| 4-oxo-β-lactams | exogenous/synthetic | - selective irreversivel NE inhibitor | COPD | [85] |
| MK0339 | exogenous/synthetic | - cell survival enhancer - stimulator of formation of mature neutrophils | ELANE‐associated neutropenia | [87] |
| Cinnoline derivatives | exogenous/synthetic | - reversible competitive inhibitors of NE | anti-inflammatory | [88] |
| Camostat mesilate | exogenous/synthetic | - anti-oxidant | chronic pancreatitis , renoprotection in patients with salt-sensitive hypertension, slows the progression of CRF, improvement of CKD progression | [92, 130, 131] |
| SB- 656933 | exogenous/synthetic | - prevention of neutrophil infiltration | CF | [99] |
| - modulation of airway inflammation | ||||
| MK-7123 | exogenous/synthetic | - antagonist of the cytokine receptor CXCR2 | COPD | [100] |
| Azithromycin | exogenous/synthetic | - inhibition of CXCL8 levels and bacterial load | COPD | [101] |
| Roflumistat | exogenous/synthetic | - inhibition of the AcPG pathway | COPD | [102] |
AATD: alpha1-antitrypsin deficiency; AcPGP: acetylated proline-glycine-proline; ALI: acute lung injury; ARDS: acute respiratory distress syndrome; CF: Cystic fibrosis; COPD: chronic obstructive pulmonary disease; CRF: chronic renal failure; CXCL8: chemokine ligand 8: CXCR2: chemokine receptor type2; NE: neutrophil elastase; SIRS systemic inflammatory response syndrome.
NET, if not removed by macrophages or cleaved by circulating DNAse activity, can contribute for tissue damage (via histones), for the pathology of several diseases, thrombosis, inhibition of apoptosis, release of autoantibodies, cancer progression and metastization [21, 53]. The exogenous administration of SLPI can reduce NET release, as showed by in vitro studies with this protease inhibitor [53, 104]. The signal inhibitory receptor on leukocytes-1 (SIRL-1) is a member of the transmembrane receptor Ig superfamily of immune inhibitory receptors, exclusively expressed on monocytes and granulocytes, that can suppress NET formation without compromising neutrophil antimicrobial functions [105].
Nowadays, epigenetic regulation modulates gene expression through DNA methylation, non-coding RNAs (ncRNA), such as long non-coding RNAs (lncRNAs) and microRNAs (miRNAs or miR), and histone post-translational modifications and histone variants. MicroRNAs are approximately 20-nucleotide, single-stranded RNA molecules that target mRNA, by partial or total complementarity, and regulate gene expression through inhibition of translation or transcript degradation. They also play a role in many biological processes, such as homeostasis, proliferation, development, apoptosis, metabolism or cell cycle [106]. When deregulated, they interfere with upstream and downstream gene network and signaling pathways, contributing for disease [106, 107]. In human bronchial epithelial (16HBE) cell line, neutrophil elastase induces miR-146a and MUC5AC (mucin 5AC, oligomeric mucus/gel-forming) mucin expression in a dose- and time-dependent stimulation. Ectopic expression miR-146a in 16HBE cells downregulated the stimulation of MUC5AC by NE, while depletion of endogenous miR-146a enhanced the MUC5AC production, which is associated with c-Jun N-terminal kinase (JNK) and nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) signaling pathway. These results suggest a therapeutic potential of manipulating miR-146a in the control of mucus overproduction in inflammatory airway diseases [108]. Also, in the B6.MRLc1 CKD mice that spontaneously develop renal inflammation with age, miR-146a renal expression was high in interstitial lesions containing inflammatory cells; the silencing of miR-146a in a human cell line from monocytes, increased the expression of inflammatory mediators, suggesting that the urinary miRNA146a excretion might be related with renal dysfunction [109]. Thus, miR-146a might regulate local inflammatory mediators and signal pathways, and could contribute for new therapeutic approaches for CKD [109]. Cathepsin S (CTSS) upregulation and activity, in CF patients airway epithelium, is dependent of miRNA-31 expression [110]. Many other miRNAs and lncRNAs are associated with the pathogenesis of respiratory diseases and useful in their diagnosis [111]. In the kidney, miRNAS deregulation contribute for kidney fibrosis and dysfunction; however, they can be used as non-invasive biomarkers for monitoring the severity and progression of renal damage and for therapeutic interventions [107, 109, 112]. Modulation of miRNAs, in vitro and in vivo, represents a novel approach in therapeutics since miRNAs can be overexpressed or inhibit. The miR-mimics are used when there is a downregulation of a miRNA, because of a disease, and its administration increases a specific miRNA. When pathological conditions result from the upregulation of a specific miRNA its inhibition is accomplished with antagomirs, as miRNAs inhibitors or blockers, like antisense oligonucleotide (morpholinos), sponges, masking, and erasers [106, 107, 112, 113].
Chronic kidney disease, elastase and NSP inhibitors
The guidelines from National Kidney-Foundation Kidney Disease Outcomes Quality Initiative (NKF-KDOQI) defined CKD as an abnormality of kidney structure or function, regardless of cause or specific clinical presentation, and proposed a staging system based on the level of glomerular filtration rate (GFR) [114]. The criteria for CKD classification is based on: 1) cause of kidney damage (albuminuria, urinary sediment abnormalities, electrolytes and other anomalies due to tubular disorders; defects detected by histology; structural aberrations identified by imaging, history of kidney transplantation); 2) the five stages GFR classification (with G3 category subdivided into G3a and G3b); 3) the three categories according to albuminuria-creatinine ratio and albumin-excretion rate [115]. The diagnosis and follow-up of patients with CDK is, thus, established according to the grade of renal dysfunction, proteinuria levels, renal filtration and markers of renal lesions [116].
CKD is accepted as a common worry for public health, contributing to increase morbidity and mortality in developed, low- and middle-income countries [117, 118]. In addition, morbidity and mortality rates are higher in ESRD patients under hemodialysis, and cardiovascular disease has been considered the leading cause of death in these patients [5]. Kidney failure requires renal replacement therapy (RRT), by kidney transplantation or hemodialysis [118, 119].
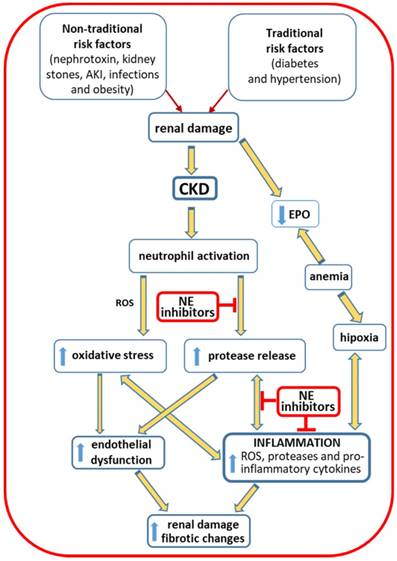
The prevalence of CKD is estimated to be 10-15% in the adult population [117, 120]. There are “traditional, conventional or classic” and “non-traditional, non-conventional or non-classical” risk factors that contribute to the global burden of the disease [118, 121, 122]. The traditional risks factors include hypertension and diabetes; obesity, a low-grade inflammatory condition, often associated to diabetes and hypertension, also favors the development of CKD. Non-traditional risks factors involve nephrotoxins, acute kidney injury (AKI), kidney stones, fetal and maternal exposures, infections and environmental exposures [118]. These risk factors are associated with renal damage, leading to kidney dysfunction and triggering an inflammatory response. The activation of neutrophils, with enhanced release of ROS, contributes to the development of oxidative stress; the release of proteases contributes to endothelial dysfunction and to inflammatory response that is further enhanced by the release of proinflammatory cytokines. All these mechanisms contribute to worsening of kidney injury and to the development of fibrotic changes. Anemia is a common complication in CKD patients that results from a reduced production of erythropoietin by the failing kidneys and may contribute to enhance inflammation. Neutrophil elastase inhibitors, as therapeutic agents, can be used to inhibit these proteases released by the activated neutrophils at renal inflammatory sites, preventing worsening of renal damage and, thus, the development of CKD and the associated complications (Figure 3) [10, 11, 123-125].
Risk factors effects in CKD. Traditional and non-traditional risk factors lead to renal damage that supports the development of chronic renal disease. The activation of neutrophils, with release of ROS and proteases, leads to oxidative stress and renal cell damage, contributing to endothelial dysfunction and to inflammation that will be further enhanced by the release of pro-inflammatory cytokines. These changes favor worsening of renal damage and the development of fibrotic alterations. Anemia, resulting from the reduced production of erythropoietin by the injured kidneys, leads to hypoxia, favoring inflammation. The inhibition of elastase, released by activated neutrophils, may be achieved by therapeutic intervention with neutrophil elastase inhibitors, preventing worsening of renal damage and, thus, the development of CKD and the associated complications. AKI: acute kidney injury; CKD: chronic kidney disease; EPO: erythropoietin; NE: neutrophil elastase; ROS: reactive oxygen species.
CKD patients under hemodialysis present high elastase levels that might be related with the enhanced inflammatory process, common in these patients [6]. Moreover, patients on HD, non-responders to recombinant human erythropoietin (rhEPO) therapy, showed higher neutrophils levels and more inflammation [6, 7]. Actually, inflammation is one of the major features in CKD patients, in the pre-dialysis period and on regular hemodialysis. The long-term intradialytic contact of blood with large surfaced artificial materials during the dialysis procedure may lead to leukocyte activation, with release of ROS and granule constituents, namely, elastase [123, 124]. In accordance, the use of vitamin E-coated cellulose acetate (CAE) membranes for dialysis procedure has been associated with a decrease in the oxidative stress and in biomarkers of inflammation [126, 127], without affecting dialysis adequacy [127]. Moreover, the elevated levels of elastase and cathepsin G, continuously released during hemodialysis session, can initiate vascular injury by cleavage of vascular endothelial cadherin in HD patients [128]. In line, it was reported that higher serum levels of free elastase, together with lower levels of AAT, contribute for the endothelial dysfunction [129].
The chronic inflammation in patients with end-stage renal disease may be associated with leukocyte activation and, thus, with increased proteolytic enzymes, such as NE, oxidative stress, showed by the imbalance oxidant and anti-oxidant defenses, as well as with an increase in pro-inflammatory cytokines [125]. Moreover, considering the high mean age of patients on dialysis and that neutrophils from older adults show increased degranulation and higher NE activity, tissue damage and inflammation is probably enhanced in ESRD patients [33]. NE and lipopolysaccharides stimulate the production of IL-8 and IL-6 [125]. IL-8 is an important neutrophil chemoattractant and the major chemokine responsible for neutrophil transmigration to inflammatory sites and neutrophil degranulation [14]. In HD patients, IL-8 is a good indicator of risk for cardiovascular complications, while IL-6 appears as a good marker of myocardial infarction. Serum concentrations of IL-8, α1-PI and of the complex NE-α1-PI are significantly higher in HD adult patients, as compared to controls. The NE-α1-PI was positively correlated with α1-PI and seems to be a better predictor of the inflammatory state in CKD patients than α1-PI and IL-8 [125]. NE-α1PI and IL-8 levels were also raised in non-infected children and young adults with CKD on continuous ambulatory peritoneal dialysis (CAPD), and the interaction between NE, α1-PI and IL-8 seems to enable the recruitment of inflammatory cells and tissue damage [10].
Studies using (5/6) nephrectomized rats treated with camostat mesilate showed that it was able to decrease oxidative stress, protease levels, inflammation and the severity of renal fibrosis [130]. When CM therapy was combined with telmisartan, a renin-angiotensin-aldosterone system (RAS) inhibitor, the effects on the reduction of oxidative stress and renal fibrosis were improved, when compared with the results for each monotherapy [131].
Sivelestat is a low molecular weight reversible competitive inhibitor of NE [55] that promotes the survival of rats with sepsis, by restoring the impairment in arterial pressure and glomerular filtration rate (GFR), and by inhibiting the increase in urea nitrogen (BUN) and neutrophil gelatinase associated lipocalin (NGAL) [132].
NETs are composed of cfDNA and DNA strands associated with histones and several other protein structures [15, 20, 22, 58]. HD patients, compared to controls, have high levels of IL-6 and DNA-histone complexes; thus, these complexes might be used as markers of risk for cardiovascular events in hemodialysis patients [133], contributing to decrease morbidity and mortality due to cardiovascular incidents, that are high in CKD and ESRD patients. In addition, it has been reported that patients with CKD present increased levels of circulating cfDNA [134, 135] and different types of DNA injury [136, 137]. cfDNA is able to selectively induce IL-6 production by monocytes that may explain the observed positive correlation of circulating cfDNA levels with the inflammatory grade in ESRD patients [134]. The correlations between the levels of DNA damage and the risk of mortality, suggest that genomic damage can be predictive for the outcome of CKD patients [136]. DNA damage has been measured in CKD patients using the micronucleus test (micronucleus frequency), the only that showed predictive potential, the comet assay (evaluation of DNA strand breaks), and the quantification of the 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodG) [137]. Coimbra et al. reported that genomic damage in ESRD patients, evaluated by comet assay and by quantification of cfDNA, was associated with lower CRP levels, when damaged DNA was still within the cell, and with higher CRP values, when injured DNA was already released into plasma. Moreover, the levels of DNA damage were correlated with age and inflammatory state and may, therefore, contribute to increase the risk for cancer and cardiovascular mortality [138].
Neutrophil elastase polymorphisms in ELANE gene may contribute to the development of several pathologies [139-141]. The presence of mutations and single nucleotide polymorphisms (SNPs) in the coding region and in the six repetitive tandem motifs of the promoter appear to influence the level of elastase expression, promoting proteolytic disturbances [139]. Severe congenital neutropenia (SCN) can be caused by mutations in ELANE gene, which have been associated with severe neutropenia and serious clinical manifestations [142]. ELANE mutations may also underlie cyclic neutropenia (CyN) [143]. Heterozygous mutations in ELANE gene are highly frequent and are the main causes for both CyN and SCN [144]. The correlations clinical genotype-phenotype of some mutations in ELANE gene seem to be linked to different prognosis [145]. Recently, a new pathogenic mutation (G201R) was reported in a patient without neutropenia, that leads to expression of a misfolded neutrophil elastase with reduced elastase activity and defective NETosis [146]. In ESRD patients, the heterozygosity for the c.-903T>G polymorphism and the homozygosity for the -741G>A polymorphism in the ELANE did not influence the circulating levels of elastase; however, the homozygous -741G>A polymorphism was associated with higher neutrophil count and with an enhanced inflammation that are usually associated with a poor outcome [147].
Anemia is a common complication in CKD, particularly in end-stage renal disease patients on HD. Erythropoietin (EPO) regulates erythropoiesis, promoting the proliferation and differentiation of erythroid cells. Recombinant human erythropoietin (rHuEPO) therapy has been used to correct CKD associated-anemia, but has potential to increase adverse cardiovascular effects, by increasing arterial blood tension and, thus, the risk of stroke, venous thromboembolism and mortality [148-150]. Some patients (5-10%) treated with rHuEPO develop resistance, needing higher rHuEPO doses to achieve target hemoglobin levels. Inflammation, oxidative stress and iron deficiency are the main factors that have been associated to resistance to rHuEPO therapy [148], and CRP was proposed as a predictor of ESA resistance in HD patients [151, 152]. Hyporesponsive patients to rHuEPO present higher levels of neutrophil activation, as showed by higher elastase levels, as compared to responders patients [6]. Currently, high doses of ESA are not recommended in anemic patients with CKD or ERSD, as epidemiologic studies showed an association with higher mortality and increased cardiovascular risk [149, 150, 153]. However, it has been described a renoprotector effect of ESA in the progression of CKD, when used in lower therapeutic doses [154, 155]. Accordingly, methoxy polyethylene glycol-epoetin beta (MPG-EPO) used to treat anemia in non-dialysis CKD patients showed to have nephroprotective effects in early stages of CKD and to reduce the CVD risk [155].
According to KDIGO guidelines, to treat anemia of CKD, the therapy with ESA require an iron supplementation to achieve a more adequate erythropoietic response [156-158]. Recent data alerts to the risk of iron overload in dialysis patients, which is associated with iron toxicity, infection/inflammation, and may contribute to a higher mortality risk. Actually, the Dialysis Outcomes and Practice Patterns Study (DOPPS) reported an increase in hemodialysis patients on intravenous iron supplementation in most countries, and the study of Bailie and collaborators also showed a rise in the prescribed iron doses over the past 10-15 years [159].
During hemodialysis procedure, red blood cells (RBCs) are mechanically stressed, due to blood flow through dialysis system, and metabolically stressed by the accumulation of uremic toxins and products of leukocyte activation, namely, ROS, elastase and other granule contents [160]. These changes contribute to membrane remodeling of RBCs, involving loss and modifications in band 3 (aggregation, fragmentation and carbonylation) and spectrin [161, 162], leading to premature aging and removal [3, 160, 163]. Thus, the products released by leukocyte activation and uremic toxins contribute to worsening of anemia, reducing the RBC life span, and may contribute to increase mortality risk in ESRD patients [3, 163].
Hemodiafiltration showed an improvement in redox potential and no exovesiculation of blood cells, compared to conventional hemodialysis, although the adverse short-term temporary but acute, oxidative-stress-driven increase in hemolysis suggest the use of antioxidant treatment during hemodiafiltration [164]. The RBCs in ESRD RhEPO responders, compared to non-responders (with enhanced inflammation), showed a predisposition to hemolysis under mechanical stress, more susceptibility to erythrophagocytosis favored by the excess of membrane-bound IgGs, a severe deficiency in CD47 protein in the membrane, and a lower expression of peroxiredoxin 2 induced by oxidative stress [165-167].
Conclusions and perspectives
Exogenous neutrophil elastase inhibitors are used in clinical practice in some specific diseases, namely when extracellular neutrophil elastase concentration goes beyond the protecting capacity of endogenous inhibitors, preventing the development of signs, symptoms and disease progression, through its role in the inhibition of the inflammatory process.
Inflammation is a common feature in CKD patients, particularly in those on dialysis, which is enhanced by the release of NE and other pro-inflammatory factors, worsening anemia and increasing the risk of morbidity and mortality. Thus, it would be important to search for new molecules to treat and prevent the adverse effects of neutrophil activation, without alteration in the antimicrobial function of neutrophils. Moreover, a deeper understanding of the mechanisms underlying NET formation, NETosis, production of ROS, cytokine activation, genomic damage, the role of ELANE polymorphisms, impaired neutrophil clearance, circulating cell free DNA, anemia, and the hemodialytic process, might give new insights to target the inflammatory process.
In summary, preservation of neutrophil elastase activity can be accomplished using endogenous and exogenous serine inhibitors. In that way, to improve the health conditions of ESRD patients, the development of new NE inhibitors with chemical stability and selectivity will mitigate the inflammatory process and can allow the identification of new predictors of inflammation and cardiovascular events.
Abbreviations
AcPGP: acetylated proline-glycine-proline; AAT or α1-PI: alfa1-anti-tripsin or alfa1-proteinase inhibitor; AATD: alpha1-antitrypsin deficiency; α1-ACT: alpha1-antichymiotrypsin; AKI: acute kidney injury; ALI: acute lung injury; ANCA: anti-neutrophil cytoplasmic antibodies; ARDS: acute respiratory distress syndrome; BM: bone marrow; BP1: bactericidal/permeability-increasing protein 1; BUN: blood urea nitrogen; CAE: coated cellulose acetate; CAPED: continuous ambulatory peritoneal dialysis; CD: cluster of differentiation; CDKs: cyclin-dependent kinases; CF: cystic fibrosis; cfDNA: cell-free DNA; CG: cathepsin G; CM: camostat mesilate; CR1: complement receptor 1; CRF: chronic renal failure; CRISP3: cysteine-rich secretory protein 3; CyN: cyclic neutropenia; COPD: chronic obstructive pulmonary disease; CTSS: cathepsin S; CVD: cardiovascular disease; CXC: chemokine receptor; CXCL: chemokine ligand; CXCR: chemokine receptor type; DAMPs: damage associated molecular pattern molecules; DC(s): dendritic cell(s); DPPI: dipeptidyl peptidase I; DOPPS: dialysis outcomes and practice patterns study; ECM: extracellular matrix; ERK kinase: extracellular signal-regulated kinase; ENA78: epithelial-derived neutrophil-activating peptide 78 or CXCL5; ERK: extracellular signal-regulated kinase; ESA: erythropoiesis stimulating agents; ESRD: end-stage renal disease; FcγRIIIb: Fc gamma receptor IIIb; GCP-2: granulocyte chemotactic protein-2 or CXCL6; G-CSF: granulocyte colony-stimulating-factor; GFR: glomerular filtration rate; GM-CSF: granulocyte-macrophage colony-stimulating-factor; GMPs: granulocyte-monocyte progenitor cells; H: histone; 16HBE: human bronchial epithelial cells; hCAP-18: human cationic antimicrobial protein-18; HD: hemodialysis; HMGB1: high mobility group protein B1; HSCs: hematopoietic stem cells; Ig: immunoglobulin; IL: interleukin; INF-γ: interferon-gamma; INFI: interferon type I; Jak /STAT: janus kinase signal transducers and activators of transcription; IR: ischemia-reperfusion; JNK: c-Jun N-terminal kinase; lncRNAs: long non-coding RNAs; LMPPs: lymphoid-primed multipotent progenitors; LPS: lipopolysaccharide; MAPK: mitogen-activated protein kinase; MIP-2: macrophage inflammatory protein-2; miRNAs or miRs: microRNAs; MLKL: mixed lineage kinase domain-like; MMP: matrix metalloprotease; MNDA: myeloid nuclear differentiation antigen; MNEI: monocyte neutrophil elastase inhibitor or SerpinB1; MPG-EPO: methoxy polyethylene glycol-epoetin beta; MPO: myeloperoxidase; MSU: monosodium urate; MUC5AC: mucin 5AC, oligomeric mucus/gel-forming; NADPH: nicotinamide adenine dinucleotide phosphate; ncRNAs: non-coding RNAs; NE: neutrophil elastase; NET(s): neutrophil extracellular trap(s); NF-κB: nuclear factor kappa-light-chain-enhancer of activated B cells; NGAL: neutrophil gelatinase associated lipocain; NKF-KDOQI: national kidney-foundation kidney disease outcomes quality initiative; NRAMP1: natural resistance associated macrophage protein-1, NSP(s): neutrophil serine protease(s); 8-oxodG: 8-oxo-7,8-dihydro-2′-deoxyguanosine; oxLDL: oxidized low-density lipoprotein; PAD4: peptidyl arginase desaminase 4; PAMPs: pathogen associated molecular pattern; PAR: protease activated receptors; PCNA: proliferating cell nuclear antigen; pDC(s): plasmocytic dendritc cell(s); PICD: phagocytosis-induce apoptotic; PKB: protein kinase B; PKC: protein kinase C; PMA: phorbol myristate acetate; PMN: polymorphonuclear; PR3: proteinase 3; PTX3: Pentraxin 3; Raf-MEK: rapidly accelerated fibrosarcoma-mitogen-activated kinase; RA: rheumatoid arthritis; RAS: renin-angiotensin-aldosterone system; RBC: red blood cell; rHuEPO: recombinant human erythropoietin; RIPK3: receptor-interacting protein kinase-3; ROS: reactive oxygen species; RRT: real replacement therapy; SCN: severe congenital neutropenia; SDF-1: stromal-derived factor-1; SerpinB1: monocyte neutrophil elastase inhibitor; SIRL-1: signal inhibitory receptor on leukocytes-1; SIRS: systemic inflammatory response syndrome; SLE: systemic lupus erythematosus; SMOL: small molecule; SLPI: secretory leukocyte protease inhibitor; TLR: toll like receptor; TNF-α: tumor necrosis factor-alfa; TRAIL: tumor necrosis factor-related apoptosis-inducing ligand; VCAM-1: vascular cell adhesion molecule-1; VLA-4: very late antigen-4; WHIM: warts, hypogammaglobulinemia, immunodeficiency and myelokathexis.
Acknowledgements
This work had the financial support from FCT/MEC through national funds and co-financed by FEDER, under the Partnership Agreement PT2020 from UCIBIO (UID/MULTI/04378/2013 - POCI/01/0145//FEDER/007728) and Norte Portugal Regional Coordination and Development Commission (CCDR-N) NORTE2020/Portugal 2020 (NORTE-01-145-FEDER-000024).
Competing Interests
The authors have declared that no competing interest exists.
References
1. Levey AS, Coresh J. Chronic kidney disease. Lancet. 2012;379:165-80
2. Levey AS, Eckardt KU, Tsukamoto Y, Levin A, Coresh J, Rossert J. et al. Definition and classification of chronic kidney disease: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO). Kidney international. 2005;67:2089-100
3. Antonelou MH, Georgatzakou HT, Tzounakas VL, Velentzas AD, Kokkalis AC, Kriebardis AG. et al. Blood modifications associated with end stage renal disease duration, progression and cardiovascular mortality: a 3-year follow-up pilot study. Journal of proteomics. 2014;101:88-101
4. Gungor O, Unal HU, Guclu A, Gezer M, Eyileten T, Guzel FB. et al. IL-33 and ST2 levels in chronic kidney disease: Associations with inflammation, vascular abnormalities, cardiovascular events, and survival. PloS one. 2017;12:e0178939
5. do Sameiro-Faria M, Ribeiro S, Costa E, Mendonca D, Teixeira L, Rocha-Pereira P. et al. Risk factors for mortality in hemodialysis patients: two-year follow-up study. Disease markers. 2013 a; 35: 791-8
6. Costa E, Rocha S, Rocha-Pereira P, Nascimento H, Castro E, Miranda V. et al. Neutrophil activation and resistance to recombinant human erythropoietin therapy in hemodialysis patients. American journal of nephrology. 2008 a; 28: 935-40
7. Pereira R, Costa E, Goncalves M, Miranda V, do Sameiro Faria M, Quintanilha A. et al. Neutrophil and monocyte activation in chronic kidney disease patients under hemodialysis and its relationship with resistance to recombinant human erythropoietin and to the hemodialysis procedure. Hemodialysis international International Symposium on Home Hemodialysis. 2010;14:295-301
8. Pereira R, Rocha S, Borges A, Nascimento H, Reis F, Miranda V. et al. Elastase release during the hemodialysis procedure seems to induce changes in red blood cell membrane proteins. Hemodialysis international International Symposium on Home Hemodialysis. 2011;15:429-31
9. Jager KJ, Lindholm B, Goldsmith D, Fliser D, Wiecek A, Suleymanlar G. et al. Cardiovascular and non-cardiovascular mortality in dialysis patients: where is the link? Kidney international supplements. 2011;1:21-3
10. Polanska B, Augustyniak D, Makulska I, Niemczuk M, Jankowski A, Zwolinska D. Elastase, alpha1-proteinase inhibitor, and interleukin-8 in children and young adults with end-stage kidney disease undergoing continuous ambulatory peritoneal dialysis. Archivum immunologiae et therapiae experimentalis. 2014;62:239-45
11. Castillo-Rodriguez E, Pizarro-Sanchez S, Sanz AB, Ramos AM, Sanchez-Nino MD, Martin-Cleary C. et al. Inflammatory Cytokines as Uremic Toxins: "Ni Son Todos Los Que Estan, Ni Estan Todos Los Que Son". Toxins. 2017:9
12. El Nahas M. Cardio-Kidney-Damage: a unifying concept. Kidney international. 2010;78:14-8
13. Ortiz A, Covic A, Fliser D, Fouque D, Goldsmith D, Kanbay M. et al. Epidemiology, contributors to, and clinical trials of mortality risk in chronic kidney failure. Lancet. 2014;383:1831-43
14. Korkmaz B, Horwitz MS, Jenne DE, Gauthier F. Neutrophil elastase, proteinase 3, and cathepsin G as therapeutic targets in human diseases. Pharmacological reviews. 2010;62:726-59
15. Kruger P, Saffarzadeh M, Weber AN, Rieber N, Radsak M, von Bernuth H. et al. Neutrophils: Between host defence, immune modulation, and tissue injury. PLoS pathogens. 2015;11:e1004651
16. Massberg S, Grahl L, von Bruehl ML, Manukyan D, Pfeiler S, Goosmann C. et al. Reciprocal coupling of coagulation and innate immunity via neutrophil serine proteases. Nature medicine. 2010;16:887-96
17. Pham CT. Neutrophil serine proteases fine-tune the inflammatory response. The international journal of biochemistry & cell biology. 2008;40:1317-33
18. von Nussbaum F, Li VM. Neutrophil elastase inhibitors for the treatment of (cardio)pulmonary diseases: Into clinical testing with pre-adaptive pharmacophores. Bioorganic & medicinal chemistry letters. 2015 a; 25: 4370-81
19. Delgado-Rizo V, Martinez-Guzman MA, Iniguez-Gutierrez L, Garcia-Orozco A, Alvarado-Navarro A, Fafutis-Morris M. Neutrophil Extracellular Traps and Its Implications in Inflammation: An Overview. Frontiers in immunology. 2017;8:81
20. Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS. et al. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532-5
21. van der Linden M, Meyaard L. Fine-tuning neutrophil activation: Strategies and consequences. Immunology letters. 2016;178:3-9
22. Yang H, Biermann MH, Brauner JM, Liu Y, Zhao Y, Herrmann M. New Insights into Neutrophil Extracellular Traps: Mechanisms of Formation and Role in Inflammation. Frontiers in immunology. 2016;7:302
23. Zahran N, Sayed A, William I, Mahmoud O, Sabry O, Rafaat M. Neutrophil apoptosis: impact of granulocyte macrophage colony stimulating factor on cell survival and viability in chronic kidney disease and hemodialysis patients. Archives of medical science: AMS. 2013;9:984-9
24. Benarafa C, Simon HU. Role of granule proteases in the life and death of neutrophils. Biochemical and biophysical research communications. 2017;482:473-81
25. Summers C, Rankin SM, Condliffe AM, Singh N, Peters AM, Chilvers ER. Neutrophil kinetics in health and disease. Trends in immunology. 2010;31:318-24
26. Hong CW. Current Understanding in Neutrophil Differentiation and Heterogeneity. Immune Netw. 2017;17:298-306
27. Lahoz-Beneytez J, Elemans M, Zhang Y, Ahmed R, Salam A, Block M. et al. Human neutrophil kinetics: modeling of stable isotope labeling data supports short blood neutrophil half-lives. Blood. 2016;127:3431-8
28. Sadik CD, Kim ND, Luster AD. Neutrophils cascading their way to inflammation. Trends in immunology. 2011;32:452-60
29. Fortin CF, Larbi A, Dupuis G, Lesur O, Fulop T Jr. GM-CSF activates the Jak/STAT pathway to rescue polymorphonuclear neutrophils from spontaneous apoptosis in young but not elderly individuals. Biogerontology. 2007;8:173-87
30. Geering B, Simon HU. Peculiarities of cell death mechanisms in neutrophils. Cell death and differentiation. 2011;18:1457-69
31. Kim D, Haynes CL. The role of p38 MAPK in neutrophil functions: single cell chemotaxis and surface marker expression. Analyst. 2013;138:6826-33
32. Xu M, Tchkonia T, Kirkland JL. Perspective: Targeting the JAK/STAT pathway to fight age-related dysfunction. Pharmacol Res. 2016;111:152-4
33. Sapey E, Greenwood H, Walton G, Mann E, Love A, Aaronson N. et al. Phosphoinositide 3-kinase inhibition restores neutrophil accuracy in the elderly: toward targeted treatments for immunosenescence. Blood. 2014;123:239-48
34. Mendelson A, Frenette PS. Hematopoietic stem cell niche maintenance during homeostasis and regeneration. Nature medicine. 2014;20:833-46
35. Eash KJ, Greenbaum AM, Gopalan PK, Link DC. CXCR2 and CXCR4 antagonistically regulate neutrophil trafficking from murine bone marrow. The Journal of clinical investigation. 2010;120:2423-31
36. Petty JM, Lenox CC, Weiss DJ, Poynter ME, Suratt BT. Crosstalk between CXCR4/stromal derived factor-1 and VLA-4/VCAM-1 pathways regulates neutrophil retention in the bone marrow. Journal of immunology. 2009;182:604-12
37. Hernandez PA, Gorlin RJ, Lukens JN, Taniuchi S, Bohinjec J, Francois F. et al. Mutations in the chemokine receptor gene CXCR4 are associated with WHIM syndrome, a combined immunodeficiency disease. Nature genetics. 2003;34:70-4
38. Ma Q, Jones D, Springer TA. The chemokine receptor CXCR4 is required for the retention of B lineage and granulocytic precursors within the bone marrow microenvironment. Immunity. 1999;10:463-71
39. Nademanee AP, DiPersio JF, Maziarz RT, Stadtmauer EA, Micallef IN, Stiff PJ. et al. Plerixafor plus granulocyte colony-stimulating factor versus placebo plus granulocyte colony-stimulating factor for mobilization of CD34(+) hematopoietic stem cells in patients with multiple myeloma and low peripheral blood CD34(+) cell count: results of a subset analysis of a randomized trial. Biology of blood and marrow transplantation: journal of the American Society for Blood and Marrow Transplantation. 2012;18:1564-72
40. Uy GL, Rettig MP, Cashen AF. Plerixafor, a CXCR4 antagonist for the mobilization of hematopoietic stem cells. Expert opinion on biological therapy. 2008;8:1797-804
41. Lanza F, Gardellini A, Laszlo D, Martino M. Plerixafor: what we still have to learn. Expert opinion on biological therapy. 2015;15:143-7
42. Pelus LM, Bian H, Fukuda S, Wong D, Merzouk A, Salari H. The CXCR4 agonist peptide, CTCE-0021, rapidly mobilizes polymorphonuclear neutrophils and hematopoietic progenitor cells into peripheral blood and synergizes with granulocyte colony-stimulating factor. Experimental hematology. 2005;33:295-307
43. Ratajczak MZ, Kim C. The use of chemokine receptor agonists in stem cell mobilization. Expert opinion on biological therapy. 2012;12:287-97
44. Jiao J, Dragomir AC, Kocabayoglu P, Rahman AH, Chow A, Hashimoto D. et al. Central role of conventional dendritic cells in regulation of bone marrow release and survival of neutrophils. Journal of immunology. 2014;192:3374-82
45. McCracken JM, Allen LA. Regulation of human neutrophil apoptosis and lifespan in health and disease. Journal of cell death. 2014;7:15-23
46. Wallach D, Kang TB, Dillon CP, Green DR. Programmed necrosis in inflammation: Toward identification of the effector molecules. Science. 2016;352:aaf2154
47. Amini P, Stojkov D, Wang X, Wicki S, Kaufmann T, Wong WW. et al. NET formation can occur independently of RIPK3 and MLKL signaling. European journal of immunology. 2016;46:178-84
48. Borregaard N. Neutrophils, from marrow to microbes. Immunity. 2010;33:657-70
49. Kettritz R. Neutral serine proteases of neutrophils. Immunol Rev. 2016;273:232-48
50. Amulic B, Cazalet C, Hayes GL, Metzler KD, Zychlinsky A. Neutrophil function: from mechanisms to disease. Annual review of immunology. 2012;30:459-89
51. Cowland JB, Borregaard N. Granulopoiesis and granules of human neutrophils. Immunol Rev. 2016;273:11-28
52. Perera NC, Schilling O, Kittel H, Back W, Kremmer E, Jenne DE. NSP4, an elastase-related protease in human neutrophils with arginine specificity. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:6229-34
53. Majewski P, Majchrzak-Gorecka M, Grygier B, Skrzeczynska-Moncznik J, Osiecka O, Cichy J. Inhibitors of Serine Proteases in Regulating the Production and Function of Neutrophil Extracellular Traps. Frontiers in immunology. 2016;7:261
54. Segal AW. How neutrophils kill microbes. Annual review of immunology. 2005;23:197-223
55. Alam SR, Newby DE, Henriksen PA. Role of the endogenous elastase inhibitor, elafin, in cardiovascular injury: from epithelium to endothelium. Biochemical pharmacology. 2012;83:695-704
56. Metzler KD, Goosmann C, Lubojemska A, Zychlinsky A, Papayannopoulos V. A myeloperoxidase-containing complex regulates neutrophil elastase release and actin dynamics during NETosis. Cell reports. 2014;8:883-96
57. Clancy DM, Henry CM, Sullivan GP, Martin SJ. Neutrophil extracellular traps can serve as platforms for processing and activation of IL-1 family cytokines. The FEBS journal. 2017;284:1712-25
58. Masuda S, Nakazawa D, Shida H, Miyoshi A, Kusunoki Y, Tomaru U. et al. NETosis markers: Quest for specific, objective, and quantitative markers. Clinica chimica acta; international journal of clinical chemistry. 2016;459:89-93
59. Yipp BG, Kubes P. NETosis: how vital is it? Blood. 2013;122:2784-94
60. O'Donoghue AJ, Jin Y, Knudsen GM, Perera NC, Jenne DE, Murphy JE. et al. Global substrate profiling of proteases in human neutrophil extracellular traps reveals consensus motif predominantly contributed by elastase. PloS one. 2013;8:e75141
61. Papayannopoulos V, Metzler KD, Hakkim A, Zychlinsky A. Neutrophil elastase and myeloperoxidase regulate the formation of neutrophil extracellular traps. The Journal of cell biology. 2010;191:677-91
62. Daigo K, Takamatsu Y, Hamakubo T. The Protective Effect against Extracellular Histones Afforded by Long-Pentraxin PTX3 as a Regulator of NETs. Frontiers in immunology. 2016;7:344
63. Lewis HD, Liddle J, Coote JE, Atkinson SJ, Barker MD, Bax BD. et al. Inhibition of PAD4 activity is sufficient to disrupt mouse and human NET formation. Nature chemical biology. 2015;11:189-91
64. Kusunoki Y, Nakazawa D, Shida H, Hattanda F, Miyoshi A, Masuda S. et al. Peptidylarginine Deiminase Inhibitor Suppresses Neutrophil Extracellular Trap Formation and MPO-ANCA Production. Frontiers in immunology. 2016;7:227
65. Yousefi S, Mihalache C, Kozlowski E, Schmid I, Simon HU. Viable neutrophils release mitochondrial DNA to form neutrophil extracellular traps. Cell death and differentiation. 2009;16:1438-44
66. Fitch PM, Roghanian A, Howie SE, Sallenave JM. Human neutrophil elastase inhibitors in innate and adaptive immunity. Biochemical Society transactions. 2006;34:279-82
67. Shaw L, Wiedow O. Therapeutic potential of human elafin. Biochemical Society transactions. 2011;39:1450-4
68. Wang T, Zhu Z, Liu Z, Yi L, Yang Z, Bian W. et al. Plasma Neutrophil Elastase and Elafin as Prognostic Biomarker for Acute Respiratory Distress Syndrome: A Multicenter Survival and Longitudinal Prospective Observation Study. Shock. 2017;48:168-74
69. Wang Z, Beach D, Su L, Zhai R, Christiani DC. A genome-wide expression analysis in blood identifies pre-elafin as a biomarker in ARDS. Am J Respir Cell Mol Biol. 2008;38:724-32
70. Majchrzak-Gorecka M, Majewski P, Grygier B, Murzyn K, Cichy J. Secretory leukocyte protease inhibitor (SLPI), a multifunctional protein in the host defense response. Cytokine & growth factor reviews. 2016;28:79-93
71. Klimenkova O, Ellerbeck W, Klimiankou M, Unalan M, Kandabarau S, Gigina A. et al. A lack of secretory leukocyte protease inhibitor (SLPI) causes defects in granulocytic differentiation. Blood. 2014;123:1239-49
72. Loison F, Zhu H, Karatepe K, Kasorn A, Liu P, Ye K. et al. Proteinase 3-dependent caspase-3 cleavage modulates neutrophil death and inflammation. The Journal of clinical investigation. 2014;124:4445-58
73. Baumann M, Pham CT, Benarafa C. SerpinB1 is critical for neutrophil survival through cell-autonomous inhibition of cathepsin G. Blood. 2013;121:3900-7 S1-6
74. Lande R, Ganguly D, Facchinetti V, Frasca L, Conrad C, Gregorio J. et al. Neutrophils activate plasmacytoid dendritic cells by releasing self-DNA-peptide complexes in systemic lupus erythematosus. Science translational medicine. 2011;3:73ra19
75. Lood C, Blanco LP, Purmalek MM, Carmona-Rivera C, De Ravin SS, Smith CK. et al. Neutrophil extracellular traps enriched in oxidized mitochondrial DNA are interferogenic and contribute to lupus-like disease. Nature medicine. 2016;22:146-53
76. von Nussbaum F, Li VM, Allerheiligen S, Anlauf S, Barfacker L, Bechem M. et al. Freezing the Bioactive Conformation to Boost Potency: The Identification of BAY 85-8501, a Selective and Potent Inhibitor of Human Neutrophil Elastase for Pulmonary Diseases. ChemMedChem. 2015 b; 10: 1163-73
77. Elborn JS, Perrett J, Forsman-Semb K, Marks-Konczalik J, Gunawardena K, Entwistle N. Efficacy, safety and effect on biomarkers of AZD9668 in cystic fibrosis. The European respiratory journal. 2012;40:969-76
78. Stevens T, Ekholm K, Granse M, Lindahl M, Kozma V, Jungar C. et al. AZD9668: pharmacological characterization of a novel oral inhibitor of neutrophil elastase. The Journal of pharmacology and experimental therapeutics. 2011;339:313-20
79. Delbosc S, Rouer M, Alsac JM, Louedec L, Philippe M, Meilhac O. et al. Elastase inhibitor AZD9668 treatment prevented progression of experimental abdominal aortic aneurysms. Journal of vascular surgery. 2016;63:486-92 e1
80. Vergelli C, Schepetkin IA, Crocetti L, Iacovone A, Giovannoni MP, Guerrini G. et al. Isoxazol-5(2H)-one: a new scaffold for potent human neutrophil elastase (HNE) inhibitors. Journal of enzyme inhibition and medicinal chemistry. 2017;32:821-31
81. von Nussbaum F, Li VM, Meibom D, Anlauf S, Bechem M, Delbeck M. et al. Potent and Selective Human Neutrophil Elastase Inhibitors with Novel Equatorial Ring Topology: in vivo Efficacy of the Polar Pyrimidopyridazine BAY-8040 in a Pulmonary Arterial Hypertension Rat Model. ChemMedChem. 2016;11:199-206
82. Lucas SD, Costa E, Guedes RC, Moreira R. Targeting COPD: advances on low-molecular-weight inhibitors of human neutrophil elastase. Med Res Rev. 2013 a; 33 Suppl 1: E73-101
83. Sjo P. Neutrophil elastase inhibitors: recent advances in the development of mechanism-based and nonelectrophilic inhibitors. Future Med Chem. 2012;4:651-60
84. Mulchande J, Simoes SI, Gaspar MM, Eleuterio CV, Oliveira R, Cruz ME. et al. Synthesis, stability, biochemical and pharmacokinetic properties of a new potent and selective 4-oxo-beta-lactam inhibitor of human leukocyte elastase. Journal of enzyme inhibition and medicinal chemistry. 2011;26:169-75
85. Ruivo EF, Goncalves LM, Carvalho LA, Guedes RC, Hofbauer S, Brito JA. et al. Clickable 4-Oxo-beta-lactam-Based Selective Probing for Human Neutrophil Elastase Related Proteomes. ChemMedChem. 2016;11:2037-42
86. Makaryan V, Kelley ML, Fletcher B, Bolyard AA, Aprikyan AA, Dale DC. Elastase inhibitors as potential therapies for ELANE-associated neutropenia. Journal of leukocyte biology. 2017;102:1143-51
87. Lucas SD, Goncalves LM, Carvalho LA, Correia HF, Da Costa EM, Guedes RA. et al. Optimization of O3-acyl kojic acid derivatives as potent and selective human neutrophil elastase inhibitors. Journal of medicinal chemistry. 2013 b; 56: 9802-6
88. Giovannoni MP, Schepetkin IA, Crocetti L, Ciciani G, Cilibrizzi A, Guerrini G. et al. Cinnoline derivatives as human neutrophil elastase inhibitors. Journal of enzyme inhibition and medicinal chemistry. 2016;31:628-39
89. Crocetti L, Bartolucci G, Cilibrizzi A, Giovannoni MP, Guerrini G, Iacovone A. et al. Synthesis and analytical characterization of new thiazol-2-(3H)-ones as human neutrophil elastase (HNE) inhibitors. Chem Cent J. 2017;11:127
90. Inuzuka T, Sato S, Baba H, Miyatake T. Suppressive effect of camostat mesilate (FOY 305) on acute experimental allergic encephalomyelitis (EAE). Neurochem Res. 1988;13:225-8
91. Ohta T, Futagami F, Arakawa H, Tsukioka Y, Kitagawa H, Kayahara M. et al. [Inhibitory effect of FOY-305 on liver metastasis of the pancreatic cancer]. Gan To Kagaku Ryoho. 1996;23:1669-72
92. Maekawa A, Kakizoe Y, Miyoshi T, Wakida N, Ko T, Shiraishi N. et al. Camostat mesilate inhibits prostasin activity and reduces blood pressure and renal injury in salt-sensitive hypertension. J Hypertens. 2009;27:181-9
93. Tang Y, Wang Y, Pei Z, Li W, Zhang D, Liu L. et al. A serine protease inhibitor from Musca domestica larva exhibits inhibitory activity against elastase and chymotrypsin. Biotechnology letters. 2016;38:1147-53
94. Wan H, Lee KS, Kim BY, Yuan M, Zhan S, You H. et al. A spider (Araneus ventricosus) chymotrypsin inhibitor that acts as an elastase inhibitor and a microbial serine protease inhibitor. Comparative biochemistry and physiology Part B, Biochemistry & molecular biology. 2013;165:36-41
95. Guillaume D, Huynh TN, Clement D, Nguyen KP, Belaaouaj A. Triterpenoids as neutrophil elastase inhibitors. Nat Prod Commun. 2015;10:167-70
96. Thang TD, Kuo PC, Hwang TL, Yang ML, Ngoc NT, Han TT. et al. Triterpenoids and steroids from Ganoderma mastoporum and their inhibitory effects on superoxide anion generation and elastase release. Molecules. 2013;18:14285-92
97. Okubo K, Kamiya M, Urano Y, Nishi H, Herter JM, Mayadas T. et al. Lactoferrin Suppresses Neutrophil Extracellular Traps Release in Inflammation. EBioMedicine. 2016;10:204-15
98. Iwata K, Doi A, Ohji G, Oka H, Oba Y, Takimoto K. et al. Effect of neutrophil elastase inhibitor (sivelestat sodium) in the treatment of acute lung injury (ALI) and acute respiratory distress syndrome (ARDS): a systematic review and meta-analysis. Intern Med. 2010;49:2423-32
99. Moss RB, Mistry SJ, Konstan MW, Pilewski JM, Kerem E, Tal-Singer R. et al. Safety and early treatment effects of the CXCR2 antagonist SB-656933 in patients with cystic fibrosis. Journal of cystic fibrosis: official journal of the European Cystic Fibrosis Society. 2013;12:241-8
100. Rennard SI, Dale DC, Donohue JF, Kanniess F, Magnussen H, Sutherland ER. et al. CXCR2 Antagonist MK-7123. A Phase 2 Proof-of-Concept Trial for Chronic Obstructive Pulmonary Disease. American journal of respiratory and critical care medicine. 2015;191:1001-11
101. Simpson JL, Powell H, Baines KJ, Milne D, Coxson HO, Hansbro PM. et al. The effect of azithromycin in adults with stable neutrophilic COPD: a double blind randomised, placebo controlled trial. PloS one. 2014;9:e105609
102. Wells JM, Jackson PL, Viera L, Bhatt SP, Gautney J, Handley G. et al. A Randomized, Placebo-controlled Trial of Roflumilast. Effect on Proline-Glycine-Proline and Neutrophilic Inflammation in Chronic Obstructive Pulmonary Disease. American journal of respiratory and critical care medicine. 2015;192:934-42
103. Robertson AL, Holmes GR, Bojarczuk AN, Burgon J, Loynes CA, Chimen M. et al. A zebrafish compound screen reveals modulation of neutrophil reverse migration as an anti-inflammatory mechanism. Science translational medicine. 2014;6:225ra29
104. Zabieglo K, Majewski P, Majchrzak-Gorecka M, Wlodarczyk A, Grygier B, Zegar A. et al. The inhibitory effect of secretory leukocyte protease inhibitor (SLPI) on formation of neutrophil extracellular traps. Journal of leukocyte biology. 2015;98:99-106
105. Van Avondt K, van der Linden M, Naccache PH, Egan DA, Meyaard L. Signal Inhibitory Receptor on Leukocytes-1 Limits the Formation of Neutrophil Extracellular Traps, but Preserves Intracellular Bacterial Killing. Journal of immunology. 2016;196:3686-94
106. Bronze-da-Rocha E. MicroRNAs expression profiles in cardiovascular diseases. Biomed Res Int. 2014;2014:985408
107. Lorenzen JM, Haller H, Thum T. MicroRNAs as mediators and therapeutic targets in chronic kidney disease. Nat Rev Nephrol. 2011;7:286-94
108. Zhong T, Perelman JM, Kolosov VP, Zhou XD. MiR-146a negatively regulates neutrophil elastase-induced MUC5AC secretion from 16HBE human bronchial epithelial cells. Mol Cell Biochem. 2011;358:249-55
109. Ichii O, Otsuka S, Sasaki N, Namiki Y, Hashimoto Y, Kon Y. Altered expression of microRNA miR-146a correlates with the development of chronic renal inflammation. Kidney international. 2012;81:280-92
110. Weldon S, McNally P, McAuley DF, Oglesby IK, Wohlford-Lenane CL, Bartlett JA. et al. miR-31 dysregulation in cystic fibrosis airways contributes to increased pulmonary cathepsin S production. American journal of respiratory and critical care medicine. 2014;190:165-74
111. Narozna B, Langwinski W, Szczepankiewicz A. Non-Coding RNAs in Pediatric Airway Diseases. Genes. 2017:8
112. Petrillo F, Iervolino A, Zacchia M, Simeoni A, Masella C, Capolongo G. et al. MicroRNAs in Renal Diseases: A Potential Novel Therapeutic Target. Kidney Dis (Basel). 2017;3:111-9
113. Gomez IG, Nakagawa N, Duffield JS. MicroRNAs as novel therapeutic targets to treat kidney injury and fibrosis. Am J Physiol Renal Physiol. 2016;310:F931-44
114. Inker LA, Astor BC, Fox CH, Isakova T, Lash JP, Peralta CA. et al. KDOQI US commentary on the 2012 KDIGO clinical practice guideline for the evaluation and management of CKD. American journal of kidney diseases: the official journal of the National Kidney Foundation. 2014;63:713-35
115. Andrassy KM. Comments on 'KDIGO 2012 Clinical Practice Guideline for the Evaluation and Management of Chronic Kidney Disease'. Kidney international. 2013;84:622-3
116. Said A, Desai C, Lerma EV. Chronic kidney disease. Disease-a-month: DM. 2015;61:374-7
117. Levin A. TM, Bonventre J, Coresh J, Donner J. A, Fogo A. B, Fox C. S, Gansevoort R.T, Heerspink H. J. L, Jardine M, Kasiske B, Köttgen A, Kretzler M, Levey A. S, Luyckx V. A., Mehta R, Moe O, Obrador G, Pannu N19, Parikh C. R, Perkovic V, Pollock C, Stenvinkel P, Tuttle K. R, Wheeler D. C, Eckardt K. U.; ISN Global Kidney Health Summit participants. Global kidney health 2017 and beyond: a roadmap for closing gaps in care, research, and policy. Lancet. 2017;390:1888-917
118. Luyckx VATKR, Garcia-Garcia G, Gharbi M.B, Heerspink H. J. L, Johnson D. W, Liu Z.-H, Massy Z. A, Moe O, Nelson R. G, Sola L, David C. Wheeler D. C, White S. L. Reducing major risk factors for chronickidney disease. Kidney international supplements. 2017;7:71-87
119. Collaborators GMaCoD. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016:388 1459-544
120. Hill N. R. FST, Oke JL1, Hirst J. A, O'Callaghan C. A, Lasserson D. S, Hobbs F. D. Global Prevalence of Chronic Kidney Disease - A Systematic Review and Meta-Analysis. PloS one. 2016;11:e0158765
121. Fan J. SH. Impact of Chronic Kidney Disease on Risk for Vascular Events. Curr Vasc Pharmacol. 2016;14:409-14
122. Siasos G. TD,. Michalea S, Oikonomou E, Kolia C, Kioufis S, Synetos A, Vlasis K, Papavassiliou A. G, Stefanadis C. Biomarkers determining cardiovascular risk in patients with kidney disease. Curr Med Chem. 2012;19:2555-71
123. Costa E, Lima M, Alves JM, Rocha S, Rocha-Pereira P, Castro E. et al. Inflammation, T-cell phenotype, and inflammatory cytokines in chronic kidney disease patients under hemodialysis and its relationship to resistance to recombinant human erythropoietin therapy. Journal of clinical immunology. 2008 b; 28: 268-75
124. Costa E, Rocha S, Rocha-Pereira P, Castro E, Reis F, Teixeira F. et al. Cross-talk between inflammation,coagulation/fibrinolysis and vascular access in hemodialysis patients. The journal of vascular access. 2008 c; 9: 248-53
125. Polanska B, Augustyniak D, Makulska I, Niemczuk M, Zwolinska D, Jankowski A. Elastase, alpha1-proteinase inhibitor, and interleukin-8 in pre-dialyzed and hemodialyzed patients with chronic kidney disease. Pediatrics international: official journal of the Japan Pediatric Society. 2010;52:735-43
126. Takouli L, Hadjiyannakos D, Metaxaki P, Sideris V, Filiopoulos V, Anogiati A. et al. Vitamin E-coated cellulose acetate dialysis membrane: long-term effect on inflammation and oxidative stress. Renal failure. 2010;32:287-93
127. Yang SK, Xiao L, Xu B, Xu XX, Liu FY, Sun L. Effects of vitamin E-coated dialyzer on oxidative stress and inflammation status in hemodialysis patients: a systematic review and meta-analysis. Renal failure. 2014;36:722-31
128. Cohen-Mazor M, Mazor R, Kristal B, Sela S. Elastase and cathepsin G from primed leukocytes cleave vascular endothelial cadherin in hemodialysis patients. Biomed Res Int. 2014;2014:459640
129. Khatib-Massalha E, Michelis R, Trabelcy B, Gerchman Y, Kristal B, Ariel A. et al. Free circulating active elastase, contributes to chronic inflammation in hemodialysis patients. Am J Physiol Renal Physiol. 2017: ajprenal 00070. 2017
130. Ueda M, Uchimura K, Narita Y, Miyasato Y, Mizumoto T, Morinaga J. et al. The serine protease inhibitor camostat mesilate attenuates the progression of chronic kidney disease through its antioxidant effects. Nephron. 2015;129:223-32
131. Narita Y, Ueda M, Uchimura K, Kakizoe Y, Miyasato Y, Mizumoto T. et al. Combination therapy with renin-angiotensin-aldosterone system inhibitor telmisartan and serine protease inhibitor camostat mesilate provides further renoprotection in a rat chronic kidney disease model. Journal of pharmacological sciences. 2016;130:110-6
132. Li G, Jia J, Ji K, Gong X, Wang R, Zhang X. et al. The neutrophil elastase inhibitor, sivelestat, attenuates sepsis-related kidney injury in rats. International journal of molecular medicine. 2016;38:767-75
133. Jeong JC, Kim JE, Gu JY, Yoo HJ, Ryu JW, Kim DK. et al. Significance of the DNA-Histone Complex Level as a Predictor of Major Adverse Cardiovascular Events in Hemodialysis Patients: The Effect of Uremic Toxin on DNA-Histone Complex Formation. Blood purification. 2016;41:64-71
134. Kohlova M, Ribeiro S, do Sameiro-Faria M, Rocha-Pereira P, Fernandes J, Reis F. et al. Circulating cell-free DNA levels in hemodialysis patients and its association with inflammation, iron metabolism, and rhEPO doses. Hemodialysis international International Symposium on Home Hemodialysis. 2013;17:664-7
135. Tovbin D, Novack V, Wiessman MP, Abd Elkadir A, Zlotnik M, Douvdevani A. Circulating cell-free DNA in hemodialysis patients predicts mortality. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2012;27:3929-35
136. Corredor Z, Stoyanova E, Rodriguez-Ribera L, Coll E, Silva I, Diaz JM. et al. Genomic damage as a biomarker of chronic kidney disease status. Environmental and molecular mutagenesis. 2015;56:301-12
137. Schupp N, Stopper H, Heidland A. DNA Damage in Chronic Kidney Disease: Evaluation of Clinical Biomarkers. Oxidative medicine and cellular longevity. 2016;2016:3592042
138. Coimbra S, Santos-Silva A, Costa E, Bronze-da-Rocha E. DNA damage in end-stage renal disease patients. Assessment by in vitro comet assay and by cell-free DNA quantification. INTECH, editor. Genotoxicity: Open Access Publisher. 2017
139. Nikolic A, Milosevic K, Boskovic S, Nestorovic B. Neutrophil elastase gene polymorphisms: modulators of response to therapy in childhood bronchiectasis? Lung. 2014;192:595-9
140. Park JY, Chen L, Lee J, Sellers T, Tockman MS. Polymorphisms in the promoter region of neutrophil elastase gene and lung cancer risk. Lung cancer. 2005;48:315-21
141. Taniguchi K, Yang P, Jett J, Bass E, Meyer R, Wang Y. et al. Polymorphisms in the promoter region of the neutrophil elastase gene are associated with lung cancer development. Clinical cancer research: an official journal of the American Association for Cancer Research. 2002;8:1115-20
142. Cho HK, Jeon IS. Different clinical phenotypes in familial severe congenital neutropenia cases with same mutation of the ELANE gene. Journal of Korean medical science. 2014;29:452-5
143. Newburger PE, Pindyck TN, Zhu Z, Bolyard AA, Aprikyan AA, Dale DC. et al. Cyclic neutropenia and severe congenital neutropenia in patients with a shared ELANE mutation and paternal haplotype: evidence for phenotype determination by modifying genes. Pediatric blood & cancer. 2010;55:314-7
144. Kurnikova M, Maschan M, Dinova E, Shagina I, Finogenova N, Mamedova E. et al. Four novel ELANE mutations in patients with congenital neutropenia. Pediatric blood & cancer. 2011;57:332-5
145. Makaryan V, Zeidler C, Bolyard AA, Skokowa J, Rodger E, Kelley ML. et al. The diversity of mutations and clinical outcomes for ELANE-associated neutropenia. Curr Opin Hematol. 2015;22:3-11
146. Thanarajasingam U, Jensen MA, Dorschner JM, Wampler Muskardin T, Ghodke-Puranik Y, Purmalek M. et al. Brief Report: A Novel ELANE Mutation Associated With Inflammatory Arthritis, Defective NETosis, and Recurrent Parvovirus Infection. Arthritis Rheumatol. 2017;69:2396-401
147. Fernandes R, Freitas B, Miranda V, Costa E, Santos-Silva A, Bronze-da-Rocha E. Polymorphisms of the ELANE Gene Promoter Region in End-Stage Chronic Kidney Disease Patients. Genes. 2016:7
148. do Sameiro-Faria M, Ribeiro S, Rocha-Pereira P, Fernandes J, Reis F, Bronze-da-Rocha E. et al. Body mass index and resistance to recombinant human erythropoietin therapy in maintenance hemodialysis patients. Renal failure. 2013 b; 35: 1392-8
149. Seliger SL, Zhang AD, Weir MR, Walker L, Hsu VD, Parsa A. et al. Erythropoiesis-stimulating agents increase the risk of acute stroke in patients with chronic kidney disease. Kidney international. 2011;80:288-94
150. Cherng YG, Lin CS, Shih CC, Hsu YH, Yeh CC, Hu CJ. et al. Stroke risk and outcomes in patients with chronic kidney disease or end-stage renal disease: Two nationwide studies. PloS one. 2018;13:e0191155
151. Yilmaz I, Ozkok A, Kostek O, Kolukisa A, Duran I, Odabas AR. et al. C-reactive protein but not hepcidin, NGAL and transferrin determines the ESA resistance in hemodialysis patients. Renal failure. 2016;38:89-95
152. Kimachi M, Fukuma S, Yamazaki S, Yamamoto Y, Akizawa T, Akiba T. et al. Minor Elevation in C-Reactive Protein Levels Predicts Incidence of Erythropoiesis-Stimulating Agent Hyporesponsiveness among Hemodialysis Patients. Nephron. 2015;131:123-30
153. Hung SC, Lin YP, Tarng DC. Erythropoiesis-stimulating agents in chronic kidney disease: what have we learned in 25 years? J Formos Med Assoc. 2014;113:3-10
154. Tsuruya K, Yoshida H, Suehiro T, Fujisaki K, Masutani K, Kitazono T. Erythropoiesis-stimulating agent slows the progression of chronic kidney disease: a possibility of a direct action of erythropoietin. Renal failure. 2016;38:390-6
155. Piotr B, Mariusz S, Jacek R. Methoxy Polyethylene Glycol-Epoetin Beta as a Novel Erythropoiesis Stimulating Agent with Possible Nephroprotective and Cardiovascular Protective Effects in Non-Dialysis Chronic Kidney Disease Patients. Curr Pharm Biotechnol. 2017;18:303-8
156. Chateauvieux S, Grigorakaki C, Morceau F, Dicato M, Diederich M. Erythropoietin, erythropoiesis and beyond. Biochemical pharmacology. 2011;82:1291-303
157. Ribeiro S, Belo L, Reis F, Santos-Silva A. Iron therapy in chronic kidney disease: Recent changes, benefits and risks. Blood reviews. 2016;30:65-72
158. Tanaka T, Nangaku M. Recent advances and clinical application of erythropoietin and erythropoiesis-stimulating agents. Experimental cell research. 2012;318:1068-73
159. Bailie GR, Larkina M, Goodkin DA, Li Y, Pisoni RL, Bieber B. et al. Variation in intravenous iron use internationally and over time: the Dialysis Outcomes and Practice Patterns Study (DOPPS). Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2013;28:2570-9
160. Antonelou MH, Kriebardis AG, Velentzas AD, Kokkalis AC, Georgakopoulou SC, Papassideri IS. Oxidative stress-associated shape transformation and membrane proteome remodeling in erythrocytes of end stage renal disease patients on hemodialysis. Journal of proteomics. 2011;74:2441-52
161. Costa E, Rocha S, Rocha-Pereira P, Castro E, Miranda V, do Sameiro Faria M. et al. Altered erythrocyte membrane protein composition in chronic kidney disease stage 5 patients under haemodialysis and recombinant human erythropoietin therapy. Blood purification. 2008 e; 26: 267-73
162. Costa E, Rocha S, Rocha-Pereira P, Castro E, Miranda V, Faria Mdo S. et al. Changes in red blood cells membrane protein composition during hemodialysis procedure. Renal failure. 2008 d; 30: 971-5
163. Georgatzakou HT, Antonelou MH, Papassideri IS, Kriebardis AG. Red blood cell abnormalities and the pathogenesis of anemia in end-stage renal disease. Proteomics Clinical applications. 2016;10:778-90
164. Georgatzakou HT, Tzounakas VL, Kriebardis AG, Velentzas AD, Kokkalis AC, Antonelou MH. et al. Short-term effects of hemodiafiltration versus conventional hemodialysis on erythrocyte performance. Can J Physiol Pharmacol. 2017 b: 1-9
165. Rocha S, Gomes D, Lima M, Bronze-da-Rocha E, Santos-Silva A. Peroxiredoxin 2, glutathione peroxidase, and catalase in the cytosol and membrane of erythrocytes under H2O2-induced oxidative stress. Free Radic Res. 2015;49:990-1003
166. Rocha S, Costa E, Coimbra S, Nascimento H, Catarino C, Rocha-Pereira P. et al. Linkage of cytosolic peroxiredoxin 2 to erythrocyte membrane imposed by hydrogen peroxide-induced oxidative stress. Blood Cells Mol Dis. 2009;43:68-73
167. Georgatzakou HT, Tzounakas VL, Kriebardis AG, Velentzas AD, Papageorgiou EG, Voulgaridou AI. et al. Pathophysiological aspects of red blood cells in end-stage renal disease patients resistant to recombinant human erythropoietin therapy. Eur J Haematol. 2017 a; 98: 590-600
Author contact
![]() Corresponding author: Professor Elsa Bronze-da-Rocha, UCIBIO/REQUIMTE, Laboratório de Bioquímica, Departamento de Ciências Biológicas, Faculdade de Farmácia, Universidade do Porto, 4050-313 Porto, Portugal. Email: elsa.rochaup.pt; Tel.: +351 220 428 568; Fax: +351 226 093 500
Corresponding author: Professor Elsa Bronze-da-Rocha, UCIBIO/REQUIMTE, Laboratório de Bioquímica, Departamento de Ciências Biológicas, Faculdade de Farmácia, Universidade do Porto, 4050-313 Porto, Portugal. Email: elsa.rochaup.pt; Tel.: +351 220 428 568; Fax: +351 226 093 500