Impact Factor ISSN: 1449-2288
- Issue 13; 2026
- Issue 12; 2026
- Issue 11; 2026
- Issue 10; 2026
- Issue 9; 2026
- Volume 22; 2026
- Past Issues
- Advance Articles
- Editorial Board
- Cover Images
- Index & Coverage
- Cover Suggestion
- Special Issues
Introduction
Materials and Methods
Results
Discussion
Abbreviations
Supplementary Material
Acknowledgements
References

 Global reach, higher impact
Global reach, higher impactInt J Biol Sci 2018; 14(11):1437-1444. doi:10.7150/ijbs.27043 This issue Cite
Research Paper
Overexpression of human mitochondrial alanyl-tRNA synthetase suppresses biochemical defects of the mt-tRNAAla mutation in cybrids
Xiaoxu Zhao1,2#, Jiamin Han1,2#, Ling Zhu1, Yun Xiao2, Chenghui Wang1,2, Fang Hong1, Pingping Jiang1,2 ![]() , Min-Xin Guan1,2
, Min-Xin Guan1,2
1. Division of Medical Genetics and Genomics, The Children's Hospital, Zhejiang University School of Medicine, Hangzhou, 310058, China.
2. Institute of Genetics Zhejiang University, and Department of Human Genetics, Zhejiang University School of Medicine, Hangzhou, 310058, China.
# These two authors contributed equally to this work.
Received 2018-5-3; Accepted 2018-7-17; Published 2018-8-6
Abstract

Mutations of mitochondrial transfer RNAs (mt-tRNAs) play a major role in a wide range of mitochondrial diseases because of the vital role of these molecules in mitochondrial translation. It has previously been reported that the overexpression of mitochondrial aminoacyl tRNA synthetases is effective at partially suppressing the defects resulting from mutations in their cognate mt-tRNAs in cells. Here we report a detailed analysis of the suppressive activities of mitochondrial alanyl-tRNA synthetase (AARS2) on mt-tRNAAla 5655 A>G mutant. Mitochondrial defects in respiration, activity of oxidative phosphorylation complexes, ATP production, mitochondrial superoxide, and membrane potential were consistently rescued in m.5655A>G cybrids upon AARS2 expression. However, AARS2 overexpression did not result in a detectable increase in mutated mt-tRNAAla but caused an increase incharged mt-tRNAAla in mutant cybrids, leading to enhanced mitochondrial translation. This indicated that AARS2 improved the aminoacylation activity in the case of m.5655A>G, rather than having a stabilizing effect on the tRNA structure. The data presented in this paper deepen our understanding of the pathogenesis of mt-tRNA diseases.
Keywords: mitochondrial tRNA, mitochondrial alanyl-tRNA synthetase, cybrid, oxidative phosphorylation
Introduction
Mitochondria are ubiquitous in eukaryotes and essential for cell survival. Disorder of mitochondrial DNA (mtDNA) causes a wide spectrum of diseases (MITOMAP, https://www.mitomap.org/MITOMAP). Most pathogenic mutations in mitochondria occur in mt-tRNA genes, leading to reduced levels in the availability of steady-state mt-tRNA for aminoacylation and mitochondrial translation and, in turn, causing oxidative phosphorylation (OXPHOS) defects [1, 2]. As mitochondrial protein synthesis is dependent on the coordinated action of mitochondrial aminoacyl-tRNA synthetases (mt-aaRSs) and their cognate mitochondrial tRNAs (mt-tRNAs), evidence has highlighted that mt-aaRS overexpression can restore some mitochondrial functions reduced by mt-tRNA mutations in human cells [3, 4] and yeast [5, 6]. One of such mt-aaRSs is the class I human leucyl-tRNA synthetase (LARS2), the overexpression of which attenuates the defect of m.3243 A>G mutations in transmitochondrial cybrids with an increase in the steady-state level of tRNALeu (UUR) 3243A>G [3]. Similarly, the overexpression of mitochondrial valyl-tRNA synthetase (VARS2, class I enzyme) has been shown to restore the steady-state levels of tRNAVal resulting from 1624 C>T in Leigh disease [7]. Moreover, the non-cognate LARS2 can partially rescue the pathological phenotype caused by the destabilizing mutations in tRNAVal [8, 9]. However, mitochondrial class II human alanyl-tRNA synthetase (AARS2), as another non-cognate, fails to rescue the biochemical deficiency of 1624 C>T mutant tRNAVal due to its different structural and functional properties, which vary between the two aaRS classes. Based on the findings obtained to date, the rescuing affects on mutations are dominated by Class I mt-aaRSs. Previous observations have also suggested that mt-aaRS suppress the defects resulting from mt-tRNA mutations due to stabilization of the correct tRNA folding rather than increase in the mt-aaRS catalytic activity [3, 8, 10].
The function of mt-tRNA in mitochondrial translation is mostly maintained by factors, such as its structural features, nucleotide modifications, and precursor processing. The above mutations, m.1624C>T and m.3243A>G in D-loop, and m.8344 A>G in TΨC-loop are important for the tRNA L-shaped structure and modification [11, 12]. The mutation m.7511 A>G (A37 in tRNA numbering) associated with deafness has been revealed to introduce m1G37 modification of the mt-tRNAAsp, leading to the impairment of mt-tRNA metabolism [1]. Recently, we found that m.5655A>G mutation (A1 at the aminoacyl acceptor stem) reduced the efficiency of tRNAAla processing at the 5′-end by RNase P, resulting in aminoacylation deficiency and reduction in the steady-state level of mt-tRNAAla [2]. Thus, it is anticipated that human AARS2 overexpression would ameliorate the defect of mt-tRNA metabolism and mitochondrial function in cybrids carrying m.5655A>G. Furthermore, we hypothesized that compared with the mechanism underlying m.3243A>G or m.8344 A>G, the major role of AARS2 overexpression in the mechanism underlying m.5655A>G is to increase the aminoacylation activity rather than tRNAAla stability and subsequently improve the biochemical function of mitochondria. To test this hypothesis, stable transfectants were constructed by transferring a human AARS2 cDNA into a cybrid carrying the homoplasmic m.5655A>G mutation and an isogenic control cybrid with the homoplasmic wild-type version of tRNAAla. These stable transfectants were analyzed for their capacity to aminoacylate tRNAs, the associated stability of tRNAAla, the mitochondrial respiratory chain function, and the mitochondrial membrane potential (Δψm).
Materials and Methods
Cell lines and culture conditions
The cybrid cell lines with the homoplasmic tRNAAla 5655A>G mutation (T) and control cell lines (C) with the homoplasmic wild-type version of tRNAAla were derived respectively from one proband harboring the m.5655A>G mutation and one control with the same mtDNA haplogroup as described previously [2], and cultured in Dulbecco's modified Eagle's medium (Hyclone) with 10% fetal bovine serum (Gemini). ApEGFP-N1-AARS2 vector was provided as a gift from Dr. Suomalainen and is described elsewhere [13]. AARS2 cDNA was inserted in Xhol I/EcoR I sites of pcDNA3.1 zeo (-) (InvitrogenTM, Life Technologies). Cybrids were transfected with pcDNA3.1-AARS2 (CVA or TVA) and pcDNA3.1 zeo (-) as a vehicle control (CV0 or TV0) for parallel analysis and then selected using 300-μg/mL/zeocin (InvitrogenTM, Life Technologies).
Northern blot analysis of mt-tRNA
Total mitochondrial RNA (mtRNA) was obtained from mitochondria isolated from mutant and control cybrid cell lines using the Totally RNA kit (Ambion) with approximately 4.0 × 107 cells. Two micrograms of total mtRNA was electrophoresed through a 15% polyacrylamide-8 M urea gel in Tris-borate-EDTA buffer and then electroblotted onto a positively charged nylonmembrane (Roche) for hybridization analysis with digoxin-labeled specific tRNA probes (Invitrogen): tRNAAla, tRNAGlu, tRNALys, tRNAHis, and 5S rRNA. The density of each band was quantified using Image J software.
mt-tRNA aminoacylation analysis
Total RNA was extracted by Trizol (Sangon Biotech, B511311-0100) from various cell lines (~5.0 × 107 cells). For the tRNA northern blotting analysis, 15-μg total RNA was electrophoresed through a urea denaturing 8% polyacrylamide-7 M urea gel to separate the charged and uncharged tRNAs, as detailed elsewhere [14]. The gels were electroblotted onto a positively charged nylon membrane (Roche) for the hybridization analysis for tRNAAla, tRNASer (AGY), tRNAThr and tRNALys with DIG-labeled oligodeoxynucleotide probes, which were synthesized with a 3′-digoxigenin modification by Invitrogen. The hybridization and density of each band were analyzed using Image J software and calculated as described previously [14].
Western blotting and AARS2 expression analysis
SDS-PAGE analysis of mitochondrial lysate was performed for mtDNA-coded proteins and of whole-cell lysate for Total OXPHOS Human WB Antibody Cocktail. After electrophoresis, gels were transferred onto a PVDF membrane (Thermo Fisher Scientific, Shanghai, China) and processed for immunoblotting. The following commercially available antibodies were used: TOM20 (ab56783), ND1 (19703-1-AP), ATP6 (55313-1-AP), ND5 (ab92624), CYTB (55090-1-AP), CO2 (ab110258), and total OXPHOS Human WB Antibody Cocktail (ab110411). All secondary antibodies were HRP-conjugated (Molecular Probes). The density of each band was quantified using Image J software.
Oxygen consumption measurements
An XF96 extracellular flux analyzer (Seahorse Bioscience) was used to measure the oxygen consumption rate (OCR) [15]. Briefly, 3 × 104 cells of each cell line were seeded in three wells each in an XF96 cell culture plate. The OCR of each well was measured over the course of programmed injections of oligomycin (1.5 μM), carbonyl cyanide-p-(trifluoromethoxy) phenylhydrazone (FCCP, 1μM), and rotenone/antimycin A (1 μM/1 μM).
Enzymatic assays for respiratory chain complexes
The enzymatic activities of complexes I (NADH-CoQ reductase) to IV (cytochrome C oxidase) were assayed as described elsewhere [16, 17]. ATP synthase activity was analyzed in the presence and absence of the specific inhibitor antimycin A (1mg/mL) in mitochondrial fractions, as described elsewhere [18]. The activity was spectrophotometrically monitored at 340 nm using Synergy H1 Hybrid Reader (BioTeK).
Mitochondrial ATP measurements
A CellTiter-Glo luminescent cell viability assay kit (Promega) was used for measuring cellular and mitochondrial ATP levels using a Synergy H1 Hybrid Reader (BioTeK), in accordance with a modified version of the manufacturer's instructions and a protocol reported elsewhere [19].
Assessment of Δψm
Δψm was assessed using a JC-10 assay kit (Abcam), in accordance with the manufacturer's recommendations, and analyzed using a Novocyte flow cytometer (ACEA Biosciences) as detailed elsewhere [19].
Mitochondrial superoxide measurement
Quantification of mitochondrial superoxide was performed using MitoSOX-Red (Invitrogen), as described elsewhere [15]. Briefly, 3×105 cells were stained with 5 μM MitoSOX-Red for 25 min at 37 °C in the dark. Subsequently, samples were processed using a Novocyte flow cytometer (ACEA Biosciences) in PE channel. The cell population and the relative fluorescence intensity were evaluated by three independent experiments.
Statistics
Results are given as mean ± standard deviation (SD) and were compared using unpaired t-tests assuming an unequal distribution. Data were analyzed using GraphPad Prism (v5.04, www.graphpad.com). Values of *P < 0.05 and **P < 0.01 were considered to be statistically significant. All reactions were performed in triplicate or quadruplicate.
Results
The steady-state levels of mutant mt-tRNAAla
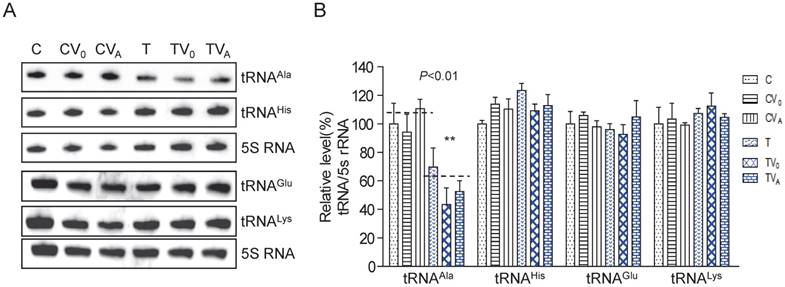
To examine whether AARS2 overexpression increases the steady-state level of mt-tRNAAla, we constructed stable transfectants based on cybrids carrying m.5655A>G (T-cell line) and the wild-type version (C-cell line) using pcDNA3.1-AARS2 or pcDNA3.1 along as a vehicle control. The level and location of AARS2 protein in cell lines were confirmed, as shown in Supplementary Figure 1. Total tRNAs were isolated from transfectants, control cybrids (CV0 and CVA), mutant cybrids (TV0 and TVA), and parental cybrids (C and T). DIG-labeled oligodeoxynucleotide probes specific for mt-tRNAAla and mt-tRNAGlu from the L-strand as well as tRNALys and tRNAHis from the H-strand were hybridized and normalized to the mitochondrial 5S rRNA. The relative levels of steady-state mt-tRNAAla in the T, TV0, TVA, C, CV0, and CVA cell lines were70%, 43%, 52%, 100%, 94%, and 111%, respectively (Figure 1). Overexpression AARS2 didn't alter the steady-state level of tRNAAla in the mutant cell lines. No significant difference was observed in the relative steady-state levels of tRNAGlu, tRNALys, or tRNAHis upon AARS2 expression between the control and mutant cybrids.
Analysis of the steady-state levels of mt-tRNAAla. (A) Northern blot analysis of the steady-state levels of mt-tRNA hybridized with the DIG-labeled oligonucleotide probes specific for tRNAAla, tRNAGlu, tRNALys, tRNAHis, and 5S RNA. (B) Quantification of the relative mt-tRNA levels normalized to the mean level of 5S RNA in its corresponding cybrids.
Aminoacylation capacity of mt-tRNAAla
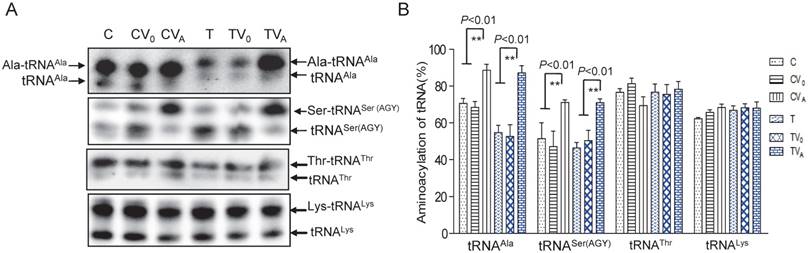
As AARS2 failed to restore the steady-state levels of mt-tRNAAla in mutant cybrids, we next investigated its effects on the aminoacylation defect of mt-tRNAAla caused by 5655A>G. For this purpose, the charged tRNA (upper band) and uncharged tRNA (lower band) were separated in acidic denaturing polyacrylamide-urea gels and hybridized with specific probes for the tRNAAla, tRNASer (AGY), tRNAThr and tRNALys. Marked increases in the amount of charged mt-tRNAAla and mt-tRNASer (AGY) in the mutant cybrids were observed (Figure 2). The rate of aminoacylation was presented as percentage by the charged mt-tRNA to the total mt-tRNA each. The aminoacylation rates of the T cybrids were clearly improved from 53% ± 6.4% in TV0 to 87% ± 4.0% in TVA, which was approximately 123% of the level in the C-cell line (Figure 2). No increase in the rate was observed in vehicle cybrids (TV0 and CV0) compared with the ratios in the parental T- and C-cell lines, respectively. Surprisingly, AARS2 overexpression also increased the aminoacylation efficiency of mt-tRNASer (AGY) from 47% ± 8.4% in CV0 to 71% ± 1.4% in CVA and 51% ± 4.2% in TV0 to 70% ± 2.1% in TVA. Whereas, no changes of aminoacylation levels were found in mt-tRNAThr and mt-tRNALys when AARS2 overexpression.
Aminoacylationassay of mt-tRNAAla and other mt-tRNAs. (A) Charged and uncharged mt-tRNAAla and other mt-tRNAs separated on an 8% polyacrylamide-7 M urea gel. (B) Quantification of the aminoacylation rates of mt-tRNAs
Increased OXPHOS protein levels
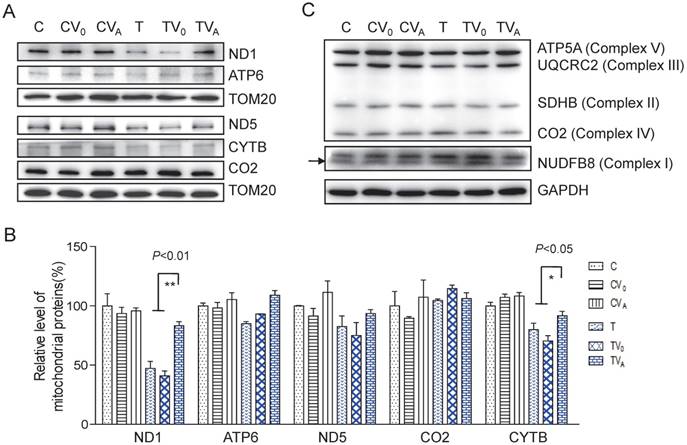
To determine whether the increase in aminoacylation led to an improvement in the translation of respiratory proteins, we measured complex protein levels. As shown in Figure 3, the levels of mitochondrial encoded ND1, ATP6, ND5, CO2, and CYTB increased following AARS2 overexpression, from the mean levels of 41%, 93%, 75%, 114%, and 70% in TV0 cell lines to 83%, 109%, 93%, 106%, and 91% in TVA cell lines, which were comparable with those in wild-type control cell lines. A modest increase was also observed in the level of ATP5A of Complex V as an assembly marker. No significant changes were detected in Complexes I, II, and III when probed for NDUFB8, SDHB, and UQCRC2, respectively, which are encoded by nuclear genes.
Enhanced OXPHOS proteins after AARS2 induction. (A)Western blot analysis of OXPHOS subunits (ND1, ATP6, ND5, CO2, and CYTB) encoded by mtDNA with TOM20 as a loading control. (B) Quantification of the relative levels of ND1, ATP6, ND5, CO2, and CYTB. (C) Western blot analysis of OXPHOS subunits of ATP5A, UQCRC2, SDHB, and NDUFB8 encoded by nuclear genes.
Improvement in respiration and activity of the OXPHOS complexes
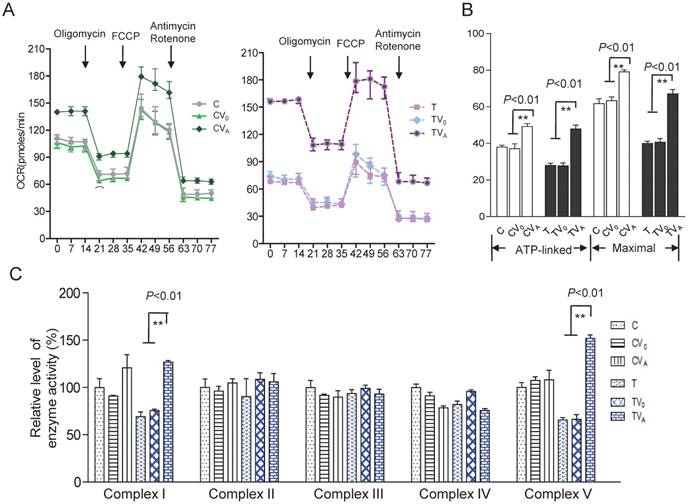
To investigate the effect of AARS2 overexpression on oxidative phosphorylation, the OCR was directly assessed as an indicator of mitochondrial respiration. AARS2 overexpression resulted in the recovery of ATP-linked and maximal respiration OCR to levels comparable to those of the vehicle controls (Figure 4A and B), from 27.7 ± 1.6 (pmoles/min) in TV0 to 48 ± 2.1(pmoles/min) in TVA (P<0.01) and from 41 ± 2.0 (pmoles/min) in TV0 to 67 ± 2.3 (pmoles/min) in TVA (P<0.01). In addition, to more accurately evaluate the recovery of respiration, OXPHOS complex activities were measured using mitochondria isolated from cybrids. No changes were observed in Complex II, III, and IV activities due to AARS2 overexpression, whereas AARS2 overexpression significantly increased Complex I and V activities (Figure 4C). The defect of Complex I caused by 5655A>G increased from 69% ± 5% of the level of wild-type control C to 127% ± 1.7% in TVA (P<0.01). Similarly, the defect of Complex V in mutant cybrids increased from 65% ± 0.8% of the level of wild-type control C to 151% ± 3.6% in TVA (P<0.01).
Respiratory chain function and activity of the OXPHOS complexes. (A) The real-time record of O2 consumption in cybrids over the course of programmed injections of oligomycin (1.5 μM), FCCP (1μM), and rotenone/antimycin A (1 μM/1 μM) for 3 × 104 cells per well. (B) Graphs present the ATP-linked OCR and maximal OCR. ATP-linked OCR was determined as the OCR before oligomycin administration minus the OCR after it. Maximal was determined as the OCR after FCCP administration minus non-mitochondrial OCR, as described previously. (C)Enzymatic activities of respiratory chain complexes I-V. Data were calculated based on at least three independent determinations.
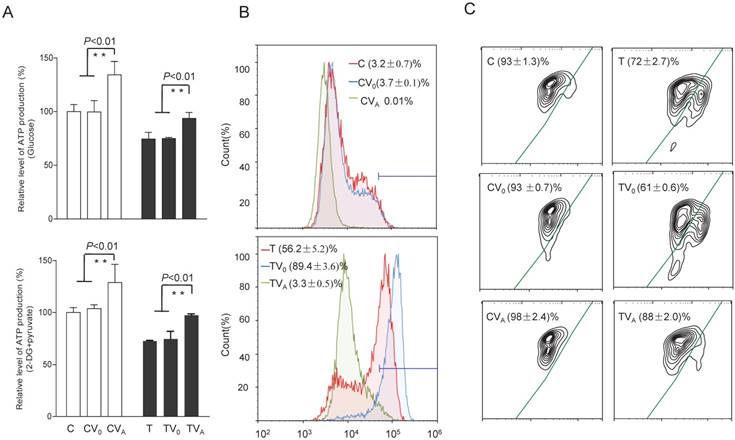
Alterations in ATP, superoxide production, and Δψm
To test whether the increases in OCR and complex activities were reflected in the ATP production of the cell lines expressing AARS2, the relative levels of cellular and mitochondrial ATP production were measured using the luciferin/luciferase assay. As shown in Figure 5, the level of total cellular ATP production (in the presence of glucose) in mutant cells improved from 74% ± 6.3% to 94% ± 5.6% upon AARS2 expression (P<0.01). The same trend was found for the level of mitochondrial ATP (in the presence of 2-deoxy-D-glucose and pyruvate), which increased from 72% ± 0.8% in T cells to 97% ± 1.3% in TVA (P<0.01), relative to the mean value in the C-cell lines. However, a significant reduction in the detectable mitochondrial superoxide population in TVA was found relative to that in the mutant T-cell line, i.e. a decrease from 56.2% ± 5.2% of positive population in the T-cell line to only 3.3% ± 0.5% in TVA (P<0.01), which was comparable to the 3.2% ± 0.7% positive population in C cybrids. ΔΨm is a sensitive marker for mitochondrial function. JC-10 fluorescent assay via flow cytometry revealed that the population with a normal level of ΔΨm in mutant cybrids had notably increased from 72% ± 2.7% in T cybrids to 88% ± 2.0% (P<0.01) upon AARS2 expression, a population level similar to that in the wild-type control C cybrids (93% ± 1.3%).
Alterations in ATP, superoxide production, and mitochondrial membrane potential. (A) ATP production in total cells (glucose) and in mitochondria (2-DG and pyruvate). (B) Mitochondrial superoxide production detected by MitoSOX-Red. (C) Mitochondrial membrane potential analysis by JC-10.
Discussion
In the present study, we showed that the mitochondrial class II AARS2 can ameliorate the mitochondrial biochemical defects associated with the 5655A>G mutation in mt-tRNAAla. Pathogenic mt-tRNA mutations usually lead to mitochondrial dysfunction for which no effective treatments are currently available. Perli et al. [4] reported that a patient with pathogenic 4277T>C mutation in the dihydrouridine loop of mt-tRNAIle remained clinically unaffected because she had a naturally higher IARS2 expression. In addition, the overexpression of class I mt-aaRSs could suppress the defects caused by mt-tRNA mutations, for example, LARS2 overexpression for the defect of mt-tRNALeu(UUR) 3243A>G [3,20], IARS2 overexpression for mt-tRNAIle 4277T>C [4], VARS2 overexpression for mt-rRNAVal 1624C>T [7], and LARS2 overexpression for non-cognate mt-tRNAVal 1624C>T [8]. The aaRSs are divided into two classes (I and II) based on the architecture of the active site [21].
Although the AARS2 differs from the other mt-aaRSs with a conserved editing domain for proofreading [22], overexpression of the entire AARS2 here typically improved the OXPHOS respiration with increased mitochondrial OCR for ATP production and reserve capacity in m.5655A>G cybrids, as previously reported [8]. The OCR results were consistent with the corrected complex I and V enzyme activities and the increased level of ATP production induced by AARS2 in the mutant cybrids. Fortunately, other biochemical defects, such as the ROS and ΔΨm in mutant cybrids were found to be improved upon AARS2 expression, relative to the wild-type control. Our results highlight that this class II mt-aaRS also has a suppressive effect on the mitochondrial defect caused by its cognate mt-tRNA mutation. However, the molecular mechanism underlying this suppression is complicated, for which conflicting findings have been reported [23]. The increased steady-state level of mt-tRNAVal in cybrids harboring 1624 C>T was observed by Rorbach et al. [7] in the presence of the entire VARS2. On the other hand, both steady-state level and aminoacylation of mt-tRNALeu in cybrids carrying 3243 A>G were increased by LARS2 overexpression [3]. Meanwhile, no change in the stability level of mt-tRNALeu 3243 A>G has been detected after the expression of the carboxy-terminal domain of LARS2 [9, 24]. In this study, AARS2 overexpression did not result in a detectable increase of the mutated mt-tRNAAla but caused an increase incharged mt-tRNAAla in mutant cybrids, leading to enhanced mitochondrial translation. This strongly suggested that AARS2 improved the aminoacylation activity rather than preventing the degradation of mt-tRNAAla 5655A>G. Guo et al. reported that AARS2 has the highest tRNA-binding affinity to enhance the rate of aminoacylation [25]. However, we hypothesized that the rescue mechanism is more likely dependent on the mt-tRNA mutations, leading to different molecular mechanisms responsible for the reduction in the level of steady-state mt-tRNA. In cases of 5′-end mutations in mt-tRNA, such as 5655A>G and 4263A>G, the inefficiency of 5′-end cleavage by RNase P for the processing of mt-tRNA precursor accounted for the marked decrease in the levels of mt-tRNAAla [2] and mt-tRNAIle [26]. In contrast, the alteration in the mt-tRNA structure caused by mutations in the D-loop or TΨC-loop was responsible for the reduction in mt-tRNA levels [11]. Considering the recognition and binding function of the mt-tRNA, mt-aaRS was thought to have a stabilizing effect on the tRNA substrate, particularly on destabilizing mutations, which in turn promotes more efficient aminoacylation [7, 20]. Therefore, our data indicate that the suppression of mitochondrial defect by AARS2 was related more to the enzyme activities than to the stabilizing effect on the mt-tRNA structure in the case of the 5655A>G mutation. Another interesting facet of the AARS2 expression is that it caused increases in the amount and rate of aminoacylation of mt-tRNASer (AGY). Beebe, et al. reported that bacterial alanyl-tRNA synthetase can misactivate glycine or serine [27]. Current evidence also found that the mammalian AARS2 can proofread the mischarged Ser-tRNAAla in vitro [28]. However, whether AARS2 overexpression facilitated the aminoacylation of mt-tRNASer (AGY) in this study requires confirmation in further studies.
In summary, the results obtained in this study demonstrate that class II AARS2 can rescue the biochemical defects of mitochondria caused by 5655A>G mutation in mt-tRNAAla, with improvement in the mitochondrial respiratory chain function, including increases in OXPHOS complex activities and ATP production, reduction in mitochondrial superoxide levels, and restoration of Δψm. Our results also showed that the rescue by AARS2 is dependent on its aminoacylation activity rather than its stabilizing effect on mt-tRNAAla 5655A>G, leading to enhanced mitochondrial translation. Finally, we suggest that the complicated mechanism of mt-aaRS rescue is mainly due to the different pathogenesis for the reduction of steady-state level of mt-tRNA. The data presented in this paper deepen our understanding of the pathogenesis of mt-tRNA disease and will enable the formulation of potential therapeutic approaches for these devastating disorders.
Abbreviations
mt: mitochondrial; aaRS: aminoacyl-tRNA synthetase; AARS2: mitochondrial alanyl-tRNA synthetase; VARS2: mitochondrial valyl-tRNA synthetase; LARS2: mitochondrial leucyl-tRNA synthetase; OXPHOS: oxidative phosphorylation; ROS: reactive oxygen species; OCR: oxygen consumption rate.
Supplementary Material
Supplementary figure.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (31371270) and the Fundamental Research Funds for the Central Universities. We thank Dr. Anu Suomalainen for generously providing the pEGFP-N1-AARS2 vector.
Competing Interests
The authors have declared that no competing interest exists.
References
1. Wang M, Peng Y, Zheng J, Zheng B, Jin X, Liu H, Wang Y, Tang X, Huang T, Jiang P, Guan MX. A deafness-associated tRNAAsp mutation alters the m1G37 modification, aminoacylation and stability of tRNAAsp and mitochondrial function. Nucleic Acids Res. 2016;44(22):10974-10985 doi: 10.1093/nar/gkw726
2. Jiang P, Wang M, Xue L, Xiao Y, Yu J, Wang H, Yao J, Liu H, Peng Y, Liu H, Li H, Chen Y, Guan MX. A Hypertension-Associated tRNAAla Mutation Alters tRNA Metabolism and Mitochondrial Function. Mol Cell Biol. 2016;36(14):1920-1930 doi: 10.1128/MCB. 00199-16
3. Li R, Guan MX. Human mitochondrial leucyl-tRNA synthetase corrects mitochondrial dysfunctions due to the tRNALeu(UUR) A3243G mutation, associated with mitochondrial encephalomyopathy, lactic acidosis, and stroke-like symptoms and diabetes. Mol Cell Biol. 2010;30:2147-2154 doi: 10.1128/MCB.01614-09
4. Perli E, Giordano C, Tuppen HA, Montopoli M, Montanari A, Orlandi M, Pisano A, Catanzaro D, Caparrotta L, Musumeci B, Autore C, Morea V, Di Micco P, Campese AF, Leopizzi M, Gallo P, Francisci S, Frontali L, Taylor RW, d'Amati G. Isoleucyl-tRNA synthetase levels modulate the penetrance of a homoplasmic m.4277T>C mitochondrial tRNA(Ile) mutation causing hypertrophic cardiomyopathy. Hum Mol Genet. 2012;21(1):85-100 doi: 10.1093/hmg/ddr440
5. De Luca C, Besagni C, Frontali L, Bolotin-Fukuhara M, Francisci S. Mutations in yeast mt tRNAs: specific and general suppression by nuclear encoded tRNA interactors. Gene. 2006;377:169-176 doi: 10.1016/j.gene.2006.04.003
6. Montanari A, De Luca C, Frontali L, Francisci S. Aminoacyl-tRNA synthetases are multivalent suppressors of defects due to human equivalent mutations in yeast mt tRNA genes. BBA-MCR. 2010;1803:1050-1057 doi: 10.1016/j.bbamcr.2010.05.003
7. Rorbach J, Yusoff AA, Tuppen H, Abg-Kamaludin DP, Chrzanowska-Lightowlers ZM, Taylor RW, Turnbull DM, McFarland R, Lightowlers RN. Overexpression of human mitochondrial valyl tRNA synthetase can partially restore levels of cognate mt-tRNAVal carrying the pathogenic C25U mutation. Nucleic Acids Res. 2008;36:3065-3074 doi: 10.1093/nar/gkn147
8. Hornig-Do HT, Montanari A, Rozanska A, Tuppen HA, Almalki AA, Abg-Kamaludin DP, Frontali L, Francisci S, Lightowlers RN, Chrzanowska-Lightowlers ZM. Human mitochondrial leucyl tRNA synthetase can suppress non cognate pathogenic mt-tRNA mutations. EMBO Mol Med. 2014;6(2):183-193 doi: 10.1002/emmm.201303202
9. Perli E, Giordano C, Pisano A, Montanari A, Campese AF, Reyes A, Ghezzi D, Nasca A, Tuppen HA, Orlandi M, Di Micco P, Poser E, Taylor RW, Colotti G, Francisci S, Morea V, Frontali L, Zeviani M, d'Amati G. The isolated carboxy-terminal domain of human mitochondrial leucyl-tRNA synthetase rescues the pathological phenotype of mitochondrial tRNA mutations in human cells. EMBO Mol Med. 2014;6(2):169-182 doi: 10.1002/emmm.201303198
10. Francisci S, Montanari A, De Luca C, Frontal L. Peptides from aminoacyl-tRNA synthetases can cure the defects due to mutations in mt tRNA genes. Mitochondrion. 2011;11:919-923 doi: 10.1016/j.mito.2011.08.006
11. Suzuki T, Nagao A, Suzuki T. Human mitochondrial tRNAs: biogenesis, function, structural aspects, and diseases. Annu Rev Genet. 2011;45:299-329 doi: 10.1146/annurev-genet-110410-132531
12. Tamaki S, Tomita M, Suzuki H, Kanai A. Systematic Analysis of the Binding Surfaces between tRNAs and Their Respective Aminoacyl tRNA Synthetase Based on Structural and Evolutionary Data. Front Genet. 2018;8:227. doi: 10.3389/fgene.2017.00227
13. Götz A, Tyynismaa H, Euro L, Ellonen P, Hyötyläinen T, Ojala T, Hämäläinen RH, Tommiska J, Raivio T, Oresic M, Karikoski R, Tammela O, Simola KO, Paetau A, Tyni T, Suomalainen A. Exome sequencing identifies mitochondrial alanyl-tRNA synthetase mutations in infantile mitochondrial cardiomyopathy. Am J Hum Genet. 2011;88(5):635-642 doi: 10.1016/j.ajhg.2011.04.006
14. Jiang P, Jin X, Peng Y, Wang M, Liu H, Liu X, Zhang Z, Ji Y, Zhang J, Liang M, Zhao F, Sun YH, Zhang M, Zhou X, Chen Y, Mo JQ, Huang T, Qu J, Guan MX. The exome sequencing identified the mutation in YARS2 encoding the mitochondrial tyrosyl-tRNA synthetase as a nuclear modifier for the phenotypic manifestation of Leber's hereditary optic neuropathy-associated mitochondrial DNA mutation. Hum Mol Genet. 2016;25(3):584-596 doi: 10.1093/hmg/ddv498
15. Yu J, Xiao Y, Liu J, Ji Y, Liu H, Xu J, Jin X, Liu L, Guan MX, Jiang P. Loss of MED1 triggers mitochondrial biogenesis in C2C12 cells. Mitochondrion. 2014;14(1):18-25 doi: 10.1016/j.mito.2013.12.004
16. Birch-Machin MA, Turnbull DM. Assaying mitochondrial respiratory complex activity in mitochondria isolated from human cells and tissues. Methods Cell Biol. 2001;65:97-117 doi: 10.1016/s0091-679x(01)65006-4
17. Meng F, He Z, Tang X, Zheng J, Jin X, Zhu Y, Ren X, Zhou M, Wang M, Gong S, Mo JQ, Shu Q, Guan MX. Contribution of the tRNA(Ile) 4317A→G mutation to the phenotypic manifestation of the deafness-associated mitochondrial 12S rRNA 1555A→G mutation. J Biol Chem. 2018;293(9):3321-3334 doi: 10.1074/jbc.RA117.000530
18. Wilmer MJ, van den Heuvel LP, Rodenburg RJ, Vogel RO, Nijtmans LG, Monnens LA, Levtchenko EN. Mitochondrial complex V expression and activity in cystinotic fibroblasts. Pediatr Res. 2008;64(5):495-497 doi: 10.1203/PDR.0b013e318183fd67
19. Gong S, Peng Y, Jiang P, Wang M, Fan M, Wang X, Zhou H, Li H, Yan Q, Huang T, Guan MX. A deafness-associated tRNAHis mutation alters the mitochondrial function, ROS production and membrane potential. Nucleic Acids Res. 2014;42(12):8039-8048 doi: 10.1093/nar/gku466
20. Park H, Davidson E, King MP. Overexpressed mitochondrial leucyl-tRNA synthetase suppresses the A3243G mutation in the mitochondrial tRNALeu(UUR) gene. RNA. 2008;14:2407-2416 doi: 10.1261/rna.1208808
21. Ribas de Pouplana L, Schimmel P. Two classes of tRNA synthetases suggested by sterically compatible dockings on tRNA acceptor stem. Cell. 2001;104:191-193 doi: 10.1016/s0092-8674(01)00204-5
22. Hilander T, Zhou XL, Konovalova S, Zhang FP, Euro L, Chilov D, Poutanen M, Chihade J, Wang ED, Tyynismaa H. Editing activity for eliminating mischarged tRNAs is essential in mammalian mitochondria. Nucleic Acids Res. 2018;46(2):849-860 doi: 10.1093/nar/gkx1231
23. Tyynismaa H, Schon EA. Mixing and matching mitochondrial aminoacyl synthetases and their tRNAs: a new way to treat respiratory chain disorders? EMBO Mol Med. 2014;6(2):155-157 doi: 10.1002/emmm.201303586
24. Perli E, Fiorillo A, Giordano C, Pisano A, Montanari A, Grazioli P, Campese AF, Di Micco P, Tuppen HA, Genovese I, Poser E, Preziuso C, Taylor RW, Morea V, Colotti G, d'Amati G. Short peptides from leucyl-tRNA synthetase rescue disease-causing mitochondrial tRNA point mutations. Hum Mol Genet. 2016;25(5):903-915 doi: 10.1093/hmg/ddv619
25. Guo M, Chong YE, Beebe K, Shapiro R, Yang XL, Schimmel P. The C-Ala domain brings together editing and aminoacylation functions on one tRNA. Science. 2009;325(5941):744-747 doi: 10.1126/science.1174343
26. Wang S, Li R, Fettermann A, Li Z, Qian Y, Liu Y, Wang X, Zhou A, Mo JQ, Yang L, Jiang P, Taschner A, Rossmanith W, Guan MX. Maternally inherited essential hypertension is associated with the novel 4263A>G mutation in the mitochondrial tRNAIle gene in a large Han Chinese family. Circ Res. 2011;108(7):862-870 doi: 10.1161/CIRCRESAHA.110.231811
27. Beebe K, Ribas De Pouplana L, Schimmel P. Elucidation of tRNA-dependent editing by a class II tRNA synthetase and significance for cell viability. EMBO J. 2003;22(3):668-675 doi: 10.1093/emboj/cdg065
28. Hilander T, Zhou XL, Konovalova S, Zhang FP, Euro L, Chilov D, Poutanen M, Chihade J, Wang ED, Tyynismaa H. Editing activity for eliminating mischarged tRNAs is essential in mammalian mitochondria. Nucleic Acids Res. 2018;46(2):849-860 doi: 10.1093/nar/ gkx1231
Author contact
![]() Corresponding author: Pingping Jiang, Ph.D., Institute of Genetics Zhejiang University, and Department of Human Genetics, Zhejiang University School of Medicine, Hangzhou 310058, China. Telephone: 86-571-88982356; FAX: 86-571-88982377; E-mail: ppjiangedu.cn
Corresponding author: Pingping Jiang, Ph.D., Institute of Genetics Zhejiang University, and Department of Human Genetics, Zhejiang University School of Medicine, Hangzhou 310058, China. Telephone: 86-571-88982356; FAX: 86-571-88982377; E-mail: ppjiangedu.cn